

Abstract
Proteolysis‐targeting chimeras (PROTACs) have emerged as a promising technology for inducing targeted protein degradation by leveraging the intrinsic ubiquitin‐proteasome system (UPS). While the potential druggability of PROTACs toward undruggable proteins has accelerated their rapid development and the wide‐range of applications across diverse disease contexts, off‐tissue effects and side‐effects of PROTACs have recently received attentions to improve their efficacy. To address these issues, spatial or temporal target protein degradation by PROTACs has been spotlighted. In this review, we explore chemical strategies for modulating protein degradation in a cell type‐specific (spatio−) and time‐specific (temporal−) manner, thereby offering insights for expanding PROTAC applications to overcome the current limitations of target protein degradation strategy.
Keywords: PROTAC, Spatial PROTAC, Spatiotemporal PROTAC, Targeted Protein Degradation (TPD), Temporal PROTAC
To overcome the limitations of conventional PROTACs, conditional PROTACs have been developed. This review highlights recent strategies for modulating targeted protein degradation in spatio‐, temporal‐, or spatiotemporal way to enhance the efficacy, specificity, and delivery of PROTACs. These advancements broaden their applicability, improve therapeutic outcomes, and pave the way for more effective treatments across a wide range of diseases.

1. Introduction
Proteostasis, the maintenance of protein homeostasis, is a vital process for preserving the integrity of the cellular proteome through protein regulation [1] Given the essential role of the proteome in cell function and survival, a number of pathways are involved in maintaining proteostasis.[ 2 , 3 , 4 , 5 , 6 , 7 ] For instance, the unfolded protein response (UPR) is activated to lower the level of unfolded proteins under cellular stress conditions to maintain proteostasis in the endoplasmic reticulum by facilitating chaperonin proteins.[ 2 , 3 ] Furthermore, cells control the lifetime of proteins through the ubiquitin‐proteasome pathway and the autophagy‐lysosomal pathway.[ 4 , 5 , 6 , 7 ] Disruption of proteostasis results in the accumulation of aberrant proteins, contributing to various diseases such as cancer,[ 8 , 9 , 10 , 11 ] neurodegenerative diseases,[ 12 , 13 ] immune‐related diseases [14] and infectious diseases.[ 15 , 16 , 17 ]
Recently, researchers in the field of drug discovery have turned their attention to protein degradation to eliminate disease‐related proteins, primarily utilizing the ubiquitin‐proteasome system (UPS). [18] In the cellular UPS, E3 ubiquitin ligases facilitate the ubiquitination of target proteins, leading to their subsequent proteasomal degradation. To achieve selective target protein degradation by harnessing the endogenous UPS, researchers have developed a novel chemical tool known as proteolysis‐targeting chimeras (PROTACs). [19] PROTACs are composed of heterobifunctional molecules containing two moieties responsible for recruiting E3 ubiquitin ligase and the protein of interest (POI), respectively, conjugated with diverse chemical linkers. [20] PROTACs promote the formation of a ternary complex involving E3 ubiquitin ligase, PROTAC, and POI, thereby triggering the ubiquitination of POI followed by UPS‐mediated degradation.
In comparison to conventional drug discovery strategies, such as small molecule inhibitors or monoclonal antibodies (mAbs), [21] PROTACs offer an exquisite feature from a perspective of druggability. In conventional drug discovery, drug molecules are required to bind active sites and control the function of the POI, [22] which make proteins lacking these sites as “undruggable proteins”. [23] For this reason, the number of druggable proteins is restricted to a small part of the entire proteome and most proteins still remain undruggble. However, PROTAC can effectively target any type of protein regardless of presence of active sites. By conjugation of E3 ligase binders and POI binders, induced the proximity of E3 ligase and POI offer the POI degradation for disease treatment. Considering the advantages of PROTACs, a number of PROTACs have been reported using well‐known E3 ligase ligands (von Hippel‐Lindau (VHL) ligands and Cereblon (CRBN) ligands).[ 24 , 25 ] Recently, PROTACs have been developed for targeting disease‐related proteins, including STAT3, [26] tau, [27] androgen receptor (AR), [28] and estrogen receptor (ER) [29] . Currently, over 20 PROTACs were in clinical trials by the end of 2022.[ 30 , 31 , 32 ]
Despite recent great interest in PROTAC development, several limitations of PROTACs have been reported such as poor pharmacokinetic properties resulting from not satisfying Lipinski's “rule of five” (RO5). [33] Another major limitation of PROTAC is the undesired on‐target off‐tumor protein degradation, resulting in off‐target effects and unexpected toxicity.[ 34 , 35 ] In response to these challenges, various strategies have been developed to overcome these limitations by controlling spatial and temporal target protein degradation (Figure 1). Herein, we review recent strategies for conditional protein degradation and provide a future perspective for overcoming current limitations of PROTACs.
Figure 1.

Conditional PROTACs. Overview of recent strategies for modulating targeted protein degradation. Conditional PROTACs are categorized into three parts. Spatial PROTACs utilize cellular abundant components or cell‐specific membrane receptors to induce cell type‐specific target protein degradation. Temporal PROTACs facilitate click chemistry to activate or quench the TPD at time‐specific manner. Spatiotemporal PROTACs is based on optical or sonodynamic control for precise degradation of target protein both spatial and temporal manners.
2. Spatial PROTAC
One of the limitations of PROTACs is the undesired normal cell toxicity resulting from their off‐site protein degradation.[ 34 , 35 ] In efforts to mitigate the on‐target off‐tumor toxicity of PROTACs, spatially controllable PROTACs (Spatial PROTAC) have been developed. The spatial PROTACs leverage cell‐specific features, such as membrane receptors and cellular environments–particularly tumor microenvironments–or utilize bioorthogonal chemical reactions for spatial protein degradation (Figure 2, Table 1).
Figure 2.

Spatial PROTACs. Overview of the mechanism of action for Spatial PROTACs and their applications. These applications include the modification of ligands by conjugating them with small molecules, monoclonal antibodies (mABs), and aptamers that target cancer‐associated membrane proteins. Spatial PROTACs can form nano‐spherical assemblies via specific assembly motifs, such as lipids or peptides conjugated to either the ligand for the POI or the E3 ligase ligand of the PROTAC. Tumor‐targeting motifs, such as folate, facilitate cell‐specific target degradation. Furthermore, various caging groups can be incorporated to conditionally activate the PROTACs in cells exhibiting elevated levels of reactive oxygen species (ROS) or glutathione (GSH), as well as in hypoxic states. Finally, tumor‐selective click reactions can be utilized, taking advantage of either elevated copper (Cu) levels or the overexpression of integrin within tumor tissues.
Table 1.
Spatial PROTAC.
|
No. |
Class |
Structure |
Protein ligand |
E3 ligase ligand |
Control moiety |
Ref |
||
|---|---|---|---|---|---|---|---|---|
|
1 |
DAC |
3 a
|
PROTAC
|
BRD4 |
VHL |
STEAP1 CLL1 HER2 |
[53] |
|
|
2 |
DAC |
3 b
|
PROTAC
|
BRD4 |
VHL |
STEAP1 CLL1 HER2 |
[53] |
|
|
3 |
DAC |
5 b
|
PROTAC
|
BRD4 |
VHL |
STEAP1 CLL1 HER2 |
[53] |
|
|
4 |
DAC |
11
|
PROTAC
|
ERα |
XIAP |
HER2 B7H4 |
[56] |
|
|
5 |
APC |
APR
|
BRD4 |
VHL |
AS1411 |
[69] |
||
|
6 |
APC |
PS‐ApTCs
|
nucleolin |
CRBN |
PS‐AS1411 |
[70] |
||
|
7 |
Folate conjugated PROTAC |
Folate‐ARV‐771
|
BRD4 |
VHL |
|
[77] |
||
|
8 |
Folate conjugated PROTAC |
FA‐S2‐MS4048
|
ALK |
CRBN |
|
[78] |
||
|
9 |
Split & Mix PROTAC |
VHL+TMXF‐θθ
(1 : 1) |
4‐Hydroxyltamoxifen
|
VHL ligand 2
|
ERα |
VHL |
– |
[80] |
|
10 |
Split & Mix PROTAC |
Tha+Enza‐θθ
(1 : 3) |
Enzalutamide
|
3‐COOH‐Thalidomide
|
AR |
CRBN |
– |
[80] |
|
11 |
Split & Mix PROTAC |
Thal+JQ1‐θθ
(1 : 5) |
(+)‐JQ1
|
3‐COOH‐Thalidomide
|
BRD4 |
CRBN |
– |
[80] |
|
12 |
Split & Mix PROTAC |
Tha+Pal‐θθ
(1 : 3) |
Palbociclib
|
3‐COOH‐Thalidomide
|
CDK6 |
CRBN |
– |
[80] |
|
13 |
Split & Mix PROTAC |
VHL+Mird‐θθ
(1 : 3) |
Mirdametinib
|
VHL ligand 2
|
MEK1/2 |
VHL |
– |
[80] |
|
14 |
Split & Mix PROTAC |
VHL‐Dasa‐θθ
(1 : 2) |
Dasatinib
|
VHL ligand 2
|
BCR‐ABL |
VHL |
– |
[80] |
|
15 |
Split & Mix PROTAC |
Tha+Gefi‐θθ
(1 : 3) |
Gefitinib
|
3‐COOH‐Thalidomide
|
EGFR |
CRBN |
– |
[80] |
|
16 |
Split & Mix PROTAC |
FA‐V‐Tam‐L
(1 : 9 : 2) |
4‐Hydroxyltamoxifen
|
VHL ligand 2
|
ERα |
VHL |
– |
[82] |
|
17 |
ROS PROTAC |
Pre‐PROTAC 7
|
BRD3 |
CRBN |
|
[101] |
||
|
18 |
ROS PROTAC |
2
|
BRD4 |
VHL |
|
[102] |
||
|
19 |
ROS PROTAC |
5
|
ER |
VHL |
|
[102] |
||
|
20 |
ROS PROTAC |
ROS‐HaloPROTAC
|
HaloGFP |
VHL |
|
[104] |
||
|
21 |
ROS PROTAC |
ROS‐BRD‐PROTAC
|
BRD4 |
VHL |
|
[104] |
||
|
22 |
ROS PROTAC |
Na‐PRO
|
HDAC |
CRBN |
|
[105] |
||
|
23 |
ROS PROTAC |
C‐srPHP
|
BRD4 |
CRBN |
|
[94] |
||
|
24 |
Hypoxia PROTAC |
8 a
|
ERα |
VHL |
|
[108] |
||
|
25 |
Hypoxia PROTAC |
8 b
|
ERα |
VHL |
|
[108] |
||
|
26 |
Hypoxia PROTAC |
8 c
|
ERα |
VHL |
|
[108] |
||
|
27 |
Hypoxia PROTAC |
8 d
|
ERα |
VHL |
|
[108] |
||
|
28 |
Hypoxia PROTAC |
8 e
|
ERα |
VHL |
|
[108] |
||
|
29 |
Hypoxia PROTAC |
12
|
EGFRDel19 |
CRBN |
|
[107] |
||
|
30 |
Hypoxia PROTAC |
13
|
EGFRDel19 |
CRBN |
|
[107] |
||
|
31 |
Hypoxia PROTAC |
9
|
EGFRDel19 |
CRBN |
|
[109] |
||
|
32 |
Hypoxia PROTAC |
10
|
EGFRDel19 |
CRBN |
|
[109] |
||
|
33 |
Hypoxia PROTAC |
C‐srPNTR
|
BRD4 |
CRBN |
|
[94] |
||
|
34 |
Hypoxia PROTAC |
17‐1
|
EGFR |
VHL |
|
[110] |
||
|
35 |
Hypoxia PROTAC |
JW4
|
– |
CRBN |
|
[111] |
||
|
|
||||||||
|
36 |
Hypoxia PROTAC |
JQ1‐CBT
|
BRD4 |
‐ |
– |
[111] |
||
|
37 |
Hypoxia PROTAC |
J252(PROTAC)
|
BRD4 |
CRBN |
– |
[111] |
||
|
38 |
GSH PROTAC |
GSH‐ER‐P1
|
ERα |
VHL |
|
[116] |
||
|
39 |
GSH PROTAC |
1 a
|
BRD4 |
VHL |
|
[117] |
||



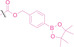
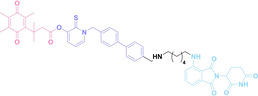
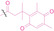
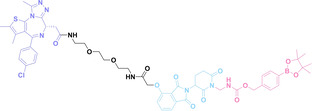







2.1. Ligand Modification
Certain type of tumor cells can be characterized by a high abundance of membrane proteins, including anchoring proteins, receptors, and transporter proteins, [36] which can be the targets of various modalities such as mAbs, [37] aptamers, [38] and receptor binding small molecules.[ 39 , 40 ] The combination of the cancer‐targeting moieties with PROTAC technology enables tumor type‐selective PROTAC delivery and subsequent protein degradation. [41]
2.1.1. Degrader‐Antibody Conjugates (DACs)
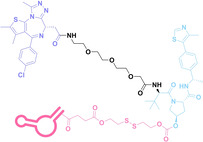
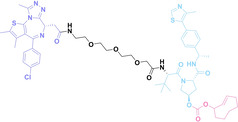
Antibody‐drug conjugates (ADCs) consist of tumor‐targeting mAbs, which are conjugated to cytotoxic payloads via chemical linkers. [42] Each component plays a crucial role in the efficacy and safety profile of ADCs. The mAb part of ADCs contributes to cancer cell selectivity, stability, and pharmacokinetic properties, while the payloads of ADCs exert anti‐tumor effects by inducing cytotoxicity such as DNA damage or microtubule inhibition. For the chemical linker in ADCs, both cleavable and non‐cleavable chemical structures have been utilized. Cleavable linkers enable controlled release of the cytotoxic payload intracellularly or within the tumor microenvironment. The cleavable ADC linkers can be categorized into three types: protease‐sensitive linkers utilizing protease cleavable peptide sequences like cathepsin B sensitive valine‐citrulline (VC) dipeptide, pH‐sensitive linkers attached to acid‐labile groups such as hydrazone, and glutathione‐sensitive linkers containing disulfide bonds. Whereas, non‐cleavable linkers, including thioether or maleimidocaproyl (MC) groups, exhibit enhanced plasma stability, and potentially improving the therapeutic index. They can offer a larger therapeutic window and reduce off‐target toxicity compared to cleavable linkers due to greater stability and tolerance.[ 42 , 43 , 44 ] The extensive ADC research and its notable efficacy have resulted in the FDA approval for 12 ADCs, with over 260 undergoing clinical trials as of 2022. [45] However, limitations of ADCs have arisen from their uniform payloads, which are driving ADC research towards payload diversification and prompting the development of degrader‐antibody conjugates (DACs). [46]
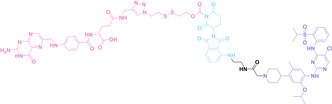
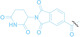
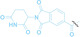
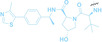
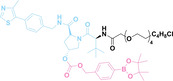
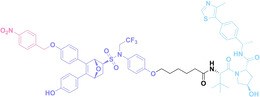
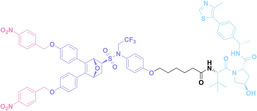
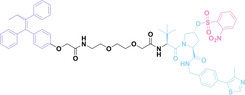
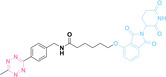
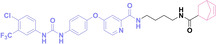
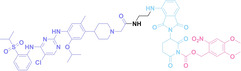
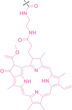
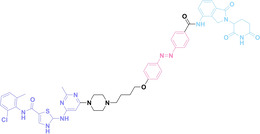
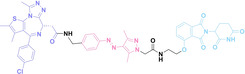
Despite the promising potential of PROTACs, their development remains challenging due to their metabolism and pharmacokinetics (DMPK). [47] To overcome these limitations, DACs have been developed by harnessing mAb‐mediated PROTAC delivery. Given the pivotal role of BET family proteins in various cancers,[ 48 , 49 , 50 ] bromodomain‐containing protein 4 (BRD4) targeting DACs have been developed.[ 34 , 51 ] VHL‐based BRD4 degraders, such as GNE‐987 and MZ1, conjugated to antibodies have shown the promising results in acute myeloid leukemia (AML) treatment using anti‐CLL1 antibody, as well as in prostate and breast cancer treatment using anti‐STEAP1 and anti‐HER2 antibodies (trastuzumab or Herceptin®) (1–3), respectively.[ 52 , 53 , 54 , 55 ] Among them, STEAP1‐targeting DACs demonstrated significantly higher efficacy, with DC50 values ranging from 1 to 1000 nM. These DACs were three to ten times more effective than CLL1 or Her2 targeting DACs. Additionally, DACs utilizing a GSH‐responsive linker demonstrated significant BRD4 degradation compared to those employing protease‐responsive or non‐cleavable linkers. Similarly, estrogen receptor (ER) degraders conjugated to anti‐HER2, anti‐B7‐H4, and CD22 antibodies through protease‐responsive linker (4) exhibited enhanced in vivo stability. [56] These examples demonstrate that DACs can improve pharmacokinetic properties of PROTACs. Moreover, proper linker design is critical for the efficacy of DACs.
Recently, Antibody neoDegrader Conjugates (AnDC™) have been reported to deliver neodegraders specifically to cancer cells toward clinical applications. For instance, ORM‐5029, designed to target HER2‐expressing advanced solid tumors via pertuzumab, is in phase I clinical trials.[ 57 , 58 ] This trial serves as a pioneering example of DACs in clinical settings.
2.1.2. Aptamer‐PROTAC Conjugates (APCs)
Aptamers, which are single‐stranded nucleic acids with intricate three‐dimensional structures, exhibit high specificity and affinity for their target proteins through various physiochemical interactions, including shape complementarity, hydrogen bonding, hydrophobic, electrostatic, van der Waals interactions and base stacking.[ 59 , 60 , 61 ] With their binding properties toward target proteins, aptamers are often referred to as “chemical antibodies” and offer distinct advantages over antibodies, such as their small size, synthetic accessibility, and tunability to enhance water solubility. [62] Notably, aptamers demonstrate excellent tissue penetration, superb in vivo stability, and lack of immunogenicity.[ 63 , 64 ] Given these advantages, aptamers have been widely used in targeted cancer therapy. [63]
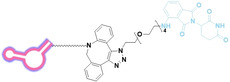
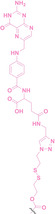
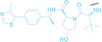
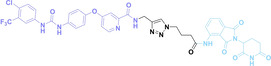
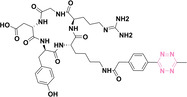
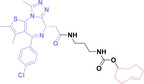
For instance, AS1411, a guanosine‐rich oligonucleotide aptamer, specifically recognizes and binds to nucleolin, which is highly expressed on the membranes of tumor cells.[ 65 , 66 ] Due to its potent inhibitory effect on nucleolin‐overexpressing tumors, AS1411 is currently in phase II clinical trial.[ 67 , 68 ] Furthermore, AS1411 has been employed in aptamer‐drug conjugates for the tumor‐specific delivery of drugs. Capitalizing on aptamers’ potential as tumor‐selective binding agents, He et al. developed the first aptamer‐PROTAC conjugate (APC), named as APR (5) by conjugating AS1411 with MZ1 via a disulfide cleavable linker. [69] The APR selectively internalized into nucleolin‐overexpressing breast cancer cells, where it was cleaved by endogenous glutathione (GSH), releasing the BRD4 degrader MZ1 and inducing subsequent BRD4 degradation. The APR demonstrated tumor‐targeting ability and potent antitumor efficacy without adverse effects on normal tissues in a mouse xenograft model.
Similarly, Liu et al. developed PS‐ApTCs (6), which is phosphorothioate‐modified AS1411 (PS‐AS1411) with CRBN via a non‐cleavable linker for UPS‐mediated nucleolin degradation. [70] The phosphorothioate modification enhanced PS‐ApTCs’ stability against nuclease‐mediated degradation. PS‐ApTCs preferentially degraded nucleolin in targeted tumor cells and exhibited enhanced antiproliferative potency compared to PS−A1411. Furthermore, PS‐ApTCs were combined with GSH‐responsive AS1411‐paclitaxel conjugates (ApDCs). The mechanism of action of ApDCs is similar to the first APC, wherein ApDCs internalize into nucleolin‐positive cells via AS1411 and undergo disulfide cleavage by GSH, releasing the cytotoxic microtubule‐targeting drug. Combination therapy with PS‐ApTCs and ApDCs demonstrated enhanced synergistic effects on tumor inhibition in a mouse xenograft model. Consequently, the APC strategy presents a promising approach to address the limitations of traditional PROTACs by enhancing their tumor‐targeting capability, antitumor efficacy, and water solubility.
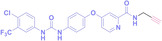
2.1.3. Folate‐Conjugated PROTAC
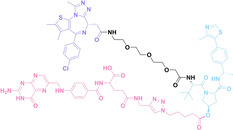
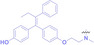
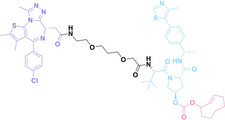
Folate, the natural form of vitamin B9, is essential for eukaryotic cell proliferation and differentiation. [71] Folate is internalized into the cells upon its binding to folate receptors (FRs). [72] Among the four isoforms of FRs, folate receptor α (FOLR1) is notably overexpressed in cancer compared to normal tissues.[ 73 , 74 ] The site‐selective expression and high‐affinity folate binding property of FOLR1 have led to its applications in targeted drug delivery.[ 75 , 76 ]
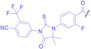
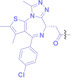
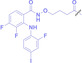
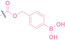
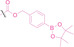
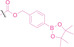
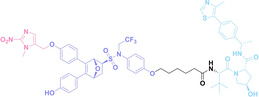
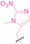
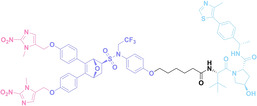
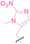
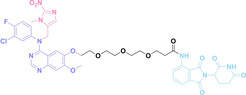
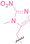
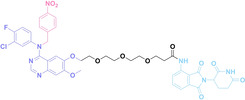
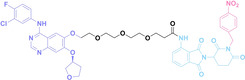
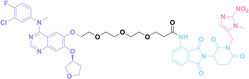
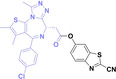
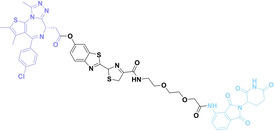
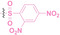
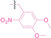
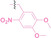
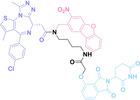
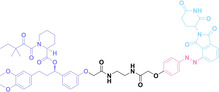
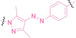
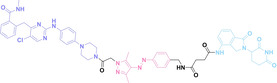
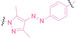
Liu et al. developed three folate‐conjugated PROTACs capable of selectively degrading BRDs, MEKs, and ALKs in cancer cells. [77] VHL‐based PROTACs targeting BRD4 (ARV‐771), MEK1/2 (MS432), and ALK (MS99) were conjugated with folate via a triazole embedded cleavable linker. The folate‐conjugated PROTACs were internalized into cancer cells via FOLR1 binding. Upon entry into the cells, the folate group was cleaved by endogenous hydrolases, releasing the active PROTAC to degrade the POI. Among them, BRD4 targeting folate‐conjugated PROTAC (7) showed high FOLR1 selective targeting ability, FOLR1 positive cancer cell specific protein degradation and excellent potency. Similarly, Chen et al. devised a CRBN‐based folate‐conjugated PROTAC by linking pomalidomide‐ALK PROTAC (MS4048) with folate via a cleavable disulfide bond (FA−S2‐MS4048, 8). [78] Specifically, the folate group was incorporated onto the glutarimide moiety of pomalidomide to restrain protein degradation by MS4048. Following folate‐mediated internalization, the disulfide bond of FA−S2‐MS4048 underwent cleavage by endogenous glutathione (GSH) and subsequent intramolecular cyclization, leading to the release of active MS4048 for the degradation of ALK.
Despite the pronounced tumor selectivity demonstrated by folate‐conjugated PROTACs, the conjugation of the folate moiety increased the molecular weight of PROTACs by over 1,000 Da, potentially compromising their oral bioavailability and pharmacokinetics. Thus, further in vivo validation is highly required to confirm the stability and cancer‐selective delivery of folate‐conjugated PROTACs.
2.2. Split & Mix PROTAC
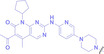
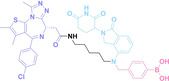
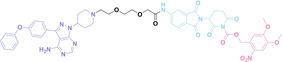
As PROTACs form a ternary complex with E3 ligase and the POI, the linker structure of PROTACs is critical. Therefore, meticulous design and optimization of the linker structure of PROTACs are necessary, which can be tedious and labor‐intensive. [79] To address these time‐consuming challenges, Yang et al. introduced a novel nanoplatform based PROTAC approach, termed split‐and‐mix PROTAC (SM‐PROTAC). [80] Instead of utilizing a chemical linker, peptides were attached to each E3 ligand recruiter and POI binder to induce self‐assembly. The diphenylglycine (Phg‐Phg; δδ) motif, known for its stability and ease of modification, was employed to facilitate self‐assembly into nanoparticles. [81] Initial testing with δδ‐conjugated fluorophores, including fluorescein (FAM) and rhodamine B (Rho), successfully resulted in the formation of fluorescent spherical nanoparticles. However, due to their negative surface potential, these nanoparticles were unable to penetrate the cellular membrane. To enhance membrane penetration and shift the surface zeta potential, the δδ motif was elongated with two positively charged arginine residues (RR). The resulting δδRR‐based nano‐spherical assemblies exhibited improved cell permeability. Nano‐spherical assemblies based on δδRR, incorporating E3 ligase recruiters such as VHL and CRBN, and POI binders including ER (9), AR (10), BRD4 (11), cyclin‐dependent kinases 4 and 6 (CDK 4/6, 12), mitogen‐activated protein kinases 1 and 2 (MEK1/2, 13), BCL‐ABL (14), and epidermal growth factor receptor (EGFR, 15), successfully induced the degradation of their target proteins. Notably, regardless of the mixing ratio of E3 ligase recruiters and POI binders, nanoparticles with a consistent 1 : 3 ratio of E3 ligase recruiters and POI binders were observed, indicating that linker optimization is not necessary.
Furthermore, a liposome‐based self‐assembly SM‐PROTAC system (LipoSM‐PROTAC, 16) was developed, incorporating folate onto FA−V‐Tam‐L to enhance tumor‐targeting ability. [82] LipoSM‐PROTAC exhibited selective uptake into FOLR1‐positive cancer cells and induced successful POI degradation.
In summary, nano‐spherical self‐assembly‐based PROTAC offers promising potential for cell‐selective PROTACs without time‐consuming linker optimization.
2.3. Caged PROTAC
The caging techniques originates from photolabile protecting groups (PPGs) which can be cleaved from a bioactive molecule upon photo‐irradiation. [83] PPGs offer spatiotemporal control of the activity of various bioactive molecules. [84] However, their light‐induced toxicity and limited tissue penetration depth of the light have prompted the exploration of alternative approaches,[ 85 , 86 , 87 , 88 ] including the development of x‐ray‐responsive caging groups. [89] Subsequently, a series of caging groups responsive to physical, chemical, and biological stimuli have been developed.[ 90 , 91 , 92 ] Researchers have conjugated stimuli‐responsive groups to PROTACs and demonstrated their precise control for desired protein degradation.[ 93 , 94 ]
2.3.1. ROS PROTAC
Reactive oxygen species (ROS) are chemically reactive molecules containing oxygen atoms and unpaired electrons, including hydrogen peroxide (H2O2), superoxide radicals (O2•−), hydroxyl radicals (•OH), and singlet oxygen (1O2). It is widely observed that ROS levels are elevated within tumor environments.[ 95 , 96 ] To exploit this characteristic, boronic acid moieties have been employed for the conditional activation of PROTACs in the presence of ROS. [97] This approach facilitates targeted PROTAC activation specifically within ROS‐rich tumor microenvironments.
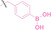
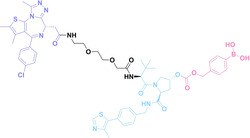
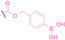
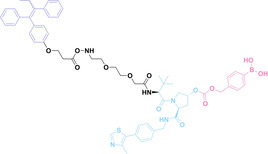
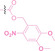
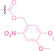
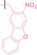
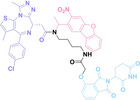
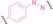
Boronic acid is renowned for its significant biological applications, owing to its facile interconversion between sp2 and sp3 forms, robust interaction with diol‐containing compounds, and notable Lewis acidity. [98] When exposed to specific ROS, particularly hydrogen peroxide (H2O2), aryl boronic acid undergoes conversion into a phenol‐derived intermediate, followed by detachment via 1,6‐benzyl elimination.[ 99 , 100 ] Recently, Liu et al. introduced ROS‐responsive pre‐PROTAC 7 (17), efficiently degrading the target protein BRD3, utilizing aryl boronic acid as a caging group. Upon exposure to H2O2, the aryl boronic acid is cleaved, activating the PROTAC. [101] Similarly, The Sun group has devised an H2O2‐responsive PROTAC (18–19), targeting BRD4 and ER, employing aryl boronic acid as a caging group, and has extended this approach to design a PROTAC targeting the estrogen receptor. [102]
Pinacol phenylboronate has also been employed in ROS‐responsive PROTACs,[ 99 , 103 ] functioning via cleavage under H2O2 conditions, triggering 1,6‐benzyl elimination and hydrolysis. [94] The Wang group utilized this mechanism in their development of ROS‐PROTACs (20–21), yielding two distinct variants capable of regulating intracellular BRD4 protein levels and selectively modulating the degradation of a fluorescent reporter protein, HaloGFP. [104] Similarly, Jia et al. demonstrated that ROS‐responsive PROTACs could selectively target histone deacetylase (Na‐PRO, 22) for degradation in response to H2O2. [105] Furthermore, the Zhang group synthesized a series of stimuli‐responsive PROTACs (sr‐PROTACs), caging thalidomide with pinacol phenylboronate (C‐srPHP, 23). These sr‐PROTACs, known as ddBET1, exhibit responsiveness to various stimuli, including H2O2, [94] underscoring the potential of ROS‐activated PROTACs for conditionally targeted protein degradation.
2.3.2. Hypoxia PROTAC
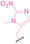
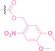
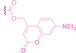
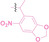
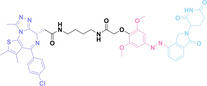
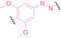
Hypoxia, a prevalent characteristic of most of solid tumors, [106] fosters an environment where Nitroreductase (NTR) activity is notably augmented. However, NTR, an enzyme capable of converting nitro groups into amino groups using NAD(P)H as an electron donor, is barely expressed in normal cells and tissues. Considering the high expression level of NTR in tumors, a hypoxia PROTAC has been designed by using a hypoxia‐activated leaving group (HALG). [107] The HALG, featuring nitroimidazole and nitrobenzene groups, can be cleaved by NTR in the presence of NAD(P)H, leading to the reduction of its nitro groups to amino groups. This cleavage of HALG releases the PROTAC molecule, enabling its therapeutic action specifically under hypoxic conditions.
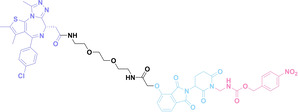
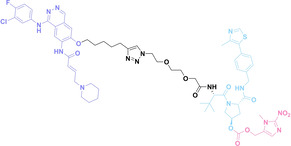
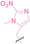
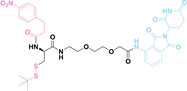
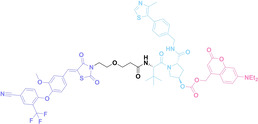
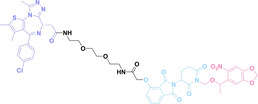
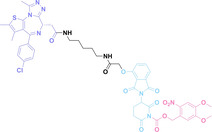
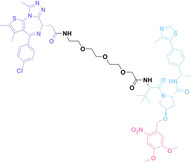
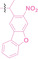
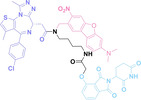
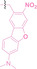
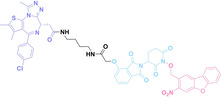
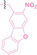
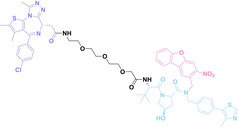
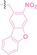
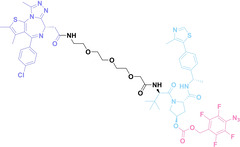
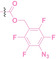
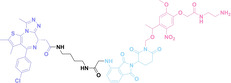
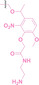
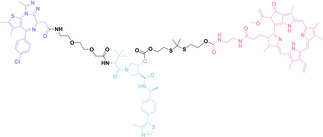
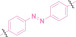
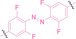
In the design of Hypoxia PROTACs, a common strategy involves conjugating the HALG either to the ligand of the POI or the E3 ligase ligand (24–37).[ 94 , 107 , 108 , 109 , 110 ] Alternatively, the HALG can be used as a caging group either for an E3 ligase ligand or the ligand of POI. Under hypoxic conditions, the HALG is cleaved and the resulting PROTAC molecules can trigger targeted protein degradation. Xie et al. incorporated HALG to ERα‐targeting PROTAC (8a–e, 24–28). [108] They used nitrobenzene (PROTAC 8a–c, 24–26) and nitroimidazole (8d–e, 27–28) as HALG. In 8a (24), one phenol hydroxyl group of the ER ligand is caged by the nitrobenzene group. In 8b (25), two phenol hydroxyl groups of the ER ligand are conjugated to the nitrobenzene group. In the case of 8c (26), the nitrobenzene group is attached to the hydroxy group on the VHL ligand. 8d (27) and 8e (28) are conjugated nitroimidazole to the ER ligands. Similarly, Cheng et al. also attached nitrobenzene or nitroimidazole to EGFRDel19‐targeting PROTAC (29–32).[ 107 , 109 ] In this case, nitroimidazole‐conjugated PROTAC showed enhanced IC50 value in hypoxic conditions compared to nitrobenzene‐conjugated PROTAC. Furthermore, the conjugation of HALG to the POI ligand exhibited better efficacy compared to its attachment to the E3 ligase ligand. An et al. compared PROTACs with various caging groups including nitrobenzene (33) and found that HALG‐conjugated PROTAC is only responsive to hypoxia condition, not by other stimuli. [94] Shi et al. reported HALG conjugated EGFR‐targeting PROTAC (34), which demonstrated an IC50 value of 4 nM and a DC50 value of 36.51 nM. [110] The Xing group recently reported enzyme‐derived clicking PROTACs (ENCTACs), tailored for stimulus‐responsive self‐assembly of PROTAC in hypoxic conditions (35–37). [94] ENCTACs are activated by the reduction of nitrobenzyl chloroformate and tert‐butyl dithiol, mediated by NTR and glutathione, respectively. This reduction process exposes a cysteine moiety of an E3 ligase binder (35), enabling its conjugation to 2‐cyanobenzothiazole moiety of a POI binder (36), which generates J252 for BRD4 degradation. [111] This strategy enhances the specificity and efficacy of PROTACs by reducing the molecular size, using an environment‐sensitive chemical handle, and ensuring activation only under specific cellular conditions.
2.3.3. GSH PROTAC
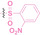
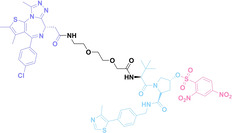
GSH is an antioxidant that plays a crucial role in protecting cells from ROS such as free radicals, and peroxides. [112] In normal cells, GSH concentration typically ranges from 0.5 to 10 mM, while in the tumor microenvironment, it is often more than 1000 times higher than that of normal cells, serving to mitigate elevated oxidative stress.[ 113 , 114 ] Capitalizing on this characteristic of tumor cells, GSH‐responsive prodrugs have been developed for cancer‐selective activation. [115] As demonstrated in ROS and hypoxia PROTACs, the exploitation of GSH‐activated disulfide bonds has also shown promising results for targeted protein degradation in cancer.[ 69 , 78 ]
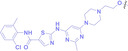
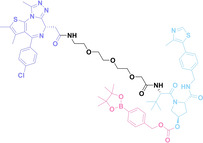
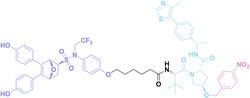
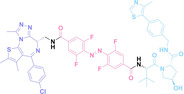
The Sun group utilized the nitrobenzenesulfonyl group as a caging group in GSH PROTACs.[ 116 , 117 ] They synthesized GSH PROTAC by introducing nitrobenzenesulfonyl group to the hydroxyl group of VHL‐based PROTAC. The nitrobenzenesulfonyl group can be cleaved by the nucleophilic attack of GSH and VHL‐based PROTAC is released for targeted protein degradation. ER (38) and BRD4 (39) targeting GSH PROTACs showed effective GSH mediated targeted protein degradation in cancer cells, while remaining minimally activated in normal cells. GSH PROTACs can now be suggested as one of the promising approaches for increasing cancer selectivity and lowering off‐tumor effects of conventional PROTACs, thereby enhancing their potentials in clinical applications.
2.4. Click Reaction
The concept of click chemistry was originally introduced by K. Barry Sharpless in 2001. [118] This reaction exhibits distinctive features, including irreversibility, one‐pot synthesis, minimal generation of by‐products, and the production of a singular product with high yield. Moreover, click chemistry can proceed without undesired side‐reactions with cellular components in the biological environment, allowing it to be called ‘Bioorthogonal Click Chemistry’. Representative examples include Copper‐catalyzed Azide‐Alkyne Cycloaddition (CuAAC), Strain‐Promoted Azide‐Alkyne Cycloaddition (SPAAC), and the Inverse Electron‐Demand Diels‐Alder reaction (IEDDA), which have been widely used in various applications in the chemical biology field.[ 119 , 120 ]
To date, the majority of developed PROTACs exhibit their molecular weight exceeding 500, failing to meet RO5 and consequently limiting their potential in drug discovery. Additionally, spatial control of targeted protein degradation is highly demanded to solve the undesired cytotoxicity issue of PROTACs. In response, integrating click reactions into PROTACs has recently been explored to address these challenges.
2.4.1. Cu PROTAC
The copper concentration in tumor tissues is notably higher compared to that of normal tissues.[ 121 , 122 ] Harnessing this characteristic of tumor tissues, various tumor‐specific therapies have been developed utilizing copper mediated chemical reactions, including CuAAC.[ 123 , 124 ]
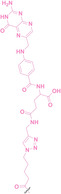
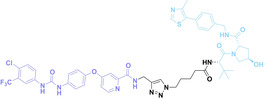
In 2023, Si et al. adopted CuAAC into PROTACs for targeted protein degradation specifically within tumor tissues. [125] This PROTAC comprised two parts: one part is a target protein ligand functionalized with an alkyne moiety (40), and the other part is an E3 ligase ligand functionalized with an azide group (41, 43). These two parts undergo self‐assembly within the tumor cells (42, 44), catalyzed by intracellular copper ions. Another advantage of this approach is the low molecular weight of the two parts, which showed their higher cellular permeability compared to conventional PROTACs.
2.4.2. Click‐to‐Release PROTAC
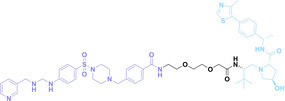
The click‐to‐release strategy has recently emerged in prodrug design, leveraging the fast reaction kinetics and high bioorthogonality of the IEDDA reaction between trans‐cyclooctene (TCO) and tetrazine.[ 126 , 127 ] Once TCO and tetrazine react, the carbamate group in TCO is cleaved into CO2 and an amine. This cleavage leads to the release of the TCO‐caged drug. Click‐to‐release PROTAC (crPROTAC) harness this concept for conditional PROTAC activation.
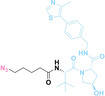
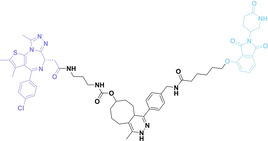
The α v β 3 integrin is widely known for its high expression on the surface of specific cancer cells and its crucial role in tumor angiogenesis, maintenance, cell invasion and proliferation. [128] In 2023, Chang et al. reported crPROTAC, which utilized the TCO‐caged PROTAC (45) and a α v β 3 integrin‐targeting ligand c(RGDyK) conjugated to tetrazine (46) to target cancer cells overexpressing α v β 3 integrin. [129] The c(RGDyK)‐conjugated tetrazine accumulates in cancer cells and releases PROTAC from crPROTAC for cancer specific targeted protein degradation.
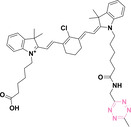
In parallel, Bi et al. developed BT‐PROTAC (47) and IR808‐conjugated tetrazine (IR808‐Tz, 48). [130] The near‐infrared (NIR) dye IR808 is a fluorescent heptamethine carbocyanine exhibiting high selectivity towards tumors.[ 131 , 132 ] IR808‐Tz selectively accumulates in tumor and selectively releases PROTAC from BT‐PROTAC. The releasing event can be monitored by click reaction dependent fluorescent emission of IR808. Initially, the fluorescence of IR808‐Tz is quenched by tetrazine before the click reaction. However, after the click reaction, tetrazine transforms into dihydrotetrazine, resulting in an increment of fluorescent signal from IR808. Therefore, IR808 can be used not only for tumor‐specific PROTAC release, but also for fluorescent PROTAC release event monitoring.
3. Temporal PROTAC
Undesired protein degradation by PROTACs within normal cells is one of the drawbacks of the current targeted protein degradation strategy. [133] To address this issue, temporal regulation of the activity of PROTACs (Temporal PROTACs) has recently been spotlighted. Current research on temporal PROTACs harnesses click reaction, which either enables the self‐assembly of PROTAC in vivo at the desired timepoint or employs a quencher to inhibit its activities (Figure 3, Table 2).
Figure 3.

Temporal PROTACs. Overview of the mechanism of Temporal PROTACs. PROTACs can be conditionally activated through a linker connection via a click reaction between tetrazine and TCO. In contrast, PROTACs can be conditionally deactivated by the addition of a poly(amidoamine) dendrimer that contains a TCO motif, which can scavenge the PROTACs with a tetrazine motif.
Table 2.
Temporal PROTAC.
|
No. |
Class |
Structure |
Protein ligand |
E3 ligase ligand |
Control moiety |
Ref |
|---|---|---|---|---|---|---|
|
40 |
Cu PROTAC |
SA
|
VEGFR‐2 PDGFR‐β EphB4 BRAF |
– |
|
[125] |
|
41 |
Cu PROTAC |
VA
|
– |
VHL |
|
[125] |
|
42 |
Cu PROTAC |
SA‐VA
|
VEGFR‐2 PDGFR‐β EphB4 BRAF |
VHL |
– |
[125] |
|
43 |
Cu PROTAC |
PA
|
‐ |
CRBN |
|
[125] |
|
44 |
Cu PROTAC |
SA‐PA
|
VEGFR‐2 PDGFR‐β EphB4 BRAF |
CRBN |
– |
[125] |
|
45 |
Click to release PROTAC |
TCO‐ARV‐771
|
BRD4 |
VHL |
|
[129] |
|
46 |
Click to release PROTAC |
c (RGDyK)‐Tz
|
‐ |
– |
|
[129] |
|
47 |
Click to release PROTAC |
BT‐PROTAC
|
BRD4 |
VHL |
|
[130] |
|
48 |
Click to release PROTAC |
IR808‐Tz
|
– |
– |
|
[130] |
|
49 |
CLIPTAC |
JQ1‐TCO
|
BRD4 |
‐ |
|
[135] |
|
50 |
CLIPTAC |
Probe 1
|
– |
CRBN |
|
[135] |
|
51 |
CLIPTAC |
JQ1‐CLIPTAC
|
BRD4 |
CRBN |
– |
[135] |
|
52 |
CLIPTAC |
S4N‐1
|
VEGFR‐2 PDGFR‐β EphB4 BRAF |
– |
|
[136] |
|
53 |
CLIPTAC |
TzB
|
– |
VHL |
|
[136] |
|
54 |
CLIPTAC |
S4N‐1 TzB
|
VEGFR‐2 PDGFR‐β EphB4 BRAF |
VHL |
[136] |
|
|
55 |
Scavenging PROTAC |
TCB‐2
|
BRD4 |
CRBN |
|
[137] |
|
56 |
Scavenging PROTAC |
PAMAM‐G5‐TCO
|
– |
– |
|
[137] |
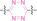
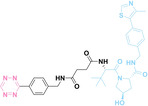
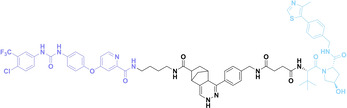
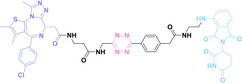
3.1. CLIPTAC
In contrast to the ‘click‐to‐release strategy’, the click reaction between tetrazine and dienophiles such as TCO, cyclopropene and norbornene can be used for conjugating two molecules. [134]
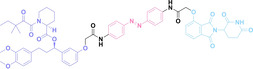
The Heightman group pioneered the development of click‐formed proteolysis targeting chimera (CLIPTAC), as an approach to address the limitations of PROTAC such as cell permeability and solubility. [135] In this study, they chose TCO as a dienophile. They synthesized a TCO‐conjugated BRD4 binder (JQ1‐TCO, 49) and tetrazine‐conjugated thalidomide derivatives (Tz‐thalidomide or Probe1, 50). These components successfully self‐assembled into JQ1‐CLIPTAC (51) in cells and promoted the degradation of target proteins. Moreover, in 2023, the Zhang group introduced CLIPTAC employing norbornene as a dienophile (52–54). [136] Norbornene‐conjugated sorafenib (52) was self‐assembled with a tetrazine‐conjugated VHL ligand (53) to generate S4 N1 TzB (54) to target multiple kinases. Despite its slow reaction kinetics compared to TCO, norbornene offers enhanced molecular stability and cost‐effectiveness in synthesis. Their comparative analysis of CLIPTACs with different tetrazine derivatives revealed that diene structure affects protein degradation, which provides new lessons for CLIPTAC design.
3.2. Scavenging PROTAC
To control the activity of PROTAC and minimize its side‐effects, scavenging PROTAC in cells has recently been suggested. In 2023, Jin et al. developed an approach for sequestering PROTAC molecules using tetrazine‐TCO click chemistry. They designed TCB‐2 (55), a PROTAC having a CRBN ligand and a BRD4 ligand with a tetrazine embedded linker. After targeted protein degradation with TCB‐2, a TCO‐conjugated Poly(amidoamine) dendrimer (PAMAM−G5‐TCO, 56) was treated to cell. This dendrimer effectively scavenges the PROTAC through tetrazine‐TCO click chemistry resulting in the inhibition of BRD4 degradation by TCB‐2. [137] Under optimal conditions, this system demonstrates complete inhibition of PROTAC activity in less than 5 min.
4. Spatiotemporal PROTAC
Prodrug strategy is based on the spatiotemporal control of drug release through optical control or specific delivery method,[ 138 , 139 , 140 ] thereby mitigating potential side effects of drugs by modulating their activities. These approaches have been applied to PROTACs to overcome their current limitations. Spatiotemporal modulation of PROTAC has been accomplished through the introduction of caging groups, nanoparticles, or diazo bridges, which can be controlled by various stimuli such as light, radiation, or ultrasound (Figure 4, Table 3).
Figure 4.

Spatiotemporal PROTACs. Overview of the mechanism of Spatiotemporal PROTACs. The active form of PROTACs can be conditionally released from nanoparticles or caging groups upon exposure to external stimuli such as light, radiation, or ultrasound. Additionally, diazo bridges can be employed to reversibly activate or inactivate PROTAC function with different light sources.
Table 3.
Spatiotemporal PROTAC.
|
No. |
Class |
Structure |
Protein ligand |
E3 ligase ligand |
Control moiety |
Ref |
|---|---|---|---|---|---|---|
|
57 |
Photocaged PROTAC |
pc‐PROTAC1
|
BRD4 |
CRBN |
|
[148] |
|
58 |
Photocaged PROTAC |
pc‐PROTAC3
|
BTK |
CRBN |
|
[148] |
|
59 |
Photocaged PROTAC |
DEACM‐caged 2
|
ERα |
VHL |
|
[149] |
|
60 |
Photocaged PROTAC |
NPOM‐caged 4
|
BRD4 |
CRBN |
|
[149] |
|
61 |
Photocaged PROTAC |
Opto‐dBET1
|
BRD4 |
CRBN |
|
[150] |
|
62 |
Photocaged PROTAC |
Opto‐dALK
|
EML4‐ALK |
CRBN |
|
[150] |
|
63 |
Photocaged PROTAC |
Photo‐caged 3
|
BRD4 |
VHL |
|
[151] |
|
64 |
Photocaged PROTAC |
Compound 9
|
BRD4 |
CRBN |
|
[152] |
|
65 |
Photocaged PROTAC |
Compound 10
|
BRD4 |
CRBN |
|
[152] |
|
66 |
Photocaged PROTAC |
Compound 11
|
BRD4 |
CRBN |
|
[152] |
|
67 |
Photocaged PROTAC |
Compound 12
|
BRD4 |
CRBN |
|
[152] |
|
68 |
Photocaged PROTAC |
Compound 17
|
BRD4 |
VHL |
|
[152] |
|
69 |
Radio triggered PROTAC |
RT‐PRO
|
BRD4 |
VHL |
|
[165] |
|
70 |
Near infrared nano PROTAC |
phoBET1
|
BRD4 |
VHL |
|
[184] |
|
71 |
Near infrared nano PROTAC |
NIR‐NAP
|
BRD4 |
VHL |
|
[185] |
|
72 |
Ultrasound nano PROTAC |
SPNFeP
|
NAMPT |
VHL |
– |
[195] |
|
73 |
Photo switchable PROTAC |
PHOTAC‐ I‐3
|
BET (BRD2,3,4) |
CRBN |
|
[200] |
|
74 |
Photo switchable PROTAC |
PHOTAC‐II‐5
|
FKBP12 |
CRBN |
|
[200] |
|
75 |
Photo switchable PROTAC |
PHOTAC‐II‐6
|
FKBP12 |
CRBN |
|
[200] |
|
76 |
Photo switchable PROTAC |
Azo‐PROTAC‐4 C
|
BCL‐ABL |
VHL |
|
[201] |
|
77 |
Photo switchable PROTAC |
photoPROTAC‐1
|
BRD2 |
VHL |
|
[199] |
|
78 |
Photo switchable PROTAC |
AP‐PROTAC‐1
|
BRD2,4 |
CRBN |
|
[205] |
|
79 |
Photo switchable PROTAC |
AP‐PROTAC‐2
|
FAK, AURORA−A, TBK |
CRBN |
|
[205] |
4.1. Caged PROTAC
4.1.1. Photocaged PROTAC
The non‐invasive and spatiotemporally controllable feature of light renders it an attractive tool for prodrug activation. [141] In this context, various photo‐caging groups including ortho‐nitrobenzyl, [142] para‐hydroxyphenacyl, [143] and Coumarin, [144] have been developed and employed for numerous prodrugs.[ 145 , 146 , 147 ]. For the synthesis of prodrugs, the photo‐caging groups are conjugated to the drug molecule and interfere with the binding between the drugs and target proteins. Light irradiation prompts the cleavage of the caging groups and releases the active drug.
Recently, the photo‐caging strategy has been applied to PROTACs.[ 148 , 149 , 150 , 151 , 152 ] In 2019, the Pan group reported a photo‐caged PROTAC (pc‐PROTAC1, 57), [148] using 4,5‐dimethoxy‐2‐nitrobenzyl (DMNB) group as a photo‐caging moiety for dBET1, [153] a well‐known BRD4 degrader. Upon 365 nm light irradiation, pc‐PROTAC successfully released dBET1 and BRD4 was degraded both in vitro assays and in vivo zebrafish experiments. Moreover, they reported pc‐PROTAC3 (58) by conjugating a photo‐caging moiety to MT‐802, [154] a well known BTK degrader, resulting in successful light‐induced BTK protein degradation.
In 2020, three additional studies on pc‐PROTACs were reported.[ 149 , 150 , 151 ] The Deiters group utilized diethylamino coumarin (DEACM) and 6‐nitropiperonyloxymethyl (NPOM) as photo‐caging groups for the development of pc‐PROTAC. [149] DEACM was introduced into a PROTAC having a VHL ligand and an ERα ligand (DEACM‐caged 2, 59). They conjugated DEACM to the hydroxyl group of the VHL ligand of PROTAC to interfere with its interaction with VHL. They also conjugated NPOM to another PROTAC having a BRD4 ligand, JQ1 and a CRBN ligand, thalidomide (NPOM‐caged 4, 60). Both pc‐PROTACs showed effective light‐mediated targeted protein degradation activities. The Wei group developed two pc‐PROTACs using DMNB. They designed opto‐dBET1 (61), which targets the BRD4 protein, and opto‐dALK (62), which targets the EML4‐ALK protein. Similarly, the Tate group developed a pc‐PROTAC utilizing DMNB, [151] employing JQ1 as a target protein binder and VHL ligand for E3 ligase recruiter (63). These three pc‐PROTACs showed excellent targeted protein degradation upon light irradiation in a spatiotemporal manner.
The above mentioned pc‐PROTACs utilize UV light for activation. Although UV light has high‐energy and efficiently cleave photocaging groups, it can damage biological systems by altering the DNA structures.[ 155 , 156 ] To tackle this problem, the Pan group developed a second generation of pc‐PROTAC employing nitrodibenzofuran (NDBF) as a photo‐caging group. [152] NDBF is responsive to less cytotoxic long wavelength blue light (405 nm) and can be cleaved from pc‐PROTAC. [157] To optimize spatiotemporal modulation of the activity of PROTACs, they compared different NDBF groups (compound 9–12, 64–67; compound 17, 68). Compound 9 (64) and 10 (65) exhibited rapid decaging initiation in less than a minute and released the PROTAC molecule effectively. On the other hand, compound 11 (66), employing dimethylamino‐nitrodibenzofuran (DMA‐NDBF), remained incompletely decaged even after 10 minutes of light irradiation, but its ability to degrade BRD4 protein was nearly as good as the other two compounds. Additionally, two PROTACs are reported with NDBF conjugated to a CRBN ligand (compound 12, 67) and to VHL ligand (compound 17, 68). Both compounds underwent the decaging process in less than a minute, exhibiting near‐complete release of PROTAC molecules and degradation of the BRD4 protein.
4.1.2. Radio‐Triggered PROTAC
X‐ray or gamma‐ray has been widely used in radiotherapy, serving as a cornerstone in cancer treatment.[ 158 , 159 ] X‐ray or gamma‐ray generates reactive species like hydrogen radicals (•H), hydroxyl radicals (•OH), and hydrated electrons (ehyd −), initiating a cascade of destructive biochemical reactions.[ 158 , 160 ] Consequently, radiotherapy induces DNA damage, cellular apoptosis and tumor necrosis. [158] In recent years, clinically relevant doses of X‐ray radiation have been used for prodrug activation by controlling the release of chemically modified caging groups.[ 161 , 162 , 163 , 164 ] Precision irradiation and deep tissue penetration of X‐ray offer promising prospects for achieving spatiotemporal activation of prodrugs in real clinics.
In 2021, Geng et al. reported a caging group that undergoes cleavage in response to X‐ray irradiation. [140] They discovered the 4‐azido‐2,3,5,6‐tetrafluorophenyl group as a X‐ray responsive moiety, which showed a 60 % decaging yield upon 60 Gy irradiation.
Yang et al. employed the 4‐azido‐2,3,5,6‐tetrafluorophenyl group to develop a Radiotherapy‐Triggered PROTAC (RT‐PROTAC). [165] They synthesized a PROTAC targeting the BRD4 protein for degradation using the VHL E3 ligase. Conjugating the radioactive caging group to the hydroxyl group of the VHL ligand part, they synthesized the RT‐PRO compound (69). Upon X‐ray irradiation, RT‐PRO effectively released the PROTAC, leading to the degradation of the BRD4 protein in vitro. Moreover, in vivo studies demonstrated its ability to inhibit tumor growth when combined with radiotherapy. However, considering the typical dose of radiotherapy (5 Gy) in real clinics, [166] the necessity of high dose X‐ray irradiation (60 Gy) is the current limitation of this approach for the real clinical applications.
4.2. Nano‐PROTAC
The utilization of a drug delivery system for target protein degradation can offer a promising way to control the local distribution of PROTACs in our body, thereby reducing their potential toxicity. Moreover it can also help PROTACs to cross biological membranes more efficiently and enable the spatiotemporal release of PROTACs in target tissues. [167] For this purpose, encapsulation of PROTACs using polymeric,[ 168 , 169 ] lipid‐based,[ 170 , 171 ] and inorganic nanoparticles, [172] results in nanoformulations, termed nano‐PROTACs. These nano‐PROTACs exhibit a substantial increase in accumulation within target tissues, leading to enhanced therapeutic efficacy and reduced side effects compared to conventional PROTACs. Furthermore, the utilization of deep tissue‐penetrating modalities such as near‐infrared (NIR) and ultrasound enables precise spatiotemporal regulation of protein degradation by nano‐PROTAC.
4.2.1. Near‐Infrared Nano‐PROTAC
NIR light, with its reduced absorption and scattering relative to ultraviolet and visible light, can penetrate into deeper tissue in our body. [173] This property is the reason for the widespread application of NIR light in vivo studies.[ 174 , 175 , 176 , 177 ] One of the most well‐known applications of NIR in cancer treatment is photothermal therapy, where it is not easy to achieve the complete eradication of tumor tissues.[ 178 , 179 ] Hence, there is a growing interest in combining NIR light with other therapeutic modalities such as nanomedicines to enhance therapeutic effects.[ 180 , 181 , 182 , 183 ] Application of NIR to nano‐PROTAC can also contribute to enhancing the efficacy of PROTACs.
He et al. employed lanthanide‐doped upconversion nanoparticles (UCNPs) capable of converting 980 nm NIR excitation to UV emission for a controllable nano‐PROTAC system. [184] They loaded a photocaged‐PROTAC (phoBET1, 70) to UCNPs to construct a NIR‐activable PROTAC nanocage (UMSNs@phoBET1). Upon irradiation with 980 nm NIR light, the upconverted UV emission from the cores of UMNSn@phoBET1 photolyzed phoBET1, releasing the BRD4 targeting PROTAC and inducing BRD4 degradation both in vitro and in vivo.
Wang et al. introduced a self‐assembling NIR‐activable nanoformulated PROTAC (NAP). [185] They conjugated BRD4 targeting PROTAC with an amphiphilic NIR photosensitizer via a singlet oxygen (1O2)‐cleavable linker (71), leading to molecular self‐assembly. Before NIR irradiation, the PROTAC remains inactive due to covalent cross‐linking. Upon systemic administration, the NAP accumulates in tumor tissue through enhanced permeability due to the NIR photosensitizer. After NIR irradiation at tumor sites, the generation of 1O2 is triggered, resulting in linker cleavage and subsequent release of PROTAC, along with the photodynamic effect by the activated NIR photosensitizer. NAP demonstrated synergistic cancer suppression through BRD4 degradation and the photodynamic effect.
The NIR‐activable PROTAC nanoplatform addresses current limitations of short‐wavelength light‐dependent PROTAC modulation strategy by reducing phototoxicity and enhancing tissue penetration of light. This innovative approach presents a promising avenue for achieving precise regulation of PROTAC in vivo.
4.2.2. Ultrasound Nano‐PROTAC
Ultrasound plays a crucial role in ultrasound imaging for disease diagnosis[ 186 , 187 , 188 ] and sonodynamic therapy (SDT) for disease treatments.[ 189 , 190 ] Conventional SDT faces several challenges due to the properties of current sonosensitizers including inherent hydrophobicity, tendency to aggregate, and poor pharmacokinetics. [191] However, recent advancements in nanotechnology have revolutionized SDT by utilizing sonosensitizer‐conjugated nanoparticles for selective tumor eradication.[ 192 , 193 ] This strategy addresses the limitations of traditional SDT by providing advantages, such as enhanced tissue penetration, and improved tumor targeting capacity. [194]
Based on the advancement of SDT, Wang et al. recently designed a semiconducting polymer nano‐PROTAC (SPNFeP, 72). [195] SPNFeP was synthesized through a self‐assemble nano‐precipitation process by integrating a sonodynamic semiconducting polymer, a ferroptosis inducer (ferrocene), a ROS‐cleavable amphiphilic polymer, and a nicotinamide phosphoribosyl transferase (NAMPT) PROTAC. Upon ultrasound irradiation to the tumor, the semiconducting polymer works as a sonosensitizer to generate singlet oxygen (1O2), while the ferroptosis inducer reacts with intratumoral hydrogen peroxide (H2O2) to produce hydroxyl radicals (•OH). This dual‐ROS generating programmable process cleaves the ROS‐cleavable polymer and induces the release of NAMPT PROTAC within the tumor. This spatiotemporal control of NAMPT degradation leads to an effective suppression of tumor infiltration of myeloid‐derived suppressive cells, which enhances antitumor immunity. The ultrasound nanoplatform enables precise regulation of PROTAC release through a dual‐programmable activation mechanism, thereby increasing antitumor efficacy in deep‐tissue tumors.
4.3. Photo‐Switchable PROTAC
Azobenzene, a derivative of diazene (HN=NH), has a unique photo‐response property, called photoisomerization. [196] Light irradiation to azobenzene triggers cis–trans or trans–cis conformational changes. [197] Leveraging its photo‐responsive properties, azobenzene has been widely utilized as a light‐triggered molecular switch in various biomolecular applications. [198] Photo‐switchable PROTACs harness the photo‐response property of azobenzene for the spatiotemporal control of targeted protein degradation. [199] For this purpose, photo‐switchable PROTACs are designed to embed azobenzene in their linker structure. Upon light irradiation, cis‐trans photoisomerization induces the conformational change of the PROTACs. As a result, the distance between the E3 ligase ligand and the ligand of the POI is significantly changed, which could affect the appropriate ternary complex formation for ubiquitination of the POI.
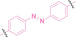
The Trauner group has developed a photo‐switchable PROTAC (PHOTAC) with an azobenzene linker (73–75). PHOTACs targeting the BET family (73) and targeting FKBP12 (74–75) demonstrated successful degradation of their POI. They reported a cis and trans rich photo‐stationary state (PSS) of PHOTAC at 70–90 %. With this excellent PSS property, they could control the targeted protein degradation precisely. Despite its efficient photostability, this PHOTAC requires continuous light pulses to sustain its degradation activity. [200] The Jiang group reported a lenalidomide‐azobenzene‐dasatinib trifunctional system, known as Azo‐PROTAC, to achieve conditional degradation of BCL‐ABL and BCR‐ABL (76), demonstrating clear switchability. [201]
However, the utilization of azobenzene in photo‐switchable systems is limited by its incomplete reversibility in cis–trans photoisomerization. [202] To achieve a 100 % on‐off system for spatiotemporal targeted protein degradation, complete reversibility in cis–trans photoisomerization should be pursued. In this context, the Carreira group developed ortho‐F4‐azobenzene (77),[ 199 , 202 ] a derivative of azobenzene, and used it for designing bistable photoPROTACs. Ortho‐F4‐azobenzene is characterized by well‐separated n→π* absorption bands at 415 nm and 530 nm, facilitating the achievement of highly efficient PSSs: 95 % trans at 415 nm and 68 % cis at 530 nm. This innovative discovery significantly enhanced the performance and reliability of photo‐switchable PROTAC systems.
Arylazopyrazole, another class of azo‐containing molecule, exhibits excellent photo‐switching capabilities and high thermal stability of the cis form.[ 203 , 204 ] This molecule undergoes cis–trans switching upon 457 nm light irradiation, resulting in the formation of 75 % trans isomer. Moreover, the trans–cis switching is achieved by 365 nm light irradiation, generating 99 % cis isomer. The Tate group used an arylazopyrazole moiety for a photo‐switchable PROTAC (AP‐PROTAC, 78–79), which has the following advantageous features: good PSS isomer abundance, rapid cis‐trans switching, an extended half‐life of the cis isomer, and excellent reversible switching yield over multiple cycles. The trans form of AP‐PROTAC is biologically active with an optimal topology for the successful E3 ligase‐PROTAC‐POI ternary complex formation. In contrast, the cis form of AP‐PROTAC remains inactive. This property of AP‐PROTAC was effectively utilized for the conditional degradation of BRD2 and multiple kinases. [205]
5. Summary and Outlook
PROTAC offers a promising advancement over conventional drugs due to its ability to target previously undruggable proteins. This unique feature of PROTAC has accelerated the drug discovery community to study undruggable proteins as novel drug targets for the treatment of diseases. However, PROTACs also have their own limitations such as high molecular weight, poor pharmacokinetic properties, and undesired cytotoxicity in normal tissues.
To address these limitations, researchers have developed various strategies termed conditional PROTACs, which enable spatial, temporal and spatiotemporal control of targeted protein degradation. For spatial control, modifications of ligands or conjugation of caging groups to PROTACs enable them to target specific proteins expressed on tumor cell membranes. Additionally, the utilization of tumor microenvironment abundant components, such as folate, ROS, NTR, and GSH, can activate PROTACs specifically in tumor cells. For temporal control, click reaction based CLITAC and scavenging PROTAC have been developed for precise control of targeted protein degradation. For spatiotemporal control, optical methods such as photocaging, radiocaging, and photoswitchable PROTACs have been employed. These PROTACs can be activated by localized optical stimuli in tumor, but the penetration depth of the light in tissue is limited. Nanoparticle‐based delivery methods have also been harnessed to enhance the protein degradation efficiency. Nano‐PROTACs have good pharmacokinetic properties and superb cell permeability. By utilizing NIR and ultrasound, nano‐PROTAC enables spatiotemporal control with deeper tissue penetration. Split&Mix PROTACs streamline the optimization process by removing laborious linker optimization steps and can also offer spatial control through targeting specific proteins.
Since the PROTAC strategy has been spotlighted for targeting undruggable proteins, addressing the limitations of PROTAC through innovative approaches is crucial for its clinical application in treating various diseases. In this review, the recent development of conditional PROTAC approaches is discussed to address the current limitations of conventional PROTACs and broaden the scope of their applicability. This review also highlights innovative strategies that enhance the efficacy, specificity, and delivery of PROTACs for targeted protein degradation. Furthermore, this review explores emerging technologies and interdisciplinary collaborations that contribute to the advancement of PROTAC research. Although a number of conditional PROTACs have been reported, there are still numerous unsolved issues for effective targeted protein degradation. We believe that future interdisciplinary research in chemistry, biology, and medicine will find a way to control the protein degradation more precisely and more efficiently.
Conflict of Interests
The authors declare no conflict of interest.
Biographical Information
Junhyeong Yim received his D.V.M (2016) in the college of veterinary medicine at Seoul National University and Ph.D (2023) in the Department of Biophysics and Chemical Biology at Seoul National University. After postdoctoral work at Seoul National University, he is working at the Multi‐dimensional Genomics Research Center at Kangwon National University. His research interests are immunomodulation, targeted protein modulation and small molecule‐target protein identification.

Biographical Information
Ala Jo received her B.S. (2011) and Ph.D. (2017) from the Department of Chemistry at Seoul National University. After postdoctoral work at Harvard Medical School, she is working at the Center for Nanomedicine of Institute for Basic Science at Yonsei University, Korea. Her research interest is focused on the extracellular vesicle analysis for disease diagnosis.

Biographical Information
Junyoung Park received his B.S. (2021) and M.S. (2023) from the Department of Chemistry at Kangwon National University. He is currently a Ph.D. candidate supervised by Prof. Jongmin Park at Kangwon National University. His research interests are focused on the development of novel bioprobes and exploration of targeted protein degradation (TPD) strategies.

Biographical Information
Minseob Koh completed his B.S. (2007) and Ph.D. (2013) in the Department of Chemistry at Seoul National University. After postdoctoral work at Scripps Research, he began his academic career in 2020 in the Department of Chemistry at Pusan National University, Korea. His research is primarily focused on the design of genetic circuits for biological logic gates, expanding the genetic code, and engineering proteins for use in biomedical applications.

Biographical Information
Gabin Kim received her B.S. in chemistry from Pusan National University in 2024. She is currently a M.S. student under the supervision of Prof. Minseob Koh at Pusan National University. Her research focuses genetic code expansion and directed evolution.

Biographical Information
Jongmin Park received his B.S. (2005) and Ph.D. (2012) from the Department of Chemistry at Seoul National University. After postdoctoral work at Harvard Medical School, he began his academic career in 2018 in the Department of Chemistry at Kangwon National University, Korea. His research interests are focused on the development of novel bioprobes, small molecule‐target protein identification and extracellular vesicle analysis for disease diagnosis.

Acknowledgments
Junhyeong Yim, Junyoung Park, and Gabin Kim contributed equally to this work. This work was supported by the Creative Challenging Research Grant (RS‐2023‐00250887 to J.Y.), the Basic Science Research Program (2023R1A2C1007899 to J.P., 2021R1C1C1005766 to M.K., and 2022R1C1C2008563 to A.J.) and Priority Research Institute Program (RS‐2023‐00271205 to J.P.) through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning. This work was also supported by Korea Basic Science Institute (KBSI) National Research Facilities & Equipment Center (NFEC) grant (2019R1A6C1010006 to J.P.) and Global‐Learning & Academic research institution for Master's⋅PhD students, and Postdocs (G‐LAMP) program (RS‐2023‐00301850 to J.P.) from the Ministry of Education, South Korea. This work is supported by 2019 Research Grant from Kangwon National University. This work was supported by the National Cancer Center Grant (NCC‐2211720) Figures were created with BioRender.com.
Yim J., Park J., Kim G., Lee H. H., Chung J. S., Jo A., Koh M., Park J., ChemMedChem 2024, 19, e202400326. 10.1002/cmdc.202400326
Contributor Information
Dr. Ala Jo, Email: chemko88@gmail.com.
Prof. Minseob Koh, Email: minseob.koh@pusan.ac.kr.
Prof. Jongmin Park, Email: jpark@kangwon.ac.kr.
References
- 1. Hoppe T., Cohen E., Genetics 2020, 215(4), 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hetz C., Chevet E., Oakes S. A., Nat. Cell Biol. 2015, 17(7), 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Read A., Schröder M., Biology 2021, 10(5), 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li Y., Li S., Wu H., Cells 2022, 11(5), 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bett J. S., Essays Biochem. 2016, 60(2), 143–151. [DOI] [PubMed] [Google Scholar]
- 6. Aman Y., Schmauck-Medina T., Hansen M., Morimoto R. I., Simon A. K., Bjedov I., Palikaras K., Simonsen A., Johansen T., Tavernarakis N., Nat. Aging 2021, 1(8), 634–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li J., Zhang D., Wiersma M., Brundel B. J., Cells 2018, 7(12), 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brancolini C., Iuliano L., Cancers 2020, 12(9), 2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen X.-Q., Shen T., Fang S.-J., Sun X.-M., Li G.-Y., Li Y.-F., Front Cell Dev. Biol. 2023, 11, 1143532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oakes S. A., Am. J. Physiol. Cell Physiol. 2017, 312(2), C93–C102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mercier R., LaPointe P., J. Biol. Chem. 2022, 298(5), 101930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Höhn A., Tramutola A., Cascella R., Oxid. Med. Cell. Longev. 2020, 2020, 5497046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sonninen T.-M., Goldsteins G., Laham-Karam N., Koistinaho J., Lehtonen Š., Cells 2020, 9(10), 2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papendorf J. J., Krüger E., Ebstein F., Cells 2022, 11(9), 1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marques M., Ramos B., Soares A. R., Ribeiro D., Cells 2019, 8(3), 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khomari F., Nabi-Afjadi M., Yarahmadi S., Eskandari H., Bahreini E., Biol. Proced. Online 2021, 23, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Neves-da-Rocha J., Santos-Saboya M. J., Lopes M. E., Rossi A., Martinez-Rossi N. M., Microorganisms 2023, 11(8), 1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alabi S. B., Crews C. M., J. Biol. Chem. 2021, 296, 100647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sakamoto K. M., Kim K. B., Kumagai A., Mercurio F., Crews C. M., Deshaies R. J., Proc. Natl. Acad. Sci. USA 2001, 98(15), 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Modell A. E., Lai S., Nguyen T. M., Choudhary A., Cell Chem. Biol. 2021, 28(7), 1081–1089. [DOI] [PubMed] [Google Scholar]
- 21. Johnson D. S., Weerapana E., Cravatt B. F., Future Med. Chem. 2010, 2(6), 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lai A. C., Crews C. M., Nat. Rev. Drug Discov. 2017, 16(2), 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xie X., Yu T., Li X., Zhang N., Foster L. J., Peng C., Huang W., He G., Signal Transduct. Target Ther. 2023, 8(1), 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K., Hines J., Winkler J. D., Crew A. P., Coleman K., Chem. Biol. 2015, 22(6), 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bondeson D. P., Mares A., Smith I. E., Ko E., Campos S., Miah A. H., Mulholland K. E., Routly N., Buckley D. L., Gustafson J. L., Nat. Chem. Biol. 2015, 11(8), 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou H., Bai L., Xu R., Zhao Y., Chen J., McEachern D., Chinnaswamy K., Wen B., Dai L., Kumar P., J. Med. Chem. 2019, 62(24), 11280–11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kargbo R. B., ACS Med. Chem. Lett. 2019, 10, 699–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Neklesa T., Snyder L. B., Willard R. R., Vitale N., Pizzano J., Gordon D. A., Bookbinder M., Macaluso J., Dong H., Ferraro C., J. Clin. Oncol. 2019, 37(259), 10.1200 [Google Scholar]
- 29. Hu J., Hu B., Wang M., Xu F., Miao B., Yang C.-Y., Wang M., Liu Z., Hayes D. F., Chinnaswamy K., J. Med. Chem. 2019, 62(3), 1420–1442. [DOI] [PubMed] [Google Scholar]
- 30. Sun X., Gao H., Yang Y., He M., Wu Y., Song Y., Tong Y., Rao Y., Signal Transduct. Target Ther. 2019, 4(1), 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mullard A., Nat. Rev. Drug Discov. 2021, 20(4), 247–250. [DOI] [PubMed] [Google Scholar]
- 32. Liu Z., Hu M., Yang Y., Du C., Zhou H., Liu C., Chen Y., Fan L., Ma H., Gong Y., Mol. Biomed. 2022, 3(1), 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lipinski C. A., Drug Discov. Today: Technol. 2004, 1(4), 337–341. [DOI] [PubMed] [Google Scholar]
- 34. Li X., Song Y., J. Hematol. Oncol. 2020, 13, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Békés M., Langley D. R., Crews C. M., Nat. Rev. Drug Discov. 2022, 21(3), 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boonstra M. C., De Geus S. W., Prevoo H. A., Hawinkels L. J., Van De Velde C. J., Kuppen P. J., Vahrmeijer A. L., Sier C. F., Biomark. Cancer 2016, 8, BIC. S38542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsao L.-C., Force J., Hartman Z. C., Cancer Res. 2021, 81(18), 4641–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Han J., Gao L., Wang J., Wang J., J. Cancer 2020, 11(23), 6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhong L., Li Y., Xiong L., Wang W., Wu M., Yuan T., Yang W., Tian C., Miao Z., Wang T., Signal Transduct. Target. Ther. 2021, 6(1), 1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fernández M., Javaid F., Chudasama V., Chem. Sci. 2018, 9(4), 790–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moon Y., Jeon S. I., Shim M. K., Kim K., Pharmaceutics 2023, 15(2), 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fu Z., Li S., Han S., Shi C., Zhang Y., Signal Transduct. Target Ther. 2022, 7(1), 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bargh J. D., Isidro-Llobet A., Parker J. S., Spring D. R., Chem. Soc. Rev. 2019, 48(16), 4361–4374. [DOI] [PubMed] [Google Scholar]
- 44. Joubert N., Beck A., Dumontet C., Denevault-Sabourin C., Pharmaceuticals 2020, 13(9), 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maecker H., Jonnalagadda V., Bhakta S., Jammalamadaka V., Junutula J. R., mAbs 2023, 15(1), 2229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Conilh L., Sadilkova L., Viricel W., Dumontet C., J. Hematol. Oncol. 2023, 16(1), 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cantrill C., Chaturvedi P., Rynn C., Schaffland J. P., Walter I., Wittwer M. B., Drug Discov. Today 2020, 25(6), 969–982. [DOI] [PubMed] [Google Scholar]
- 48. Wu S.-Y., Chiang C.-M., J. Biol. Chem. 2007, 282(18), 13141–13145. [DOI] [PubMed] [Google Scholar]
- 49. Donati B., Lorenzini E., Ciarrocchi A., Mol. Cancer 2018, 17(1), 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheung K. L., Kim C., Zhou M.-M., Front. Mol. Biosci. 2021, 8, 728777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang C.-Y., Qin C., Bai L., Wang S., Drug Discov. Today: Technol. 2019, 31, 43–51. [DOI] [PubMed] [Google Scholar]
- 52. Pillow T. H., Adhikari P., Blake R. A., Chen J., Del Rosario G., Deshmukh G., Figueroa I., Gascoigne K. E., Kamath A. V., Kaufman S., ChemMedChem 2020, 15(1), 17–25. [DOI] [PubMed] [Google Scholar]
- 53. Dragovich P. S., Pillow T. H., Blake R. A., Sadowsky J. D., Adaligil E., Adhikari P., Bhakta S., Blaquiere N., Chen J., dela Cruz-Chuh J., J. Med. Chem. 2021, 64(5), 2534–2575. [DOI] [PubMed] [Google Scholar]
- 54. Dragovich P. S., Pillow T. H., Blake R. A., Sadowsky J. D., Adaligil E., Adhikari P., Chen J., Corr N., dela Cruz-Chuh J., Del Rosario G., J. Med. Chem. 2021, 64(5), 2576–2607. [DOI] [PubMed] [Google Scholar]
- 55. Maneiro M. A., Forte N., Shchepinova M. M., Kounde C. S., Chudasama V., Baker J. R., Tate E. W., ACS Chem. Biol. 2020, 15(6), 1306–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dragovich P. S., Adhikari P., Blake R. A., Blaquiere N., Chen J., Cheng Y.-X., den Besten W., Han J., Hartman S. J., He J., Bioorg. Med. Chem. Lett. 2020, 30(4), 126907. [DOI] [PubMed] [Google Scholar]
- 57.J. Palacino, C. Bai, Y. Yi, A. Skaletskaya, K. Takrouri, W. Wong, M.-S. Kim, D.-K. Choi, D.-Y. Kim, Y. Yang, Cancer Res. 2022, 82(12_Supplement), 3933–3933.
- 58.S. A. Hurvitz, E. P. Hamilton, A. I. Spira, P. R. Pohlmann, A. Giordano, K. Clifton, B. D. Anderson, S. Dutta, U. Mangipudi, S. Saini, J. Clin. Oncol. 2023, 41(16_Supplement), TPS1114.
- 59. Keefe A. D., Pai S., Ellington A., Nat. Rev. Drug Discov. 2010, 9(7), 537–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bunka D. H., Stockley P. G., Nat. Rev. Microbiol. 2006, 4(8), 588–596. [DOI] [PubMed] [Google Scholar]
- 61. Kong H. Y., Byun J., Biomol. Ther. 2013, 21(6), 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim D.-H., Seo J.-M., Shin K.-J., Yang S.-G., Biomater. Res. 2021, 25, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhou J., Rossi J., Nat. Rev. Drug Discov. 2017, 16(3), 181–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Thiel K. W., Giangrande P. H., Ther. Deliv. 2010, 1(6), 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mongelard F., Bouvet P., Curr. Opin. Mol. Ther. 2010, 12(1), 107–114. [PubMed] [Google Scholar]
- 66. Bates P. J., Laber D. A., Miller D. M., Thomas S. D., Trent J. O., Exp. Mol. Pathol. 2009, 86(3), 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rosenberg J. E., Bambury R. M., Van Allen E. M., Drabkin H. A., Lara P. N., Harzstark A. L., Wagle N., Figlin R. A., Smith G. W., Garraway L. A., Invest. New Drugs 2014, 32, 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yazdian-Robati R., Bayat P., Oroojalian F., Zargari M., Ramezani M., Taghdisi S. M., Abnous K., Int. J. Biol. Macromol. 2020, 155, 1420–1431. [DOI] [PubMed] [Google Scholar]
- 69. He S., Gao F., Ma J., Ma H., Dong G., Sheng C., Angew. Chem. 2021, 133(43), 23487–23493. [DOI] [PubMed] [Google Scholar]
- 70. Liu R., Liu Z., Chen M., Xing H., Zhang P., Zhang J., Chem. Sci. 2024, 15(1), 134–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhao R., Goldman I. D., Mol. Asp. Med. 2013, 34(2–3), 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhao R., Diop-Bove N., Visentin M., Goldman I. D., Annu. Rev. Nutr. 2011, 31, 177–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yi Y.-S., Immune Netw. 2016, 16(6), 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Elnakat H., Adv. Drug Deliv. Rev. 2004, 56(8), 1067–1084. [DOI] [PubMed] [Google Scholar]
- 75. Sudimack J., Lee R. J., Adv. Drug Deliv. Rev. 2000, 41(2), 147–162. [DOI] [PubMed] [Google Scholar]
- 76. Zhao X., Li H., Lee R. J., Expert Opin. Drug Deliv. 2008, 5(3), 309–319. [DOI] [PubMed] [Google Scholar]
- 77. Liu J., Chen H., Liu Y., Shen Y., Meng F., Kaniskan H. U., Jin J., Wei W., J. Am. Chem. Soc. 2021, 143(19), 7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chen H., Liu J., Kaniskan H. U. M., Wei W., Jin J., J. Med. Chem. 2021, 64(16), 12273–12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Paiva S.-L., Crews C. M., Curr. Opin. Chem. Biol. 2019, 50, 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yang F., Luo Q., Wang Y., Liang H., Wang Y., Hou Z., Wan C., Wang Y., Liu Z., Ye Y., J. Am. Chem. Soc. 2023, 145(14), 7879–7887. [DOI] [PubMed] [Google Scholar]
- 81. Reches M., Gazit E., Nano Lett. 2004, 4(4), 581–585. [Google Scholar]
- 82. Song C., Jiao Z., Hou Z., Wang R., Lian C., Xing Y., Luo Q., An Y., Yang F., Wang Y., J. Am. Chem. Soc. 2023, 145(40), 21860–21870. [DOI] [PubMed] [Google Scholar]
- 83. Ellis-Davies G. C., Nat. Methods 2007, 4(8), 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mayer G., Heckel A., Angew. Chem. Int. Ed. 2006, 45(30), 4900–4921. [DOI] [PubMed] [Google Scholar]
- 85. Klán P., Solomek T., Bochet C. G., Blanc A., Givens R., Rubina M., Popik V., Kostikov A., Wirz J., Chem. Rev. 2013, 113(1), 119–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cadet J., Douki T., J. Invest. Dermatol. 2011, 131(5), 1005–1007. [DOI] [PubMed] [Google Scholar]
- 87. Mouret S., Baudouin C., Charveron M., Favier A., Cadet J., Douki T., Proc. Natl. Acad. Sci. USA 2006, 103(37), 13765–13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Oikawa S., Tada-Oikawa S., Kawanishi S., Biochemistry 2001, 40(15), 4763–4768. [DOI] [PubMed] [Google Scholar]
- 89. Petit M., Bort G., Doan B. T., Sicard C., Ogden D., Scherman D., Ferroud C., Dalko P. I., Angew. Chem. Int. Ed. 2011, 41(50), 9708–9711. [DOI] [PubMed] [Google Scholar]
- 90. Cao Z. Q., Wang G. J., Chem. Rec. 2016, 16(3), 1398–1435. [DOI] [PubMed] [Google Scholar]
- 91. Cabane E., Zhang X., Langowska K., Palivan C. G., Meier W., Biointerphases 2012, 7(1). [DOI] [PubMed] [Google Scholar]
- 92. Wei M., Gao Y., Li X., Serpe M. J., Polym. Chem. 2017, 8(1), 127–143. [Google Scholar]
- 93. Gao J., Yang L., Lei S., Zhou F., Nie H., Peng B., Xu T., Chen X., Yang X., Sheng C., Sci. Bull. 2023, 68, 1069–1085. [DOI] [PubMed] [Google Scholar]
- 94. An K., Deng X., Chi H., Zhang Y., Li Y., Cheng M., Ni Z., Yang Z., Wang C., Chen J., Angew. Chem. Int. Ed. 2023, 62(39), e202306824. [DOI] [PubMed] [Google Scholar]
- 95. Szatrowski T. P., Nathan C. F., Cancer Res 1991, 51(3), 794–798. [PubMed] [Google Scholar]
- 96. Winterbourn C. C., Biochem. Soc. Trans. 2020, 48(3), 745–754. [DOI] [PubMed] [Google Scholar]
- 97. Kuivila H. G., Armour A. G., J. Am. Chem. Soc. 1957, 79(21), 5659–5662. [Google Scholar]
- 98. Silva M. P., Saraiva L., Pinto M., Sousa M. E., Molecules 2020, 25(18), 4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Alouane A., Labruere R., Le Saux T., Schmidt F., Jullien L., Angew. Chem. Int. Ed. Engl. 2015, 54(26), 7492–7509. [DOI] [PubMed] [Google Scholar]
- 100. Saxon E., Peng X., ChemBioChem 2022, 23(3), e202100366. [DOI] [PubMed] [Google Scholar]
- 101. Liu H., Ren C., Sun R., Wang H., Zhan Y., Yang X., Jiang B., Chen H., Chem. Commun. 2022, 58(72), 10072–10075. [DOI] [PubMed] [Google Scholar]
- 102. Yu D., Fan H., Zhou Z., Zhang Y., Sun J., Wang L., Jia Y., Tian J., Campbell A., Mi W., ChemBioChem 2023, 24(17), e202300422. [DOI] [PubMed] [Google Scholar]
- 103. Lippert A. R., Van de Bittner G. C., Chang C. J., ACC Chem. Res. 2011, 44(9), 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Liang C. J., Zheng Q. Z., Luo T. L., Cai W. Q., Mao L. Q., Wang M., CCS Chem. 2022, 4(12), 3809–3819. [Google Scholar]
- 105. Jia Q., Zhang Y., Liu F., Dong W., Zhu L., Wang F., Jiang J. H., Anal. Chem. 2023, 95(45), 16474–16480. [DOI] [PubMed] [Google Scholar]
- 106. Zeng W., Liu P., Pan W., Singh S. R., Wei Y., Cancer Lett. 2015, 356(2), 263–267. [DOI] [PubMed] [Google Scholar]
- 107. Cheng W., Li S., Wen X., Han S., Wang S., Wei H., Song Z., Wang Y., Tian X., Zhang X., Chem. Commun. 2021, 57(95), 12852–12855. [DOI] [PubMed] [Google Scholar]
- 108. Xie B., Xu B., Xin L., Wei Y., Guo X., Dong C., Bioorg. Chem. 2023, 137, 106590. [DOI] [PubMed] [Google Scholar]
- 109. Cheng W., Li S., Han S., Miao R., Wang S., Liu C., Wei H., Tian X., Zhang X., Bioorg. Med. Chem. 2023, 82, 117237. [DOI] [PubMed] [Google Scholar]
- 110. Shi S., Du Y., Zou Y., Niu J., Cai Z., Wang X., Qiu F., Ding Y., Yang G., Wu Y., Xu Y., Zhu Q., J. Med. Chem. 2022, 65(6), 5057–5071. [DOI] [PubMed] [Google Scholar]
- 111. Do T. C., Lau J. W., Sun C., Liu S., Kha K. T., Lim S. T., Oon Y. Y., Kwan Y. P., Ma J. J., Mu Y., Liu X., Carney T. J., Wang X., Xing B., Sci. Adv. 2022, 8(50), eabq2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Pompella A., Visvikis A., Paolicchi A., De Tata V., Casini A. F., Biochem. Pharmacol. 2003, 66(8), 1499–1503. [DOI] [PubMed] [Google Scholar]
- 113. Meister A., Anderson M. E., Annu. Rev. Biochem. 1983, 52(1), 711–760. [DOI] [PubMed] [Google Scholar]
- 114. Xue L., Yu D., Sun J., Guan L., Xie C., Wang L., Jia Y., Tian J., Fan H., Sun H., Analyst 2023, 148(3), 532–538. [DOI] [PubMed] [Google Scholar]
- 115. Ling X., Tu J., Wang J., Shajii A., Kong N., Feng C., Zhang Y., Yu M., Xie T., Bharwani Z., ACS Nano 2018, 13(1), 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Zhou Z., Fan H., Yu D., Shi F., Li Q., Zhang Z., Wang X., Zhang X., Dong C., Sun H., Bioorg. Med. Chem. 2023, 96, 117526. [DOI] [PubMed] [Google Scholar]
- 117. Fan H., Zhou Z., Yu D., Sun J., Wang L., Jia Y., Tian J., Mi W., Sun H., Bioorg. Chem. 2023, 140, 106793. [DOI] [PubMed] [Google Scholar]
- 118. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. Engl. 2001, 40(11), 2004–2021. [DOI] [PubMed] [Google Scholar]
- 119. Baskin J. M., Bertozzi C. R., Qsar. Comb. Sci. 2007, 26(11–12), 1211–1219. [Google Scholar]
- 120. Zeng D., Zeglis B. M., Lewis J. S., Anderson C. J., J. Nucl. Med. 2013, 54(6), 829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kamiya T., J. Clin. Biochem. Nutr. 2022, 71(1), 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Margalioth E. J., Schenker J. G., Chevion M., Cancer 1983, 52(5), 868–872. [DOI] [PubMed] [Google Scholar]
- 123. Xue X., Qian C., Tao Q., Dai Y., Lv M., Dong J., Su Z., Qian Y., Zhao J., Liu H. K., Guo Z., Natl. Sci. Rev. 2021, 8(9), nwaa286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liu H., Zhang Y., Zheng S., Weng Z., Ma J., Li Y., Xie X., Zheng W., Biochem. Biophys. Res. Commun. 2016, 477(4), 1031–1037. [DOI] [PubMed] [Google Scholar]
- 125. Si R., Hai P., Zheng Y., Wang J., Zhang Q., Li Y., Pan X., Zhang J., Eur. J. Med. Chem. 2023, 257, 115497. [DOI] [PubMed] [Google Scholar]
- 126. Rossin R., van Duijnhoven S. M., Ten Hoeve W., Janssen H. M., Kleijn L. H., Hoeben F. J., Versteegen R. M., Robillard M. S., Bioconjug. Chem. 2016, 27(7), 1697–1706. [DOI] [PubMed] [Google Scholar]
- 127. Khan I., Agris P. F., Yigit M. V., Royzen M., Chem. Commun. 2016, 52(36), 6174–6177. [DOI] [PubMed] [Google Scholar]
- 128. Brooks P. C., Cell 1994, 79(7), 1157–1164. [DOI] [PubMed] [Google Scholar]
- 129. Chang M., Gao F., Pontigon D., Gnawali G., Xu H., Wang W., J. Am. Chem. Soc. 2023, 145(25), 14155–14163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Bi T., Liang P., Zhou Y., Wang H., Huang R., Sun Q., Shen H., Yang S., Ren W., Liu Z., J. Med. Chem. 2023, 66(21), 14843–14852. [DOI] [PubMed] [Google Scholar]
- 131. Wu J. B., Lin T. P., Gallagher J. D., Kushal S., Chung L. W., Zhau H. E., Olenyuk B. Z., Shih J. C., J. Am. Chem. Soc. 2015, 137(6), 2366–2374. [DOI] [PubMed] [Google Scholar]
- 132. Choi P. J., Park T. I. H., Cooper E., Dragunow M., Denny W. A., Jose J., Bioconjug. Chem. 2020, 31(7), 1724–1739. [DOI] [PubMed] [Google Scholar]
- 133. Mingchao Xiao J. Z., Qiang Wang, Jia Liu, Ma Leina, Biomolecules 2022, 12(9), 1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Knall A. C., Slugovc C., Chem. Soc. Rev. 2013, 42(12), 5131–5142. [DOI] [PubMed] [Google Scholar]
- 135. Lebraud H., Wright D. J., Johnson C. N., Heightman T. D., ACS Cent. Sci. 2016, 2(12), 927–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Si R., Zhu H., Wang J., Zhang Q., Li Y., Pan X., Zhang J., Bioorg. Chem. 2023, 135, 106497. [DOI] [PubMed] [Google Scholar]
- 137. Jin Y., Fan J., Wang R., Wang X., Li N., You Q., Jiang Z., J. Am. Chem. Soc. 2023, 145(13), 7218–7229. [DOI] [PubMed] [Google Scholar]
- 138. Yoon M. S., Lee Y. J., Shin H. J., Park C. W., Han S. B., Jung J. K., Kim J. S., Shin D. H., Pharmaceutics 2020, 12(12), 1156.33261219 [Google Scholar]
- 139. Han H. H., Wang H. M., Jangili P., Li M., Wu L., Zang Y., Sedgwick A. C., Li J., He X. P., James T. D., Kim J. S., Chem. Soc. Rev. 2023, 52(3), 879–920. [DOI] [PubMed] [Google Scholar]
- 140. Geng J., Zhang Y., Gao Q., Neumann K., Dong H., Porter H., Potter M., Ren H., Argyle D., Bradley M., Nat. Chem. 2021, 13(8), 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Silva J. M., Silva E., Reis R. L., J. Control Release 2019, 298, 154–176. [DOI] [PubMed] [Google Scholar]
- 142. Aujard I., Benbrahim C., Gouget M., Ruel O., Baudin J. B., Neveu P., Jullien L., Chemistry 2006, 12(26), 6865–6879. [DOI] [PubMed] [Google Scholar]
- 143. Givens R. S., Stensrud K., Conrad, 2nd P. G., Yousef A. L., Perera C., Senadheera S. N., Heger D., Wirz J., Can. J. Chem. 2011, 89(3), 364–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hammer C. A., Falahati K., Jakob A., Klimek R., Burghardt I., Heckel A., Wachtveitl J., J. Phys. Chem. Lett. 2018, 9(6), 1448–1453. [DOI] [PubMed] [Google Scholar]
- 145. Horbert R., Pinchuk B., Davies P., Alessi D., Peifer C., ACS Chem. Biol. 2015, 10(9), 2099–2107. [DOI] [PubMed] [Google Scholar]
- 146. Skwarczynski M., Noguchi M., Hirota S., Sohma Y., Kimura T., Hayashi Y., Kiso Y., Bioorg. Med. Chem. Lett. 2006, 16(17), 4492–4496. [DOI] [PubMed] [Google Scholar]
- 147. Wong P. T., Tang S., Cannon J., Mukherjee J., Isham D., Gam K., Payne M., Yanik S. A., J. R. Baker, Jr. , Choi S. K., ChemBioChem 2017, 18(1), 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Xue G., Wang K., Zhou D., Zhong H., Pan Z., J. Am. Chem. Soc. 2019, 141(46), 18370–18374. [DOI] [PubMed] [Google Scholar]
- 149. Naro Y., Darrah K., Deiters A., J. Am. Chem. Soc. 2020, 142(5), 2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Liu J., Chen H., Ma L., He Z., Wang D., Liu Y., Lin Q., Zhang T., Gray N., Kaniskan H. U., Jin J., Wei W., Sci. Adv. 2020, 6(8), eaay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Kounde C. S., Shchepinova M. M., Saunders C. N., Muelbaier M., Rackham M. D., Harling J. D., Tate E. W., Chem. Commun. 2020, 56(41), 5532–5535. [DOI] [PubMed] [Google Scholar]
- 152. Weng W., Xue G., Pan Z., Eur. J. Med. Chem. 2024, 265, 116062. [DOI] [PubMed] [Google Scholar]
- 153. Winter G. E., Buckley D. L., Paulk J., Roberts J. M., Souza A., Dhe-Paganon S., Bradner J. E., Science 2015, 348(6241), 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Buhimschi A. D., Armstrong H. A., Toure M., Jaime-Figueroa S., Chen T. L., Lehman A. M., Woyach J. A., Johnson A. J., Byrd J. C., Crews C. M., Biochemistry 2018, 57(26), 3564–3575. [DOI] [PubMed] [Google Scholar]
- 155. Kim K., Park H., Lim K. M., Toxicol. Res. 2015, 31(2), 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kciuk M., Marciniak B., Mojzych M., Kontek R., Int. J. Mol. Sci. 2020, 21(19), 7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Momotake A., Lindegger N., Niggli E., Barsotti R. J., Ellis-Davies G. C., Nat. Methods 2006, 3(1), 35–40. [DOI] [PubMed] [Google Scholar]
- 158. Schaue D., McBride W. H., Nat. Rev. Clin. Oncol. 2015, 12(9), 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Allen C., Her S., Jaffray D. A., Adv. Drug Deliv. Rev. 2017, 109, 1–2. [DOI] [PubMed] [Google Scholar]
- 160. Lu Q.-B., Mutat. Res. 2010, 704(1–3), 190–199. [DOI] [PubMed] [Google Scholar]
- 161. Geng J., Zhang Y., Gao Q., Neumann K., Dong H., Porter H., Potter M., Ren H., Argyle D., Bradley M., Nat. Chem. 2021, 13(8), 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Cao Y., Si J., Zheng M., Zhou Q., Ge Z., Chem. Commun. 2023, 59(54), 8323–8331. [DOI] [PubMed] [Google Scholar]
- 163. Ding Z., Guo Z., Zheng Y., Wang Z., Fu Q., Liu Z., J. Am. Chem. Soc. 2022, 144(21), 9458–9464. [DOI] [PubMed] [Google Scholar]
- 164. Guo Z., Hong H., Zheng Y., Wang Z., Ding Z., Fu Q., Liu Z., Angew. Chem. Int. Ed. 2022, 61(34), e202205014. [DOI] [PubMed] [Google Scholar]
- 165. Yang C., Yang Y., Li Y., Ni Q., Li J., J. Am. Chem. Soc. 2023, 145(1), 385–391. [DOI] [PubMed] [Google Scholar]
- 166. Isacsson B. G., Ulf, Acta Oncol. 2001, 40(8), 958–967. [DOI] [PubMed] [Google Scholar]
- 167. Chen Y., Tandon I., Heelan W., Wang Y., Tang W., Hu Q., Chem. Soc. Rev. 2022, 51(13), 5330–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Luo T., Zheng Q., Shao L., Ma T., Mao L., Wang M., Angew. Chem. 2022, 134(39), e202206277. [DOI] [PubMed] [Google Scholar]
- 169. Zhang C., He S., Zeng Z., Cheng P., Pu K., Angew. Chem. Int. Ed. 2022, 61(8), e202114957. [DOI] [PubMed] [Google Scholar]
- 170. Zhang C., Zeng Z., Cui D., He S., Jiang Y., Li J., Huang J., Pu K., Nat. Commun. 2021, 12(1), 2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Chen J., Qiu M., Ma F., Yang L., Glass Z., Xu Q., J. Control. Release 2021, 330, 1244–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Wang Y., Han L., Liu F., Yang F., Jiang X., Sun H., Feng F., Xue J., Liu W., Colloids Surf., B 2020, 188, 110795. [DOI] [PubMed] [Google Scholar]
- 173.S. L. Jacques, Advances in Optical Imaging and Photon Migration, 1996, OPC364.
- 174. Zhu S., Tian R., Antaris A. L., Chen X., Dai H., Adv. Mater. 2019, 31(24), 1900321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Vankayala R., Hwang K. C., Adv. Mater. 2018, 30(23), 1706320. [DOI] [PubMed] [Google Scholar]
- 176. Sasaki Y., Oshikawa M., Bharmoria P., Kouno H., Hayashi-Takagi A., Sato M., Ajioka I., Yanai N., Kimizuka N., Angew. Chem. Int. Ed. 2019, 58(49), 17827–17833. [DOI] [PubMed] [Google Scholar]
- 177. Vahrmeijer A. L., Hutteman M., Van Der Vorst J. R., Van De Velde C. J., Frangioni J. V., Nat. Rev. Clin. Oncol. 2013, 10(9), 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Salimi M., Mosca S., Gardner B., Palombo F., Matousek P., Stone N., Nanomaterials 2022, 12(6), 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Deng X., Shao Z., Zhao Y., Adv. Sci. 2021, 8(3), 2002504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Denkova A. G., de Kruijff R. M., Serra-Crespo P., Adv. Healthcare Mater. 2018, 7(8), 1701211. [DOI] [PubMed] [Google Scholar]
- 181. Gu Z., Zhu S., Yan L., Zhao F., Zhao Y., Adv. Mater. 2019, 31(9), 1800662. [DOI] [PubMed] [Google Scholar]
- 182. Liu Y., Crawford B. M., Vo-Dinh T., Immunotherapy 2018, 10(13), 1175–1188. [DOI] [PubMed] [Google Scholar]
- 183. Sun H., Feng M., Chen S., Wang R., Luo Y., Yin B., Li J., Wang X., J. Mater. Chem. B 2020, 8(32), 7149–7159. [DOI] [PubMed] [Google Scholar]
- 184. He Q., Zhou L., Yu D., Zhu R., Chen Y., Song M., Liu X., Liao Y., Ding T., Fan W., J. Med. Chem. 2023, 66(15), 10458–10472. [DOI] [PubMed] [Google Scholar]
- 185. Wang W., Zhu C., Zhang B., Feng Y., Zhang Y., Li J., J. Am. Chem. Soc. 2023, 145(30), 16642–16649. [DOI] [PubMed] [Google Scholar]
- 186. Rumack C. M., Levine D., Diagnostic Ultrasound, Amsterdam: Elsevier Health Sciences, 2023. [Google Scholar]
- 187. Ter Haar G., Prog. Biophys. Mol. Biol. 2007, 93(1–3), 111–129. [DOI] [PubMed] [Google Scholar]
- 188. Ouyang J., Xie A., Zhou J., Liu R., Wang L., Liu H., Kong N., Tao W., Chem. Soc. Rev. 2022, 51(12), 4996–5041. [DOI] [PubMed] [Google Scholar]
- 189. Wells P. N., Phys. Med. Biol. 2006, 51(13), R83. [DOI] [PubMed] [Google Scholar]
- 190. Rosenthal I., Sostaric J. Z., Riesz P., Ultrason. Sonochem. 2004, 11(6), 349–363. [DOI] [PubMed] [Google Scholar]
- 191. Konan Y. N., Gurny R., Allémann E., J. Photochem. Photobiol., B 2002, 66(2), 89–106. [DOI] [PubMed] [Google Scholar]
- 192. Qian X., Zheng Y., Chen Y., Adv. Mater. 2016, 28(37), 8097–8129. [DOI] [PubMed] [Google Scholar]
- 193. Shibaguchi H., Tsuru H., Kuroki M., Kuroki M., Anticancer Res. 2011, 31(7), 2425–2429. [PubMed] [Google Scholar]
- 194. Xu H., Zhang X., Han R., Yang P., Ma H., Song Y., Lu Z., Yin W., Wu X., Wang H., RSC Adv. 2016, 6(56), 50697–50705. [Google Scholar]
- 195. Wang F., Dong G., Ding M., Yu N., Sheng C., Li J., Small 2024, 20(8), 2306378. [DOI] [PubMed] [Google Scholar]
- 196. Brieke C., Rohrbach F., Gottschalk A., Mayer G., Heckel A., Angew. Chem. Int. Ed. Engl. 2012, 51(34), 8446–8476. [DOI] [PubMed] [Google Scholar]
- 197. Bandara H. M., Burdette S. C., Chem. Soc. Rev. 2012, 41(5), 1809–1825. [DOI] [PubMed] [Google Scholar]
- 198. Beharry A. A., Woolley G. A., Chem. Soc. Rev. 2011, 40(8), 4422–4437. [DOI] [PubMed] [Google Scholar]
- 199. Pfaff P., Samarasinghe K. T. G., Crews C. M., Carreira E. M., ACS Cent. Sci. 2019, 5(10), 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Reynders M., Matsuura B. S., Berouti M., Simoneschi D., Marzio A., Pagano M., Trauner D., Sci. Adv. 2020, 6(8), eaay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Jin Y. H., Lu M. C., Wang Y., Shan W. X., Wang X. Y., You Q. D., Jiang Z. Y., J. Med. Chem. 2020, 63(9), 4644–4654. [DOI] [PubMed] [Google Scholar]
- 202. Bleger D., Schwarz J., Brouwer A. M., Hecht S., J. Am. Chem. Soc. 2012, 134(51), 20597–20600. [DOI] [PubMed] [Google Scholar]
- 203. Calbo J., Weston C. E., White A. J., Rzepa H. S., Contreras-Garcia J., Fuchter M. J., J. Am. Chem. Soc. 2017, 139(3), 1261–1274. [DOI] [PubMed] [Google Scholar]
- 204. Weston C. E., Richardson R. D., Haycock P. R., White A. J., Fuchter M. J., J. Am. Chem. Soc. 2014, 136(34), 11878–11881. [DOI] [PubMed] [Google Scholar]
- 205. Zhang Q., Kounde C. S., Mondal M., Greenfield J. L., Baker J. R., Kotelnikov S., Ignatov M., Tinworth C. P., Zhang L., Conole D., De Vita E., Kozakov D., McCluskey A., Harling J. D., Fuchter M. J., Tate E. W., Chem. Commun. 2022, 58(78), 10933–10936. [DOI] [PMC free article] [PubMed] [Google Scholar]