

Abstract
Epithelioid inflammatory myofibroblastic sarcoma (EIMS) is a rare and aggressive subtype of inflammatory myofibroblastic tumor. The disease is associated with rearrangements of the anaplastic lymphoma kinase (ALK). In this paper, we present the clinicopathological features and treatment of a female patient diagnosed with EIMS. In 2019, an 18-year-old female patient was admitted to the hospital with abdominal pain. Radiological examinations confirmed a large pelvic mass which was subsequently resected. After re-evaluation of the initial histologic diagnosis, the final diagnosis of EIMS was established. Consequently, due to the lack of response to chemotherapy and deteriorating clinical condition, she began the therapy with ALK inhibitors. In total, the patient was treated with crizotinib, alectinib, and lorlatinib. As a result, after over 4 years since the initial diagnosis, she is still alive with significantly improved clinical condition and quality of life. This paper demonstrates the clinical benefits of sequential therapy of ALK inhibitors and an exceptionally long response to lorlatinib, a third-generation tyrosine kinase inhibitor.
Keywords: ALK inhibitors, ALK rearrangement, epithelioid inflammatory myofibroblastic sarcoma, lorlatinib
Introduction
Inflammatory myofibroblastic tumor (IMT) is a neoplasm of mesenchymal origin, which typically occurs in children and adolescents. 1 Epithelioid inflammatory myofibroblastic sarcoma (EIMS) is a rare and aggressive variant of IMT. 2 It is associated with rearrangement of the anaplastic lymphoma kinase (ALK), which often shows nuclear membrane or perinuclear staining.2,3 Studies have demonstrated that patients with EIMS could be treated with tyrosine kinase inhibitors (TKIs) targeting ALK. Crizotinib, a first-generation ALK-TKI, has been approved for the treatment of ALK-positive neoplasms, such as non-small-cell lung cancer (NSCLC), anaplastic large-cell lymphoma (ALCL), and IMT. Nevertheless, the use of three generations of ALK-TKIs has been rarely described in the treatment of EIMS. In this paper, we present a case of an ALK-positive EIMS patient with a complex diagnostic pathway, who was treated with crizotinib, alectinib, and lorlatinib. The reporting of this case report conforms to the CARE guidelines (Supplemental Materials).
Case report
In July 2019, a previously healthy 18-year-old female was admitted to the emergency department due to abdominal pain. Ultrasonographic examination showed an 11 × 9 cm lesion in the lower abdomen, which was displacing the uterus. The patient was consulted by a gynecologist and then referred to the surgical department. Laboratory examination revealed microcytic anemia, together with an elevated concentration of C-reactive protein (CRP, 228.93 mg/L (normal value <5) and thrombocytosis (669,000/μL (normal range 1.5–4.0 × 105)). Abdominal and pelvic computed tomography (CT) confirmed a heterogeneous mass measuring 11.5 × 9.8 × 7.8 cm with solid and cystic components, located above the urinary bladder and anterior to the uterus. The lesion extended to the inner surface of the anterior wall of the pelvis. On the right side, it was adjacent to the external iliac vessels and the appendix, while on the left side to the external iliac vessels and the sigmoid loop. The lesion adhered laterally to the distal segments of the ureters. A loop of the small intestine was “embedded” in the pathological mass (Figure 1). Consequently, the tumor was resected, together with approximately 50 cm of small intestine. It was irregular, ulcerative, and infiltrated the serosa. The tumor was composed of a 5.5 × 2.5 cm fragment located inside the intestinal lumen and a 12.5 × 6.5 × 9 cm subserosal fragment. It was fused to the omentum and had a ruptured capsule. The tumor was solid, gray-brown with a gelatinous consistency. A second 2 cm brown tumor was found in the serosa 7 cm from one of the incisions. Histopathological examination of the main mass and smaller nodule confirmed a non-epithelial malignant neoplasm, which was composed of spindle-shaped epithelioid cells that infiltrated the entire intestinal wall (from the serosa to the mucosa) and omentum. Tumor cells had vesicular nuclei, prominent large nucleoli, and amphophilic cytoplasm. Numerous neutrophils and eosinophils in myxoid tumor stroma were observed (Figure 2(a)). No tumor necrosis was found. Mitotic activity was 7/10 HPF. Numerous neutrophils and eosinophils in the tumor stroma were observed. According to the immunohistochemical analysis, neoplastic cells were positive for CD68, CD31, and CD4 antigens. Immunostaining for leukocyte common antigen was nonspecific (+/−). The expression of pancytokeratin (panCK), S100, desmin, SMA, CD3, CD20, CD30, CD34, EMA, CD117, MelanA, CD56, CD99, EpCAM, MPO, CD14, CD19, TdT, CD1a, CD23, CD43, CD7, CD79a, and PAX5 were negative. The proliferation index Ki67 ranged from 20% to 30%. Consequently, a diagnosis of histiocytic sarcoma was established. In August 2019, an interdisciplinary team referred the patient to the National Institute of Oncology, where the diagnosis was evaluated and confirmed. During September and October, CT revealed tumors measuring 6.4 × 6.3 × 4.6 and 9.6 × 5.2 × 4.1 cm adjacent to the uterus and peritoneal implants measuring 2.5 × 1.2 and 1.6 × 1.8 cm. Clinically, ascites and pathological abdominal mass were found, while laboratory examinations confirmed rising CRP levels from 190.69 to 476.27 mg/L, together with leukocytosis (11,900/μL) and thrombocytosis (612,000/μL). During histopathological verification of the primary specimen, immunohistochemical staining with the ALK antibody revealed a nuclear membrane reaction in tumor cells (Figure 2(b)). ALK rearrangement was confirmed by fluorescence in situ hybridization. Consequently, the final diagnosis of EIMS was established. During hospitalization, a bone marrow biopsy was performed and did not show neoplastic infiltration. Whole body CT confirmed innumerable neoplastic implants in the abdominal cavity, which were surrounding the liver and spleen. Furthermore, they were located on the visceral surface of both domes of the diaphragm, in the omentum, and in the paracolic recesses. The right lower region of the abdominal cavity and small pelvis were almost completely filled with neoplastic implants. The largest masses (up to 28 mm) were located in the subdiaphragmatic region. These implants distorted the contour of the aorta at the bifurcation, iliac vessels, urinary bladder, and the uterus. A 2 mm sclerotic lesion was detected in the vertebral body of C3, suspected of representing metastatic disease. Overall, CT confirmed intra-abdominal sarcoma dissemination. Subsequently, the patient was transferred to the Department of Clinical Oncology, Chemotherapy and Cancer Immunotherapy of the Pomeranian Medical University in Szczecin. Due to the progression of the disease, the patient was qualified for chemotherapy according to the AD protocol (doxorubicin 15 mg/m² (days 1–4 i.v.) + dacarbazine 187.5 mg/m² (days 1–4 i.v.)). 4 After the first course of chemotherapy, grade 3 hematological toxicities were observed in addition to a persistently high concentration of CRP (399 mg/L) and ferritin (776 μg/L (normal range 10–200 µg/L)). The patient received a second course of AD and subsequently, her general condition further deteriorated and she required supportive treatment. Due to high concentrations of inflammatory parameters and procalcitonin (9.99 ng/mL (normal range <0.5 ng/mL), the patient received empirical antibiotic therapy (tazobactam + piperacillin). Repeated collection of blood cultures did not show bacterial growth. Therefore, we could exclude sepsis and confirm that the escalation of inflammatory parameters is caused by the progression of neoplasm not responding to the oncological treatment. Due to the poor condition (Eastern Cooperative Oncology Group (ECOG) performance status 3), the patient was not qualified for another line of chemotherapy. In December 2019, clinical and radiological progression of the disease was confirmed. After analyzing the available scientific literature and approval of the university ethics committee, the patient received crizotinib (Xalkori; Pfizer, New York, USA) as a rescue therapy (2 × 250 mg). Due to poor tolerance, 4 weeks later, the drug was changed to alectinib (Alecensa; Roche, Basel, Switzerland), a second-generation ALK inhibitor (2 × 600 mg). CT examination performed in February 2020 confirmed partial regression throughout the abdominal cavity, and complete regression of pathological foci in the lower half of the abdominal cavity and pelvis. In March 2020, laboratory examinations demonstrated normal values of leukocytes (6.57 × 103/μL), platelets (260 × 103/μL), and CRP (<0.3 mg/L). In August 2020, CT showed the progression of the disease in the abdominal cavity with an infiltration of the small intestine. Two larger implants measuring 22 × 15 mm and 6 mm were found in the abdomen, while a 30 × 35 mm implant was revealed in the pelvis. In September 2020, the treatment with alectinib was completed. However, in October 2020, the patient started experiencing clinical symptoms of accelerated disease progression, which included gastrointestinal obstruction and effusion in the abdominal cavity. Deteriorating laboratory parameters reflected clinical progression and the patient’s condition decreased significantly (ECOG 3), which led to an attempt to implement lorlatinib (Lorviqua; Pfizer) 1 × 100 mg after approval of the university ethics committee. The tolerance was good and after 4 weeks of treatment, a reduction in abdominal circumference and abdominal pain was observed. Furthermore, the clinical condition stage improved to ECOG 2. Consequently, treatment with lorlatinib was continued at the appropriate dose. CT scan from February 2021 confirmed a partial regression of abdominal and pelvic implants. In addition, diagnostic parameters were normalized. The patient continued to receive lorlatinib with monthly monitoring of blood and biochemical parameters, as well as a lipid profile. Throughout the treatment, we observed elevated concentrations of triglycerides and total cholesterol, which was resolved using fibrates and statins. In the follow-up CT examinations performed in 2021, an almost complete regression of peritoneal implants was observed (Figure 3). Despite the episodes of elevated blood pressure and persistent peripheral edema (fluid retention) which were managed with diuretics, the patient tolerated the treatment. In June 2023, CT examinations showed continuous stabilization of the disease. In the following months, once again elevated levels of triglycerides were detected, which were stabilized using lipid-lowering medications. The most recent CT performed in May 2024 showed no signs of progression (Figure 4). Up to this date, we observe a good tolerance to lorlatinib. The application of ALK inhibitors significantly improved the patient’s quality of life, which allowed her to be socially active and continue undergraduate education.
Figure 1.

(a and b) CT of the patient showing primary pelvic lesion (white arrows).
CT, computed tomography.
Figure 2.
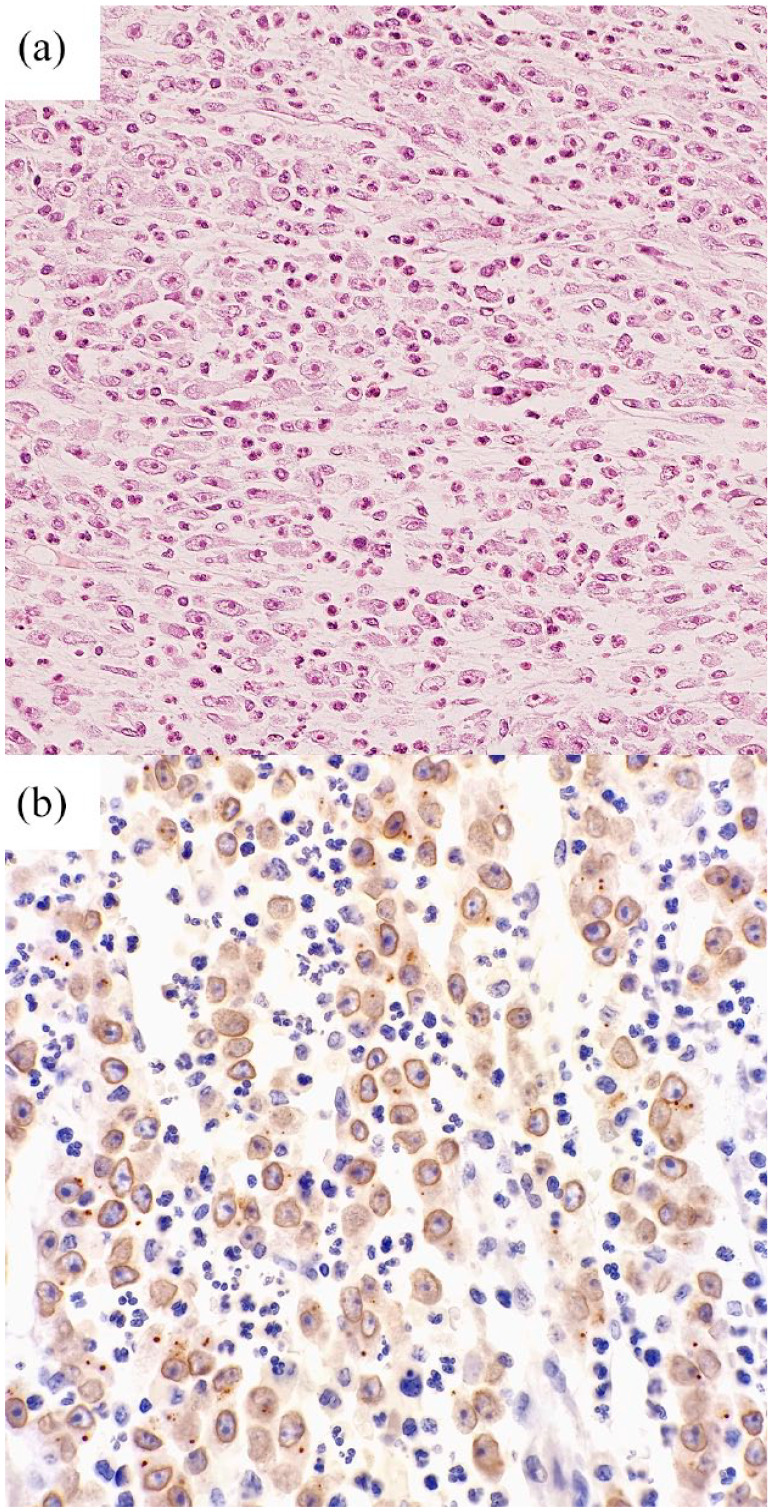
Histopathological specimen of EIMS. (a) The tumor was composed of epithelioid cells and prominent inflammatory infiltrate by neutrophils. (b) Immunohistochemistry for ALK showed a nuclear membrane staining pattern.
ALK, anaplastic lymphoma kinase; EIMS, epithelioid inflammatory myofibroblastic sarcoma.
Figure 3.

(a–f) CT of the patient throughout treatment with crizotinib, alectinib, and lorlatinib demonstrated decreasing diameters of abdominal and pelvic lesions (white arrows) and accumulated fluid (black arrows).
CT, computed tomography.
Figure 4.
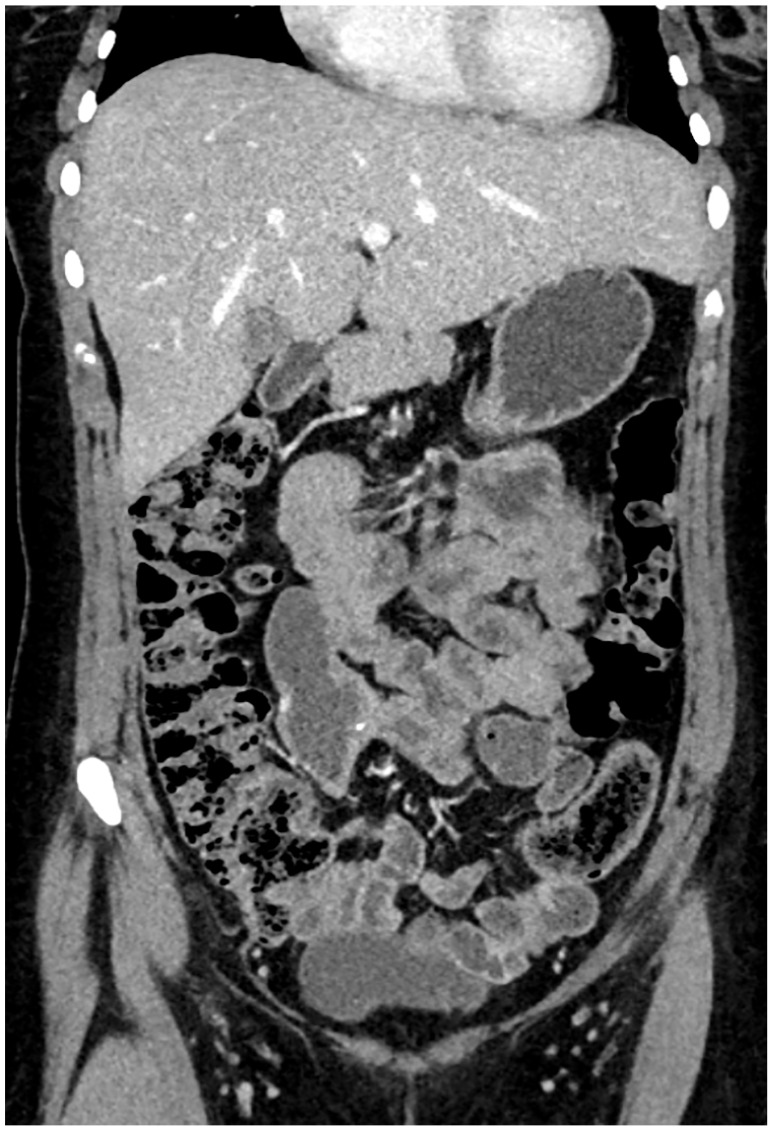
The most recent CT scan showed an almost complete response during treatment with lorlatinib.
CT, computed tomography.
Discussion
EIMS is a rare and relatively novel entity recognized in 2011. 2 As a result, case reports represent the majority of studies describing EIMS in the literature. These papers demonstrate that EIMS typically develops in the abdominal or pelvic regions. 5 Nevertheless, the tumor has been reported to occur in other sites as well, such as the thoracic region, 6 maxilla, 7 and central nervous system, 8 among others. EIMS is typically associated with the rearrangement of ALK, a tyrosine kinase that stimulates several signaling pathways, including MAPK, PI3K/AKT, JAK/STAT, and PLC-γ. Similarly to other tyrosine kinases (e.g., EGFR), its abnormal activation may promote survival and inhibit apoptosis, which is the hallmark of tumorigenesis. Pathological activation of ALK can occur through point mutations, as in neuroblastoma, or due to a fusion with a partner gene, like in NSCLC or ALCL. 9 Some rearrangements are considered typical for particular diseases. For instance, EML4-ALK is classically associated with NSCLC, while NPM-ALK fusion suggests ALCL. In the case of EIMS, RANBP2-ALK is considered as a typical rearrangement.10,11 Nevertheless, other fusion partners have been found in patients with EIMS as well, such as EML4-ALK 12 or PRRC2B-ALK. 13 Recent studies showed that ALK can form fusions with multiple genes. It is considered that over 90 fusion partners exist just in NSCLC. 14
In the presented case, the patient rapidly underwent surgical resection of the tumor. Other authors also showed surgical resection as the first choice of treatment. 3 However, patients with EIMS may present with already unresectable disease, as demonstrated by Liu et al. In this study, all seven patients with EIMS could not be treated surgically due to large lesions. 15 In the described case, surgery was the initial therapeutic modality, but follow-up CT revealed disseminated disease.
EIMS is an aggressive disease and rapid postoperative recurrence may occur. 2 Importantly, systemic treatment with classic cytostatic agents might not provide clinical benefits. Kurihara-Hosokawa et al. reported an EIMS patient, who underwent two surgical resections, but large tumors were observed 1 month after the last surgery. Doxorubicin treatment did not inhibit tumor growth. 16 A similar lack of response to doxorubicin was also described in a paper by Kimbara et al. 11 where new tumors and intraperitoneal implants were promptly detected postoperatively and did not shrink with initial treatment. These clinical conditions (recurrences, peritoneal dissemination, severe symptoms, lack of response to chemotherapy) indicated a poor prognosis. In the abovementioned reports, crizotinib remarkably relieved symptoms and induced tumor shrinkage. However, as demonstrated by Jiang et al., 12 degradation of neoplastic cells induced by crizotinib may lead to tumor lysis syndrome. In our report, treatment with crizotinib was poorly tolerated and a positive response was observed after substitution to alectinib. Importantly, ALK inhibitors are associated with acquired resistance, which requires another agent targeting ALK or a different treatment strategy. In this report, post-alectinib progression resulted in an introduction of lorlatinib, a third-generation ALK-TKI. Acquired resistance to ALK-TKIs develops through several mechanisms, including resistance mutations or bypass signaling, among others. 17 In the group of lung cancer patients treated with ALK inhibitors, resistance mutations seem to be the most common mechanism driving the resistance. 18 Numerous resistance ALK mutations have been identified so far, including L1196M, G1269A, and F1174V, among many others. These mutations drive resistance to different generations of ALK inhibitors and thus, different ALK inhibitors could restore treatment response. 17 Searching for ALK mutations and alterations of other genes at the time of progression would allow for a targeted substitution therapy. Interestingly, another strategy to increase the efficacy of ALK inhibitors would be their combination with other agents. For instance, Fordham et al. 19 demonstrated that a combination of crizotinib or ceritinib (a second-generation inhibitor) with brentuximab-vedotin (anti-CD30) provided favorable survival in CD30-positive EIMS xenografts.
Our patient has been treated with three different ALK inhibitors, crizotinib, alectinib, and lorlatinib (Figure 5). Nevertheless, limited information regarding the sequential use of ALK-TKIs has been reported. Wang and colleagues described an EIMS patient that was treated with four ALK inhibitors. Initially, the patient underwent a cytoreductive surgery, which was followed by a quick relapse. After the detection of the PRRC2B-ALK fusion, it was decided to introduce crizotinib, which resulted in a progression-free survival (PFS) of 5 months. After the detection of the secondary ALK mutation R1192P, crizotinib was replaced with alectinib, which led to a partial response, and a PFS of 5.5 months. At the time of progression, ALK L1196M mutation was identified. Therefore, a third-line treatment with ceritinib was started. The PFS was 6.6 months and a partial response was achieved. Subsequently, the patient was given lorlatinib and achieved the PFS of 5 months. 13 This case demonstrates the clinical benefits of testing for resistance mutations and opportunities for targeted next-line treatment agents. To the best of our knowledge, only a few studies described the use of lorlatinib in patients with inflammatory myofibroblastic sarcomas. 20 Importantly, our study demonstrates an exceptionally long response throughout 45 months of lorlatinib treatment. Furthermore, since the initial diagnosis of histiocytic sarcoma, the patient survived for approximately 59 months, which is extraordinary in the case of this aggressive malignancy. Nonetheless, long survival of EIMS patients treated with ALK inhibitors has been demonstrated in other reports as well. 21
Figure 5.

Timeline of introduced therapies in the described patient.
Table 1 summarizes clinical data from case report studies published in the last 5 years (2019–2024) that describe the experience of ALK inhibitors in EIMS patients.8,10,13,22–27 A thorough literature search in the PubMed database was performed using the following search terms: “EIMS and ALK inhibitor,” “EIMS ALK,” “Epithelioid inflammatory myofibroblastic sarcoma ALK,” and their combinations. Pediatric cases and studies with little clinical data were not included in the table. In the last 5 years, researchers demonstrated the importance of sequential use of ALK therapeutics. Increasing access to ALK inhibitors, together with greater knowledge about the management of adverse events and resistance mechanisms lead to enhanced survival of patients suffering from this rare and aggressive malignancy.
Table 1.
Summary of clinical data from case reports published between 2019 and 2024 describing the use of ALK inhibitors in patients with EIMS.
| First author | Year of publication | Age at diagnosis | Involved region/organ | Treatment methods | Survival/follow-up information | Reference |
|---|---|---|---|---|---|---|
| Xiaojing Xu | 2019 | 28 | Abdomen and pelvis | - Crizotinib - Brigatinib |
3 years (alive) | Xu et al. 25 |
| Shijie Zhang | 2019 | 46 | Abdomen | - Surgical resection - Crizotinib - Anlotinib |
16 months (dead) | Zhang and Wang 26 |
| Shefali Chopra | 2021 | 72 | Central nervous system | - Surgical resection - Alectinib |
4 months (alive) | Chopra et al. 8 |
| Zhan Wang | 2022 | 42 | Pelvis, peritoneal metastasis | - Surgical resection - Crizotinib - Alectinib - Ceritinib - Lorlatinib |
3 years (alive) | Wang et al. 13 |
| Priyanka Singh | 2022 | 25 | Lungs | - Crizotinib - Surgical resection |
3 years (alive) | Singh et al. 24 |
| Mengmeng Li | 2023 | 31 | Abdomen | - Surgical resection - Ensartinib |
1 year (alive) | Li et al. 10 |
| XiaoQing Li | 2023 | Middle-aged | Gastric | - Surgical resection - Chemotherapy - Anlotinib - Ensartinib |
2 year (alive) | Li et al. 23 |
| Soheila Aminimoghaddam | 2023 | 20 | Tight ovary, uterus, and peritoneum | - Surgical resection - Crizotinib - Chemotherapy |
8 months (dead) |
Aminimoghaddam and Pourali 22 |
| Yang Zheng | 2024 | 34 | Mediastinum | - Lorlatinib, - Surgical resection |
2 years (alive) | Zheng et al. 27 |
| Our patient | 2024 | 18 | Abdomen and pelvis | - Surgical resection - Chemotherapy - Crizotinib - Alectinib - Lorlatinib |
59 months since the initial diagnosis (alive) | – |
ALK, anaplastic lymphoma kinase; EIMS, epithelioid inflammatory myofibroblastic sarcoma.
Conclusion
To conclude, we report the clinicopathological features and treatment course of a patient with rare and aggressive EIMS. We strongly believe that our report significantly contributes to the literature by demonstrating the clinical benefits of sequential ALK treatment and exceptionally long response to lorlatinib. The use of ALK inhibitors significantly improved patient’s quality of life.
Supplemental Material
Supplemental material, sj-pdf-1-tam-10.1177_17588359241298489 for Epithelioid inflammatory myofibroblastic sarcoma with exceptionally long response to lorlatinib—a case report by Rafał Becht, Kajetan Kiełbowski, Justyna Żychowska, Wojciech Poncyljusz, Aleksandra Łanocha, Katarzyna Kozak, Ewa Gabrysz-Trybek and Paweł Domagała in Therapeutic Advances in Medical Oncology
Acknowledgments
None.
Footnotes
ORCID iDs: Rafał Becht  https://orcid.org/0000-0003-4820-0629
https://orcid.org/0000-0003-4820-0629
Justyna Żychowska
https://orcid.org/0009-0007-3477-3430
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Rafał Becht, Rafał Becht Department of Clinical Oncology, Chemotherapy and Cancer Immunotherapy, Pomeranian Medical University in Szczecin, Unii Lubelskiej 1, Szczecin 71-252, Poland.
Kajetan Kiełbowski, Department of Clinical Oncology, Chemotherapy and Cancer Immunotherapy, Pomeranian Medical University in Szczecin, Szczecin, Poland.
Justyna Żychowska, Department of Clinical Oncology, Chemotherapy and Cancer Immunotherapy, Pomeranian Medical University in Szczecin, Szczecin, Poland.
Wojciech Poncyljusz, Department of Diagnostic Imaging and Interventional Radiology, Pomeranian Medical University in Szczecin, Szczecin, Poland.
Aleksandra Łanocha, Department of Hematology and Transplantology, Pomeranian Medical University in Szczecin, Szczecin, Poland.
Katarzyna Kozak, Department of Soft Tissue/Bone Sarcoma and Melanoma, The Maria Sklodowska-Curie National Research Institute of Oncology, Warsaw, Poland.
Ewa Gabrysz-Trybek, Department of Diagnostic Imaging and Interventional Radiology, Pomeranian Medical University in Szczecin, Szczecin, Poland.
Paweł Domagała, Department of Pathology, Pomeranian Medical University in Szczecin, Szczecin, Poland.
Declarations
Ethics approval and consent to participate: Not applicable.
Consent for publication: Written consent was obtained.
Author contributions: Rafał Becht: Conceptualization; Investigation; Supervision; Writing – original draft; Writing – review & editing.
Kajetan Kiełbowski: Writing – original draft; Writing – review & editing.
Justyna Żychowska: Writing – original draft; Writing – review & editing.
Wojciech Poncyljusz: Investigation; Writing – original draft; Writing – review & editing.
Aleksandra Łanocha: Investigation; Writing – original draft; Writing – review & editing.
Katarzyna Kozak: Investigation; Writing – original draft; Writing – review & editing.
Ewa Gabrysz-Trybek: Investigation; Writing – original draft; Writing – review & editing.
Paweł Domagała: Investigation; Writing – original draft; Writing – review & editing.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Publication fee has been funded by Pomeranian Medical University in Szczecin, Poland.
The authors declare that there is no conflict of interest.
Availability of data and materials: Not applicable.
References
- 1. Du X, Gao Y, Zhao H, et al. Clinicopathological analysis of epithelioid inflammatory myofibroblastic sarcoma. Oncol Lett 2018; 15(6): 9317–9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mariño-Enríquez A, Wang WL, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol 2011; 35(1): 135–144. [DOI] [PubMed] [Google Scholar]
- 3. Yu L, Liu J, Lao IW, et al. Epithelioid inflammatory myofibroblastic sarcoma: a clinicopathological, immunohistochemical and molecular cytogenetic analysis of five additional cases and review of the literature. Diagn Pathol 2016; 11(1): 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adkins KE, Solimando DA, Waddell JA. Doxorubicin and dacarbazine (AD) regimen for soft tissue sarcomas. Hosp Pharm 2015; 50(3): 194–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dou W, Guan Y, Liu T, et al. Epithelioid inflammatory myofibroblastic sarcoma: a case report and brief literature review. Front Oncol 2023; 13: 1212529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pan T, Sun X, Wu X, et al. Mediastinal epithelioid inflammatory myofibroblastic sarcoma with the EML4-ALK fusion: a case report and literature review. Respirol Case Rep 2023; 12(1): e01267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nayyar V, Bhutia O, Kakkar A, et al. Epithelioid inflammatory myofibroblastic sarcoma arising in the maxilla: first reported case in the jaws. Oral Surg Oral Med Oral Pathol Oral Radiol 2023; 136(1): e15–e19. [DOI] [PubMed] [Google Scholar]
- 8. Chopra S, Maloney N, Wang WL. Epithelioid inflammatory myofibroblastic sarcoma with VCL-ALK fusion of central nervous system: case report and brief review of the literature. Brain Tumor Pathol 2022; 39(1): 35–42. [DOI] [PubMed] [Google Scholar]
- 9. Hallberg B, Palmer RH. The role of the ALK receptor in cancer biology. Ann Oncol 2016; 27(Suppl. 3): iii4–iii15. [DOI] [PubMed] [Google Scholar]
- 10. Li M, Xing R, Huang J, et al. Case report: epithelioid inflammatory myofibroblastic sarcoma treated with an ALK TKI ensartinib. Front Oncol 2023; 13: 1084456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kimbara S, Takeda K, Fukushima H, et al. A case report of epithelioid inflammatory myofibroblastic sarcoma with RANBP2-ALK fusion gene treated with the ALK inhibitor, crizotinib. Jpn J Clin Oncol 2014; 44(9): 868–871. [DOI] [PubMed] [Google Scholar]
- 12. Jiang Q, Tong HX, Hou YY, et al. Identification of EML4-ALK as an alternative fusion gene in epithelioid inflammatory myofibroblastic sarcoma. Orphanet J Rare Dis 2017; 12(1): 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Z, Geng Y, Yuan LY, et al. Durable clinical response to ALK tyrosine kinase inhibitors in epithelioid inflammatory myofibroblastic sarcoma harboring PRRC2B-ALK rearrangement: a case report. Front Oncol 2022; 12: 761558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiang Y, Zhang S, Fang X, et al. Therapeutic advances of rare ALK fusions in non-small cell lung cancer. Curr Oncol 2022; 29(10): 7816–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu X, Gong C, Zhang J, et al. Clinicopathological analysis and treatment of adult patients with inflammatory myofibroblastic tumor: a 15-year single-center study. Cancer Res Treat 2023; 55(3): 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kurihara-Hosokawa K, Kawasaki I, Tamai A, et al. Epithelioid inflammatory myofibroblastic sarcoma responsive to surgery and an ALK inhibitor in a patient with panhypopituitarism. Intern Med 2014; 53(19): 2211–2214. [DOI] [PubMed] [Google Scholar]
- 17. Kielbowski K, Zychowska J, Becht R. Anaplastic lymphoma kinase inhibitors—a review of anticancer properties, clinical efficacy, and resistance mechanisms. Front Pharmacol 2023; 14: 1285374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu L, Zou Z, Li Y, et al. Progression patterns, resistant mechanisms and subsequent therapy for ALK-positive NSCLC in the era of second-generation ALK-TKIs. J Transl Med 2024; 22(1): 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fordham AM, Xie J, Gifford AJ, et al. CD30 and ALK combination therapy has high therapeutic potency in RANBP2-ALK-rearranged epithelioid inflammatory myofibroblastic sarcoma. Br J Cancer 2020; 123(7): 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yuan C, Ma MJ, Parker JV, et al. Metastatic anaplastic lymphoma kinase-1 (ALK-1)-rearranged inflammatory myofibroblastic sarcoma to the brain with leptomeningeal involvement: favorable response to serial ALK inhibitors: a case report. Am J Case Rep 2017; 18: 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trahair T, Gifford AJ, Fordham A, et al. Crizotinib and surgery for long-term disease control in children and adolescents with ALK-positive inflammatory myofibroblastic tumors. JCO Precis Oncol 2019; 3: PO.18.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aminimoghaddam S, Pourali R. Epithelioid inflammatory myofibroblastic sarcoma with poor response to crizotinib: a case report. Clin Med Insights Case Rep 2023; 16: 11795476231163954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li X, Zheng J, Li X, et al. Case report: ensartinib for gastric epithelioid inflammatory myofibrosarcoma with STRN-ALK fusion. Front Oncol 2023; 13: 1252221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Singh P, Nambirajan A, Gaur MK, et al. Primary pulmonary epithelioid inflammatory myofibroblastic sarcoma: a rare entity and a literature review. J Pathol Transl Med 2022; 56(4): 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu X, Li H, Peng K, et al. ALK-G1269A mutation in epithelioid inflammatory myofibroblastic sarcoma after progression on crizotinib: a case report. Oncol Lett 2019; 17(2): 2370–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang S, Wang Z. A case report on epithelioid inflammatory myofibroblastic sarcoma in the abdominal cavity. Int J Clin Exp Pathol 2019; 12(10): 3934–3939. [PMC free article] [PubMed] [Google Scholar]
- 27. Zheng Y, Zhao F, Ren Y, et al. A case report: pathological complete response to neoadjuvant lorlatinib for epithelioid inflammatory myofibroblastic sarcoma with EML4-ALK rearrangement. Front Pharmacol 2024; 15: 1401428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-pdf-1-tam-10.1177_17588359241298489 for Epithelioid inflammatory myofibroblastic sarcoma with exceptionally long response to lorlatinib—a case report by Rafał Becht, Kajetan Kiełbowski, Justyna Żychowska, Wojciech Poncyljusz, Aleksandra Łanocha, Katarzyna Kozak, Ewa Gabrysz-Trybek and Paweł Domagała in Therapeutic Advances in Medical Oncology