

Abstract
Complex amyloid aggregation of amyloid-β (1–40) (Aβ1–40) in terms of monomer structures has not been fully understood. Herein, we report the microscopic mechanism and pathways of Aβ1–40 aggregation with macroscopic viewpoints through tuning its initial structure and solubility. Partial helical structures of Aβ1–40 induced by low solvent polarity accelerated cytotoxic Aβ1–40 amyloid fibrillation, while predominantly helical folds did not aggregate. Changes in the solvent polarity caused a rapid formation of β-structure-rich protofibrils or oligomers via aggregation-prone helical structures. Modulation of the pH and salt concentration transformed oligomers to protofibrils, which proceeded to amyloid formation. We reveal diverse molecular mechanisms underlying Aβ1–40 aggregation with conceptual energy diagrams and propose that aggregation-prone partial helical structures are key to inducing amyloidogenesis. We demonstrate that context-dependent protein aggregation is comprehensively understood using the macroscopic phase diagram, which provides general insights into differentiation of amyloid formation and phase separation from unfolded and folded structures.
Keywords: Alzheimer’s disease, amyloid β, amyloid fibril, protein misfolding and aggregation, aggregation pathway, helical structure, phase diagram
Graphical Abstract
Destabilized soluble proteins misfold and self-assemble to insoluble aggregates, which triggers a loss of biological function and progression of disease. In the last two decades, protein misfolding and aggregation have attracted considerable attention in many research fields. A number of studies have been conducted to elucidate the role of protein aggregation in the pathogenesis of more than 50 disorders, including diverse neurodegenerative diseases and systematic amyloidosis, as well as in homeostasis in various cells. The biologically beneficial functions of protein aggregates have also been revealed in fungal coat formation, biofilm formation, melanin biosynthesis, and the immune system.1,2 Innovative attempts to design and generate protein aggregate-based biomaterials have recently increased in terms of biotechnology.3,4 Phase separation is one type of protein aggregation for biological functions and has garnered significant interest in recent years.5
Proteins have shown the capability to form various types of aggregates, including oligomers, amorphous aggregates, and fibrillar aggregates such as immature and mature amyloid fibrils, depending on the conditions. Significant efforts have been made to characterize the structural and morphological properties of individual aggregates at atomic and molecular levels.6–9 Oligomers and amorphous aggregates generally show spherical morphologies with or without defined secondary structures, while immature and mature amyloid fibrils exhibit fibrillar morphologies with a β-structure-rich core and unstructured flanking regions.8
Among various protein aggregates, the formation of amyloid fibrils and their structures have been well examined. Amyloid fibrils exhibit hierarchical suprastructures. They consist of bundles of several protofilaments that are further composed of β-sheets with hydrogen bond networks among monomers.1 Although the basic mechanisms responsible for amyloid formation have been uncovered, obtaining more detailed information on the sophisticated mechanisms underlying amyloidogenesis remains elusive. Amyloid fibrillation has shown crystal-like formation. Productive nucleation limits the entire process by introducing a lag time, and a rapid fibril growth phase subsequently emerges by consuming precursor proteins.10 Sonication has shown to be highly effective in inducing amyloid formation in vitro.11–13 Effects of sonication are rationalized by the generation and burst of cavitation microbubbles. Bubbles are created and collapsed in our body during or after decompression from pressure exposures such as those undergone by scuba divers, astronauts, and caisson and tunnel workers.14 Cavitation bubbles provide air (hydrophobic)–water (hydrophilic) interfaces for condensing precursor proteins to promote aggregation. Of note, large mechanical forces have been shown to cause the formation of cavitation bubbles in the brain, which may be responsible for Alzheimer’s disease.15 Burst of bubbles is accompanied by large shearing forces as well as high temperature and pressure, which may be efficient for the primary nucleation and secondary nucleation pathway. Physiological fluids at the interface between hydrophilic and hydrophobic surfaces generate shearing forces.16 Thus, sonication can mimic physiological and pathological conditions for aggregation-related diseases.
Pre-amyloid aggregates are often detected during amyloidogenesis. Small oligomers of amyloid-β (Aβ),17 α-synuclein (αSN),18 and human islet amyloid polypeptide (hIAPP)19 grow to larger oligomers that are ultimately converted to amyloid fibrils. The self-assembly of immature fibrils of Aβ,20 polyglutamine,21 and tau,22 i.e., protofibrils, precedes amyloid fibrillation. These nonmature amyloid aggregates have been identified under certain conditions as end products that do not proceed to amyloid fibrillation;23 therefore, these aggregated species have been characterized as off-pathway intermediates.
Biophysical studies on the aggregation-competent structures of precursor proteins at the atomic and secondary structure levels have markedly increased our understanding of the mechanisms underlying amyloid fibrillation. Native-like intermediates of the SH3 domain with an extended C-terminus showed higher amyloidogenicity than native-state structures.24 Native-like β2-microglobulin (β2m) intermediates with transPro 32 and frataxin in the folding transition state have been suggested to be amyloidogenic.25,26 αSN, with a rigid and compact C-terminus, was found to increase amyloidogenicity.27 These findings suggest that microscopic structural properties in the initial states are important for governing aggregation pathways.
The macroscopic natures of proteins in solution, solubility and supersaturation, have recently been applied to explain protein aggregation.11–13,28–31 Proteins dissolved over their solubility form insoluble aggregates in equilibrium with residual monomers. Protein solubility is controlled by solvent polarity using inorganic solvents such as alcohols and also adjusted by salts based on Hofmeister series or temperatures.12,13,28,32 Supersaturation delays nucleation-dependent phase transition. Supersaturation is a universal definition used to describe the kinetically trapped soluble states of molecules before nucleation when molecules dissolved over their thermodynamic solubility (i.e., threshold concentrations).11 Powerful agitation, such as sonication, is required to efficiently indicate the most thermodynamically stable aggregation type through the promotion of nucleation with the disruption of supersaturated states. Recent studies have also reported that supersaturation is key to understanding protein aggregation in vivo and its role in the pathogenesis of disorders. Supersaturated proteins are common among a number of neurodegenerative diseases,29 and the remodeling of supersaturated proteome during aging predominantly regulates protein aggregation in Caenorhabditis elegans.33 We previously demonstrated that a phase diagram of protein aggregation was effective for the macroscopic description of protein aggregation, and the combination of alcohol and sonication was markedly useful for examining supersaturation-limited protein aggregation.28
Unstructured (poly)peptides with alcohols transform to α-helical conformations, for instance, corticotropin-releasing factor,34 C-terminus of histones H1 and H5,35 and αSN36 due primarily to the stabilization of intramolecular hydrogen bonds. Alcohols have been widely used for diverse biological membrane-mimicking studies of proteins and peptides, as they provide clear and simple in vitro experimental conditions that biological membrane systems in vivo cannot provide due to technical difficulties.37 In the last two decades, alcohols have shown their superiority to induce amyloidogenesis of proteins such as acylphosphatase (AcP),38,39 β2m,40 and Aβ peptides41,42 and to trap intermediates for investigations using multiple biophysical techniques. Previous results exhibited that 2,2,2-trifluoroethanol (TFE) is effective for generating Aβ amyloidogenesis.41 More recently, Aβ peptides have been suggested to self-assemble into amyloid fibrils or nonfibrillar aggregates depending on the concentration of fluorinated alcohols.42 However, much remains to be understood about how alcohol-induced structural variations of Aβ monomers impact their complex aggregation. In addition, phase diagram-based solubility and supersaturation, key concepts for the macroscopic understanding of aggregation, have not been systematically introduced to the study of alcohol-induced Aβ aggregation.
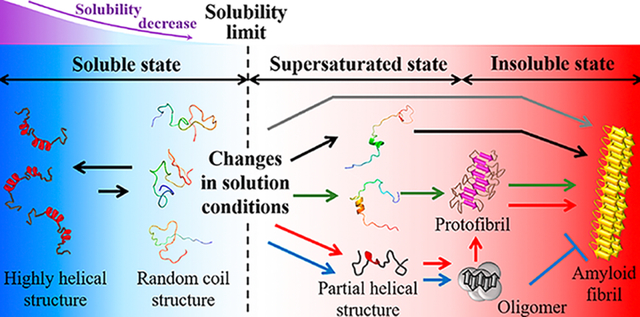
Collectively, these findings prompted us to perform an in-depth investigation on the complex protein aggregation process under various conditions based on microscopic and macroscopic viewpoints in order to obtain a more complete understanding of the mechanisms and principles of protein aggregation. Herein, we report our findings regarding diverse structural conversions of Aβ (1–40) (Aβ1–40) and their relation to aggregation pathways. Fluorinated alcohols and sonication were used to modulate the structure and solubility of Aβ1–40 monomers and to identify the final molecular species, respectively. We illustrate the importance of helical structures formed at early stages that determine the microscopic Aβ1–40 aggregation pathway (Figure 1A) and present the mechanisms by which Aβ1–40 aggregates into the final aggregated species with distinct solvent polarity. We further introduce macroscopic viewpoints, solubility and supersaturation, to understand the monomer structure-dependent amyloid formation (Figure 1A). We confirm that hybrid phase diagrams containing microscopic structural information on precursor proteins and macroscopic phase transition are useful for elucidating the general properties of the aggregation behaviors of folded and unfolded precursor proteins as well as phase separation. Our overall results would also be useful to investigate other amyloid proteins and the aggregation process under diverse conditions. Such studies would be valuable in the development of inhibitors against toxic aggregates and potentially to engineer compounds as potential therapeutics to suppress the pathology of amyloid-related diseases.
Figure 1.
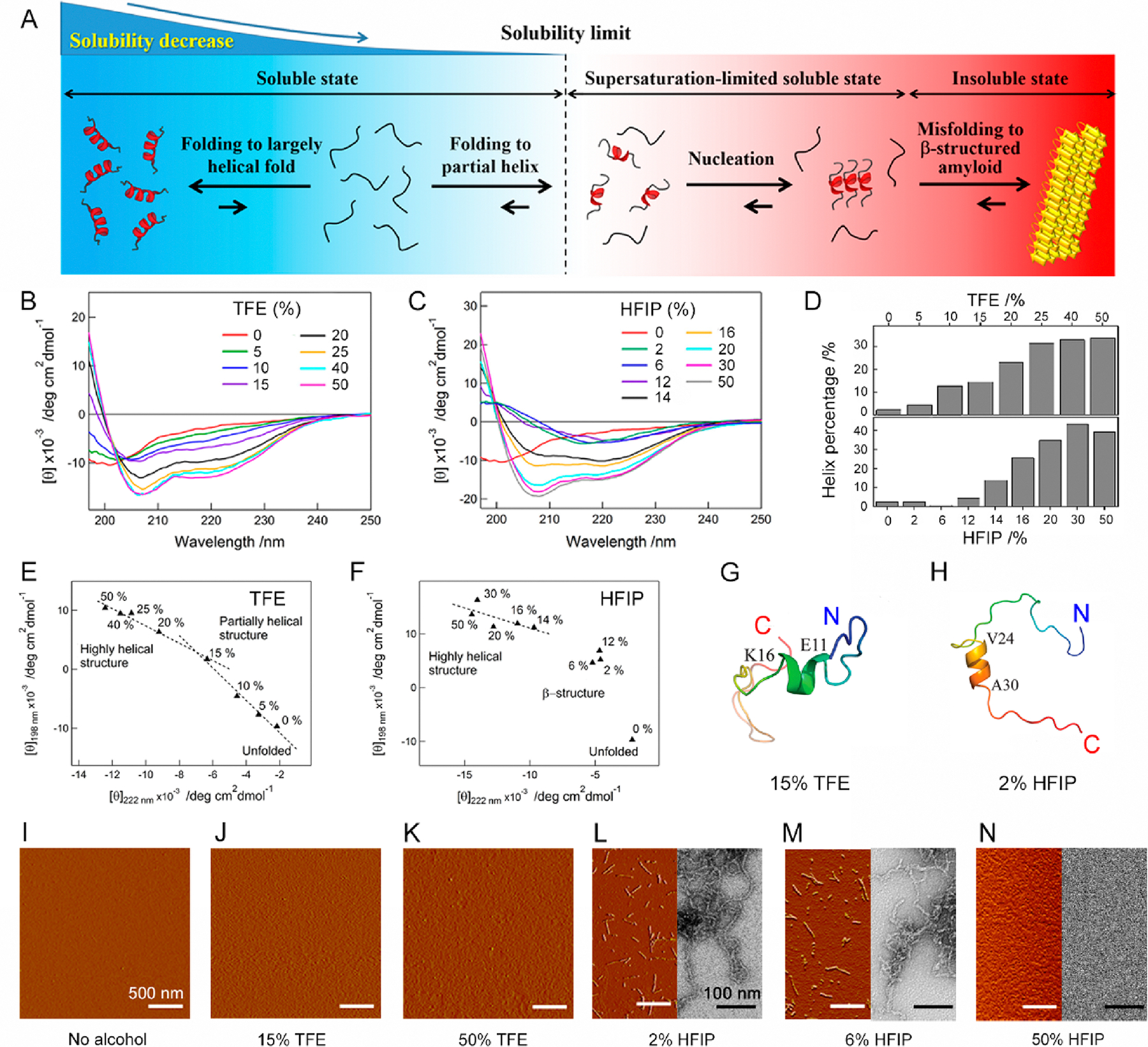
Conformational transition of Aβ1–40 depending on solvent polarity. (A) Schematic representation of the structural change of random coil-like Aβ1–40 (left, light blue region) to a largely (left, blue region) and partially (right, light red region) helical fold, which increases and decreases solubility, respectively. Partial helical Aβ1–40 is in a kinetically soluble state due to supersaturation, and insoluble β-structure-rich amyloid fibrils (right, red region) are thermodynamically formed after the disruption of supersaturation. (B and C) Far-UV CD spectra of Aβ1–40 at various concentrations of (B) TFE and (C) HFIP. (D) The content of α-helix structures of Aβ1–40 at each TFE (upper panel) and HFIP (lower panel) concentration was analyzed using the corresponding CD spectra and BeStSel algorithm.44 (E and F) Transition diagrams of Aβ1–40 in (E) TFE and (F) HFIP constructed based on ellipticity values at 222 and 198 nm in B and C. Linear fits of points (E) from 0 to 15% and from 15 to 50% TFE and (F) from 14 to 50% HFIP are represented by dotted lines. (G and H) Structures of Aβ1–40 in (G) 85% H2O and 15% TFE as well as (H) 98% H2O and 2% HFIP after 100 ns simulation are shown. The color of the peptide structures changes from blue to red at the N- and C-termini, respectively. (I–N) AFM and TEM images of the Aβ1–40 solution at the distinct type and concentration of alcohol were obtained immediately after sample preparation. The scale bars in the AFM and TEM images represent 500 and 100 nm, respectively.
RESULTS AND DISCUSSION
Characterization of Initial Conformational States of Aβ1–40 in Water/Alcohol Mixtures.
In order to examine initial structural states of Aβ1–40, a series of far-UV circular dichroism (CD) spectra of Aβ1–40 were recorded with increases in the concentrations of TFE (Figure 1B) and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (Figure 1C). In the absence of alcohol, the Aβ1–40 solution showed a minimum at ~200 nm, suggestive of the random-coil-like structures as reported by previous studies.41,43 A narrow distribution of sharp NMR peaks also suggested largely unstructured Aβ1–40 monomers (Figure S1). Elevations in the concentration of TFE induced changes in far-UV CD spectra with increases in CD signal intensities at shorter wavelengths (<~203, ~208, and ~222 nm, Figure 1B), indicating a structural transition to an α-helix-rich structure and the formation of largely helical structures at 50% TFE. The prediction of the content of secondary structures using the BeStSel algorithm44 supported TFE-concentration-dependent increases in the α-helix from ~3 to ~35% (Figure 1D, upper panel). The content of the α-helix (33%) in 40% TFE was well consistent with that of the NMR structure (35%).45 Although all spectra at 0–50% TFE appeared to intersect at ~203 nm, they did not share a single isodichroic point. CD spectra at 0–15% and 15–50% showed isodichroic points at ~201 and ~204 nm, respectively. Likewise, the transition diagram constructed by plotting CD intensities at 198 and 222 nm obtained at distinguished TFE concentrations exhibited two linear dependences that intersected at ~15% TFE (Figure 1E), suggesting the existence of different helical structures and partial helical structures at ~15% TFE. Atomic force microscopy (AFM) images confirmed the absence of aggregation from 0 to 50% TFE (Figure 1I–K), suggesting that the structural transition from disordered Aβ1–40 to different helical conformations did not cause Aβ aggregation at the initial stage.
The change in the CD spectra at various HFIP concentrations was more dynamic (Figure 1C). The AFM of HFIP at low concentrations ranging from 2% to 12% induced the alteration of the random coil-like spectrum to a characteristic spectrum with a single minimum at ~220 nm and the enhancement in the CD intensity at shorter wavelengths, which indicated a β-structure-rich conformational state. Upon further increasing concentrations of HFIP to 14% and 16%, both the negative and positive CD signals were intensified, resulting in the conversion of the overall spectrum to an α-helix-dominated shape. At higher HFIP concentrations of more than 20%, a typical far-UV spectrum of the α-helix-rich structure was obtained. The predicted content of α-helix was as low as ~0.5 to ~4.5% at low HFIP concentrations (0–12%) and was increased up to ~40% at 50% HFIP (Figure 1D, lower panel). All CD spectra showed one isodichroic point at ~200 nm, except for the spectrum without HFIP, suggesting a transition from β to α. The transition diagram displayed a linear dependence between 14% and 50% HFIP (Figure 1F). Transmission electron microscopy (TEM) and AFM images visualized short, curvilinear fibrillar aggregates at 2%, 6%, and 12% HFIP (Figures 1L,M and S2A–G), indicating the rapid formation of β-structured protofibrils. Although differences in CD spectra suggested distinct secondary structures of protofibrils at the different concentration of HFIP, all protofibrils showed similar morphologies (Figures 1C,L,M and S2A–G). High-resolution TEM images revealed that protofibrils at all conditions consisted of a single strand with a width of ~5 nm (Figure S2A–G). At greater than 20% HFIP, no aggregate was detected in TEM and AFM images (Figures 1N and S2H), suggesting Aβ1–40 with highly helical structures remained soluble at high HFIP concentrations.
Next, we performed a MD simulation to determine the mechanistic and structural arrangement of Aβ1–40 in mixtures of two different solvents containing 85% H2O and 15% TFE, where a partially helical intermediate was observed, as well as 98% H2O and 2% HFIP, where rapid β-rich aggregation occurred at the atomic resolution. Largely unfolded Aβ1–40 was structurally diverged depending on conditions (Figures 1G,H and S3). In 15% TFE solution, Aβ1–40 rearranged with the induction of a short helical structure (residues E11–K16) (Figures 1G and S3A). The helical content of 12.5% observed was in good agreement with the CD analysis. Meanwhile, the addition of 2% HFIP also induced a partial helical structure at the residues V24–A30 from largely unfolded structures (Figures 1H and S3B). MD snapshots revealed that the homogeneously distributed TFE/HFIP molecules aggregated surrounding Aβ1–40 (Figure S4). Therefore, the discerning intermolecular interaction between Aβ1–40 and organic solvent molecules plays a central role in mediating the initial partial helical conformations of Aβ by reinforcing intramolecular hydrogen bonds.46
Observation of Aβ1–40 Aggregation in Water/Alcohol Mixtures.
The real-time monitoring of Aβ1–40 aggregation was first performed by the thioflavin T (ThT) assay under quiescent conditions without TFE (Figure 2A). Even after incubation for ~2 days, no change was observed in the ThT fluorescence intensity. The solution of Aβ1–40 after incubation showed the same far-UV CD spectrum recorded prior to incubation (Figure 2B). These results indicated that amyloid fibrils were not formed. On one hand, sonication rapidly increased the ThT fluorescence intensity, with a lag time of ~1 h and an elongation rate constant of ~10 h−1 (Figure 2L and M), and triggered a far-UV CD spectrum of a typical cross-β structure of amyloid fibrils by showing a minimum at ~220 nm, indicative of the nucleation-dependent amyloid fibrillation of Aβ1–40 (Figure 2B). The formation of amyloid fibrils was also confirmed by TEM (Figure 2G) and one-dimensional proton NMR spectroscopy based on the disappearance of NMR peaks (Figure S1).
Figure 2.
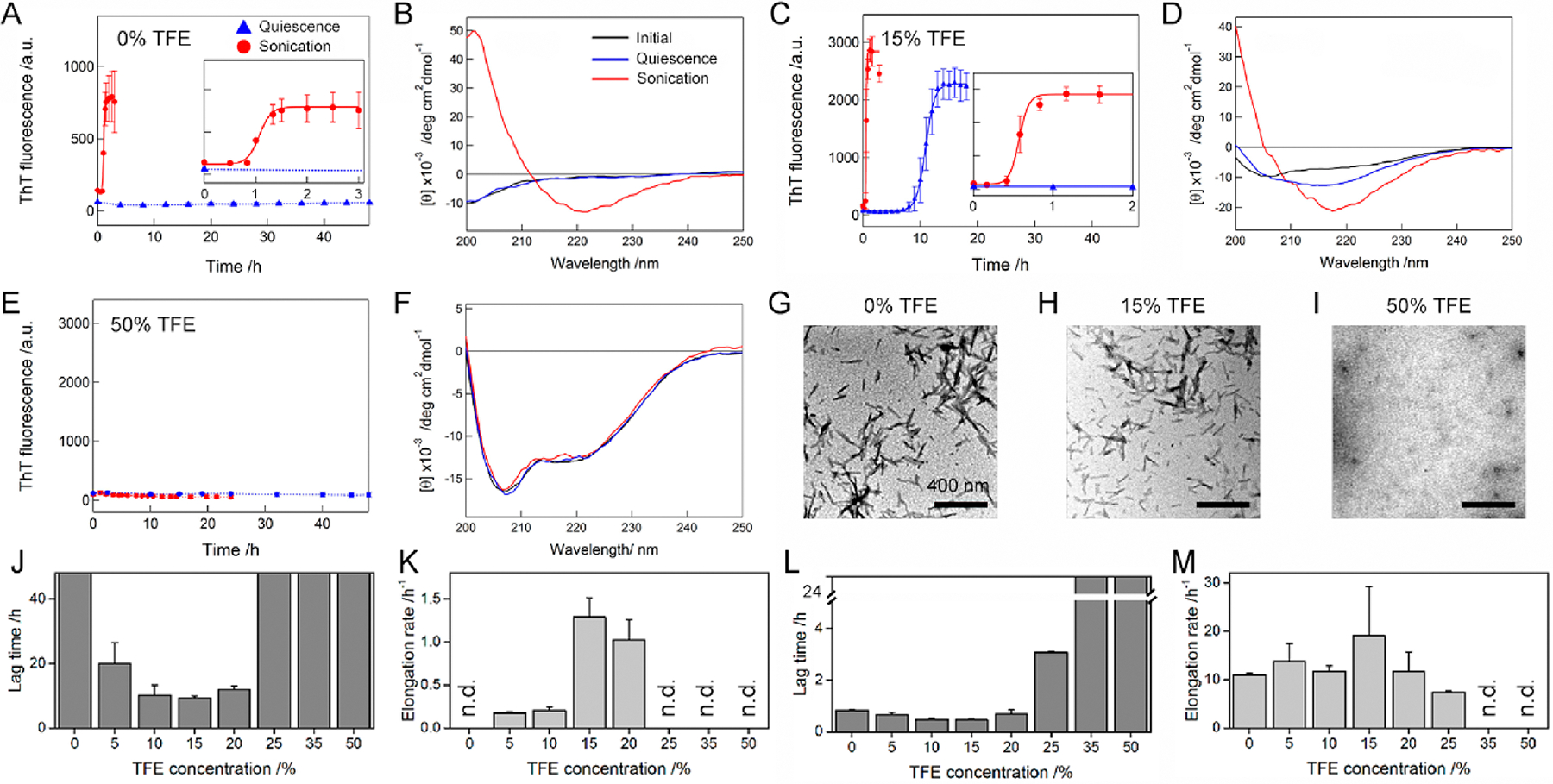
TFE concentration-dependent amyloid formation of Aβ1–40 under quiescent and stimulated conditions. (A–I) The aggregation of Aβ1–40 was traced by (A, C, and E) ThT fluorescence, (B, D, and F) far-UV CD, and (G, H, and I) TEM at various TFE concentrations. The ThT intensities of the Aβ1–40 solution without agitation (blue triangle) and with sonication (red circle) were plotted as a function of time. Solid lines represent the fit curves of the kinetics of Aβ1–40 amyloid fibrillation. Dotted lines were drawn as an eye-guide only. Inserts in A and C are the magnified profiles of rapid kinetics of Aβ1–40 fibrillation with sonication. Far-UV CD spectra of Aβ1–40 were recorded after incubation without agitation (blue line) and with sonication (red line). The CD spectra of Aβ1–40 measured soon after sample preparation are shown as a comparison (black line). TEM images were taken from Aβ1–40 samples after incubation with sonication. The concentration of TFE used was displayed above the TEM images. Scale bar = 400 nm. (J–M) The lag time (J and L) and elongation rate constant (K and M) of Aβ1–40 fibrillation in TFE/water mixtures without agitation (J and K) and with sonication (L and M) are displayed. “n.d.” denotes the TFE concentrations at which no enhancement in the ThT fluorescence intensity was observed. Error bars indicate the standard deviation of three independent measurements.
An increase in the ThT fluorescence intensity and the reduced lag time were broadly observed with the far-UV CD spectra of cross-β amyloid architectures when the concentration of TFE was elevated from 0 to 20% under quiescent conditions (Figures 2C,D and S5A–C), indicating the acceleration of amyloid formation. Further enhancement in the TFE concentration to more than 25% caused no change in the ThT-based kinetics and CD spectra, which suggested that amyloid generation did not occur (Figures 2E,F and S5D,E). Thus, our results presented the minimum lag time and maximum elongation rate at ~15% TFE (Figure 2J and K).
Aβ1–40 in the 5–25% TFE solution, subjected to sonication, showed a noticeable reduction in the lag times and a steeper elongation phase with the far-UV CD spectra of the cross-β structures of amyloid fibrils as well as fibrillar aggregates in the TEM image (Figures 2C,D,H and S5A–D), supporting the acceleration of amyloid formation from that under alcohol-free conditions. Although the effects of sonication lowered the difference between lag times or elongation rates, the TFE-concentration dependence of two kinetic parameters was similar to that under quiescent conditions with indication of minimum lag times at 0–20% TFE and a maximum elongation rate at 15% TFE (Figure 2L and M). In addition, amyloid fibrils formed both with and without 15% TFE decreased the viability of human neuroblastoma cells by ~20%, suggesting the cytotoxicity of amyloid fibrils (Figure S6). At greater than 40% TFE, sonication did not affect the ThT fluorescence (Figure 2E).
It is commonly accepted that ThT binds to surface side-chain grooves of amyloid fibrils largely through hydrophobic interactions.47 Thus, the increased hydrophobicity at higher TFE concentrations may disrupt the interaction between ThT and Aβ1–40 amyloid fibrils, although large enhancements of the ThT fluorescence intensity have been detected when fibrils of lysozyme,12 insulin,13 and AcP38 formed in solutions containing more than 40% TFE. Far-UV CD results showed that Aβ1–40 monomers remained largely unstructured throughout the incubation, which was in good agreement with the absence of aggregates in the TEM micrograph (Figures 2F,I and S5E). These results supported that Aβ1–40 amyloid fibrillation was blocked at the concentrations of TFE larger than 40%.
The addition of HFIP led to a characteristic aggregation profile of Aβ1–40 (Figure 3). The ThT fluorescence at 2% HFIP, recorded immediately after the initiation of the ThT assay under quiescent conditions, showed the high intensity, proposing the formation of ThT-positive aggregates (Figure 3A). Moreover, there was no increase in the ThT fluorescence intensities of a 2% HFIP solution without Aβ1–40 (Figure S7), further suggesting that the initial high ThT intensity in Figure 3A was attributed to Aβ1–40 aggregation. Based on short rod-like curvilinear fibrillar morphologies (Figure 1L) and the far-UV CD spectrum of a β-structure, which exhibited a minimum at ~218 nm (Figure 3B), β-structured protofibrils were generated. A sigmoidal increase in the ThT fluorescence was observed after a lag time of ~6 h. After incubation, long and thick mature amyloid fibrils were shown in the AFM image (Figure S8A) with the far-UV CD spectrum of a cross-β structure (Figure 3B). Strong agitation with sonication significantly accelerated the kinetic process of the transition from protofibrils to mature amyloid fibrils, resulting in its completion within ~2 h (Figure 3A). The sonicated samples displayed the signature of a β-structure molecular species with a minimum at ~215 to ~220 nm (Figure 3B). Both TEM (Figure 3C) and AFM images (Figure S8B) presented that fibrillar aggregates were produced and sonication clearly generated short aggregates. Taken together, these results indicated the formation of amyloid fibrils. The conversion rate constants from proto- to mature fibrils were 0.15 ± 0.05 and 2.49 ± 0.70 h−1 in the absence and presence of sonication, respectively.
Figure 3.
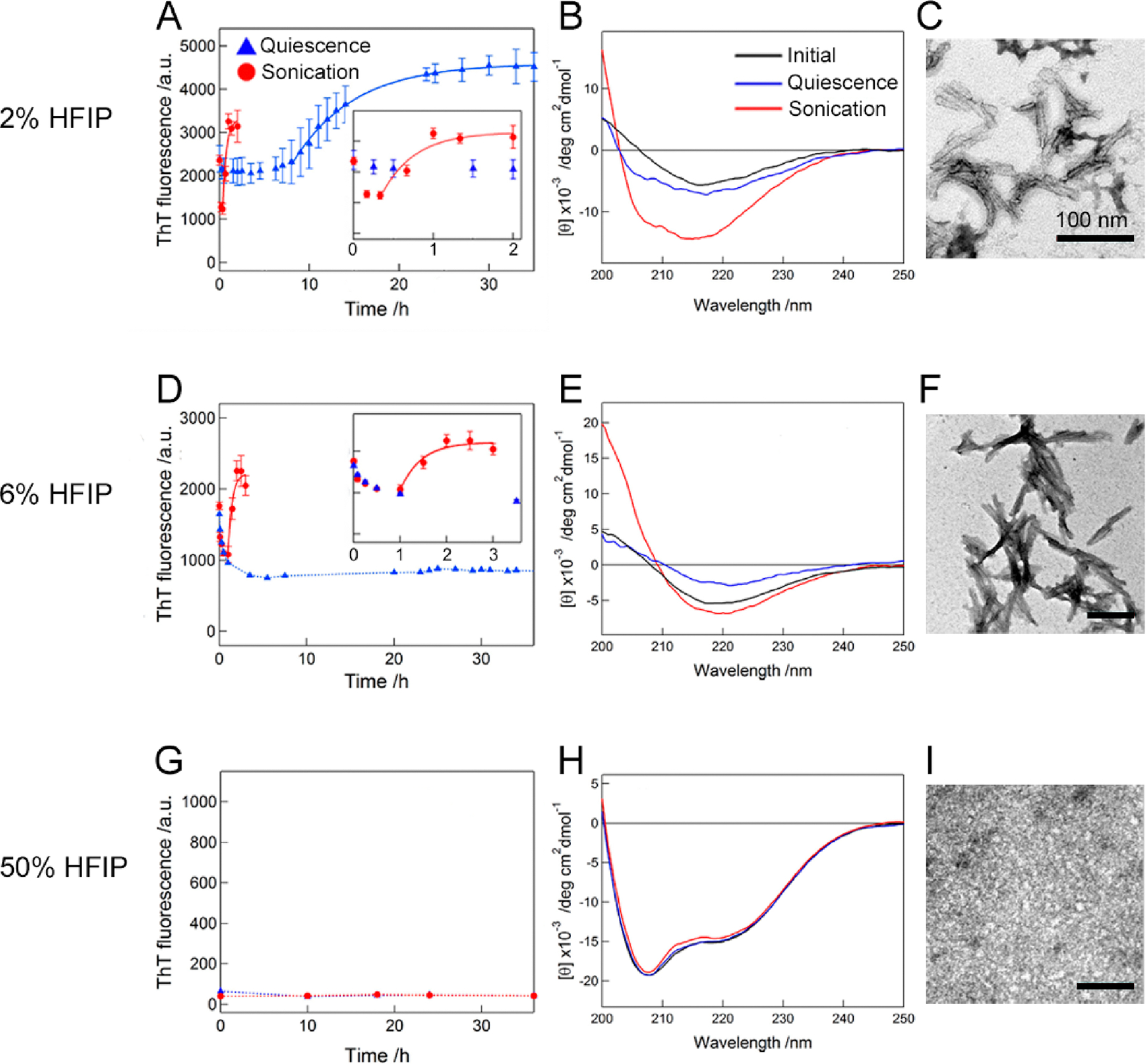
HFIP concentration-dependent amyloid formation of Aβ1–40 under quiescent and stimulated conditions. (A–I) The aggregation of Aβ1–40 was traced by (A, D, and G) ThT fluorescence, (B, E, and H) far-UV CD, and (C, F, and I) TEM at various HFIP concentrations. The ThT intensities of the Aβ1–40 solution without agitation (blue triangle) and with sonication (red circle) were plotted as a function of time. Solid lines represent the fit curves of the conversion of protofibrils to amyloid fibrils. Dotted lines were drawn as an eye-guide only. Inserts in A and D are the magnified profiles of rapid kinetics of Aβ1–40 fibrillation with sonication. Far-UV CD spectra of Aβ1–40 were recorded after incubation without agitation (blue line) and with sonication (red line). The CD spectra of Aβ1–40 measured after sample preparation are shown for a comparison (black line). TEM images were taken from the Aβ1–40 samples after incubation with sonication. Scale bar = 100 nm.
At 6% HFIP without sonication, the initial high ThT fluorescence intensity (Figure 3D) with rod-like curvilinear fibrillar aggregates (Figure 1M) and the far-UV CD spectrum of a β-structured pattern (Figure 3E) are shown, supporting the presence of ThT-positive β-structured protofibrils. In addition, similar to the result at 2% HFIP, the sample solution containing 6% HFIP without Aβ1–40 did not impact the initial ThT fluorescence (Figure S7). The intensity of ThT fluorescence did not change even after ~35 h. The far-UV CD spectrum of a β-structured pattern was very similar regardless of incubation (Figure 3E), and the AFM image exhibited small aggregates (Figure S8C), which suggested that protofibrils still remained. However, sonication caused a rapid and marked increase in the ThT intensity and completed the reaction at ~2 h with the conversion rate constant of 2.21 ± 0.85 h−1 (Figure 3D). The CD spectrum of the sonicated sample solution showed a β-structured pattern with an increased positive CD signal at a low wavelength (<210 nm) (Figure 3E). TEM (Figure 3F) and AFM images (Figure S8D) at the ThT end point states presented short amyloid fibrils due to sonication.
Although strong ultrasonic effects concealed the difference in aggregation kinetics, faster amyloid fibrillation at 2% HFIP than that at 6% HFIP under quiescent conditions suggested that the colloidal and kinetic stabilities of protofibrils at 2% HFIP were lower than those at 6% HFIP. At a high HFIP concentration of 50%, the ThT fluorescence without sonication maintained a very low intensity during the incubation (Figure 3G). In addition, an α-helical far-UV CD spectrum did not change its pattern (Figure 3H). Any aggregates were not found in the TEM image (Figure 3I).
Characterization of Aβ1–40 Oligomers and Their Conversion to Amyloid Fibrils.
In order to determine various types and the interconversion of aggregates, we prepared an additional type of aggregates, Aβ1–40 oligomers, based on previous studies (Figure 4).48,49 Aβ1–40, which was freshly dissolved in 100% HFIP, was diluted to 10% HFIP using deionized water (see details in Methods). The far-UV CD measurement of the sample solution showed a β-structure-representing spectrum, which was similar to that of protofibrils, but distinguishable from that of mature amyloid fibrils (Figure 4A). The ThT fluorescence intensity of aggregates was noticeably lower than those of protofibrils and amyloid fibrils (Figure 4B). The TEM and AFM images indicated the production of spherical aggregates (Figure 4C), which were also morphologically different from fibrillar aggregates, protofibrils, and amyloid fibrils (Figures 1–3 and S2 and S8). High-resolution TEM images identified diverse types of oligomers with different morphologies and sizes (Figure S9) that would not be detected using AFM. For example, a donut-shaped oligomer that was supposed to contain β-barrel-like structures50 was observed (Figure S9C). Oligomers with a spheroid morphology (Figure S9G) or irregular bulges on the surface (Figure S9H) were also detected, consistent with previous findings that different types of oligomers were detected both in vitro and in vivo.51,52 The ELISA using an oligomer-specific antibody (A11)49 unambiguously confirmed the formation of Aβ1–40 oligomers showing a high intensity for the antibody reaction at 450 nm (Figure 4D). Taken together, these results indicated the oligomerization of Aβ1–40, which was rationalized by the following two reasons: (1) solubility of Aβ1–40 monomers in 100% HFIP decreased due to the dilution to 10% HFIP, which triggered aggregation such as oligomerization; (2) intermolecular hydrogen bonding for β-structured Aβ1–40 oligomers at 10% HFIP became more thermodynamically favorable than intramolecular hydrogen bonding for helical Aβ1–40.
Figure 4.
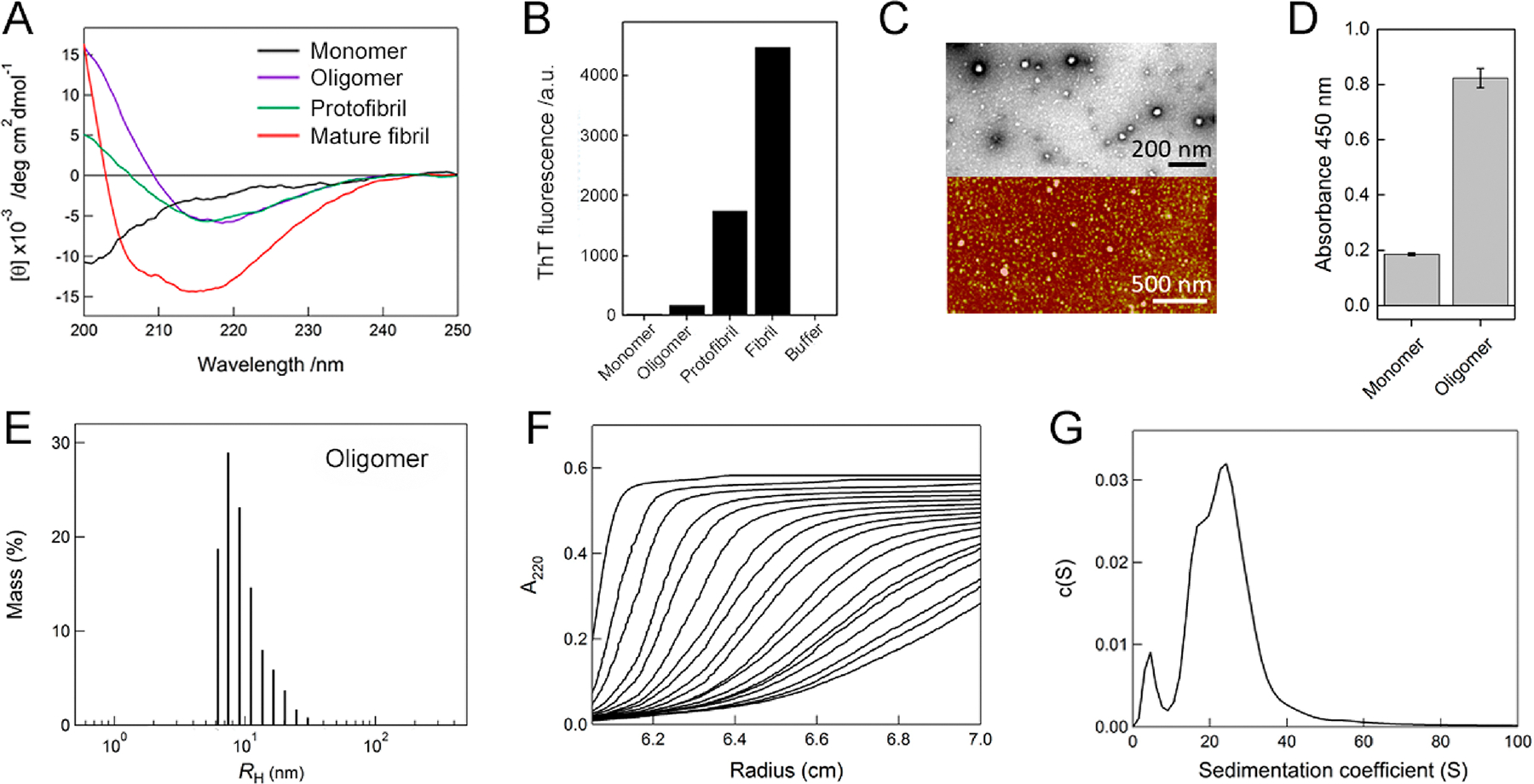
Characterization of Aβ1–40 oligomers. (A) Far-UV CD spectra of Aβ1–40 monomers (black line), oligomers (magenta line), protofibrils (green line), and mature amyloid fibrils (red line). (B) Fluorescence intensities of ThT at 485 nm for Aβ1–40 monomers, oligomers, protofibrils, mature amyloid fibrils, and buffer alone. (C) TEM (upper panel) and AFM (lower panel) images of Aβ1–40 oligomers. The scale bars of TEM and AFM represent 200 and 500 nm, respectively. (D) Detection of Aβ1–40 oligomers using the sandwich ELISA based on absorbance at 450 nm. Absorbance of monomers was recorded for the control. (E) Distribution of the hydrodynamic radii of Aβ1–40 oligomers, obtained by dynamic light scattering measurements. (F) Sedimentation velocity profiles of Aβ1–40 oligomers optically detected at 220 nm. Fitted sedimentation boundaries measured at an interval of 15 min are shown. (G) Distribution of the sedimentation coefficient of Aβ1–40 oligomers derived from the sedimentation velocity data in F.
The sizes of Aβ1–40 oligomers were then analyzed (Figure 4E–G). Dynamic light scattering measurements exhibited a narrow distribution of the hydrodynamic radius of Aβ1–40 oligomers with a diameter of ~10 to ~50 nm (Figure 4E), which was consistent with the sizes of oligomers observed in the TEM and AFM images. High molecular weights of oligomers induced NMR peak broadening (Figure S1). A clear sedimentation profile indicated the absence of large Aβ1–40 aggregates (Figure 4F). Sedimentation coefficient (s) values obtained by the sedimentation velocity measurement of analytical ultracentrifugation were distributed from 0 to ~40 S with indication of one small and two large peaks (Figure 4G). Two s-values, ~5.2 and ~24.7 S, were representatively selected to assess the molecular weights and number of Aβ1–40 monomers consisting of oligomers. The molecular weights of Aβ1–40 oligomers were calculated to be ~70 and ~740 kDa. Therefore, ~17 and ~170 Aβ1–40 peptides formed small and large oligomers, respectively.
The solutions of Aβ1–40 oligomers were incubated for ~2 days without agitation, and no changes were observed in the ThT fluorescence intensity (Figure 5A). We also did not detect any significant differences in the far-UV CD spectra that depended on incubation (Figure 5B). Extensive sonication was introduced; however, no significant alteration was observed in ThT fluorescence over incubation time despite a small initial decrease (Figure 5A). Additionally, a similar pattern for the CD spectrum with the reduced intensity was obtained (Figure 5B). All TEM images exhibited Aβ1–40 oligomers with spherical morphologies (Figure 5C–E). Clustered oligomers that may decrease the CD intensity were shown (Figure 5E), and this may be due to sonication.
Figure 5.
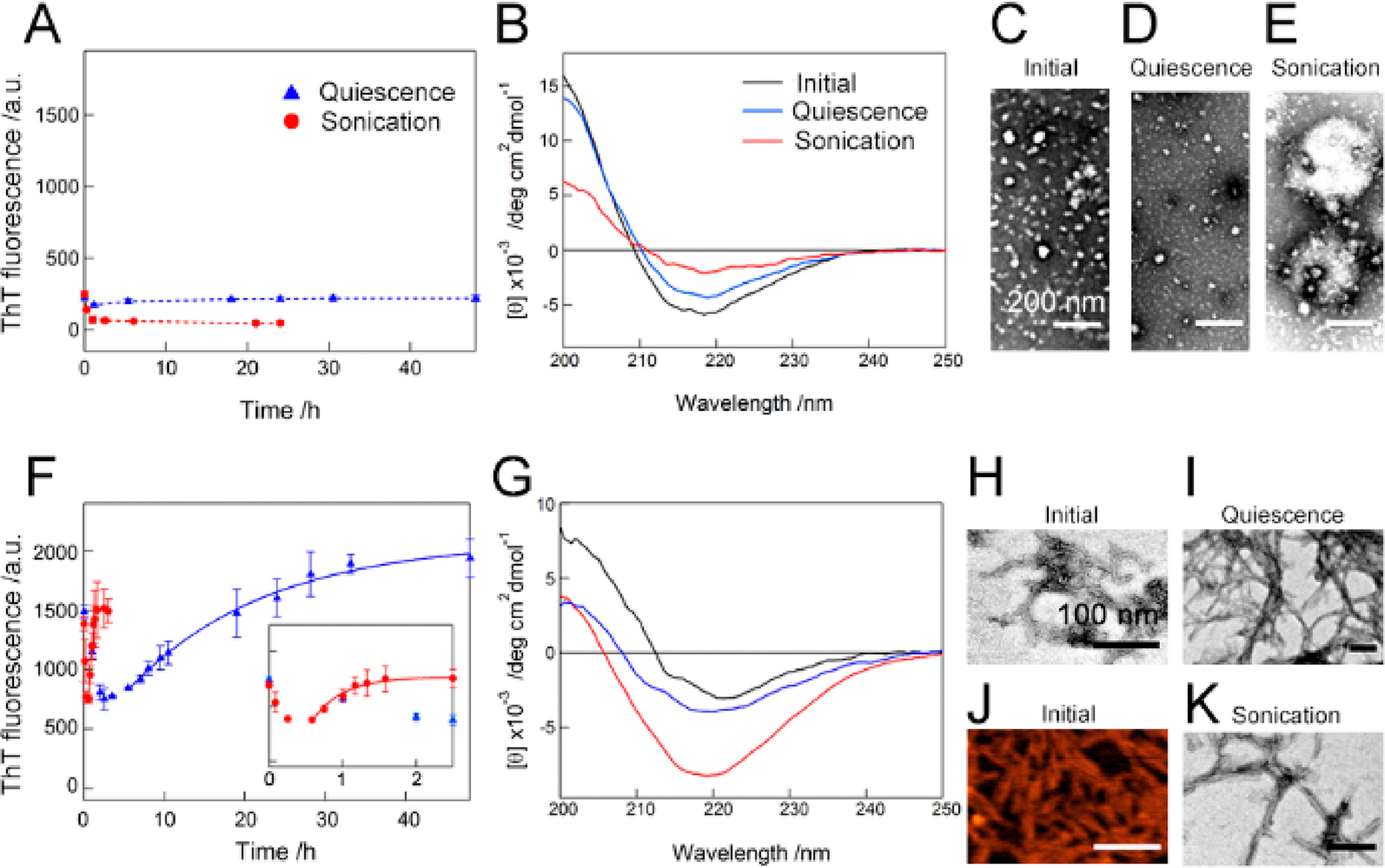
Context-dependent aggregation of Aβ1–40 oligomers. (A–K) The aggregation behaviors of Aβ1–40 oligomers were observed by (A and F) ThT fluorescence, (B and G) far-UV CD spectra, (C–E, H and I, and K) TEM, and (J) AFM in the (A–E) absence and (F–K) presence of 10 mM sodium phosphate buffer (pH 7.5) containing 100 mM NaCl. The ThT intensities of Aβ1–40 oligomers without agitation (blue triangle) and with sonication (red circle) were plotted as a function of time. Dotted lines were drawn as an eye-guide. The inset in F represents a magnified kinetic trace of the rapid increase in the ThT fluorescence intensity with sonication. The far-UV CD spectra of Aβ1–40 oligomers were recorded after incubation without agitation (blue line) and with sonication (red line). The CD spectra of Aβ1–40 oligomers measured before incubation (black line) are displayed for a comparison. The scale bars in C–E and H–K indicate 200 and 100 nm, respectively.
The context-dependent transformation of Aβ1–40 oligomers was then examined (Figure 5F–K). We added 10 mM sodium phosphate buffer (pH 7.5) containing 100 mM NaCl to the solution of oligomers and observed the time-course of variation of the ThT fluorescence (Figure 5F). The ThT fluorescence intensity at time zero was more than 8-fold larger than that of oligomers before changing the condition, which indicated that the change to a physiological condition rapidly increased the ThT fluorescence intensity of the oligomer solution within the experimental dead time (~5 min) (Figure 5A and F). In contrast, the addition of 10 mM sodium phosphate buffer (pH 7.5) containing 100 mM NaCl to solution without Aβ1–40 oligomers did not increase the ThT fluorescence intensity, which ruled out the possibility that a solvent effect caused the enhancement in the ThT fluorescence value (Figure S10). The pattern of the far-UV CD spectrum was similar to that of oligomers with a decrease in the CD intensity (Figure 5G). The TEM and AFM images exhibited short curvilinear fibrillar aggregates (Figure 5H and J), which indicated the burst formation of Aβ1–40 protofibrils, i.e., the conformational conversion from oligomers to protofibrils.
Incubation under quiescent conditions presented a gradual increase in the ThT fluorescence intensity with saturation at ~35 h (Figure 5F), and the final product displayed the CD spectrum of β-rich structures (Figure 5G). The TEM image clearly showed the production of mature Aβ1–40 amyloid fibrils (Figure 5I). Sonication strongly promoted the reaction within ~2 h with indication of the increased fluorescence intensity of ThT and the CD signal (Figure 5F and G). Initial decreases in the ThT fluorescence intensity were observed in the presence and absence of sonication (Figure 5F). Amyloid fibrils were detected in the TEM image of the samples after incubation with sonication (Figure 5K). The conversion rate constants of prototo mature fibrils were 0.06 ± 0.02 and 2.26 ± 0.50 h−1 without and with sonication, respectively, which were similar to those in water/HFIP mixtures.
In order to examine effects of environmental factors on the conversion of oligomers to other types of aggregates in more detail, the aggregation process of Aβ1–40 oligomers, formed at pH ~4, with and without 10 mM sodium phosphate buffer (pH 7.5) at the various concentration of NaCl (0–2000 mM) was investigated (Figure 6). The addition of 10 mM sodium phosphate buffer (pH 7.5) without NaCl induced a rapid increase in the ThT fluorescence intensity at the initial stage (Figure 6A), which indicated the formation of protofibrils as observed in Figure 5. Incubation with continuous shaking led to the gradual increase in the ThT intensity with saturation at ~20 h. The far-UV CD spectrum showed a typical pattern of β-structure-rich conformations (Figure 6B), and the TEM image exhibited the formation of mature amyloid fibrils (Figure 6G), indicating that the pH jump from ~4 to 7.5 is enough to cause the conversion from oligomers to mature amyloids via protofibrils.
Figure 6.
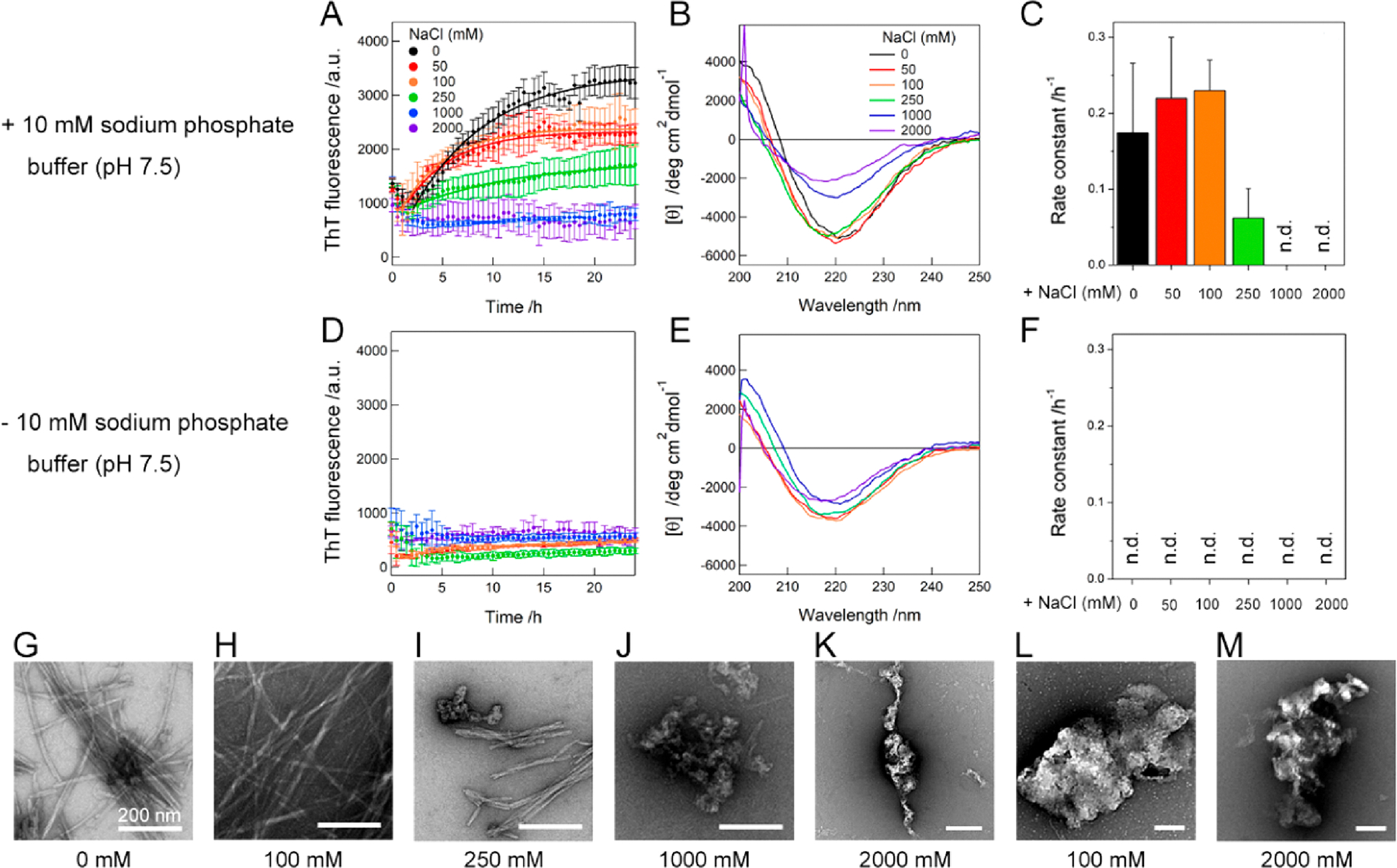
Effects of the pH and NaCl concentration on the transition of Aβ1–40 oligomers. (A–M) Aggregation behaviors of Aβ1–40 oligomers at the various concentrations of NaCl were observed using (A, C, D, and F) ThT fluorescence, (B and E) far-UV CD spectra, and (G–M) TEM in the (A–C and G–K) presence and (D–F, L, and M) absence of 10 mM sodium phosphate buffer (pH 7.5). Solid lines in A represent the fit curves for the kinetic analysis of the conversion to amyloid fibrils. The rate constant for the conversion is shown in C and F. Error bars represent the standard deviation of three independent measurements. “n.d.” denotes the NaCl concentrations where the protofibril-to-amyloid fibril conversion did not occur. The far-UV CD spectra of Aβ1–40 oligomers were recorded after the 24 h incubation. The concentrations of NaCl used are displayed in A, B, and G–M. The scale bars in G–M indicate 200 nm.
At the concentration of NaCl ranging from 50 to 250 mM, the initial ThT fluorescence was similar to that in the absence of NaCl, implicating the formation of protofibrils, and the gradual enhancement in the ThT fluorescence intensity was observed. CD spectra showed the formation of β-structure-rich aggregates (Figure 6B), and TEM images exhibited long fibrillar aggregates (Figure 6H and I), indicating that the final product is amyloid fibrils. The addition of the high concentrations of NaCl (1000 and 2000 mM) resulted in no increase in the ThT fluorescence even after ~24 h (Figure 6A), and large aggregates of protofibrils that may decrease the CD intensity (Figure 6B) were predominant without amyloid fibrils in the TEM images (Figure 6J and K). The kinetic analyses revealed an optimum concentration of NaCl, i.e., ~100 mM, to convert oligomers to amyloid fibrils through protofibrils (Figure 6C). We speculated that the initial decrease in the ThT fluorescence was related to a conversion of protofibrils to form amyloid fibrils.
On the other hand, no increase in the ThT intensity was observed throughout the incubation with the addition of NaCl (50–2000 mM NaCl) alone (Figure 6D) by showing β-structured CD spectra (Figure 6E). TEM images visualized large aggregates without any morphology of protofibrils and amyloid fibrils at both 100 and 2000 mM NaCl (Figure 6L and M). All these results indicated that the addition of NaCl to oligomer samples without a pH shift induced the formation of large amorphous aggregates, which may consist of small β-structured oligomers instead of amyloidogenic aggregation via protofibrillation.
Amyloid Plaque Deposition in Vivo by the Injection of Aβ1–40 Oligomers and Protofibrils.
In order to investigate whether Aβ1–40 oligomers or protofibrils formed in vitro are involved in Aβ amyloid deposition, i.e., amyloid plaque formation, in vivo, we bilaterally injected these two types of Aβ1–40 aggregates and vehicle controls into the hippocampus CA1 regions of wild-type adult mice (Figure 7A). At 24 h after intrahippocampal injections, the mice were perfused and fixed, and immunohistochemistry was performed with anti-4G8 antibodies, which have been used to visualize Aβ plaques in vivo (Figure 7B).53 Interestingly, we found that oligomer- or protofibril-injected mice significantly increased 4G8-positive immunoreactivity in the hippocampus CA1 region compared with vehicle-injected mice (Figure 7B), implicating the formation of amyloid plaques. The quantitative analysis revealed that 4G8 fluorescence intensities of mice with the injections of oligomers and protofibrils were ~10 fold higher than those with vehicle injections (Figure 7C). The 4G8 fluorescence intensity of protofibril-injected mice was higher than that of oligomer-injected mice. These results may suggest that oligomers and protofibrils formed in vitro can be changed to amyloid fibrils for Aβ plaque deposition in vivo. The lower fluorescence intensity of oligomer-injected mice than that of protofibril-injected mice suggested that protofibrils might convert to amyloid fibrils more easily than oligomers in vivo, as oligomers undergo the conversion to amyloid fibrils via protofibrils. We speculate that complicated environments in brains may impair the oligomer-to-protofibril transition to some extent.
Figure 7.
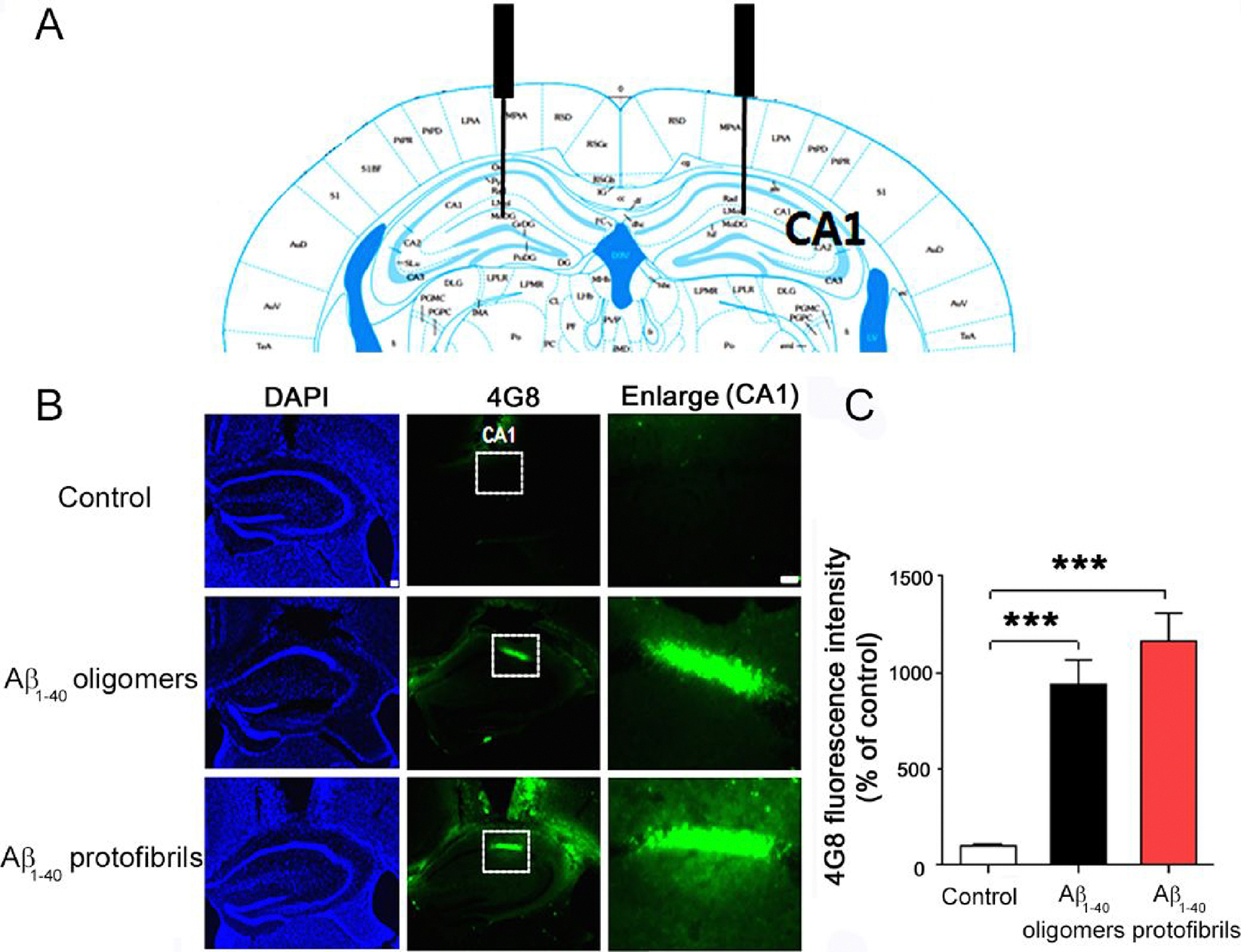
Immunohistochemical examination of the deposition of amyloid plaques in the hippocampal CA1 region. (A) Illustration of the cross section of the mouse brain to show the injection site (CA1). (B) Immunohistochemical results at 24 h after the injection of vehicles (top row), Aβ1–40 oligomers (middle row), and protofibrils (bottom row) to the bilateral hippocampal CA1 regions. DAPI (left column), 4G8 immunostaining (middle column), and magnification of CA1 regions (right column) are shown. (C) Quantification of 4G8-positive amyloid plaques in the hippocampal CA1 region (n = 4 mice per group). Data represent mean ± SEM (***p < 0.001).
Microscopic Characterization of Aβ1–40 Aggregation Pathways.
Amyloid formation has been extensively studied for many different amyloid proteins and peptides; however, the different mechanisms and pathways underlying the aggregation process have not been well established. Recent studies have demonstrated the use of variables and conditions, such as temperature,54 lipid bilayer/nanodiscs,55,56 and chemical molecules57–59 such as phthalocyanine tetrasulfonate, 2,8-bis(2,4-dihydroxyphenyl)-7-hydroxyphenoxazin-3-one, and heparin sulfate to probe the aggregation pathways. In our present studies, we aimed to comprehensively reveal the aggregation mechanisms and pathways employing Aβ1–40 as a model system and a variety of biophysical experiments and MD simulation.
Six possible microscopic pathways are revealed for Aβ1–40 aggregation (pathways 1 to 5) at the atomic and molecular level (Figures 8 and S11) (Table 1). In aqueous media, the formation of amyloid fibrils (βAF) from random-coil conformations of Aβ1–40 (RC) was slow under quiescent conditions due to a high energy barrier, and sonication accelerated cytotoxic fibrillation (Figure S11) via a decrease in this energy barrier (pathway 1, RC-βAF transition). At moderate concentrations of TFE, partial helical structures (αP) with relatively high stability accumulated as a kinetic intermediate that proceeded to β-structured amyloid fibrillation (pathway 2, RC-αP-βAF transition) with acceleration by sonication. Aβ1–40 with a helical content of ~15 to ~20% at 15–20% TFE may be optimal for toxic amyloid formation, which is consistent with previous findings.41 The MD simulation in 15% TFE revealed a partial helical structure of Aβ1–40 composed of E11, V12, H13, H14, Q15, and K16, which may be responsible for the productive nucleation for amyloidogenesis through helix–helix interactions.60 Notably, the introduction of proline, known as a helix breaker, at residue 14 significantly retarded the lag time for nucleation.61 The extension of this partial helix from H13 to D23 induced the generation of high molecular weight assemblies.62 The aggregation of α-helical structures in order to proceed to amyloid formation was observed in polyQ fibrillation.63 Aggregation-prone partial helical structures of αSN,31,36 hIAPP,64 a fragment of β2m,65 and apoA-I66 have also been suggested.60,67
Figure 8.
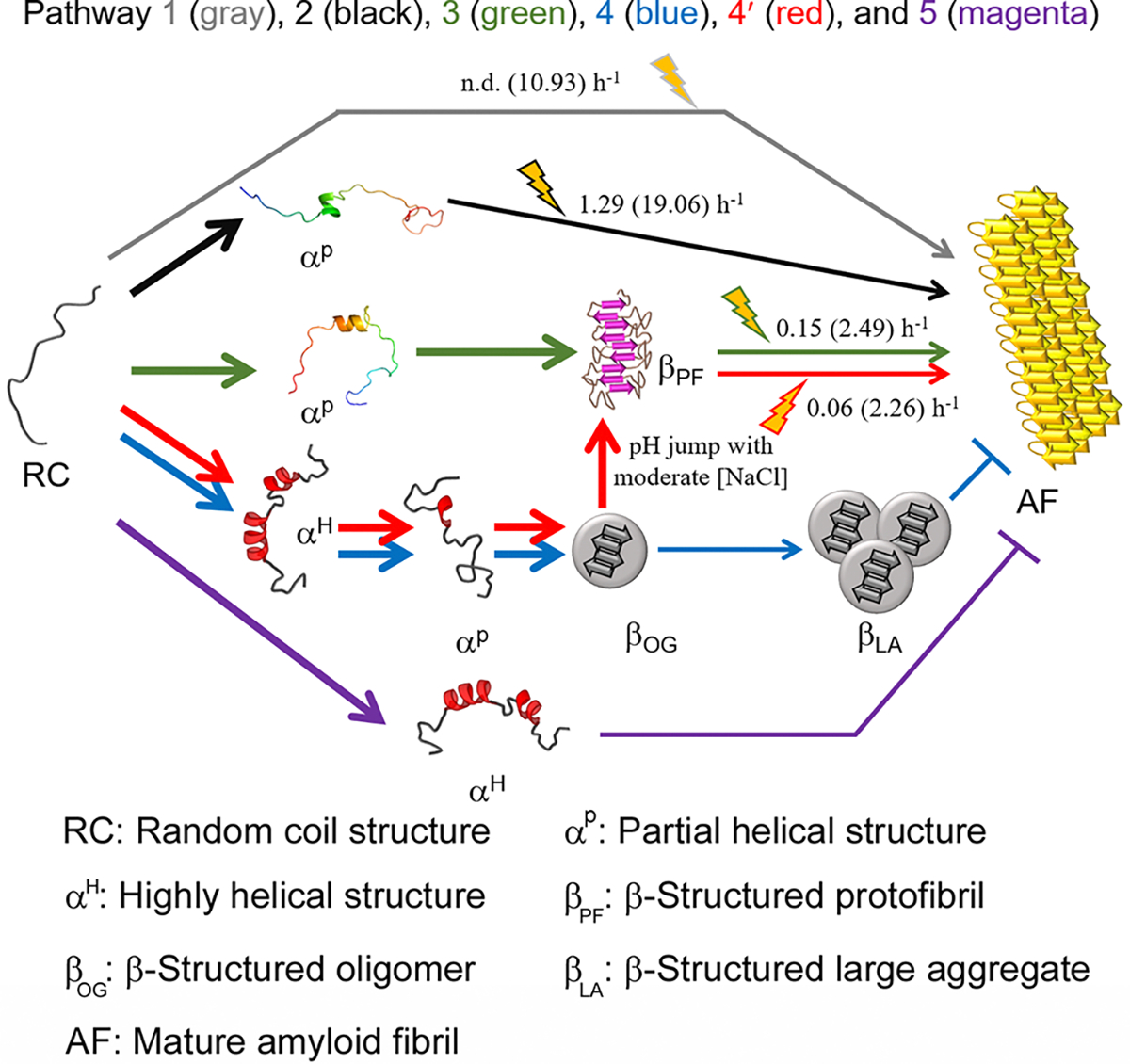
Scheme of the microscopic aggregation pathway of Aβ1–40. Various aggregation pathways are shown and guided by arrowed curves in distinct colors: pathway 1 (gray), pathway 2 (black), pathway 3 (green), pathway 4 (blue), pathway 4′ (red), and pathway 5 (magenta). αP in pathways 2 and 3 obtained from MD simulations was used. αP in pathways 4 and 4′ and αH in pathways 4, 4′, and 5 were conceptually drawn. αP in pathways 3, 4, and 4′ is too transient to be detected using general experimental tools used here. The markedly rapid reactions of which kinetic parameters could not be obtained are represented using bold arrowed curves. The reactions accelerated by sonication are highlighted with cartoons. Kinetic parameters (i.e., the rate constant of fibril growth and conversion) in the absence and presence (values in parentheses) of sonication are shown. “n.d.” denotes the reaction that did not show an increase in the ThT fluorescence during incubation time used here.
Table 1.
Summary of Various aβ1–40 Aggregation Pathways
| pathway | structural transition and aggregation scheme |
| pathway 1 | random coil (RC) → β-structured amyloid fibril (βAF) (RC-βAF transition) |
| pathway 2 | random coil → partial α-helical structure (αP) → β-structured amyloid fibril |
| (RC-αP-βAF transition) | |
| pathway 3 | random coil → partial α-helical structure → β-structured protofibril (βPF) → β-structured amyloid fibril |
| (RC-αP-βPF-βAF transition) | |
| pathway 4 | random coil → highly α-helical structure (αH) → partial α-helical structure → β-structured oligomer (βOG) → β-structured large aggregate (βLA) (RC-αH-αP-βOG-βLA transition) |
| pathway 4′ | random coil → highly α-helical structure → partial α-helical structure → β-structured oligomer → β-structured protofibril → β-structured amyloid fibril (RC-αH-αP-βOG-βPF-βAF transition) |
| αH and αP indicate highly and partially α-helical structures at 100% and 10% HFIP, respectively; italics indicate molecular species at a physiological pH value (e.g., pH 7.5) with NaCl in the range 0–250 mM | |
| pathway 5 | random coil → highly α-helical structure (RC-αH transition) |
| αH represents highly α-helical structures at high alcohol concentrations. |
In pathway 3 (RC-αP-βPF-βAF transition), the addition of small amounts of HFIP (2%) triggered a dynamic structural transition from RC. αP, which was undetectable in a general experimental time scale, must be unstable (i.e., transient) and aggregation-prone with a shallow energy minimum. αP showed one short helical region (V24, G25, S26, N27, K28, G29, and A30) (Figure 1H, right panel) which corresponds to the turn region (V24–A30) between two β-strands (V12–V24 and A30–V40) in the hydrophobic core of amyloid fibrils.68 Destabilization of turn structures of Aβ monomers (V24–K28) induced largely disordered and helical structures which promoted oligomerization or protofibrillation,69,70 implicating the importance of short helical region (V24–K28) in determining an aggregation pathway. The enhanced hydrophobicity of the partial helical structure in 2% HFIP, relative to 15% TFE, might strengthen the helix–helix interaction, causing rapid aggregation. ThT-positive β-structured protofibrils (βPF) are apparent kinetic intermediates of misfolded aggregates. No similar structure of αP in 2% HFIP is available up to now due probably to this significant aggregation propensity. Cytotoxic amyloid fibrils (Figure S6) emerged as a final molecular species with the disappearance of βPF (Figure 3), and the transition was again promoted by sonication. Since similar rapid formation of β-structured aggregates and ThT kinetic profiles were observed at 6% and 12% HFIP (Figures 1 and 3), we speculate that sticky helical conformations may also transiently exist at 6–12% HFIP and follow pathway 3.
In pathway 4 (RC-αH-αP-βOG-βLA transition) and pathway 4′ (RC-αH-αP-βOG-βPF-βAF transition), the RC underwent a transition to ThT-negative β-structured oligomers (βOG) via αP at 10% HFIP, which might be distinguished from a highly α-helical structure at 100% HFIP (αH) (Figure S12). In the absence of pH jump, further incubation with sonication or modulation of ionic strength led to the production of nonfibrillar large aggregates accommodating β structures (βLA). A pH jump from ~4.0 to 7.5 with moderate concentrations of NaCl changed the aggregation mechanism from pathway 4 to pathway 4′. βOG transformed rapidly to ThT-positive βPF, which eventually formed βAF. High concentrations of NaCl with a pH jump blocked pathway 4′, while the absence of a pH jump kept pathway 4.
It has been long debated whether oligomers represent on-pathway intermediates to amyloid fibril formation or off-pathway products.71,72 Aβ peptides have shown the formation of both on- and off-pathway oligomers depending on environmental conditions such as metal ions73 and small molecules59 as well as preparation methods.49,74 Herein, βOG was unable to convert to amyloid fibrils even with extensive treatment of sonication, which indicated that βOG is an off-pathway aggregate, as it represents a deep minimum in the energy landscape (Figure S11). Previous studies suggested that a significant structural conversion was required for the transition from oligomers to amyloid fibrils,75 and this transition was determined by the shape, compactness, and amount of β-structures in oligomers.76 Increases in stability of βOG due to the formation of β structures might decrease an energy level (i.e., thermodynamic stabilization) by creating a high energy barrier (i.e., kinetic stabilization) for the structural reorganization of oligomers toward amyloid aggregation. However, the shift to physiological conditions led to a pathway toward amyloid formation of βOG via protofibrillation in vitro (Figure 6) and possibly in vivo (Figure 7).
pH has been considered to play a crucial role in determining the aggregation pathways of proteins. It has been observed that pH jumps triggered amyloid aggregation of hIAPP,77 human procarboxypeptidase A2,78 and the SH3 domain.79 We herein demonstrated that the pH jump to 7.5 was a prerequisite for the transition from the end product of Aβ1–40 oligomers to mature amyloid fibrils via intermediates of protofibrils. On the other hand, studies of the salt effects on protein aggregation have been extensively performed.80–82 Changes in β2m aggregation pathways from amyloid formation to amorphous aggregation due to the increase in the concentration of NaCl have been observed.11 These observations suggested that neutral salts such as NaCl may be important for determining the aggregation pathway. In this study, we showed that a physiological condition (neutral pH, ionic strength of 125 mM, and 37 °C) was critical for amyloid formation of Aβ1–40. The injection of Aβ1–40 oligomers and protofibrils to brains of mice might cause the formation of amyloid plaques (Figure 7).
Pathways 3 and 4′ demonstrated that Aβ1–40 amyloid formation was in equilibrium because the final product of both was amyloid fibrils irrespective of the distinct aggregation pathway. On the other hand, pathway 5 (RC-αH transition) prevailed at high alcohol concentrations (>40% TFE and >30% HFIP). Predominantly α-helical structures (αH) were highly stable and did not form any aggregates. Highly helical structures of Aβ peptides in the high concentration of alcohol (e.g., 40% TFE45 or 80% HFIP83) or the presence of sodium dodecyl sulfate (SDS)84 showed no aggregation, thereby providing sufficient time for structural determination using NMR spectroscopy not like αP in pathway 3.
Aβ1–40 exhibited a similar pathway alteration in a manner that depended on the concentration of SDS.85 In the absence of SDS, Aβ1–40 was fibrillated with pathway 1. An increase in the concentration of SDS resulted in pathway 4′ via pathway 3. Similar findings of multiple pathways were also reported for the aggregation of acid-induced disordered β2m and intrinsically disordered αSN.36,40,86,87 Unstructured β2m indicated pathway 1 without TFE and pathway 2 with an increase in the concentration of TFE. αSN was previously shown to self-assemble with variations in the concentrations of SDS, TFE, or lipids in model membranes.31,36,67,86,87 αSN presented the following pathway transition with an increase in the amount of SDS or lipids: pathway 1 → pathway 3 → pathway 5.
A recent study reported that the distinct structural states of familial mutants of αSN were responsible for different aggregation pathways,88 which suggested the importance of initial conformations for aggregation. Thus, we propose the importance of α-helical structures at the early stages of aggregation for controlling Aβ1–40 aggregation pathways. Amyloid-competent partial helical structures predominated at moderate and low concentrations of TFE and HFIP, respectively. These helices may be less prominent in aqueous media, which results in slow fibrillation. It is worth noting that the position of partial helical structures in Aβ peptides is also important for the aggregation pathway and amyloidogenicity as suggested in pathways 2 (αP in 15% TFE) and 3 (αP in 2% HFIP). Highly helical structures that are markedly less active may be present at high alcohol concentrations, resulting in the absence of amyloidogenesis.
It is worth noting that fluorinated alcohols also have the capability to stabilize the intermolecular β-sheet structures.39,89 Enhancements in intermolecular hydrogen bonds due to the stabilization of β-sheet structures have been suggested to be a driving force for the promotion of AcP self-assembly at TFE concentrations ranging from 5% to 25%39 and αSN amyloid generation in 2.5–10% TFE.89 Thus, it was concluded that fluorinated alcohols stabilize first aggregation-prone partial helical structures with the enhancement of intramolecular hydrogen bonds in terms of kinetics, however, thermodynamically stabilize the final end product of amyloid fibrils with β-sheet structures due to enhanced intermolecular hydrogen bonds as shown in pathway 2.
Sonication has been shown to produce cavitation bubbles that provide air–water interfaces.11 The helical folding of amphiphilic peptides and the deformation of amyloid fibrils have been observed at the air–water interface.90,91 Therefore, the fibrillation-promoting effects of sonication may be dually attributed to the accumulation and condensation of active helical conformations for nucleation (pathways 1 and 2) and to the remodeling of intermediate aggregates to amyloid fibrils (pathways 3 and 4′).
Fast helix folding has been observed in short peptides and globular proteins for the formation of intramolecular hydrogen bonds.92 An α–β transition during protein folding has been demonstrated in a limited number of cases, such as β-lactoglobulin93 and the intestinal fatty acid-binding protein.94 Random coil-α–β transition, however, may be a generic property of intermolecular protein aggregation when forces to form intramolecular hydrogen bonds are strong, for example, under conditions of low solvent polarity or strong electrostatic interactions such as membrane binding. Our microscopic mechanical models may be useful for understanding Aβ1–40 aggregation mechanisms on membranes in nonpolar environments. Fast folding to partial helical states coupled to rapid aggregation may be beneficial for Aβ1–40 in order to protect it from proteolysis; however, this impairs proteostasis.
A large number of studies on structural details and toxic natures of protein aggregates have been reported in the past decade with advances in methods and techniques.6–9,95,96 However, the fundamental question on whether aggregates generated in vitro display structural and/or toxic features similar to those accumulated in patients is still open to discussion. Recent structural studies revealed that heparin-induced tau filaments had a larger core region97 than those obtained from patients of Alzheimer’s and Pick’s diseases.6,95,96 Our in vivo results implied that the injection of either Aβ oligomers or protofibrils generated in in vitro systems induced the formation of amyloid plaque in brains of mice, although the relevance of in vitro aggregates to aggregates formed in patients of Alzheimer’s disease still needs to be elucidated. Aβ1–40 aggregation and resulting plaque formation in vivo are much more complicated processes than those in our in vitro system due to the complex intra- and extracellular environments. Thus, future investigation on complicated Aβ1–40 aggregation pathways in vivo with the advances in biological or physiological techniques will help us clarify the mechanism underlying Aβ1–40 self-assembly.
Phase Diagrams of Aβ1–40 Aggregation.
We previously demonstrated that phase diagrams were very useful for comprehensively illustrating the protein aggregation of lysozyme,12 insulin,13 and β2m30 in a macroscopic manner and further showed a potential link between macroscopic views of phase equilibrium and microscopic views of structural states using phase diagrams of cytochrome c and αSN aggregation.28,31,67 Through our current studies, we expanded the phase diagram-based general description of protein aggregation in terms of both viewpoints.
The sonication of water/TFE mixtures broadened the amyloid-forming region of Aβ1–40 (0–25% TFE) (Figure 9A) over that under quiescent conditions (5–20%) (Figure 9B), indicating the high metastability of supersaturation at 0 to ~5% and ~20 to ~25% TFE. Aβ1–40 slowly formed amyloid fibrils at 2% HFIP and protofibrils at 6–12% HFIP without agitation (Figure 9C). Sonication rapidly transformed protofibrils to mature amyloid fibrils (Figure 9D). These results suggested the high metastability of supersaturation at 0 to ~2% HFIP and possibly the absence of apparent supersaturation at ~6% HFIP. Low colloidal stability of protofibrils caused transformation to thermodynamically stable amyloid fibrils. At high concentrations of alcohol (>40% TFE and >30% HFIP), Aβ1–40 remained soluble regardless of extensive sonication. These phase behaviors of Aβ1–40 may be macroscopically explained by the metastability of supersaturation with solubility.28 The metastability of supersaturation may be similar to an energy barrier for protein aggregation such as amyloid formation (Figure S11). Thus, strong supersaturation prolongs amyloid formation due to a high energy barrier for productive nucleation. Sonication induces effectively the collapse of supersaturated states, revealing amyloidogenicity.
Figure 9.
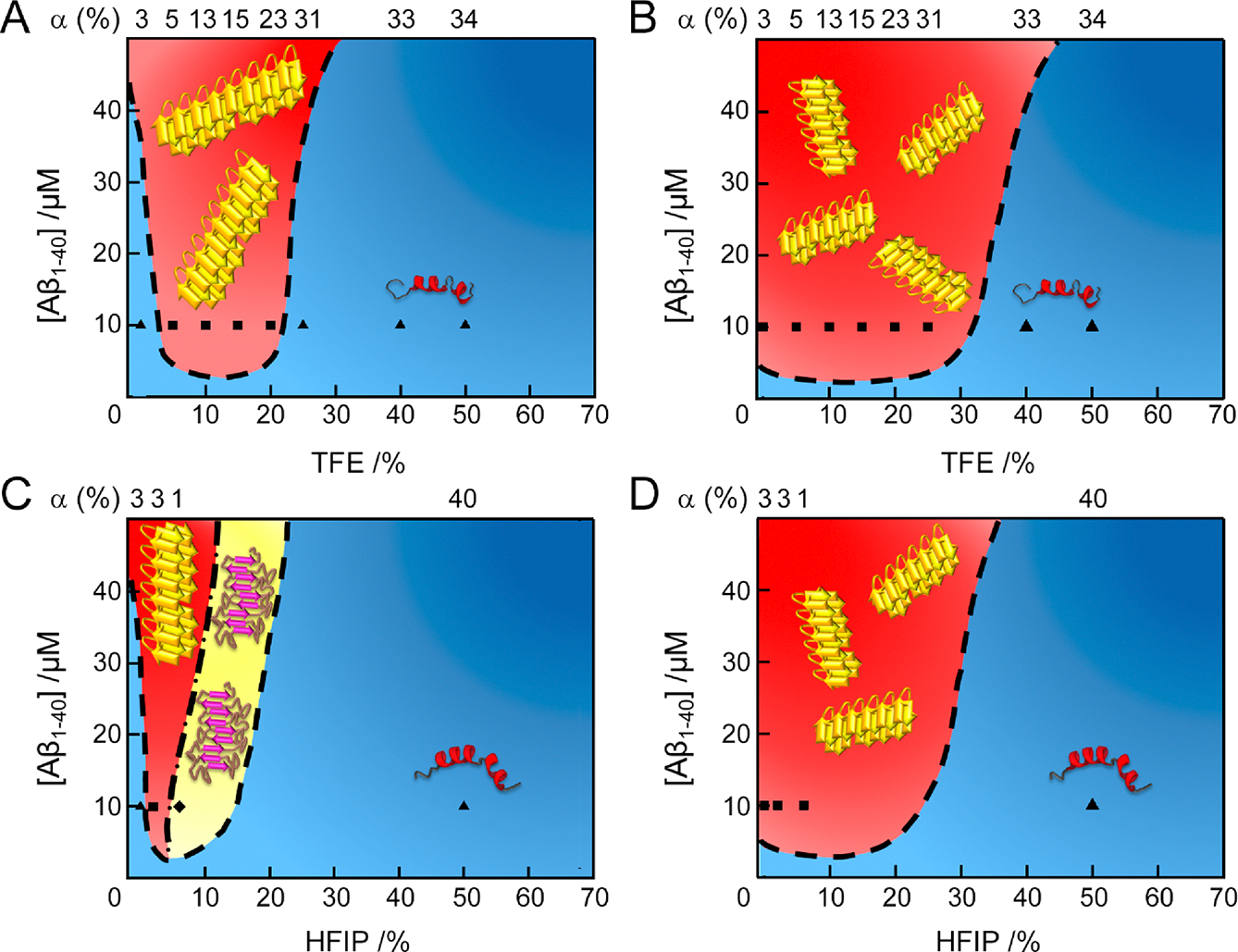
Macroscopic phase diagrams of Aβ1–40 aggregation in water/alcohol mixtures. (A–D) Phase diagrams of Aβ1–40 in the (A and C) absence and (B and D) presence of sonication. Cartoons of helical monomers, amyloid fibrils, and protofibrils are illustrated. Colors and symbols represent molecular species: (blue region and ▲) soluble monomers; (red region and ■) mature amyloid fibrils; (yellow region and ◆) protofibrils. Each symbol indicates the concentration of Aβ1–40 and alcohol used. The α-helical contents of the initial states of Aβ1–40 before aggregation, analyzed by the BeStSel algorithm,44 are displayed at the top of the phase diagrams. The dashed and dash-dotted lines at each phase diagram represent conceptual solubility and boundary curves, respectively. Aβ1–40 aggregation in the presence of 100 mM NaCl at pH 7.5 with (A and B) TFE and (C and D) HFIP.
Higher solubility greater than 25% TFE protects Aβ1–40 from aggregation and is not under the control of supersaturation and agitation. Soluble helical folds at high alcohol concentrations may be stabilized by enthalpic gains from the formation of intramolecular hydrogen bonds. Consequently, the low aggregation propensity of largely helical structures will also be ascribed to high solubility as a macroscopic thermodynamic property or to an amyloid-protected helical conformation as a microscopic structural feature.
The incorporation of the initial helical percentage in phase diagrams provided insights into the aggregation behaviors of unfolded and structured proteins (Figures 9 and S13). Phase diagrams with sonication showed that Aβ1–40 with 3–31% initial helical structures at 0–25% TFE (Figure 9B) and acid-denatured β2m with 0–38% helical structures at 0–50% TFE aggregated to amyloid fibrils (Figure S13A and B).40 As shown by phase diagrams of the aggregation of native globular proteins, unfolded structures with helix contents of ~20 to ~50% for lysozyme12 (Figure S13C and D) and ~40 to ~50% for insulin13 (Figure S13E and F) formed amyloid fibrils. These results indicate that native globular proteins exhibited a higher propensity for amyloid fibrillation when the contents of α-helix were generally higher (~20% to ~50%) than those of initially disordered proteins (~0 to ~40%) due to the alcohol-induced unfolding of native structures. Detailed information on helical states at the atomic and residue levels is needed in order to obtain a deeper understanding of the interplay between helical structures and aggregation propensities as well as aggregate types. Further case studies based on the phase diagram, which also includes detailed structural information, are required to identify a more general relationship among protein aggregation, structures, and solubilities. Finally, currently available phase diagrams will be applicable to the macroscopic and microscopic understanding of phase separation, which is a weak protein aggregation.5
CONCLUSIONS
Understanding of the molecular mechanisms of Aβ1–40 aggregation under varying conditions is of significant importance for controlling amyloidogenesis of Aβ peptides and for developing effective therapeutic modalities. We herein demonstrated that Aβ1–40 underwent diverse aggregation pathways depending on the initial structure and solubility at the residue and molecular level. We proposed a general concept for amyloid aggregation: partially helical structures are aggregation prone and amyloidogenic with a decrease in solubility, while largely helical structures maintain monomeric states due to high solubility. In addition to these microscopic molecular mechanisms, macroscopic phase diagrams of aggregation of Aβ1–40 and several other amyloid proteins with information on initial structures further provided broad insights into supersaturation-limited amyloid generation of unfolded and structured proteins. Future studies on context-dependent aggregation based on microscopic and macroscopic viewpoints will improve our understanding of complicated Aβ1–40 aggregation in vivo and proteopathy.
METHODS
Chemicals.
Lyophilized Aβ1–40 purchased from Peptide Institute Inc. (Osaka, Japan) (purity ≥95.0%) was dissolved in 0.05% (w/w) ammonia solution at a concentration of 100 μM and stored at −80 °C, as described previously.55 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was obtained from Sigma-Aldrich. ThT was obtained from Wako Pure Chemical Industries, Ltd. TFE, HFIP, and other reagents were purchased from Nacalai Tesque (Kyoto, Japan).
Aβ1–40 Oligomer Preparation.
Aβ1–40 oligomers were prepared as described previously.48,49 Briefly, we dissolved lyophilized Aβ1–40 in 100% HFIP (v/v) at 2.5 mg mL−1 (~580 μM) and incubated Aβ1–40 at room temperature for 15 min. Samples were then diluted 10-fold with doubly deionized water. After a further incubation at room temperature for 15 min, samples were centrifuged at 14000g for 15 min to remove large aggregates.
Monitoring of Aβ1–40 Aggregation in Microplates by the ThT Fluorescence Assay.
Sample solutions (200 μL) were transferred to each well of a 96-well microplate (Greiner-Bio-One, Tokyo, Japan), and a sealing film was affixed to prevent evaporation (PowerSeal Cristal View, Greiner-Bio-One). The microplate, set on a water bath-type ultrasonic transmitter with a temperature controller (Elestein SP070-PG-M, Elekon Sci. Inc., Chiba, Japan), was subjected to repetitive sonication pulses for 1 min at 9 min intervals from three directions. Temperature was maintained at 37 °C throughout the experiment. The frequency and power output of the ultrasonic waves were set to 17–20 kHz and 350 W, respectively. Unless otherwise stated, the following experimental conditions were used to produce amyloid fibrils: 10 μM Aβ1–40, 10 mM sodium phosphate buffer (pH 7.5), 100 mM NaCl, 5 μM ThT, and alcohols at various concentrations. The context-dependent aggregation process of Aβ1–40 oligomers in 2% HFIP (pH ~4) to other types of aggregates was first investigated at 37 °C with and without sonication and 10 mM sodium phosphate buffer (pH 7.5) containing 100 mM NaCl. For more detailed information, another experimental condition with continuous shaking (425 cpm) was next used with and without 10 mM sodium phosphate buffer (pH 7.5) at the various concentrations of NaCl (0–2 M). ThT fluorescence intensity for all experiments was monitored using a microplate reader (MTP-810, Corona Electric Co. Ibaraki, Japan, or Synergy Neo2 Hybrid Multi-Mode, Biotek, VT, USA) with excitation and emission wavelengths of 450 and 490 nm, respectively.
Kinetic parameters of Aβ1−40 fibrillation in TFE were obtained by fitting fibrillation curves to the sigmoid equation given below.
| (1) |
where and are the initial and final baselines, respectively. and indicate the rate constant of elongation and time, respectively. is the half-time when ThT fluorescence reaches 50% of its maximum intensity. The lag time was calculated with the following relationship: .98
The apparent rate constant of the transition from protofibrils to mature amyloid fibrils in a water/HFIP mixture was obtained by fitting the transition time course to the following exponential function:
| (2) |
where is the offset and represents the amplitude of the change in the ThT intensity. is the rate constant for the protofibril–amyloid fibril transition and indicates the incubation time. All kinetic parameters were obtained using the averaged values of three independent data.
Monitoring of Aβ1–40 Aggregation in Test Tubes by the ThT Fluorescence Assay.
Since sealing films are dissolved at high HFIP concentrations (>10%), Aβ1–40 aggregation was monitored using test tubes in 10 mM sodium phosphate buffer (pH 7.5) containing 100 mM NaCl, 5 μM ThT, and 50% HFIP (v/v). The ultrasonic transmitter and experimental temperature were identical to those for measurements in microplates. Sample solution was transferred to a 0.5 cm light-path cuvette at the desired time points, and ThT fluorescence intensity was recorded using a Hitachi F4500 fluorescence spectrophotometer (Hitachi, Tokyo, Japan) with excitation and emission wavelengths of 450 and 490 nm, respectively.
CD Spectroscopy.
The far-UV CD spectra of Aβ1–40 sample solutions were recorded using a Jasco J820 spectropolarimeter (Tokyo, Japan) equipped with a Peltier temperature cell holder controller. A cuvette with a light-path length of 1 mm was used. Sixteen scans of individual samples were accumulated and averaged. All spectra were expressed as the mean residue ellipticity, [θ] (deg cm2 dmol−1), after subtracting the signal from a solution without Aβ1–40.
AFM and TEM.
AFM images were obtained using a Digital Instruments Nanoscope IIIa scanning microscope (Veeco Instruments Inc., NY, USA). Sample solutions with aliquots of 20 μL were spotted on freshly cleaved mica plates and incubated for 10–15 min. Samples were then gently rinsed three times with 20 μL of doubly deionized water to wash off salt crystals. Residual water was blown off with compressed air and then air-dried. TEM images were measured using a Bio-High voltage EM system (JEM-1400 Plus at 120 kV and JEM-1000 BEF at 1000 kV; JEOL Ltd., Tokyo, Japan), a monochromated Cs-corrected TEM (Mono ARM200, JEOL Ltd., Tokyo, Japan), and a Hitachi H-7650 transmission microscope (Hitachi, Tokyo, Japan) with a voltage of 80 kV, as reported previously.55 For TEM analyses, we applied 4–5 μL of sample solution to a collodion-coated copper grid (Nisshin EM Co., Tokyo, Japan) and incubated it for 1 min. The remaining solution was then removed with filter paper, and 5 μL of doubly deionized water containing 2% (w/w) ammonium molybdate or 2% (w/w) uranyl acetate was spotted onto collodion-coated copper grids. After 1 min, the remaining solution was removed in the same manner.
Analytical Ultracentrifugation.
The size distribution of Aβ1–40 oligomers in 2% HFIP (v/v) was investigated by sedimentation velocity measurements using a Beckman-Coulter Optima XL-A analytical ultracentrifuge (Beckman Coulter, FL, USA). Samples were centrifuged at 3000 rpm (700g) for 5 min to stabilize absorbance and temperature. The rotor speed was increased to 18 000 rpm (26000g). Absorbance data at 220 nm were recorded at 25 °C. Radial increments were set to 0.003 cm. The continuous scanning mode was selected with one scan every 15 min. We analyzed absorbance data using SEDFIT (http://www.analyticalultracentrifugation.com/default.htm), and parameters were calculated by SEDNTERP (http://bitcwiki.sr.unh.edu/index.php/Main_Page).
Dynamic Light Scattering.
Dynamic light scattering measurements were performed for an Aβ1–40 oligomer solution containing 2% HFIP (v/v) using a DynaPro NanoStar instrument (Wyatt Technology Co., CA, USA) and a 100 μL plastic cuvette. Dust particles were removed from the sample before measurements by spinning down at 10000g for 5 min. The cuvette was placed in the sample chamber maintained at 25 °C. An average of scattering data was obtained using 20 separate scans collected over 20 s. We processed data using the manufacturer’s software (Dynamics; Wyatt Technology Co.).
MD Simulation.
The MD simulation systems were set up in reference to our experimental conditions. The Aβ1–40 peptide (PDB ID: 1AML45) was simulated in solution containing either 85% H2O/15% TFE or 98% H2O/2% HFIP. The topology files for the TFE and HFIP compounds were generated from the Automated Topology Builder.99 SPC water model was employed. All simulations were performed using the GROMACS simulation suite version 5.0.7 and the Gromos53a6 force field. Aβ1–40 was submerged in a cubic box with a minimum distance of 12 Å from the box boundary. The MD systems were subjected to a NaCl concentration of 100 mM and neutralized by adding requisite counterions. The equilibration of MD systems was achieved using NVT and NPT ensembles at 310 K and 1 bar using typical parameters described in our prior report.100 A production MD run of 100 ns was considered for structural and dynamic interpretation. In order to generate an unfolded Aβ1–40 conformation, a helical Aβ1–40 molecule was simulated at 373 K for 100 ns and then gradually annealed to 310 K. The conformational transition of Aβ1–40 from unfolded to helically folded states in the presence of either TFE or HFIP was thus investigated on a time scale of 100 ns. The protein structure visualizations were carried out using PyMOL (https://pymol.org/) and Discovery Studio Visualizer 3.5 (http://accelrys.com/products/collaborative-science/biovia-discovery-studio/). MD trajectory analysis was done using Visual Molecular Dynamics (www.ks.uiuc.edu/Research/vmd/).
Cytotoxicity Experiments.
Human neuroblastoma SH-SY5Y cells were purchased from the American Type Cell Collection (ATCC, VA, USA). Cells were maintained in media containing 50% minimum essential medium (Gibco, NY, USA) and 50% F12 (Gibco), supplemented with 5% (v/v) fetal bovine serum (Gibco) and 100 U/mL penicillin (Gibco). The cells were grown in a humidified atmosphere with 5% CO2 at 37 °C. Cell viability was determined by the MTT assay. The cells were seeded in a 96-well microplate (15 000 cells in 100 μL per well). In order to identify the cytotoxicity of Aβ1–40 amyloid fibrils, Aβ1–40 (100 μM) in three different solvents, 0% alcohol, 15% TFE, or 10% HFIP, was incubated overnight at 37 °C. Formation of amyloid fibrils was confirmed using the ThT assay. Aβ1–40 amyloid fibrils were diluted by 10-fold to obtain the final concentration of 10 μM and then treated with cells. After a 24 h incubation, MTT of 25 μM in PBS (pH 7.4) (Gibco) was incubated in each well, and the plate was incubated for 3 h at 37 °C. Formazan produced by the cells was solubilized by the addition of an acidic solution of N,N-dimethylformamide (50% (v/v)) (pH 4.5) and SDS (20% (w/v)) overnight at room temperature in the dark. The absorbance was measured at 600 nm using a SpectraMax M5e microplate reader (Molecular Devices, CA, USA). Cell viability was calculated relative to the absorbance of cells themselves. Error bars were calculated as standard errors from three independent experiments.
Enzyme-Linked Immunosorbent Assay.
A 96-well ELISA plate kit (Sumitomo Bakelite, Japan) was used. Each well was coated with 100 μL of an anti-amyloid oligomer antibody (1:100) (Merck, Germany) dissolved in the coating buffer supplied by the manufacturer. After wells were washed three times with washing buffer (10 mM phosphate buffer (pH 7.5) containing 100 mM NaCl and 0.05% Tween 20), 100 μL of 5.8 μM Aβ1–40 monomers or oligomers formed with the same Aβ1–40 concentration as well as 10 mM phosphate buffer (pH 7.5) containing 100 mM NaCl and 1% HFIP were added to the wells and incubated at 25 °C for 1 h. After wells were washed three times with washing buffer, bound Aβ1–40 was detected with an anti-human Aβ antibody (Dako, CA, USA) and horseradish peroxidase-conjugated anti-mouse immunoglobulin antibody (1:1000) (Dako) followed by color development using 3,3′,5,5′-tetramethylbenzidine as the peroxidase substrate (Bio-Rad, CA, USA). Absorbance was measured at 450 nm using a SpectraMax M5 microplate reader (Molecular Devices, CA, USA). The absorbance of the well, which was not coated with the anti-amyloid oligomer antibody, was subtracted from each data point.
Intrahippocampal Injection of Aβ1–40 Aggregates and Immunohistochemistry.
Aβ1–40 oligomers and protofibrils formed in vitro were separately injected to wild-type mice. All experimental procedures were performed in accordance with approved animal protocols and guidelines established by the Korea Brain Research Institute (IACUC-2016–0013). Male C57BL6/N mice (8 weeks old and 25–30 g) were purchased from Orient-Bio Company (Gyeonggido, Korea) and housed in a pathogen-free facility with 12 h of light and dark per day at 22 °C. Mice were anaesthetized with avertin (2,2,2 tribromoethanol in 2-methyl-2-butanol and 250 mg/kg, i.p.) and placed in a stereotaxic apparatus (Stoelting, IL, USA). Oligomers, protofibrils, or vehicle controls were injected bilaterally into the hippocampus CA1 regions in a volume of 1.5 μL at a flow rate of 0.2 μL/min by using a Hamilton 10 μL syringe and a 26 G needle. The stereotaxical coordinates of the hippocampus CA1 injection site were −2.5 mm anterior/posterior, ±2.0 mm medial/lateral, and −2.0 mm dorsal/ventral from the bregma. At 24 h after injection, mice treated with injection of oligomers, protofibrils, or vehicles were perfused and fixed for immunohistochemistry.
Briefly, each brain section was rinsed with phosphate-buffered saline (PBS) three times and incubated with blocking solution containing 0.2% Triton X-100, 1% bovine serum albumin (BSA), and 4% goat serum in PBS for 90 min at room temperature. The brain sections were then washed twice with 0.5% BSA in PBST and incubated at 4 °C with anti-4G8 antibodies (1:200, Biolegend, CA, USA) overnight. The next day, the brain sections were washed with 0.5% BSA in PBST three times and incubated with Alexa 488-conjugated goat anti-mouse IgG (1:200, Life Technologies, CA, USA) for 90 min at room temperature. The brain sections were then rinsed with PBS three times, mounted on slide glass, and covered with DAPI-containing mounting solution (Vector Laboratories, CA, USA). Images of the stained tissues were captured using fluorescence microscopy (DMi8, Leica, Wetzlar, Germany), and fluorescence intensity was measured using ImageJ (NIH) software.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Yuji Goto (Osaka University, Japan) for providing instruments. We also thank Dr. Mayu Terakawa (Columbia University, USA) for support of TEM measurements. This work was supported by the Grant-in-Aid for Young Scientists (B) (15K18518 and 25870407, Japan) (Y.-H.L.), the Korea Basic Science Institute grant (T38624) (Y.-H.L.), the National Research Foundation of Korea (NRF) grants funded by the Korean government (NRF-PG2018123 and NRF-PG2019046) (Y.-H.L.), a National Research Council of Science & Technology (NST) grant funded by the Korean government (MSIP) (CAP-17–05-KIGAM) (Y.-H.L.), the National Institutes of Health grant (AG048934) (A.R.), an NRF grant funded by the Korean government (NRF-2017R1A2B3002585) (M.H.L.), the Samsung Science and Technology Foundation (SSTF-BA1401–13) (S.H.), and the basic research program of Korea Brain Research Institute funded by the Ministry of Science, ICT & Future Planning (19-BR-02–02) (H.S.H.).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.9b01578.
Additional figures (PDF)
REFERENCES
- (1).Chiti F; Dobson CM Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [DOI] [PubMed] [Google Scholar]
- (2).Eisenberg D; Jucker M The Amyloid State of Proteins in Human Diseases. Cell 2012, 148, 1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tao K; Makam P; Aizen R; Gazit E Self-Assembling Peptide Semiconductors. Science 2017, 358, eaam9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wei G; Su Z; Reynolds NP; Arosio P; Hamley IW; Gazit E; Mezzenga R Self-Assembling Peptide and Protein Amyloids: From Structure to Tailored Function in Nanotechnology. Chem. Soc. Rev. 2017, 46, 4661–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shin Y; Brangwynne CP Liquid Phase Condensation in Cell Physiology and Disease. Science 2017, 357, eaaf4382. [DOI] [PubMed] [Google Scholar]
- (6).Fitzpatrick AWP; Falcon B; He S; Murzin AG; Murshudov G; Garringer HJ; Crowther RA; Ghetti B; Goedert M; Scheres SHW Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Guerrero-Ferreira R; Taylor NMI; Mona D; Ringler P; Lauer ME; Riek R; Britschgi M; Stahlberg H Cryo-EM Structure of α-Synuclein Fibrils. eLife 2018, 7, No. e36402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tuttle MD; Comellas G; Nieuwkoop AJ; Covell DJ; Berthold DA; Kloepper KD; Courtney JM; Kim JK; Barclay AM; Kendall A; Wan W; Stubbs G; Schwieters CD; Lee VMY; George JM; Rienstra CM Solid-State NMR Structure of a Pathogenic Fibril of Full-length Human α-Synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Walti MA; Ravotti F; Arai H; Glabe CG; Wall JS; Bockmann A; Guntert P; Meier BH; Riek R Atomic-Resolution Structure of a Disease-Relevant Aβ(1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4976–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Harper JD; Lansbury PT Jr. Models of Amyloid Seeding in Alzheimer’s Disease and Scrapie: Mechanistic Truths and Physiological Consequences of the Time-dependent Solubility of Amyloid Proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [DOI] [PubMed] [Google Scholar]
- (11).Yoshimura Y; Lin YX; Yagi H; Lee YH; Kitayama H; Sakurai K; So M; Ogi H; Naiki H; Goto Y Distinguishing Crystal-Like Amyloid Fibrils and Glass-Like Amorphous Aggregates from Their Kinetics of Formation. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 14446–14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lin Y; Lee YH; Yoshimura Y; Yagi H; Goto Y Solubility and Supersaturation-Dependent Protein Misfolding Revealed by Ultrasonication. Langmuir 2014, 30, 1845–1854. [DOI] [PubMed] [Google Scholar]
- (13).Muta H; Lee YH; Kardos J; Lin Y; Yagi H; Goto Y Supersaturation-Limited Amyloid Fibrillation of Insulin Revealed by Ultrasonication. J. Biol. Chem. 2014, 289, 18228–18238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Papadopoulou V; Tang MX; Balestra C; Eckersley RJ; Karapantsios TD Circulatory Bubble Dynamics: From Physical to Biological Aspects. Adv. Colloid Interface Sci. 2014, 206, 239–249. [DOI] [PubMed] [Google Scholar]
- (15).Wu YT; Adnan A Effect of Shock-Induced Cavitation Bubble Collapse on the Damage in the Simulated Perineuronal Net of the Brain. Sci. Rep. 2017, 7, 5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Stoppini M; Bellotti V Systemic Amyloidosis: Lessons from β2-Microglobulin. J. Biol. Chem. 2015, 290, 9951–9958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Benilova I; Karran E; De Strooper B The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [DOI] [PubMed] [Google Scholar]
- (18).Iljina M; Garcia GA; Horrocks MH; Tosatto L; Choi ML; Ganzinger KA; Abramov AY; Gandhi S; Wood NW; Cremades N; Dobson CM; Knowles TP; Klenerman D Kinetic Model of the Aggregation of α-Synuclein Provides Insights into Prion-Like Spreading. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E1206–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Brender JR; Salamekh S; Ramamoorthy A Membrane Disruption and Early Events in the Aggregation of the Diabetes Related Peptide IAPP from a Molecular Perspective. Acc. Chem. Res. 2012, 45, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Harper JD; Wong SS; Lieber CM; Lansbury PT Jr. Assembly of Aβ Amyloid Protofibrils: An In Vitro Model for a Possible Early Event in Alzheimer’s Disease. Biochemistry 1999, 38, 8972–8980. [DOI] [PubMed] [Google Scholar]
- (21).Poirier MA; Li H; Macosko J; Cai S; Amzel M; Ross CA Huntingtin Spheroids and Protofibrils as Precursors in Polyglutamine Fibrilization. J. Biol. Chem. 2002, 277, 41032–41037. [DOI] [PubMed] [Google Scholar]
- (22).Ramachandran G; Udgaonkar JB Understanding the Kinetic Roles of the Inducer Heparin and of Rod-Like Protofibrils during Amyloid Fibril Formation by Tau Protein. J. Biol. Chem. 2011, 286, 38948–38959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lee YH; Chatani E; Sasahara K; Naiki H; Goto Y A Comprehensive Model for Packing and Hydration for Amyloid Fibrils of β2-Microglobulin. J. Biol. Chem. 2009, 284, 2169–2175. [DOI] [PubMed] [Google Scholar]
- (24).Neudecker P; Robustelli P; Cavalli A; Walsh P; Lundstrom P; Zarrine-Afsar A; Sharpe S; Vendruscolo M; Kay LE Structure of an Intermediate State in Protein Folding and Aggregation. Science 2012, 336, 362–366. [DOI] [PubMed] [Google Scholar]
- (25).Jahn TR; Parker MJ; Homans SW; Radford SE Amyloid Formation under Physiological Conditions Proceeds via a Native-Like Folding Intermediate. Nat. Struct. Mol. Biol. 2006, 13, 195–201. [DOI] [PubMed] [Google Scholar]
- (26).Gianni S; Camilloni C; Giri R; Toto A; Bonetti D; Morrone A; Sormanni P; Brunori M; Vendruscolo M Understanding the Frustration Arising from the Competition between Function, Misfolding, and Aggregation in a Globular Protein. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 14141–14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cho MK; Nodet G; Kim HY; Jensen MR; Bernado P; Fernandez CO; Becker S; Blackledge M; Zweckstetter M Structural Characterization of α-Synuclein in an Aggregation Prone State. Protein Sci. 2009, 18, 1840–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lin Y; Kardos J; Imai M; Ikenoue T; Kinoshita M; Sugiki T; Ishimori K; Goto Y; Lee YH Amorphous Aggregation of Cytochrome c with Inherently Low Amyloidogenicity Is Characterized by the Metastability of Supersaturation and the Phase Diagram. Langmuir 2016, 32, 2010–2022. [DOI] [PubMed] [Google Scholar]
- (29).Ciryam P; Tartaglia GG; Morimoto RI; Dobson CM; Vendruscolo M Widespread Aggregation and Neurodegenerative Diseases Are Associated with Supersaturated Proteins. Cell Rep. 2013, 5, 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ikenoue T; Lee YH; Kardos J; Yagi H; Ikegami T; Naiki H; Goto Y Heat of Supersaturation-Limited Amyloid Burst Directly Monitored by Isothermal Titration Calorimetry. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 6654–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Terakawa MS; Lee YH; Kinoshita M; Lin Y; Sugiki T; Fukui N; Ikenoue T; Kawata Y; Goto Y Membrane-Induced Initial Structure of α-Synuclein Control Its Amyloidogenesis on Model Membranes. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 757–766. [DOI] [PubMed] [Google Scholar]
- (32).Retailleau P; Ducruix A; Ries-Kautt M Importance of the Nature of Anions in Lysozyme Crystallisation Correlated with Protein Net Charge Variation. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 1576–1581. [DOI] [PubMed] [Google Scholar]
- (33).Walther DM; Kasturi P; Zheng M; Pinkert S; Vecchi G; Ciryam P; Morimoto RI; Dobson CM; Vendruscolo M; Mann M; Hartl FU Widespread Proteome Remodeling and Aggregation in Aging C. elegans. Cell 2015, 161, 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Pallai PV; Mabilia M; Goodman M; Vale W; Rivier J Structural Homology of Corticotropin-Releasing Factor, Sauvagine, and Urotensin I: Circular Dichroism and Prediction Studies. Proc. Natl. Acad. Sci. U. S. A. 1983, 80, 6770–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Clark DJ; Hill CS; Martin SR; Thomas JO Alpha-Helix in the Carboxy-Terminal Domains of Histones H1 and H5. EMBO J. 1988, 7, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Anderson VL; Ramlall TF; Rospigliosi CC; Webb WW; Eliezer D Identification of a Helical Intermediate in Trifluoroethanol-Induced α-Synuclein Aggregation. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18850–18855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Otzen DE Amyloid Formation in Surfactants and Alcohols: Membrane Mimetics or Structural Switchers? Curr. Protein Pept. Sci. 2010, 11, 355–371. [DOI] [PubMed] [Google Scholar]
- (38).Plakoutsi G; Taddei N; Stefani M; Chiti F Aggregation of the Acylphosphatase from Sulfolobus Solfataricus: The Folded and Partially Unfolded States can both be Precursors for Amyloid Formation. J. Biol. Chem. 2004, 279, 14111–14119. [DOI] [PubMed] [Google Scholar]
- (39).Calamai M; Chiti F; Dobson CM Amyloid Fibril Formation can Proceed from Different Conformations of a Partially Unfolded Protein. Biophys. J. 2005, 89, 4201–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Chatani E; Yagi H; Naiki H; Goto Y Polymorphism of β2-Microglobulin Amyloid Fibrils Manifested by Ultrasonication-Enhanced Fibril Formation in Trifluoroethanol. J. Biol. Chem. 2012, 287, 22827–22837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fezoui Y; Teplow DB Kinetic Studies of Amyloid β-Protein Fibril Assembly. Differential Effects of α-Helix Stabilization. J. Biol. Chem. 2002, 277, 36948–36954. [DOI] [PubMed] [Google Scholar]
- (42).Pachahara SK; Adicherla H; Nagaraj R Self-Assembly of Aβ40, Aβ42 and Aβ43 Peptides in Aqueous Mixtures of Fluorinated Alcohols. PLoS One 2015, 10, No. e0136567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Roche J; Shen Y; Lee JH; Ying J; Bax A Monomeric Aβ1–40 and Aβ1–42 Peptides in Solution Adopt Very Similar Ramachandran Map Distributions That Closely Resemble Random Coil. Biochemistry 2016, 55, 762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Micsonai A; Wien F; Kernya L; Lee YH; Goto Y; Refregiers M; Kardos J Accurate Secondary Structure Prediction and Fold Recognition for Circular Dichroism Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E3095–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sticht H; Bayer P; Willbold D; Dames S; Hilbich C; Beyreuther K; Frank RW; Rosch P Structure of Amyloid A4-(1–40)-Peptide of Alzheimer’s Disease. Eur. J. Biochem. 1995, 233, 293– 298. [DOI] [PubMed] [Google Scholar]
- (46).Roccatano D; Colombo G; Fioroni M; Mark AE Mechanism by Which 2,2,2-Trifluoroethanol/Water Mixtures Stabilize Secondary-Structure Formation in Peptides: A Molecular Dynamics Study. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12179–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Biancalana M; Koide S Molecular Mechanism of Thioflavin-T Binding to Amyloid Fibrils. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Morgado I; Wieligmann K; Bereza M; Ronicke R; Meinhardt K; Annamalai K; Baumann M; Wacker J; Hortschansky P; Malesevic M; Parthier C; Mawrin C; Schiene-Fischer C; Reymann KG; Stubbs MT; Balbach J; Gorlach M; Horn U; Fandrich M Molecular Basis of β-Amyloid Oligomer Recognition with a Conformational Antibody Fragment. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 12503–12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kayed R; Head E; Thompson JL; McIntire TM; Milton SC; Cotman CW; Glabe CG Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [DOI] [PubMed] [Google Scholar]
- (50).Sun Y; Kakinen A; Xing Y; Faridi P; Nandakumar A; Purcell AW; Davis TP; Ke PC; Ding F Amyloid Self-Assembly of hIAPP8–20 via the Accumulation of Helical Oligomers, α-Helix to β-Sheet Transition, and Formation of β-Barrel Intermediates. Small 2019, 15, No. e1805166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Winner B; Jappelli R; Maji SK; Desplats PA; Boyer L; Aigner S; Hetzer C; Loher T; Vilar M; Campioni S; Tzitzilonis C; Soragni A; Jessberger S; Mira H; Consiglio A; Pham E; Masliah E; Gage FH; Riek R In Vivo Demonstration that α-Synuclein Oligomers Are Toxic. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Gosal WS; Morten IJ; Hewitt EW; Smith DA; Thomson NH; Radford SE Competing Pathways Determine Fibril Morphology in the Self-Assembly of β2-Microglobulin into Amyloid. J. Mol. Biol. 2005, 351, 850–864. [DOI] [PubMed] [Google Scholar]
- (53).Alafuzoff I; Pikkarainen M; Arzberger T; Thal DR; Al-Sarraj S; Bell J; Bodi I; Budka H; Capetillo-Zarate E; Ferrer I; Gelpi E; Gentleman S; Giaccone G; Kavantzas N; King A; Korkolopoulou P; Kovacs GG; Meyronet D; Monoranu C; Parchi P; et al. Inter-Laboratory Comparison of Neuropathological Assessments of β-Amyloid Protein: A Study of the BrainNet Europe Consortium. Acta Neuropathol. 2008, 115, 533–546. [DOI] [PubMed] [Google Scholar]
- (54).Chatani E; Inoue R; Imamura H; Sugiyama M; Kato M; Yamamoto M; Nishida K; Kanaya T Early Aggregation Preceding the Nucleation of Insulin Amyloid Fibrils as Monitored by Small Angle X-ray Scattering. Sci. Rep. 2015, 5, 15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Terakawa MS; Yagi H; Adachi M; Lee YH; Goto Y Small Liposomes Accelerate the Fibrillation of Amyloid β (1–40). J. Biol. Chem. 2015, 290, 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Korshavn KJ; Satriano C; Lin Y; Zhang R; Dulchavsky M; Bhunia A; Ivanova MI; Lee YH; La Rosa C; Lim MH; Ramamoorthy A Reduced Lipid Bilayer Thickness Regulates the Aggregation and Cytotoxicity of Amyloid-β. J. Biol. Chem. 2017, 292, 4638–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Motamedi-Shad N; Monsellier E; Torrassa S; Relini A; Chiti F Kinetic Analysis of Amyloid Formation in the Presence of Heparan Sulfate: Faster Unfolding and Change of Pathway. J. Biol. Chem. 2009, 284, 29921–29934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Lamberto GR; Binolfi A; Orcellet ML; Bertoncini CW; Zweckstetter M; Griesinger C; Fernandez CO Structural and Mechanistic Basis behind the Inhibitory Interaction of PcTS on α-Synuclein Amyloid Fibril Formation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 21057–21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bieschke J; Herbst M; Wiglenda T; Friedrich RP; Boeddrich A; Schiele F; Kleckers D; Lopez del Amo JM; Gruning BA; Wang Q; Schmidt MR; Lurz R; Anwyl R; Schnoegl S; Fandrich M; Frank RF; Reif B; Gunther S; Walsh DM; Wanker EE Small-Molecule Conversion of Toxic Oligomers to Nontoxic β-Sheet-Rich Amyloid Fibrils. Nat. Chem. Biol. 2012, 8, 93–101. [DOI] [PubMed] [Google Scholar]
- (60).Abedini A; Raleigh DP A Role for Helical Intermediates in Amyloid Formation by Natively Unfolded Polypeptides? Phys. Biol. 2009, 6, No. 015005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Williams AD; Portelius E; Kheterpal I; Guo JT; Cook KD; Xu Y; Wetzel R Mapping Aβ Amyloid Fibril Secondary Structure Using Scanning Proline Mutagenesis. J. Mol. Biol. 2004, 335, 833–842. [DOI] [PubMed] [Google Scholar]
- (62).Vivekanandan S; Brender JR; Lee SY; Ramamoorthy A A Partially Folded Structure of Amyloid-β(1–40) in an Aqueous Environment. Biochem. Biophys. Res. Commun. 2011, 411, 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Jayaraman M; Kodali R; Sahoo B; Thakur AK; Mayasundari A; Mishra R; Peterson CB; Wetzel R Slow Amyloid Nucleation via α-Helix-Rich Oligomeric Intermediates in Short Polyglutamine-Containing Huntingtin Fragments. J. Mol. Biol. 2012, 415, 881–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Nanga RP; Brender JR; Vivekanandan S; Ramamoorthy A Structure and Membrane Orientation of IAPP in Its Natively Amidated Form at Physiological pH in a Membrane Environment. Biochim. Biophys. Acta, Biomembr. 2011, 1808, 2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Yamaguchi K; Naiki H; Goto Y Mechanism by Which the Amyloid-Like Fibrils of a β2-Microglobulin Fragment Are Induced by Fluorine-Substituted Alcohols. J. Mol. Biol. 2006, 363, 279–288. [DOI] [PubMed] [Google Scholar]
- (66).Mizuguchi C; Ogata F; Mikawa S; Tsuji K; Baba T; Shigenaga A; Shimanouchi T; Okuhira K; Otaka A; Saito H Amyloidogenic Mutation Promotes Fibril Formation of the N-Terminal Apolipoprotein A-I on Lipid Membranes. J. Biol. Chem. 2015, 290, 20947–20959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Terakawa MS; Lin Y; Kinoshita M; Kanemura S; Itoh D; Sugiki T; Okumura M; Ramamoorthy A; Lee YH Impact of Membrane Curvature on Amyloid Aggregation. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1741–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Petkova AT; Ishii Y; Balbach JJ; Antzutkin ON; Leapman RD; Delaglio F; Tycko R A Structural Model for Alzheimer’s β-Amyloid Fibrils Based on Experimental Constraints from Solid State NMR. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Grant MA; Lazo ND; Lomakin A; Condron MM; Arai H; Yamin G; Rigby AC; Teplow DB Familial Alzheimer’s Disease Mutations Alter the Stability of the Amyloid β-Protein Monomer Folding Nucleus. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 16522–16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Straub JE; Guevara J; Huo S; Lee JP Long Time Dynamic Simulations: Exploring the Folding Pathways of an Alzheimer’s Amyloid Aβ-Peptide. Acc. Chem. Res. 2002, 35, 473–481. [DOI] [PubMed] [Google Scholar]
- (71).Bemporad F; Chiti F Protein Misfolded Oligomers: Experimental Approaches, Mechanism of Formation, and Structure-Toxicity Relationships. Chem. Biol. 2012, 19, 315–327. [DOI] [PubMed] [Google Scholar]
- (72).Fandrich M Oligomeric Intermediates in Amyloid Formation: Structure Determination and Mechanisms of Toxicity. J. Mol. Biol. 2012, 421, 427–440. [DOI] [PubMed] [Google Scholar]
- (73).Hane F; Leonenko Z Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation. Biomolecules 2014, 4, 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Barghorn S; Nimmrich V; Striebinger A; Krantz C; Keller P; Janson B; Bahr M; Schmidt M; Bitner RS; Harlan J; Barlow E; Ebert U; Hillen H Globular Amyloid β-peptide1–42 Oligomer - a Homogenous and Stable Neuropathological Protein in Alzheimer’s Disease. J. Neurochem. 2005, 95, 834–847. [DOI] [PubMed] [Google Scholar]
- (75).Sarroukh R; Cerf E; Derclaye S; Dufrene YF; Goormaghtigh E; Ruysschaert JM; Raussens V Transformation of Amyloid β(1–40) Oligomers into Fibrils Is Characterized by a Major Change in Secondary Structure. Cell. Mol. Life Sci. 2011, 68, 1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Yu X; Zheng J Polymorphic Structures of Alzheimer’s βAmyloid Globulomers. PLoS One 2011, 6, No. e20575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Cao P; Raleigh DP Ester to Amide Switch Peptides Provide a Simple Method for Preparing Monomeric Islet Amyloid Polypeptide under Physiologically Relevant Conditions and Facilitate Investigations of Amyloid Formation. J. Am. Chem. Soc. 2010, 132, 4052–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Cerda-Costa N; Esteras-Chopo A; Aviles FX; Serrano L; Villegas V Early Kinetics of Amyloid Fibril Formation Reveals Conformational Reorganisation of Initial Aggregates. J. Mol. Biol. 2007, 366, 1351–1363. [DOI] [PubMed] [Google Scholar]
- (79).Guijarro JI; Sunde M; Jones JA; Campbell ID; Dobson CM Amyloid Fibril Formation by an SH3 Domain. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 4224–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Campioni S; Mannini B; Lopez-Alonso JP; Shalova IN; Penco A; Mulvihill E; Laurents DV; Relini A; Chiti F Salt Anions Promote the Conversion of HypF-N into Amyloid-like Oligomers and Modulate the Structure of the Oligomers and the Monomeric Precursor State. J. Mol. Biol. 2012, 424, 132–149. [DOI] [PubMed] [Google Scholar]
- (81).Marek PJ; Patsalo V; Green DF; Raleigh DP Ionic Strength Effects on Amyloid Formation by Amylin Are a Complicated Interplay among Debye Screening, Ion Selectivity, and Hofmeister Effects. Biochemistry 2012, 51, 8478–8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Buell AK; Hung P; Salvatella X; Welland ME; Dobson CM; Knowles TP Electrostatic Effects in Filamentous Protein Aggregation. Biophys. J. 2013, 104, 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Crescenzi O; Tomaselli S; Guerrini R; Salvadori S; D’Ursi AM; Temussi PA; Picone D Solution Structure of the Alzheimer Amyloid β-Peptide (1–42) in an Apolar Microenvironment. Similarity with a Virus Fusion Domain. Eur. J. Biochem. 2002, 269, 5642–5648. [DOI] [PubMed] [Google Scholar]
- (84).Coles M; Bicknell W; Watson AA; Fairlie DP; Craik DJ Solution Structure of Amyloid β-Peptide(1–40) in a Water-Micelle Environment. Is the Membrane-Spanning Domain Where We Think It Is? Biochemistry 1998, 37, 11064–11077. [DOI] [PubMed] [Google Scholar]
- (85).Abelein A; Kaspersen JD; Nielsen SB; Jensen GV; Christiansen G; Pedersen JS; Danielsson J; Otzen DE; Graslund A Formation of Dynamic Soluble Surfactant-Induced Amyloid β Peptide Aggregation Intermediates. J. Biol. Chem. 2013, 288, 23518–23528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Giehm L; Oliveira CL; Christiansen G; Pedersen JS; Otzen DE SDS-Induced Fibrillation of α-Synuclein: An Alternative Fibrillation Pathway. J. Mol. Biol. 2010, 401, 115–133. [DOI] [PubMed] [Google Scholar]
- (87).Galvagnion C; Buell AK; Meisl G; Michaels TC; Vendruscolo M; Knowles TP; Dobson CM Lipid Vesicles Trigger α-Synuclein Aggregation by Stimulating Primary Nucleation. Nat. Chem. Biol. 2015, 11, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Sahay S; Ghosh D; Dwivedi S; Anoop A; Mohite GM; Kombrabail M; Krishnamoorthy G; Maji SK Familial Parkinson Disease-Associated Mutations Alter the Site-Specific Microenvironment and Dynamics of α-Synuclein. J. Biol. Chem. 2015, 290, 7804–7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Munishkina LA; Phelan C; Uversky VN; Fink AL Conformational Behavior and Aggregation of α-Synuclein in Organic Solvents: Modeling the Effects of Membranes. Biochemistry 2003, 42, 2720–2730. [DOI] [PubMed] [Google Scholar]
- (90).Wang C; Shah N; Thakur G; Zhou F; Leblanc RM α-Synuclein in α-Helical Conformation at Air-Water Interface: Implication of Conformation and Orientation Changes during Its Accumulation/Aggregation. Chem. Commun. 2010, 46, 6702–6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Noda S; So M; Adachi M; Kardos J; Akazawa-Ogawa Y; Hagihara Y; Goto Y Thioflavin T-Silent Denaturation Intermediates Support the Main-Chain-Dominated Architecture of Amyloid Fibrils. Biochemistry 2016, 55, 3937–3948. [DOI] [PubMed] [Google Scholar]
- (92).Kritzer JA; Tirado-Rives J; Hart SA; Lear JD; Jorgensen WL; Schepartz A Relationship between Side Chain Structure and 14-Helix Stability of β3-Peptides in Water. J. Am. Chem. Soc. 2005, 127, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Hamada D; Segawa S; Goto Y Non-Native α-Helical Intermediate in the Refolding of β-Lactoglobulin, a Predominantly β-Sheet Protein. Nat. Struct. Mol. Biol. 1996, 3, 868–873. [DOI] [PubMed] [Google Scholar]
- (94).Sarkar-Banerjee S; Chowdhury S; Paul SS; Dutta D; Ghosh A; Chattopadhyay K The Non-Native Helical Intermediate State May Accumulate at Low pH in the Folding and Aggregation Landscape of the Intestinal Fatty Acid Binding Protein. Biochemistry 2016, 55, 4457–4468. [DOI] [PubMed] [Google Scholar]
- (95).Swuec P; Lavatelli F; Tasaki M; Paissoni C; Rognoni P; Maritan M; Brambilla F; Milani P; Mauri P; Camilloni C; Palladini G; Merlini G; Ricagno S; Bolognesi M Cryo-EM Structure of Cardiac Amyloid Fibrils from an Immunoglobulin Light Chain AL Amyloidosis Patient. Nat. Commun. 2019, 10, 1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Falcon B; Zhang W; Murzin AG; Murshudov G; Garringer HJ; Vidal R; Crowther RA; Ghetti B; Scheres SHW; Goedert M Structures of Filaments from Pick’s Disease Reveal a Novel Tau Protein Fold. Nature 2018, 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Zhang W; Falcon B; Murzin AG; Fan J; Crowther RA; Goedert M; Scheres SH Heparin-Induced Tau Filaments Are Polymorphic and Differ from Those in Alzheimer’s and Pick’s Diseases. eLife 2019, 8, e43584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Nielsen L; Khurana R; Coats A; Frokjaer S; Brange J; Vyas S; Uversky VN; Fink AL Effect of Environmental Factors on the Kinetics of Insulin Fibril Formation: Elucidation of the Molecular Mechanism. Biochemistry 2001, 40, 6036–6046. [DOI] [PubMed] [Google Scholar]
- (99).Malde AK; Zuo L; Breeze M; Stroet M; Poger D; Nair PC; Oostenbrink C; Mark AE An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [DOI] [PubMed] [Google Scholar]
- (100).Sahoo BR; Maruyama K; Edula JR; Tougan T; Lin Y; Lee YH; Horii T; Fujiwara T Mechanistic and Structural Basis of Bioengineered Bovine Cathelicidin-5 with Optimized Therapeutic Activity. Sci. Rep. 2017, 7, 44781. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.