

Abstract
It is nowadays well accepted that chronic inflammation plays a pivotal role in tumor initiation and progression. Under this aspect, the oral cavity is predestined to examine this connection because periodontitis is a highly prevalent chronic inflammatory disease and oral squamous cell carcinomas are the most common oral malignant lesions. In this review, we describe how particular molecules of the human innate host defense system may participate as molecular links between these two important chronic noncommunicable diseases (NCDs). Specific focus is directed toward antimicrobial polypeptides, such as the cathelicidin LL‐37 and human defensins, as well as S100 proteins and alarmins. We report in which way these peptides and proteins are able to initiate and support oral tumorigenesis, showing direct mechanisms by binding to growth‐stimulating cell surface receptors and/or indirect effects, for example, inducing tumor‐promoting genes. Finally, bacterial challenges with impact on oral cancerogenesis are briefly addressed.
Keywords: alarmins, chronic inflammation, defensins, innate host defense, oral tumorigenesis, S100 proteins
1. INTRODUCTION
Oral squamous cell carcinomas (OSCCs) represent the majority (90%) of intra‐oral malignant tumors and belong to the large group of head and neck cancers. 1 Although their etiology is multifactorial, chronic inflammatory conditions due to poor oral hygiene, as found in most of the OSCC patients, are suggested to be carcinogenic cofactors. Periodontitis is the most common chronic inflammatory noncommunicable disease of humans 2 and has intensively been discussed as an important cofactor, together with tobacco and alcohol, in initiating and promoting oral cancerogenesis. 1 , 3 , 4 , 5 , 6 , 7 An association of periodontitis with cancer, particularly with epithelial head and neck tumors such as OSCC, 8 , 9 has already been described since many years. 10 , 11 Today, this link is substantiated by a large number of epidemiological studies showing an increased risk for head and neck tumors in patients with periodontitis. 12 , 13 , 14 , 15 , 16 , 17 , 18 However, the mechanisms by which oral cancer may develop among individuals who have periodontal disease are not entirely clear, even though a variety of plausible mechanisms have been suggested. 18 , 19 , 20 , 21
Associations between inflammatory markers (CCL2, IL‐6, IL‐8, and Cox‐2) and tumor formation have been shown. 22 , 23 , 24 Also, interactions between the innate and adaptive immune systems play an important role. 25 The human host defense system consists of innate and adaptive components. The adaptive immune system specifically reacts against pathogens and generates immunological memories with the help of specialized cells such as T and B lymphocytes. In contrast, the innate immune system responds with a broad specificity for conserved features of microorganisms. 26 , 27 Nearly all tissues participate in innate host defense, especially as barrier surfaces such as mucosal epithelia in the oral cavity, the gastrointestinal tract, the respiratory tract, or the urogenital tract. 28 Pathogens are recognized via special receptors, so‐called pattern recognition receptors (PRRs), such as toll‐like receptors (TLRs). The innate immune system responds to microbial challenges very rapidly through cellular mechanisms (phagocytosis and cytotoxicity) or secretion of specific factors, such as antimicrobial polypeptides (AMPs), complement factors, cytokines, and proteases. Hence, host epithelia function as physical and also chemical barriers against microorganisms. 26 , 27 , 28 , 29 , 30 AMPs play an outstanding role among those molecules mentioned above since they can be proposed as a molecular link between infection and oral tumorigenesis (Figure 1). The reason for this is based upon their cellular functions, namely first as compounds with very distinct antimicrobial activity and secondly as molecules affecting various other processes (maturation, differentiation, wound healing, and proliferation) of host cells. AMPs have in common that they are endogenous proteins of low molecular weight with a broad spectrum of antimicrobial activities against fungi, bacteria, and viruses and, thus, serve as the first biochemical barrier of the host against microbial challenges. 31 , 32 , 33 , 34 , 35 The AMP family consists of a wide variety of different proteins, such as defensins, S100 proteins, cathelicidins, members of the ribonuclease A family, such as RNase7, and Dermcidin. 33 , 35 A lot of progress has been made during the last years regarding the discovery of more cellular functions of AMPs other than just antimicrobial functions, particularly functions associated with tumorigenesis. 34 , 35 , 36 , 37
FIGURE 1.
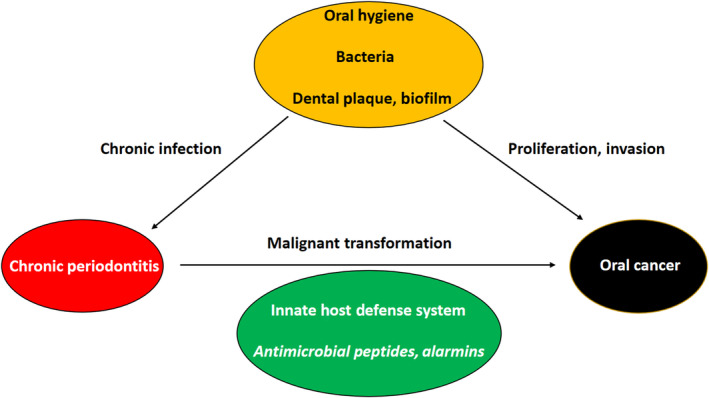
Risk factors, effects, and molecular participants in the axis of chronic infection–inflammation (CP)–oral tumorigenesis. Persisting bacterial challenges in form of dental plaque or biofilm lead to a manifestation of CP and permanent activation of the innate host defense system, which affects malignant transformation and hence oral tumorigenesis. Thereby, antimicrobial peptides and alarmins serve as molecular links between these cellular processes. Furthermore, pathogens also have a direct impact on oral cancer.
This review will mainly focus on molecular factors of the innate host defense system that could serve as a link between chronic periodontal inflammation and oral tumorigenesis (Figure 1).
2. INNATE HOST DEFENSE SYSTEM
Microbial infections resulted in the evolutionary development of an efficient host defense system, consisting of an innate part and an adaptive part. 26 The innate immune system recognizes pathogens by PRRs, which are germline encoded and exhibit a broad specificity against conserved moieties of microbes. 38 In contrast, the adaptive immune response is based upon antigen receptors, which are transcribed by assembling gene segments via the process of somatic recombination resulting in a huge variety of narrow specificities. 39 Thus, the innate immune system and the adaptive immune system use fundamentally different repertoires to respond to the molecular diversity of microbial pathogens. 26 Due to different modes of reacting against pathogenic challenges, responses of the innate immune system are far more rapid (within minutes) than of the adaptive immune system which requires up to several days. 27 Detection of pathogens by the innate host defense system is based upon recognition of microbial unique molecular structures. 38 Ligands for PRRs are so‐called pathogen‐associated molecular patterns (PAMPs). They are invariant in microorganisms and are essential metabolites of physiological pathways unique to microbes. PAMPs are mainly members of cell wall components, for example, bacterial lipopolysaccharides (LPS), lipoteichoic acids (LTAs), peptidoglycans, or fungal β‐glucan. Viral PAMPs derive from virus nucleic acids, since all viral compounds are synthesized by host cells and are unique to viruses for specific RNA or DNA modifications, allowing the discrimination between host and pathogens. 40 There are several classes of PRRs with the family of toll‐like receptors (TLRs) as the best characterized group. These receptors are transmembrane proteins which can bind cell‐wall compounds and nucleic acids of bacterial, fungal, and viral origin. The TLR family consists of eleven members which exhibit narrow specificities and affinities to various different microbial PAMPs. For example, TLR‐4 binds LPS from gram‐negative bacteria, TLR‐2 recognizes lipopeptides from gram‐positive bacteria, flagellins are ligands for TLR‐5, and bacterial DNA is a ligand for TLR‐9. 41 Signal transduction after ligand binding to TLRs is executed through intracellularly translocating the transcription factor nuclear factor‐κB (NF‐κB) into the nucleus binding to genes involved in inflammatory processes. 41 As a consequence, TLR activation leads to an enhanced production and secretion of pro‐inflammatory cytokines, such as interleukin‐1β (IL‐1β) and IL‐6, which in turn organize inflammatory responses. 26 , 42 Also, in parallel, antimicrobial proteins (AMPs) are liberated from epithelial cells to directly degrade microbial structures and thus function as the first line of defense or biochemical barrier. 28 , 30 The second line in innate host defense is the recruitment of immune cells, such as neutrophils, to the site of infection via diapedesis and positive chemotactic cellular migration caused by secreted chemotactic cytokines and chemokines. 43 , 44 Neutrophils belong to the first cells, which are migrating to the infectious lesion. This cell type is responsible for destroying microbial pathogens by phagocytosis, production of reactive oxygen species (ROS), degranulation, intracellular killing, and formation of extracellular traps (NETs). Neutrophils accumulate at the site of infection and die, as a consequence, of attacking the pathogens, which then leads to the development of NETs. 45 , 46 , 47 , 48 This molecular network consists of a scaffold of chromatin fibers with various degrading proteins which are involved in microbial disintegration. 49 Since this process of cell degradation shows a broad specificity, an accumulation of a high number of neutrophils will cause formation and destruction of host tissues. 44
This leads us to a further group which has been identified to play a key role in innate host defense. Members of this protein family were found to be released after endogenous tissue injury, cell damage, or stress and are thus named damage‐associated molecular pattern (DAMP) molecules or alarmins. 50 Gene expression of several of these molecules, for example, defensins, cathelicidin, S100 proteins, and high mobility group protein B1 (HMGB1), is up‐regulated by PAMPs and/or pro‐inflammatory stimuli. 50 An important sensor for DAMP recognition is the receptor for advanced glycation end products (RAGE), a multi‐ligand receptor with a binding capacity for structurally unrelated, diverse molecules. 51 , 52 RAGE expression is under the control of the transcription factor NF‐κB (nuclear factor‐κB), hence affected by inflammatory stimuli. 52 RAGE signaling occurs via a variety of different down‐stream targets, such as NF‐κB, 53 mitogen‐activated protein kinases (MAPKs), 54 phosphatidylinositol 3‐kinases (PI3Ks), 55 Rho GTPases, 56 Janus kinases/signal transducer and activator of transcription (JAKs/STAT) 57 , and Src kinases 58 implicating RAGE to also have a prominent role in cancer. 52
In summary, microbial infection leads to liberation of PAMPs which, in consequence, can bind to host PRRs. Activation of these receptors causes a rapid response of the innate defense system. Hence, pro‐inflammatory cytokines and AMPs are secreted in order to initiate the “fight” against microbial invasion. In case of tissue destruction, stress, and injury, a second system comes to life with RAGE as a major player. Both systems are responsible for releasing soluble factors which are on the one hand necessary to enable the host to protect it against pathogen attacks. On the other hand, these cell‐derived factors are also involved in tumor initiation and progression. In this context, the innate host defense system plays a remarkable role in tumorigenesis (Figure 2).
FIGURE 2.
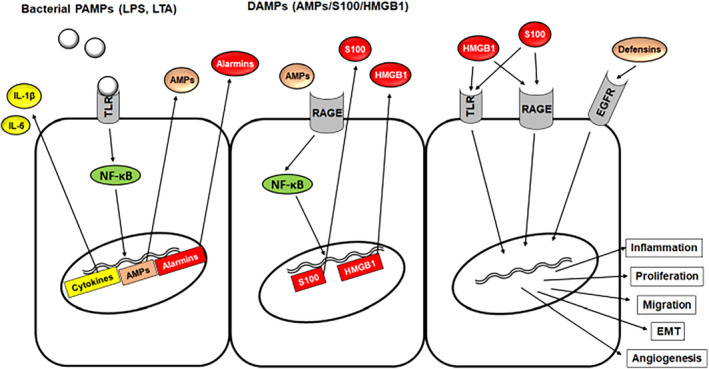
From bacterial challenge to tumor development. Schematic description of how the innate host defense system affects tumorigenesis. PAMPs bind and activate PRRs, such as TLRs. Subsequently, NF‐κB is translocated into the nucleus enhancing gene activation of cytokines, AMPs, and alarmins. These molecules are secreted and exert their work, partly as DAMPs, outside of the cell by acting as ligands for TLRs or RAGE. Hence, these molecules function as signal amplifiers. Moreover, cell proliferation can be promoted by AMPs serving as ligands for EGFR. AMP, antimicrobial peptide; DAMP, damage‐associated molecular pattern; EMT, epithelial–mesenchymal transition; EGFR, epidermal growth factor receptor; HMGB1, high mobility group protein B1; LPS, lipopolysaccharides; LTAs, lipoteichoic acids; NF‐κB, nuclear factor‐κB; PAMPs, pathogen‐associated molecular patterns; RAGE, receptor for advanced glycation end products; TLR, toll‐like receptor.
2.1. Antimicrobial polypeptides
In 1981, Boman and coworkers described for the first time inducible antimicrobial peptides discovered in the moth Hyalophora cecropia. 59 This observation led to the identification of several hundred host defense peptides participating in innate immunity of various multicellular organisms with defensins as outstanding among these peptides. 60 In humans, two classes of defensins are known consisting of six α‐defensins (HNP1‐4 and HD5‐6) and four β‐defensins (hBD1‐4). 61 , 62 Another important member of human AMPs, LL‐37, belongs to the cathelicidins. 63 Finally, the third relevant group contains members of the S100 family, a class of calcium‐/metal‐ion‐binding proteins with a multitude of various cellular functions in addition to their antimicrobial moiety. 35 , 37 , 64 , 65 , 66 , 67 , 68 The molecular structures of these AMPs are shown in Figure 3 with one representative member for each group including their typical characteristic features (Figure 3). 69 , 70 , 71 The following paragraphs will focus particularly on cathelicidins, S100 proteins, and human defensins regarding their significance as key players on the interface of inflammation and tumorigenesis.
FIGURE 3.
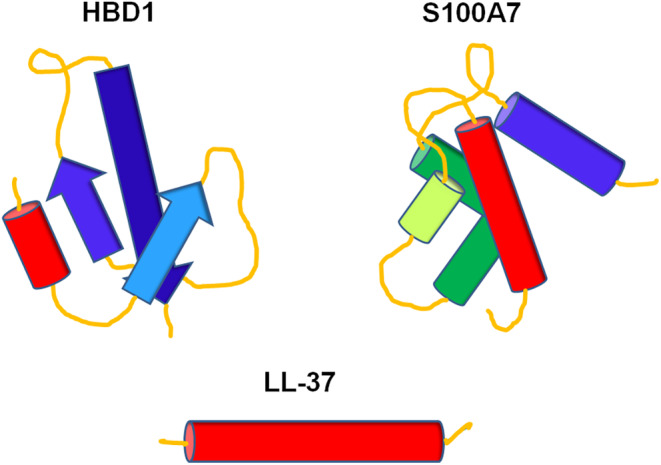
Schematic molecular structures of AMPs. HBD1 representing the group of defensins with the typical β‐sheet motifs, 69 S100A7 with multiple α‐helices, 70 characteristic for S100 proteins, and the cathelicidin LL‐37 consisting of one single α‐helix. 71 Rod‐like shapes represent α‐helical structures, and arrows stand for β‐sheet strands.
2.1.1. Cathelicidins
Cathelicidins are a group of AMPs with numerous members in various eukaryotic organisms. However, only one cathelicidin, namely LL‐37, exists in humans. It is a C‐terminal proteolytically cleaved fragment of its precursor “human cationic antimicrobial peptide‐18” (hCAP‐18) and consists of 37 amino acids with two leucine residues at its mature amino‐terminus. LL‐37 is a peptide with an α‐helical motif. 71 It is biologically active against bacteria, viruses, fungi, and microbial parasites. Human CAP‐18 is transcribed as a precursor protein with a signal sequence responsible for vesicle storage or secretion. 72 It is expressed in various types of immune cells, 63 , 73 , 74 , 75 , 76 , 77 such as neutrophils, monocytes, macrophages, dendritic cells, and also epithelial cells. 72 , 78 The expression pattern is differently regulated depending on cell types with either exogenous microbial or endogenous stimulating molecules. 79 Transcription of hCAP‐18 is up‐regulated by various PAMPs, such as bacterial LPS or LTA, and microbial metabolites, such as butyrate. 79 , 80 Induction and/or release occurs either directly by activation of PRRs or indirectly through cytokines which are secreted after TLR stimulation. 75 , 81 Vitamin D3 is another inducer for hCAP‐18 expression, modulates its serum level, and plays an important role in host defense of epithelial cells. 82 , 83 , 84 , 85 In addition, hypoxia positively affects hCAP‐18 transcript levels. 86 The hCAP‐18 protein is stored in cellular granules after translation. When secreted, the N‐terminal signal sequence is proteolytically removed. 87 Afterward, mature LL‐37 is extracellularly released by proteinase 3 or kallikrein from its pro‐peptide. 88 , 89 LL‐37 is a cationic peptide with a helical conformation. 90 These structural features allow this AMP to interact with bacterial membranes as pore‐forming toxin leading to membranolytic bacterial lysis. 72 , 91 , 92 Besides its antimicrobial activity, LL‐37 possesses numerous other cellular functions in host protection: It can act as an alarmin, as a chemoattractant to immune cells, and as a modulator of inflammatory responses and can also promote wound healing. 72 LL‐37's role in regenerative processes is due to its ability to bind to various different receptors: Activation of formyl peptide receptor‐like‐1 (FPRL‐1) leads to enhanced angiogenesis 93 and transactivation of epidermal growth factor receptor (EGFR) to elevated migration and proliferation of keratinocytes. 94 , 95 Both processes belong to the hallmarks of cancer. 96 , 97 Since LL‐37 has been found in periodontal diseases, 98 , 99 this cathelicidin is a potent player in the inflammation–tumorigenesis axis.
2.1.2. Defensins
Human defensins belong to a family of AMPs which contribute to host defense against various microbes, such as bacteria, fungi, or membrane‐coated viruses. There are two subfamilies in humans with six α‐defensins, or human neutrophil peptides (HNP1‐4), mainly present in neutrophils and intestinal Paneth cells (HD5‐6), whereas four β‐defensins (hBD1‐4) appear primarily in epithelial cell layers. But besides that, human defensins have also been detected in body fluids and premalignancies and even various tumors. Defensins are structurally similar cationic peptides of low molecular weight (3–5 kDa) with characteristic triple‐stranded β‐sheet motifs 69 stabilized by six cysteine residues forming three intramolecular disulfide bonds. These chemical properties are responsible for their amphiphilic character which enables the peptides not only to disrupt microbial membranes and thus kill the pathogens, but also to establish various additional functions in different cellular processes, either as extra‐/intracellular molecules, or even by their ability to cross cellular membranes. Both defensin groups differ in their characteristic pairing of cystine bridges: While the six cysteines in α‐defensins are connected between 1–6/2–4/3–5, hBDs form 1–5/2–4/3–6 cystine bridges. HNP1‐4 are mainly expressed in polymorphonuclear leukocytes (PMNs) and monocytes. They are produced as functionally inactive precursor forms and stored as mature peptides in primary azurophil granules. When neutrophils phagocyte microbes into vacuoles, HNP‐containing granules fuse with these vacuoles and thus bring the defensin molecules into contact with the target microorganisms. Gene expression of HNPs is constitutive but can be up‐regulated by inflammatory cytokines, such as IL‐1β, IL‐6, or TNF‐α. 31 , 32 , 100 , 101 , 102 Although more than twenty‐eight human β‐defensin genes have been found in a computational genome search, 103 , 104 only four proteins have been studied in detail. Human β‐defensins are mainly localized in various epithelia (skin, urogenital, respiratory, and nasolacrimal tracts, eye, testes, oral tissues, and salivary and mammary glands) and are also present in body fluids, such as urine, saliva, and lacrimal fluid. HBDs are synthesized as precursor peptides and stored in cellular granules. They are secreted and subsequently processed by extracellular proteases into their mature and active form. Although hBD1 is constitutively expressed, its transcript level can be up‐regulated by LPS or γ‐interferon. In contrast, gene activities of hBD2‐4 are inducible by IL‐1β, LPS, TLRs, TNF‐α, and various growth factors. 31 , 32 , 34 , 100 , 101 Human defensins are involved in a wide variety of cellular functions other than antimicrobial functions. They show immunomodulatory and chemotactic effects on immune cells by cross‐regulation of pro‐inflammatory cytokines and chemokines in dendritic cells 105 and by serving as chemoattractant for leukocytes and T cells. 106 , 107 , 108 , 109 , 110 Furthermore, defensins play an important role in wound healing and regeneration by driving cell migration, proliferation, and differentiation. HBD1‐4 have been shown to enhance the migration of dermal keratinocytes. Additionally, these AMPs are also able to positively affect the proliferation rate by phosphorylation of EGFR. 111 HBD2 enhances maturation/differentiation/mineralization, and in addition, hBD3 enhances the proliferation rate of osteoblast‐like cells, hence may participate in bone tissue regeneration. 112 In lung, HNPs increase wound closure, fibroblast proliferation, and collagen synthesis via β‐catenin pathway. 113 , 114 Human defensins are shown to be involved in tissue regeneration and wound healing: They have positive impacts on the proliferation and differentiation of keratinocytes, fibroblasts, osteoblasts, and even endothelial cells. 115 HBD1 has been detected in nuclei of keratinocytes of burned skin. 116 It is proposed that intranuclear hBD1 might regulate keratinocyte‐protecting processes. 117
The oral cavity is an anatomical structure which possesses ideal conditions for establishing a persistent oral microbial flora: It is humid, provides a growth‐promoting temperature, a nearly constant presentation of nutrients, and has hard tissues for developing microbial biofilms. 118 Its innate host defense system consists of the following components: (i) saliva with AMPs, (ii) neutrophils containing HNPs with the capability for establishing NETs, and (iii) epithelia with a layer of secreted AMPs serving as biochemical and physical barrier against microbes. Defensins have been found in salivary glands, saliva, gingival and mucosal epithelia, tongue, dentogingival junctions, and various oral lesions, including tumors. 34 , 119 , 120 , 121 , 122 , 123 , 124 , 125
Saliva and gingival crevicular fluid (GCF) contain α‐defensins as well as hBDs. It has been shown that peptide levels of HNP1‐4 and hBD2 are enhanced in crevicular fluids of patients suffering from inflammatory gingival and periodontal diseases. 124 , 126 Hence, saliva and GCF possess a striking tool for host defense.
As already mentioned above, HNPs are mainly expressed in neutrophils. These peptides can either act directly against microbes in fused cellular phagocytic vesicles or by deploying their antimicrobial activity through degranulation and via NETs. HNP‐containing cells occur in the dentogingival junction region and junctional epithelia of both healthy and diseased patients. HNPs are active against the oral gram‐positive and gram‐negative pathogens Streptococcus mutans, Fusobacterium nucleatum, Treponema denticola, Porphyromonas gingivalis, and Aggregatibacter actinomycetemcomitans. But besides their antimicrobial effects, α‐defensins can also play an additional important role in supporting the maintenance of periodontal health by immunomodulatory activities and positive effects on wound healing. 118 , 122 , 127 , 128 In summary, HNPs have a significant impact on host defense mechanisms in inflammatory tissues. Nevertheless, α‐defensins have also been found in oral lesions and salivary tumors: HNP1‐4 transcript level is enhanced in oral irritation fibromas and leukoplakias, whereas these genes are up‐regulated in preferentially malignant salivary cancer entities, in contrast to benign pleomorphic adenoma. 34 , 129 , 130
Human β‐defensins are mainly present in oral epithelia, but also in tongue, and salivary glands including saliva. HBD1 is constitutively expressed in oral differentiated keratinocytes of both healthy and inflamed gingival specimens and predominantly found in the upper epithelial cell layer. However, it has been demonstrated that gene expression of hBD1 can be up‐regulated by a penta‐acylated LPS from Porphyromonas gingivalis. HBD1 itself is active against Fusobacterium nucleatum, Aggregatibacter actinomycetemcomitans, and Porphyromonas gingivalis. 34 , 120 , 122 , 123 , 128 In addition to its antimicrobial activity, hBD1 is also a chemoattractant for monocytes, 131 it is able to affect proliferation rates, positively for keratinocytes, 111 but negatively for oral cancer cells, and it cross‐regulates gene expression of hBD3 in OSCC cells. 132 Cancer‐specific loss of hBD1 expression was first described in tumors of the urogenital tract, suggesting hBD1 as a potential tumor‐suppressor gene. 133 , 134 Later, this observation of a decreased hBD1 expression has also been reported in oral squamous cell carcinoma 5 , 135 and salivary gland tumors. 136 Moreover, hBD1 has been found in various oral lesions, for example, irritation fibromas, leukoplakias, and salivary glands, and accumulated in the nucleus of different tumor entities of salivary glands. 5 , 130 , 137 , 138 HBD2 is also, like hBD1, expressed in keratinocytes of the upper layer of gingival epithelia from both healthy and diseased patients. Its gene expression is inducible by pro‐inflammatory cytokines, such as IL‐1β, TNF‐α, or various oral bacteria (Fusobacterium nucleatum, Aggregatibacter actinomycetemcomitans, Prevotella intermedia, and Porphyromonas gingivalis). HBD2 is biologically active against gram‐negative bacteria and the yeast Candida albicans. 34 , 120 , 122 , 123 , 128 Furthermore, hBD2 is able to activate dendritic cells through activation of TLR4, 139 positively affects the proliferation rates of keratinocytes 111 and OSCC cells, 132 and is involved in cell growth and differentiation of osteoblasts 112 and tube formation of endothelial cells. 115 HBD2 has been detected in the same oral tissues as already described for hBD1, with slightly reduced transcript levels in premalignant leukoplakias and malignant OSCCs compared to gingiva and salivary gland tumors. 5 , 136 HBD3 is different compared to the other two β‐defensins, hBD1 and hBD2. It is not only expressed in the basal part of the epithelial layer in health, while in disease it has also been found in the spinous layer, but shows a higher net charge with +9 due to a higher content of basic amino acids and maintains its biological activity in the presence of high salt concentrations. 32 HBD3 gene expression is inducible by inflammatory cytokines and various oral pathogens (Fusobacterium nucleatum, Aggregatibacter actinomycetemcomitans, Prevotella intermedia, and Porphyromonas gingivalis) but down‐regulated by Treponema denticola and Tannerella forsythia. 128 Furthermore, hBD3's microbial targets are the same bacteria mentioned above serving as bacterial inducers with additionally Streptococcus mutans and sanguinis. 140 HBD3, as chemoattractant, is involved in immune cell migration of monocytes, macrophages, and dendritic cells. 105 , 141 Furthermore, hBD3 participates in wound healing processes by enhancing cell proliferation and migration of keratinocytes and osteoblast‐like cells. 111 , 112 In cancer, hBD3 has positive effects on tumor cell growth and additionally cross‐regulates hBD1 and hBD2. 132 Gene products of hBD3 have been found in numerous oral tissues other than gingiva: remarkably higher expressed in irritation fibromas, leukoplakias, and OSCCs 5 , 142 but down‐regulated in salivary gland tumors. 136 In contrast to the three hBDs described above, little is known about hBD4 in oral tissues. It is expressed in epithelia of healthy and inflamed gingiva, predominantly in the basal layer of CP specimens. 143 Its gene activity can be induced by phorbol 12‐myristate 13‐acetate (PMA) and TNF‐α in gingival epithelial cells in vitro. 144 There has been no difference in transcript levels observed in healthy or CP tissue samples. But it is noteworthy that hBD4 was present in only 30% of healthy and 40% of CP patients. 143 To summarize, human defensins have been described to be involved in chronic periodontal inflammation as well as in oral tumorigenesis. On the one hand, DEFB1 encoding for hBD1 has been identified as a relevant periodontitis‐associated gene 145 with important functions in local host defense, but on the other hand, hBD1 has also been examined as a substantial factor in proliferation control of oral cancers. 5 , 132 , 137 , 138 , 143 In modulating OSCC cell proliferation, hBD1 down‐regulates proliferation as a potential tumor suppressor, 5 , 132 , 133 , 134 , 135 whereas hBD3 enhances the proliferation rate as a putative protooncogene. 5 , 132 , 135 , 142 Hence, we suggest hBDs act as key molecules in the inflammation–tumor axis. 5 , 34 , 129 , 137 , 138 , 142 , 146 , 147
2.1.3. S100 proteins
Human S100 proteins belong to a gene family clustered on chromosome 1q21 containing more than 20 calcium‐binding EF‐hand‐type members. Gene expression of the different S100 proteins is dependent on the cell type, various cellular factors, such as growth factors or cytokines, and activated PRRs. Although they share a high degree of structural similarities, they, however, show a wide variety of different intra‐ and extracellular functions. S100 proteins are involved in proliferation, differentiation, apoptosis, calcium homeostasis, migration, invasion, and inflammation and act as alarmins. They interact intracellularly with cytoskeletal components, transcription factors, and nucleic acids. If present in the extracellular space, S100 proteins modulate cellular functions by activating various cell surface receptors, for example, RAGE, TLR4, or EGFR.. 36 , 64 , 65 , 66 , 68 , 148 , 149 , 150 , 151 Hence, some of these polypeptides are participants in human host defense and tumorigenesis. Moreover, S100A7/A8/A9/A12 yet show antimicrobial properties 35 , 68 , 151 which are based upon their potential to bind calcium and metal ions, thus leading to nutrient metal limitations, so‐called nutritional immunity. 67 , 151 , 152 Besides that, some members of the S100 family are involved in interactions between tumor cells and their microenvironment 153 , 154 : It is important for tumor cells in order to survive, to develop primary cancers, or to metastasize to modify their surrounding tissue conditions into a tumor‐beneficial microenvironment. 155 There is a ping‐pong interplay between tumor cells and neighboring stroma cells affecting each other by secretion of various stimulatory factors, such as growth factors, cytokines, degrading enzymes, or chemoattractants. 154 , 156 , 157 Several S100 proteins have been shown to be actors participating in these processes for developing and supporting a tumor‐beneficial microenvironment. 153 Since this review is dealing with the role of AMPs in cancerogenesis, only the abovementioned four antimicrobial S100 members S100A7/A8/A9/A12 will be described here in more detail, and in addition, S100A4 will also be described, because this protein plays a predominant role in the axis of host defense–cancerogenesis.
S100A7, also named “psoriasin,” was first isolated from psoriatic skin lesions exhibiting antimicrobial activity. 158 , 159 , 160 Furthermore, this calcium‐binding protein has been identified as an alarmin, a chemoattractant, and an amplifier of inflammation. It is expressed in epithelial cells and acts both intra‐ and extracellularly. The mechanism of how S100 proteins are secreted is still not clear, because they lack signal sequences specific for membrane trafficking. Released S100A7 binds to RAGE, a cell surface receptor involved in inflammatory processes and also tumorigenesis. Cytoplasmic psoriasin can interfere with β‐catenin, translocate into the nucleus, activate the oncogene c‐Myc, and thus affect proliferation. Moreover, S100A7 plays an important role in matrix degradation by regulating matrix metalloproteinases (MMPs), in angiogenesis through vascular endothelial growth factor (VEGF), and in establishing a tumor‐tolerogenic microenvironment by modifying immune cell subpopulations into a tumor‐supportive phenotype. 64 , 161 , 162 , 163 Psoriasin has been examined in crevicular fluids of periodontal pockets 99 and also in various oral lesions 130 , 136 , 164 , 165 which makes this protein a potential player in the molecular link between host defense and tumorigenesis.
S100A8, also termed calgranulin A, is constitutively expressed in neutrophils and inducible in monocytes and epithelial cells 160 , 166 and is associated with inflammatory diseases 167 and cancer. 153 Its expression is induced by LPS, tumor necrosis factor‐α (TNF‐α), interleukin‐1β (IL‐β), and ultraviolet radiation. 155 , 166 , 168 S100A8 is a modulator in inflammation, involved in immune cell chemotaxis, a scavenger molecule for reactive oxygen, and serves as AMP. It is a natural ligand for RAGE. 68 , 151 , 166 S100A8 has been detected in gingival crevicular fluid from periodontitis sites 99 and in oral and head and neck squamous cell carcinoma. 169 , 170 , 171 , 172 Furthermore, calgranulin A has a strong tendency to form a heterocomplex with S100A9. 66
S100A9, calgranulin B, affects immune cell migration and shows antimicrobial activities. 66 , 68 It is often co‐expressed with its partner protein S100A8 forming a heterocomplex, called calprotectin. S100A8/A9 is released by neutrophils and monocytes, 173 affects pro‐inflammatory cytokine production, modulates TLR4 activity, and promotes tumor cell development by binding to RAGE. 66 Since calprotectin is able to bind divalent cations, it can modify enzyme activities of MMPs by sequestering Zn2+. 166 Furthermore, S100A8/A9 participates in the formation of a tumor‐supportive microenvironment, hence plays a role in metastasis. 153 Calprotectin has been identified in oral keratinocytes, inflamed oral tissues, and oral lesions. 99 , 169 , 170 , 171 , 172 , 174 , 175
S100A12, also termed calgranulin C, is constitutively expressed in neutrophils. Its gene is induced by TNF‐α and IL‐6 in monocytes and macrophages, respectively. Through interaction with RAGE, it is itself able to induce pro‐inflammatory cytokines in mast cells, which are relevant for chemoattractant recruitment of neutrophils and monocytes. Furthermore, it has been detected in keratinocytes in psoriatic lesions. 66 , 160 S100A12 shows also antimicrobial potential by entrapping Zn2+ ions, but only in the presence of excess Ca2+. S100A8/A9 has also been shown to sequester Zn2+ ions. Nevertheless, S100A12 exhibits a more selective specificity or preference for Zn2+, whereas calprotectin can bind other divalent metal ions, such as Fe2+, Mn2+, or Ni2+, too. Hence, it is suggested that S100A8/A9 and S100A12 act together in the proximity of neutrophils. 152 S100A12 is higher expressed in gingival monocytes from patients suffering periodontitis. 176 An enhanced transcript level of calgranulin C has been observed in a tongue squamous cell carcinoma cell line overexpressing the putative tumor suppressor brain‐expressed X‐linked 4 (BEX4). 169
Although S100A4 lacks antimicrobial activities, this protein should also be introduced at this point, since it is a key player between inflammation and tumorigenesis. S100A4, also termed fibroblast‐specific protein‐1 (FSP‐1) or metastasin, has effects on numerous cell types and various cellular processes. Intracellularly, S100A4 drives cell migration, apoptosis, and stemness maintenance. Cytoskeletal proteins are interacting targets for S100A4, but also the tumor suppressor p53. Present in the extracellular space, S100A4 affects various pro‐inflammatory processes and metastasis‐promoting activities. It is involved in cell motility, epithelial–mesenchymal transition (EMT), differentiation, angiogenesis, and autophagy. S100A4 is a multi‐receptor ligand exhibiting the ability to bind to RAGE, TLR4, EGFR, and annexin A2. Probably the most important effect of this S100 protein is its role as extracellular player in immune modulation under pro‐inflammatory conditions: acting as stimulator for specific cytokine production leading to the formation of a tumor‐supportive microenvironment. Thus, S100A4 can connect host defense with tumorigenesis on a molecular level. 66 , 177 , 178 In oral tissues, S100A4 has been found in periodontal ligament cells, 179 gingiva, irritation fibromas, leukoplakias, and OSCCs. 170
2.2. Alarmin HMGB1
Alarmins are dual‐function proteins, which exert their biological activity within cells and also in the extracellular space. Many of these molecules are released after cell death due to inflammation or stress and are described as DAMPs. Alarmins play a central role in inflammatory diseases and cancer development. 150 The prototype of this protein group is HMGB1. It is expressed in almost all eukaryotic cells. HMGB1 expression is modulated by cytokines, such as interferon‐γ (IFN‐γ), TNF‐α, and transforming growth factor‐β (TGF‐β). As a nuclear protein, HMGB1 participates in stabilizing nucleosomal DNA and in regulating gene activities. Additionally, HMGB1 is also present in the cytoplasm and can actively be secreted after stimulation due to infection, injury, and inflammation or passively be liberated by cell death. Typical receptors for HMGB1 are TLRs (TLR2 and TLR4) and RAGE. Extracellular HMGB1 is involved in promoting angiogenesis, inflammation, proliferation, metastasis, and cell migration. 52 , 180 , 181 , 182 HMGB1 has been detected in gingival tissues and GCF from patients suffering CP or peri‐implantitis. Soluble HMGB1 induces pro‐inflammatory cytokines leading to extended inflammation. Its gene is activated in oral cells, such as gingival fibroblasts, periodontal ligament cells, or OSCC‐derived cells, by butyric acid, a metabolite and virulence factor of Porphyromonas gingivalis, and by LPS from periopathogens or IL‐1β. 183 Hence, HMGB1 is also a mediator molecule connecting the axis inflammation–cancer development.
3. CANCER
An established tumor is a complex tissue which consists of a symbiotic relationship between malignant, transformed tumor cells, and normal stromal cells that support tumor growth and, in case of metastasis, help tumor cells to spread to distant tissues. Tumor cells possess specific features which characterize them as malignant cancerous cells. 184 These molecular and cellular properties are brilliantly summarized in “hallmarks of cancer” 96 , 97 :
Sustaining proliferative signaling: Normal cells need growth signals prior to achieve the state of proliferation. These exogenous signals are transduced into the cells via transmembrane receptors and hence positively affect cell cycle. It is of vital importance for normal tissues to carefully control this process to maintain normal architecture and function. In contrast, cancer tissues contain tumor cells with the potential to chronically proliferate. These cells develop sovereignty over their own determination. This is achieved by activation of specific pro‐proliferative down‐stream signaling pathways (overexpression of growth factors (GFs) and growth signal receptors; formation of truncated constitutively active receptors; and hyperactivation of intracellular kinases) or antagonizing attenuation of proliferative signaling (compromising Ras activity).
Evading growth suppressors: It is essential for cancer cells to not only positively affect growth‐promoting processes but also overcome negative feedback programs, for example, tumor suppressors or contact inhibition mechanisms. The most common tumor suppressor genes encode the RB (retinoblastoma‐associated) protein and tumor protein TP53 (p53). RB is involved in transmitting particularly extracellular signals into the cell and is responsible for activating or not activating the cell's signaling cascade for growth or division. TP53 participates in cell‐cycle regulation by integrating endogenous stress and damage signals and is thus able to initiate a halt in cell‐cycle progression, if the state of the cell shows suboptimal conditions, such as genome damage, low energy status, low levels of nucleotides, glucose, or oxygen. It is obvious that a disturbance or loss in functions of these proteins can cause severe problems in controlling the cell cycle, thus having a strong impact of uncontrollable proliferation. Contact inhibition is a pivotal process in life enabling cells to develop and form tissues of definite architecture and size. It is based upon coupling cell surface adhesion molecules, such as E‐cadherin, to growth factor receptors (e.g., EGFR), leading to an attenuation of growth stimulatory effects.
Resisting cell death: Apoptosis‐based programmed cell death is an essential mechanism in tissue development. The cellular apoptotic system consists of extracellular death‐inducing signals, such as Fas ligand/Fas receptor, and an intrinsic machinery of endogenous intracellular signals. Apoptotic triggers lead to inhibition of antiapoptotic proteins (Bcl‐2), which in turn activates proapoptotic proteins, such as Bax, which, as a consequence, disrupts the outer mitochondrial membrane and thus releases cytochrome c. This process initiates a cascade of different cellular changes culminating ultimately in cell disassembling. Despite this, tumor cells have evolved various strategies to overcome apoptosis which is necessary for cancerogenesis: loss of function of TP53, up‐regulation of proapoptotic genes, and down‐regulation of antiapoptotic genes.
Enabling replicative immortality: Normal cells are intrinsically limited with respect to multiplication. Cells which have gone through a certain number of cell divisions enter the state of senescence, in which cells are viable but not able to proliferate. Senescence is followed by the state of crisis leading to cell death. Occasionally, some cells succeed in these processes and stay vital and proliferative, thus gaining immortalization. Unlimited proliferation is due to telomeres which are involved in the protection of chromosomal ends. Successive cell cycles with chromosomal replication lead to an erosion of telomeres in normal cells. Unprotected chromosomal ends are involved in end‐to‐end fusions yielding karyotypic disarrays. This event forces cells to enter the state of cellular crisis. Tumor cells circumvent the shortening of telomeres by enhanced gene expression of the telomerase, an enzyme which catalyzes the addition of hexanucleotide repeats to the ends of telomeric DNA, thus stabilizing the length of telomeres.
Inducing angiogenesis: Tumors, such as normal tissues, are dependent on the delivery of nutrients and oxygen on the one hand and the evacuation of unfavorable metabolites and carbon dioxide on the other hand. These needs are covered by angiogenesis. The development of vessels is driven by two different processes, namely the differentiation into endothelial cells followed by tube formation (vasculogenesis) and sprouting of new vessels (angiogenesis). After finishing organogenesis and body growth, angiogenesis is physiologically transitioning into a state of quiescence. Under specific conditions, for example, wound healing, vascularization is transiently turned on again. But during tumor initiation and progression, this process of vessel formation is permanently activated. This angiogenic switch is affected by various different factors that stimulate angiogenesis. 185 VEGFs are well‐known ligands which are involved in vascularization. Their gene expression is up‐regulated by hypoxia and oncogenes. 186 Other proangiogenic factors belong to the family of fibroblast growth factors (FGFs). 187 Angiogenesis is induced at a very early stage in tumor development. Expression of proangiogenic factors can be stimulated by oncogenes such as Ras and Myc or indirectly by immune–inflammatory cells.
Activating invasion and metastasis: Metastases are the reason for the high death rate of cancer. 188 Invasion and metastasis are complex processes, called invasion–metastasis cascade 189 in which cells of primary tumors spread out of their cancer origin to disseminate, invade, and colonize new areas in the body with favorable microenvironments to form new cancer tissues. This multi‐step process consists of a change in primary tumor cell phenotype gaining cells with an increased capability of motility, invasiveness, and modification of the extracellular matrix (ECM) based upon epithelial–mesenchymal transition (EMT). A prominent alteration is the loss of E‐cadherin, an important molecule of cell–cell adhesion, connected to the simultaneous up‐regulation of N‐cadherin, a protein which is associated with migration. Thus, carcinoma cells achieve the ability to disseminate and migrate out of their original cancer tissue. One of the next steps in metastasis is the invasion of individual tumor cells or multicellular clumps into the vascular system. By this process, these circulating tumor cells (CTCs) become able to travel to distant sites to form new metastases. But to become successful in entering new tissues, it is necessary for CTCs to leave the vascular system. This extravasation through the vessels, also called transendothelial migration (TEM), is initiated and controlled by the carcinoma cells themselves, which secrete factors, such as MMPs and VEGF, with the ability to disrupt vascular integrity. Having reached the new tissue site, the disseminated tumor cells (DTCs) have to cope with a cancer‐unfavorable microenvironment which is lacking cell growth‐promoting factors, supportive stromal cells, and ECM constituents. Hence, DTCs fall into a state of growth arrest. In the same way, as primary tumor cells are dependent on their surrounding cells and tissue, DTCs also need a beneficial microenvironment which is achieved by secreted factors, such as TGF‐β1, or pro‐inflammatory cells. Eventually, dormant DTCs leave their state of growth arrest and enter the state of proliferation which is the beginning of new metastatic colonization. 188 The above‐described six hallmarks of cancer are still the epitome of understanding the biology of cancer. 96 , 97 Nevertheless, some more “enabling characteristics and emerging hallmarks” of tumorigenesis have been mentioned 97 with “tumor‐promoting inflammation” as the most important one with respect to the topic of the present review. In the middle of the 19th century, Virchow discovered leukocytes in cancerous tissues and drew a connection between cancer and inflammation. 190 Some decades ago, it was found that some tumors were infiltrated by various cells of the innate and adaptive immune systems. Hence, inflammatory conditions within the tumor were thereby assumed. This observation led to a new view on cancer, namely that tumors could be considered as never healing wounds. 191 In the following decades, research on the impact of inflammation on cancer became more into focus showing tumor‐promoting effects by members of the innate immune system. 192 Very many tumors induce an endogenous inflammatory response resulting in a tumor‐supporting microenvironment. Growing tumors reach at certain points a state at which they become short of nutrients and oxygen due to strained vascularization and alteration of tissue homeostasis. This leads to necrotic cell deaths with liberation of pro‐inflammatory mediators, such as IL‐1 or HMGB1, resulting, as a consequence, in neo‐vascularization with support of additional growth factors supplied by inflammatory and immune cells. 184 , 193 , 194 , 195 Tumor‐associated macrophages (TAMs) are a major constituent of tumor stroma. Although these immune cells participate in tumor cell and tissue destruction, TAMs are also involved in malignant progression due to secretion of growth factors and cytokines. Today, it is generally accepted that cancer predisposition and development are affected by chronic inflammation. Two pathways, namely the extrinsic pathway (oncogenic‐driven) and the intrinsic pathway (microenvironment‐dependent), connect inflammation and cancerogenesis. 190 The key player in this cellular process is the transcription factor NF‐κB. This protein is present in almost all cells and possesses the ability to bind to over 150 down‐stream genes. Besides its role in the inflammatory response, activated NF‐κB also affects target genes involved in proliferation, such as cyclin D1 or c‐Myc. In addition, NF‐κB contributes to EMT and metastasis by regulating inter alia MMPs, E‐cadherin, or VEGF. 196 The immune system does not only respond to microbial attacks, but also to signal molecules derived from stressed or dying cells. As already mentioned above, the organism reacts on this challenge with the secretion of alarmins. One of the key players, which participates in connecting inflammation to cancer, is HMGB1. This alarmin is able to bind to TLR4 and RAGE. Both cell surface receptors activate the abovementioned transcription factor NF‐κB. Consequently, HMGB1 and further alarmins, such as S100 proteins, contribute to maintain an inflammatory state resulting in a tumor‐promoting microenvironment and thus driving tumorigenesis. 52 , 182 In summary, chronic inflammation is an important factor to establish and maintain malignancies, even participating in invasion and metastasis.
4. EFFECTS OF INNATE HOST DEFENSE MOLECULES ON ORAL CANCEROGENESIS
As already mentioned above, elements of the innate host defense system are involved in the development of cancer, filling the molecular link between chronic inflammation and cancerogenesis (Figures 1, 2, 4). This chapter is dealing particularly with their role in the formation of oral tumors. It will be described in which way antimicrobial peptides, S100 proteins, and alarmins execute initiation and progression of oral malignancies. These molecules exert their role with primary effects, such as binding to EGFR with associated direct impacts, and secondary effects, such as amplifier functions via EGFR or RAGE (Figures 2, 4).
FIGURE 4.
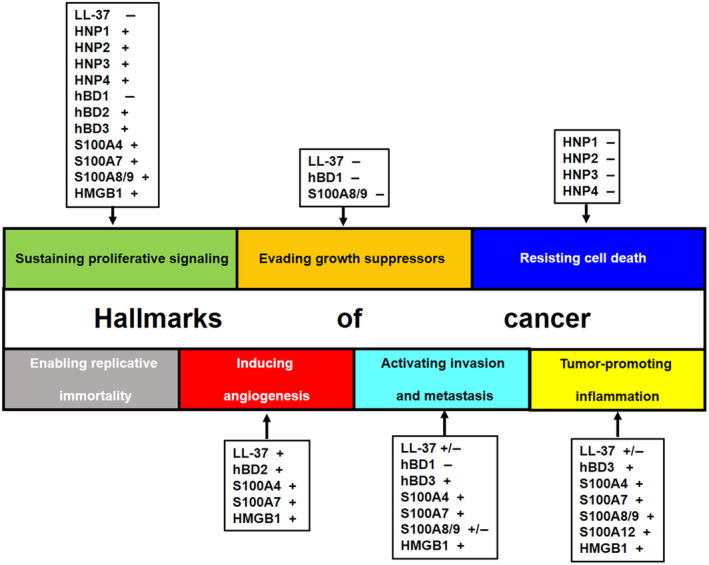
Impact of molecules of the innate host defense system on “hallmarks of cancer”. 96 , 97 The effects of LL‐37, α‐ and β‐defensins, S100 proteins (metastasin, psoriasin, and calgranulin A, B, and C), and HMGB1 on the different hallmarks of cancer are described. hBD, human β‐defensin; HMGB1, high mobility group protein B1; HNP, human neutrophil peptide.
Recently, it has been suggested that an imbalance of human microbiome homeostasis can also cause malignant transformation by promoting chronic inflammation. 197 , 198 , 199
As described above, antimicrobial peptides play a pivotal role in connecting infection and chronic inflammation to oral carcinogenesis. LL‐37 is mainly expressed in neutrophils. Moreover, an increased peptide level has been observed in gingival crevicular fluid and saliva of patients suffering from periodontitis. LL‐37 acts in two antagonistic ways on tumorigenesis: (1) On the one hand, it is able to bind LPS, which leads to inhibition of inflammatory processes and hence counteracts tumor‐promoting inflammation. (2) On the other hand, LL‐37 is a ligand to the formyl peptide receptor‐like 1, which is involved in angiogenesis, a typical tumor‐supporting process. Furthermore, LL‐37 positively affects the production of IL‐6, a tumor‐promoting cytokine with the capability of regulating several genes involved in proliferation through signaling via JAK/STAT3 pathways. 124 , 128 , 192 LL‐37 expression levels have been shown to be down‐regulated in OSCC tissues compared to normal oral mucosa. Additionally, LL‐37 overexpression suppresses proliferation, migration, and invasion of an OSCC cell line. These observations suggest that LL‐37 might act as a putative tumor suppressor. 200 , 201 , 202 Exogenously applied LL‐37 on three oral tongue squamous cell carcinoma cell lines led to reduced proliferation, but also in contrast to enhanced migration and invasion 203 (Figure 4).
Higher protein levels of the defensin HNP1 were detected in saliva of patients suffering from oral lichen planus, leukoplakias, and OSCCs compared to healthy individuals. 204 , 205 This defensin was also found in submandibular glands of OSCC patients. 206 Additionally, HNP1 expression levels were reported to be enhanced in benign and premalignant oral lesions, such as irritation fibromas 130 and leukoplakias 129 , and also in different entities of salivary gland tumors. 207 Stimulation of oral keratinocyte and OSCC cells with HNP1 led to significant cytotoxic effects on both cell types. 208 , 209 However, in contrast, it has also been shown that HNP1 promotes proliferation of OSCC cells by affecting cyclin D1 expression and EGFR activation. 210 These contrasting results might be due to different concentrations used in the experiments of both groups. HNP2 and HNP3 are overexpressed in oral squamous tongue carcinomas 211 ; additionally, HNP2, HNP3, and HNP4 are overexpressed in oral irritation fibromas, 130 leukoplakias, 129 and various salivary gland tumors. 207 These human defensins also positively affect OSCC cell proliferation in the same way as shown for HNP1 due to their molecular three‐dimensional similarities with EGF, hence serving as structure analog ligands for EGFR. 210 Whether HNPs exert more cytotoxic or pro‐proliferative effects seems to be concentration‐dependent 212 (Figure 4).
Human β‐defensin 1 has been found in oral irritation fibromas, leukoplakias, and OSCCs with reduced expression levels correlating to the grade of malignancy 5 , 123 and in various OSCC cell lines. 135 A down‐regulation of hBD1 has also been observed in salivary gland tumors 136 , 138 associated with a nuclear accumulation of this peptide. 137 A decreased expression level of this human β‐defensin has also been reported for other tumor entities, as mentioned above, assuming hBD1 to be a putative tumor suppressor protein. 5 , 132 , 133 , 134 , 135 , 136 , 138 Stimulation of an OSCC cell line (BHY) with hBD1 led to a reduced proliferation rate associated with a simultaneous initial up‐regulation of hBD1 itself in combination with an up‐regulation of hBD2 and hBD3 which might be due to EGFR activation. 132 , 210 Hence, hBD1 seems to have two functions with opposite effects: (1) It has tumor suppressor activity, and (2) it activates EGFR. Thus, the proliferation rate with respect to hBD1 seems to be dependent on which of the two effects prevails. The fact that exogenous hBD1 is able to decrease the proliferation rate can be explained with the assumption that hBD1 has the ability to re‐enter cells by crossing cytoplasm membranes due to its chemical properties and hence functions intracellularly as tumor suppressor. 102 , 132 Another impact of hBD1 is an inhibition of invasion and migration in a transgenic hBD1‐expressing OSCC cell line. 213 HBD2 has been reported to be also down‐regulated in OSCCs as well as in salivary gland tumors but to a lesser extent compared to hBD1. 5 , 123 , 136 , 138 Besides its occurrence in oral malignancies, hBD2 has been detected in oral irritation fibromas and leukoplakias and in intratumoral endothelial cells, too. 5 , 214 Stimulation of BHY cells with hBD2 led to an increase in proliferation which could be explained by EGFR activation. 132 , 210 HBD3 shows a more inconsistent picture in oral cancer: It is reported to be up‐regulated in OSCC tissue specimens by two groups 5 , 142 , whereas another group showed a decrease in expression; however, only OSCC cell lines were used instead in this study. 135 In contrast to OSCCs, hBD3 expression is decreased in salivary gland tumors. 136 It has been suggested that hBD3 might serve as a mitogen‐responsive gene in oral tumor initiation since this defensin is positively affected by EGF stimulation in oral epithelial cells. 215 The same observation has been made in an OSCC cell line which exhibits an enhanced proliferation rate after hBD3 stimulation. 132 Moreover, hBD3 binds to EGFR, like the four HNPs, hBD1, and hBD2. 210 Recently, it has been observed that hBD3 and NF‐κB synergistically promote proliferation and invasion of OSCCs. 216 Furthermore, it has been demonstrated that the three hBDs show cross‐effects on each of their gene expression 132 (Figure 4; Table 1). The effects of human β‐defensins are summarized in Figure 5.
TABLE 1.
Summary of effects with corresponding mechanisms on oral tumorigenesis exerted by molecules of the innate host defense system.
| Protein | Effects | Mechanisms |
|---|---|---|
| LL‐37 |
Angiogenesis (+) Proliferation (+) Proliferation (−) |
Formyl peptide receptor‐like 1 IL‐6 (+) Tumor suppressor |
|
HNP1 HNP2 HNP3 HNP4 |
Proliferation (+) Proliferation (−) |
EGFR; cyclin D (+) Cytotoxicity (+): dose dependent |
| hBD1 |
Proliferation (+) Proliferation (−) |
EGFR; cyclin D (+); hBD1/2/3 (+) Tumor suppressor |
|
hBD2 hBD3 |
Proliferation (+) | EGFR; cyclin D (+); hBD1/2/3 (+) |
| S100A7 |
Proliferation (+) Invasion (+) |
Unknown ITGβ6; β‐catenin; p38/MAPK |
| S100A8/A9 |
Proliferation (−) Proliferation (+) Invasion (−) Migration (−) |
EGFR cleavage (+) IL‐6 (+) Unknown Unknown |
| S100A4 |
Invasion (+) Metastasis (+) |
E‐cadherin (−) E‐cadherin (−) |
| HMGB1 |
Proliferation (+) Angiogenesis (+) |
IL‐6 (+) RAGE (+) |
Abbreviations: (+), up‐regulated/activated; (−), down‐regulated; EGFR, epidermal growth factor receptor; hBD, human β‐defensin; HMGB1, high mobility group protein B1; HNP, human neutrophil peptide; ITGβ6, integrin β6; MAPK, mitogen‐activated protein kinase; RAGE, receptor for advanced glycation end products.
FIGURE 5.
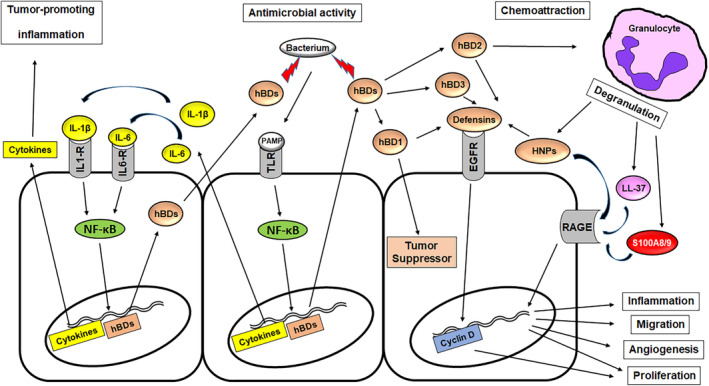
Overview of effects initiated by hBDs after TLR activation. Middle part: Bacterial PAMPs are binding to TLR leading to activation, NF‐κB translocation, and induction of genes encoding inflammatory cytokines and hBDs. These proteins are secreted to the extracellular matrix. HBDs attack as AMPs the present bacteria. Left: Cytokines (IL‐1β and IL‐6) induce a tumor‐promoting microenvironment. Additionally, pro‐inflammatory cytokines also induce the hBD expression and thus function as amplifiers. Right: HBD1 re‐enters the cells and can function as tumor suppressor. HBD2 chemoattracts granulocytes. HBD1, hBD2, and hBD3 can bind to EGFR and promote proliferation via cyclin D induction. Degranulation of granulocytes leads to release of HNPs, which can bind to EGFR and RAGE. Also, LL‐37 and calprotectin (S100A8/A9) are liberated and work as RAGE ligands. Consequentially, RAGE activation affects inflammation, migration, angiogenesis, and proliferation. EGFR, epidermal growth factor receptor; hBD, human β‐defensin; IL‐1‐R, IL‐1 receptor; IL‐6‐R, IL‐6 receptor; NF‐κB, nuclear factor‐κB; PAMPs, pathogen‐associated molecular patterns; RAGE, receptor for advanced glycation end products; TLR, toll‐like receptor.
Psoriasin or S100A7 is up‐regulated in oral irritation fibromas, leukoplakias, and OSCCs. The transcript level correlates with the stage of malignancy. Also, nuclear S100A7 occurrence is associated with poor prognosis. 129 , 130 , 164 , 165 , 217 , 218 , 222 , 223 In contrast to the abovementioned oral lesions, S100A7 expression is decreased in salivary gland tumors. 136 S100A7 has been identified as a protein partner for integrin β6 (ITGβ6) subunit and is involved in integrin‐dependent carcinoma cell invasion. 219 Also, psoriasin plays a role in tumor cell progression and invasion through signaling via the β‐catenin 220 and p38/MAPK pathways. 221 Overexpression of S100A7 leads to promotion of proliferation and simultaneous suppression of differentiation. 222 S100A8 and S100A9 are down‐regulated in OSCCs and head and neck squamous cell carcinomas (HNSCCs). 170 , 223 , 224 Calprotectin (S100A8/A9) expression inhibits proliferation, invasion, and migration and is thus supposed to function as a tumor suppressor. 224 Furthermore, intracellular calprotectin promotes EGFR cleavage 171 and increases apoptosis as a response to DNA damage in HNSCCs 225 leading to a prolonged survival rate of patients with high‐calprotectin tumors. 171 , 225 Patients suffering CP show an enhanced level of calprotectin in saliva and GCF. 226 , 227 Stimulation of periodontal ligament cells with S100A8 and S100A9 causes an increase in tumor‐promoting pro‐inflammatory IL‐6 expression via NF‐κB activation, 227 in gingival fibroblast via TLR4 signaling. 228 These observations demonstrate how calprotectin, as a member of the innate host defense system, is able to modulate and establish a tumor‐promoting microenvironment by maintaining a pro‐inflammatory status. S100A4, also named metastasin, shows an inconsistent expression pattern on OSCCs: It has been found to be down‐regulated in two studies, 170 , 223 whereas other groups reported up‐regulated S100A4 expression which was associated with metastases, invasion, and poor diagnosis. 229 , 230 High S100A4 transcript levels have been demonstrated to be related to the occurrence of inflammatory cells in the tumor stroma. 231 Overexpression of S100A4 in OSCC cells causes a down‐regulation of E‐cadherin, 232 which is an important step in metastatic and invasive mechanisms (Figure 4). A recently published review describes the roles of S100 proteins in HNSCCs very nicely. 233
High levels of the alarmin HMGB1 have been detected in inflamed gingival tissues of CP patients. Secreted HMGB1 causes an inflammatory response by inducing pro‐inflammatory cytokine expression, for example, of IL‐6. Furthermore, it is involved in neovascularization by activating RAGE which expression is associated with angiogenesis in OSCCs. 181 , 183 Hence, HMGB1 enhances a tumor‐promoting microenvironment (Figure 4).
A summary of the effects of members of the innate host defense system including the corresponding mechanisms is listed in Table 1.
5. SUMMARY
In summary, the goal of the present review was to illustrate possible molecular pathways explaining the link of infection to inflammation and eventually to cancer development. Persisting bacterial challenges based upon the establishment of a dental biofilm is a source of chronic periodontal infection which causes a permanent activation of the innate host defense system leading to chronic periodontal inflammation, driven by molecules, such as defensins, S100 proteins, and alarmins. This response appears to have a strong impact on malignant transformation associated with oral tumor initiation and cancerogenesis (Figures 1, 5). Most of the current studies are limited to in vitro experiments. In the future, further evidence from well‐planned longitudinal epidemiological, mechanistic, and intervention studies will be needed to elucidate the possible causative relationship between activation of the innate host defense by periodontal infection/inflammation and oral cancer development.
AUTHOR CONTRIBUTIONS
All authors have made substantial contributions to the design and conception of the manuscript, drafted the work or revised it critically for important intellectual content, and approved the version to be published.
FUNDING INFORMATION
This research received no external funding.
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflict of interest.
ACKNOWLEDGMENTs
Open Access funding enabled and organized by Projekt DEAL.
Winter J, Jepsen S. Role of innate host defense proteins in oral cancerogenesis. Periodontol 2000. 2024;96:203‐220. doi: 10.1111/prd.12552
DATA AVAILABILITY STATEMENT
Supporting data and materials are available on request to the corresponding author.
REFERENCES
- 1. Silverman S Jr, Dillon W, Fischbein N. Diagnosis. In: Silverman S Jr, ed. Oral Cancer. 4th ed. BC Decker Inc.; 1998:25‐33. [Google Scholar]
- 2. Chen MX, Zhong YJ, Dong QQ, Wong HM, Wen YF. Global, regional, and national burden of severe periodontitis, 1990–2019: an analysis of the global burden of disease study 2019. J Clin Periodontol. 2021;48:1165‐1188. [DOI] [PubMed] [Google Scholar]
- 3. Chaambers MJ, Jacob RJ. How carcinogens cause cancer. Tex Dent J. 1994;111:13‐19. [PubMed] [Google Scholar]
- 4. Wenghoefer M, Pesch B, Harth V, et al. Association between head and neck cancer and microsomal epoxide hydrolase genotypes. Arch Toxicol. 2003;77:37‐41. [DOI] [PubMed] [Google Scholar]
- 5. Wenghoefer M, Pantelis A, Dommisch H, et al. Decreased gene expression of human beta‐defensin‐1 in the development of squamous cell carcinoma of the oral cavity. Int J Oral Maxillofac Surg. 2008;37:660‐663. [DOI] [PubMed] [Google Scholar]
- 6. Ogbureke KUE, Bingham C. Overview of oral cancer. In: Ogbureke KUE, ed. Oral cancer. InTech; 2012:3‐20. [Google Scholar]
- 7. Tonetti MS, Jepsen S, Jin L, Otomo‐Corgel J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: a call for global action. J Clin Periodontol. 2017;44:456‐462. [DOI] [PubMed] [Google Scholar]
- 8. Tezal M, Sullivan MA, Reid ME, et al. Chronic periodontitis and the risk of tongue cancer. Arch Otolaryngol Head Neck Surg. 2007;133:450‐454. [DOI] [PubMed] [Google Scholar]
- 9. Tezal M, Sullivan MA, Hyland A, et al. Chronic periodontitis and the incidence of head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2009;18:2406‐2412. [DOI] [PubMed] [Google Scholar]
- 10. Meyer MS, Joshipura K, Giovannucci E, Michaud DS. A review of the relationship between tooth loss, periodontal disease, and cancer. Cancer Causes Control. 2008;19:895‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fitzpatrick SG, Katz J. The association between periodontal disease and cancer: a review of the literature. J Dent. 2010;38:83‐95. [DOI] [PubMed] [Google Scholar]
- 12. Zeng XT, Deng AP, Li C, Xia LY, Niu YM, Leng WD. Periodontal disease and risk of head and neck cancer: a meta‐analysis of observational studies. PloS One. 2013;8:e79017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yao QW, Zhou D‐S, Peng H‐J, Ji P, Liu DC. Association of periodontal disease with oral cancer: a meta‐analysis. Tumour Biol. 2014;35:7073‐7077. [DOI] [PubMed] [Google Scholar]
- 14. Ye L, Jiang Y, Liu W, Tao HB. Correlation between periodontal disease and oral cancer risk: a meta‐analysis 1. J Cancer Res Ther. 2016;12(Supplement):C237‐C240. [DOI] [PubMed] [Google Scholar]
- 15. Javed F, Warnakulasuriya S. Is there a relationship between periodontal disease and oral cancer? A systematic review of currently available evidence. Crit Rev Oncol Hematol. 2016;97:197‐205. [DOI] [PubMed] [Google Scholar]
- 16. Chung SD, Tsai MC, Huang CC, Kao LT, Chen CH. A population‐based study on the associations between chronic periodontitis and the risk of cancer. Int J Clin Oncol. 2016;21:219‐223. [DOI] [PubMed] [Google Scholar]
- 17. Shin YJ, Choung HW, Lee JH, Rhyu IC, Kim HD. Association of Periodontitis with Oral cancer: a case‐control study. J Dent Res. 2019;98:526‐533. [DOI] [PubMed] [Google Scholar]
- 18. Nwizu N, Wactawski‐Wende J, Genco RJ. Periodontal disease and cancer: epidemiologic studies and possible mechanisms. Periodontol 2000. 2020;83:213‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoare A, Soto C, Rojas‐Celis V, Bravo D. Chronic inflammation as a link between periodontitis and carcinogenesis. Mediators Inflamm. 2019;2019:1029857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Irani S, Barati I, Badiei M. Periodontitis and oral cancer – current concepts of the etiopathogenesis. Oncol Rev. 2020;14:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hajishengallis G, Chavakis T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. 2021;21:426‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. St John MA, Li Y, Zhou X, et al. Interleukin 6 and interleukin 8 as potential biomarkers for oral cavity and oropharyngeal squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2004;130:929‐935. [DOI] [PubMed] [Google Scholar]
- 23. Michiels K, Schutyser E, Conings R, et al. Carcinoma cell‐derived chemokines and their presence in oral fluid. Eur J Oral Sci. 2009;117:362‐368. [DOI] [PubMed] [Google Scholar]
- 24. Pitiyage G, Tilakaratne WM, Tavassoli M, Warnakulasuriya S. Molecular markers in oral epithelial dysplasia: review. J Oral Pathol Med. 2009;38:737‐752. [DOI] [PubMed] [Google Scholar]
- 25. Porta C, Larghi P, Rimoldi M, et al. Cellular and molecular pathways linking inflammation and cancer. Immunobiology. 2009;214:761‐777. [DOI] [PubMed] [Google Scholar]
- 26. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819‐826. [DOI] [PubMed] [Google Scholar]
- 27. Netea MG, Quintin J, van der Meer JW. Trained immunity: a memory for innate host defense. Cell Host Microbe. 2011;9:355‐361. [DOI] [PubMed] [Google Scholar]
- 28. Gasteiger G, D'Osualdo A, Schubert DA, Weber A, Bruscia EM, Hartl D. Cellular innate immunity: an old game with new players. J Innate Immun. 2017;9:111‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meyle J, Dommisch H, Groeger S, Giacaman RA, Costalonga M, Herzberg M. The innate host response in caries and periodontitis. J Clin Periodontol. 2017;44:1215‐1225. [DOI] [PubMed] [Google Scholar]
- 30. Paludan SR, Pradeu T, Masters SL, Mogensen TH. Constitutive immune mechanisms: mediators of host defence and immune regulation. Nat Rev Immunol. 2021;21:137‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710‐720. [DOI] [PubMed] [Google Scholar]
- 32. Lehrer R. Primate defensins. Nature. Rev Microbiol. 2004;2:727‐738. [DOI] [PubMed] [Google Scholar]
- 33. Harder J, Gläser R, Schröder JM. Human antimicrobial proteins effectors of innate immunity. J Endotoxin Res. 2007;13:317‐338. [DOI] [PubMed] [Google Scholar]
- 34. Winter J, Wenghoefer M. Human Defensins: potential tools for clinical applications. Polymers. 2012;4:691‐709. [Google Scholar]
- 35. Niyonsaba F, Kiatsurayanon C, Chieosilapatham P, Ogawa H. Friends or foes? Host defense (antimicrobial) peptides and proteins in human skin diseases. Exp Dermatol. 2017;26:989‐998. [DOI] [PubMed] [Google Scholar]
- 36. Heizmann CW. S100 proteins: diagnostic and prognostic biomarkers in laboratory medicine. Biochim Biophys Acta Mol Cell Res. 2019;1866:1197‐1206. [DOI] [PubMed] [Google Scholar]
- 37. Gonzalez LL, Garrie K, Turner MD. Role of S100 proteins in health and disease. Biochim Biophys Acta Mol Cell Res. 2020;1867:118677. [DOI] [PubMed] [Google Scholar]
- 38. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1‐13. [DOI] [PubMed] [Google Scholar]
- 39. Schatz DG, Oettinger MA, Schlissel MS. V(D)J recombination: molecular biology and regulation. Annu Rev Immunol. 1992;10:359‐383. [DOI] [PubMed] [Google Scholar]
- 40. Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak‐Rothstein A. Toll‐like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27‐42. [DOI] [PubMed] [Google Scholar]
- 41. Kawai T, Akira S. Pathogen recognition with toll‐like receptors. Curr Opin Immunol. 2005;17:338‐344. [DOI] [PubMed] [Google Scholar]
- 42. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783‐801. [DOI] [PubMed] [Google Scholar]
- 43. Lewkowicz N, Mycko MP, Przygodzka P, et al. Induction of human IL‐10‐producing neutrophils by LPS‐stimulated Treg cells and IL‐10. Mucosal Immunol. 2016;9:364‐378. [DOI] [PubMed] [Google Scholar]
- 44. Becerra‐Ruiz JS, Guerrero‐Velázquez C, Martínez‐Esquivias F, Martínez‐Pérez LA, Guzmán‐Flores JM. Innate and adaptive immunity of periodontal disease. From etiology to alveolar bone loss. Oral Dis. 2022;28:1441‐1447. [DOI] [PubMed] [Google Scholar]
- 45. Hirschfeld J. Dynamic interactions of neutrophils and biofilms. J Oral Microbiol. 2014;6:26102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hirschfeld J, Dommisch H, Skora P, et al. Neutrophil extracellular trap formation in supragingival biofilms. Int J Med Microbiol. 2015;305:453‐463. [DOI] [PubMed] [Google Scholar]
- 47. Hirschfeld J, Roberts HM, Chapple IL, et al. Effects of Aggregatibacter actinomycetemcomitans leukotoxin on neutrophil migration and extracellular trap formation. J Oral Microbiol. 2016;8:33070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deschner J, Jepsen S. Polymorphonuclear cells (neutrophils). In: Gruber B, Stadlinger H, Terheyden T, eds. Cell‐to‐cell communication. Cell atlas – visual biology in oral medicine. Quintessence Publishing; 2022:165‐174. [Google Scholar]
- 49. Moonen CGJ, Hirschfeld J, Cheng L, Chapple ILC, Loos BG, Nicu EA. Oral neutrophils characterized: chemotactic, phagocytic, and neutrophil extracellular trap (NET) formation properties. Front Immunol. 2019;10:635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359‐365. [DOI] [PubMed] [Google Scholar]
- 51. Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage‐associated molecular pattern molecules. J Leukoc Biol. 2007;81:28‐37. [DOI] [PubMed] [Google Scholar]
- 52. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367‐388. [DOI] [PubMed] [Google Scholar]
- 53. Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889‐901. [DOI] [PubMed] [Google Scholar]
- 54. Palumbo R, De Marchis F, Pusterla T, Conti A, Alessio M, Bianchi ME. Src family kinases are necessary for cell migration induced by extracellular HMGB1. J Leukoc Biol. 2009;86:617‐623. [DOI] [PubMed] [Google Scholar]
- 55. Touré F, Zahm J‐M, Garnotel R, et al. Receptor for advanced glycation end‐products (RAGE) modulates neutrophil adhesion and migration on glycoxidated extracellular matrix. Biochem J. 2008;416:255‐261. [DOI] [PubMed] [Google Scholar]
- 56. Hudson BI, Kalea AZ, Del Mar AM, et al. Interaction of the RAGE cytoplasmic domain with diaphanous‐1 is required for ligand‐stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem. 2008;283:34457‐34468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kim JY, Park HK, Yoon JS, et al. Advanced glycation end product (AGE)‐induced proliferation of HEL cells via receptor for AGE‐related signal pathways. Int J Oncol. 2008;33:493‐501. [PubMed] [Google Scholar]
- 58. Reddy MA, Li S‐L, Sahar S, et al. Key role of Src kinase in S100B‐induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J Biol Chem. 2006;281:13685‐13693. [DOI] [PubMed] [Google Scholar]
- 59. Steiner H, Hultmark D, Engström A, Bennich H, Boman HG. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature. 1981;292:246‐248. [DOI] [PubMed] [Google Scholar]
- 60. Hoffmann JA, Kafatos FC, Janeway CA Jr, Ezekowitz RAB. Phylogenetic perspectives in innate immunity. Science. 1999;284:1313‐1317. [DOI] [PubMed] [Google Scholar]
- 61. Zasloff M. Innate immunity, antimicrobial peptides, and protection of the oral cavity. Lancet. 2002;360:1116‐1117. [DOI] [PubMed] [Google Scholar]
- 62. Cederlund A, Gudmundsson GH, Agerberth B. Antimicrobial peptides important in innate immunity. FEBS J. 2011;278:3942‐3951. [DOI] [PubMed] [Google Scholar]
- 63. Agerberth B, Charo J, Werr J, et al. The human antimicrobial and chemotactic peptides LL‐37 and alpha‐defensins are expressed by specific lymphocyte and monocyte populations. Blood. 2000;96:3086‐3093. [PubMed] [Google Scholar]
- 64. Donato R. Functional roles of S100 proteins, calcium‐binding proteins of the EF‐hand type. Biochim Biophys Acta. 1999;1450:191‐231. [DOI] [PubMed] [Google Scholar]
- 65. Donato R. S100: a multigenic family of calcium‐modulated proteins of the EF‐hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol. 2001;33:637‐668. [DOI] [PubMed] [Google Scholar]
- 66. Donato R, Cannon BR, Sorci G, et al. Functions of S100 proteins. Curr Mol Med. 2013;13:24‐57. [PMC free article] [PubMed] [Google Scholar]
- 67. Zackular JP, Chazin WC, Skaar EP. Nutritional immunity: S100 proteins at the host‐pathogen interface. J Biol Chem. 2015;290:18991‐18998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pirr S, Richter M, Fehlhaber B, et al. High amounts of S100‐alarmins confer antimicrobial activity on human breast milk targeting pathogens relevant in neonatal sepsis. Front Immunol. 2017;8:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schibli DJ, Hunter HN, Aseyev V, et al. The solution structures of the human beta‐defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus . J Biol Chem. 2002;277:8279‐8289. [DOI] [PubMed] [Google Scholar]
- 70. Brodersen DE, Etzerodt M, Madsen P, et al. EF‐hands at atomic resolution: the structure of human psoriasin (S100A7) solved by MAD phasing. Structure. 1998;6:477‐489. [DOI] [PubMed] [Google Scholar]
- 71. Wang G. Structures of human host defense cathelicidin LL‐37 and its smallest antimicrobial peptide KR‐12 in lipid micelles. J Biol Chem. 2008;283:32637‐32643. [DOI] [PubMed] [Google Scholar]
- 72. Xhindoli D, Pacor S, Benincasa M, Scocchi M, Gennaro R, Tossi A. The human cathelicidin LL‐37 – a pore‐forming antibacterial peptide and host‐cell modulator. Biochem Biophys Acta. 2016;1858:546‐566. [DOI] [PubMed] [Google Scholar]
- 73. Sørensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N. The human antibacterial cathelicidin, hCAP‐18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood. 1997;90:2796‐2803. [PubMed] [Google Scholar]
- 74. Dürr UNH, Sudheendra US, Ramamoorthy A. LL‐37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta. 2006;1758:1408‐1425. [DOI] [PubMed] [Google Scholar]
- 75. Rivas‐Santiago B, Hernandez‐Pando R, Carranza C, et al. Expression of cathelicidin LL‐37 during mycobacterium tuberculosis infection in human alveolar macrophages, monocytes, neutrophils, and epithelial cells. Infect Immun. 2008;76:935‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yang D, de la Rosa G, Tewary P, Oppenheim JJ. Alarmins link neutrophils and dendritic cells. Trends Immunol. 2009;30:531‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sonawane A, Santos JC, Mishra BB, et al. Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell Microbiol. 2011;13:1601‐1617. [DOI] [PubMed] [Google Scholar]
- 78. Afshar M, Gallo RL. Innate immune defense system of the skin. Vet Dermatol. 2013;24:32‐38.e8‐9. [DOI] [PubMed] [Google Scholar]
- 79. Schauber J, Dorschner RA, Yamasaki K, Brouha B, Gallo RL. Control of the innate epithelial antimicrobial response is cell‐type specific and dependent on relevant microenvironmental stimuli. Immunology. 2006;118:509‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nell MJ, Tjabringa GS, Vonk MJ, Hiemstra PS, Grote JJ. Bacterial products increase expression of the human cathelicidin hCAP‐18/LL‐37 in cultured human sinus epithelial cells. FEMS Immunol Med Microbiol. 2004;42:225‐231. [DOI] [PubMed] [Google Scholar]
- 81. Lai Y, Gallo RL. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009;30:131‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wang TT, Nestel FP, Bourdeau V, et al. Cutting edge: 1,25‐dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol. 2004;173:2909‐2912. [DOI] [PubMed] [Google Scholar]
- 83. Schauber J, Gallo RL. The vitamin D pathway: a new target for control of the skin's immune response? Exp Dermatol. 2008;17:633‐639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up‐regulated in myeloid cells by 1,25‐dihydroxyvitamin D3. FASEB J. 2005;19:1067‐1077. [DOI] [PubMed] [Google Scholar]
- 85. Dixon BM, Barker T, McKinnon T, et al. Positive correlation between circulating cathelicidin antimicrobial peptide (hCAP18/LL‐37) and 25‐hydroxyvitamin D levels in healthy adults. BMC Res Notes. 2012;5:575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Peyssonnaux C, Boutin AT, Zinkernagel AS, Datta V, Nizet V, Johnson RS. Critical role of HIF‐1alpha in keratinocyte defense against bacterial infection. J Invest Dermatol. 2008;128:1964‐1968. [DOI] [PubMed] [Google Scholar]
- 87. Shinnar AE, Butler KL, Park HJ. Cathelicidin family of antimicrobial peptides: proteolytic processing and protease resistance. Bioorg Chem. 2003;31:425‐436. [DOI] [PubMed] [Google Scholar]
- 88. Sørensen OE, Follin P, Johnsen AH, et al. Human cathelicidin, hCAP‐18, is processed to the antimicrobial peptide LL‐37 by extracellular cleavage with proteinase 3. Blood. 2001;97:3951‐3959. [DOI] [PubMed] [Google Scholar]
- 89. Yamasaki K, Schauber J, Coda A, et al. Kallikrein‐mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 2006;20:2068‐2080. [DOI] [PubMed] [Google Scholar]
- 90. Johansson J, Gudmundsson GH, Rottenberg ME, Berndt KD, Agerberth B. Conformation‐dependent antibacterial activity of the naturally occurring human peptide LL‐37. J Biol Chem. 1998;273:3718‐3724. [DOI] [PubMed] [Google Scholar]
- 91. Lee CC, Sun Y, Qian S, Huang HW. Transmembrane pores formed by human antimicrobial peptide LL‐37. Biophys J. 2011;100:1688‐1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Xhindoli D, Morgera F, Zinth U, Rizzo R, Pacor S, Tossi A. New aspects of the structure and mode of action of the human cathelicidin LL‐37 revealed by the intrinsic probe p‐cyanophenylalanine. Biochem J. 2015;465:443‐457. [DOI] [PubMed] [Google Scholar]
- 93. Koczulla R, von Degenfeld G, Kupatt C, et al. An angiogenic role for the human peptide antibiotic LL‐37/hCAP‐18. J Clin Invest. 2003;111:1665‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tokumaru S, Sayama K, Shirakata Y, et al. Induction of keratinocyte migration via transactivation of the epidermal growth factor receptor by the antimicrobial peptide LL‐37. J Immunol. 2005;175:4662‐4668. [DOI] [PubMed] [Google Scholar]
- 95. Yin J, Yu FS. LL‐37 via EGFR transactivation to promote high glucose‐attenuated epithelial wound healing in organ‐cultured corneas. Invest Ophthalmol Vis Sci. 2010;51:1891‐1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57‐70. [DOI] [PubMed] [Google Scholar]
- 97. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 98. Türkoglu O, Kandiloglu G, Berdeli A, Emingil G, Atilla G. Antimicrobial peptide hCAP‐18/LL‐37 protein and mRNA expressions in different periodontal disease. Oral Dis. 2011;17:60‐67. [DOI] [PubMed] [Google Scholar]
- 99. Kido J, Bando M, Hiroshima Y, et al. Analysis of proteins in human gingival crevicular fluid by mass spectrometry. J Periodontal Res. 2012;47:488‐499. [DOI] [PubMed] [Google Scholar]
- 100. Hazlett L, Wu M. Defensins in innate immunity. Cell Tissue Res. 2011;343:175‐188. [DOI] [PubMed] [Google Scholar]
- 101. Zasloff M. Antimicrobial peptides of multicellular organisms: my perspective. Adv Exp Med Biol. 2019;1117:3‐6. [DOI] [PubMed] [Google Scholar]
- 102. Yeasmin R, Brewer A, Fine LR, Zhang L. Molecular dynamics simulations of human Beta‐Defensin type 3 crossing different lipid bilayers. ACS Omega. 2021;6:13926‐13939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Schutte BC, Mitros JP, Bartlett JA, et al. Discovery of five conserved beta ‐defensin gene clusters using a computational search strategy. Proc Natl Acad Sci U S A. 2002;99:2129‐2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yamaguchi Y, Ouchi Y. Antimicrobial peptide defensin: identification of novel isoforms and the characterization of their physiological roles and their significance in the pathogenesis of diseases. Proc Jpn Acad Ser B Phys Biol Sci. 2012;88:152‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Presicce P, Giannelli S, Taddeo A, Villa ML, Della Bella S. Human defensins activate monocyte‐derived dendritic cells, promote the production of proinflammatory cytokines, and up‐regulate the surface expression of CD91. J Leukoc Biol. 2009;86:941‐948. [DOI] [PubMed] [Google Scholar]
- 106. Territo MC, Ganz T, Selsted ME, Lehrer RI. Monocyte‐chemotactic activity of defensins from human neutrophils. J Clin Invest. 1989;84:2017‐2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yang D, Chen Q, Chertov O, Oppenheim JJ. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J Leukoc Biol. 2000;68:9‐14. [PubMed] [Google Scholar]
- 108. Niyonsaba F, Iwabuchi K, Matsuda H, Ogawa H, Nagaoka I. Epithelial cell‐derived human beta‐defensin‐2 acts as a chemotaxin for mast cells through a pertussis toxin‐sensitive and phospholipase C‐dependent pathway. Int Immunol. 2002;14:421‐426. [DOI] [PubMed] [Google Scholar]
- 109. Niyonsaba F, Ogawa H, Nagaoka I. Human beta‐defensin‐2 functions as a chemotactic agent for tumour necrosis factor‐alpha‐treated human neutrophils. Immunology. 2004;111:273‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Funderburg N, Lederman MM, Feng Z, et al. Human ‐defensin‐3 activates professional antigen‐presenting cells via toll‐like receptors 1 and 2. Proc Natl Acad Sci USA. 2007;104:18631‐18635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Niyonsaba F, Ushio H, Nakano N, et al. Antimicrobial peptides human beta‐defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J Invest Dermatol. 2007;127:594‐604. [DOI] [PubMed] [Google Scholar]
- 112. Kraus D, Deschner J, Jäger A, et al. Human β‐defensins differently affect proliferation, differentiation, and mineralization of osteoblast‐like MG63 cells. J Cell Physiol. 2012;227:994‐1003. [DOI] [PubMed] [Google Scholar]
- 113. Aarbiou J, Verhoosel RM, Van Wetering S, et al. Neutrophil defensins enhance lung epithelial wound closure and mucin gene expression in vitro. Am J Respir Cell Mol Biol. 2004;30:193‐201. [DOI] [PubMed] [Google Scholar]
- 114. Han W, Wang W, Mohammed KA, Su Y. Alpha‐defensins increase lung fibroblast proliferation and collagen synthesis via the beta‐catenin signaling pathway. FEBS J. 2009;276:6603‐6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Baroni A, Donnarumma G, Paoletti I, et al. Antimicrobial human beta‐defensin‐2 stimulates migration, proliferation and tube formation of human umbilical vein endothelial cells. Peptides. 2009;30:267‐272. [DOI] [PubMed] [Google Scholar]
- 116. Bick RJ, Poindexter BJ, Buja LM, Lawyer CH, Milner SM, Bhat S. Nuclear localization of HBD‐1 in human keratinocytes. J Burns Wounds. 2007;7:e3. [PMC free article] [PubMed] [Google Scholar]
- 117. Prado‐Montes de Oca E. Human beta‐defensin 1: a restless warrior against allergies, infections and cancer. Int J Biochem Cell Biol. 2010;42:800‐804. [DOI] [PubMed] [Google Scholar]
- 118. De Sousa Gomes P, Fernandes MH. Defensins in the oral cavity: distribution and biological role. J Oral Pathol Med. 2010;39:1‐9. [DOI] [PubMed] [Google Scholar]
- 119. Mathews M, Jia HP, Guthmiller JM, et al. Production of beta‐defensin antimicrobial peptides by the oral mucosa and salivary glands. Infect Immun. 1999;67:2740‐2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dale BA, Krisanprakornkit S. Defensin antimicrobial peptides in the oral cavity. J Oral Pathol Med. 2001;30:321‐327. [DOI] [PubMed] [Google Scholar]
- 121. Dunsche A, Açil Y, Siebert R, Harder J, Schröder JM, Jepsen S. Expression profile of human defensins and antimicrobial proteins in oral tissues. J Oral Pathol Med. 2001;30:154‐158. [DOI] [PubMed] [Google Scholar]
- 122. Marshall RI. Gingival defensins: linking the innate and adaptive immune responses to dental plaque. Periodontol 2000. 2004;35:14‐20. [DOI] [PubMed] [Google Scholar]
- 123. Abiko Y, Saitoh M, Nishimura M, Yamazaki M, Sawamura D, Kaku T. Role of β‐defensins in oral health and disease. Med Mol Morphol. 2007;40:179‐184. [DOI] [PubMed] [Google Scholar]
- 124. Gorr S‐U. Antimicrobial peptides in periodontal innate defense. Front Oral Biol. 2012;15:84‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Dommisch H, Jepsen S. Diverse functions of defensins and other antimicrobial peptides in periodontal tissues. Periodontol 2000. 2015;69:96‐110. [DOI] [PubMed] [Google Scholar]
- 126. Preianò M, Savino R, Villella C, Pelaia C, Terracciano R. Gingival Crevicular fluid Peptidome profiling in healthy and in periodontal diseases. Int J Mol Sci. 2020;21:5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Scott DA, Krauss J. Neutrophils in periodontal inflammation. Front Oral Biol. 2012;15:56‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Greer A, Zenobia C, Darveau RP. Defensins and LL‐37: a review of function in the gingival epithelium. Periodontol 2000. 2013;63:67‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wenghoefer M, Pantelis A, Najafi T, et al. Gene expression of oncogenes, antimicrobial peptides, and cytokines in the development of oral leucoplakia. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;110:351‐356. [DOI] [PubMed] [Google Scholar]
- 130. Winter J, Pantelis A, Allam JP, et al. High α‐defensin and S100A7 expression and missing DOC‐1 down‐regulation characterize irritation fibromas of the oral cavity and may counteract malignant transformation. J Craniofac Surg. 2011;22:100‐104. [DOI] [PubMed] [Google Scholar]
- 131. McDermott AM. Defensins and other antimicrobial peptides at the ocular surface. Ocul Surf. 2004;2:229‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Winter J, Pantelis A, Reich R, et al. Human beta‐defensin‐1, ‐2, and ‐3 exhibit opposite effects on oral squamous cell carcinoma cell proliferation. Cancer Invest. 2011;29:196‐201. [DOI] [PubMed] [Google Scholar]
- 133. Donald CD, Sun CQ, Lim SD, et al. Cancer‐specific loss of beta‐defensin 1 in renal and prostatic carcinomas. Lab Invest. 2003;83:501‐505. [DOI] [PubMed] [Google Scholar]
- 134. Sun CQ, Arnold R, Fernandez‐Golarz C, et al. Human beta‐defensin‐1, a potential chromosome 8p tumor suppressor: control of transcription and induction of apoptosis in renal cell carcinoma. Cancer Res. 2006;66:8542‐8549. [DOI] [PubMed] [Google Scholar]
- 135. Joly S, Compton LM, Pujol C, Kurago ZB, Guthmiller JM. Loss of human beta‐defensin 1, 2, and 3 expression in oral squamous cell carcinoma. Oral Microbiol Immunol. 2009;24:353‐360. [DOI] [PubMed] [Google Scholar]
- 136. Kesting MR, Stoeckelhuber M, Kuppek A, et al. Human β‐defensins and psoriasin/S100A7 expression in salivary glands: anti‐oncogenic molecules for potential therapeutic approaches. BioDrugs. 2012;26:33‐42. [DOI] [PubMed] [Google Scholar]
- 137. Wenghoefer M, Pantelis A, Dommisch H, et al. Nuclear hBD‐1 accumulation in malignant salivary gland tumours. BMC Cancer. 2008;8:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Pantelis A, Wenghoefer M, Haas S, et al. Down regulation and nuclear localization of human beta‐defensin‐1 in pleomorphic adenomas of salivary glands. Oral Oncol. 2009;45:526‐530. [DOI] [PubMed] [Google Scholar]
- 139. Biragyn A, Ruffini PA, Leifer CA, et al. Toll‐like receptor 4‐dependent activation of dendritic cells by beta‐defensin 2. Science. 2002;298:1025‐1029. [DOI] [PubMed] [Google Scholar]
- 140. Gorr SU, Abdolhosseini M. Antimicrobial peptides and periodontal disease. J Clin Periodontol. 2011;38(Suppl 11):126‐141. [DOI] [PubMed] [Google Scholar]
- 141. Pingel LC, Kohlgraf KG, Hansen CJ, et al. Human beta‐defensin 3 binds to hemagglutinin B (rHagB), a non‐fimbrial adhesin from Porphyromonas gingivalis, and attenuates a pro‐inflammatory cytokine response. Immunol Cell Biol. 2008;86:643‐649. [DOI] [PubMed] [Google Scholar]
- 142. Kesting MR, Loeffelbein DJ, Hasler RJ, et al. Expression profile of human beta‐defensin 3 in oral squamous cell carcinoma. Cancer Invest. 2009;27:575‐581. [DOI] [PubMed] [Google Scholar]
- 143. Li X, Duan D, Yang J, et al. The expression of human β‐defensins (hBD‐1, hBD‐2, hBD‐3, hBD‐4) in gingival epithelia. Arch Oral Biol. 2016;66:15‐21. [DOI] [PubMed] [Google Scholar]
- 144. Vankeerberghen A, Nuytten H, Dierickx K, Quirynen M, Cassiman JJ, Cuppens H. Differential induction of human beta‐defensin expression by periodontal commensals and pathogens in periodontal pocket epithelial cells. J Periodontol. 2005;76:1293‐1303. [DOI] [PubMed] [Google Scholar]
- 145. Schaefer AS, Richter GM, Nothnagel M, et al. A 3' UTR transition within DEFB1 is associated with chronic and aggressive periodontitis. Genes Immun. 2010;11:45‐54. [DOI] [PubMed] [Google Scholar]
- 146. Dommisch H, Açil Y, Dunsche A, Winter J, Jepsen S. Differential gene expression of human beta‐defensins (hBD‐1, ‐2, ‐3) in inflammatory gingival diseases. Oral Microbiol Immunol. 2005;20:186‐190. [DOI] [PubMed] [Google Scholar]
- 147. Kompuinen J, Keskin M, Yilmaz D, Gürsoy M, Gürsoy UK. Human β‐Defensins in diagnosis of head and neck cancers. Cell. 2023;12:830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Chen H, Xu C, Jin Q, Liu Z. S100 protein family in human cancer. Am J Cancer Res. 2014;4:89‐115. [PMC free article] [PubMed] [Google Scholar]
- 149. Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat Rev Cancer. 2015;15:96‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Berheloot D, Latz E. HMGB1, IL1α, IL‐33 and S100 proteins: dual‐function alarmins. Cell Mol Immunol. 2017;14:43‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Kozlyuk N, Monteith AJ, Garcia V, Damo SM, Skaar EP, Chazin WJ. S100 proteins in the innate immune response to pathogens. Methods Mol Biol. 2019;1929:275‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Cunden LS, Nolan EM. Bioinorganic explorations of Zn(II) sequestration by human S100 host‐defense proteins. Biochemistry. 2018;57:1673‐1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Salama I, Malone PS, Mihaimeed F, Jones JL. A review of the S100 proteins in cancer. Eur J Surg Oncol. 2008;34:357‐364. [DOI] [PubMed] [Google Scholar]
- 154. Lukanidin E, Sleeman JP. Building the niche: the role of the S100 proteins in metastatic growth. Semin Cancer Biol. 2012;22:216‐225. [DOI] [PubMed] [Google Scholar]
- 155. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904‐5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Langley RR, Fidler IJ. The seed and soil hypothesis revisited–the role of tumor‐stroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527‐2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Watson PH, Leygue ER, Murphy LC. Psoriasin (S100A7). Int J Biochem Cell Biol. 1998;30:567‐571. [DOI] [PubMed] [Google Scholar]
- 159. Gläser R, Harder J, Lange H, Bartels J, Christophers E, Schröder JM. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat Immunol. 2005;6:57‐64. [DOI] [PubMed] [Google Scholar]
- 160. Halawi A, Abbas O, Mahalingam M. S100 proteins and the skin: a review. J Eur Acad Dermatol Venereol. 2014;28:405‐414. [DOI] [PubMed] [Google Scholar]
- 161. Gebhardt C, Riehl A, Durchdewald M, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205:275‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Wolf R, Howard OM, Dong HF, et al. Chemotactic activity of S100A7 (Psoriasin) is mediated by the receptor for advanced glycation end products and potentiates inflammation with highly homologous but functionally distinct S100A15. J Immunol. 2008;181:1499‐1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Hattinger E, Zwicker S, Ruzicka T, Yuspa SH, Wolf R. Opposing functions of psoriasin (S100A7) and koebnerisin (S100A15) in epithelial carcinogenesis. Curr Opin Pharmacol. 2013;13:588‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kesting MR, Sudhoff H, Hasler RJ, et al. Psoriasin (S100A7) up‐regulation in oral squamous cell carcinoma and its relation to clinicopathologic features. Oral Oncol. 2009;45:731‐736. [DOI] [PubMed] [Google Scholar]
- 165. Winter J, Pantelis A, Reich R, et al. Risk estimation for a malignant transformation of oral lesions by S100A7 and doc‐1 gene expression. Cancer Invest. 2011;29:478‐484. [DOI] [PubMed] [Google Scholar]
- 166. Goyette J, Geczy CL. Inflammation‐associated S100 proteins: new mechanisms that regulate function. Amino Acids. 2011;41:821‐842. [DOI] [PubMed] [Google Scholar]
- 167. Foell D, Frosch M, Sorg C, Roth J. Phagocyte‐specific calcium‐binding S100 proteins as clinical laboratory markers of inflammation. Clin Chim Acta. 2004;344:37‐51. [DOI] [PubMed] [Google Scholar]
- 168. Kido J‐I, Hayashi N, Kataoka M, Nagata T. Calprotectin expression in human monocytes: induction by Porphyromonas gingivalis lipopolysaccharide, tumor necrosis factor‐α, and interleukin‐1β. J Periodontol. 2005;76:437‐442. [DOI] [PubMed] [Google Scholar]
- 169. Gao W, Li JZ, Chen SQ, Chu CY, Chan JY, Wong TS. Decreased brain‐expressed X‐linked 4 (BEX4) expression promotes growth of oral squamous cell carcinoma. J Exp Clin Cancer Res. 2016;35:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Reckenbeil J, Kraus D, Probstmeier R, et al. Cellular distribution and gene expression pattern of Metastasin (S100A4), Calgranulin a (S100A8), and Calgranulin B (S100A9) in Oral lesions as markers for molecular pathology. Cancer Invest. 2016;34:246‐254. [DOI] [PubMed] [Google Scholar]
- 171. Argyris PP, Slama Z, Malz C, et al. Intracellular calprotectin (S100A8/A9) controls epithelial differentiation and caspase‐mediated cleavage of EGFR in head and neck squamous cell carcinoma. Oral Oncol. 2019;95:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Probstmeier R, Kraus D, Wenghoefer M, Winter J. S100 proteins as biomarkers in risk estimations for malignant transformation in Oral lesions. Methods Mol Biol. 2019;1929:763‐771. [DOI] [PubMed] [Google Scholar]
- 173. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol. 2018;9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Eversole LR, Miyasaki KT, Christensen RE. The distribution of the antimicrobial protein, calprotectin, in normal oral keratinocytes. Arch Oral Biol. 1992;37:963‐968. [DOI] [PubMed] [Google Scholar]
- 175. Eversole LR, Miyasaki KT, Christensen RE. Keratinocyte expression of calprotectin in oral inflammatory mucosal diseases. J Oral Pathol Med. 1993;22:303‐307. [DOI] [PubMed] [Google Scholar]
- 176. Lira‐Junior R, Holmström SB, Clark R, et al. S100A12 expression is modulated during monocyte differentiation and reflects periodontitis severity. Front Immunol. 2020;11:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Fei F, Qu J, Zhang M, Li Y, Zhang S. S100A4 in cancer progression and metastasis: a systematic review. Oncotarget. 2017;8:73219‐73239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Ambartsumian N, Klingelhöfer J, Grigorian M. The multifaceted S100A4 protein in cancer and inflammation. Methods Mol Biol. 2019;1929:339‐365. [DOI] [PubMed] [Google Scholar]
- 179. Chen K, Xiong H, Huang Y, Liu C. Comparative analysis of in vitro periodontal characteristics of stem cells from apical papilla (SCAP) and periodontal ligament stem cells (PDLSCs). Arch Oral Biol. 2013;58:997‐1006. [DOI] [PubMed] [Google Scholar]
- 180. Ellerman JE, Brown CK, de Vera M, et al. Masquerader: high mobility group box‐1 and cancer. Clin Cancer Res. 2007;13:2836‐2848. [DOI] [PubMed] [Google Scholar]
- 181. Sasahira T, Kirita T, Kuniyasu H. Update of molecular pathobiology in oral cancer: a review. Int J Clin Oncol. 2014;19:431‐436. [DOI] [PubMed] [Google Scholar]
- 182. Tesarova P, Kalousova M, Zima T, Tesar V. HMGB1, S100 proteins and other RAGE ligands in cancer – markers, mediators and putative therapeutic targets. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2016;160:1‐10. [DOI] [PubMed] [Google Scholar]
- 183. Yamashiro K, Ideguchi H, Aoyagi H, et al. High mobility group box 1 expression in Oral inflammation and regeneration. Front Immunol. 2020;11:1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol. 2012;30:677‐706. [DOI] [PubMed] [Google Scholar]
- 185. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353‐364. [DOI] [PubMed] [Google Scholar]
- 186. Ferrara N. Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol. 2009;29:789‐791. [DOI] [PubMed] [Google Scholar]
- 187. Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009;19:329‐337. [DOI] [PubMed] [Google Scholar]
- 188. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168:670‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453‐458. [DOI] [PubMed] [Google Scholar]
- 190. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539‐545. [DOI] [PubMed] [Google Scholar]
- 191. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650‐1659. [DOI] [PubMed] [Google Scholar]
- 192. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer‐related inflammation. Nature. 2008;454:436‐444. [DOI] [PubMed] [Google Scholar]
- 194. Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol. 2004;4:641‐648. [DOI] [PubMed] [Google Scholar]
- 195. Karin M. Nuclear factor‐kappaB in cancer development and progression. Nature. 2006;441:431‐436. [DOI] [PubMed] [Google Scholar]
- 196. Naugler WE, Karin M. NF‐kappaB and cancer‐identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Montero PH, Patel SG. Cancer of the oral cavity. Surg Oncol Clin N Am. 2015;24:491‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Dzutsev A, Badger JH, Perez‐Chanona E, et al. Microbes and cancer. Annu Rev Immunol. 2017;35:199‐228. [DOI] [PubMed] [Google Scholar]
- 199. Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Chen X, Qi G, Qin M, et al. DNA methylation directly downregulates human cathelicidin antimicrobial peptide gene (CAMP) promoter activity. Oncotarget. 2017;8:27943‐27952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Chen X, Zou X, Qi G, et al. Roles and mechanisms of human cathelicidin LL‐37 in cancer. Cell Physiol Biochem. 2018;47:1060‐1073. [DOI] [PubMed] [Google Scholar]
- 202. Chen X, Ji S, Si J, et al. Human cathelicidin antimicrobial peptide suppresses proliferation, migration and invasion of oral carcinoma HSC‐3 cells via a novel mechanism involving caspase‐3 mediated apoptosis. Mol Med Rep. 2020;22:5243‐5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Vierthaler M, Rodrigues PC, Sundquist E, Siponen M, Salo T, Risteli M. Fluctuating role of antimicrobial peptide hCAP18/LL‐37 in oral tongue dysplasia and carcinoma. Oncol Rep. 2020;44:325‐338. [DOI] [PubMed] [Google Scholar]
- 204. Mizukawa N, Sugiyama K, Fukunaga J, et al. Defensin‐1, a peptide detected in the saliva of oral squamous cell carcinoma patients. Anticancer Res. 1998;18:4645‐4649. [PubMed] [Google Scholar]
- 205. Mizukawa N, Sugiyama K, Ueno T, Mishima K, Takagi S, Sugahara T. Defensin‐1, an antimicrobial peptide present in the saliva of patients with oral diseases. Oral Dis. 1999;5:139‐142. [DOI] [PubMed] [Google Scholar]
- 206. Mizukawa N, Sugiyama K, Kamio M, et al. Immunohistochemical staining of human alpha‐defensin‐1 (HNP‐1), in the submandibular glands of patients with oral carcinomas. Anticancer Res. 2000;20:1125‐1127. [PubMed] [Google Scholar]
- 207. Winter J, Pantelis A, Kraus D, et al. Human α‐defensin (DEFA) gene expression helps to characterise benign and malignant salivary gland tumours. BMC Cancer. 2012;12:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. McKeown ST, Lundy FT, Nelson J, et al. The cytotoxic effects of human neutrophil peptide‐1 (HNP1) and lactoferrin on oral squamous cell carcinoma (OSCC) in vitro. Oral Oncol. 2006;42:685‐690. [DOI] [PubMed] [Google Scholar]
- 209. Musrati AA, Tervahartiala T, Gürsoy M, et al. Human neutrophil peptide‐1 affects matrix metalloproteinase‐2, ‐8 and ‐9 secretions of oral squamous cell carcinoma cell lines in vitro. Arch Oral Biol. 2016;66:1‐7. [DOI] [PubMed] [Google Scholar]
- 210. Hoppe T, Kraus D, Novak N, et al. Oral pathogens change proliferation properties of oral tumor cells by affecting gene expression of human defensins. Tumour Biol. 2016;37:13789‐13798. [DOI] [PubMed] [Google Scholar]
- 211. Lundy FT, Orr DF, Gallagher JR, et al. Identification and overexpression of human neutrophil alpha‐defensins (human neutrophil peptides 1, 2 and 3) in squamous cell carcinomas of the human tongue. Oral Oncol. 2004;40:139‐144. [DOI] [PubMed] [Google Scholar]
- 212. Nishimura M, Abiko Y, Kurashige Y, et al. Effect of defensin peptides on eukaryotic cells: primary epithelial cells, fibroblasts and squamous cell carcinoma cell lines. J Dermatol Sci. 2004;36:87‐95. [DOI] [PubMed] [Google Scholar]
- 213. Han Q, Wang R, Sun C, et al. Human beta‐defensin‐1 suppresses tumor migration and invasion and is an independent predictor for survival of oral squamous cell carcinoma patients. PloS One. 2014;9:e91867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Kawsar HI, Ghosh SK, Hirsch SA, Koon HB, Weinberg A, Jin G. Expression of human beta‐defensin‐2 in intratumoral vascular endothelium and in endothelial cells induced by transforming growth factor beta. Peptides. 2010;31:195‐201. [DOI] [PubMed] [Google Scholar]
- 215. Kawsar HI, Weinberg A, Hirsch SA, et al. Overexpression of human beta‐defensin‐3 in oral dysplasia: potential role in macrophage trafficking. Oral Oncol. 2009;45:696‐702. [DOI] [PubMed] [Google Scholar]
- 216. Du Y, Yang Y, Zhang W, Yang C, Xu P. Human β‐defensin‐3 and nuclear factor‐kappa B p65 synergistically promote the cell proliferation and invasion of oral squamous cell carcinoma. Transl Oncol. 2023;27:101582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Tripathi SC, Matta A, Kaur J, et al. Nuclear S100A7 is associated with poor prognosis in head and neck cancer. PloS One. 2010;5:e11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Kaur J, Matta A, Kak I, et al. S100A7 overexpression is a predictive marker for high risk of malignant transformation in oral dysplasia. Int J Cancer. 2014;134:1379‐1388. [DOI] [PubMed] [Google Scholar]
- 219. Morgan MR, Jazayeri M, Ramsay AG, et al. Psoriasin (S100A7) associates with integrin β6 subunit and is required for αvβ6‐dependent carcinoma cell invasion. Oncogene. 2011;30:1422‐1435. [DOI] [PubMed] [Google Scholar]
- 220. Zhou G, Xie TX, Zhao M, et al. Reciprocal negative regulation between S100A7/psoriasin and beta‐catenin signaling plays an important role in tumor progression of squamous cell carcinoma of oral cavity. Oncogene. 2008;27:3527‐3538. [DOI] [PubMed] [Google Scholar]
- 221. Dey KK, Bharti R, Dey G, et al. S100A7 has an oncogenic role in oral squamous cell carcinoma by activating p38/MAPK and RAB2A signaling pathway. Cancer Gene Ther. 2016;23:382‐391. [DOI] [PubMed] [Google Scholar]
- 222. Qi Z, Li T, Kong F, et al. The characteristics and function of S100A7 induction in squamous cell carcinoma: heterogeneity, promotion of cell proliferation and suppression of differentiation. PloS One. 2015;10:e0128887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Sapkota D, Bruland O, Bøe OE, et al. Expression profile of the S100 gene family members in oral squamous cell carcinomas. J Oral Pathol Med. 2008;37:607‐615. [DOI] [PubMed] [Google Scholar]
- 224. Argyris PP, Slama ZM, Ross KF, Khammanivong A, Herzberg MC. Calprotectin and the initiation and progression of head and neck cancer. J Dent Res. 2018;97:674‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Argyris PP, Saavedra F, Malz C, et al. Intracellular calprotectin (S100A8/A9) facilitates DNA damage responses and promotes apoptosis in head and neck squamous cell carcinoma. Oral Oncol. 2023;137:106304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Holmström SB, Lira‐Junior R, Zwicker S, et al. MMP‐12 and S100s in saliva reflect different aspects of periodontal inflammation. Cytokine. 2019;113:155‐161. [DOI] [PubMed] [Google Scholar]
- 227. Zheng Y, Hou J, Peng L, et al. The pro‐apoptotic and pro‐inflammatory effects of calprotectin on human periodontal ligament cells. PloS One. 2014;9:e110421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Nishikawa Y, Kajiura Y, Lew JH, Kido JI, Nagata T, Naruishi K. Calprotectin induces IL‐6 and MCP‐1 production via toll‐like receptor 4 signaling in human gingival fibroblasts. J Cell Physiol. 2017;232:1862‐1871. [DOI] [PubMed] [Google Scholar]
- 229. Moriyama‐Kita M, Endo Y, Yonemura Y, et al. Correlation of S100A4 expression with invasion and metastasis in oral squamous cell carcinoma. Oral Oncol. 2004;40:496‐500. [DOI] [PubMed] [Google Scholar]
- 230. Natarajan J, Hunter K, Mutalik VS, Radhakrishnan R. Overexpression of S100A4 as a biomarker of metastasis and recurrence in oral squamous cell carcinoma. J Appl Oral Sci. 2014;22:426‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Wetting HL, Hadler‐Olsen E, Magnussen S, et al. S100A4 expression in xenograft tumors of human carcinoma cell lines is induced by the tumor microenvironment. Am J Pathol. 2011;178:2389‐2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Moriyama‐Kita M, Endo Y, Yonemura Y, et al. S100A4 regulates E‐cadherin expression in oral squamous cell carcinoma. Cancer Lett. 2005;230:211‐218. [DOI] [PubMed] [Google Scholar]
- 233. Hu Y, Han Y, He M, Zhang Y, Zou X. S100 proteins in head and neck squamous cell carcinoma (review). Oncol Lett. 2023;26:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Supporting data and materials are available on request to the corresponding author.