

Abstract
Survivin, a homodimeric protein and a member of the IAP family, plays a vital function in cell survival and cycle progression by interacting with various proteins and complexes. Its expression is upregulated in cancers but not detectable in normal tissues. Thus, it has been regarded and validated as an ideal cancer target. However, survivin is “undruggable” due to its lack of enzymatic activities or active sites for small molecules to bind/inhibit. Academic and industrial laboratories have explored different strategies to overcome this hurdle over the past two decades, with some compounds advanced into clinical testing. These strategies include inhibiting survivin expression, its interaction with binding partners and homodimerization. Here, we provide comprehensive analyses of these strategies and perspective on different small molecule survivin inhibitors to help drug discovery targeting “undruggable” proteins in general and survivin specifically with a true survivin inhibitor that will prevail in the foreseeable future.
Graphical Abstract
1. INTRODUCTION
1.1. Inhibitor of Apoptosis Proteins.
Inhibitor of apoptosis proteins (IAPs) are major antiapoptotic regulators that inhibit cysteine proteases and caspases.1,2 Humans have eight IAPs, including neuronal apoptosis inhibitory protein (NAIP), cellular IAP 1 (cIAP1), cellular IAP 2 (cIAP2), X-linked IAP (XIAP), survivin, Baculovirus IAP Repeat (BIR)-containing ubiquitin-conjugating enzyme (BRUCE), melanoma IAP (ML-IAP or Livin), and IAP-like Protein 2 (ILP2) (Figure 1A and Table 1).3 The IAPs are characterized by the presence of one to three BIR domains and, hence, their corresponding genes are named BIRC (BIR containing) 1 through 8 (Figure 1A and Table 1). Five IAPs, including XIAP, cIAP1/2, Livin, and ILP2, contain a Really Interesting New Gene (RING) domain. While BRUCE has a ubiquitin-conjugating (UBC) domain, cIAP1/2, XIAP and ILP2 contain a ubiquitin-associated domain (UBA). In addition, NAIP has two leucine-rich repeat (LRR) domains, and one nucleotide-binding and oligomerization (NACHT) domain, and cIAP1/2 also contains a caspase recruitment domain (CARD).4 Although the BIR domain was thought to mediate the binding of IAPs to caspases, XIAP is the only IAP that has been shown to interact with and inhibit caspase activity and ubiquitinate them to promote their degradation.5,6
Figure 1.

IAP family members and survivin. (A) Linear domain structures of all eight IAP family members. (B) Crystal structure of survivin as a homodimer (PDB code: 1F3H). (C) Survivin function in extrinsic and intrinsic apoptosis. Fas L, Fas ligand; TRAIL, TNF-related apoptosis-inducing ligand; Cyto c, cytochrome c.
Table 1.
Members of the IAPs, and the Status of Reported Inhibitors
| gene name | protein name | year identified | molecular mass (kDa) | inhibitorsa | development stage |
|---|---|---|---|---|---|
| BIRC1 | NAIP | 1995 | 160 | none reported | |
| BIRC2 | cIAP1 | 1995 | 69 | SM-1295 | Preclinical7 |
| AZD5582 | Preclinical8 | ||||
| AT-406 | Phase I9 | ||||
| ASTX660 | Phase I/II10 | ||||
| Birinapant | Phase I/II11 | ||||
| BIRC3 | cIAP2 | 1995 | 68 | SM-1295 | Preclinical7 |
| AZD5582 | Preclinical8 | ||||
| AT-406 | Phase I9 | ||||
| LCL161 | Phase I12 | ||||
| BIRC4 | XIAP | 1996 | 55 | MX69 | Preclinical13 |
| AZD5582 | Preclinical8 | ||||
| GDC-0152 | Phase I14 | ||||
| LCL161 | Phase I12 | ||||
| ASTX660 | Phase I/II10 | ||||
| BIRC5 | Survivin | 1997 | 16.5 | YM155 | Phase II15 |
| EM-1421 | Phase I16 | ||||
| FL118 | INDb,17 | ||||
| UC-112/MX106 | Preclinical18 | ||||
| LQZ-7F/7F1/7I | Preclinical19–21 | ||||
| BIRC6 | BRUCE | 1998 | 528 | None reported | |
| BIRC7 | ML-IAP or Livin | 2000 | 33 | None reported | |
| BIRC8 | ILP2 | 2001 | 27 | None reported |
Small molecule agents.
IND, investigational new drug.
Recent findings have shown that IAPs could serve as potential biomarkers and, more importantly, as therapeutic targets in cancer treatment due to their dysregulation and overexpression in cancer and association with poor prognosis and drug resistance.22,23 Generally, IAPs exert their functions by interacting with vital players of both the intrinsic and extrinsic apoptosis pathways to thwart cell death, including (1) the initiators such as cytochrome c, and the apoptosome consisting of cytochrome c, apoptotic peptidase activating factor 1 (Apaf-1) and pro-caspase 9, and (2) the signal transducers/executioners such as caspase 3/8/9.24,25 Because of their functions in antiapoptosis and association with cancer, small molecule inhibitors targeting IAPs have been identified, with some under clinical study as summarized in Table 1.
1.2. Survivin.
Human survivin, the smallest member of the IAP family with a molecular weight of 16.5 kDa, consists of 142 amino acid residues and exists as a homodimer with a single BIR domain in the N-terminus, a zinc-finger fold, and an extended C-terminal helical coiled coil (Figure 1B).26 Unlike other IAPs, which are found mainly in the cytosol and to a lesser extent in the nucleus,27 survivin is present in multiple subcellular locations, including the nucleus, the cytosol, mitochondria, and extracellular exosomes.28–30 However, similar to other IAPs, survivin has been shown to inhibit both extrinsic and intrinsic apoptosis pathways in cell lines and animal models.31,32
Several mechanisms have been proposed to delineate how survivin inhibits apoptosis (Figure 1C). It may prevent apoptosis induction by interacting with pro-apoptotic proteins to inhibit caspase activation. For example, the second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI (SMAC/DIABLO), a pro-apoptotic protein, promotes cytochrome c-dependent apoptosis by binding to and neutralizing IAPs following release from mitochondria and activating caspase 3.33 Survivin binds directly to SMAC/DIABLO in mitochondria and prevents it from being released into the cytosol to activate apoptosis.34 Indeed, disrupting the interaction between survivin and SMAC/DIABLO induces apoptosis.35
While survivin has been suggested to bind directly to and inhibit caspases 3 and 7 using recombinant proteins,36 one study showed that the chemically synthesized survivin failed to inhibit caspase 3 activity even at 10 μM.37 It is unclear if the chemically synthesized survivin is functional; however, another study using mouse and human survivin also failed to show caspase 3 inhibition.38 Survivin has also been thought to bind to caspase 9 and inhibit its activation.39 However, it was later shown that survivin alone does not bind to caspase 9 but rather works cooperatively with hepatitis B X-interacting protein (HBXIP) to bind to pro-caspase 9 and inhibits its activation.40 Survivin may also form the survivin-XIAP complex that stabilizes XIAP against ubiquitination and proteasomal destruction to synergize the inhibition of caspase 9.41 It appears that multiple BIR domains are required for caspase binding, evidenced by cIAP1/242 and XIAP.43,44 Thus, it is more likely that survivin inhibits apoptosis by serving as a protein–protein interaction coordinator among IAPs. In addition, survivin also acts as a critical mitotic regulator in the nucleus, playing an essential role in proper mitosis and cytokinesis by interacting with the chromosomal passenger complex (CPC).25,45–47
Despite the progress in understanding the mechanism of survivin function, its detailed action modes at molecular and atomic levels remain largely unclear, including the functions of each domain and the critical residues in coordinating with or binding to its partners, and the signaling pathways orchestrated by survivin. While further studies are required in this aspect to assist drug discovery targeting survivin, inhibitors generated thus far may also be used as chemical probes to dissect the biology of survivin function at the molecular and atomic levels.
1.3. Survivin as a Therapeutic Target.
Survivin is overexpressed in almost all human cancers,48,49 but undetectable in most normal adult tissues.50 The high survivin expression level is predictive of poor clinical outcome and is associated with tumor relapse.51,52 Survivin has been consistently demonstrated to be a pivotal contributor to radiotherapy and chemotherapy resistance due to its role in antiapoptosis.53–55 It has been shown that survivin knockdown in glioma and breast cancer cells decreased angiogenesis.56,57 Silencing or downregulating survivin expression using ribozyme or siRNAs also led to increased cell death and chemosensitivity.58,59 These findings together indicate that survivin is clinically relevant and perhaps an ideal target for anticancer drug discovery.25
2. MEDICINAL CHEMISTRY OF SMALL MOLECULE SURVIVIN INHIBITORS
It has been very challenging to target survivin, which has been historically considered “undruggable” due to the lack of known enzymatic activities or active sites to be targeted by small molecule compounds.60 To bypass this obstacle, several strategies have been tested. The first one was to inhibit transcription of the survivin gene. The small molecule inhibitors identified to do so include compounds 1 (YM155), 2 (EM-1421), and 3 (FL118) (Figure 2A). As discussed above, survivin exerts most of its functions by interacting with other proteins. Thus, inhibiting survivin interaction with these proteins using small molecules has been attempted, with representative inhibitors including compound 4 (UC-112) and its analogs 5 (MX106) and 6 (12b). Finally, survivin exists and functions as a homodimer and, thus, inhibiting its homodimerization to induce its degradation in the proteasome has also exhibited some success, as exemplified by compounds 7 (LQZ-7F), 8 (7F1), 9 (LQZ-7I), 10 (LLP3), and 11 (4a′). These inhibitors (Figure 2A) are at different stages of development, with most in preclinical studies for their efficacies, safety, pharmacodynamics (PD), and pharmacokinetics (PK) profiles, and only 1 and 2 advanced into clinical testing (Table 2).61,62
Figure 2.

Structures of small molecule inhibitors that are included (A) in or excluded (B) from discussion in this work.
Table 2.
Representative Compounds
| compounds | mechanism of action | in vitro IC50 (μM) | application/clinical trial (year initiated) | SARa status |
|---|---|---|---|---|
| 1 (YM155) | Inhibiting Sp1 and RIPK270,71 | 0.008 (PC-3), 0.002 (PPC-1), 0.004 (DU145) | Phase I for advanced cancer (2009) and NSCLC (2010) | Limited SAR without better compound identified |
| Disrupting ILF3-p54nrb complex72 | 0.008 (TSU-Pr1), 0.011 (22Rv1), 0.004 (SK-MEL-5), 0.006 (A375)74 | Phase II for CRPC (2007), DLBCL (2007), NHL (2009), melanoma (2009), metastatic breast cancer (2009), high-grade B-cell lymphoma (2022) | ||
| Inducing DNA damage73 | 0.0005 (NKF-NB-3), 0.0007 (NKF-NB-3)75 | |||
| 2 (EM-1421) | Inhibiting Sp176 | 16.6 (A375), 61.5 (HT-29), 42.4 (MCF-7) 85.0 (HaCat), 44.5 (HepG2)77 |
Phase I or II for head and neck tumors (2003), CIN (2005), leukemia (2008), refractory solid tumors (2008), lymphoma and rHGG (2015), and high-grade glioma (NCT02575794) | Systematic SARb with a more potent compound 37 identified |
| 3 (FL118) | Inhibiting expression of survivin, Mcl-1, XLAP, cIAP278 | 0.0003 (H460), 0.0006 (EKVX), 0.0009 (A549) | Preclinical for cancers of head and neck, colon, and pancreas, as well as metastatic lung cancer and RRMM | Limited SAR with a more potent compound 52 (9c) identified |
| Inhibiting | 0.0004 (HCT116)80 | Under IND | ||
| DDX579 | 0.0005 (HCT-8)81 0.037 (A2008), 0.009 (SW620)82 |
|||
| 4 (UC-112) | Disrupting SMAC and survivin interaction83 | 1.6 (A375), 2.5 (M14) | Preclinical for P-glycoprotein- overexpressing colorectal and cervical cancers | Systematic SAR with several leads identified |
| Inhibiting expression of other IAPs83 | 3.2 M14/LCC6MDR1), 0.7 (PC-3), 3.4 (DU145)84 | |||
| 7 (LQZ-7F) | Inhibiting survivin dimerization and inducing survivin degradation20 | 4.3 (PC-3), 1.9 (C4–2)20 | Preclinical for CRPC | Limited SAR with 7–9 as candidates |
| 10 (FFP3) | Binding to the dimerization domain85 | 31.2 (GBM528)c,85 | Preclinical for glioma | Systematic SAR with 10 and 11 as leads |
Abbreviations use: CIN, cervical intraepithelial neoplasia. CRPC, castration-resistant prostate cancer. DLBCL, diffuse large B-cell lymphoma. ILF3, interleukin enhancer-binding factor 3. NHL, non-Hodgkin’s lymphoma. NSCLC, nonsmall-cell lung cancer. rHGG, recurrent high-grade glioma. RIPK2, receptor-interacting protein kinase 2. RRMM, relapsed/refractory multiple myeloma. SAR, structure–activity relationship. Sp1, specific protein 1.
Systematic SAR is defined as available SAR for each functional group.
Determined by sphere formation assay.
However, because 1 and 2 do not act directly on the survivin protein but inhibit its upstream transcription factors (see below), they are not survivin inhibitors per se and readers are advised to be cautious when citing these compounds as survivin inhibitors. Nevertheless, because these compounds inhibit survivin expression, selectively target cancer cells, and have been studied in detail and are being tested in clinical trials without major toxicity, critically examining these compounds may provide valuable perspectives on developing inhibitors to selectively inhibit survivin expression.
In this perspective, we will focus on the medicinal chemistry of the well-characterized small molecule compounds by examining their chemical structures, pharmacological properties, and provide insights and perspectives on their future improvements or developments. Additionally, we will critically discuss the different strategies in targeting survivin and place these in perspective for future investigations. Biologics, including dominant negative survivin and antisense oligonucleotides as well as vaccines, are out of scope and, thus, are excluded from discussion. In addition to the small molecule inhibitors shown in Figure 2A that have been the subject of extensive medicinal chemistry studies and analyses, a few others that are nonselective and for which there is a limited understanding of mechanism of action and lack of associated pharmacological or medicinal chemistry studies, are excluded. These inhibitors include compounds 12 (indinavir),63 13 (nelfinavir),64 14 (GDP366),65 15 (withanone),66 16 (S12),67 17 (SF002–96–1),68 18 (ZINC2243688), and 19 (ZINC057885)69 (Figure 2B).
Here, we attempt to interpret the drug-likeness of the prominent inhibitors using the well-defined Lipinski’s Rule of Five (RO5) as a guide,86 and by analyzing their drug design, structure–activity relationship (SAR), key medicinal chemistry and pharmacological properties from published work. Note that RO5 is used with caution as these rules were established based on the orally administered drugs approved prior to 199786 and the drug-like properties are constantly changing with a growing number of approved drugs that do not strictly follow these rules.87
2.1. Compound 1 (YM155).
Compound 1 (sepantronium bromide) is an ammonium salt with a positively charged imidazolium ring (Figure 3A), termed an onium compound. It was first identified in 2007 by Astellas Pharma using a high throughput screening (HTS) assay of survivin promoter-driven luciferase reporter expression aiming for small molecule inhibitors that may bind to the promoter sequence of the survivin gene and inhibit its expression.74 Compound 1 indeed suppressed survivin expression at both mRNA and protein levels in various cancer cells with nanomolar potency after a 6-h treatment.88,89 It had favorable selective profiles over other IAPs including cIAP2 and XIAP, and had no effect on the expression of antiapoptotic Bcl-2-related genes such as Bcl-2, Bcl-xL, and Bad.74 More importantly, 1 preferably accumulates in tumors, with ~20-fold higher concentration than that in plasma.74
Figure 3.

Medicinal chemistry of compound 1 and its derivatives. (A) Chemical structure of compound 1. (B–C) Analogs of compound 1 with substitutions on N1 (B) and N3 (C) as highlighted in cyan and pink, respectively. (D) Proposed lead derivatives 20–23.
2.1.1. Biological Properties.
Compound 1 inhibited cancer cell proliferation with an average IC50 of ~15 nM in 119 human cancer cell lines, and an IC50 range of 0.49 nM to 248 nM in a series of drug-resistant neuroblastoma and pancreatic cancer cell lines, suggesting that it is a highly potent anticancer agent.74,90,91 Although 1 is toxic to normal cells such as primary human astrocytes, WA09 human ES cell-derived neural progenitor cells, and primary human lung fibroblasts cells,70 with IC50 values ranging from 5 to 90 nM, it requires much lower concentrations (~1–100 fold) to kill glioblastoma (GBM) cells, with IC50 values ranging from 0.78 to 4.5 nM, suggesting the potential for a safety margin.70 In orthotopic xenograft PC-3 prostate tumor and ectopic xenograft tumors of breast, lung, and bladder as well as melanoma, administration of 1 via a continuous 3- or 7-day infusion significantly suppressed the tumor growth, without significant toxicity as indicated by low body weight loss in the animals.74,91
Importantly, 1 is well tolerated by humans, with a maximum tolerated dose (MTD) of 4.8 mg/m2.92 However, limited or modest efficacy at best was observed in clinical trials against various solid tumors. No improvement in response rate was observed for nonsmall cell lung cancer (NSCLC) patients in two phase II trials of 1, either as a single agent or in combination with carboplatin or paclitaxel.15,93 In another phase II clinical trial, modest efficacy was observed with 25% of castration-resistant prostate cancer (CRPC) patients displaying prolonged stable disease.94 In a study testing 1 in combination with erlotinib in patients with advanced NSCLC that are refractory to EGFR inhibitors, a favorable safety profile with moderate clinical efficacy and little significant drug–drug interactions were observed.61 Currently, there are no ongoing or active clinical trials on 1 in the U.S. but it is still active in China and Japan.
One of the major issues with compound 1 is its poor PK profile due to rapid elimination from plasma at a rate of 34–48 L/h (Table 3),92,96 requiring it to be continuously infused intravenously (IV) 24 h a day in a 7-day dosing cycle (168 h).92,96 Another issue is the lack of understanding in the precise mechanism of its action. As previously mentioned, although 1 was identified using a HTS assay for inhibitors that bind to and inhibit the survivin promoter and it indeed inhibits survivin transcription,74 it was later shown that 1 does not bind to the survivin promoter as anticipated but rather inhibits the transcription factor Sp197 and disrupts the interaction between interleukin enhancer-binding protein factor 3 (ILF3) and p54nrb, a critical complex for survivin transcription.72 These findings are not surprising because the cell-based HTS assay used did not eliminate the potential inhibition of transcription factors that may activate the survivin promoter. Furthermore, there is compelling evidence suggesting that 1 may be a DNA damage-inducing agent and its inhibition of survivin expression is secondary to this event.98 However, a later study showed that 1-induced DNA damage in neuroblastoma UKF-NB-3 cells may be secondary to survivin down-regulation since survivin knockdown using siRNA induced DNA damage without 1.75 The latter observation is consistent with previous studies that provided indirect evidence of survivin-depletion leading to DNA damage.99,100 These findings indicate that survivin may play a role in protecting against DNA damage.
Table 3.
Pharmacokinetic Profiles of Compounds 1, 2 and 3
| compounds | species | route | Tmaxa(h) | T1/2(h) | Cmax(ng/mL) | Cl(mL/h/kg) | AUC (ng × h/mL) | F (%) |
|---|---|---|---|---|---|---|---|---|
| 1 (YM155) | human92 | IV | ND | 26.3 | 10.7 | 47 700 | 1439 | ND |
| human61 | IV | 45.98 | 17.18 | 11.95 | 34 030 | 1832 | ND | |
| 2 (EM-1421) | mouse95 | IV | 0.08 | ND | 1064.60 | ND | 2984.54 | ND |
| mouse95 | Oral | 8.0 | ND | 441.51 | ND | 2635.47 | 88.29 | |
| human16 | IV | ND | 17.1 | ND | 54 000 | ND | ND | |
| 3 (FL118) | mouse81 | IV | 0.17 | 1.8 | 43 | 14 287 | 82 | ND |
Abbreviations used: Tmax, the time it takes for a drug to reach the maximum concentration. T1/2, the time required for plasma concentration of a drug to decrease by 50%. Cmax, the maximum concentration in plasma. Cl, clearance rate. AUC, the aera under the curve. F, fraction absorbed (bioavailability). ND, not determined. IV, intravenously.
Most recently, it was shown that 1 inhibits the receptor-interacting protein kinase 2 (RIPK2), which also mediates its effect on survivin expression.70,101 Considering that Sp1 is a ubiquitous transcription factor with multiple downstream target genes, RIPK2 also has multiple target proteins, and compound 1 does not bind to survivin, designating 1 as a survivin inhibitor is inappropriate and misleading. Nevertheless, as discussed above the fact that it continues to be actively investigated calls for perspective insight and discussion on future directions of its study, and we believe that there is some value in thoroughly examining its medicinal chemistry and pharmacology. Hence, we provide our thoughts on its medicinal chemistry and pharmacology for readers with interest in 1 and perhaps also in targeting gene transcription in general and survivin transcription specifically.
2.1.2. Medicinal Chemistry.
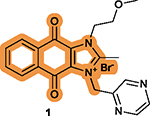
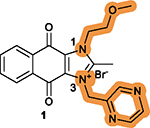
While hundreds of 1 analogs have been disclosed in the patent application,102 the SARs for the benzene ring (I) and imidazolium ring (II) of the dioxonaphthoimidazolium scaffold and the relationship to anticancer activity have not been clearly defined.102 There are two recent reports on SAR studies of 1, including a study showing DNA intercalation and cytotoxicity using the clear cell renal cell carcinoma (ccRCC) cell lines RCC786–0 and RCC4/VA, NSCLC cell lines H1299 and H1666, and nonmalignant human lung fibroblast IMR-90,103 and another study showing anticancer activity of 1 derivatives using human pluripotent stem cells (hPSCs).104 In these two studies, all its derivatives have the intact dioxonaphthoimidazolium scaffold, the essential pharmacophore of 1, but with various substituents on N1 (Figure 3B) and N3 (Figure 3C) of ring II. The key findings associated with the SARs of 1 are summarized in Table 4.103,104
Table 4.
SARs Associated with Compound 1a
| position/scaffold | SAR |
|---|---|
|
| |
|
The intact dioxonaphthoimidazolium scaffold is
required; The critical motif on the tricyclic scaffold has an order of quinone > imidazolium > benzene.104 |
|
Substitutions at the N1 and
N3 positions are tolerable; Short, unbranched, and cyclized alkyl substitutions at both N1 and N3 are favorable; A pyrazine moiety at N3 is necessary and usually optimal; Substituting pyrazine with benzene decreases the potency; The combination of substitutions at N1 and N3 is complex, resulting in no compounds with higher potency.103,104 |
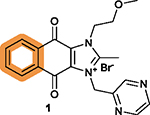
|
Substitutions with electron-withdrawing groups on the benzene ring are optimal, without differences for electron-withdrawing groups at different positions.103,104 |
The highlighted elements are the points of discussion.
Currently, about three hundred analogs of compound 1 have been synthesized and tested. However, only several derivatives, including 20 (AB7), 21, and 22 (Figure 3D), exhibited favorable in vitro and in vivo tumor-inhibiting activities.103,104 Unfortunately, none of these derivatives showed superior activity compared to 1 in either inhibiting survivin expression or suppressing cancer cell proliferation. Compound 23 (UFSHR, Figure 3D) also exhibited lower potency than 1 in suppressing survivin expression in the pancreatic cancer cell lines PPCL-46 and PPCL-LM1, which were generated from patient-derived xenograft tumors of stage III or IV pancreatic ductal adenocarcinoma (PDAC). However, 0.1 mg/kg 23 via intraperitoneal (IP) injection achieved similar and better activity than 0.01 mg/kg 1 (IP) in inhibiting PPCL-LM1 and PPCL-46 xenograft tumors, respectively, but at a 10-fold higher dose.48
In addition, compound 1 was confirmed to be a substrate of the ATP-binding cassette (ABC) transporter subfamily B member 1 (ABCB1, also known as P-glycoprotein) of which overexpression facilitated its efflux from cancer cells and thereby conferred its resistance.105,106 Thus, it cannot be utilized for cancer patients overexpressing ABCB1.
2.1.3. Key Structural Properties.
Compound 1 is an ionized and positively charged molecule. Ionized molecules account for only ~4% of all approved drugs, and most of these drugs are for external use to treat infectious diseases.107,108 While in most cases, cationic molecules are more potent cytotoxins toward cancer cells than the anionic molecules,109,110 ionized molecules when used in vivo usually have absorption issues due to lower membrane permeability than neutral molecules.111 However, positively charged molecules may enter cancer cells using the organic cation transporters, as has been suggested for 1.74,112 Solute carrier family 35 member F2 (SLC35F2) has also been suggested to contribute to 1’s uptake and its expression may predict the in vitro and in vivo efficacy of 1.73,75
Intriguingly, many tool compounds are cationic, including the commercially available fluorescent tracking/imaging MitoTracker probes.113,114 These compounds typically possess a chemical formula of (R3) N+(halogen)− or (Ph3) P+(halogen)−, which may favorably target and accumulate in the negatively charged inner mitochondrial membranes.113 Since 1 also contains the (R3) N+(halogen)− motif, it may also accumulate in mitochondria. Indeed, a recent study showed that it accumulates in mitochondria and causes mitochondrial dysfunction, suggesting another potential mechanism of action for compound 1.115
Compound 1 has a 1,4-benzoquinone containing dual carbonyl groups which is prevalent in both biological molecules and approved drugs.116 The two carbonyl groups together with the vinyl group can form a conjugated system that appears to be stable. However, the 1,4-benzoquinone motif is oxidatively active under physiological conditions. It can undergo specific redox reactions with reductive biomolecules or by metabolic enzymes, which may contribute to its poor PK profile.116 Indeed, it was shown recently that the carbonyl groups on dioxonaphthoimidazoliums analogs with the same pharmacophore as 1 underwent redox reaction catalyzed by respiratory enzyme type II NADH dehydrogenase (NDH2), leading to increased production of reactive oxygen species (ROS).117,118 Future studies to modify the two carbonyl groups may help reduce their chemical reactivity and improve the PK profile.
2.1.4. Perspective.
Compound 1 has a favorable molecular weight of 443, zero H-bond donors (HBDs), and seven H-bond acceptors (HBAs), but an unfavorable cLog P of −3.58. The negative cLog P value indicates a strong hydrophilicity that may cause absorption issues following oral administration. We used a Radar Chart to visualize the overall properties of 1 (Figure 4A), which shows that it is an extremely unbalanced candidate without an optimal cLog P value. The low cLog P value renders it a highly polar and soluble compound that can only be administered in clinical trials via infusion. The high polarity along with the fact that its uptake may rely on transporters may exclude it from being developed for tumors in the central nervous system due to the need to traverse the blood-brain barrier (BBB).70,119
Figure 4.

Perspectives of compound 1. (A) Radar Chart analysis of 1. The scale of cLog P (calculated by ChemDraw 20.0) and HBD is equivalent to the original value multiplied by 100 and that of HBA multiplied by 50. (B) Proposed modifications of 1. (C) The structures of compounds 32 (G-555) and 33 (doxofylline). (D) Ketal moiety is used in the synthesis of 34 (lactonamycin).
Given that 1 is a highly potent candidate and has been tested in clinical trials, future studies should be focused on correcting its PK profiles by modifying its structure or developing pharmaceutical formulations that may eliminate the need for a 3- or 7-day continuous infusion.15,94 With the above medicinal chemistry and key structural characteristics of 1, we envision a few studies to improve it based on several candidates, including compounds 20 and 23. It has been shown that minor structural modifications on lead compounds can have meaningful impacts on pharmacological profiles due to significant effects on molecular and physicochemical properties and intra- and intermolecular interactions.120 Thus, minor structural modifications to eliminate the positive charge may help improve the PK profile of 1. For example, a minor modification by reducing the C=N in ring II to CH–N or replacing the nitrogen atom (N) with a carbon atom (C), as shown in compounds 24–29 (Figure 4B), respectively, is expected to lead to an electrically neutral molecule that retains all of the essential pharmacophore of 1 and may possess comparable anticancer potencies with possible better PK/PD profiles than 1.
Next, structural modifications to increase the in vivo stability should be considered. For example, compound 30 with specific halogen substitution on the benzene ring, including fluoro (−F), chloro (−Cl), bromo (−Br), iodo (−I), or trifluoromethyl (−CF3) (Figure 4B) may help to stabilize the benzene ring, leading to enhanced resistance to metabolism.121,122
Also, protection of the carbonyl group(s) by ethylene glycol (Figure 4B) may generate 31 with a ketal group for development of a new chemical entity or a potential prodrug although such a moiety is stable in neutral or even acidic conditions and, thus, cannot convert back into 1.123 The ketal moiety is used as a common chemical strategy to protect a carbonyl group in organic chemistry124 and also as a motif in drug structures, as in the compound 32 (G-5555), a p21activated kinases (PAKs) inhibitor,125 and 33, the approved drug doxofylline (Figure 4C).126 A quinone to ketal molecular edit has been successfully exemplified in the synthesis of 34 (lactonamycin),127 an antimicrobial drug that has also been shown to be a potent anticancer agent with nanomolar IC50 values (Figure 4D)128. However, a ketal derivative of a quinone-based molecule has yet to be developed or approved. Nevertheless, a compound with a ketal derived from the quinone may be worthy of consideration for testing whether it can function as a new chemical entity.
The successful enforcement of these proposed modifications may help further develop 1 into an effective anticancer drug with an improved PK/PD profile.
2.2. Compound 2 (EM-1421).
Compound 2 (Figure 5A), tetra-O-methyl-nordihydroguaiaretic acid, also known as terameprocol or M4N, is a semisynthetic compound derived from 35, the natural product nordihydroguaiaretic acid (NDGA, Figure 5A) from Larrea tridentata.129 Compound 35, a poly phenolic antioxidative compound categorized as a lignan, was discovered over one century ago and has been used in food chemistry as a preservative for more than 70 years.130 As the main active component, 35, developed by the University of Arizona Cancer Center under the trade name Actinex, was approved by the FDA in 1992 for skin-related damage/cancer due to sun exposure. However, it was withdrawn in 1996 due to unexpected side effects and a small market. Compound 2 is generated directly by the methylation of the four phenolic hydroxy groups of 35.77 Similar to 1, 2 is also a transcriptional repressor of the survivin gene by inhibiting the ubiquitous transcription factor Sp1.76,131 As such, it also inhibits the expression of cyclin dependent kinase 1 (CDK1), a downstream gene of Sp1,132 and the expression of vascular endothelial growth factor (VEGF).133 It has also been shown to inhibit AKT phosphorylation and activation.134,135 Similar also to 1, new targets may continue to be identified with continued study. Thus, designating 2 as a specific survivin inhibitor is also inappropriate and misleading since it does not bind to survivin. Nevertheless, 2 is now under evaluation in multiple phase I/II clinical studies sponsored by Erimos Pharmaceuticals.
Figure 5.

Medicinal chemistry of 2. (A) Chemical structures of 35 (NDGA) and 2. (B–E) Ether bond (B), ester bond (C), end-ring (D), and linear or cycled carbon chain bridge (E) modifications on compound 2. (F) Chemical structure of 36 (NDGA-Cl2). (G) Compound 2 analogs with different length of the carbon-bridge or number of hydroxyl groups on benzene rings.
2.2.1. Biological Properties.
Compound 2 has IC50 values ranging from 16 to 85 μM in various cancer cell lines, including A375, HT-29, MCF-7, HaCat, and HepG2 cells, and suppresses the growth of xenografts derived from breast, prostate, colorectal, and liver tumors in mouse models.77,95,136 In addition, it was able to sensitize radiotherapy in NSCLC cells.137 The bioavailability of orally administered 2 in mice is very good, reaching 88% (Table 3).95 While 2 shares similarity with 1 in that it requires IV administration in clinical trials, the infusion time is shorter and dosing is of a lower frequency than for 1, with a cycle of 1500 mg dosing over 6 h, three times/week for 2 weeks followed by a week of rest.134 The first clinical trial with 2 was conducted in 2003 for refractory malignant head and neck tumors (NCT00057512). However, the results of this trial have not been posted. In 2007 and 2008, the safety profile of 2 was reported as a vaginal ointment ultimately intended as treatment for cervical intraepithelial neoplasia caused by human papilloma virus (HPV) in a phase I/II trial in healthy adult women.138,139 The ointment formulated with 1–2% compound 2 showed a promising safety profile with no severe adverse effects in healthy subjects.138,139 Among advanced leukemia patients recruited, compound 2 at 1000, 1500, or 2200 mg, three times/week via IV, demonstrated a safe profile with partial responses in a few patients with acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) in a phase I clinical study conducted in 2015.140 In another phase I clinical study in 2012 for high-grade gliomas, 2 did not display any significant response, although 32% of patients showed stable disease.16 In 2019, a phase I/II clinical trial (NCT00404248) to study the side effects and to optimize the dose of 2 in patients with recurrent high-grade glioma was completed, with the results showing that 1700 mg/day for five consecutive days was safe. Another phase I clinical trial (NCT02575794) to test MTD in patients with high-grade glioma has been scheduled but is inactive in recruiting patients. As disclosed in the pipeline of Erimos Pharmaceuticals, 2 is proposed for multiple phase I clinical trials for solid tumors, advanced leukemia, glioma, and head and neck cancer.16,134
2.2.2. Medicinal Chemistry.
Compound 2 is a relatively simple compound with two major chemical groups, the linker and the two methyl groups to protect the two hydroxy groups (−OH) of each catechol (Figure 5A). Most of the known derivatives and analogs have the two benzene rings but vary in the identity of substituents or protective groups on the catechol moieties, and the linker between the two catechols (Figure 5B–F). These derivatives include substitutions of four methyl groups on the two catechols using other functional groups via the ether bond (Figure 5B), ester bond (Figure 5C), or end-ring (Figure 5D), and modification of the linear carbon chain linker using different length and ring substitution motifs (Figure 5E). Although hundreds of 2 derivatives and analogs have been synthesized and characterized, no derivative with better drug-likeness than the parent drug 2 has been identified.141,142 While one study showed that the introduction of chlorine on each benzene ring increased anticancer potency as compared to 2, the compound 36 (NDGA-Cl2, Figure 5F) was eventually abandoned due to its high toxicity in vivo.77 Table 5 shows a summary of key SAR information associated with 2.
Table 5.
SARs Associated with Compound 2a
| position/scaffold | SAR |
|---|---|
|
| |
|
|
End-ring modification (ester or ether bond)
decreases activity;77,141 Short chain phenol esters show comparable activity to 2;77 The effect of longer chain or other ether bond modifications on activity is unknown; Two phenolic groups on each benzene ring (e.g., 37) are better than either one (38) or three (39) such groups.141 |

|
A linear carbon chain linker is better than a
cyclized linker;142 The two methyl groups are dispensable;141 The optimum length of the carbon linker is 4 (37) and shorter (40) or longer (41) one reduces activity.143,144 |
The highlighted elements are the points of discussion.144
2.2.3. Key Structural Properties.
There are several potential key/interesting modification strategies of the natural product 35, the parent compound of 2. First, phenolic hydroxy groups (PHGs) have long been regarded as free radical traps that can capture various forms of free radicals, including ROS and reactive nitrogen species (RNS), leading to reduced oxidative stress and acting as antioxidants that can be applied as cytoprotective agents.145 However, PHGs, especially the poly phenolic groups, are unstable and easily oxidized into quinones.146,147 Therefore, PHGs in small molecules are usually protected by other functional groups through an ester or ether bond and these simple modifications may lead to a more potent drug candidates.148,149 Ester bond modification of PHGs often results in prodrugs that can be hydrolyzed into its active form in vivo, mediated by mild base/acidic conditions or carboxylesterases enriched in the liver and blood.150,151 In contrast, ethers are a much more stable form and the product usually acts as an intact molecule to bind to its target or exert its therapeutic function.149 In the case of compound 2, methylation of PHGs in NDGA resulted in a promising product that is currently being studied in clinical trials. However, the phenol ether could be metabolized to the free phenol by cytochrome P450 (CYP) as shown previously152 although no such study has been conducted for 2. Future PK studies are necessary to test this possibility.
Second, the length of the carbon chain in ether or ester derivatives of PHGs will affect lipophilicity.153,154 While generally the longer the side chain, the more lipophilic the molecule, the correlation with bioactivity is usually more complex and unpredictable.153,154 In addition, as the side chains grow longer and bigger, the molecules tend to be more stable against hydrolysis due to steric hindrance.155,156 Given that the essential pharmacophore of 2 is actually the NDGA, bulkier side chains may reduce potency and should be avoided.77
Third, 2 does not contain a nitrogen atom while many of its ester or ether derivatives do (Figure 5B/C).141 Such nitrogen-containing groups (NCGs) are one class of solubilizing moieties commonly adopted in medicinal chemistry to help address solubility issues of lead compounds.157 NCGs in biologically active compounds usually exist in different forms including amine salt, neutral amide, or heterocyclic compounds.158 It is estimated that nearly 75% of FDA-approved small molecule drugs contain at least one nitrogen atom, and ~60% have a nitrogen-containing heterocyclic, suggesting that NCGs are prevalent in approved drugs.158,159 Thus, the addition of NCGs to NDGA was speculated to increase its drug-likeness by protecting PHGs, forming a hydrogen bond with its target, and improving water solubility. Unfortunately, no better derivative than 2 using this approach has been identified (Figure 5B). More such compounds may need to be synthesized to further test this possibility.
2.2.4. Perspective.
Structurally, 2 is not an ideal drug since it offers extremely limited space for further improvement and possesses a high cLog P value, as shown in the Radar Chart (Figure 6A). Since all four polar hydroxy groups are protected as methyl ethers, compound 2 is hydrophobic with poor solubility. However, this hydrophobicity may help it penetrate the BBB, as suggested by its trials for glioma.16 Similar to 1, we offer below some perspective on future development since 2 is being actively investigated in clinical trials. In this regard, modifying 37 with four free PHGs (Figure 5G) and an IC50 value of 0.3 μM against a small cell lung cancer cell line H69 may provide a better option for development.143
Figure 6.

Perspective of 2. (A) Radar Chart analysis of 2. The scale of cLog P and HBDs is equivalent to the original value multiplied by 100 and that of HBAs multiplied by 50. (B) Chemical structure of proposed derivatives 42–45.
First, given that the four free PHGs in 37 are unstable, they likely need to remain protected similar to that in 2. Esterification or etherification with short side chains as proposed in 42 or 43 (Figure 6B) is preferred based on previous findings.
Second, the liner linker appears to be more favorable for cytotoxicity although the data for this conclusion are limited. However, several other types of linkers such as butanediols in secoisolariciresinol,160 and tetrahydrofuran in taxiresinol161 have been used and shown to have promising activities. Thus, further studies testing additional linkers such as those in the proposed 44 and 45 may result in better derivatives.
Finally, considering that poly phenolic compounds may possess cytoprotective activity, 37, the hydrolysis product of 42/43 (Figure 6B), could be explored for its ability to alleviate adverse effects induced by toxic chemotherapeutics in combination therapy.
Similar to 1, 2 also inhibits the transcription factor Sp1, which indirectly inhibits survivin expression. Interestingly, these two compounds are safe for use in animal models and in clinical trials despite the fact that they inhibit a ubiquitous transcription factor Sp1 with multiple downstream target genes,25,60 indicating that Sp1 may be a potential target without major toxicity. However, their limited clinical efficacy suggests that targeting Sp1 to suppress survivin expression along with other Sp1 downstream genes may not be feasible and represents an inappropriate strategy for therapeutic development. Nevertheless, the information accumulated from more than ten years of preclinical study and clinical trials may be useful to transform 1 and 2 into potent candidates that are not only safe but also effective.
2.3. Compound 3 (FL118).
Compound 3 (Figure 7A) was identified via HTS in 2012 using a reporter assay of the survivin gene promoter in an attempt to identify novel inhibitors that disrupt the promoter activity, similar to the strategy used in the discovery of 1.78,162 However, 3 also inhibits the expression of Mcl-1, and two other IAPs including XIAP, and cIAP2.163 Thus, 3 is also a nonselective inhibitor of survivin expression. Interestingly, 3 structurally resembles the topoisomerase I inhibitors, 46 (camptothecin), 47 (irinotecan), and 48 (topotecan), which have been approved by FDA for cancer treatments (Figure 7A/B).
Figure 7.

Medicinal chemistry of 3. (A) Chemical structures of 3 and 46 (camptothecin). (B) Chemical structures of camptothecin-based topoisomerase 1 inhibitors 47 (irinotecan), 48 (topotecan), and irinotecan metabolite 49 (SN-38). (C) Derivatives 50–58 with modified R7. (D) Derivatives 59 (9-Q6) and 60 (9-Q20) with modified R9. (E) Derivative 61 (Val-FL118) with addition of valine and 62–65 with other modifications at the hydroxyl group. The positions that are tolerable to modification are highlighted as R7 or R9 in claret.
2.3.1. Biological Properties.
Compound 3 displayed nanomolar IC50 values in a series of cancer cell lines with strong antitumor activity in xenografts of squamous cell carcinoma, colon cancer, multiple myeloma cell lines, as well as a patient-derived head and neck cancer xenograft when combined with the first-line therapeutic gemcitabine.17,164,165 It was also found that SW620 and HCT-8 cancer cells are more sensitive to 3 than normal adult human dermal fibroblast and human gingival fibroblast cells, suggesting the existence of a therapeutic window.78 However, 3 appears to have a short half-life in mice, with a t1/2 of only 1.8 h following IV administration (Table 3). A recent study on its mechanism of action showed that 3 may work as a molecular glue to directly target and bind to DEAD-box helicase 5 (DDX5), an ATP-dependent RNA helicase with transcription coactivator function, with a Kd of 34.4 nM, leading to dephosphorylation and degradation of DDX5 in the proteasome.79 DDX5 promotes cancer progression by interacting with transcription factors including signal transducer and activator of transcription 3 (STAT3), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), cyclin D1, c-Myc, and β-catenin.166 Compound 3-induced degradation of DDX5 is expected to down-regulate the expression of many downstream target genes of these transcription factors, including survivin.79 Indeed, in addition to inhibiting XIAP and cIAP2 expression,163 3 has been shown to inhibit the expression of c-Myc, a known target gene of DDX5-β-catenin coactivation.79 Furthermore, 3 with oral dosing at 2.5 mg/kg (four times a week for five to 7 weeks) is more effective on PDAC patient-derived xenograft (PDX) tumors with high DDX5 level than those with low-level.79 DDX knockdown in colon cancer and PDAC cells also caused insensitivity to 3. These results suggest that DDX5 is indeed a target of 3. Currently, 3 is the basis of IND application although clinical studies do not appear to have been scheduled.
Many 3 analogs have been reported, including nine new derivatives (50–58) recently synthesized with different R7 substituents on ring II (Figure 7C),82 two compounds with different R9 substituents on ring I (Figure 7D), and five compounds with modifications on the hydroxy group (Figure 7E).167 Compounds 51 and 52 (Figure 7C) have a similar IC50 values of 11 and 10 nM in A2008 cells, respectively, and 0.17 and 0.11 nM in SW620 cells, respectively, and both are ~3- to 6-fold more potent than 3. 52 was selected for further evaluation over 51 due to convenience of synthesis and purification and because it consistently showed higher potency than 3 in a series of cancer cell lines with IC50 values ranging from 0.78 to 19 nM.82 Compound 52 (Figure 7C) was also more potent than 3 in inducing apoptosis of NCI-H446 lung cancer cells and in drug-resistant H69AR cells. It also appears to be more efficient in reducing both mRNA and protein levels of survivin in H69AR cells than in NCI-H445 cells.82 However, similar to 3, 52 also reduced the levels of XIAP in both cell lines, suggesting that it too is nonselective in inhibiting survivin expression. In addition, the irinotecan-resistant NCI-H446/Iri and etoposide/cisplatin combination (EP)-resistant NCI-H446/EP cells are more sensitive to 52 than their respective parental cells, suggesting that the drug-resistant cancer cells may have gained collateral sensitivity to inhibition of survivin expression. More importantly, intragastric (IG) dosing of 52 at 0.75, 1.5, and 3 mg/kg (once/week for 4 weeks) dose-dependently inhibited NCI-H446 xenograft growth, and at 3 mg/kg it was more potent than 3, consistent with the in vitro results.82 At 3 mg/kg, 52 also inhibited the growth of xenograft tumors derived from drug-resistant cancer cells NCI-H446/Iri and NCI-H446/EP. However, significant weight loss was observed in test animals, indicating that 52 may have toxicity.82
2.3.2. Medicinal Chemistry.
Preliminary SAR studies of the R7 and R9 substituents and the hydroxy group on ring V have been reported (Figure 7).82,164,167,168 Compounds 59 (9-Q6) and 60 (9-Q20) as shown in Figure 7D were well-absorbed across 2D and 3D CaCo-2 cell models. The IC50 values for 60 in HCT116, MCF-7, HepG2, HeLa, and A549 cells range from 0.78 to 3.58 μM, indicating that it is 16 to 30-fold less potent than 3.164 While the activity of 59 has not been reported, it is reasonable to predict that it may possess cytotoxicity comparable to 60 considering their structural similarity. The cytotoxicity of 61 (Val-FL118) in Figure 7E is weaker but comparable to 3 in HepG2 and HCT116 cells.169 However, 61 may be a good candidate with improved water solubility due to the hydrophilic valine motif. Four other derivatives, 62–65, with modifications of the hydroxy group (Figure 7E) all are much less potent than 3 in FaDu, SW620, and HCT-8 cells, suggesting that the free hydroxy group is important.167 Recently, it was found that a nitrogen at the para site of pyridine ring (50) or a bis-trifluoromethyl phenyl group at R7 (58) in Figure 7C were less favorable than the other rings.82 On the basis of these limited medicinal chemistry studies and the patent published in 2015,170 we performed a preliminary SAR analysis of 3 (Table 6).
Table 6.
SARs Associated with Compound 3a
| position/scaffold | SAR |
|---|---|
|
| |
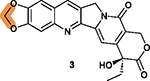
|
Methylene group is essential for inhibiting DDX5, down-regulating survivin expression and dispensing of topoisomerase 1 inhibition.78 |
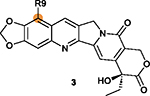
|
R9 position on ring I is tolerant of modifications with the substituted benzene (R-Ph) ring decreasing cytotoxicity (e.g., 59 and 60).164,168 |
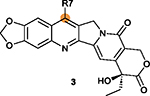
|
R7 on ring II is tolerant of modifications with
different rings; Five-membered heterocyclic rings are generally better than a six-member pyridine or benzene ring.82 |
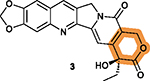
|
The lactone ring V may be unstable under acidic or basic conditions that may undermine 3’s activity and stability.170 |
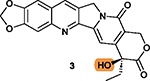
|
The free hydroxy group on ring V is critical for
cytotoxicity;167 Protection by hydrophilic groups via ester bond such as that in 61 and compounds 62–65 reduces cytotoxicity.167,169 |
The highlighted elements are the points of discussion.
2.3.3. Key Structural Properties.
Although 3 is structurally related to topoisomerase 1 inhibitors including 46 (camptothecin), 47 (irinotecan), 48 (topotecan), and 49 (SN-38, the active metabolite of irinotecan), it has been shown that 3 works differently78 and that its anticancer activity is unrelated to topoisomerase 1 inhibition.171 Thus, it would be interesting to analyze the structural differences between 3 and these topoisomerase 1 inhibitors, which may confer different biological events in cancer cells.
Compound 3 is 100- to 1000-fold more active in inhibiting survivin expression/transcription than inhibiting topoisomerase 1 activity.78 However, 48 and 49 exhibited 10 to 100-fold lower activity than 3 in inhibiting the expression of Mcl-1, XIAP, and cIAP2.81 Furthermore, 3 is not a substrate of ABC transporters such as ABCB1 or ABCG2,81 which differentiates it from 48 and 49.172 These findings suggest that although structurally similar, 3 is more selective than topoisomerase 1 inhibitors in suppressing survivin expression and less potent toward topoisomerase 1 inhibition with an ability to bypass ABC transporter-mediated drug efflux as a source of resistance.
The cocrystal structure and computer-assisted docking analysis of 46 and 48 with topoisomerase 1 showed that the active groups for topoisomerase 1 binding were the lactone (ring V), the hydroxy adjacent to the lactone, the hydroxy on ring I, and the carbonyl group on the pyridine ring (IV), while no direct binding interactions were found for the quinolone motif (I and II).173,174 Considering that the only structural differences among these topoisomerase 1 inhibitors are the substituents on rings I and II (Figure 7B) and that 3 differs from them also on ring I, it would be of interest to examine these differences in relationship to their target selectivity. While the hydroxy group on ring I of topotecan contributes to binding to topoisomerase 1, as discussed above, and hydrolysis of 47 results in the free hydroxy group in 49, 3 cannot be hydrolyzed to give the free hydroxy group on ring I with both oxygen atoms protected by the intervening methylene group, which is much more stable than the ester bond in 47. This particular entity maybe the major factor that prevents 3 from inhibiting topoisomerase 1 and may also be responsible for selective binding to and inhibition of DDX5.
2.3.4. Perspective.
Both compounds 3 and 52 are drug-like molecules given that several topoisomerase 1 inhibitors with similar structures have been approved by the FDA. The Radar Charts presented in Figure 8A/B suggest that both compounds follow the RO5. However, 3 has very low water solubility of less than 0.001 mg/mL. Due to its hydrophobicity, FL118 can permeate cells, as determined using 2D and 3D CaCo-2 cell models.164
Figure 8.

Perspective of 3. (A-B) Radar Chart analysis of compounds 3 (A) and 52 (B). The scale of cLog P and HBDs is equivalent to the original value multiplied by 100 and that of HBAs multiplied by 50. (C) Proposed modifications/optimizations of 3. Compound 67 is derived from topotecan; 68 and 69 are inspired by compound 2, and the discovery of 74 (icotinib), respectively. (D) 74 (icotinib) and 75 (erlotinib).
Considering that rings I/II are tolerant of structural modifications and that the lack of a hydrophilic motif as compared with 47 and 48 may cause poor water solubility, we envision a few minor modifications on rings I or II of 3 to improve its solubility and/or potency without undermining its drug-likeness (Figure 8C).
First, the nitrogen atom in the pyridine ring may form a salt with either hydrochloric acid (HCl) or methanesulfonic acid (MSA), two commonly used acids among FDA-approved drugs,175 that should possess improved water solubility (66).
Second, introducing a hydrophilic dimethylaminomethyl group on ring I (67), similar to 48, may improve water solubility significantly. The resulting compound 67 could also be converted into a salt by exposing to HCl or MSA to further improve solubility.
Third, the protective methylene group could be replaced by two separate methyl groups (68) or a crown ether (69). The two methyl groups linked via ether bonds are also resistant to hydrolysis (see discussion above for 2) and may resemble the effects of 3 on survivin expression. The crown ether-modified 3 may not only represent a new chemical entity, but also possess better bioavailability from modified membrane permeability.176 A successful example of such a molecule is 74 (icotinib) compared with its predecessor 75 (erlotinib) as shown in Figure 8D.177
Fourthly, it is possible to combine the modifications in 67–69 to create 70 and 71 with advantages from 67–69 (Figure 8C). While 70 has a suitable molecular weight, 71 may be too big with a molecular weight 551.6. In addition, the synthetic method to create 70 and 71 may be technically challenging.
Finally, with higher molecular weight (Figure 8B) and better potency than 3,82 52 can be further developed by introducing a hydrophilic tail, such as a dimethylaminomethyl group, at the R9 position on ring I, generating 72 (Figure 8C), which may increase its drug-likeness. Additionally, a crown ether can also be used to replace the mathelene on ring I of 52, generating 73. However, caution needs to be exercised due to the toxicity observed with 52 in mice.
2.4. Compound 4 (UC-112) and Its Analogs.
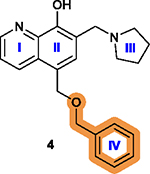
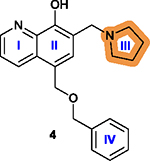
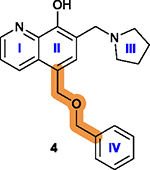
Survivin functions by binding to and interacting with many other essential cellular proteins. Thus, there have been various efforts in targeting survivin interaction with its binding partners using small molecule inhibitors. In this regard, a series of compounds containing a 7-(pyrrolidin-1-ylmethyl)quinolin-8-ol motif constructed with rings I–III (Figure 9A) were identified in a shape-based structural screen for SMAC mimetics that inhibit the interaction between SMAC and IAPs inspired by the SMAC-XIAP complex structure.83 One of these compounds, 4 (Figure 9B), inhibited the expression of XIAP, cIAP1, cIAP2, and survivin. It possessed IC50 values ranging from 0.7 to 3.4 μM in suppressing melanoma A375 and M14 cell proliferation as well as prostate cancer PC-3 and DU145 cell lines by activating caspases.83 Varying the linker between the 7-(pyrrolidin-1-ylmethyl)quinolin-8-ol motif and the benzene ring IV (Figure 9B–D) and substituents on the benzene ring IV or its replacement by other aromatic rings (Figure 9E–G) led to several second generation analogs including 5 (Figure 9F),18 76, and 77 (Figure 9D), as well as 78–80 (Figure 9E) with structural extension by varying substituents on the indole moiety.84,178 Compound 6 is a third generation of 4 analog discovered recently (Figure 9G).179
Figure 9.

Medicinal chemistry of 4. (A–B) Chemical structures of 7-(pyrrolidin-1-ylmethyl)quinolin-8-ol motif (A) and 4 (B). Replacement of the linker and benzene ring IV in 4 with different groups led to 76 and 77 (C and D). (E) Using methyl as a linker and replacing benzene with indole motif led to the discovery of 78 and its analogs 79 and 80. Further study revealed another lead 5 (F) and 6 (G).
2.4.1. Biological Properties.
Compound 4 is ~2.5-fold more effective against cancer cell lines than normal human keratinocyte Hacat cells or dermal fibroblast adult cells.83 Thus, there may be a small therapeutic window. Nevertheless, treatment using 20 and 40 mg/kg of 4 (IP, 5 days/week for 3 weeks) significantly inhibited the growth of A375 melanoma xenografts, with little reduction in the body weight of the mice,83 suggesting that it may be tolerable at the dose used, at least in mice. Although the initial intention was to inhibit SMAC binding to all IAPs, 4 dose-dependently inhibited the expression of survivin the most in a proteasome-dependent manner and its effect on other IAPs, including cIAP1/2 and XIAP, is relatively weaker.83 A docking analysis suggested that 4 might bind to the BIR domain of survivin,84 a proposal that needs experimental validation. Since cIAP1/2 and XIAP all have multiple BIR domains, it is intriguing that 4 is less effective to these proteins than survivin, which has only one BIR domain. It is also unclear how 4 binding to the BIR domain leads to proteasome-dependent survivin degradation.
Compound 5 (Figure 9F), a second generation of 4 analog which has an isopropyl group on benzene ring IV, showed an average of 4-fold increase in activity in inhibiting cancer cell proliferation and an increase in selectivity to survivin when compared to 4.18 Compound 5 at 20 and 40 mg/kg via IP injection (5 days/week for 3 weeks) was effective at inhibiting the A375 melanoma xenograft growth in mice.18 The third generation compound 6 (Figure 9G) had an average IC50 value of 1.4 μM in a variety of cancer cell lines and showed antitumor effects in an A375 melanoma xenograft model and orthotopic ovarian cancer model at 20 and 40 mg/kg via IP administration (3 days/week for 15 days).179
2.4.2. Medicinal Chemistry.
7-(Pyrrolidin-1-ylmethyl)-quinolin-8-ol has been shown to be the essential pharmacophore that is critical for anticancer effects, and any other structural modifications/changes on this scaffold led to reduced or lost bioactivity.178 The major structural modifications of 4 were on the ring IV and the linker between the ring IV and II (Figure 9) with key findings summarized in Table 7.
Table 7.
SARs Associated with Compound 4a
| position/scaffold | SAR |
|---|---|
|
| |
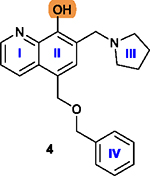
|
The free hydroxy group is essential and its removal leads to a loss of cytotoxicity.178 |
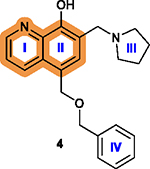
|
The quinolone ring is essential; Adding a halogen atom, replacing ring I with a benzene ring, or removing ring I significantly decreases cytotoxicity.178 |
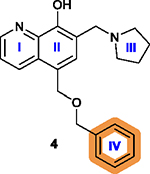
|
Introduction of an electron-withdrawing group
increases activity and the increase in the size of substituent from –F
to –Br generally increases the activity, although bulky substitutions
are unfavorable; Introduction of a hydrophobic group to the para-position is favorable;18 Replacing the isopropyl group with an azide group or an ethynyl group on the para-position is less favorable while a more lipophilic group is slightly more favorable;178 Replacing the phenyl ring IV with heterocyclic rings is less favorable.18 |
|
Replacing the benzyloxy with substituted indole is favorable, with monosubstitution better than di-substitution on the indole moiety and analogs substituted at the 5- or 6-position offering improved anticancer potency.84 |
|
Pyrrolidine is the best and most favorable among many substitutions.83 |
|
Increasing the length between the oxygen atom and
ring IV is not favorable;18 Replacing the oxygen atom of the linker with sulfur is favorable, while a methylamine linker is unfavorable;18,178 A simple methylene as a linker is favorable;84 Limited structural modifications to the linker are tolerated in maintaining the efficiency of disrupting protein-protein interactions.178 |
The highlighted elements are the points of discussion.
2.4.3. Key Structural Properties.
Compound 4 has two key structural properties. First, the quinoline ring is a prevalent scaffold among synthetic drug candidates and approved drugs,180 and from this perspective, 4 and its analogs are drug-like. Furthermore, 4 and its analogs appear to have higher cytotoxicity toward drug-resistant cancer cells that overexpress specific ABC transporters, including ABCB1, ABCC1, and ABCG2, which are known to cause resistance to anticancer drugs by actively exporting them from cells.181,182 Interestingly, 4, 5, 76, and 79 were all found to inhibit survival of ABCB1-overexpressing cancer cells ~3- to 10-fold more efficiently than cells without ABCB1 overexpression.84,178,179 However, no interactions between these compounds and ABCB1 have been demonstrated and the mechanism for this potential collateral sensitivity is unclear.
The second structural property that is worthy of discussion is the relatively low metabolic stability of 4 and 5, as compared to compound 6. The free and exposed benzene ring in 4 or the iso-propyl benzene in 5 usually undergoes oxidative reaction quickly in phase-I metabolism in vivo.183 Indeed, a recent stability study using human liver microsomes showed that 6 was metabolically more stable with a t1/2 of 1.5 h and clearance rate of 15.7 μL/min/mg than 5 with a t1/2 of 0.85 h and clearance rate of 136.6 μL/min/mg.179 It is noteworthy that both 4 and 5 require daily administration while 6 could be administered every other day (all three dosed at 20 and 40 mg/kg) for in vivo studies using melanoma A375 xenograft models, consistent with the notion that 6 is metabolically more stable and slower in clearance than 5, as discussed above. It is tempting to speculate that the trifluoromethyl group on the benzene ring as well as the triazole linker in 6 may contribute to its improved stability.
Furthermore, the core pharmacophore 7-(pyrrolidin-1-ylmethyl)quinolin-8-ol was synthesized via a Mannich reaction.179 It is possible that this moiety may undergo a retro-Mannich reaction, generating a reactive Michael acceptor that may cause stability issue or off-target effects. This speculation needs to be tested and, if proven true, medicinal chemistry work is necessary to stabilize it.
2.4.4. Perspectives.
Compound 4 appears to have all critical drug-like properties (Figure 10A) and no predicted major pitfalls including solubility and bioavailability were identified except the possible retro-Mannich reaction as discussed above. Structural modifications on the quinolone moiety and its substitutions without changing the R1 (a–f) are possible to further improve 5, 6, 76, 77, 79, and 80 (Figure 10B).
Figure 10.

Perspective of 4 and its analogs. (A) Radar Chart analysis of 4. The scale of cLog P and HBDs is equivalent to the original value multiplied by 100 and that of HBAs multiplied by 50. (B) Proposed modification/optimization of 4. Quinoxaline (UC-P1 series) or quinazoline (UC-P2 series) are used to replace quinolone and −NH2, −SH, or CN are used to replace −OH in R2. Residues in R1 (a–f) are adopted from 5, 6, 76, 77, 79, and 80.
First, quinolone may be replaced with either quinoxaline (UC-P1 series) or quinazoline (UC-P2 series), leaving R1 (a–f) of each compound unchanged (Figure 10B). Quinoxaline and quinazoline represent another two common moieties among approved drugs especially tyrosine kinase inhibitors (TKIs), including the epidermal growth factor receptor (EGFR) inhibitors, lapatinib, afatinib, gefitinib, dacomitinib, 74 (icotinib), and 75 (erlotinib).184,185 While quinoxaline and quinazoline possess similar physical and chemical properties, they each have one additional nitrogen atom than the quinoline in 4, which may lead to higher polarity and increase the solubility or drug-likeness.186 The replacement of quinoline with quinoxaline or quinazoline may also introduce additional activity in inhibiting ABC transporters since growing evidence supports that TKIs containing quinoxaline or quinazoline are capable of inhibiting ABCB1 or ABCG2, leading to sensitization to anticancer drugs that are substrates of these transporters.187,188
Second, the hydroxy at R2 may be replaced by bioisosteres such as HBD groups like NH2, SH, or an HBA group like CN (Figure 10B). Bioisosteres are a class of atoms or chemical groups with different structures but similar physical or chemical properties conferring similar biological activities.189,190 Generally, bioisosteric replacement is to enhance target-binding affinity, reduce toxicity and correct poor PK, or generate new chemical entities. Further, CN anchored here may also help prevent possible retro-Mannich reaction of the 7-(pyrrolidin-1-ylmethyl)quinolin-8-ol.
2.5. Compound 7 (LQZ-7F) and Its Analogs.
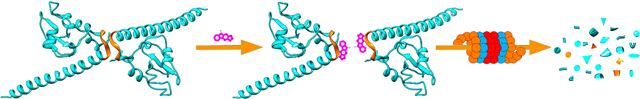
As discussed above, inhibiting survivin expression by targeting its upstream regulators such as transcription factor Sp1 or coactivator DDX5 do not selectively inhibit survivin expression. Inhibiting survivin interaction with its binding partners may also inhibit other IAPs as discussed above. Selective inhibition may limit to one-binding partner of survivin while spare other partners which bind to different sites on survivin. Thus, a specific strategy to eliminate survivin selectively is necessary to target this protein. In this regard, it was hypothesized that effective inhibition of homodimerization of survivin would expose its hydrophobic interface, similar to the exposure of the hydrophobic core in a globular protein during denaturation or misfolding, leading to recognition of the monomeric survivin as a misfolded protein by the cell quality control system and target it for destruction by the proteasome (Figure 11) and, thus, effectively eliminate survivin.20 However, unlike targeting heterodimerization of two different proteins, targeting homodimerization is more challenging since HTS assays such as the use of amplified luminescent proximity homogeneous assay (Alpha) are not readily applicable.
Figure 11.

Scheme of survivin dissociation and degradation in the proteasome. The two identical survivin subunits in a homodimer are shown in blue with the hydrophobic patch in the dimerization interface indicated in gold. The red dot represents inhibitors that bind to the hydrophobic dimerization interface, inhibiting dimerization, leading to destruction of survivin in proteasome.
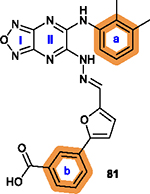
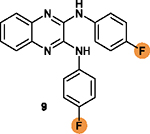
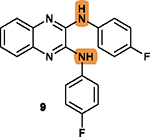
To overcome this challenge, a computer-based screen was used to target the homodimerization of survivin. Briefly, critical residues responsible for hydrophobic interactions between the two identical survivin subunits were studied using a computational analysis that identified Leu6, Pro7, Pro8, Ala9, Trp10, Phe93, Glu94, Glu95, Leu96, Thr97, Leu98, Gly99, Phe101, and Leu102 as potentially important residues.20 In silico screening of small molecules targeting these critical residues at the dimerization interface of survivin was performed with the top-scoring candidates assessed with a nondenaturing polyacrylamide gel electrophoresis (PAGE) assay of purified survivin.20 These studies led to the identification of a hit compound, 81 (LQZ-7, Figure 12A) that induced dissociation of the recombinant homodimeric survivin and inhibited homodimerization of nascent survivin newly synthesized in a cell-free system.20 It also induced proteasome-dependent survivin degradation, as expected. As shown in Figure 12B, several 81 analogs, 82 (LQZ-7A), 83 (LQZ-7B), 84 (LQZ-7C), 85 (LQZ-7D), 86 (LQZ-7E), and 7 (LQZ-7F), were also identified from the chemical library that had different activities in targeting survivin and inducing spontaneous apoptosis in prostate cancer cell lines (Table 8).20 Further medicinal chemistry study to optimize these inhibitors has also been attempted, leading to the identification of the two novel survivin inhibitors 8 (7F1, Figure 12B) and 9 (LQZ-7I, Figure 12C) that are very active in inducing survivin degradation, suppressing proliferation and inducing the spontaneous apoptosis of CRPC cells.19–21
Figure 12.

Medicinal chemistry associated with 81 (LQZ-7). (A, B) Chemical structures of the hit compound 81 and its six analogs including 7 and its hydrolysis product 8. (C) Chemical structures of 87 (LQZ-7G), 88 (LQZ-7H), 9 (LQZ-7I), 89 (LQZ-7J), and 90 (LQZ-7K) originated from 81. Labile hydrazone linker and other linkers are highlighted in cyan. 7 has a fused tetracyclic ring. 9 differs from other analogs 87–90 mainly by the substitution patterns on the two benzene rings.
Table 8.
Survivin Homodimerization Inhibitors
| interaction with survivin | |||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| compd | IC50 (μM) | apoptosis induction | binding | dimerization inhibition | degradation | synergism with docetaxel | in vivo activity |
|
|
|
|
|
|
|||
| 81 (LQZ-7) | 20 (PC-3) 25 (DU145) |
NDa | yes | yes | yes | ND | ND |
| 82 (LQZ-7A) | 4.8 (PC-3) 7.2 (DU145) |
ND | ND | ND | no | ND | ND |
| 83 (LQZ-7B) | 3.5 (PC-3) 3.3 (DU145 |
ND | ND | ND | yes | ND | ND |
| 84 (LQZ-7C) | 3.5 (PC-3) 3.5 (DU145) |
ND | ND | ND | yes | ND | ND |
| 85 (LQZ-7D) | 2.1 (PC-3) 2.6 (DU145) |
ND | ND | ND | no | ND | ND |
| 86 (LQZ-7E) | 49.8 (PC-3) 60.3 (DU145) |
ND | ND | ND | no | ND | ND |
| 7 (LQZ-7F) | 4.3 (PC-3) 1.9 (C4–2) |
yes | yes | yes | yes | yes | yes |
| 87 (LQZ-7G) | 20.7 (PC-3) 14.3 (C4–2) |
ND | ND | no | ND | ND | ND |
| 88 (LQZ-7H) | 19.7 (PC-3) 12.7 (C4–2) |
ND | ND | no | ND | ND | ND |
| 9 (LQZ-7I) | 4.8 (PC-3) 3.1 (C4–2) |
yes | ND | yes | yes | yes | yes |
| 89 (LQZ-7J) | 24.8 (PC-3) 17.6 (C4–2) |
ND | ND | no | ND | ND | ND |
| 90 (LQZ-7K) | 18.2 (PC-3) 18.6 (C4–2) |
ND | ND | no | ND | ND | ND |
| 8 (7F1) | 0.16 (PC-3) 0.17 (C4–2) |
yes | ND | yes | yes | yes | ND |
ND, not determined.
2.5.1. Biological Properties.
Compound 81 and its active derivatives inhibited homodimerization of survivin as determined using a nondenaturing PAGE analysis of purified recombinant proteins or using a mammalian two-hybrid assay.19–21 These inhibitors induced survivin degradation in proteasome and reduced survivin half-life from ~2–3 h to 25–50 min.19,20 Interestingly, the other members of the IAP family such as cIAP1/2 and XIAP were not significantly affected, suggesting that this class of inhibitors is selective for survivin. Furthermore, 7 with an amino group in its tail was immobilized using CNBr-activated Sepharose to pull down purified recombinant survivin while 81 was analyzed for its interaction with purified survivin using a fluorogenic assay, which demonstrated their direct binding to survivin.20 The affinity of 81 binding to survivin was estimated to be 0.24 μM (Kd) using the fluorogenic assay.20 The IC50 value for 81 toward the proliferation of prostate cancer cell lines DU145 and PC-3 was significantly improved from 20 to 25 μM to 1.9–4.3 μM for 7 (Table 8). The low potency of 81 compared with its active analogs was thought to be due to the presence of the carboxylic acid which may negatively impact cellular permeability.
The third-generation derivative 8, which expresses an IC50 value of ~160 nM toward CRPC cell lines, is much more potent than its predecessor 7 (Table 8).21 These inhibitors have also been shown to induce spontaneous apoptosis of prostate and other cancer cell lines and synergize with docetaxel in prostate cancer cells (Table 8).19–21 However, only 7 and 9 have been tested in vivo where, at 25 mg/kg (every 3 days for eight treatments, IP) and 100 mg/kg (every other day for ten treatments, oral), respectively, and they both significantly inhibited the growth of PC-3 xenograft tumors without any obvious toxicity (Table 8).19,20 However, the pharmacokinetics of these compounds have not been studied.
2.5.2. Medicinal Chemistry.
Among analogs 82 to 86, all have a similar range of IC50 values with the exception of 86, which has a much higher IC50 value than that of the parent 81 (Table 8). This observation suggests that a ring structure fused with benzene ring a as in 86 (Figure 12B) is unfavorable, and that the hydrophilic hydroxy group on benzene ring b of 81 is also unfavorable for cytotoxicity possibly due to its poor membrane permeability or low affinity toward the hydrophobic dimeric interface of survivin. It appears that nonpolar groups, including methyl, nitro, and halogens such as −Cl and −Br, on both benzene rings a/b are favorable (Figure 12A/B), which may direct further structural modification for optimization.
Molecular dynamic simulation analysis of 81 docked in the survivin dimerization interface suggested that benzene ring a and the secondary amine bond are two significant motifs that bind to the survivin dimeric interface engaging residues Glu94, Phe93, Phe101, and Leu98, while the furazanopyrazine ring I and II (Figure 12A) did not contribute significantly to survivin binding.20 Therefore, analogs were synthesized using a benzene ring to replace the furazan ring and using an amide (87 and 88) or secondary amine linker (9, 89, and 90) to replace the labile hydrazone linker (Figure 12C). Compound 9, a quinoxaline linked with two para-F benzyl rings via two secondary amine bonds, showed the most potent anticancer effect among the derivatives without the hydrazone linker.19
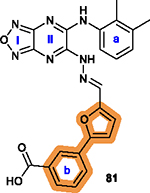
Because the hydrazone linker is labile and may undergo hydrolysis, 7 was thought to be a prodrug with its hydrolysis under acidic conditions mimicking that in the tumor microenvironment that may lead to the production of 8 (Figure 12B), which was tested for inhibition of survivin homodimerization. Interestingly, 8 was ~20-fold more potent than 7 in inhibiting PC-3 and C4–2 cells (Table 8).21 It also effectively inhibited survivin homodimerization, induced its degradation via the proteasome, and induced spontaneous apoptosis as expected.21 Furthermore, compounds 82 to 86 may be subject to redox activation due to the iminoquinone moiety. Thus, cautions should be practiced when further evaluating these compounds. The key SAR analyses of 81 and 9 are summarized in Table 9.
Table 9.
SARs Associated with Compounds 81 and 9a
| position/scaffold | SAR |
|---|---|
|
| |
|
Benzene rings a and b are tolerant of
modification with hydrophobic substituents, such as methyl, nitro, halogen or
carboxylate ester; Other fused ring structures in a are less favorable.20 |
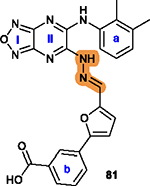
|
The hydrazone linker is unstable and may undergo
hydrolysis under acidic conditions;21 Its replacement with a secondary amine bond is generally favorable.19 |
|
The motif of phenylfuran is tolerable to be replaced.20 |
|
Two fluorine substituents are favorable for
cytotoxicity; Their replacement with electropositive substituent may decrease cytotoxicity;19 |
|
The NHs is necessary to form hydrogen bonds with
residues in the survivin dimeric interface; Its replacement with OH is less favorable.19 |
The highlighted elements are the points of discussion.
2.5.3. Key structural properties.
The survivin homodimerization inhibitors discussed above can be categorized into two groups of structurally different scaffolds, with 8 representative of one group and 9 the other. Compound 8 has a rigid structure with a locked tetracyclic ring system consisting of two 5-membered rings (I and III) and two 6-membered rings (II and IV in Figure 12B). While rigid structures are not rare among approved drugs, the absence of the flexibility provided by rotatable bonds and the deficiency of hydrophilic groups is usually problematic for solubility.191,192 Interestingly, the FDA-approved topoisomerase inhibitors 46 and doxorubicin both have rigid backbone structures but with hydrophilic tails to adjust their polarity and aqueous solubility. Indeed, 7 with a hydrophilic tail is more soluble than 8, and it was successfully administered via IP injection and validated to be effective in the prostate cancer cell PC-3 xenograft model.20 However, 8 was not studied in vivo due to low solubility.21 Thus, structural modifications of 8 to increase its solubility while maintaining its potency is desirable.
Compound 9 contains a quinoxaline ring, which is a prominent scaffold to inhibit ABC transporters as discussed above for the quinoline in 4 (Section 2.4.4). Compound 9 and its analogs may also possess similar biological activities to overcome MDR mediated by ABCB1 or ABCG2, warranting further evaluation. Interestingly, 9 was also predicted to be a substrate of ABCB1 following an analysis using an online tool that predicts drug-likeness and ADME parameters (http://www.swissadme.ch/). Further studies are necessary to determine if 9 and its analogs are substrates or inhibitors of ABC transporters and caution needs to be exercised during its development for potential ABC transporter-mediated resistance.
2.5.4. Perspectives.
Because 7 has an appropriate cLog P value of 3.92 (Figure 13A) and it has been successfully administered in an animal model, it has potential for further preclinical evaluation. On the other hand, 8 is more lipophilic (Figure 13B) with possible solubility issues due to its rigid structure as discussed above. Interestingly, 8 has a lower cLog P compared with 7, suggesting that its solubility could be easily improved. For example, the two nitrogen atoms in the pyridine with a pKa values of 1.4 and 1.1 for N1 and N2,193 respectively, could be used to convert 8 into organic salts using HCl or MSA (91 or 92, Figure 13D) without structural modification to increase its solubility. It is, however, noteworthy that both nitrogen atoms are weak bases and this approach may be difficult. Nevertheless, given its high potency and low molecular weight, 8 may serve as a lead compound for structural modification designed to increase its solubility as discussed below.
Figure 13.

Perspectives of compounds 7–9. (A–C) The radar charts of 7 (A), 8 (B) and 9 (C). (D–E) Proposed further structural modifications of 8.
Linking 8 with solubilizing moieties such as dimethylaminomethyl group (R1) may increase its polarity and allow facile transformation into organic salts using HCl or MSA (93, Figure 13E) with improved aqueous solubility.194 Additionally, introducing hydrophilic groups on the benzene ring, such as a carboxyl group (94) and its sodium salt (95) (Figure 13E) may also increase its solubility although such modifications may compromise membrane permeability.
It was shown previously that 7 can work as the prodrug of 8,21 thus, versatile solubilizing moieties can also be linked with 8 via either imine or hydrazone bond at the carbonyl group.
Although 9 is a lipophilic compound and has a cLog P of nearly 6 (Figure 13C), it was successfully solubilized in corn oil containing 10% DMSO and was successfully administered into mice via oral gavarge.19 Clearly, an optimized drug delivery system is required for this lipophilic compound. In addition, 9 has IC50 values ranging from 3 to 5 μM in prostate cancer cells. Thus, its potency needs to be improved. Considering that 9 has two strong electronegative F groups and that the other analogs of lower potency possess either one F or electro-positive groups (e.g., methylenedioxy in 88, methoxy in 89, or iso-butyl in 90), it remains to be determined if the two unsubstituted benzene rings or with other electronegative substituents could improve potency. A thorough and systematic SAR study is needed to evaluate optimal substituents on the two benzene rings. The potential modifications include substituents of electronegative groups, the types, position, and number of electronegative groups.
2.6. Compound 10 (LLP3) and Its Analogs.
In 2007, Abbott Laboratories conducted a nuclear magnetic resonance (NMR)- and affinity-based screening of their libraries for compounds that can bind to survivin and identified a hit compound 96 (Abbott 1), composed of three linked rings (I–III, Figure 14A).195 Compound 96 is insoluble and, thus, follow-up structural modifications to rings I and III were conducted, leading to the identification of 97 (Abbott 8, Figure 14B/C) which was shown to bind to the dimeric interface of survivin using NMR while a Kd of 75 μM was determined using an enzymatic assay.195
Figure 14.

Medicinal chemistry associated with 97 (Abbott 8). (A-B) Chemical structures of the hit compound 96 (Abbott 1), (A) and its analog 97 (B). (C) Functional groups linked to the two benzene rings on 96. (D) Hydrophilic moieties that are used to modify ring I. (E) The leading compound 10 (LLP3) and its analog 98 (LLP9). (F) A series of novel fused tricyclic ring analogs. (G–H) Six series of 97 analogs with different substitutions. Bn, benzyl. Key structural differences were highlighted.
A series of 97 analogs modified with NCGs as solubilizing moieties were subsequently synthesized and studied (Figure 14D), leading to the identification of several compounds with adequate water solubility and cell permeability.195 While some of these compounds possessed a high affinity for survivin with a Kd of ~40–90 nM in in vitro assays, cell-based anticancer activity and effect on survivin stability or in vivo efficacy have yet to be published.195 Using 96 and 97 as the starting scaffold, additional analogs linking various functional groups on the two benzene rings (I and II) were also synthesized and tested in cell-based assays, leading to the identification of two active compounds, including 10 (LLP3) and 98 (LLP9) (Figure 14E) with enhanced affinity toward the dimeric interface of survivin due to extended benzyloxy moieties.196,197 A series of new 97 analogs with a fused three-membered ring (Figure 14F) has also been synthesized and tested.198 However, no derivative from this round showed better binding affinity to survivin as determined by the ability to form a compound–survivin complex using recombinant L54 M mutant survivin. Recently, six series including 99–113 (4a-e, 5a-e, and 6a-e) and 11, 114–124, (4a′–d′, 5a′–d′ and 6a′–d′) of 97 analogs were reported in which the carbonyl group (C=O) was edited to either an amino (NH2) or thiocarbonyl (C=S) on ring II, as well as different modifications on ring I/III (Figure 14G/H).199,200 Of these compounds, 108 and 11 were found to be the most potent in suppressing cancer cell proliferation (see below).
2.6.1. Biological Properties.
Compounds 10 and 98 are potent in suppressing the mitosis and proliferation of prostate cancer PC-3 cells.196,197 Compounds 10 could also synergize with irinotecan in XIAP-associated factor 1 (XAF1)-positive but not p53-mutated cancer cells.201 However, no further details were provided on whether this synergism was due to targeting survivin.
Compound 108 was found to be the most effective among the 4a-6e series (Figure 14G) in suppressing cancer cell proliferation with IC50 values of 4.46 μM, 3.59 μM, and 6.01 μM for PC-3, MDA-MB-231, and HepG2 cells, respectively.199 In addition, 108 was about 17- to 29-fold more effective against these cancer cells than normal human fetal lung fibroblast WI-8 cells, suggesting a good therapeutic window.199 Compounds 99–103 possess different ring II elements when compared to 104–113, and they did not show obvious cytotoxicity at ~50 μM. With IC50 values of >100 μM for 99–101 and 103 and IC50 values ranging from 30 to 66 μM for 102 in PC-3, MDA-MB-231, and HepG2 cell lines, the anticancer potency of these compounds is poor. The IC50 values of 104–107 range from 14.4 to 52 μM and are generally more cytotoxic than 109–113 at 23 to 67 μM, suggesting that the carbonyl group on ring III is necessary for cytotoxicity. Compound 108, with IC50 values ranging from 3.59 to 6 μM, is most potent among all of the tested compounds in these three series and may be more potent than the approved drug 5-FU in these three cancer cell lines.199 Compound 108 reduced survivin expression, consistent with its disruption of survivin homodimerization as discussed above. However, no data have been provided on inhibition of survivin homodimerization or if the reduced survivin expression is due to proteasome-dependent degradation as discussed above for 7–9. Furthermore, unlike the compounds 7–9 of homodimerization inhibitors, 108 at 1–10 μM also downregulated the expression of XIAP, cIAP, and Livin, suggesting that it is unlikely a selective survivin homodimerization inhibitor.199
Interestingly, after removal of the fluorine substituent on the benzene ring, the resulting compounds 11, 114–116 (Figure 14H) showed 100-fold higher potency than 99–101, as revealed in another study.200 The most potent compound 11 possessed IC50 values of 0.42 μM, 0.66 μM and 1.22 μM, in PC-3, MDA-MB-231, and HepG2 cells, respectively, ~5- to 10-fold better than that of 108, and almost 100-fold more potent than 99.200 It is noteworthy that 11 has a much higher IC50 value of 193 μM for inhibiting normal WI-38 fibroblast cells, suggesting a good therapeutic window. Compound 11 also induced apoptosis, arrested cell cycle at G2/M and pre-G1 phases of PC-3 cells. Furthermore, 11 at 1, 5, and 10 μM downregulated survivin and Bcl-2 protein levels and induced cleavage of PARP and caspase 7, suggesting induction of apoptosis possibly via both survivin and Bcl-2.200 However, it is unclear if 11 has any effects on the expression of other IAPs and if reduction of Bcl-2 expression is indirect or an off-target effect. It also remains to be determined if 11 inhibits survivin homodimerization and whether that may contribute to 11-induced survivin loss. Furthermore, its antitumor effects remain to be validated using in vivo models.
Using NMR, 97 was found to interact with the dimeric interface of both subunits of survivin primarily via hydrophobic interaction between the ring III and Phe101 and Leu6 in one subunit and residue Leu102 and Leu98 in the other, and the ring I contacting the side chains of residues Leu96, Phe93, Val89, Leu104, and Phe86 in the same subunit which donates Phe101 and Leu6 to interaction with the ring III.195 If this prediction is true, then 97 will unlikely dissociate the survivin dimer due to its binding to both subunits; however, this binding may cause a conformational change in survivin that leads to its degradation.
The three rings on 10, including the core scaffold 3-cyano-2-pyridone and two benzyloxy groups, are predicted to form three strong π-stacking interactions with Phe86, Phe93, and Phe101 of survivin as determined by Autodock-Vina, with a docking score of −11.3 kcal/mol which is slightly lower than 98 with a score of −10.6 kcal/mol.197
2.6.2. Medicinal Chemistry.
Compound 97 derivatives constructed with various three-membered rings (Figure 14F) have been synthesized and tested.198 However, no derivative showed better binding affinity to survivin as determined by their ability in forming compound-survivin complexes using recombinant L54 M mutant survivin.198 Additional 97 analogs have been reported, but biological data have been described only for 108 and 11.199,200 Thus, we can only extract useful information from 10, 11, 99, and 108 for a critical SAR analysis of 97 as shown in Table 10.
Table 10.
SARs Associated with Compound 97a
| position/scaffold | SAR |
|---|---|
|
| |
|
The intact pyridone ring II and its carbonyl
group are necessary for anticancer activity; Replacement of the carbonyl with –NH2 or C=S is generally less favorable.199 |
|
Ring I is tolerant of modification; The position para- to the OH is more favorable to target survivin; Saturated 5- or 6-membered rings or benzene, benzyloxy ring substituents are favorable for binding affinity.195,199 |
|
Hydrophobic groups are
preferred; Electron-donating groups like methyl, methoxy groups are favorable;195,199 Benzyloxy (–OBn) on ring III is favorable.195,197 |
The highlighted elements are the points of discussion.
2.6.3. Key Structural Properties.
The relationship between the structures and related pharmacological study of the 97 series compounds are largely unavailable due to lack of consistent cell-based assays or direct evidence of on-target validation. However, 97 presented solubility issues195 and, thus, effort was made to introduce hydrophilic moieties into the molecule, as shown in Figure 14D.195 While compounds with higher binding affinity to survivin were found, no detailed physicochemical properties and biological activities of these derivatives, e.g., cytotoxicity and their effects on survivin, were reported.
The finding of significant improvement in potency from 99 to 11 by removal of the fluorine substituent is very intriguing. Usually in the structural modification of a hit compound, the addition of a fluorine substituent improves affinity and thereby bioactivity,202 as represented by 9,19 although there are also many examples of erosion due to the addition of a fluorine substituent.203 However, 11 with fluorine has a predicted binding energy of −8.5 kcal/mol,199,200 compared with −4.9 kcal/mol for 99 that has a fluorine199 as both determined using Molecular Operating Environment (MOE-Dock 2014.09). While this difference in binding energy requires practical validation, it suggests that 11 has a higher affinity toward survivin than 99. Upon examination of the docking results from these studies,199,200 we found that neither the fluorobenzyloxy in 99 nor the benzyloxy in 11 contributed significantly to survivin binding. Clearly, how the removal of fluorine generates such a major difference in cytotoxicity and whether this difference is due to off-target effect or a change in binding to survivin remains to be determined. Future studies to investigate these issues are likely important for further improving the selectivity and affinity to survivin.
It is also noteworthy that the predicted binding mode of 108 and 11 to the survivin dimeric interface differs from that of 10. While the interaction of 10 with survivin was predicted to involve mostly π-stacking,197 108 can engage in hydrogen bonding interactions with Thr97 and Leu98,199 and 11 with Glu40, Lys15, and Arg18.200 It has been predicted that four residues, Phe93, Glu94, Leu98, and Phe101, in the dimeric interface are essential for hydrophobic interactions between survivin and 108 and 11.199,200 These findings are consistent with that of the compounds 7–9 of dimerization inhibitors discussed above.20 However, structural studies of the interaction between these inhibitors and survivin is required to delineate the detailed binding mode at an atomic level.
2.6.4. Perspectives.
On the basis of the SAR information associated with 97, 108, and 11, it is reasonable to conclude that ring II is necessary for the activity and that the other two rings are tolerant of modification. Figure 15A–C shows that 97 could serve as a lead for further modification, including the addition of hydrophilic motif/solubilizing moieties to improve its solubility, and 11 appears to be a good candidate for further evaluation. However, 10, due to its high molecular weight (535.00) and unfavorable cLog P value (6.94), may have issues with solubility and PK profiles, making it a less favorable candidate for further study.
Figure 15.

Perspective of 97 and its analogs. (A–C) The Radar Charts for 97 (A), 10 (B), and 11 (C). (D) The proposed future structural optimization based on leading compounds 10, 11, and 108.
Therefore, we propose the synthesis of molecular hybrids of 10 with 108 and 11 using 11 as the core structure as described below. Molecular hybridization is a feasible and prevalent method in medicinal chemistry and drug design, which fuses two pharmacophores or key structure features of two different drugs into one molecule that usually possesses all merits of its parent drugs.204,205
First, 108 with a methoxy group is more potently cytotoxic than 104–107 with different substituent patterns on ring III. Ring III in 11, and 114–116 does not incorporate a methoxy group. Thus, we propose to add a methoxy group to ring III of 11 at different positions including para-(125), meta- (126) or ortho- (127) (Figure 15D). Similarly, other common electron-donating groups that have been used among approved drugs, such as −C(CH3)3 (128–130), or −N(CH3)2 (131–133), could also be considered.
Second, it is known that 10 with two benzyloxy groups on ring III is more potently cytotoxic than with either one or no benzyloxy group. Thus, adding two benzyloxy groups into ring III, generating 134, may increase the potency of 11.
It has been shown that Cl and OH substituents on ring I of 10 show higher cytotoxicity than those without these two substituent.195 However, in 11, no such substituents were linked on ring I. Thus, fusing the scaffold on ring I of 10 with 11 via introducing both Cl and OH to ring I, generating 135 or 136, may help further enhance the potency of 11.
Finally, because 11 has a high cLog P value, indicative of poor water solubility, transforming it into a salt with HCl (137) or MSA (138) may improve its solubility as discussed above.
3. SUMMARY AND FUTURE PERSPECTIVE
In this perspective, we have analyzed the medicinal chemistry of 1 to 11. Although 1 and 2 have been tested in phase I/II clinical studies, defining them as survivin inhibitors may be inappropriate and misleading because their true targets are not survivin nor do they selectively inhibit survivin expression. However, survivin may be used as a biomarker to determine their biological effects. Because of the unsuccessful outcome of the clinical trials with 1 or 2, it is tempting to speculate that targeting the ubiquitous transcription factors such as Sp1 may not be feasible in suppressing survivin expression for clinical therapy. One possible caveat is the simultaneous inhibition of other downstream target genes that may counteract the reduction of survivin expression and rescue cancer cells from survivin downregulation-induced apoptosis. Similarly, 3 is also a multitargeting compound that can downregulate survivin expression but does not bind directly to and inhibit survivin protein.
Compound 4, however, was identified using the crystal structure of SMAC-XIAP BIR3 domain (PDB 1G73),83 and it is reasonable to predict that 4 may directly bind to the survivin protein. However, it may also bind to the BIR domains of other IAP members. Indeed, it affects survivin expression and the expression of other IAPs although to a lesser extent compared to survivin. Compounds 7–9, however, were designed to target the hydrophobic dimeric interface of survivin to induce survivin degradation in the proteasome. They have been shown to bind directly to survivin and are selective for survivin without any effect on other IAPs. Therefore, these compounds are considered to be selective survivin inhibitors. Compounds 10 and 11 also fall into this category although it is unclear if their binding to the dimeric interface inhibits survivin dimerization and/or causes survivin degradation. Nevertheless, these inhibitors that target the dimeric interface of survivin identified using two different approaches deserve to be considered survivin inhibitor leads for more in-depth studies and development. Future studies focusing on the binding modes of these inhibitors to survivin at atomic level in structural studies in combination with medicinal chemistry optimization will likely lead to break-through findings that can assist further design and discovery of selective survivin inhibitors for development.
From the perspective of a medicinal chemist, 1 may be of little value for further laborious modification, but rather for minor changes as proposed above to improve its PK profile. One critical pitfall for 2 and its analog 37, other than promiscuity, is the poor quality as a lead compound. The simple structure, the paucity of active and functional groups, as well as a lack of feasible/available synthetic methodology for further modification render it a challenging candidate. Compound 3, a later-confirmed DDR, but not survivin, inhibitor, can be further improved in potency and solubility via medicinal chemistry optimization, as discussed above. Compound 6, a third generation 4 analog, appears to be an optimized lead for preclinical studies due to its improved potency and metabolic stability. Modifications to simplify the linker may help decrease the complexity and improve its potency. Finally, given that 8, 9 and 11 are direct survivin inhibitors, as discussed above, more medicinal chemistry effort to improve their potency, solubility, and drug-likeness along with a thorough and systematic SAR analysis of each functional group are necessary.
While targeting survivin transcription was initially thought to be appropriate if its promoter DNA sequence could be selectively targeted since survivin was considered “undruggable”, recent successes in targeting survivin dimerization or its interaction with its binding partners suggest that it is possible to develop inhibitors targeting the survivin protein directly. The approach in inhibiting survivin homodimerization leading to its degradation is interesting considering that the strategy in targeted protein degradation (TPD) including the technology of proteolysis-targeting chimeras (PROTAC)206,207 and molecular-glues208 are gaining rapid traction with nearly 50 TPD agents currently undergoing clinical evaluation.206,207 It is noteworthy that the advantage of the compounds 7–9 in targeting the homodimerization interface to induce survivin degradation is that it does not require any moiety to recruit E3 ligase to initiate proteasome-dependent degradation of survivin. Future studies in this direction are warranted to selectively induce degradation of homodimeric target proteins in general and inhibit survivin protein specifically for further development.
On the basis of the above critical evaluation of targeting survivin over the past two decades, we envision that the future discovery and development of survivin inhibitors may need to focus on in-depth systematic SAR study and structural optimization of the existing true survivin inhibitors, as described above. Other foci are envisioned to include deciphering detailed interactions between inhibitor(s) and survivin at atomic level and more biological and pharmacological studies of the roles and modes of action of survivin in cancer cells to assist drug discovery targeting survivin. Development of PROTACs that selectively induce survivin degradation is thought to be achievable and may open a new area of study. Finally, the successes in targeting survivin homodimerization may inspire similar studies using the same approach targeting other “undruggable” proteins that exist and function as homodimers.
As discussed above, while a few inhibitors targeting the survivin protein rather than its upstream transcription factors have been tested for their cytotoxicity and effect on survivin, only 7, 81, and 97 and its analogs have been shown to directly bind to survivin. However, no study has been conducted to investigate the detailed interaction between survivin and the inhibitors with the exception of 97. Thus, one of the future challenges in the discovery and development of survivin inhibitors is to generate atomic structures of survivin bound with these inhibitors using X-ray crystallography, Cryo-EM, and/or NMR. The structural information generated from these studies will likely help propel further optimization and guide optimization to more potent and selective survivin inhibitors.
There are also many open questions regarding the biology of survivin in its mechanism of action and specific residues involved in binding to its partners. The new information generated from these studies will likely assist design of appropriate assays to screen for survivin inhibitors and approaches for selectively targeting survivin. In addition, the survivin inhibitors discussed here may serve as chemical probes to facilitate decoding the biology of survivin function and its biochemistry, including its dimerization, stability, and interaction with other partners.
The finding that disrupting survivin homodimerization led to survivin destruction, as successfully shown using 7–9,20 suggests that it is reasonable to develop PROTAC or molecular glues that may eliminate the survivin protein more effectively. PROTAC or molecular glues act by hijacking a ubiquitin E3 ligase to mediate targeted protein degradation.209 These molecules have considerable potential to eliminate “undruggable” protein targets such as survivin.210,211 Many promising molecules generated using the PROTAC technology have been developed with about 50 currently in clinical trials.206,207,212–214 Given the availability of survivin-targeting small molecules, including 4 and 7 to 9, such a PROTAC targeting survivin is not out of reach in the near future.
The successful identification of 8, 9, and 4 may inspire studies on targeting survivin homodimerization and its interaction with binding partners. It may also inspire studies on other “undruggable” proteins that exist as homodimers and/or function by interacting with other proteins similarly to survivin. Because these “undruggable” proteins have no known enzymatic activity and lack active centers/pockets that could easily be targeted, inhibiting homodimerization to destabilize them or inhibiting their interaction with binding partners may represent a superior strategy to selectively eliminate them or disrupt their interaction with their partners. However, in many cases such as survivin, we lack an understanding of how these target proteins interact with their binding partners and whether they bind to different partners at different domains of the target protein. The compound that inhibits target protein binding to one partner may not inhibit its interaction with another. However, eliminating these target proteins by disrupting their homodimerization, as demonstrated here for survivin, will affect all downstream effects. It is also noteworthy that eliminating a target protein may result in compensatory changes in survival mechanisms of cancer cells, leading to resistance to such inhibitors. Considering that survivin is a member of the IAP family, eliminating survivin following long-term treatment with homodimerization inhibitors may result in resistance due to possible compensatory upregulation of other IAP family members.
HTS helps to facilitate the identification of hit compounds and may be used for targeting homodimerization or interaction of a target protein such as survivin with its binding partners. While such a HTS assay may be developed by adopting the Alpha assay which has been successful for inhibiting heterodimerization, it is very challenging to develop such a functional assay for HTS targeting homodimerization. Virtual screening to identify top-scoring homodimerization inhibitory compounds, which can then be used for testing using low throughput assays such as nondenaturing PAGE and mammalian two-hybrid assays, should be able to help overcome the lack of functional HTS assays as demonstrated for the discovery of 81.20
In conclusion, one of the major highlights among many drug-discovery lessons learned over the past half century is the difficulty in translating new targets into new drugs which are plagued with a high failure rate in every step including (1) identification of hit compounds, (2) structural modification and optimization, (3) cell- and animal-based PK/PD and therapeutic evaluation, (4) further structural modification for optimization, and (5) clinical trials.215 These challenges are further compounded when targeting an “undruggable” protein such as survivin. It is noteworthy that ~80% of the human proteome are “undruggable” despite the fact that many of them have been shown to be ideal targets using molecular approaches.216,217 Developing novel strategies targeting these proteins will likely result in breakthroughs for drug discovery and development. Different strategies attempted in targeting survivin protein or its expression regulators as discussed here provide valuable information and lessons learned on targeting “undruggable” proteins in general, which will help future drug discovery targeting other “undruggable” proteins with a true survivin inhibitor that will prevail in benefiting cancer patients in the foreseeable future.
ACKNOWLEDGMENTS
This work was supported in part by the NIH grant R01 GM127656 (JYL).
ABBREVIATIONS USED
- AKT
protein kinase B
- Alpha
amplified luminescent proximity homogeneous assay
- AML
acute myeloid leukemia
- Apaf-1
activating factor 1
- BBB
blood-brain barrier
- BIR
Baculovirus IAP Repeat
- BIRC
BIR containing
- BRUCE
BIR repeat-containing ubiquitin conjugating enzyme
- C
carbon atom
- CARD
caspase recruitment domain
- ccRCC
clear cell renal cell carcinoma
- cIAP
cellular IAP
- CDK1
cyclin-dependent kinase-1
- CIN
cervical intraepithelial neoplasia
- CML
chronic myeloid leukemia
- CPC
chromosomal passenger complex
- CRPC
castration-resistant prostate cancer
- DDX5
DEAD-box helicase 5
- DLBCL
diffuse large B-cell lymphoma
- EGFR
epidermal growth factor receptor
- GBM
glioblastoma
- γH2AX
γ-histone H2A
- HBA
H-bond acceptor
- HBD
H-bond donor
- HBXIP
hepatitis B X-interacting protein
- HCl
hydrochloric acid
- HPV
human papilloma virus
- hPSCs
human pluripotent stem cells
- IAPs
inhibitor of apoptosis proteins
- ILP2
IAP-like Protein 2
- ILF3
Interleukin enhancer-binding factor 3
- IND
Investigational New Drug
- IG
intragastric
- IP
intraperitoneal
- IV
intravenous
- LRR
leucine-rich repeat
- ML-IAP
melanoma IAP
- MDR
multidrug resistance
- MSA
methanesulfonic acid
- MTD
maximum tolerated dose
- N
nitrogen atom
- NACHT
nucleotide-binding and oligomerization
- NAIP
neuronal apoptosis inhibitory protein
- NCGs
nitrogen-containing groups
- NDGA
nordihydroguaiaretic acid
- NDH2
Type II NADH dehydrogenase
- NHL
non-Hodgkin’s lymphoma
- NMR
nuclear magnetic resonance
- NSCLC
nonsmall-cell lung cancer
- PAGE
polyacrylamide gel electrophoresis
- PAKs
p21-activated kinases
- PD
pharmacodynamics
- PDAC
pancreatic ductal adenocarcinoma
- PDX
patient-derived xenograft
- PHGs
phenolic NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells
- PK
pharmacokinetics
- PROTAC
proteolysis targeting chimera
- RING
Really Interesting New Gene
- RIPK2
receptor-interacting protein kinase 2
- rHGG
recurrent high-grade glioma
- RNS
reactive nitrogen species
- RO5
Rule of Five
- ROS
reactive oxygen species
- RRMM
relapsed/refractory multiple myeloma
- SAR
structure–activity relationship
- SLC35F2
solute carrier family 35 member F2
- SMAC/DIABLO
the second mitochondria-derived activator of caspases/direct IAP-binding protein with low pI
- STAT3
signal transducer and activator of transcription 3
- TKIs
tyrosine kinase inhibitors
- TPD
targeted protein degradation
- UBA
ubiquitin-associated domain
- UBC
ubiquitin-conjugating
- VEGF
vascular endothelial growth factors
- t 1/2
half-life
- XAF1
XIAP-associated factor 1
- XIAP
X-linked IAP
Biographies
Qingbin Cui received his Ph.D. in Medicinal Chemistry from Jinan University College of Pharmacy in 2014. Dr. Cui worked at Sun Yatsen University Cancer Center from 2014–2017 and then at St. John’s University from 2017–2019. In September 2019, Dr. Cui joined the University of Toledo College of Medicine and Life Sciences as a postdoctoral fellow and is recently appointed Research Assistant Professor in the Department of Cell and Cancer Biology. His research interests are in drug design, synthesis, and mechanism studies.
Caoqinglong Huang received his B.S. degree in Biological Science from University of Dayton in 2018, and currently a Ph.D. candidate of Cell and Cancer Biology at the University of Toledo College of Medicine and Life Sciences. His research focuses on drug discovery and development targeting survivin in prostate cancer.
Jing-Yuan Liu received her Ph.D. in Structure Biology from Indiana University School of Medicine in 2004. Dr. Liu worked as a postdoctoral fellow studying Computational Biology and Bioinformatics in the Department of Medical & Molecular Genetics followed by appointment as Assistant Professor of Computer and Information Science at Indiana University Purdue University of Indianapolis and of Pharmacology and Toxicology at Indiana University School of Medicine. She is currently Associate Professor of Medicine at the University of Toledo College of Medicine and Life Sciences. Her research mainly focuses on understanding the interactions between protein and protein or between protein and small molecules for drug discovery using bioinformatics and computational approaches and then validate those findings using biophysical and biochemical methods and/or patient data mining.
Jian-Ting Zhang obtained his Ph.D. in Molecular and Cell Biology from the State University of New York at Buffalo in 1989 followed by a postdoctoral training in Experimental Therapeutics at Ontario Cancer Institute/Princess Margret Hospital affiliated with the University of Toronto. He was appointed Assistant Professor of Physiology and Biophysics in 1993 at University of Texas Medical Branch and Associate Professor of Pharmacology and Toxicology in 1998 at Indiana University School of Medicine, and later promoted to Professor in 2003 and Andrew and Peggy Thompson Chair in Hematology/Oncology in 2007. Currently, Dr. Zhang is Professor and the Chair of the Department of Cell and Cancer Biology and the McMaster Endowed Chair in Biochemistry at the University of Toledo College of Medicine and Life Sciences. His research spans three major areas including drug resistance in cancer chemotherapy, drug discovery and pharmacology, and translational control in gene expression.
Footnotes
The authors declare no competing financial interest.
Contributor Information
Qingbin Cui, Department of Cell and Cancer Biology, University of Toledo College of Medicine and Life Sciences, Toledo, Ohio 43614, United States.
Caoqinglong Huang, Department of Cell and Cancer Biology, University of Toledo College of Medicine and Life Sciences, Toledo, Ohio 43614, United States.
Jing-Yuan Liu, Department of Medicine, University of Toledo College of Medicine and Life Sciences, Toledo, Ohio 43614, United States.
Jian-Ting Zhang, Department of Cell and Cancer Biology, University of Toledo College of Medicine and Life Sciences, Toledo, Ohio 43614, United States.
REFERENCES
- (1).Fulda S Molecular pathways: targeting inhibitor of apoptosis proteins in cancer-from molecular mechanism to therapeutic application. Clin. Cancer Res. 2014, 20, 289–295. [DOI] [PubMed] [Google Scholar]
- (2).Hay BA; Wassarman DA; Rubin GM Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell 1995, 83, 1253–1262. [DOI] [PubMed] [Google Scholar]
- (3).Salvesen GS; Duckett CS IAP proteins: blocking the road to death’s door. Nat. Rev. Mol. Cell Bio. 2002, 3, 401–410. [DOI] [PubMed] [Google Scholar]
- (4).Mace PD; Shirley S; Day CL Assembling the building blocks: structure and function of inhibitor of apoptosis proteins. Cell Death Differ. 2010, 17, 46–53. [DOI] [PubMed] [Google Scholar]
- (5).Morizane Y; Honda R; Fukami K; Yasuda H X-linked inhibitor of apoptosis functions as ubiquitin ligase toward mature caspase-9 and cytosolic Smac/DIABLO. J. Biochem. 2005, 137, 125–132. [DOI] [PubMed] [Google Scholar]
- (6).Eckelman BP; Salvesen GS; Scott FL Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. Embo Rep. 2006, 7, 988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sun H; Lu J; Liu L; Yang CY; Wang S Potent and selective small-molecule inhibitors of cIAP1/2 proteins reveal that the binding of Smac mimetics to XIAP BIR3 is not required for their effective induction of cell death in tumor cells. ACS Chem. Biol. 2014, 9, 994–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hennessy EJ; Adam A; Aquila BM; Castriotta LM; Cook D; Hattersley M; Hird AW; Huntington C; Kamhi VM; Laing NM; Li D; MacIntyre T; Omer CA; Oza V; Patterson T; Repik G; Rooney MT; Saeh JC; Sha L; Vasbinder MM; Wang H; Whitston D Discovery of a novel class of dimeric Smac mimetics as potent IAP antagonists resulting in a clinical candidate for the treatment of cancer (AZD5582). J. Med. Chem. 2013, 56, 9897–9919. [DOI] [PubMed] [Google Scholar]
- (9).DiPersio JF; Erba HP; Larson RA; Luger SM; Tallman MS; Brill JM; Vuagniaux G; Rouits E; Sorensen JM; Zanna C Oral Debio1143 (AT406), an antagonist of inhibitor of apoptosis proteins, combined with daunorubicin and cytarabine in patients with poor-risk acute myeloid leukemia-results of a phase I dose-escalation study. Cl Lymph Myelom Leuk 2015, 15, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mita MM; LoRusso PM; Papadopoulos KP; Gordon MS; Mita AC; Ferraldeschi R; Keer H; Oganesian A; Su XY; Jueliger S; Tolcher AW A phase I study of ASTX660, an antagonist of inhibitors of apoptosis proteins, in adults with advanced cancers or lymphoma. Clin. Cancer Res. 2020, 26, 2819–2826. [DOI] [PubMed] [Google Scholar]
- (11).Condon SM; Mitsuuchi Y; Deng Y; LaPorte MG; Rippin SR; Haimowitz T; Alexander MD; Kumar PT; Hendi MS; Lee YH; Benetatos CA; Yu G; Kapoor GS; Neiman E; Seipel ME; Burns JM; Graham MA; McKinlay MA; Li X; Wang J; Shi Y; Feltham R; Bettjeman B; Cumming MH; Vince JE; Khan N; Silke J; Day CL; Chunduru SK Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J. Med. Chem. 2014, 57, 3666–3677. [DOI] [PubMed] [Google Scholar]
- (12).Morita S; Minami H; Mitsuma A; Toyoda M; Kiyota N; Ando Y A phase I study of LCL161, a novel oral pan-inhibitor of apoptosis protein (IAP) antagonist, in Japanese patients with advanced solid tumors. Asia-Pac. J. Clin. Oncol. 2022, 18, No. e427–e434. [DOI] [PubMed] [Google Scholar]
- (13).Gu L; Zhang H; Liu T; Zhou S; Du Y; Xiong J; Yi S; Qu CK; Fu H; Zhou M Discovery of dual inhibitors of MDM2 and XIAP for cancer treatment. Cancer Cell 2016, 30, 623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Flygare JA; Beresini M; Budha N; Chan H; Chan IT; Cheeti S; Cohen F; Deshayes K; Doerner K; Eckhardt SG; Elliott LO; Feng B; Franklin MC; Reisner SF; Gazzard L; Halladay J; Hymowitz SG; La H; LoRusso P; Maurer B; Murray L; Plise E; Quan C; Stephan JP; Young SG; Tom J; Tsui V; Um J; Varfolomeev E; Vucic D; Wagner AJ; Wallweber HJ; Wang L; Ware J; Wen Z; Wong H; Wong JM; Wong M; Wong S; Yu R; Zobel K; Fairbrother WJ Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). J. Med. Chem. 2012, 55, 4101–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kelly RJ; Thomas A; Rajan A; Chun G; Lopez-Chavez A; Szabo E; Spencer S; Carter CA; Guha U; Khozin S; Poondru S; Van Sant C; Keating A; Steinberg SM; Figg W; Giaccone G A phase I/II study of sepantronium bromide (YM155, survivin suppressor) with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2013, 24, 2601–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Grossman SA; Ye X; Peereboom D; Rosenfeld MR; Mikkelsen T; Supko JG; Desideri S Phase I study of terameprocol in patients with recurrent high-grade glioma. Neuro-Oncology 2012, 14, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Holthof LC; van der Horst HJ; van Hal-van Veen SE; Ruiter RWJ; Li F; Buijze M; Andersen MN; Yuan H; de Bruijn J; van de Donk NWCJ; Lokhorst HM; Zweegman S; Groen RWJ; Mutis T Preclinical evidence for an effective therapeutic activity of FL118, a novel survivin inhibitor, in patients with relapsed/refractory multiple myeloma. Haematologica 2020, 105, No. e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Xiao M; Wang J; Lin Z; Lu Y; Li Z; White SW; Miller DD; Li W Design, synthesis and structure-activity relationship studies of novel survivin inhibitors with potent anti-proliferative properties. PLoS One 2015, 10, No. e129807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Peery R; Kyei-Baffour K; Dong Z; Liu J; de Andrade Horn P; Dai M; Liu J-Y; Zhang J-T Synthesis and identification of a novel lead targeting survivin dimerization for proteasome-dependent degradation. J. Med. Chem. 2020, 63, 7243–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Qi J; Dong Z; Liu J; Peery RC; Zhang S; Liu JY; Zhang JT Effective targeting of the survivin dimerization interface with small-molecule inhibitors. Cancer Res. 2016, 76, 453–462. [DOI] [PubMed] [Google Scholar]
- (21).Peery R; Cui Q; Kyei-Baffour K; Josephraj S; Huang C; Dong Z; Dai M; Zhang JT; Liu JY A novel survivin dimerization inhibitor without a labile hydrazone linker induces spontaneous apoptosis and synergizes with docetaxel in prostate cancer cells. Bioorgan. Med. Chem. 2022, 65, 116761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cong H; Xu L; Wu Y; Qu Z; Bian T; Zhang W; Xing C; Zhuang C Inhibitor of Apoptosis Protein (IAP) antagonists in anticancer agent discovery: current status and perspectives. J. Med. Chem. 2019, 62, 5750–5772. [DOI] [PubMed] [Google Scholar]
- (23).Sugihara E; Hashimoto N; Osuka S; Shimizu T; Ueno S; Okazaki S; Yaguchi T; Kawakami Y; Kosaki K; Sato TA; Okamoto S; Saya H The Inhibitor of Apoptosis Protein Livin confers resistance to Fas-mediated immune cytotoxicity in refractory lymphoma. Cancer Res. 2020, 80, 4439–4450. [DOI] [PubMed] [Google Scholar]
- (24).de Almagro MC; Vucic D The inhibitor of apoptosis (IAP) proteins are critical regulators of signaling pathways and targets for anti-cancer therapy. Exp Oncol 2012, 34, 200–211. [PubMed] [Google Scholar]
- (25).Peery RC; Liu JY; Zhang JT Targeting survivin for therapeutic discovery: past, present, and future promises. Drug Discovery Today 2017, 22, 1466–1477. [DOI] [PubMed] [Google Scholar]
- (26).Verdecia MA; Huang H; Dutil E; Kaiser DA; Hunter T; Noel JP Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat. Struct. Biol. 2000, 7, 602–608. [DOI] [PubMed] [Google Scholar]
- (27).Vischioni B; van der Valk P; Span SW; Kruyt FA; Rodriguez JA; Giaccone G Expression and localization of inhibitor of apoptosis proteins in normal human tissues. Hum. Pathol. 2006, 37, 78–86. [DOI] [PubMed] [Google Scholar]
- (28).Fortugno P; Wall NR; Giodini A; O’Connor DS; Plescia J; Padgett KM; Tognin S; Marchisio PC; Altieri DC Survivin exists in immunochemically distinct subcellular pools and is involved in spindle microtubule function. J. Cell Sci. 2002, 115, 575–585. [DOI] [PubMed] [Google Scholar]
- (29).Temme A; Rieger M; Reber F; Lindemann D; Weigle B; Diestelkoetter-Bachert P; Ehninger G; Tatsuka M; Terada Y; Rieber EP Localization, dynamics, and function of survivin revealed by expression of functional survivinDsRed fusion proteins in the living cell. Mol. Biol. Cell 2003, 14, 78–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wall N; Khan S; Ferguson Bennit H; Asuncion Valenzuela MM; Turay D; Diaz Osterman C; Moyron R; Esebanmen G; Ashok A Localization and upregulation of survivin in cancer health disparities: a clinical perspective. Biol-Targets Ther 2015, 9, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Small S; Keerthivasan G; Huang Z; Gurbuxani S; Crispino JD Overexpression of survivin initiates hematologic malignancies in vivo. Leukemia 2010, 24, 1920–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lorenzetti MA; Mosna MJ; De Matteo EN; Garcia Lombardi M; Colli SL; Preciado MV Overexpression of survivin in pediatric Hodgkin lymphoma tumor cells: Characterization of protein expression and splice-variants transcription profile. Exp. Mol. Pathol. 2019, 108, 24–31. [DOI] [PubMed] [Google Scholar]
- (33).Du C; Fang M; Li Y; Li L; Wang X Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [DOI] [PubMed] [Google Scholar]
- (34).Ceballos-Cancino G; Espinosa M; Maldonado V; Melendez-Zajgla J Regulation of mitochondrial Smac/DIABLO-selective release by survivin. Oncogene 2007, 26, 7569–7575. [DOI] [PubMed] [Google Scholar]
- (35).Song Z; Yao X; Wu M Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J. Biol. Chem. 2003, 278, 23130–23140. [DOI] [PubMed] [Google Scholar]
- (36).Shin S; Sung BJ; Cho YS; Kim HJ; Ha NC; Hwang JI; Chung CW; Jung YK; Oh BH An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and –7. Biochemistry-Us 2001, 40, 1117–1123. [DOI] [PubMed] [Google Scholar]
- (37).Li C; Wu Z; Liu M; Pazgier M; Lu W Chemically synthesized human survivin does not inhibit caspase-3. Protein Sci. 2008, 17, 1624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Banks DP; Plescia J; Altieri DC; Chen J; Rosenberg SH; Zhang H; Ng SC Survivin does not inhibit caspase-3 activity. Blood 2000, 96, 4002–4003. [PubMed] [Google Scholar]
- (39).O’Connor DS; Grossman D; Plescia J; Li F; Zhang H; Villa A; Tognin S; Marchisio PC; Altieri DC Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. P. Natl. Acad. Sci. Usa 2000, 97, 13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Marusawa H; Matsuzawa S; Welsh K; Zou H; Armstrong R; Tamm I; Reed JC HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003, 22, 2729–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Dohi T; Okada K; Xia F; Wilford CE; Samuel T; Welsh K; Marusawa H; Zou H; Armstrong R; Matsuzawa S; Salvesen GS; Reed JC; Altieri DC An IAP-IAP complex inhibits apoptosis. J. Biol. Chem. 2004, 279, 34087–34090. [DOI] [PubMed] [Google Scholar]
- (42).Eckelman BP; Salvesen GS The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J. Biol. Chem. 2006, 281, 3254–3260. [DOI] [PubMed] [Google Scholar]
- (43).Chai J; Shiozaki E; Srinivasula SM; Wu Q; Dataa P; Alnemri ES; Shi Y Structural basis of caspase-7 inhibition by XIAP. Cell 2001, 104, 769–780. [DOI] [PubMed] [Google Scholar]
- (44).Riedl SJ; Renatus M; Schwarzenbacher R; Zhou Q; Sun C; Fesik SW; Liddington RC; Salvesen GS Structural basis for the inhibition of caspase-3 by XIAP. Cell 2001, 104, 791–800. [DOI] [PubMed] [Google Scholar]
- (45).Mita AC; Mita MM; Nawrocki ST; Giles FJ Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [DOI] [PubMed] [Google Scholar]
- (46).Carmena M; Wheelock M; Funabiki H; Earnshaw WC The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Bio. 2012, 13, 789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Singh A; Spitzer MH; Joy JP; Kaileh M; Qiu X; Nolan GP; Sen R Postmitotic G1 phase survivin drives mitogen-independent cell division of B lymphocytes. P. Natl. Acad. Sci. Usa 2022, 119, No. e2115567119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Brown M; Zhang W; Yan D; Kenath R; Le L; Wang H; Delitto D; Ostrov D; Robertson K; Liu C; Pham K The role of survivin in the progression of pancreatic ductal adenocarcinoma (PDAC) and a novel survivin-targeted therapeutic for PDAC. PLoS One 2020, 15, No. e226917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).He X; Yang K; Wang H; Chen X; Wu H; Yao L; Ma S Expression and clinical significance of survivin in ovarian cancer: A meta-analysis. PLoS One 2018, 13, No. e194463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Groner B; Weiss A Targeting survivin in cancer: novel drug development approaches. Biodrugs 2014, 28, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ryan B; O’Donovan N; Browne B; O’Shea C; Crown J; Hill AD; McDermott E; O’Higgins N; Duffy MJ Expression of survivin and its splice variants survivin-2B and survivin-DeltaEx3 in breast cancer. Br. J. Cancer 2005, 92, 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hennigs JK; Minner S; Tennstedt P; Loser R; Huland H; Klose H; Graefen M; Schlomm T; Sauter G; Bokemeyer C; Honecker F Subcellular compartmentalization of survivin is associated with biological aggressiveness and prognosis in prostate cancer. Sci. Rep-Uk 2020, 10, 3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Grdina DJ; Murley JS; Miller RC; Mauceri HJ; Sutton HG; Li JJ; Woloschak GE; Weichselbaum RR A surviving-associated adaptive response in radiation therapy. Cancer Res. 2013, 73, 4418–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhang M; Latham DE; Delaney MA; Chakravarti A Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene 2005, 24, 2474–2482. [DOI] [PubMed] [Google Scholar]
- (55).Zaffaroni N; Pennati M; Colella G; Perego P; Supino R; Gatti L; Pilotti S; Zunino F; Daidone MG Expression of the anti-apoptotic gene survivin correlates with taxol resistance in human ovarian cancer. Cell. Mol. Life Sci. 2002, 59, 1406–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Wang P; Zhen H; Zhang J; Zhang W; Zhang R; Cheng X; Guo G; Mao X; Wang J; Zhang X Survivin promotes glioma angiogenesis through vascular endothelial growth factor and basic fibroblast growth factor in vitro and in vivo. Mol. Carcinogen. 2012, 51, 586–595. [DOI] [PubMed] [Google Scholar]
- (57).Li QX; Zhao J; Liu JY; Jia LT; Huang HY; Xu YM; Zhang Y; Zhang R; Wang CJ; Yao LB; Chen SY; Yang AG Survivin stable knockdown by siRNA inhibits tumor cell growth and angiogenesis in breast and cervical cancers. Cancer Biol. Ther. 2006, 5, 860–866. [DOI] [PubMed] [Google Scholar]
- (58).Vivas-Mejia PE; Rodriguez-Aguayo C; Han HD; Shahzad MM; Valiyeva F; Shibayama M; Chavez-Reyes A; Sood AK; Lopez-Berestein G Silencing survivin splice variant 2B leads to antitumor activity in taxane-resistant ovarian cancer. Clin. Cancer Res. 2011, 17, 3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Pennati M; Binda M; De Cesare M; Pratesi G; Folini M; Citti L; Daidone MG; Zunino F; Zaffaroni N Ribozyme-mediated down-regulation of survivin expression sensitizes human melanoma cells to topotecan in vitro and in vivo. Carcinogenesis 2004, 25, 1129–1136. [DOI] [PubMed] [Google Scholar]
- (60).Li F; Aljahdali I; Ling X Cancer therapeutics using survivin BIRC5 as a target: what can we do after over two decades of study? J. Exp. Clin. Canc. Res. 2019, 38, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Shimizu T; Nishio K; Sakai K; Okamoto I; Okamoto K; Takeda M; Morishita M; Nakagawa K Phase I safety and pharmacokinetic study of YM155, a potent selective survivin inhibitor, in combination with erlotinib in patients with EGFR TKI refractory advanced non-small cell lung cancer. Cancer Chemoth. Pharm. 2020, 86, 211. [DOI] [PubMed] [Google Scholar]
- (62).Grossman SA; Ye X; Peereboom D; Rosenfeld MR; Mikkelsen T; Supko JG; Desideri S Phase I study of terameprocol in patients with recurrent high-grade glioma. Neuro-Oncology 2012, 14, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Sarvagalla S; Cheung CHA; Tsai J; Hsieh HP; Coumar MS Disruption of protein-protein interactions: hot spot detection, structure-based virtual screening and in vitro testing for the anti-cancer drug target - survivin. Rsc Adv. 2016, 6, 31947–31959. [Google Scholar]
- (64).Gupta V; Samuleson CG; Su S; Chen TC Nelfinavir potentiation of imatinib cytotoxicity in meningioma cells via survivin inhibition. Neurosurg Focus 2007, 23, No. E9. [DOI] [PubMed] [Google Scholar]
- (65).Shi X; Wang D; Ding K; Lu Z; Jin Y; Zhang J; Pan J GDP366, a novel small molecule dual inhibitor of survivin and Op18, induces cell growth inhibition, cellular senescence and mitotic catastrophe in human cancer cells. Cancer Biol. Ther. 2010, 9, 640–650. [DOI] [PubMed] [Google Scholar]
- (66).Wadegaonkar VP; Wadegaonkar PA Withanone as an inhibitor of survivin: a potential drug candidate for cancer therapy. J. Biotechnol. 2013, 168, 229–233. [DOI] [PubMed] [Google Scholar]
- (67).Berezov A; Cai Z; Freudenberg JA; Zhang H; Cheng X; Thompson T; Murali R; Greene MI; Wang Q Disabling the mitotic spindle and tumor growth by targeting a cavity-induced allosteric site of survivin. Oncogene 2012, 31, 1938–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Felix S; Sandjo LP; Opatz T; Erkel G SF002–96–1, a new drimane sesquiterpene lactone from an Aspergillus species, inhibits survivin expression. Beilstein J. Org. Chem. 2013, 9, 2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Quispe PA; Lavecchia MJ; Leon IE On the discovery of a potential survivin inhibitor combining computational tools and cytotoxicity studies. Heliyon 2019, 5, No. e2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).West TJ; Bi J; Martinez-Pena F; Curtis EJ; Gazaniga NR; Mischel PS; Lairson LL A cell type selective YM155 prodrug targets receptor-interacting protein kinase 2 to induce brain cancer cell death. J. Am. Chem. Soc. 2023, 145 (15), 8355–8363, DOI: 10.1021/jacs.2c11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Cheng Q; Ling X; Haller A; Nakahara T; Yamanaka K; Kita A; Koutoku H; Takeuchi M; Brattain MG; Li F Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. Int. J. Biochem Mol. Biol. 2012, 3, 179–197. [PMC free article] [PubMed] [Google Scholar]
- (72).Yamauchi T; Nakamura N; Hiramoto M; Yuri M; Yokota H; Naitou M; Takeuchi M; Yamanaka K; Kita A; Nakahara T; Kinoyama I; Matsuhisa A; Kaneko N; Koutoku H; Sasamata M; Kobori M; Katou M; Tawara S; Kawabata S; Furuichi K Sepantronium bromide (YM155) induces disruption of the ILF3/p54(nrb) complex, which is required for survivin expression. Biochem. Bioph. Res. Co. 2012, 425, 711–716. [DOI] [PubMed] [Google Scholar]
- (73).Winter GE; Radic B; Mayor-Ruiz C; Blomen VA; Trefzer C; Kandasamy RK; Huber K; Gridling M; Chen D; Klampfl T; Kralovics R; Kubicek S; Fernandez-Capetillo O; Brummelkamp TR; Superti-Furga G The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity. Nat. Chem. Biol. 2014, 10, 768–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Nakahara T; Takeuchi M; Kinoyama I; Minematsu T; Shirasuna K; Matsuhisa A; Kita A; Tominaga F; Yamanaka K; Kudoh M; Sasamata M YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007, 67, 8014–8021. [DOI] [PubMed] [Google Scholar]
- (75).Voges Y; Michaelis M; Rothweiler F; Schaller T; Schneider C; Politt K; Mernberger M; Nist A; Stiewe T; Wass MN; Rodel F; Cinatl J Effects of YM155 on survivin levels and viability in neuroblastoma cells with acquired drug resistance. Cell Death Dis 2016, 7, No. e2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Sun Y; Giacalone NJ; Lu B Terameprocol (tetra-O-methyl nordihydroguaiaretic acid), an inhibitor of Sp1-mediated survivin transcription, induces radiosensitization in non-small cell lung carcinoma. J. Thorac Oncol 2011, 6, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Meyers RO Anticancer structure-activity relationships of semisynthetic alalogs of nordihydroguaiaretic acid. The University of Arizona, 2005. [Google Scholar]
- (78).Ling X; Cao S; Cheng Q; Keefe JT; Rustum YM; Li F A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS One 2012, 7, No. e45571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Ling X; Wu W; Aljahdali I; Liao J; Santha S; Fountzilas C; Boland PM; Li F FL118, acting as a ‘molecular glue degrader’, binds to dephosphorylates and degrades the oncoprotein DDX5 (p68) to control c-Myc, survivin and mutant Kras against colorectal and pancreatic cancer with high efficacy. Clin Transl Med. 2022, 12, No. e881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Westover D; Ling X; Lam H; Welch J; Jin C; Gongora C; Del Rio M; Wani M; Li F FL118, a novel camptothecin derivative, is insensitive to ABCG2 expression and shows improved efficacy in comparison with irinotecan in colon and lung cancer models with ABCG2-induced resistance. Mol. Cancer 2015, 14, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Ling X; Liu X; Zhong K; Smith N; Prey J; Li F FL118, a novel camptothecin analogue, overcomes irinotecan and topotecan resistance in human tumor xenograft models. Am. J. Transl Res. 2015, 7, 1765–1781. [PMC free article] [PubMed] [Google Scholar]
- (82).Zhang G; Yin R; Dai X; Wu G; Qi X; Yu R; Li J; Jiang T Design, synthesis, and biological evaluation of novel 7-substituted 10,11-methylenedioxy-camptothecin derivatives against drug-resistant small-cell lung cancer in vitro and in vivo. Eur. J. Med. Chem. 2022, 241, 114610. [DOI] [PubMed] [Google Scholar]
- (83).Wang J; Li W Discovery of novel second mitochondria-derived activator of caspase mimetics as selective inhibitor of apoptosis protein inhibitors. J. Pharmacol. Exp. Ther. 2014, 349, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Wang Q; Arnst KE; Xue Y; Lei ZN; Ma D; Chen ZS; Miller DD; Li W Synthesis and biological evaluation of indole-based UC-112 analogs as potent and selective survivin inhibitors. Eur. J. Med. Chem. 2018, 149, 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Guvenc H; Pavlyukov MS; Joshi K; Kurt H; Banasavadi-Siddegowda YK; Mao P; Hong C; Yamada R; Kwon CH; Bhasin D; Chettiar S; Kitange G; Park IH; Sarkaria JN; Li C; Shakhparonov MI; Nakano I Impairment of glioma stem cell survival and growth by a novel inhibitor for Survivin-Ran protein complex. Clin. Cancer Res. 2013, 19, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Lipinski CA Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Tox. Met. 2000, 44, 235–249. [DOI] [PubMed] [Google Scholar]
- (87).Kenny PW; Montanari CA Inflation of correlation in the pursuit of drug-likeness. J. Comput. Aid. Mol. Des. 2013, 27, 1–13. [DOI] [PubMed] [Google Scholar]
- (88).Ghadimi MP; Young ED; Belousov R; Zhang Y; Lopez G; Lusby K; Kivlin C; Demicco EG; Creighton CJ; Lazar AJ; Pollock RE; Lev D Survivin is a viable target for the treatment of malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2012, 18, 2545–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Yamanaka K; Nakahara T; Yamauchi T; Kita A; Takeuchi M; Kiyonaga F; Kaneko N; Sasamata M Antitumor activity of YM155, a selective small-molecule survivin suppressant, alone and in combination with docetaxel in human malignant melanoma models. Clin. Cancer Res. 2011, 17, 5423–5431. [DOI] [PubMed] [Google Scholar]
- (90).Yoon DH; Shin JS; Jin DH; Hong SW; Jung KA; Kim SM; Hong YS; Kim KP; Lee JL; Suh C; Lee JS; Kim TW The survivin suppressant YM155 potentiates chemosensitivity to gemcitabine in the human pancreatic cancer cell line MiaPaCa-2. Anticancer Res. 2012, 32, 1681–1688. [PubMed] [Google Scholar]
- (91).Nakahara T; Kita A; Yamanaka K; Mori M; Amino N; Takeuchi M; Tominaga F; Kinoyama I; Matsuhisa A; Kudou M; Sasamata M Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci. 2011, 102, 614–621. [DOI] [PubMed] [Google Scholar]
- (92).Tolcher AW; Mita A; Lewis LD; Garrett CR; Till E; Daud AI; Patnaik A; Papadopoulos K; Takimoto C; Bartels P; Keating A; Antonia S Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J. Clin. Oncol. 2008, 26, 5198–5203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Giaccone G; Zatloukal P; Roubec J; Floor K; Musil J; Kuta M; van Klaveren RJ; Chaudhary S; Gunther A; Shamsili S Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J. Clin. Oncol. 2009, 27, 4481–4486. [DOI] [PubMed] [Google Scholar]
- (94).Tolcher AW; Quinn DI; Ferrari A; Ahmann F; Giaccone G; Drake T; Keating A; de Bono JS A phase II study of YM155, a novel small-molecule suppressor of survivin, in castration-resistant taxane-pretreated prostate cancer. Ann. Oncol. 2012, 23, 968–973. [DOI] [PubMed] [Google Scholar]
- (95).Park R; Chang CC; Liang YC; Chung Y; Henry RA; Lin E; Mold DE; Huang RC Systemic treatment with tetra-O-methyl nordihydroguaiaretic acid suppresses the growth of human xenograft tumors. Clin. Cancer Res. 2005, 11, 4601–4609. [DOI] [PubMed] [Google Scholar]
- (96).Aoyama Y; Nishimura T; Sawamoto T; Satoh T; Katashima M; Nakagawa K Pharmacokinetics of sepantronium bromide (YM155), a small-molecule suppressor of survivin, in Japanese patients with advanced solid tumors: dose proportionality and influence of renal impairment. Cancer Chemoth. Pharm. 2012, 70, 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Cheng Q; Ling X; Haller A; Nakahara T; Yamanaka K; Kita A; Koutoku H; Takeuchi M; Brattain MG; Li F Suppression of survivin promoter activity by YM155 involves disruption of Sp1-DNA interaction in the survivin core promoter. Int. J. Biochem Mol. Biol. 2012, 3, 179–197. [PMC free article] [PubMed] [Google Scholar]
- (98).Glaros TG; Stockwin LH; Mullendore ME; Smith B; Morrison BL; Newton DL The ″survivin suppressants″ NSC 80467 and YM155 induce a DNA damage response. Cancer Chemoth. Pharm. 2012, 70, 207–212. [DOI] [PubMed] [Google Scholar]
- (99).Rodel F; Hoffmann J; Distel L; Herrmann M; Noisternig T; Papadopoulos T; Sauer R; Rodel C Survivin as a radio-resistance factor, and prognostic and therapeutic target for radio-therapy in rectal cancer. Cancer Res. 2005, 65, 4881–4887. [DOI] [PubMed] [Google Scholar]
- (100).Iwasa T; Okamoto I; Suzuki M; Nakahara T; Yamanaka K; Hatashita E; Yamada Y; Fukuoka M; Ono K; Nakagawa K Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in non-small cell lung cancer cell lines. Clin. Cancer Res. 2008, 14, 6496–6504. [DOI] [PubMed] [Google Scholar]
- (101).Lucki NC; Villa GR; Vergani N; Bollong MJ; Beyer BA; Lee JW; Anglin JL; Spangenberg SH; Chin EN; Sharma A; Johnson K; Sander PN; Gordon P; Skirboll SL; Wurdak H; Schultz PG; Mischel PS; Lairson LL A cell type-selective apoptosis-inducing small molecule for the treatment of brain cancer. P. Natl. Acad. Sci. Usa 2019, 116, 6435–6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Matsuhisa A; Kinoyama I; Yoyoshima A; Nakahara Y; Takeuchi M; Okada M Fused imidazolium derivatives. A1, 2–14–2001, 2003.
- (103).Go YH; Lim C; Jeong HC; Kwon OS; Chung S; Lee H; Kim W; Suh YG; Son WS; Lee MO; Cha HJ; Kim SH Structure-activity relationship analysis of YM155 for inducing selective cell death of human pluripotent stem cells. Front Chem. 2019, 7, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Ho SH; Sim MY; Yee WL; Yang T; Yuen SP; Go ML Antiproliferative, DNA intercalation and redox cycling activities of dioxonaphtho[2,3-d]imidazolium analogs of YM155: A structure-activity relationship study. Eur. J. Med. Chem. 2015, 104, 42–56. [DOI] [PubMed] [Google Scholar]
- (105).Lamers F; Schild L; Koster J; Versteeg R; Caron HN; Molenaar JJ Targeted BIRC5 silencing using YM155 causes cell death in neuroblastoma cells with low ABCB1 expression. Eur. J. Cancer 2012, 48, 763–771. [DOI] [PubMed] [Google Scholar]
- (106).Iwai M; Minematsu T; Li Q; Iwatsubo T; Usui T Utility of P-glycoprotein and organic cation transporter 1 double-transfected LLC-PK1 cells for studying the interaction of YM155 monobromide, novel small-molecule survivin suppressant, with P-glycoprotein. Drug Metab. Dispos. 2011, 39, 2314–2320. [DOI] [PubMed] [Google Scholar]
- (107).Manallack DT; Prankerd RJ; Yuriev E; Oprea TI; Chalmers DK The significance of acid/base properties in drug discovery. Chem. Soc. Rev. 2013, 42, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Tsarevsky NV; Slaveykova V; Manev S; Lazarov D The onium compounds. J. Chem. Educ. 1997, 74, 734–736. [Google Scholar]
- (109).Kollar J; Machacek M; Halaskova M; Lenco J; Kucera R; Demuth J; Rohlickova M; Hasonova K; Miletin M; Novakova V; Zimcik P Cationic versus anionic phthalocyanines for photodynamic therapy: what a difference the charge makes. J. Med. Chem. 2020, 63, 7616. [DOI] [PubMed] [Google Scholar]
- (110).Cui Q; Ding W; Liu P; Luo B; Yang J; Lu W; Hu Y; Huang P; Wen S Developing bi-gold compound BGC2a to target mitochondria for the elimination of cancer cells. Int. J. Mol. Sci. 2022, 23, 12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Palermo EF; Lee DK; Ramamoorthy A; Kuroda K Role of cationic group structure in membrane binding and disruption by amphiphilic copolymers. J. Phys. Chem. B 2011, 115, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Iwai M; Minematsu T; Li Q; Iwatsubo T; Usui T Utility of P-glycoprotein and organic cation transporter 1 double-transfected LLC-PK1 cells for studying the interaction of YM155 monobromide, novel small-molecule survivin suppressant, with P-glycoprotein. Drug Metab. Dispos. 2011, 39, 2314–2320. [DOI] [PubMed] [Google Scholar]
- (113).Cui Q; Wen S; Huang P Targeting cancer cell mitochondria as a therapeutic approach: recent updates. Future Med. Chem. 2017, 9, 929–949. [DOI] [PubMed] [Google Scholar]
- (114).Chazotte B Labeling mitochondria with MitoTracker dyes. Cold Spring Harb Protoc 2011, 2011, 990–992. [DOI] [PubMed] [Google Scholar]
- (115).Mondal A; Jia D; Bhatt V; Akel M; Roberge J; Guo JY; Langenfeld J Ym155 localizes to the mitochondria leading to mitochondria dysfunction and activation of AMPK that inhibits BMP signaling in lung cancer cells. Sci. Rep-UK 2022, 12, 13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Abraham I; Joshi R; Pardasani P; Pardasani RT Recent advances in 1,4-benzoquinone chemistry. J. Brazil. Chem. Soc. 2011, 22, 385–421. [Google Scholar]
- (117).Li M; Yamada Y; Rodriguez GM; Dick T; Go ML Redox cycling dioxonaphthoimidazoliums disrupt iron homeostasis in mycobacterium bovis Bacillus Calmette-Guerin. Microbiol Spectr 2022, 10, No. e197022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Fridianto KT; Li M; Hards K; Negatu DA; Cook GM; Dick T; Lam Y; Go ML Functionalized dioxonaphthoimidazoliums: a redox cycling chemotype with potent bactericidal activities against mycobacterium tuberculosis. J. Med. Chem. 2021, 64, 15991–16007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Minematsu T; Sonoda T; Hashimoto T; Iwai M; Oppeneer T; Felder L; Shirai N; Miyashita A; Usui T Pharmacokinetics, distribution and excretion of YM155 monobromide, a novel small-molecule survivin suppressant, in male and pregnant or lactating female rats. Biopharm. Drug Dispos. 2012, 33, 160–169. [DOI] [PubMed] [Google Scholar]
- (120).Pennington LD; Aquila BM; Choi Y; Valiulin RA; Muegge I Positional analogue scanning: an effective strategy for multiparameter optimization in drug design. J. Med. Chem. 2020, 63, 8956. [DOI] [PubMed] [Google Scholar]
- (121).Wilcken R; Zimmermann MO; Lange A; Joerger AC; Boeckler FM Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J. Med. Chem. 2013, 56, 1363–1388. [DOI] [PubMed] [Google Scholar]
- (122).Xu Z; Yang Z; Liu Y; Lu Y; Chen K; Zhu W Halogen bond: its role beyond drug-target binding affinity for drug discovery and development. J. Chem. Inf Model 2014, 54, 69–78. [DOI] [PubMed] [Google Scholar]
- (123).Wu YJ; Meanwell NA Geminal Diheteroatomic Motifs: Some Applications of Acetals, Ketals, and Their Sulfur and Nitrogen Homologues in Medicinal Chemistry and Drug Design. J. Med. Chem. 2021, 64, 9786–9874. [DOI] [PubMed] [Google Scholar]
- (124).Smith MB Chapter 7 - Protecting Groups. In Organic Synthesis, 3rd ed.; Smith MB, Ed.; Academic Press: Oxford, 2010; pp 587–622. [Google Scholar]
- (125).Ndubaku CO; Crawford JJ; Drobnick J; Aliagas I; Campbell D; Dong P; Dornan LM; Duron S; Epler J; Gazzard L; Heise CE; Hoeflich KP; Jakubiak D; La H; Lee W; Lin B; Lyssikatos JP; Maksimoska J; Marmorstein R; Murray LJ; O’Brien T; Oh A; Ramaswamy S; Wang W; Zhao X; Zhong Y; Blackwood E; Rudolph J Design of Selective PAK1 Inhibitor G-5555: Improving Properties by Employing an Unorthodox Low-pK a Polar Moiety. ACS Med. Chem. Lett. 2015, 6, 1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Wu YJ; Meanwell NA Geminal Diheteroatomic Motifs: Some Applications of Acetals, Ketals, and Their Sulfur and Nitrogen Homologues in Medicinal Chemistry and Drug Design. J. Med. Chem. 2021, 64, 9786–9874. [DOI] [PubMed] [Google Scholar]
- (127).Henderson DA; Collier PN; Pave G; Rzepa P; White AJ; Burrows JN; Barrett AG Studies on the total synthesis of lactonamycin: construction of model ABCD ring systems. J. Org. Chem. 2006, 71, 2434–2444. [DOI] [PubMed] [Google Scholar]
- (128).Matsumoto N; Tsuchida T; Maruyama M; Kinoshita N; Homma Y; Iinuma H; Sawa T; Hamada M; Takeuchi T; Heida N; Yoshioka T Lactonamycin, a new antimicrobial antibiotic produced by Streptomyces rishiriensis MJ773–88K4. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J. Antibiot. 1999, 52, 269–275. [DOI] [PubMed] [Google Scholar]
- (129).Lu JM; Nurko J; Weakley SM; Jiang J; Kougias P; Lin PH; Yao Q; Chen C Molecular mechanisms and clinical applications of nordihydroguaiaretic acid (NDGA) and its derivatives: an update. Med. Sci. Monitor 2010, 16, RA93–RA100. [PMC free article] [PubMed] [Google Scholar]
- (130).Manda G; Rojo AI; Martinez-Klimova E; Pedraza-Chaverri J; Cuadrado A Nordihydroguaiaretic acid: from herbal medicine to clinical development for cancer and chronic diseases. Front Pharmacol 2020, 11, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Smolewski P Terameprocol, a novel site-specific transcription inhibitor with anticancer activity. Idrugs 2008, 11, 204–214. [PubMed] [Google Scholar]
- (132).Chang CC; Heller JD; Kuo J; Huang RC Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. P. Natl. Acad. Sci. Usa 2004, 101, 13239–13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Chao A; Lin CY; Wu RC; Lee YS; Lee LY; Tsai CL; Yang LY; Liu H; Chen SJ; Wang TH; Lai CH The combination of everolimus and terameprocol exerts synergistic antiproliferative effects in endometrial cancer: molecular role of insulin-like growth factor binding protein 2. J. Mol. Med. 2018, 96, 1251–1266. [DOI] [PubMed] [Google Scholar]
- (134).Tibes R; McDonagh KT; Lekakis L; Bogenberger JM; Kim S; Frazer N; Mohrland S; Bassett D; Garcia R; Schroeder K; Shanmugam V; Carpten J; Hagelstrom RT; Beaudry C; Von Hoff D; Shea TC Phase I study of the novel Cdc2/CDK1 and AKT inhibitor terameprocol in patients with advanced leukemias. Invest. New Drug. 2015, 33, 389–396. [DOI] [PubMed] [Google Scholar]
- (135).Mak DH; Schober WD; Chen W; Heller J; Andreeff M; Carter BZ Tetra-O-methyl nordihydroguaiaretic acid inhibits growth and induces death of leukemia cells independent of Cdc2 and survivin. Leukemia Lymphoma 2007, 48, 774–785. [DOI] [PubMed] [Google Scholar]
- (136).Heller JD; Kuo J; Wu TC; Kast WM; Huang RC Tetra-O-methyl nordihydroguaiaretic acid induces G2 arrest in mammalian cells and exhibits tumoricidal activity in vivo. Cancer Res. 2001, 61 (14), 5499–5504. [PubMed] [Google Scholar]
- (137).Sun Y; Giacalone NJ; Lu B Terameprocol (tetra-O-methyl nordihydroguaiaretic acid), an inhibitor of Sp1-mediated survivin transcription, induces radiosensitization in non-small cell lung carcinoma. J. Thorac Oncol 2011, 6, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Khanna N; Dalby R; Tan M; Arnold S; Stern J; Frazer N Phase I/II clinical safety studies of terameprocol vaginal ointment. Gynecol. Oncol. 2007, 107, 554–562. [DOI] [PubMed] [Google Scholar]
- (139).Khanna N; Dalby R; Connor A; Church A; Stern J; Frazer N Phase I clinical trial of repeat dose terameprocol vaginal ointment in healthy female volunteers. Sex. Transm. Dis. 2008, 35, 577–582. [DOI] [PubMed] [Google Scholar]
- (140).Tibes R; McDonagh KT; Lekakis L; Bogenberger JM; Kim S; Frazer N; Mohrland S; Bassett D; Garcia R; Schroeder K; Shanmugam V; Carpten J; Hagelstrom RT; Beaudry C; Von Hoff D; Shea TC Phase I study of the novel Cdc2/CDK1 and AKT inhibitor terameprocol in patients with advanced leukemias. Invest. New Drug. 2015, 33, 389–396. [DOI] [PubMed] [Google Scholar]
- (141).Chen Q Nordihydroguaiaretic acid analogues: their chemical synthesis and biological activities. Curr. Top. Med. Chem. 2009, 9, 1636–1659. [DOI] [PubMed] [Google Scholar]
- (142).Li X; Jiang JH; Chen Q; Xiao SX; Li CH; Gu HW; Zhang H; Hu JL; Yao FH; Li QG Synthesis of nordihydroguaiaretic acid derivatives and their bioactivities on S. pombe and K562 cell lines. Eur. J. Med. Chem. 2013, 62, 605–613. [DOI] [PubMed] [Google Scholar]
- (143).McDonald RW; Bunjobpon W; Liu T; Fessler S; Pardo OE; Freer IK; Glaser M; Seckl MJ; Robins DJ Synthesis and anticancer activity of nordihydroguaiaretic acid (NDGA) and analogues. Anti-Cancer Drug Des. 2001, 16, 261–270. [PubMed] [Google Scholar]
- (144).Blecha JE; Anderson MO; Chow JM; Guevarra CC; Pender C; Penaranda C; Zavodovskaya M; Youngren JF; Berkman CE Inhibition of IGF-1R and lipoxygenase by nordihydroguaiaretic acid (NDGA) analogs. Bioorg. Med. Chem. Lett. 2007, 17, 4026–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Rahman MM; Rahaman MS; Islam MR; Rahman F; Mithi FM; Alqahtani T; Almikhlafi MA; Alghamdi SQ; Alruwaili AS; Hossain MS; Ahmed M; Das R; Emran TB; Uddin MS Role of Phenolic Compounds in Human Disease: Current Knowledge and Future Prospects. Molecules 2022, 27, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Tsao R Chemistry and biochemistry of dietary polyphenols. Nutrients 2010, 2, 1231–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Huo C; Wan SB; Lam WH; Li L; Wang Z; Landis-Piwowar KR; Chen D; Dou QP; Chan TH The challenge of developing green tea polyphenols as therapeutic agents. Inflammopharmacology 2008, 16, 248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Cui Q; Chen Y; Zhang M; Shan L; Sun Y; Yu P; Zhang G; Wang D; Zhao Z; Xu Q; Xu B; Wang Y Design, synthesis, and preliminary cardioprotective effect evaluation of danshensu derivatives. Chem. Biol. Drug Des 2014, 84, 282–291. [DOI] [PubMed] [Google Scholar]
- (149).Scott KA; Cox PB; Njardarson JT Phenols in pharmaceuticals: analysis of a recurring motif. J. Med. Chem. 2022, 65, 7044–7072. [DOI] [PubMed] [Google Scholar]
- (150).Casey Laizure S; Herring V; Hu Z; Witbrodt K; Parker RB The role of human carboxylesterases in drug metabolism: have we overlooked their importance? Pharmacotherapy 2013, 33, 210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Wang D; Zou L; Jin Q; Hou J; Ge G; Yang L Human carboxylesterases: a comprehensive review. Acta Pharm. Sin B 2018, 8, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Guedes J. d. S.; Carneiro TR; Pinheiro P. d. S. M.; Fraga CAM; Sant’Anna CMR; Barreiro EJ; Lima LM Methyl Effect on the Metabolism, Chemical Stability, and Permeability Profile of Bioactive N-Sulfonylhydrazones. ACS Omega 2022, 7, 38752–38765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Duangjit S; Pamornpathomkul B; Opanasopit P; Rojanarata T; Obata Y; Takayama K; Ngawhirunpat T Role of the charge, carbon chain length, and content of surfactant on the skin penetration of meloxicam-loaded liposomes. Int. J. Nanomed 2014, 9, 2005–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Tsantili-Kakoulidou A; Demopoulos VJ Drug-like properties and fraction lipophilicity index as a combined metric. Admet Dmpk 2021, 9, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Fu Q; Wang Y; Ma Y; Zhang D; Fallon JK; Yang X; Liu D; He Z; Liu F Programmed hydrolysis in designing paclitaxel prodrug for nanocarrier assembly. Sci. Rep-Uk 2015, 5, 12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Torres-Luna C; Hu N; Domszy R; Fan X; Yang J; Briber RM; Wang NS; Yang A Effect of carbon chain length, ionic strength, and pH on the in vitro release kinetics of cationic drugs from fatty-acid-loaded contact lenses. Pharmaceutics 2021, 13, 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Wermuth CG; Lesuisse D Chapter 30 - Preparation of Water-Soluble Compounds by Covalent Attachment of Solubilizing Moieties. In The Practice of Medicinal Chemistry, 4th ed.; Wermuth CG; Aldous D; Raboisson P; Rognan D, Eds.; Academic Press: San Diego, 2015; pp 723–745. [Google Scholar]
- (158).Heravi MM; Zadsirjan V Prescribed drugs containing nitrogen heterocycles: an overview. Rsc Adv. 2020, 10, 44247–44311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Vitaku E; Smith DT; Njardarson JT Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (160).Li N; Wu JL; Hasegawa T; Sakai J; Wang LY; Kakuta S; Furuya Y; Tomida A; Tsuruo T; Ando M Bioactive dibenzylbutyrolactone and dibenzylbutanediol lignans from Peperomia duclouxii. J. Nat. Prod. 2006, 69, 234–239. [DOI] [PubMed] [Google Scholar]
- (161).Chattopadhyay SK; Kumar TR; Maulik PR; Srivastava S; Garg A; Sharon A; Negi AS; Khanuja SP Absolute configuration and anticancer activity of taxiresinol and related lignans of Taxus wallichiana. Bioorgan. Med. Chem. 2003, 11, 4945–4948. [DOI] [PubMed] [Google Scholar]
- (162).Li F Discovery of survivin inhibitors and beyond: FL118 as a proof of concept. Int. Rev. Cel Mol. Bio 2013, 305, 217–252. [DOI] [PubMed] [Google Scholar]
- (163).Ling X; Li F An intravenous (i.v.) route-compatible formulation of FL118, a survivin, Mcl-1, XIAP, and cIAP2 selective inhibitor, improves FL118 antitumor efficacy and therapeutic index (TI). Am. J. Transl Res. 2013, 5, 139–154. [PMC free article] [PubMed] [Google Scholar]
- (164).Weng Q; Zhou L; Xia L; Zheng Y; Zhang X; Li F; Li Q In vitro evaluation of FL118 and 9-Q20 cytotoxicity and cellular uptake in 2D and 3D different cell models. Cancer Chemoth. Pharm. 2019, 84, 527–537. [DOI] [PubMed] [Google Scholar]
- (165).Ling X; Wu W; Fan C; Xu C; Liao J; Rich LJ; Huang RY; Repasky EA; Wang X; Li F An ABCG2 non-substrate anticancer agent FL118 targets drug-resistant cancer stem-like cells and overcomes treatment resistance of human pancreatic cancer. J. Exp. Clin. Canc. Res. 2018, 37, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Li F; Fountzilas C; Puzanov I; Attwood KM; Morrison C; Ling X Multiple functions of the DEAD-box RNA helicase, DDX5 (p68), make DDX5 a superior oncogenic biomarker and target for targeted cancer therapy. Am. J. Cancer Res. 2021, 11, 5190–5213. [PMC free article] [PubMed] [Google Scholar]
- (167).Zhao J; Ling X; Cao S; Liu X; Wan S; Jiang T; Li F Antitumor activity of FL118, a survivin, Mcl-1, XIAP, and cIAP2 selective inhibitor, is highly dependent on its primary structure and steric configuration. Mol. Pharmaceut. 2014, 11, 457–467. [DOI] [PubMed] [Google Scholar]
- (168).Zhou Y; Hu W; Zhang X; Wang Y; Zhuang W; Li F; Li Q Cellular uptake and transport characteristics of FL118 derivatives in Caco-2 cell monolayers. Chem. Pharm. Bull. 2021, 69, 1054–1060. [DOI] [PubMed] [Google Scholar]
- (169).Zhou L; Weng Q; Zheng Y; Zhou Y; Li Q; Li F Uptake and efflux of FL118 and two FL118 derivatives in 3D cell model. Cytotechnology 2019, 71, 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Ling X; Li F Use of the fl118 core chemical structure platform to generate fl118 derivatives for treatment of human disease. 3–24–2015, 2015.
- (171).Li F; Ling X; Harris DL; Liao J; Wang Y; Westover D; Jiang G; Xu B; Boland PM; Jin C Topoisomerase I (Top1): a major target of FL118 for its antitumor efficacy or mainly involved in its side effects of hematopoietic toxicity? Am. J. Cancer Res. 2017, 7, 370–382. [PMC free article] [PubMed] [Google Scholar]
- (172).Fan YF; Zhang W; Zeng L; Lei ZN; Cai CY; Gupta P; Yang DH; Cui Q; Qin ZD; Chen ZS; Trombetta LD Dacomitinib antagonizes multidrug resistance (MDR) in cancer cells by inhibiting the efflux activity of ABCB1 and ABCG2 transporters. Cancer Lett. 2018, 421, 186–198. [DOI] [PubMed] [Google Scholar]
- (173).Staker BL; Hjerrild K; Feese MD; Behnke CA; Burgin AJ; Stewart L The mechanism of topoisomerase I poisoning by a camptothecin analog. P. Natl. Acad. Sci. Usa 2002, 99, 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Redinbo MR; Stewart L; Kuhn P; Champoux JJ; Hol WG Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998, 279, 1504–1513. [DOI] [PubMed] [Google Scholar]
- (175).Gupta D; Bhatia D; Dave V; Sutariya V; Varghese Gupta S Salts of therapeutic agents: chemical, physicochemical, and biological considerations. Molecules 2018, 23, 1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Morrison P; Porfiryeva NN; Chahal S; Salakhov IA; Lacourt C; Semina II; Moustafine RI; Khutoryanskiy VV Crown Ethers: Novel Permeability Enhancers for Ocular Drug Delivery? Mol. Pharmaceut. 2017, 14, 3528–3538. [DOI] [PubMed] [Google Scholar]
- (177).Ding HX; Liu KK-C; Sakya SM; Flick AC; O’Donnell CJ Synthetic approaches to the 2011 new drugs. Bioorgan. Med. Chem. 2013, 21, 2795–2825. [DOI] [PubMed] [Google Scholar]
- (178).Xiao M; Xue Y; Wu Z; Lei ZN; Wang J; Chen ZS; Li W Design, synthesis and biological evaluation of selective survivin inhibitors. J. Biomed Res. 2019, 33, 82–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Albadari N; Deng S; Chen H; Zhao G; Yue J; Zhang S; Miller DD; Wu Z; Li W Synthesis and biological evaluation of selective survivin inhibitors derived from the MX-106 hydroxyquinoline scaffold. Eur. J. Med. Chem. 2021, 224, 113719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Martorana A; La Monica G; Lauria A Quinoline-Based Molecules Targeting c-Met, EGF, and VEGF Receptors and the Proteins Involved in Related Carcinogenic Pathways. Molecules 2020, 25, 4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (181).Wang JQ; Wu ZX; Yang Y; Teng QX; Li YD; Lei ZN; Jani KA; Kaushal N; Chen ZS ATP-binding cassette (ABC) transporters in cancer: A review of recent updates. J. Evid-Based Med. 2021, 14, 232–256. [DOI] [PubMed] [Google Scholar]
- (182).Mo W; Zhang JT Human ABCG2: structure, function, and its role in multidrug resistance. Int. J. Biochem Mol. Biol. 2012, 3, 1–27. [PMC free article] [PubMed] [Google Scholar]
- (183).Lazzara PR; Moore TW Scaffold-hopping as a strategy to address metabolic liabilities of aromatic compounds. Rsc Med. Chem. 2020, 11, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Tariq S; Somakala K; Amir M Quinoxaline: An insight into the recent pharmacological advances. Eur. J. Med. Chem. 2018, 143, 542–557. [DOI] [PubMed] [Google Scholar]
- (185).Kaushal T; Srivastava G; Sharma A; Singh Negi A An insight into medicinal chemistry of anticancer quinoxalines. Bioorgan. Med. Chem. 2019, 27, 16–35. [DOI] [PubMed] [Google Scholar]
- (186).Zhao J; Zhang Y; Wang M; Liu Q; Lei X; Wu M; Guo S; Yi D; Li Q; Ma L; Liu Z; Guo F; Wang J; Li X; Wang Y; Cen S Quinoline and quinazoline derivatives inhibit viral RNA synthesis by SARS-CoV-2 RdRp. Acs Infect Dis 2021, 7, 1535–1544. [DOI] [PubMed] [Google Scholar]
- (187).Dai CL; Tiwari AK; Wu CP; Su XD; Wang SR; Liu DG; Ashby CJ; Huang Y; Robey RW; Liang YJ; Chen LM; Shi CJ; Ambudkar SV; Chen ZS; Fu LW Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008, 68, 7905–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Gao HL; Gupta P; Cui Q; Ashar YV; Wu ZX; Zeng L; Lei ZN; Teng QX; Ashby CJ; Guan Y; Chen ZS Sapitinib reverses anticancer drug resistance in colon cancer cells overexpressing the ABCB1 transporter. Front Oncol 2020, 10, 574861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Hamada Y; Kiso Y The application of bioisosteres in drug design for novel drug discovery: focusing on acid protease inhibitors. Expert Opin Drug Dis 2012, 7, 903–922. [DOI] [PubMed] [Google Scholar]
- (190).Brown N Bioisosteres in Medicinal Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2012. [Google Scholar]
- (191).Aldeghi M; Malhotra S; Selwood DL; Chan AW Two- and three-dimensional rings in drugs. Chem. Biol. Drug Des 2014, 83, 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Shearer J; Castro JL; Lawson A; MacCoss M; Taylor RD Rings in Clinical Trials and Drugs: Present and Future. J. Med. Chem. 2022, 65, 8699–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Pan X; Wang H; Li C; Zhang J; Ji C MolGpka: A Web Server for Small Molecule pK(a) Prediction Using a Graph-Convolutional Neural Network. J. Chem. Inf Model 2021, 61, 3159–3165. [DOI] [PubMed] [Google Scholar]
- (194).Rodil A; Bosisio S; Ayoup MS; Quinn L; Cordes DB; Slawin A; Murphy CD; Michel J; O’Hagan D Metabolism and hydrophilicity of the polarised ‘Janus face’ all-cis tetrafluorocyclohexyl ring, a candidate motif for drug discovery. Chem. Sci. 2018, 9, 3023–3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Wendt MD; Sun C; Kunzer A; Sauer D; Sarris K; Hoff E; Yu L; Nettesheim DG; Chen J; Jin S; Comess KM; Fan Y; Anderson SN; Isaac B; Olejniczak ET; Hajduk PJ; Rosenberg SH; Elmore SW Discovery of a novel small molecule binding site of human survivin. Bioorg. Med. Chem. Lett. 2007, 17, 3122–3129. [DOI] [PubMed] [Google Scholar]
- (196).Xia F; Canovas PM; Guadagno TM; Altieri DC A survivin-ran complex regulates spindle formation in tumor cells. Mol. Cell. Biol. 2008, 28, 5299–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Chettiar SN; Cooley JV; Park IH; Bhasin D; Chakravarti A; Li PK; Li C; Jacob NK Design, synthesis and biological studies of survivin dimerization modulators that prolong mitotic cycle. Bioorg. Med. Chem. Lett. 2013, 23, 5429–5433. [DOI] [PubMed] [Google Scholar]
- (198).Ibrahim TM; Ernst C; Lange A; Hennig S; Boeckler FM Small-Molecule Intervention At The Dimerization Interface Of Survivin By Novel Rigidized Scaffolds. Drug Des Devel Ther 2019, 13, 4247–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Sabour R; Harras MF; Mohamed Al Kamaly O; Altwaijry N Discovery of novel 3-cyanopyridines as survivin modulators and apoptosis inducers. Molecules 2020, 25, 4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Sabour R; Harras MF; Mehany A Design, synthesis, cytotoxicity screening and molecular docking of new 3-cyanopyridines as survivin inhibitors and apoptosis inducers. Bioorg. Chem. 2020, 94, 103358. [DOI] [PubMed] [Google Scholar]
- (201).Steigerwald C; Rasenberger B; Christmann M; Tomicic MT Sensitization of colorectal cancer cells to irinotecan by the Survivin inhibitor LLP3 depends on XAF1 proficiency in the context of mutated p53. Arch. Toxicol. 2018, 92, 2645–2648. [DOI] [PubMed] [Google Scholar]
- (202).Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- (203).Meanwell NA Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- (204).Soltan OM; Shoman ME; Abdel-Aziz SA; Narumi A; Konno H; Abdel-Aziz M Molecular hybrids: A five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy. Eur. J. Med. Chem. 2021, 225, 113768. [DOI] [PubMed] [Google Scholar]
- (205).Berube G An overview of molecular hybrids in drug discovery. Expert Opin Drug Dis 2016, 11, 281–305. [DOI] [PubMed] [Google Scholar]
- (206).Ruffilli C; Roth S; Rodrigo M; Boyd H; Zelcer N; Moreau K Proteolysis targeting chimeras (PROTACs): a perspective on integral membrane protein degradation. Acs Pharmacol Transl 2022, 5, 849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Fang Y; Wang S; Han S; Zhao Y; Yu C; Liu H; Li N Targeted protein degrader development for cancer: advances, challenges, and opportunities. Trends Pharmacol. Sci. 2023, 44, 303–317. [DOI] [PubMed] [Google Scholar]
- (208).Sasso JM; Tenchov R; Wang D; Johnson LS; Wang X; Zhou QA Molecular glues: the adhesive connecting targeted protein degradation to the clinic. Biochemistry-Us 2023, 62, 601–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Gu S; Cui D; Chen X; Xiong X; Zhao Y PROTACs: an emerging targeting technique for protein degradation in drug discovery. Bioessays 2018, 40, No. e1700247. [DOI] [PubMed] [Google Scholar]
- (210).Huang X; Dixit VM Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016, 26, 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Churcher I PROTAC-induced protein degradation in drug discovery: breaking the rules or just making new ones? J. Med. Chem. 2018, 61, 444–452. [DOI] [PubMed] [Google Scholar]
- (212).Li Y; Yang J; Aguilar A; McEachern D; Przybranowski S; Liu L; Yang CY; Wang M; Han X; Wang S Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem. 2019, 62, 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Hu J; Hu B; Wang M; Xu F; Miao B; Yang CY; Wang M; Liu Z; Hayes DF; Chinnaswamy K; Delproposto J; Stuckey J; Wang S Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J. Med. Chem. 2019, 62, 1420–1442. [DOI] [PubMed] [Google Scholar]
- (214).Mullard A First targeted protein degrader hits the clinic. Nat. Rev. Drug Discovery 2019, 18, 237–239. [DOI] [PubMed] [Google Scholar]
- (215).Salazar DE; Gormley G Chapter 41 - Modern Drug Discovery and Development. In Clinical and Translational Science, 2nd ed.; Robertson D, Williams GH, Eds.; Academic Press: Cambridge, MA, 2017; pp 719–743. [Google Scholar]
- (216).Russ AP; Lampel S The druggable genome: an update. Drug Discovery Today 2005, 10, 1607–1610. [DOI] [PubMed] [Google Scholar]
- (217).Crews CM Targeting the undruggable proteome: the small molecules of my dreams. Chem. Biol. 2010, 17, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]