

Abstract
Mycobacterium tuberculosis is a lethal human pathogen, with the key flavoenzyme for catalyzing bacterial cell-wall biosynthesis, decaprenylphosphoryl-D-ribose oxidase (DprE1), considered an Achilles heal for tuberculosis (TB) progression. Inhibition of DprE1 blocks cell wall biosynthesis and is a highly promising antitubercular target. Macozinone (PBTZ169, a benzothiazinone (BTZ) derivative) is an irreversible DprE1 inhibitor that has attracted considerable attention because it exhibits an additive activity when combined with other anti-TB drugs. Herein, 754 BTZ analogs were assembled in a virtual library and evaluated against the DprE1 target using a covalent docking approach. After validation of the employed covalent docking approach, BTZ analogs were screened. Analogs with a docking score less than –9.0 kcal/mol were advanced for molecular dynamics (MD) simulations, followed by binding energy evaluations utilizing the MM-GBSA approach. Three BTZ analogs–namely, PubChem-155-924-621, PubChem-127-032-794, and PubChem-155-923-972– exhibited higher binding affinities against DprE1 compared to PBTZ169 with ΔGbinding values of –77.2, –74.3, and –65.4 kcal/mol, versus –49.8 kcal/mol, respectively. Structural and energetical analyses were performed for the identified analogs against DprE1 throughout the 100 ns MD simulations, and the results demonstrated the great stability of the identified BTZ analogs. Physicochemical and ADMET characteristics indicated the oral bioavailability of the identified BTZ analogs. The obtained in-silico results provide promising anti-TB inhibitors that are worth being subjected to in-vitro and in-vivo investigations.
Introduction
Mycobacterium tuberculosis is a slow-growing pathogenic bacterium that requires up to six months of antibiotic treatment for eradication [1]. With the development of antibiotic resistance, it persists as a health threat, especially in low- and middle-income nations [2–4]. The resurgence of tuberculosis (TB) as a global pandemic has been influenced in part by a lack of an effective vaccine [5] as well as by a significant increase in TB drug resistance [6]. At the same time, there has been a global refocusing over the last twenty years by health organizations that have resulted in a significant reduction in TB-related mortality [7].
A principal flavoenzyme in M. tuberculosis cell-wall formation is decaprenyl-phosphoryl-D-ribose oxidase (DprE1); indeed, several potent covalent and noncovalent DprE1 inhibitors have been identified [8,9], including isoniazid, rifampin, pyrazinamide, ethambutol, and streptomycin that showed promise as anti-TB agents [10,11]. Currently, four DprE1 inhibitors have entered clinical trials–namely, BTZ043, TBA-7371, OPC-167832, and macozinone (PBTZ169). OPC-167832 and TBA-7371 are noncovalent inhibitors that function as highly effective antimycobacterial agents [12,13]. Among the covalent-specific DrpE1 inhibitors, 8-nitrobenzothiazinone (BTZ) analogs, including macozinone (PBTZ169), demonstrated promising inhibitory activity in in-vitro tests [14–16]. The electrophilic nitro group of BTZ is the apparent covalent warhead, which forms an irreversible covalent bond with the nucleophilic CYS387 residue in the enzyme active site [17]. According to a previous study, PBTZ169 was three to seven times more effective than BTZ043 against M. tuberculosis [18]. A previous study indicated that PBTZ169 could significantly improve patient survivorship in cases suffering from multidrug-resistant TB [19]. In fact, PBTZ169 is currently undergoing phase I/II clinical trials for the treatment of TB [20], and a PBTZ169, pyrazinamide, bedaquiline cocktail is being considered as a prospective anti-TB regime [18].
In the current study, 754 BTZ analogs were obtained from the PubChem database and virtually screened as prospective DprE1 inhibitors using covalent docking computations. Based on the covalent docking scores, molecular dynamics (MD) simulations for potent BTZ analogs in complex with DprE1 were examined. Furthermore, post-MD analyses were inspected, and the corresponding binding energies were computed utilizing the MM-GBSA approach. Physicochemical and pharmacokinetic features of the identified BTZ analogs were also predicted. A schematic diagram of the in-silico methods employed for screening BTZ analogs is shown in Fig 1. Such in-silico computations provide insight with regard to the suitability of the identified BTZ analogs for the future development of potential anti-TB drug candidates.
Fig 1. Schematic diagram of the utilized in-silico approaches in the virtual screening strategy of BTZ analogs.

Computational methodology
DprE1 preparation
The crystal structure of M. tuberculosis DprE1 in complex with PBTZ169 ligand (PDB code: 4NCR [18]) was utilized as a template for all covalent molecular docking and molecular dynamics computations. The non-terminal missing residues were constructed using the Modeller software [21], which are located at 272–283 and 315–330. DprE1 enzyme was prepared by eliminating all ions, inhibitor, and water molecules. The protonation states were then examined using the H++ web server [22]. Besides, all missing hydrogen atoms were added with the following parameters: external dielectric = 80, salinity = 0.15, internal dielectric = 10, and pH = 7.0. The quality of the modeled structure was evaluated based on the Ramachandran plot occupancy of residues, utilizing the PROCHECK server [23].
Covalent inhibitors preparation
In the current study, a total of 785 BTZ analogs were retrieved from the PubChem database (https://pubchem.ncbi.nlm.nih.gov) in an SDF format. The duplicated compounds were eliminated according to the International Chemical Identifier key (InChIKey), giving 754 BTZ analogs [24]. Subsequently, the three-dimensional (3D) structures of BTZ analogs were created using Omega2 software with a maximum of 200 conformers generated within a 10 kcal/mol energy window [25,26]. The ionization state of each BTZ analog was examined using the Fixpka tool within the QUACPAC software [27]. The geometrical structures were subsequently minimized by the MMFF94S force field within SZYBKI software [28,29]. The charges of the investigated BTZ analogs were estimated using the Gasteiger-Marsili method [30].
Covalent docking
All covalent docking calculations were conducted using AutoDock4.2.6 software [31]. The applied docking protocol keeps the macromolecule rigid while permitting flexibility in the ligand. For preparation purposes, the DprE1 enzyme was converted into a pdbqt file [32]. Except for the number of genetic algorithm (GA) runs and the maximum number of energy evaluations (eval), all covalent docking parameters were adjusted to their default settings. Fast and expensive accuracy levels of calculations were employed to virtually screen the BTZ analogs. For docking computations, the GA and eval parameters were increased from 25 and 2,500,000 to 250 and 25,000,000 for fast and expensive levels of accuracy, respectively. In both levels of calculations, the grid box was placed at 40 Å × 40 Å × 40 Å. The grid spacing value of 0.375 Å was used. The grid box was centered on 17.176, −20.119, and 1.875 (XYZ coordinates). The predicted docking modes for each BTZ analog were processed using a built-in clustering analysis with an RMSD tolerance of 1.0 Å. The lowest energy conformation from the largest cluster was chosen as the representative docking mode.
MD simulations
Molecular dynamics (MD) simulations of the most potent BTZ analogs complexed with the DprE1 enzyme were carried out using AMBER20 software [33]. The details of the utilized MD simulations are described in Ref. [34–38]. DprE1 enzyme and BTZ analogs were characterized using the AMBER force field of 14SB and the General AMBER force field (GAFF2), respectively [39,40]. For atomic charges calculations, the irreversible covalent inhibitors with CYS387 residue were capped using acetyl and methylamide groups and subjected to geometrical optimization at B3LYP/6-31G* level of theory using Gaussian09 software [41]. A restrained electrostatic potential (RESP) approach at HF/6-31G* level was then utilized to assign the atomic partial charges of the optimized inhibitors [42]. The parameters of covalent inhibitors with CYS387 residue (included in the covalent bond exhibition) and atom types were characterized and defined utilizing an antechamber module within the AMBER package. The docked BTZ analog-DprE1 complexes were solvated in an octahedron box of TIP3P water molecules. Sodium or chloride counterions were inserted to neutralize all solvated systems. Moreover, the ionic strength of the solution was also tuned to 0.15 M NaCl. Nevertheless, all prepared DprE1-analogs complexes were minimized for 5000 cycles. The minimized systems were gently heated up to 310 K for 50 ps. The DprE1-analogs complexes were equilibrated with a simulation time of 10 ns. After that, the production stages were run on the equilibrated systems throughout 1, 10, 25, and 100 ns. The snapshots were assembled each 10 ps through all production stages. The Particle Mesh Ewald (PME) method was employed to handle long-range electrostatic interactions under periodic boundary conditions, with a cutoff of 12 Å [43]. To maintain the temperature at 298 K, Langevin dynamics were used with a collision frequency (gamma_ln) set to 1.0. Pressure control was achieved using a Berendsen barostat with a relaxation time of 2 ps [44]. Bonds involving hydrogen atoms were constrained using the SHAKE algorithm, with a time step of 2 fs [45]. All calculations, including MD simulations, quantum mechanics computations, and covalent molecular docking estimations, were undertaken using the CompChem GPU (pmemd.cuda)/CPU hybrid cluster (hpc.compchem.net). All graphical representations were generated with the assistance of BIOVIA Discovery Studio Visualizer [46].
Binding energy computations
The binding energy of the most promising BTZ analogs in complex with DprE1 enzyme was computed using the MM-GBSA (molecular mechanics-generalized Born surface area) approach [47]. The modified GB model proposed by Onufriev et al. (igb = 2) was used to calculate the polar solvation energy [48]. The following equation can be used to compute the MM-GBSA binding energy:
| Eq (1) |
where energy term (G) is given as follows:
| Eq (2) |
| Eq (3) |
| Eq (4) |
EMM stands for molecular mechanics gas-phase energy. Gsolv indicates the solvation energy. Eint is the internal MM energy involving angle, bond, and dihedral energies. EvdW indicates van der Waals energy. Eele is electrostatic energy. Entropy contributions were disregarded owing to the high computational cost [49,50].
Physicochemical features
The online SWISS-ADME (https://www.swissadme.ch) server was employed to investigate the physicochemical features of the most potent BTZ analogs as DprE1 irreversible inhibitors. These physicochemical characteristics included MW (molecular weight), HBD (number of H-bond donors), TPSA (topological polar surface area), HBA (number of H-bond acceptors), and MlogP (n-octanol/water partition coefficient).
Anticipation of the pharmacokinetic and toxicity characteristics
The online pkCSM server was used to estimate the pharmacokinetic features (http://biosig.unimelb.edu.au/pkcsm/prediction). Pharmacokinetic characteristics involve absorption, distribution, metabolism, and excretion. Besides, the toxicity feature was predicted. Absorption (A) is evaluated based on water solubility (logS). The distribution (D) was assessed based on Log BB (blood-brain barrier) and CNS (central nervous system) permeability. The Cytochromes P450 (CYPs) models were used to predict metabolism (M). For excretion (E) property, total clearance was evaluated. The toxicity of the identified BTZ analogs was predicted according to AMES toxicity.
Results and discussion
Validation test
Before the virtual screening of the BTZ analogs against DprE1, an assessment of the efficiency of the utilized covalent docking protocol to predict the correct docking mode and binding affinity of DprE1 inhibitors was initially evaluated based on test sets I and II, respectively. As well, the DprE1 enzyme was validated based on the Ramachandran plot (S1 Fig). Notably, the Ramachandran plot predicts the structural stereochemical properties of the protein. As illustrated in S1 Fig, 92.1% of the residues fall within the most favored regions, 7.7% in the additionally allowed regions, and 0.3% in the generously allowed regions.
Test set I
The co-crystallized PBTZ169 inhibitor was re-docked utilizing covalent docking. The predicted docking mode was then examined and compared to the native structure to validate the efficiency of the employed docking technique to predict the docking mode of the DprE1 inhibitor (Fig 2). The predicted docking pose of PBTZ169 inside the DprE1 active site was similar to its native binding mode with an RMSD value of 0.73 Å. The computed PBTZ169 docking score in a complex with DprE1 was −7.8 kcal/mol. The good score is attributed to PBTZ169’s ability to form a covalent bond with the SH group of CYS387 (1.16 Å) (Fig 2). In addition, the oxygen of the thiazine-4-one and NO2 group exhibited two H-bonds with the NH3 group of LYS134 (2.52 Å) and LYS418 (2.00 Å). As well, PBTZ169 demonstrated carbon-hydrogen bonds with HIS132, GLY117, and SER228 (Fig 2). PBTZ169 also displayed halogen, pi-sigma, alkyl, and pi-alkyl interactions with GLY133, VAL362, LYS367, and HIS132 residues, respectively (Fig 2).
Fig 2.

(a) Superimposition illustration of the predicted docking pose (in pink) and the native binding mode of PBTZ169 (in dark cyan), (b) 3D and (c) 2D molecular interactions of the expected docking pose of PBTZ169 complexed with DprE1 enzyme.
Test set II
Test set II included six irreversible covalent inhibitors–namely BTZ043, PBTZ169, DNB1, VI-9376, BTO, and cBT–with known MIC values with DprE1 [9]. For test set II inhibitors, covalent docking calculations were performed to assess the efficiency of the utilized docking protocol to predict the binding affinity of DprE1 inhibitors. The predicted covalent docking scores were then compared with the corresponding experimental ΔGexp (S1 Table and Fig 3A). These docked complexes were subjected to MD simulations over 100 ns. Besides, the corresponding binding energies were computed using the MM-GBSA approach. The estimated binding affinities were compared to ΔGexp values (S1 Table and Fig 3B).
Fig 3.

(a) Predicted covalent docking scores and (b) estimated binding energy of test Set II inhibitors complexed with DprE1 enzyme relative to the comparable to the experimental binding energy (ΔGexp).
Favorable correlations between calculated covalent docking scores and binding affinities as well as ΔGexp were observed with correlation coefficient (R2) values of 0.93 and 0.98, respectively (S1 Table and Fig 3). These findings imply that AutoDock4.2.6 software precisely portends inhibitor-DprE1 binding score and docking pose. Thus, the validated covalent docking protocol was employed to screen potential DprE1 inhibitors.
Virtual screening of BTZ analogs
Computational techniques are used in various scientific fields, especially in drug discovery and molecular biology [51,52]. Virtual screening (VS) is a powerful computational technique used in the early stages of drug discovery. VS allows researchers to filter large libraries of chemical compounds to identify potential bioactive molecules that could interact with specific biological targets [53–55]. In the current study, an in-house database containing 754 BTZ analogs was visually screened using AutoDock4.2.6 software (Fig 1). To reduce computational time and cost, the covalent docking computations of the corrected analogs were executed using fast covalent parameters (i.e., GA = 25 and eval = 2,500,000). The fast docking scores of these analogs were computed and gathered in S2 Table. As enrolled in S2 Table, 349 out of 754 BTZ analogs demonstrated covalent docking scores less than that of PBTZ169 (calc. −7.8 kcal/mol). These BTZ analogs were selected and subjected to expensive covalent docking computations (i.e., GA = 250 and eval = 25,000,000). The computed expensive docking scores are listed in S3 Table. According to expensive docking computations, only 94 BTZ analogs manifested covalent docking scores < −9.0 kcal/mol. 2D representations of the most promising 94 BTZ analogs complexed with the DprE1 enzyme are depicted in S2 Fig. Besides, the 2D chemical structures, docking scores, and binding features of the three most potent BTZ analogs with the DprE1 enzyme are summarized in Table 1. It is worth noting that these three analogs were selected based on the calculated MM-GBSA binding energy during the 100 ns MD course detailed in the following sections.
Table 1. Predicted covalent docking scores (in kcal/mol) and binding features of PBTZ169 and the most potent BTZ analogs within the DprE1 active sitea.
| No. | PubChem Code | 2D Chemical Structure | Covalent Docking Score (kcal/mol) | Binding Featuresb | |
|---|---|---|---|---|---|
| Fast | Expensive | ||||
|
PBTZ169
(PubChem-573-313-86) |
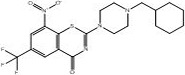
|
−7.8 | −7.8 | CYS387 (1.16Å: Covalent bond), LYS134 (2.52 Å: H-bond), LYS418 (2.00 Å: H-bond) |
|
| 1 |
PubChem-155-924-621 |
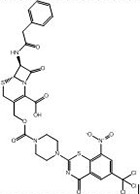
|
−15.0 | −15.7 | CYS387 (1.41 Å: Covalent bond), TRP17 (2.76 Å: H-bond), GLN334 (2.63 Å: H-bond), ASP389 (2.23 Å: H-bond), LYS418 (2.11 Å: H-bond) |
| 2 |
PubChem-127-032-794 |
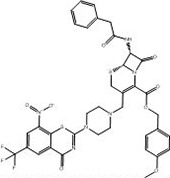
|
−14.3 | −14.7 | CYS387 (1.42 Å: Covalent bond), THR118 (2.68 Å: H-bond), HIS132 (2.85 Å: H-bond), LYS418 (1.80 Å: H-bond) |
| 3 |
PubChem-155-923-972 |
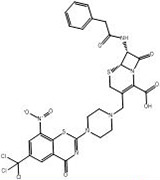
|
−12.8 | −13.3 |
CYS387 (1.16 Å: Covalent bond), ARG58 (2.08 Å: H-bond), THR118 (2.16 Å: H-bond), GLN326 (2.08 Å: H-bond), GLY328 (2.55 Å: H-bond), LYS418 (2.40 Å: H-bond) |
a Data ranked in accordance with the expensive covalent docking scores.
b Intermolecular H-bonds (in Å) and covalent bonds are only mentioned.
According to data listed in S2 Fig and Table 1, most of the examined BTZ analogs showed nearly identical docking poses within the active site, forming an irreversible covalent bond with CYS387 and multiple H-bonds with THR118, HIS132, and LYS418 residues.
Fig 4 illustrates 2D and 3D representations of PubChem-155-924-621, PubChem-127-032-794, and PubChem-155-923-972; the nitro (NO2) group of the three BTZ analogs formed an irreversible covalent bond with the SH group of CYS387 (1.41, 1.42, and 1.16 Å, respectively) (Fig 4).
Fig 4.

3D and 2D representations of (a) PubChem-155-924-621, (b) PubChem-127-032-794, and (c) PubChem-155-923-972 complexed with DprE1 enzyme.
PubChem-155-924-621 showed a great binding affinity with DprE1 with a covalent docking score of −15.7 kcal/mol, demonstrating three H-bonds with proximal residues within the active site (Table 1). Examining the docking pose indicated that carbonyl oxygen formed an H-bond with the NH group of TRP17 (2.76 Å) (Table 1 and Fig 4). In addition, the hydroxyl group of the carboxylic acid group exhibited two H-bonds with carbonyl and carboxylic groups of GLN334 (2.63 Å) and ASP389 (2.23 Å) (Table 1 and Fig 4).
PubChem-127-032-794 also demonstrated a promising binding affinity with DprE1 with a covalent docking score of −14.7 kcal/mol. The strong binding of PubChem-127-032-794 with the enzyme is ascribed to the presence of three H-bonds (Table 1 and Fig 4). More precisely, the oxygen of azetidine-one formed an H-bond with the hydroxyl of THR118 (2.86 Å) (Fig 4). An H-bond was predicted between fluorine and the NH group of the HIS132 (2.85 Å) (Fig 4). Furthermore, the oxygen of the anisole ring established an H-bond with the NH3 group of LYS418 (1.80 Å) (Table 1 and Fig 4).
Lastly, PubChem-155-923-972 displayed a good binding affinity with the DprE1 with a covalent docking score of −13.3 kcal/mol. The oxygen of the nitro group contributed to two H-bonds with the NH3 of LYS418 (2.40 Å) and the carbonyl group of GLN326 (2.08 Å) (Table 1 and Fig 4). The NH group established an H-bond with the carbonyl group of GLY328 (2.55 Å) (Table 1 and Fig 4). In addition, the hydroxyl of the carboxylic group formed two H-bonds with the NH group of ARG58 (2.08 Å) and the OH group of THR118 (2.16 Å).
Molecular dynamics (MD) simulations
MD simulations probe the constancy of ligand-target complexes, the reliability of ligand-target binding affinities, and the conformational elasticity [56,57]. 94 BTZ analogs with covalent docking scores < −9.0 kcal/mol were chosen and submitted to MD simulations. It is worth noting that the docking score of –9.0 kcal/mol was chosen as the threshold value to shortlist the potent BTZ analogs. To minimize time and computational costs, the MD simulations were run over 1 ns. The corresponding binding affinities are listed in S4 Table. From S4 Table, 33 out of 94 BTZ analogs demonstrated binding energies (ΔGbinding) less than −50.0 kcal/mol. To obtain more credible DprE1 binding affinities, these 33 BTZ analogs were further submitted to MD simulations throughout 10 ns. The corresponding binding affinities were evaluated and collected in S5 Table. Notably, 23 BTZ analogs manifested binding energies < −50.0 kcal/mol (S5 Table). The MD simulations of these analogs complexed with DprE1 were prolonged to 25 ns. Additionally, the corresponding binding affinities were calculated (S6 Table). Interestingly, out of these 23 BTZ analogs, PubChem-155-924-621, PubChem-127-032-794, and PubChem-155-923-972 exposed binding energies (ΔGbinding) with values of −73.1, −69.7, and −62.8 kcal/mol, respectively, compared to PBTZ169 (ΔGbinding = −42.9 kcal/mol). In order to obtain more rigorous binding energies, MD simulations for those three analogs were elongated to 100 ns. Moreover, the corresponding binding affinities were calculated (see Fig 5). Interestingly, there was no noticeable difference in the estimated binding energies for the identified BTZ analogs complexed with DprE1 throughout the 25 and 100 ns MD simulations. Compared to PBTZ169 (ΔGbinding = −49.8 kcal/mol), the computed binding energies of PubChem-155-924-621, PubChem-127-032-794, and PubChem-155-923-972 were −77.2, −74.3, and −65.4 kcal/mol, respectively.
Fig 5. Calculated binding energies of PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 complexed with DprE1 enzyme over 1, 10, 25, and 100 ns MD simulations.

To examine the binding for these selected BTZ analogs, the computed binding affinities were further decomposed (Fig 6). From Fig 6, ΔEvdW was the predominant contributor for PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 complexed with DprE1 with an average value of −89.3, −91.3, −79.6, and −50.7 kcal/mol, respectively (Fig 6). In addition, ΔEele was appropriate with values of −66.4, −19.9, −35.6, and −29.8 kcal/mol for PubChem-155-924-621-, PubChem-127-032-794-, PubChem-155-923-972-, and PBTZ169-DprE1 complexes, respectively (Fig 6). By comparing the average values of ΔEvdW and ΔEele, it was indicated that ΔEvdW approximately was one and a half-fold of ΔEele. Notably, ΔEint has a tiny contributor for the investigated BTZ analogs complexed with the enzyme, with values ranging from 2.8 to 4.3 kcal/mol (Fig 6).
Fig 6. Binding energy decomposition for PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 complexed with DprE1 enzyme throughout 100 ns MD simulations.

In addition, a per-residue energy decomposition analysis was implemented to investigate the amino acid residues that exhibit prominent participation with DprE1. Only residues with binding energy contributions lower than −0.50 kcal/mol were considered and are displayed in Fig 7A. MET319, PHE320, GLN334, VAL365, and CYS387 interact with PubChem-155-924-621, PubChem-127-032-794, and PubChem-155-923-972, and PBTZ169. CYS387 had a significant role in ΔGbinding with values of −0.69, −6.95, −4.88, and −1.12 kcal/mol for PubChem-155-924-621-, PubChem-127-032-794-, PubChem-155-923-972-, and PBTZ169-DprE1 complexes, respectively (Fig 7A). Inspecting the final trajectory of the BTZ analogs and PBTZ169 complexed with DprE1 indicated preserved H-bonds with key residues over the MD simulations (Fig 7B). Noteworthy, the three investigated BTZ analogs complexed with DprE1 have approximately identical interaction patterns with proximal residues, implying propinquity in the binding mode of these studied complexes.
Fig 7.

(a) The energy contribution of the most significant residues to the overall binding energy and (b) 2D molecular interactions of binding modes of (i) PubChem-155-924-621, (ii) PubChem-127-032-794, (iii) PubChem-155-923-972, and (iv) PBTZ169 complexed with DprE1 enzyme according to the final snapshot throughout a 100 ns MD simulation.
Post-MD analyses
To inspect the conformational changes and the stability of the most promising BTZ analogs complexed with DprE1, post-MD analyses were employed during the 100 ns MD course. Post-MD analyses involved the binding energy per trajectory, the number of H-bonds, CoM distance, RMSD, RMSF, Rg, and SASA [58,59].
Binding energy per trajectory
To estimate the constancy of PBTZ169, PubChem-155-924-621, PubChem-127-032-794, and PubChem-155-923-972 complexed with the DprE1 active site, the correlation between MM-GBSA binding affinity and time was executed throughout the simulation time of 100 ns (Fig 8A). The most intriguing aspect of Fig 8A is the high stability of PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 complexed with the enzyme with ΔGbinding values of −77.2, −74.3, −65.4, and −49.8 kcal/mol, respectively. Based on these outcomes, the three BTZ analogs and PBTZ169 complexed with DprE1 kept immutability over 100 ns MD course.
Fig 8.

(a) Estimated binding energy per trajectory, (b) CoM distances, and (c) RMSD of the backbone atoms from the initial conformational of PubChem-155-924-621 (in dark cyan), PubChem-127-032-794 (in blue), PubChem-155-923-972 (in violet), and PBTZ169 (in black) towards DprE1 enzyme during the 100 ns MD course.
CoM distance
The center-of-mass (CoM) distance between the selected BTZ analogs and CYS387 was adopted throughout a 100 ns MD simulation to understand the constancy of DprE1-analog complexes (Fig 8B). As depicted in Fig 8B, the gauged CoM distance was steady for PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 complexed with DprE1 with an average value of 7.3, 11.1, 6.8, and 5.7 Å, respectively. These outcomes demonstrated that PBTZ169 and the investigated BTZ analogs bind tightly with the DprE1 enzyme.
Root-mean-square deviation (RMSD)
To examine the structural and positional variations inside the DprE1 active site and inspect the structural stabilization of the complexes, RMSD was measured (Fig 8C). The average RMSD values for PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 complexed with DprE1 enzyme were 0.23, 0.32, 0.23, and 0.63 nm, respectively (Fig 8C). The RMSD analysis displayed that the investigated complexes tended to exist in an equilibrium state after 20 ns until the end of the 100 ns MD simulations, and the proposed analogs were tightly bonded without changing the overall topology of DprE1.
Root-mean-square fluctuation (RMSF)
To assess the conformational variation and stability of the backbone in apo-, PubChem-155-924-621-, PubChem-127-032-794-, PubChem-155-923-972-, and PBTZ169-DprE1, the RMSF analysis of Cα atoms was measured (Fig 9A). As shown in Fig 9A, the residues remained stable for the investigated systems throughout the 100 ns MD simulations. Nevertheless, a greater fluctuation was observed at residues from 260 to 290 and from 310 to 330, indicating great flexibility in these regions. The average RMSF values for apo-, PubChem-155-924-621-, PubChem-127-032-794-, PubChem-155-923-972-, and PBTZ169-DprE1 were found to be 0.14, 0.13, 0.15, 0.14, and 0.14 nm, respectively (Fig 9A).
Fig 9.

Estimated (a) RMSF, (b) Rg, and (c) SASA for apo- (in grey), PubChem-155-924-621- (in dark cyan), PubChem-127-032-794- (in blue), PubChem-155-923-972- (in violet), and PBTZ169-DprE1 (in black) over 100 ns MD simulations.
Radius of gyration (Rg)
The Rg analysis was executed to inspect the compactness of DprE1 in its apo form and complex form with identified BTZ analogs over 100 ns MD simulation. The Rg analysis provided insights into the overall folding and unfolding behavior of the DprE1 structure upon binding with the identified BTZ analogs (Fig 9B). The average Rg values were 2.22, 2.26, 2.25, 2.28, and 2.24 nm for apo-, PubChem-155-924-621-, PubChem-127-032-794-, PubChem-155-923-972-, and PBTZ169-DprE1, respectively (Fig 9B). The Rg results indicated that DprE1 maintained its compactness when bound to the BTZ analogs and PBTZ169 throughout 100 ns MD simulations. These findings unveiled that the binding of PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 significantly stabilized the DprE1 structure.
Solvent-accessible surface area (SASA). SASA analysis was performed to gain a deeper understanding of the interactions between the complexes and the solvent over the course of the 100 ns MD simulations. Fig 9C illustrates the graph for SASA vs. simulation time for apo-, PubChem-155-924-621-, PubChem-127-032-794-, PubChem-155-923-972-, and PBTZ169-DprE1. As depicted in Fig 9C, the average SASA values were found to be 198.91, 207.56, 199.90, 207.46, and 202.04 nm2 for the apo-, PubChem-155-924-621-, PubChem-127-032-794-, PubChem-155-923-972-, and PBTZ169-DprE1, respectively. These findings demonstrated that no significant changes in the SASA values were observed for DprE1 due to its complexation with BTZ analogs. These results revealed that the BTZ analogs did not notably affect the solvent exposure of the DprE1 enzyme.
H-bond number
H-bond analysis was performed to estimate the number of H-bonds between the identified BTZ analogs and DprE1 enzyme over a 100 ns MD course (Fig 10). Interestingly, PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 complexed with DprE1 revealed an average number of H-bonds was 2, 1, 1, and 2, respectively. Generally, these post-dynamics studies endorsed the inherent steadiness of the investigated BTZ analogs in complex with DprE1 enzyme over a 100 ns MD course.
Fig 10. Number of H-bonds for PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 complexed with DprE1 enzyme during the 100 ns MD simulations.

Physicochemical features
The druggability of each compound was determined by its physicochemical characteristics [60]. To predict the bioavailability and physicochemical features of the examined BTZ analogs as DprE1 inhibitors, the SWISS-ADME server was used. Fig 11 illustrates the anticipated physicochemical properties of the examined BTZ analogs. MLogP values for PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 were 2.75, 3.25, 2.89, and 3.49, respectively, demonstrating that these analogs exhibited high lipophilicity. Molecular weight (MW) ranged from 750 to 850 dalton (Fig 11). However, PBTZ169 demonstrated MW with a value of 456.5 dalton. Additionally, the number of H-bond acceptors (HBA) for PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 was 10, 13, 9, and 8, respectively (Fig 11). Notably, this increase in molecular weight and hydrogen bond acceptors is unlikely to significantly impact molecule transmission and diffusion, as many FDA-approved drugs have deviated from the conventional molecular weight limit of 500 and the number of hydrogen bond acceptors of 10 [61]. The number of HBD for PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 was less than 5 (Fig 11). The TPSA values of the identified analogs ranged from 110 to 200 Å2, demonstrating their good oral absorption and membrane permeability [62].
Fig 11. Estimated physiochemical features of PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 as DprE1 inhibitors.

Pharmacokinetic characteristics
The pkCSM server was employed to evaluate the pharmacokinetic properties of identified BTZ analogs. ADMET properties of the investigated analogs are compiled in Table 2. LogS values of PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 were −2.99, −4.1, −2.9, and −5.6, respectively, and were considered to be reasonably soluble (Table 2). PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 demonstrated log BB with values of −2.4, −2.1, −2.0, and −0.9, respectively. The log PS value for CNS permeability ranged from −4 to −2, indicating impenetrability. The majority of the identified analogs were predicted to be unable to cross the CNS or permeate the BBB barrier (Table 2). In terms of metabolism (M), PBTZ169 and PubChem-127-032-794 were inhibitors of CYP3A4 enzyme, whereas PubChem-155-924-621 and PubChem-155-923-972 were non-inhibitors of CYP3A4 enzyme (Table 2). Additionally, the result of the metabolism test indicates that PBTZ169 and the identified analogs act as substrates for the CYP3A4 enzyme (Table 2). For excretion (E) property, the estimated total clearance for PubChem-155-924-621, PubChem-127-032-794, PubChem-155-923-972, and PBTZ169 was 0.26, −0.3, −0.53, and 0.02, respectively (Table 2). According to toxicity (T) analysis, PubChem-155-924-621 and PubChem-155-923-972 were non-toxic, while PBTZ169 and PubChem-127-032-794 were toxic (Table 2). All the examined BTZ analogs revealed satisfied outcomes, with some values even better than PBTZ169, as indicated in Table 2.
Table 2. Pharmacokinetic and toxicity profile for the identified BTZ analogs and PBTZ169.
| PubChem Code |
Absorption (A) |
Distribution (D) | Metabolism (M) | Excretion (E) | Toxicity (T) | ||
|---|---|---|---|---|---|---|---|
| LogS | Blood Brain Barrier (BBB) | CNS Permeability | CYP3A4 Substrate | CYP3A4 Inhibitor |
Total Clearance |
AMES Toxicity |
|
| PBTZ169 | −5.6 | −0.9 | −2.1 | Yes | Yes | 0.02 | Yes |
| PubChem-155-924-621 | −2.99 | −2.4 | −3.6 | Yes | No | 0.26 | No |
| PubChem-127-032-794 | −4.1 | −2.1 | −3.5 | Yes | Yes | −0.3 | Yes |
| PubChem-155-923-972 | −2.9 | −2.0 | −3.6 | Yes | No | −0.53 | No |
Conclusions
DprE1 is reported to be a crucial and very effective target for inhibiting tuberculosis (TB). As a prospective DprE1 irreversible covalent inhibitor, BTZ analogs have received increased attention because they demonstrate additive activity when combined with other anti-TB drugs. To identify effective DprE1 inhibitors, a library of 754 BTZ analogs was constructed and screened against DprE1 using covalent docking computations. Based on covalent docking scores, the most promising BTZ analogs with docking scores < −9.0 kcal/mol were subjected to MD simulations, followed by binding energy evaluations using the MM-GBSA approach. In accordance with the MM-GBSA computations, PubChem-155-924-621, PubChem-127-032-794, and PubChem-155-923-972 demonstrated promising ΔGbinding with values of −77.2, −74.3, and −65.4 kcal/mol, respectively, compared to PBTZ169 (ΔGbinding = −49.8 kcal/mol). Post-dynamics analyses showed that the identified BTZ analogs demonstrated high stability over 100 ns MD simulations. The predicted physicochemical and ADMET properties of the identified BTZ analogs proposed the promising oral bioavailability of PubChem-155-924-621, Pub-Chem-127-032-794, and PubChem-155-923-972 as potential tuberculosis drug candidates. These findings indicated that PubChem-155-924-621, PubChem-127-032-794, and PubChem-155-923-972 may be potent DprE1 inhibitors that warrant additional in-vitro and in-vivo assays. The current in-silico results established that these compounds are recommended for clinical investigations against tuberculosis.
Supporting information
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2024R678), King Saud University, Riyadh, Saudi Arabia. The computational work was completed with resources provided by the Center for High-Performance Computing (Cape Town, South Africa, http://www.chpc.ac.za), and Bibliotheca Alexandrina (http://hpc.bibalex.org).
Abbreviations
- DprE1
Decaprenylphosphoryl-D-ribose oxidase
- TB
Tuberculosis
- M. tuberculosis
Mycobacterium tuberculosis
- BTZ
Benzothiazinone
- InChIKey
International Chemical Identifier key
- 3D
Three-dimensional
- GA
Genetic algorithm
- eval
Energy evaluations
- MD
Molecular dynamics
- GAFF2
General AMBER force field
- RESP
Restrained electrostatic potential
- MM-GBSA
Molecular mechanics-generalized Born surface area
- G
Energy term
- E MM
Molecular mechanics gas-phase energy
- G solv
Solvation energy
- E int
Internal molecular mechanics energy
- E vdW
Van der Waals energy
- E ele
Electrostatic energy
- MW
Molecular weight
- HBD
H-Bond donors
- TPSA
Topological polar surface area
- HBA
H-Bond acceptors
- MlogP
n-Octanol/water partition coefficient
- ADMET
Absorption distribution metabolism excretion toxicity
- logS
Water solubility
- BBB
Blood-brain barrier
- CNS
Central nervous system
- CYP
Cytochromes P450
- ΔGexp
Experimental binding energy
- R 2
Correlation coefficient
- CoM
Center-of-mass
- RMSD
Root-mean-square deviation
- RMSF
Root-mean-square fluctuation
- Rg
Radius of gyration
- SASA
Solvent accessible surface area
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
The author(s) received no specific funding for this work.
References
- 1.Smith I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev. 2003;16: 463–96. Epub 2003/07/15. doi: 10.1128/CMR.16.3.463-496.2003 ; PubMed Central PMCID: PMC164219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandhu GK. Tuberculosis: current situation, challenges and overview of its control programs in India. J Glob Infect Dis. 2011;3: 143–50. Epub 2011/07/07. doi: 10.4103/0974-777X.81691 ; PubMed Central PMCID: PMC3125027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Togun T, Kampmann B, Stoker NG, Lipman M. Anticipating the impact of the COVID-19 pandemic on TB patients and TB control programmes. Ann Clin Microbiol Antimicrob. 2020;19: 21. Epub 2020/05/25. doi: 10.1186/s12941-020-00363-1 ; PubMed Central PMCID: PMC7245173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc Natl Acad Sci USA. 2004;101: 4871–6. doi: 10.1073/pnas.0305627101 WOS:000220761200029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dheda K, Gumbo T, Maartens G, Dooley KE, McNerney R, Murray M, et al. The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respir Med. 2017;5: 291–360. Epub 2017/03/28. doi: 10.1016/S2213-2600(17)30079-6 . [DOI] [PubMed] [Google Scholar]
- 6.Dey R, Nandi S, Samadder A, Saxena A, Saxena AK. Exploring the potential inhibition of candidate drug molecules for clinical investigation based on their docking or crystallographic analyses against M. tuberculosis enzyme targets. Curr Top Med Chem. 2020;20: 2662–80. doi: 10.2174/1568026620666200903163921 WOS:000592667300003. [DOI] [PubMed] [Google Scholar]
- 7.Organization WH. Gear up to end TB: introducing the end TB strategy. World Health Organization, 2015. [Google Scholar]
- 8.Chikhale RV, Barmade MA, Murumkar PR, Yadav MR. Overview of the development of DprE1 inhibitors for combating the Menace of Tuberculosis. J Med Chem. 2018;61: 8563–93. Epub 2018/06/01. doi: 10.1021/acs.jmedchem.8b00281 . [DOI] [PubMed] [Google Scholar]
- 9.Piton J, Foo CS, Cole ST. Structural studies of Mycobacterium tuberculosis DprE1 interacting with its inhibitors. Drug Discov Today. 2017;22: 526–33. Epub 2016/09/27. doi: 10.1016/j.drudis.2016.09.014 . [DOI] [PubMed] [Google Scholar]
- 10.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307: 223–7. Epub 2004/12/14. doi: 10.1126/science.1106753 . [DOI] [PubMed] [Google Scholar]
- 11.Mikusova K, Huang H, Yagi T, Holsters M, Vereecke D, D’Haeze W, et al. Decaprenylphosphoryl arabinofuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J Bacteriol. 2005;187: 8020–5. Epub 2005/11/18. doi: 10.1128/JB.187.23.8020-8025.2005 ; PubMed Central PMCID: PMC1291263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shirude PS, Shandil RK, Manjunatha MR, Sadler C, Panda M, Panduga V, et al. Lead optimization of 1,4-azaindoles as antimycobacterial agents. J Med Chem. 2014;57: 5728–37. Epub 2014/05/31. doi: 10.1021/jm500571f . [DOI] [PubMed] [Google Scholar]
- 13.Hariguchi N, Chen X, Hayashi Y, Kawano Y, Fujiwara M, Matsuba M, et al. OPC-167832, a novel carbostyril derivative with potent antituberculosis activity as a DprE1 inhibitor. Antimicrob Agents Chemother. 2020;64: 10.1128/aac.02020-19. Epub 2020/04/02. doi: 10.1128/AAC.02020-19 ; PubMed Central PMCID: PMC7269503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richter A, Rudolph I, Mollmann U, Voigt K, Chung CW, Singh OMP, et al. Novel insight into the reaction of nitro, nitroso and hydroxylamino benzothiazinones and of benzoxacinones with Mycobacterium tuberculosis DprE1. Sci Rep. 2018;8: 13473. Epub 2018/09/09. doi: 10.1038/s41598-018-31316-6 ; PubMed Central PMCID: PMC6128881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makarov V, Manina G, Mikusova K, Mollmann U, Ryabova O, Saint-Joanis B, et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324: 801–4. Epub 2009/03/21. doi: 10.1126/science.1171583 ; PubMed Central PMCID: PMC3128490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong L, Gao C, Shi YJ, Tao X, Rong J, Liu KL, et al. Identification of a new series of benzothiazinone derivatives with excellent antitubercular activity and improved pharmacokinetic profiles. RSC Adv. 2018;8: 11163–76. Epub 2018/03/21. doi: 10.1039/c8ra00720a ; PubMed Central PMCID: PMC9078972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grover S, Alderwick LJ, Mishra AK, Krumbach K, Marienhagen J, Eggeling L, et al. Benzothiazinones mediate killing of Corynebacterineae by blocking decaprenyl phosphate recycling involved in cell wall biosynthesis. J Biol Chem. 2014;289: 6177–87. Epub 2014/01/22. doi: 10.1074/jbc.M113.522623 ; PubMed Central PMCID: PMC3937683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med. 2014;6: 372–83. Epub 2014/02/07. doi: 10.1002/emmm.201303575 ; PubMed Central PMCID: PMC3958311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma XC, Han B, Wang AY, Yang L, Huang MH, Chowdhury K, et al. Identification of benzothiazones containing a hexahydropyrrolo[3,4-c]pyrrol moiety as antitubercular agents against MDR-MTB. RSC Adv. 2020;10: 14410–4. doi: 10.1039/d0ra00750a WOS:000528043700059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bendre AD, Peters PJ, Kumar J. Tuberculosis: Past, present and future of the treatment and drug discovery research. Curr Res Pharmacol Drug Discov. 2021;2: 100037. Epub 2021/12/16. doi: 10.1016/j.crphar.2021.100037 ; PubMed Central PMCID: PMC8663960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinform. 2016;54: 5 6 1–5 6 37. Epub 2016/06/21. doi: 10.1002/cpbi.3 ; PubMed Central PMCID: PMC5031415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gordon JC, Myers JB, Folta T, Shoja V, Heath LS, Onufriev A. H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005;33: W368–W71. Epub 2005/06/28. doi: 10.1093/nar/gki464 ; PubMed Central PMCID: PMC1160225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck—a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr. 1993;26: 283–91. doi: 10.1107/S0021889892009944 WOS:A1993KY85100021. [DOI] [Google Scholar]
- 24.Heller SR, McNaught A, Pletnev I, Stein S, Tchekhovskoi D. InChI, the IUPAC International Chemical Identifier. J Cheminform. 2015;7: 23. Epub 2015/07/03. doi: 10.1186/s13321-015-0068-4 ; PubMed Central PMCID: PMC4486400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.OMEGA. 2.5.1.4 ed. Santa Fe, NM, USA: OpenEye Scientific Software; 2013.
- 26.Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model. 2010;50: 572–84. Epub 2010/03/20. doi: 10.1021/ci100031x ; PubMed Central PMCID: PMC2859685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.QUACPAC. 1.7.0.2 ed. Santa Fe, NM, USA: OpenEye Scientific Software; 2016.
- 28.Halgren TA. MMFF VI. MMFF94s option for energy minimization studies. J Comput Chem. 1999;20: 720–9. Epub 1999/05/01. doi: . [DOI] [PubMed] [Google Scholar]
- 29.SZYBKI. 1.9.0.3 ed. Santa Fe, NM, USA: OpenEye Scientific Software; 2016.
- 30.Gasteiger J, Marsili M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron. 1980;36: 3219–28. doi: 10.1016/0040-4020(80)80168-2 [DOI] [Google Scholar]
- 31.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30: 2785–91. Epub 2009/04/29. doi: 10.1002/jcc.21256 ; PubMed Central PMCID: PMC2760638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11: 905–19. Epub 2016/04/15. doi: 10.1038/nprot.2016.051 ; PubMed Central PMCID: PMC4868550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Case DA, Belfon K, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE, et al. AMBER 2020. University of California, San Francisco; 2020. [Google Scholar]
- 34.Ibrahim MAA, Abdelrahman AHM, Atia MAM, Mohamed TA, Moustafa MF, Hakami AR, et al. Blue biotechnology: Computational screening of sarcophyton cembranoid diterpenes for SARS-CoV-2 main protease inhibition. Mar Drugs. 2021;19: 391. Epub 2021/08/07. doi: 10.3390/md19070391 ; PubMed Central PMCID: PMC8308023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ibrahim MAA, Badr EAA, Abdelrahman AHM, Almansour NM, Mekhemer GAH, Shawky AM, et al. In Silico targeting human multidrug transporter ABCG2 in breast cancer: Database screening, molecular docking, and molecular dynamics study. Mol Inform. 2022;41: e2060039. Epub 2021/09/08. doi: 10.1002/minf.202060039 . [DOI] [PubMed] [Google Scholar]
- 36.Ibrahim MAA, Badr EAA, Abdelrahman AHM, Almansour NM, Shawky AM, Mekhemer GAH, et al. Prospective drug candidates as human multidrug transporter ABCG2 inhibitors: An in silico drug discovery study. Cell Biochem Biophys. 2021;79: 189–200. Epub 2021/05/07. doi: 10.1007/s12013-021-00985-y . [DOI] [PubMed] [Google Scholar]
- 37.Ibrahim MAA, Abdeljawaad KAA, Abdelrahman AHM, Alzahrani OR, Alshabrmi FM, Khalaf E, et al. Non-beta-lactam allosteric inhibitors target methicillin-resistant staphylococcus aureus: An in silico drug discovery study. Antibiotics (Basel). 2021;10: 934–56. Epub 2021/08/28. doi: 10.3390/antibiotics10080934 ; PubMed Central PMCID: PMC8388891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ibrahim MAA, Abdelrahman AHM, Allemailem KS, Almatroudi A, Moustafa MF, Hegazy MF. In silico evaluation of prospective anti-COVID-19 drug candidates as potential SARS-CoV-2 main protease inhibitors. Protein J. 2021;40: 296–309. Epub 2021/01/03. doi: 10.1007/s10930-020-09945-6 ; PubMed Central PMCID: PMC7776322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11: 3696–713. Epub 2015/11/18. doi: 10.1021/acs.jctc.5b00255 ; PubMed Central PMCID: PMC4821407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25: 1157–74. Epub 2004/04/30. doi: 10.1002/jcc.20035 . [DOI] [PubMed] [Google Scholar]
- 41.Frisch GWT J., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., et al. , Gaussian, Inc., Wallingford CT. Gaussian 09, Revision E.01. 2009. [Google Scholar]
- 42.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—the RESP model. J Phys Chem. 1993;97: 10269–80. doi: 10.1021/j100142a004 WOS:A1993MA74100004. [DOI] [Google Scholar]
- 43.Darden T, York D, Pedersen L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98: 10089–92. doi: 10.1063/1.464397 [DOI] [Google Scholar]
- 44.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-dynamics with coupling to an external bath. J Chem Phys. 1984;81: 3684–90. doi: 10.1063/1.448118 WOS:A1984TQ73500045. [DOI] [Google Scholar]
- 45.Miyamoto S, Kollman PA. Settle—an analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem. 1992;13: 952–62. doi: 10.1002/jcc.540130805 WOS:A1992JL80700004. [DOI] [Google Scholar]
- 46.Dassault Systèmes BIOVIA DSV, Version 2019; Dassault Systèmes: San Diego, CA, USA. 2019.
- 47.Massova I, Kollman PA. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect Drug Discov. 2000;18: 113–35. doi: 10.1023/A:1008763014207. WOS:000086013500006. [DOI] [Google Scholar]
- 48.Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55: 383–94. Epub 2004/03/30. doi: 10.1002/prot.20033 . [DOI] [PubMed] [Google Scholar]
- 49.Hou T, Wang J, Li Y, Wang W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J Comput Chem. 2011;32: 866–77. Epub 2010/10/16. doi: 10.1002/jcc.21666 ; PubMed Central PMCID: PMC3043139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang E, Sun H, Wang J, Wang Z, Liu H, Zhang JZH, et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem Rev. 2019;119: 9478–508. doi: 10.1021/acs.chemrev.9b00055 [DOI] [PubMed] [Google Scholar]
- 51.Amin MR, Yasmin F, Dey S, Mahmud S, Saleh MA, Emran TB, et al. Methyl beta-D-galactopyranoside esters as potential inhibitors for SARS-CoV-2 protease enzyme: synthesis, antimicrobial, PASS, molecular docking, molecular dynamics simulations and quantum computations. Glycoconj J. 2022;39: 261–90. Epub 2022/01/18. doi: 10.1007/s10719-021-10039-3 ; PubMed Central PMCID: PMC8761875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anowar Hosen M, Sultana Munia N, Al-Ghorbani M, Baashen M, Almalki FA, Ben Hadda T, et al. Synthesis, antimicrobial, molecular docking and molecular dynamics studies of lauroyl thymidine analogs against SARS-CoV-2: POM study and identification of the pharmacophore sites. Bioorg Chem. 2022;125: 105850. Epub 2022/05/10. doi: 10.1016/j.bioorg.2022.105850 ; PubMed Central PMCID: PMC9065685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McInnes C. Virtual screening strategies in drug discovery. Curr Opin Chem Biol. 2007;11: 494–502. Epub 2007/10/16. doi: 10.1016/j.cbpa.2007.08.033 . [DOI] [PubMed] [Google Scholar]
- 54.Bajorath F. Integration of virtual and high-throughput screening. Nat Rev Drug Discov. 2002;1: 882–94. doi: 10.1038/nrd941 WOS:000179115600016. [DOI] [PubMed] [Google Scholar]
- 55.Kawsar SMA, Hossain MA, Saha S, Abdallah EM, Bhat AR, Ahmed S, et al. Nucleoside-Based Drug Target with General Antimicrobial Screening and Specific Computational Studies against SARS-CoV-2 Main Protease. Chemistryselect. 2024;9: e202304774. doi: 10.1002/slct.202304774 WOS:001202436000001. [DOI] [Google Scholar]
- 56.De Vivo M, Masetti M, Bottegoni G, Cavalli A. Role of molecular dynamics and related methods in drug discovery. J Med Chem. 2016;59: 4035–61. Epub 2016/01/26. doi: 10.1021/acs.jmedchem.5b01684 . [DOI] [PubMed] [Google Scholar]
- 57.Kerrigan JE. Molecular dynamics simulations in drug design. In: Kortagere S, editor. In Silico Models for Drug Discovery. Totowa, NJ: Humana Press; 2013. p. 95–113. [DOI] [PubMed] [Google Scholar]
- 58.Akter S, Alhatlani BY, Abdallah EM, Saha S, Ferdous J, Hossain ME, et al. Exploring Cinnamoyl-Substituted Mannopyranosides: Synthesis, Evaluation of Antimicrobial Properties, and Molecular Docking Studies Targeting H5N1 Influenza A Virus. Molecules. 2023;28: 8001. Epub 2023/12/23. doi: 10.3390/molecules28248001 ; PubMed Central PMCID: PMC10745968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.S MAK, Hosen MA, Ahmad S, El Bakri Y, Laaroussi H, Ben Hadda T, et al. Potential SARS-CoV-2 RdRp inhibitors of cytidine derivatives: Molecular docking, molecular dynamic simulations, ADMET, and POM analyses for the identification of pharmacophore sites. PLoS One. 2022;17: e0273256. Epub 2022/11/29. doi: 10.1371/journal.pone.0273256 ; PubMed Central PMCID: PMC9704642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen M, Tian S, Li Y, Li Q, Xu X, Wang J, et al. Drug-likeness analysis of traditional Chinese medicines: 1. property distributions of drug-like compounds, non-drug-like compounds and natural compounds from traditional Chinese medicines. J Cheminform. 2012;4: 31. Epub 2012/11/28. doi: 10.1186/1758-2946-4-31 ; PubMed Central PMCID: PMC3538521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mullard A. Re-assessing the rule of 5, two decades on. Nat Rev Drug Discov. 2018;17: 777–. Epub 2018/10/31. doi: 10.1038/nrd.2018.197 . [DOI] [PubMed] [Google Scholar]
- 62.Bakht MA, Yar MS, Abdel-Hamid SG, Al Qasoumi SI, Samad A. Molecular properties prediction, synthesis and antimicrobial activity of some newer oxadiazole derivatives. Eur J Med Chem. 2010;45: 5862–9. doi: 10.1016/j.ejmech.2010.07.069 WOS:000285485000038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.