

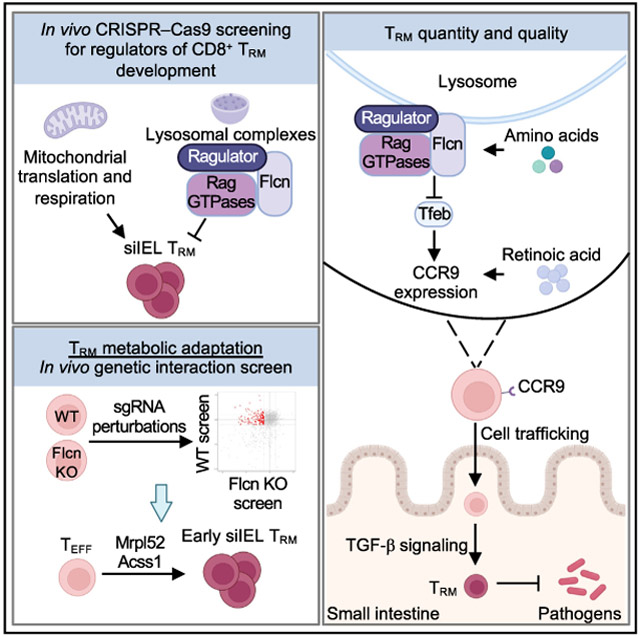
SUMMARY
Nutrient availability and organelle biology direct tissue homeostasis and cell fate, but how these processes orchestrate tissue immunity remains poorly defined. Here, using in vivo CRISPR-Cas9 screens, we uncovered organelle signaling and metabolic processes shaping CD8+ tissue-resident memory T (TRM) cell development. TRM cells depended on mitochondrial translation and respiration. Conversely, three nutrient-dependent lysosomal signaling nodes—Flcn, Ragulator, and Rag GTPases—inhibited intestinal TRM cell formation. Depleting these molecules or amino acids activated the transcription factor Tfeb, thereby linking nutrient stress to TRM programming. Further, Flcn deficiency promoted protective TRM cell responses in the small intestine. Mechanistically, the Flcn-Tfeb axis restrained retinoic acid-induced CCR9 expression for migration and transforming growth factor β (TGF-β)-mediated programming for lineage differentiation. Genetic interaction screening revealed that the mitochondrial protein Mrpl52 enabled early TRM cell formation, while Acss1 controlled TRM cell development under Flcn deficiency-associated lysosomal dysregulation. Thus, the interplay between nutrients, organelle signaling, and metabolic adaptation dictates tissue immunity.
Graphical Abstract
In brief
How organelle signaling and metabolic adaptation orchestrate tissue-resident CD8+ T (TRM) cell development remains poorly defined. Here, Raynor et al. establish three nutrient-dependent lysosomal signaling nodes as negative regulators of TRM differentiation in the small intestine. These results uncover mechanisms dictating TRM cell quality and quantity for tissue immunity.
INTRODUCTION
CD8+ T cells provide protection from infectious and malignant diseases and aid in tissue homeostasis to instruct organismal health.1 During a primary infection, CD8+ T cells differentiate into effector CD8+ T cells (TEFF) or memory CD8+ T (TMEM) cells capable of mediating protective immunity to secondary infection.2-4 TMEM subsets include central memory (TCM) and effector memory (TEM) cells that can circulate via the vasculature and lymphoid tissues (collectively called circulatory TMEM or TCIRC).3 By contrast, tissue-resident memory T (TRM) cells do not recirculate and persist in tissues.3,4 TRM and TRM-like cells have emerging immunotherapeutic potential in infection and cancer.2,4,5 Therefore, identification of regulators, especially poorly defined negative regulators, of TRM development may uncover immunotherapeutic targets to reprogram adaptive immunity in immune-mediated diseases.
Spatial and temporal events orchestrate TRM programming.6 TEFF cells from lymphoid tissues marked by low expression of KLRG1 are more prone to develop into TRM cells.7,8 TRM cells can also arise from a circulatory TEFF cell subpopulation with TRM-like transcriptional features,9 suggesting that lymphoid tissues may provide initial signals for TRM transcriptional programming. Further, TEFF cells that localize into non-lymphoid tissues acquire a TRM transcriptional program within the first week of infection,7 suggesting that tissue microenvironment-derived cues, such as transforming growth factor β (TGF-β),10,11 promote TRM development. How T cells are poised as TRM precursors for select tissue types remains underexplored.
Metabolic reprogramming orchestrates cell state and fate in immune cells.12 Further, nutrient composition is dynamically regulated in tissues during infection,13 suggesting that cells within a microenvironment must adapt to alterations in nutrient availability for their functional fitness. Although metabolic pathways are emerging as important regulators of TCIRC cells, less is known about tissue-specific metabolic regulation of TRM cells.14-18 Given the importance of TRM cells for tissue immunity, insight into the signaling and metabolic basis of their development and tissue adaptation is fundamental to our understanding of adaptive immunity. Here, we systemically targeted mitochondrial and lysosomal genes using in vivo pooled CRISPR-Cas9 screens and uncovered discrete functions of organelle signaling and its interplay with nutrients and metabolic adaptation in shaping TRM development.
RESULTS
In vivo CRISPR screening uncovers lysosome signaling nodes as negative regulators of TRM development
To dissect mitochondria and lysosome-associated regulators of CD8+ TRM development, we generated a lentiviral CRISPR library targeting 1,589 mitochondrial and lysosomal genes that contained four unique single-guide RNAs (sgRNAs) targeting each gene, along with 500 non-targeting control (NTC) sgRNAs. Ovalbumin (OVA)-specific OT-I cells expressing Cas9 were transduced with the library and transferred into C57BL/6 wild-type (WT) mice, followed by infection with Listeria monocytogenes expressing OVA (LM-OVA). Then, we sort-purified splenic or small intestinal intraepithelial lymphocyte (siIEL) OT-I TEFF cells at day 7.5 post-infection (p.i.), and splenic OT-I (called TCIRC), splenic TCM, splenic TEM, or siIEL cells (called TRM) at day 30 p.i., and determined enrichment or depletion of sgRNAs in these populations (Figure 1A). sgRNAs targeting Flcn, Lamtor1, Lamtor4, Rraga, and Rragc were among the top enriched guides in siIEL TRM cells versus input but not splenic TCIRC versus input, nominating these genes as negative regulators for TRM cells (Figure S1A; Table S1).
Figure 1. Lysosome-associated signaling nodes negatively regulate TRM development.
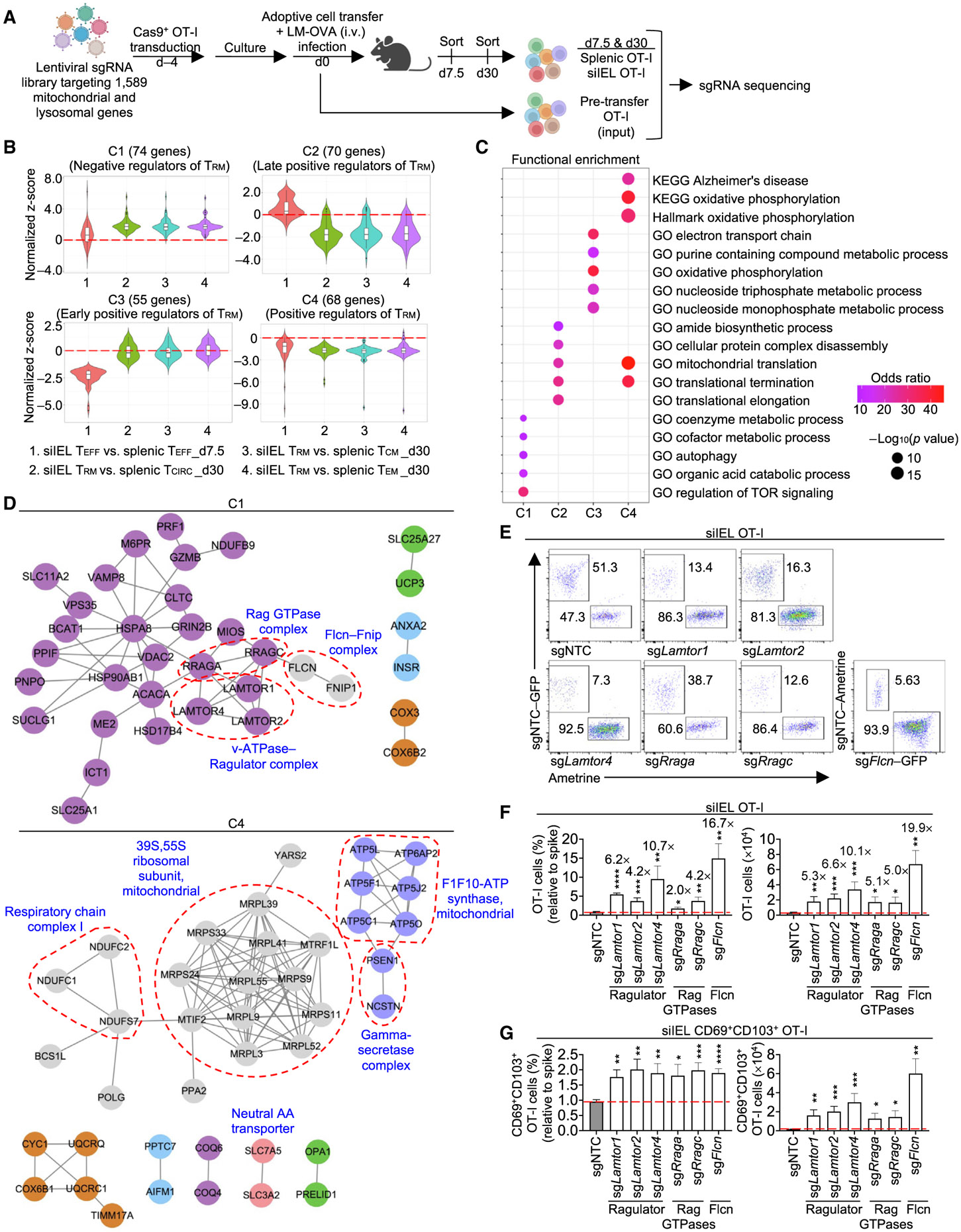
(A) Schematic for in vivo CRISPR screen. Day (d) in all figures.
(B) Indicated pairwise comparisons of sgRNA abundance between splenic and siIEL OT-I cells identified candidate genes with perturbation effects in at least one of the four comparisons, followed by hierarchical clustering into four gene clusters.
(C) Functional enrichment analysis of cluster (C)1, C2, C3, and C4 genes identified in (B).
(D) PPI networks of encoding C1 and C4 genes. Red dotted lines indicate known protein complexes.
(E–G) OT-I cells transduced with sgRNAs targeting the indicated genes (Ametrine+ or GFP+) were co-transferred at a 1:1 ratio with OT-I cells transduced with sgNTC (‘‘spike’’; Ametrine+ or GFP+) into WT mice, followed by LM-OVA infection (dual-color transfer system). Flow cytometry analysis (E) and frequencies (relative to spike) and numbers of total (F) or CD69+CD103+ OT-I cells (G) in siIEL at day 21 p.i. (n ≥ 3 per group). Fold-change relative to sgNTC is indicated in (F). Fisher’s exact test (C), two-tailed unpaired Student’s t test (F and G). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data (mean ± SEM) represent two experiments (F and G).
See also Figure S1.
To systemically evaluate pathways mediating TRM and TCIRC formation, we performed four pairwise comparisons between splenic and siIEL OT-I cells (see STAR Methods) and identified 267 sgRNA-targeted genes with enrichment or depletion in at least one of the four comparisons. These genes were hierarchically clustered into four gene clusters (Figure 1B; Table S1). Cluster 1 (C1) was composed of 74 genes and showed enrichment in siIEL relative to spleen upon genetic perturbations. Among C1 genes, 18 genes displayed modest enrichment in siIEL TEFF cells, whereas 60 genes were enriched in siIEL TRM cells (4 genes were enriched in siIEL TEFF and TRM cells) (Figure 1B; Table S1). Conversely, 55 of the 70 genes in C2 were depleted in siIEL TRM relative to all splenic TMEM populations on day 30 p.i., while 15 genes were enriched in siIEL versus spleen on day 7.5 p.i. (Figure 1B; Table S1). Additionally, all 55 genes in C3 were depleted in siIEL TEFF relative to splenic TEFF cells, but showed negligible effects on siIEL cells at the memory phase (Figure 1B; Table S1), indicating that genes in C2 and C3 serve temporal, positive effects on TRM cells. Finally, C4 had largely reciprocal effects compared with C1 and contained 68 genes that were more modestly depleted in siIEL than splenic cells at these time points (Figure 1B; Table S1). Thus, the in vivo CRISPR-Cas9 screen nominated mitochondria- and lysosome-associated negative (C1) and positive (C3 and C4, and the majority of C2 genes) regulators of TRM development in the small intestine.
Functional enrichment analysis for C1 genes revealed enrichment for regulation of mechanistic target of rapamycin (mTOR) signaling (Figures 1C and S1B; Table S2), organic acid catabolic process, and autophagy-associated catabolic pathway, while C2 genes were enriched for pathways related to mitochondrial translation (Figure 1C; Table S2). Further, C3 and C4 genes were enriched for electron transport chain (ETC) or oxidative phosphorylation (OXPHOS), and C4 genes also showed an enrichment for mitochondrial translation (Figures 1C and S1B; Table S2). Thus, mitochondrial translation and bioenergetic pathways are nominated as positive regulators of TRM development, in line with previous studies.14,16,17 Conversely, most negative regulators are associated with the lysosome, including lysosome-related signaling and catabolic pathways, which remain poorly understood in T cell biology.
To reconstruct signaling circuits, we integrated the top hits with composite protein-protein interaction (PPI) databases19,20 (see STAR Methods) to infer functional modules mediating immune cell signal transduction.19,21,22 This integrative analysis for C1 genes (TRM negative regulators) identified multiple components of Ragulator (Lamtor1, Lamtor2, and Lamtor4), Rag GTPase (Rraga and Rragc, encoding for RagA and RagC, respectively), and Flcn (Flcn and Fnip1) complexes (Figure 1D). These complexes transduce nutrient-dependent signals at lysosomes,23,24 although their roles in adaptive immunity remain poorly understood. Conversely, PPI network analysis of TRM positive regulators in C2, C3, and C4 identified mitochondrial ribosomes and ETC components (Figures 1D and S1C). Thus, Flcn, Ragulator, and Rag GTPase complexes are putative negative regulators of TRM cells, while mitochondrial ribosomes and ETC complexes are positive regulators.
Next, we validated top negative regulators of siIEL TRM differentiation, including Flcn, Ragulator, and Rag GTPase complexes (Figure S1A), using our in vivo dual-color transfer system (see STAR Methods).25 At day 21 p.i., we assessed OT-I cell accumulation in the siIEL and their co-expression of CD69 and CD103 as markers for siIEL TRM cells.6,15 Targeting of Ragulator, Rag GTPases, or Flcn increased total and CD69+CD103+ cell accumulation in siIEL (Figures 1E-1G and S1D-S1F). Fnip1 deletion had modest or trending effects on total and CD69+CD103+ accumulation in the small intestine (Figures S1G and S1H), suggesting redundancy with other homologous molecules such as Fnip2 (Hasumi et al.26). In particular, Flcn was the top negative regulator of siIEL TRM development (Figures 1F, 1G, and S1A; Table S1), and this effect was also observed at the effector phase (Figures S1I and S1J). Altogether, these results reveal selective lysosome-associated signaling complexes, especially Flcn, as negative regulators of siIEL TRM development.
Targeting Flcn promotes accumulation of functional TRM cells in the small intestine
TRM cells are maintained long term.4 At >60 days p.i., Flcn-deficient OT-I cells were enriched in siIEL and Peyer’s patches (PPs) but reduced in other tissues examined (Figure 2A). To validate these results, we bred Cd4CreFlcnfl/fl mice with OT-I mice for conditional ablation of Flcn in OT-I cells and then co-transferred naive WT and Flcn-deficient OT-I cells into WT mice, followed by LM-OVA infection. At >60 days p.i., Flcn-deficient cells showed enhanced accumulation in the siIEL and, to a lesser extent, PP, but were reduced in other tissues (Figure S2A). Next, we co-transferred naive WT and Cd4CreFlcnfl/fl P14 cells into naive mice followed by lymphocytic choriomeningitis virus Armstrong strain (LCMV-Arm) infection. At day 7 p.i., Flcn-deficient P14 cells were reduced in spleen, liver, and lung but elevated in siIEL (Figure S2B). Further, Flcn deficiency resulted in increased proportions of CD103+ and CD69+CD103+ siIEL P14 cells (Figure S2C). Flcn-deficient P14 cells were also elevated in siIEL at day >60 (Figure S2D). Altogether, these data establish a critical role for Flcn in opposing siIEL TRM but promoting TMEM formation in lymphoid and other non-lymphoid tissues.
Figure 2. Targeting Flcn promotes functional TRM cell accumulation in small intestine.
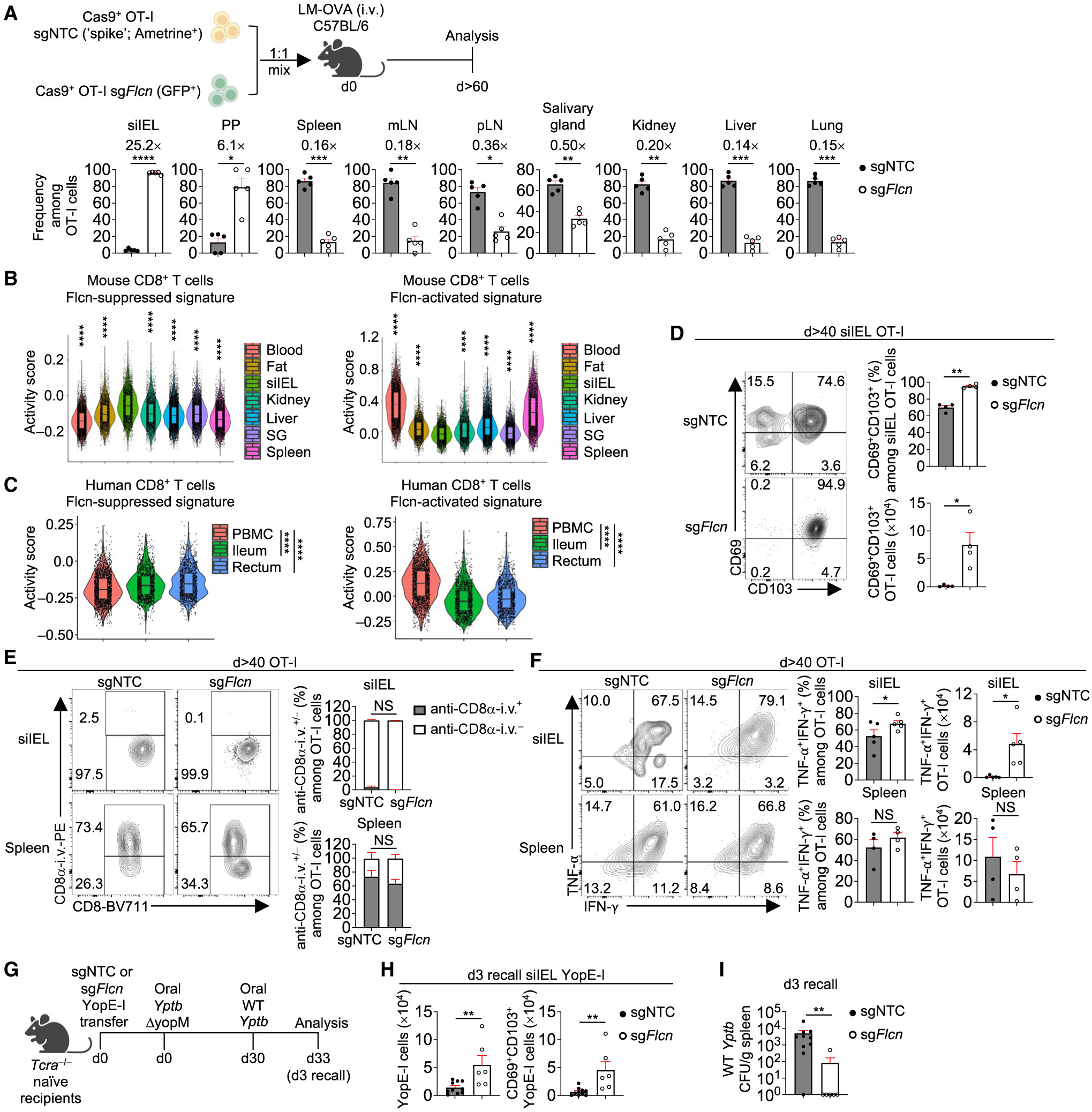
(A) Frequencies among sgRNA-transduced OT-I cells in indicated tissues at day >60 p.i. (n = 5 per group). Fold-change relative to sgNTC is indicated.
(B and C) Violin plots showing activity scores of Flcn-suppressed and Flcn-activated signatures (see STAR Methods) in public scRNA-seq datasets of mouse27 (B) or healthy human28 (C) CD8+ T cells from indicated tissues. Asterisks indicate statistical significance in siIEL versus each tissue in (B). PBMCs, peripheral blood mononuclear cells; SG, salivary gland.
(D) Frequency and number of CD69+CD103+ OT-I cells transduced with indicated sgRNAs in siIEL at day >40 p.i. (n = 4 per group).
(E) Frequencies of CD8α-i.v.+ and CD8α-i.v.− among indicated sgRNA-transduced OT-I cells in siIEL or spleen at day >40 p.i. (n = 4 per group).
(F) Frequencies and numbers of TNF-α+IFN-γ+ OT-I cells transduced with indicated sgRNAs in siIEL or spleen at day >40 p.i. (n ≥ 4 per group).
(G–I) Schematic for Yptb recall response29 (G). Total or CD69+CD103+ YopE-I cells in siIEL (H) and bacterial burden in spleen (I) on day 3 after secondary infection (n ≥ 6 per group). Colony-forming units (CFUs). Two-tailed paired Student’s t test (A and D–F), two-tailed unpaired Student’s t test (H), two-tailed Mann-Whitney test (I), Wilcoxon rank sum test (B and C). NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data (mean ± SEM) are compiled from ≥2 (A, H, and I) or represent ≥3 (D–F) experiments.
See also Figure S2.
Compared with Flcn, Ragulator and Rag GTPase complexes showed discrete effects on TMEM accumulation. Lamtor4-, RagA-, and RagC-deficient OT-I cells had increased proportions in the siIEL and most lymphoid tissues at day 30 p.i. (Figure S2E). Lamtor4-deficient cells were reduced in the salivary gland, liver, lung, and blood (Figure S2E), whereas RagA- and RagC-deficient cells were largely unaltered at day 30 p.i. (Figure S2E). At day 60 p.i., RagA-deficient cells had increased proportions in all lymphoid and non-lymphoid tissues examined, whereas Lamtort4- and RagC-deficient cells only accumulated in lymphoid tissues and siIEL (Figure S2F). Thus, Lamtor4, RagA, and RagC show discrete spatiotemporal effects on TMEM formation. These results, combined with the accumulation of Flcn-deficient cells in the siIEL and PP, highlight Flcn as a selective regulator of TRM development in the small intestine.
To examine tissue specificity and human relevance of Flcn regulation of TRM development, we examined the enrichment of Flcn-suppressed and Flcn-activated signatures (see STAR Methods) in a public dataset containing TMEM cells from multiple tissues.27 Flcn-suppressed signature was increased in TRM cells from siIEL compared with TCIRC (from the blood and spleen) or TRM cells from other non-lymphoid tissues,27 whereas Flcn-activated signature was reduced (Figure 2B). Among human CD8+ T cells,28 those from the ileum and rectum also had higher and lower activity scores of the Flcn-suppressed and Flcn-activated signatures, respectively, than those from blood (Figure 2C). These results suggest that Flcn more selectively restricts TRM generation in the small intestine.
We next tested whether Flcn-deficient cells were bona fide TRM cells. At the memory phase, the proportion of CD69+ CD103+ Flcn-deficient OT-I cells was increased in siIEL (Figures 2D and S2G), suggesting their more terminal differentiation state.30,31 By contrast, among Flcn-deficient cells in lymphoid tissues, expression of CD69 or CD103 and proportions of lymphoid-tissue-resident CD69+CD62L− CD8+ T cells32 or CD69−CD62L+ TCM cells were unaltered (Figures S2H and S2I). Next, to distinguish cells in circulation and tissue, we administered anti-CD8α antibody intravenously (i.v.) to mice at the memory phase.7,33 The majority of Flcn-deficient cells in siIEL (but not spleen) were CD8α-i.v.− (Figure 2E), supporting their classification as TRM cells. Moreover, the proportion and especially number of Flcn-deficient cells expressing tumor necrosis factor alpha (TNF-α) and interferon (IFN)-γ were enhanced in siIEL but not spleen at the memory phase (Figure 2F), suggesting their increased function.
To directly test this, we employed an oral infection model of the gastrointestinal pathogen Yptb,29,34 which disseminates into a systemic infection.35 We adoptively transferred sgNTC- or sgFlcn-transduced YopE-I cells (expressing a transgenic T cell receptor [TCR] specific to Yptb) into Tcra−/− mice, followed by oral infection with attenuated Yptb ΔyopM (Figure 2G). After rechallenge with WT Yptb at day 30 p.i.,29 we found that total and CD69+CD103+ Flcn-deficient YopE-I cell numbers were increased in the siIEL (Figure 2H). Further, these cells better controlled systemic dissemination of Yptb (Figure 2I). Thus, targeting Flcn improves both quantity and quality of the small intestinal TRM response.
Increased Tfeb activity upon Flcn, Ragulator, and Rag GTPases deficiencies or amino acid deprivation promotes TRM formation in cooperation with Tfe3
To establish molecular mechanisms, we isolated Flcn-deficient and control OT-I cells derived from the spleen or siIEL on day 7.5 p.i. and performed assay for transposase-accessible chromatin through high-throughput sequencing (ATAC-seq) to assess chromatin state, followed by transcription factor footprinting and motif enrichment analyses.36-38 Footprinting analysis unveiled increased activity of Mitf family transcription factors (includes Tfeb, Tfe3, Mitf, and Tfec) in Flcn-deficient cells (Figures 3A and S3A). Further, transcriptome profiling of Flcn-deficient siIEL cells at day 7.5 p.i., followed by gene set enrichment analysis (GSEA), showed that Tfeb-regulated genes39 were elevated (Figure 3B). Mitf family transcription factors promote lysosome-related gene expression.39-41 Accordingly, Flcn-deficient siIEL cells expressed more lysosomal-associated membrane protein 1 (LAMP-1) than control cells (Figure 3C). Also, LAMP-1 expression and Tfeb-activated signature (see STAR Methods) were increased in cells deficient for Ragulator and Rag GTPase complexes (Figures 3C and 3D). Nuclear Tfeb levels were also elevated in splenic and siIEL cells lacking Flcn, Lamtor4, RagA, or RagC (Figures 3E and S3B). Moreover, Tfeb activity was increased during TRM generation, as Tfeb-regulated genes39 and lysosome-associated signatures were increased in P14 siIEL cells compared with splenic P14 cells at days 4 and 7 post-acute LCMV infection42 (Figure S3C). Further, siIEL cells expressed higher levels of nuclear Tfeb than splenic cells at day 7.5 p.i. after LM-OVA infection (Figure S3D). Thus, aberrant Tfeb activation is a shared feature of Flcn, Ragulator, and Rag GTPase complex deficiencies, and elevated Tfeb activity at the effector phase is associated with siIEL TRM formation.
Figure 3. Flcn, Ragulator, and Rag GTPase deficiencies or amino acid starvation activates Tfeb to promote siIEL TRM development.
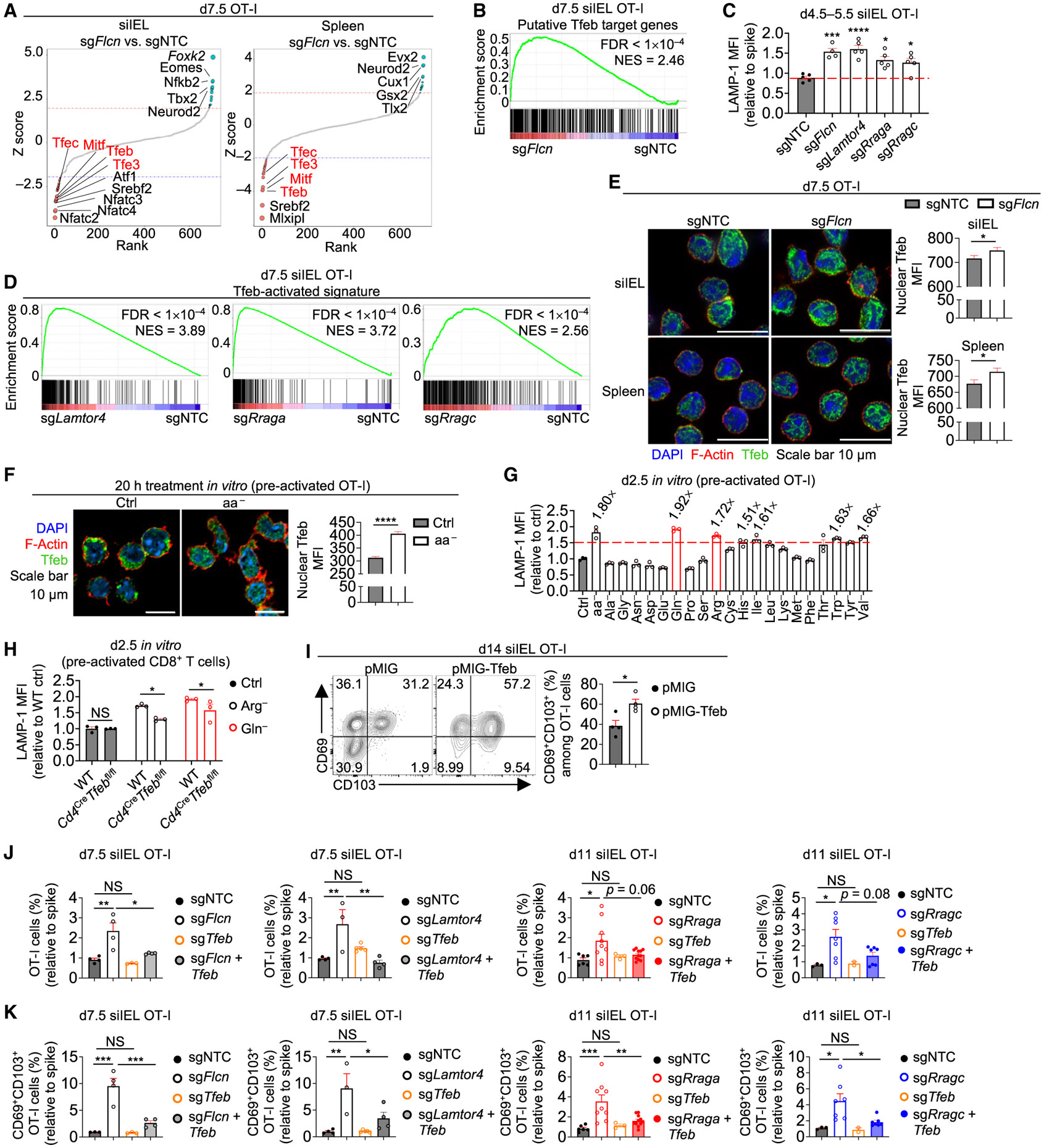
(A) sgNTC (Ametrine+)- and sgFIcn (GFP+)-transduced OT-I cells from indicated tissues of same host mice were profiled by ATAC-seq at day 7.5 p.i. Footprinting analysis in sgFIcn- versus sgNTC-transduced OT-I cells is shown.
(B) Indicated sgRNA-transduced OT-I cells in siIEL at day 7.5 p.i. were profiled for transcriptome analysis. GSEA enrichment plot of Tfeb putative target genes39 comparing sgFIcn- versus sgNTC-transduced OT-I cells. FDR, false discovery rate; NES, normalized enrichment score.
(C) Intracellular LAMP-1 expression (relative to spike; based on mean fluorescence intensity [MFI]) of indicated sgRNA-transduced OT-I cells in siIEL at days 4.5–5.5 p.i. (n ≥ 4 per group) (from dual-color transfer system).
(D) GSEA enrichment plots of Tfeb-activated signature (see STAR Methods) in indicated sgRNA-transduced versus sgNTC-transduced spike siIEL OT-I cells that were profiled for transcriptome analysis at day 7.5 p.i. (from dual-color transfer system).
(E) Nuclear Tfeb levels (based on MFI) in indicated sgRNA-transduced OT-I cells from siIEL or spleen at day 7.5 p.i. (from dual-color transfer system) (n > 140 cells per group).
(F) Confocal imaging analysis of nuclear Tfeb levels (based on MFI) in pre-activated OT-I cells cultured in control RPMI medium (Ctrl) or RPMI medium lacking all amino acids (aa−) for 20 h (see STAR Methods) (n > 160 cells per group).
(G) Intracellular LAMP-1 expression (relative to cells in Ctrl medium; based on MFI) in pre-activated OT-I cells cultured in RPMI medium lacking indicated amino acids for 2.5 days, with fold-change (if >1.5) relative to Ctrl medium indicated (n = 3 technical replicates per group).
(H) Intracellular LAMP-1 expression (relative to WT cells in Ctrl medium; based on MFI) in pre-activated WT or Cd4CreTfebfl/fl CD8+ T cells cultured in Ctrl and arginine-free (Arg−) or glutamine-free (Gln−) RPMI medium for 2.5 days (n = 3 technical replicates per group).
(I) Frequencies of CD69+CD103+ pMIG (mCherry+) and pMIG-Tfeb (GFP+)-transduced OT-I cells in siIEL at day 14 p.i. (from dual-color transfer system) (n = 4 per group).
(J and K) Frequencies (relative to spike) of total (J) or CD69+CD103+ siIEL OT-I cells (K) transduced with indicated sgRNAs at days 7.5 or 11 p.i. (from dual-color transfer system) (n ≥ 3 per group). One-way ANOVA (C, J, and K), two-tailed paired Student’s t test (E and I), two-tailed unpaired Student’s t test (F), two-way ANOVA (H). NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data (mean ± SEM) are compiled from or represent ≥2 experiments (C and E–K).
See also Figure S3.
Flcn, Ragulator, and Rag GTPases regulate nutrient-dependent signaling at lysosomes, which includes regulation of mTOR complex 1 (mTORC1) signaling.23,24 Therefore, we examined phosphorylation of ribosomal protein S6 and 4E-BP1 (two established mTORC1 targets23,24) at the early effector phase that is associated with dynamic mTORC1 signaling.25 The phosphorylation of S6 or 4E-BP1 was reduced in the absence of Lamtor4 or RagA but not Flcn (Figure S3E). Next, we examined mTORC1 function in TRM development in OT-I cells lacking Raptor. At this early time point, Flcn (but not Lamtor4 or RagA)-deficient cells were accumulated in siIEL, and Raptor-deficient siIEL cells were reduced (Figure S3F). Further, Raptor-deficient cells did not show increased CD103 expression in siIEL, in contrast to cells lacking Flcn, Lamtor4, or RagA (Figure S3G), suggesting that reduced mTORC1 signaling alone does not account for increased cell accumulation or CD103 expression in siIEL cells lacking Lamtor4 or RagA. Rather, alternative signaling events downstream of Flcn, Ragulator, or Rag GTPase complexes may be involved.
Because Tfeb is activated upon amino acid deprivation,23,43 we next examined nuclear Tfeb levels and LAMP-1 expression upon total amino acid deprivation in pre-activated CD8+ T cells and found both were increased (Figures 3F and 3G). We then tested the effects of depleting individual amino acids and found that CD8+ T cells cultured in medium lacking glutamine, arginine, histidine, isoleucine, tryptophan, and valine had a >1.5-fold increase in LAMP-1 expression than cells cultured in control medium, with arginine or glutamine-free medium having the largest effect (Figure 3G). Further, arginine or glutamine deprivation-induced LAMP-1 expression was partly dependent on Tfeb (Figure 3H). Thus, acute starvation of selective amino acids promotes Tfeb activity in activated CD8+ T cells.
The above data raised the possibility that elevated Tfeb activity promotes siIEL TRM development. Thus, we transduced OT-I cells with a retroviral vector to overexpress constitutively active Tfeb,44 followed by adoptive transfer and LM-OVA infection. Upon increasing Tfeb activity, indicated by elevated LAMP-1 expression (Figure S3H), the frequency of CD69+ CD103+ cells in siIEL was enhanced (Figure 3I). Further, Tfeb co-deletion in Flcn-, Lamtor4-, RagA-, and RagC-deficient OT-I cells partly blocked the phenotypes of enhanced accumulation of total and CD69+CD103+ cells in siIEL (Figures 3J and 3K). Therefore, enforced activation of Tfeb enhances TRM development in small intestine.
As Tfeb co-deletion in Flcn-deficient OT-I cells only partly mitigated their elevated siIEL TRM development, we asked whether elevated Tfe3 activity in Flcn-deficient cells (Figures 3A and S3A) contributes to enhanced TRM formation. We transduced OT-I cells from Cd4CreFlcnfl/flCas9+ mice (or control mice without Cre-recombinase, called Flcnfl/flCas9+) with sgRNAs targeting Tfeb or Tfe3. Tfeb or Tfe3 single deletion in Flcn-deficient OT-I cells partly rectified increased siIEL cells at the effector phase, while co-deletion of both Tfeb and Tfe3 in Flcn-deficient cells further rectified cell accumulation (Figure S3I). For mechanistic insights, we performed transcriptome profiling and weighted gene correlation network analysis (WGCNA)45 to cluster genes into nine modules (M1-M9) (Figure S3J; Table S3). WGCNA M4 and M6 contained genes with elevated and reduced expression, respectively, in the absence of Flcn, and their expression profiles were partly rectified by Tfeb and/or Tfe3 co-deletion. M4 genes included Itgae (encoding for CD103) and were enriched for the core TRM signature7 and curated siIEL TRM signatures7,8,46 (see STAR Methods), while the core TCIRC signature7 and curated TCM and TEM signatures7,46 (see STAR Methods) were enriched in M6 genes (Figure S3K; Table S3). Further, GSEA revealed decreased siIEL TRM signatures and elevated TCM and TEM signatures in Tfe3-deficient and Tfeb-Tfe3-deficient cells (Figure S3L). Moreover, Tfeb or Tfe3 deletion reduced selective siIEL TRM signature genes, including Fabp1 and Fabp2 (Frizzell et al.47) (Figure S3M). Thus, Tfeb and Tfe3 contribute to siIEL TRM programming in both WT and Flcn-deficient contexts.
Deletion of Flcn promotes CCR9 expression and T cell trafficking to small intestine
We next addressed cellular mechanisms by which Flcn-deficient OT-I cells accumulate in small intestine and found that their cell proliferation or survival was not increased (Figures S4A-S4F). To test the involvement of cell migration to the small intestine, an important step in establishing tissue residency,6,48 we utilized an in vivo migration assay.7 The proportion of Flcn-deficient versus control P14 cells was increased in siIEL but decreased in other tissues (Figures 4A and 4B), suggesting an enhanced capacity to localize to small intestine. Transcriptome profiling of Flcn-deficient TEFF cells revealed Ccr9, which contributes to small intestinal CD8+ T cell homing,49,50 among the top genes with increased expression (Figure 4C). Further, Flcn-deficient TEFF cells had increased expression of CCR9, but not small intestinal homing integrin α4β7 (Kok et al.6), in the blood, siIEL, and PP (Figures 4D, 4E, S4G, and S4H). Additionally, retinoic acid-induced CCR9 expression51 was elevated on splenic Flcn-deficient CD8+ T cells in vitro (Figure 4F). Mechanistically, Tfeb, and to a lesser extent Tfe3, contributed to increased CCR9 expression in Flcn-deficient TEFF cells (Figures 4G and S3J). Next, we co-deleted Flcn and CCR9 and found that CCR9 co-deletion blocked accumulation of Flcn-deficient total cells in siIEL (Figure 4H). Thus, accumulation of Flcn-deficient cells in small intestine requires CCR9, and Tfeb and Tfe3 contribute to elevated CCR9 expression in Flcn-deficient cells.
Figure 4. Flcn deficiency enhances CCR9 expression and CD8+ T cell trafficking to small intestine.
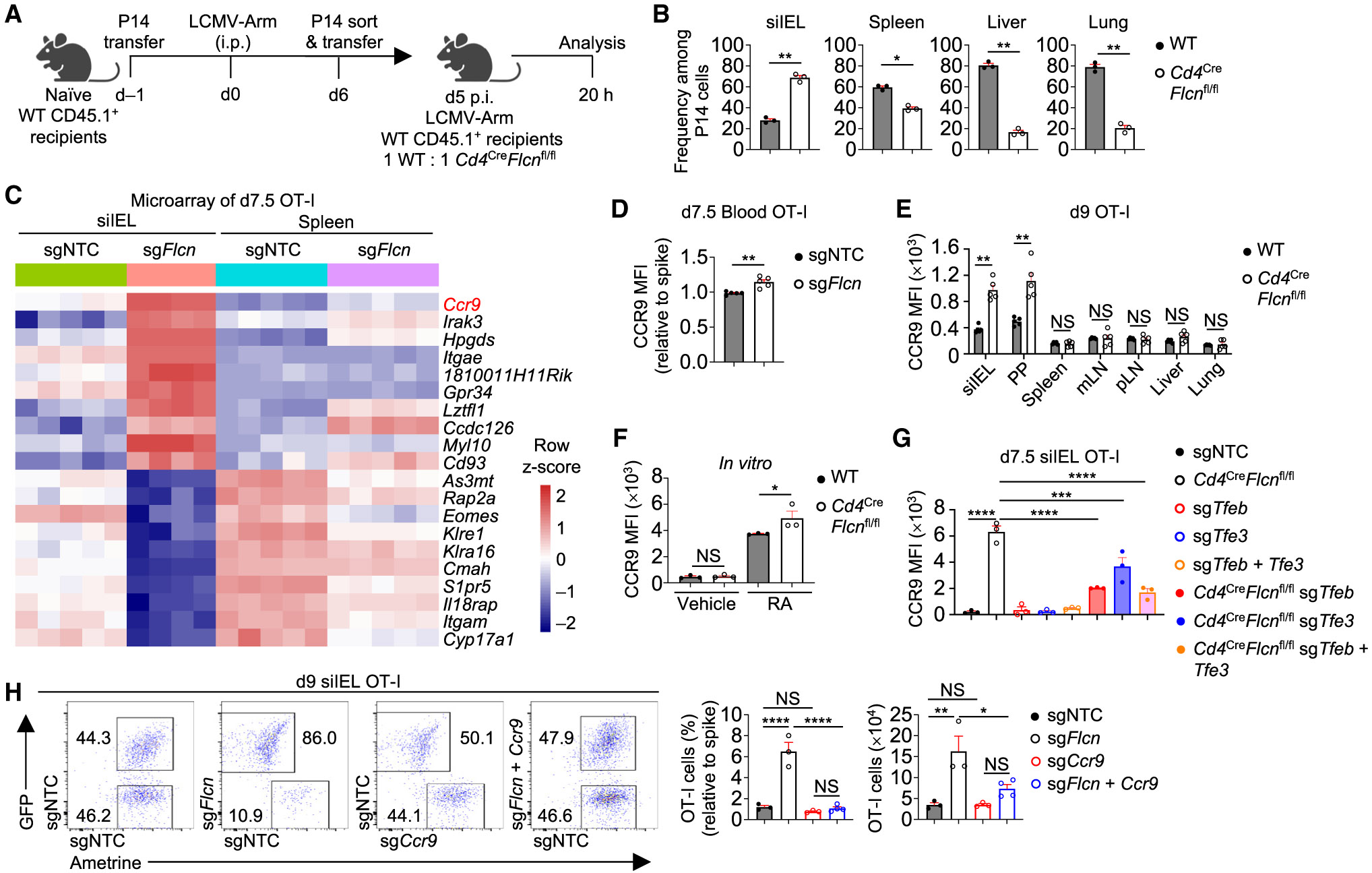
(A and B) Schematic for CD8+ T cell migration assay (A). Frequencies among transferred P14 cells in indicated tissues (B) (n = 3 per group).
(C) Transcriptome profiling of indicated sgRNA-transduced OT-I cells in siIEL or spleen at day 7.5 p.i. (from dual-color transfer system). Heatmap depicts the top 10 increased (log2FC > 0.5, FDR < 0.05; ranked by log2FC) and decreased (log2FC < −0.5, FDR < 0.05; ranked by log2FC) genes in sgFIcn- versus sgNTC- transduced siIEL OT-I cells.
(D) CCR9 expression (relative to spike; based on MFI) on indicated sgRNA-transduced OT-I cells in the blood at day 7.5 p.i. (from dual-color transfer system) (n = 5 per group).
(E) Naive WT and Flcn-deficient (from Cd4CreFlcnfl/fl mice) OT-I cells were sort-purified and co-transferred into WT mice at a 1:1 ratio, followed by LM-OVA infection 1 day later. Quantification of CCR9 expression (based on MFI) on OT-I cells from the indicated tissues at day 9 p.i. (n = 5 per group).
(F) CCR9 expression (based on MFI) on WT and Flcn-deficient (from Cd4CreFlcnfl/fl mice) CD8+ T cells cultured with retinoic acid (or vehicle) for 5 days (see STAR Methods) (n = 3 technical replicates per group).
(G) WT (from Flcnfl/flCas9+ mice) or Flcn-deficient (from Cd4CreFlcnfl/flCas9+ mice) OT-I cells were transduced or co-transduced with indicated sgRNAs. Quantification of CCR9 expression (based on MFI) on siIEL OT-I cells at day 7.5 p.i. is shown (n = 3 per group).
(H) Frequency (relative to spike) and number of indicated sgRNA-transduced OT-I cells in siIEL at day 9 p.i. (from dual-color transfer system) (n ≥ 3 per group). Two-tailed paired Student’s t test (B and E), two-tailed unpaired Student’s t test (D), two-way ANOVA (F), one-way ANOVA (G and H). NS, not significant; *p < 0.05, **p < 0.01, ****p < 0.0001. Data (mean ± SEM) represent ≥2 (B, D, E, G, and H) or three (F) experiments.
See also Figure S4.
Flcn deficiency and amino acid deprivation accelerate TRM programming by interplaying with TGF-β signaling
Beyond memory time points, Flcn-deficient OT-I cells also accumulated in siIEL (but not spleen) at earlier stages of infection based on flow cytometry and confocal imaging analyses (Figures S5A-S5C). Additionally, Flcn-deficient cells had increased proportions and numbers of CD69−CD103+ and CD69+CD103+ cells in the small intestine (Figures S5D and S5E). To examine whether Flcn-deficient cells undergo altered or accelerated TRM programming, we performed transcriptome profiling and GSEA of splenic WT and Flcn-deficient cells at day p.i. This analysis revealed that Flcn-deficient cells were enriched for the core TRM gene signature, whereas the core TCIRC signature was reduced (Figure S5F), suggesting that these cells may be predisposed for TRM development. Further, KLRG1 expression was decreased in Flcn-deficient splenic OT-I cells (Figure S5G), in line with KLRG1lo cells better developing into siIEL TRM cells than KLRG1hi cells.7,8 We next used single-cell RNA sequencing (scRNA-seq) to explore Flcn-coordinated regulation of cellular diversity and TRM programming. Uniform manifold approximation and projection (UMAP) plot analysis revealed that control and Flcn-deficient cells displayed altered transcriptional signatures at days 4.5 and 7.5 p.i. (Figure S5H). Compared with control siIEL cells, Flcn-deficient cells increased siIEL TRM and reduced TCM signatures7 at both time points (Figure S5I), suggesting that early TEFF cells in the small intestine may acquire TRM programs more rapidly in the absence of Flcn. Unbiased subclustering analysis identified three subclusters marked by high or low Itgae expression or cell cycling (based on Mki67 expression) (Figures 5A and S5J). Further, the core TRM and curated siIEL TRM signatures were elevated in the Itgae+ subcluster, while the core TCIRC and curated TCM signatures were reduced (Figure 5B), suggesting that the Itgae+ subcluster resembles TRM cells. Slingshot pseudotime analysis52 uncovered a differentiation trajectory from the cycling cluster through the Itgae− cluster to the Itgae+ cluster, with cells from day 7.5 p.i. predicted to be more terminally differentiated than those from day 4.5 p.i. (Figures 5A and 5C). Moreover, Flcn-deficient cells accumulated at the more terminal stage of pseudotime at both days 4.5 and 7.5 p.i (Figure 5C), supporting their accelerated TRM differentiation. Collectively, Flcn-deficient siIEL cells display an early induction for small intestinal TRM programs.
Figure 5. Flcn limits TGF-β signaling to control TRM programming.
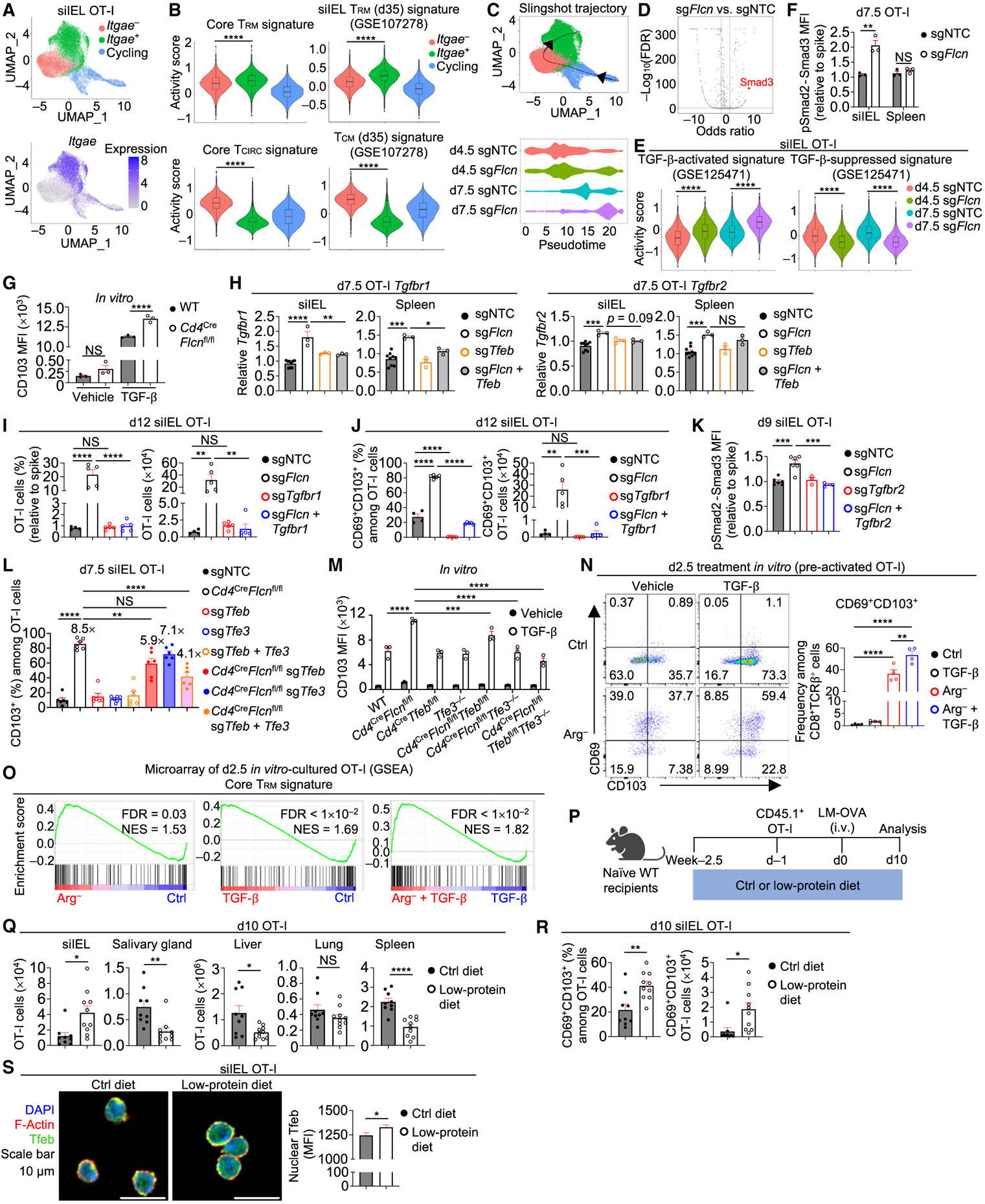
(A) sgNTC (Ametrine+)- and sgFlcn (GFP+)-transduced OT-I cells from siIEL of the same host were profiled by scRNA-seq at days 4.5 and 7.5 p.i. (n = 2 per group; pooled from 2 mice). UMAP plots depicting three clusters (cycling, Itgae−, and Itgae+) and Itgae expression.
(B) Violin plots showing activity scores of core TRM signature,7 core TCIRC signature,7 curated siIEL TRM signature,7 and curated TCM signature7 (see STAR Methods) among indicated 3 clusters.
(C) UMAP plot showing Slingshot trajectory analysis52 and pseudotime plot of indicated sgRNA-transduced siIEL OT-I cells at days 4.5 and 7.5 p.i.
(D) Plot depicting transcription factor motif enrichment analysis (including Smad3) comparing sgFlcn- versus sgNTC-transduced siIEL OT-I cells that were profiled by ATAC-seq at day 7.5 p.i.
(E) Violin plots showing activity scores of TGF-β-activated and TGF-β-suppressed signatures53 (see STAR Methods) in indicated sgRNA-transduced siIEL OT-I cells from scRNA-seq profiling in (A).
(F) pSmad2-Smad3 levels (relative to spike; based on MFI) in indicated sgRNA-transduced OT-I cells from siIEL or spleen on day 7.5 p.i. (n = 3 per group).
(G) CD103 expression (based on MFI) on WT or Flcn-deficient (from Cd4CreFlcnfl/fl mice) CD8+ T cells cultured with rhTGF-β1 (or vehicle) for 5 days (see STAR Methods) (n = 3 technical replicates per group).
(H) Real-time PCR analysis of Tgfbr1 and Tgfbr2 expression in indicated sgRNA-transduced OT-I cells from siIEL or spleen at day 7.5 p.i. (n ≥ 3 per group).
(I and J) Frequencies (relative to spike) and numbers of total (I) or CD69+CD103+ siIEL OT-I cells (J) transduced with indicated sgRNAs at day 12 p.i. (from dual-color transfer system) (n ≥ 4 per group).
(K) pSmad2-Smad3 levels (relative to spike; based on MFI) in indicated sgRNA-transduced OT-I cells from siIEL at day 9 p.i. (from dual-color transfer system) (n ≥ 3 per group).
(L) WT (from Flcnfl/flCas9+ mice) or Flcn-deficient (from Cd4CreFlcnfl/flCas9+ mice) OT-I cells that were transduced or co-transduced with indicated sgRNAs. Frequency of CD103+ siIEL OT-I cells at day 7.5 p.i. is shown. Fold-change relative to sgNTC is indicated (n = 6 per group).
(M) CD103 expression (based on MFI) on CD8+ T cells cultured with rhTGF-β1 (or vehicle) for 5 days (see STAR Methods) (n = 3 technical replicates per group).
(N) Pre-activated OT-I cells were transferred into Ctrl RPMI medium or Arg− RPMI medium containing rhTGF-β1 (or vehicle) for 2.5 days (see STAR Methods). Frequency of CD69+CD103+ OT-I cells is shown (n = 4 technical replicates per group).
(O) GSEA enrichment plots showing the core TRM signature7 in cells cultured in Arg− versus Ctrl medium, cells cultured in Ctrl medium plus rhTGF-β1 versus Ctrl medium, or cells cultured in Arg− medium plus rhTGF-β1 versus Ctrl medium plus rhTGF-β1 (as in N).
(P–R) Schematic for LM-OVA infection in mice fed control or low-protein diet (P). OT-I cell numbers in indicated tissues (Q) or frequency and number of CD69+CD103+ siIEL OT-I cells (R) at day 10 p.i. (n = 9–10 per group).
(S) Nuclear Tfeb MFI in siIEL OT-I cells from mice fed indicated diets (n > 30 cells per group). Wilcoxon rank sum test (B and E), two-tailed unpaired Student’s t test (F and Q–S), one-way ANOVA (H–L and N), two-way ANOVA (G and M). NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data (mean ± SEM) are compiled from two (K, L, Q, and R) or represent ≥2 (F), one (H–J and S), or three (G, M, and N) experiments.
See also Figure S5.
Motif enrichment analysis of aforementioned ATAC-seq data revealed enriched activity of Smad3 (Figure 5D), a downstream mediator of TGF-β signaling,54 in Flcn-deficient cells. Additionally, a TGF-β-activated signature53 was increased and a TGF-β-suppressed signature53 was reduced in Flcn-deficient siIEL cells (Figure 5E). Further, Flcn-deficient cells had markedly elevated expression of CD103, a target for TGF-β signaling4,10 (Figures S3G, S5D, and S5E). Thus, Flcn-deficient cells may be more responsive to TGF-β, which promotes TRM formation and maintenance in the small intestine.10,11,27,53 Accordingly, phosphorylated Smad2-Smad3 (pSmad2-Smad3) levels were elevated in Flcn-deficient siIEL cells (Figure 5F). Moreover, CD103 induction by TGF-β stimulation was enhanced in Flcn-deficient CD8+ T cells (Figure 5G). To establish the underlying mechanisms, we examined expression of TGF-βR1 and TGF-βR2, which dynamically control T cell sensitivity to TGF-β signaling.54-56 Flcn-deficient OT-I cells had increased expression of Tgbr1 and, to a lesser extent, Tgfbr2, than control cells (Figure S5K). Further, deletion of Tfeb in Flcn-deficient cells rectified increased expression of Tgfbrl in Flcn-deficient cells from spleen and siIEL (Figure 5H). Thus, Flcn deficiency promotes enhanced TGF-β signaling in siIEL TRM cells, and the Flcn-Tfeb axis modulates expression of TGF-β receptors.
Next, we transduced WT and Flcn-deficient OT-I cells with sgTgfbr1 or sgTgfbr2 to test their functional contribution to TRM development. TGF-βR1 or TGF-βR2 co-deletion in Flcn-deficient cells rectified the accumulation of siIEL cells (Figures 5I and S5L). Further, compared with Flcn-deficient cells, Flcn-TGF-βR1 or Flcn-TGF-βR2 double-deficient cells did not show elevated differentiation into CD69+CD103+ cells in the small intestine, associated with a blockade of excessive pSmad2-Smad3 signals (Figures 5J, 5K, and S5M). Thus, aberrant TGF-β receptor signaling contributes to altered siIEL responses occurring in the absence of Flcn. We next tested whether Flcn affects TGF-β signaling via Tfeb or Tfe3. The increased expression of Itgae in Flcn-deficient cells was partly rescued by the deletion of Tfeb and/or Tfe3 (Figure S3J). Further, the accumulation of Flcn-deficient CD103+ cells in the small intestine and elevated TGF-β-mediated induction of CD103 expression on Flcn-deficient cells were partly rectified by codeletion of Tfeb and/or Tfe3 (Figures 5L and 5M). By contrast, TGF-βR1 co-deletion in Flcn-deficient cells did not rescue elevated LAMP-1 expression (Figure S5N). These data indicate that the Tfeb-Tfe3 pathway acts upstream of TGF-β signaling to shape TRM-related programming.
To further explore the interplay between nutrient stress-sensitive Tfeb-Tfe3 pathway and TGF-β signaling, CD8+ T cells were activated and cultured in arginine- or glutamine-free medium with or without TGF-β, which can induce a TRM-like gene signature in vitro.53 Arginine or glutamine starvation, in combination with TGF-β, increased the proportions of CD69+CD103+ CD8+ T cells compared with vehicle treatment or TGF-β treatment alone (Figures 5N and S5O). Transcriptome profiling showed that arginine starvation and TGF-β treatment alone or in combination induced changes in gene expression profiles of activated CD8+ T cells (Figure S5P). Activated CD8+ T cells cultured in arginine-free medium or treated with TGF-β were enriched for core TRM and curated siIEL TRM signatures, while core TCIRC, curated TCM, and curated TEM signatures were reduced (Figures 5O and S5Q). Further, compared with CD8+ T cells treated with TGF-β alone, cells cultured under arginine starvation together with TGF-β stimulation had enriched and reduced TRM− and TCIRC-related signatures, respectively (Figures 5O and S5Q). Altogether, loss of Flcn or deprivation of selective amino acids sensitizes CD8+ T cells to TGF-β-induced TRM programming.
To test the contribution of diet-derived amino acids to TRM development, we examined TRM formation in mice fed with control or low-protein diet (Figure 5P). While OT-I cell numbers were reduced in salivary gland, liver, and spleen (but not lung) in mice fed with a low-protein diet, cell number was increased in the small intestine (Figure 5Q). CD69+CD103+ cells also accumulated in small intestine in mice fed with a low-protein diet (Figure 5R), coincident to elevated nuclear Tfeb levels in siIEL cells (Figure 5S). Together, these data show that limiting dietary protein levels supports TRM formation in small intestine, albeit at the expense of cell quantity in lymphoid and other non-lymphoid tissues.
Genetic interaction screening identifies mitochondrial functions in supporting TRM generation
We examined possible mechanisms by which CD8+ T cells adapt to nutrient availability to orchestrate tissue immunity,12,15 including under conditions of Flcn deficiency that resemble nutrient stress (based on activation of Tfeb and Tfe340,43). To this end, we performed a secondary in vivo CRISPR-Cas9 screen by transducing Flcn-deficient OT-I cells with the pooled lentiviral sgRNA library targeting mitochondria and lysosome genes, followed by adoptive transfer and LM-OVA infection (Figure 6A). First, we identified 167 genetic perturbations that selectively alleviated increased accumulation of Flcn-deficient but not WT siIEL cells (see STAR Methods; Figure S6A upper; Table S4). Mitochondrial translation was the top enriched pathway in those genes (Figure S6B; Table S5). Second, we examined sgRNA abundance in CD103+ and CD103− siIEL cells to identify positive contributors to increased CD103 expression in Flcn-deficient cells, which nominated 167 additional genes (Figure S6A lower; Table S4) that were enriched in the mitochondrial OXPHOS pathway (Figure S6B; Table S5). Thus, Flcn-deficient cells appear to require mitochondrial translation and OXPHOS for their accumulation and TRM programming, with such effects consistent with the important role for mitochondria in positively regulating TRM formation.14,17
Figure 6. Mitochondria- and lysosome-scale genetic interactions for TRM development.
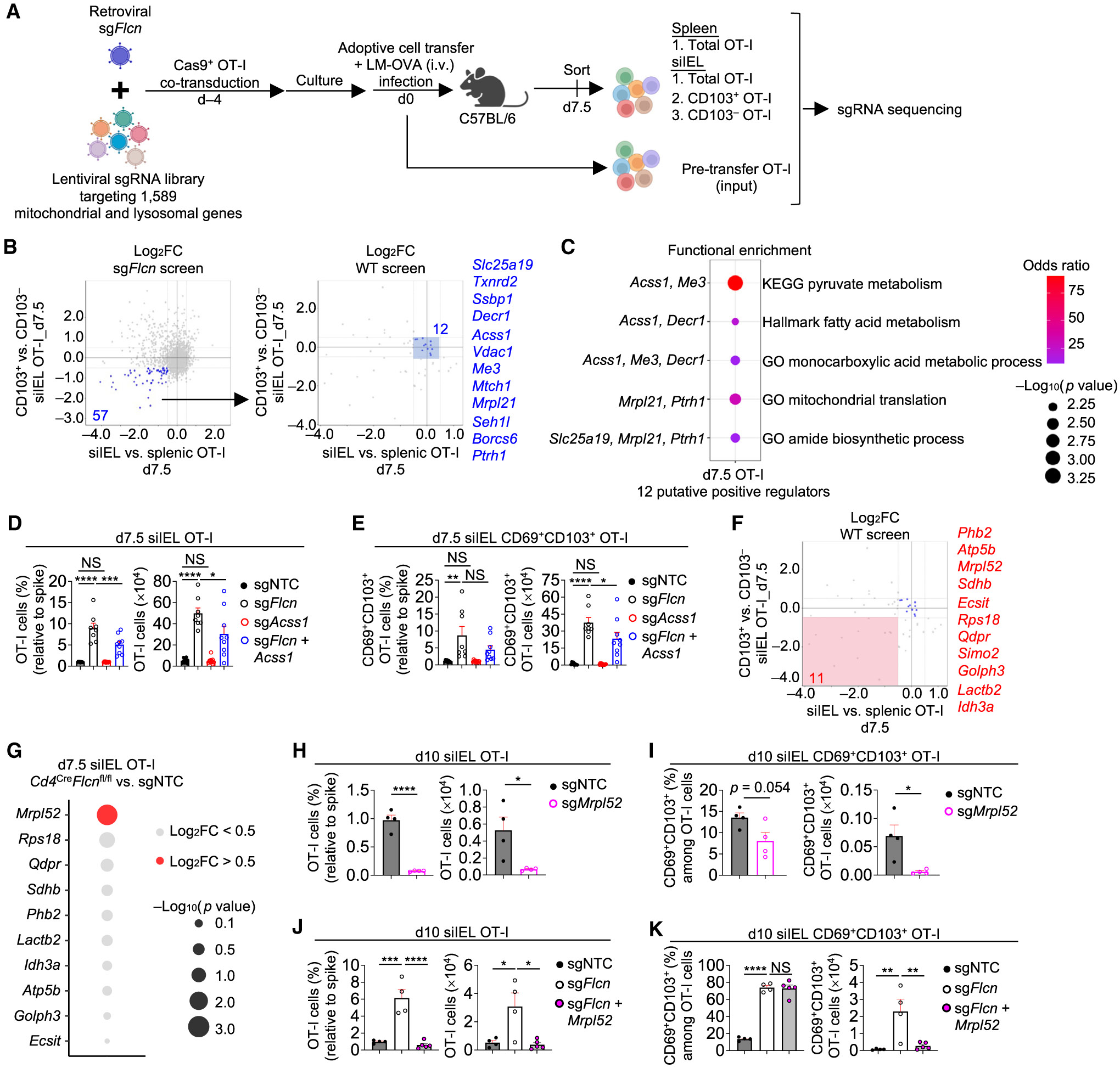
(A) Schematic for in vivo genetic interaction CRISPR screen in sgFlcn-transduced Cas9-expressing OT-I cells.
(B) Sectored scatterplots of gene-level log2FC from CRISPR screens using sgFlcn-transduced OT-I cells (described in A) or WT (Flcn-sufficient) OT-I cells (described in Figure 1A). 12 of 57 genes were nominated as positive regulators of Flcn-deficient total and CD103+ siIEL OT-I cell accumulation without effects on WT OT-I cells (see STAR Methods).
(C) Functional enrichment analysis of the 12 genes defined in (B).
(D and E) Frequencies (relative to spike) and numbers of total (D) or CD69+CD103+ (E) siIEL OT-I cells transduced with indicated sgRNAs at day 7.5 p.i. (from dual-color transfer system) (n ≥ 8 per group).
(F) Sectored scatterplot of gene-level log2FC in WTOT-I cells from the CRISPR screen as described in (B). 11 of 57 genes were nominated as positive regulators of both Flcn-deficient and WT total and CD103+ siIEL OT-I cell accumulation (see STAR Methods).
(G) Bubble plot depicting log2FCs and p values of the 11 genes from (F), comparing Flcn-deficient versus sgNTC-transduced siIEL OT-I cells that were profiled for transcriptome analysis at day 7.5 p.i. (see also Figure S3J).
(H–K) Frequencies (relative to spike) and numbers of total (H and J) or CD69+CD103+ (I and K) siIEL OT-I cells transduced with indicated sgRNAs at day 10 p.i. (from dual-color transfer system) (n ≥ 4 per group). Fisher’s exact test (C), one-way ANOVA (D, E, J, and K), or two-tailed unpaired Student’s t test (H and I). NS, not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Data (mean ± SEM) are compiled from two (D and E) or represent two (H–K) experiments.
See also Figure S6.
We next focused on putative targets that could rectify both OT-I cell accumulation and CD103+ siIEL generation by selecting for genetic perturbations that selectively lowered accumulation of Flcn-deficient siIEL relative to spleen cells and those that decreased CD103 expression in Flcn-deficient siIEL cells (Figure 6B), leading to the identification of 57 candidates. Out of these 57 genes, we excluded those that also had perturbation effects in the WT background, leading to the identification of 12 candidates with possible rescue effects on both Flcn-deficient siIEL accumulation and CD103+ cell generation (Figure 6B). Pyruvate metabolism was the top enriched pathway in these 12 genes (Figure 6C; Table S5), which included Acss1 that can convert acetate into acetyl-coenzyme A (CoA) in mitochondria.57,58 Acss1 co-deletion partly blocked the increased accumulation of Flcn-deficient siIEL cells (Figure 6D) and increased cellularity (albeit not proportion) of Flcn-deficient CD69+CD103+ siIEL cells (Figure 6E). Thus, Acss1 contributes to early accumulation of Flcn-deficient siIEL CD8+ T cells.
We next extended our analysis of putative targets whose deletions could block total and CD103+ siIEL cell accumulation in both WT and Flcn-deficient cells. Among the 57 genetic perturbations that reduced the accumulation of Flcn-deficient siIEL cells and their enhanced CD103 expression (Figure 6B), 11 of them also had perturbation effects in WT cells, thereby nominating them as positive regulators in WT and Flcn-deficient contexts (Figure 6F). Functional enrichment analysis of these 11 genes revealed OXPHOS, TCA cycle, adipogenesis, and mitochondrion organization among the top pathways (Figure S6C; Table S5). Among these 11 genes, only Mrpl52, a mitochondrial ribosomal gene elevated in TMEM cells,59 was expressed at higher levels in Flcn-deficient than control silEL cells (Figure 6G). We therefore tested the contribution of Mrpl52 to both Flcn-sufficient and Flcn-deficient siIEL TRM development. In Flcn-sufficient cells, Mrpl52 deletion reduced total and CD69+CD103+ cell accumulation in the siIEL (Figures 6H and 6I). Mrpl52 co-deletion in Flcn-deficient cells also blocked accumulation of total and CD69+CD103+ siIEL cells (Figures 6J and 6K). Thus, Mrpl52 positively regulates early accumulation and TRM-like programming of WT and Flcn-deficient CD8+ T cells in small intestine.
DISCUSSION
How nutrients and organelle biology contribute to TRM responses and tissue immunity remain understudied. Using in vivo CRISPR-Cas9 screens, we revealed mitochondrial and lysosomal processes that positively and negatively regulate TRM development in the small intestine, respectively. Further, Tfeb-Tfe3 signaling, which is induced by amino acid deprivation, contributes to TRM development, thereby linking nutrient stress to cell fate decisions. Moreover, the Flcn-Tfeb axis controls cell trafficking to the small intestine and interplays with TGF-β signaling to tune siIEL TRM programming. Genetic interaction screening identified Acss1 and Mrpl52 as critical for regulating TRM metabolic adaptation in different contexts. Altogether, these results establish three lysosome-associated signaling nodes that connect nutrient and immunological signals to cell fate decisions for orchestrating tissue immunity in the small intestine.
Beyond bioenergetic roles, mitochondria and lysosomes can regulate cell fate decisions.60,61 Further, extracellular nutrients and metabolites impact TRM formation,14,17,47,56,62 but how cells integrate nutrient availability with signaling processes remains elusive. We revealed that deletion of Flcn, Ragulator, or Rag GTPases enhances TRM development via Tfeb and/or Tfe3. Amino acid deprivation (especially arginine or glutamine) in vitro or low-protein diets in vivo recapitulates these effects, suggesting that arginine and glutamine may regulate T cell memory by both signaling25 and metabolic effects.63 Besides amino acids,23,24 whether restriction of other nutrients regulating Rag GTPase activity64,65 also contributes to TRM programming warrants further investigation. Moreover, whether cellular energetic stress signals through the AMP-activated protein kinase (AMPK)-Tfeb axis62,66 to orchestrate TRM formation could be explored in future studies.
Metabolic adaptation underlies cellular fitness in different microenvironments.12 Utilizing Flcn deficiency to model a condition of nutrient stress, we revealed a dependence on mitochondrial enzyme Acss1 for TRM development under such conditions. These results extend upon the previous findings that Acss167,68 and acetate69,70 contribute to CD8+ T cell effector functions or longevity in different contexts of nutrient restriction. Our study suggests that crosstalk between lysosomes and mitochondria integrates nutrient stress signals with cellular metabolic adaptation within different tissue microenvironments. Whether the interplay exists between these lysosome-associated complexes and autophagy, another lysosome-mediated process with roles in TCIRC71 and TRM72 development, warrants further investigation.
Negative regulators of CD8+ T cell fate decisions are powerful immunotherapeutic disease targets.73,74 Despite much emphasis on positive regulators,4,75 relatively less is known about negative or spatiotemporal regulators of TRM development. We showed that targeting Flcn enhances retinoic acid-induced CCR9 expression and cell migration to the small intestine. Flcn deficiency also sensitizes cells to TGF-β signaling for accelerated TRM programming, thus establishing Flcn as a potent and specific negative regulator of small intestinal TRM development and protective immunity. However, deficiency in Ragulator or Rag GTPases promotes the establishment of not only small intestinal TRM cells but also TMEM cells in other tissues, suggesting that increased Tfeb activity may not always skew CD8+ T cells away from the TCIRC pool to the intestinal TRM pool. Indeed, these effects on TCIRC generation may be attributed to the partial reduction of mTORC1 signaling, as impaired mTORC1 signaling is associated with enhanced TCIRC formation.25,76 Ablation of mTORC1 signaling by Raptor deletion impeded early TRM development, likely through the inhibition of cell growth and proliferation.77 Because mTORC1 promotes mitochondrial ribosome biogenesis and OXPHOS in naive T cells during quiescence exit,19 mTORC1 may orchestrate such mitochondrial processes to promote TRM development in the small intestine and possibly other non-lymphoid tissues.
Collectively, our findings establish an interplay between nutrient stress and organelle signaling in shaping TRM development and provide targets to enhance TRM formation and function to better protect against pathogen infection. We propose a multistep, temporal model of TRM development regulated by nutrient-dependent signaling, which is composed of mTORC1-mediated cell expansion (which requires nutrients22,77,78), retinoic acid-induced, CCR9-dependent cell migration to the small intestine, and tissue-specific immune signals for TRM differentiation. The gain-of-function effects achieved by targeting Flcn and potentially other negative regulators of TRM cells identified in our study will likely open avenues for immunotherapeutic intervention of infection and possibly cancer.2,4,5 These regulators, as well as mitochondrial pathways, may also contribute to our understanding and modulation of autoimmunity and other inflammatory diseases, where TRM has been implicated in exacerbating disease.2,4
Limitations of the study
While Flcn inhibits functional TRM formation at both effector and memory phases, whether Flcn contributes to the maintenance of TRM cells requires investigation. Tfeb activity is dynamically regulated during early siIEL TRM formation, although how TRM cells or their putative precursors experience nutrient restriction requires further exploration. This question is important to address, as dietary interventions to treat infection or other diseases may have broad impacts on immunity.12,13 Moreover, how Tfeb activation promotes Ccr9 and Tgfbr1 expression requires investigation. Finally, the effects of organelle and nutrient-directed signaling processes on human TRM responses await further study.
STAR★METHODS
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
Male and female mice at 6–20 weeks of age were used for the study. C57BL/6, OT-I,91 Rosa26-Cas9 knockin (Strain # 026179)92 (Cas9+), Tfe3−/− (Strain #042292), and Tcra−/− (Strain # 002116) mice were purchased from The Jackson Laboratory. Cd4Cre mice have been previously described.93 Flcnfl/fl mice94 and Tfebfl/fl mice43 were kindly provided by Laura Schmidt and Andrea Ballabio, respectively. T cell-specific deletion of Flcn or Tfeb was generated by breeding Cd4Cre mice with Flcnfl/fl mice or Tfebfl/fl mice, and these mice were further bred to mice with germline deletion of Tfe3. Rosa26-Cas9+ mice were crossed with OT-I, P14,95 or YopE-I transgenic mice to express Cas9 in antigen-specific CD8+ T cells. We also crossed Cd4CreFlcnfl/fl mice with Cas9+ OT-I, OT-I or P14 transgenic mice to generate Flcn-deficient antigen-specific CD8+ T cells. YopE-I and Tcra−/− mice were bred and maintained under specific-pathogen-free (SPF) conditions in an American Association for the Accreditation of Laboratory Animal Care (AAALAC)-accredited animal facility at the NIAID, and experiments were performed in accordance with the NIAID Animal Care and Use Committee. All other mice were housed in SPF conditions in the Animal Resource Center at St. Jude Children’s Research Hospital. Experiments and procedures were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) of St. Jude Children’s Research Hospital.
Cell lines
Retroviral packaging Plat-E cells, provided by Dr. Yun-Cai Liu, were from female origin. with the core plasmid (sgRNA plasmid or pMIG-II-overexpressing plasmid) and packaging plasmid pCL-Eco (Addgene #12371). HEK293T cells were used for lentivirus production. Cells were cultured in DMEM (catalog #11965118, Thermo Fisher Scientific) containing 10% FBS and 1× penicillin–streptomycin at 37 °C.
METHOD DETAILS
Cell isolation
Isolation of siIEL was performed as previously described.34,96 Briefly, Peyer’s patches were removed, and the intestine was cut longitudinally and washed in PBS to remove the luminal contents. The intestine was then cut laterally into 1 cm pieces and incubated in 10 ml digestion buffer [RPMI 1640 (catalog #11875085, Thermo Fisher Scientific) containing 0.154 mg/ml (1 mM) dithiothreitol (catalog #D9779, Sigma-Aldrich), 5mM EDTA (catalog #15575020, Thermo Fisher Scientific), 25 mM HEPES (catalog #15630-080, Gibco), and 55 μM β-mercaptoethanol (catalog #M6250, Sigma-Aldrich)] for 20 min at 37 °C while shaking. The digestion buffer was then strained through a 70 μm filter (Falcon), and single cell suspensions were separated using a 40/80% (vol/vol) Percoll (catalog #P1644, Sigma-Aldrich) gradient. Isolation of lymphocytes from the salivary gland or kidney was performed as previously described.33 Briefly, salivary gland or kidney was cut into small pieces and incubated in 10 ml digestion buffer [RPMI 1640 containing 5% FBS (Thermo Fisher Scientific), 2 mM MgCl2 (catalog #AM9530G, Ambion), 2 mM CaCl2 (catalog # J63122, Thermo Fisher Scientific), and 100 IU/ml type I collagenase (catalog #LS004194, Worthington)] for 45 min at 37 °C while shaking. For isolation of lymphocytes from the liver or lung, mice were perfused through the left ventricle of the heart with phosphate buffered saline (PBS, Thermo Fisher Scientific) before tissues were harvested. Liver or lung was cut into pieces and incubated in 10 ml digestion buffer [(HBSS containing 2% FBS, 0.5 mg/ml DNase I (catalog #DN25, Sigma-Aldrich), and 1 mg/ml type IV collagenase (catalog #LS004188, Worthington)] for 30 min at 37 °C while shaking. After enzymatic digestion, salivary gland, kidney, liver, or lung tissue was further mechanically dissociated over a 70 μm filter. Single cell suspension from salivary gland, kidney, liver, or lung was separated using a 40/80% (vol/vol) Percoll gradient to isolate the lymphocytes. Spleen, pLN, mLN, Peyer’s patches, and bone marrow were processed by mechanical dissociation in HBSS containing 2% FBS and straining through 70 μm nylon mesh. Blood was collected via retro-orbital route. For spleen and blood, red blood cells were lysed with ACK buffer (catalog #A1049201, Thermo Fisher Scientific).
Flow cytometry
Immune cells were isolated from blood and tissues as described above. For analysis of surface markers, cells were stained in PBS containing 2% FBS for 30 min at room temperature with the appropriate surface antibodies: anti-TCRβ (H57-597, Biolegend), anti-CD8α (53-6.7, Cytek/Tonbo), anti-CD103 (2E7, Thermo Fisher Scientific), anti-CD69 (H1.2F3, Biolegend), anti-CCR9 (CW-1.2, BD Bioscience), anti-CD45.1 (A20, Biolegend), anti-CD45.2 (104, Biolegend), anti-Thy1.1 (OX-7, Biolegend), anti-Thy1.2 (53-2.1, Biolegend), streptavidin (Thermo Fisher Scientific), anti-α4β7 (DATK32, Thermo Fisher Scientific), anti-KLRG1 (2F1, Biolegend), anti-CD62L (MEL-14, Biolegend) and fixable viability dye (catalog #65-0865, Thermo Fisher Scientific). For analysis of intracellular LAMP-1 (1D4B, Thermo Fisher Scientific) expression, cells were fixed with 4% formaldehyde (catalog #18814-20, Polysciences), followed by permeabilization with BD Perm buffer (catalog #554723, BD Biosciences). For analysis of intracellular Ki67 (SolA15, Thermo Fisher Scientific) expression, cells were fixed in 2% paraformaldehyde (Thermo Fisher Scientific) followed by permeabilization using 90% methanol. For assessment of cytokine production, OT-I cells were stimulated with OVA257–264 (SIINFEKL) peptide (1 μM; Macromolecular Synthesis Core Facility, St. Jude Children’s Research Hospital) in the presence of GolgiStop (catalog #554724, BD Bioscience) and GolgiPlug (catalog #555029, BD Bioscience) for 5 h at 37 °C. Intracellular staining of IFN-γ (XMG1.2, Biolegend) and TNF-α (MP6-XT22, Biolegend) was performed using BD CytoFix/CytoPerm fixation/permeabilization kit (catalog #554714, BD Biosciences). For detection of phosphorylated proteins, cells were fixed with Phosflow lyse/fix buffer (558049, BD Biosciences), permeabilized with Phosflow perm buffer III (558050, BD Biosciences), and stained with antibodies for phosphorylated-S6 (Ser235–Ser236, D57.2.2E, Cell Signaling Technology), phosphorylated-4EBP1 (Thr37–Thr46, 236B4, Cell Signaling Technology), or phosphorylated-Smad2-Smad3 (Ser465-Ser467 (Smad2) and Ser423-Ser425 (Smad3), 072-670, BD Biosciences). BrdU and active caspase-3 staining was performed according to the manufacturer’s instructions (catalog #552598, BD Biosciences) using anti-BrdU (catalog #552598, BD Biosciences or Bu20a, Biolegend) and anti-active caspase-3 (C92-605, BD Biosciences). Flow cytometry data were acquired on LSRII, LSR Fortessa, or Symphony A3 instruments (BD Biosciences) using FACSDiva software (version 8) and analyzed using FlowJo software (Tree Star, v10.10.0).
Viral production
The lentiviral and retroviral sgRNA vectors were previously described.25,74 Retrovirus was produced by co-transfecting Plat-E cells (provided by Y.-C. Liu) with the core plasmid (sgRNA plasmid or pMIG-II-overexpressing plasmid) and packaging plasmid pCL-Eco (Addgene #12371). Lentivirus was produced by co-transfecting the lentiviral mitochondria-lysosome library plasmids, psPAX2, and pCAG4-Eco in HEK293T cells. For both retrovirus and lentivirus, supernatant was harvested at 48 h after transfection and stored at −80 °C. For genes that were included in our mitochondria-lysosome library, we chose two guides with the highest levels of enrichment in our primary or genetic interaction screens. sgRNAs not within the mitochondria–lysosome library (e.g., Tgfbr2) were designed using an online tool from Broad Institute (https://portals.broadinstitute.org/gppx/crispick/public). Individual sgRNAs used in this study were listed in Table S6. The constitutively active Tfeb sequence (Addgene #79014) was subcloned into the pMIG-II retroviral vector (Addgene #52107).
Naive T cell isolation and viral transduction
Naive OT-I, P14, or YopE-I cells were isolated from the spleen and peripheral lymph nodes (pLN) of Cas9+ OT-I, Cas9+ P14, or Cas9+ YopE-I mice by magnetic bead purification according to the manufacturer’s instructions (naïve CD8+ T cell isolation kit, cat #130-096-543, Miltenyi Biotech). Purified naive OT-I, P14, or YopE-I cells were activated for 20 h with 5 μg/ml plate-bound anti-CD3 (2C11, Bio X Cell) and 5 μg/ml plate-bound anti-CD28 (37.51, Bio X Cell) in complete Click’s medium (catalog #9195, Irvine Scientific) containing 10% fetal bovine serum (FBS; R&D Systems), 1× penicillin–streptomycin–L-glutamine (catalog #15140122, Thermo Fisher Scientific), and 55 μM β-mercaptoethanol. Lentiviral and retroviral transductions were performed by spin-infection at 900g for 3 h with 10 μg/ml polybrene (catalog #TR-1003, Sigma-Aldrich). After spin-infection, cells were placed in fresh complete Click’s medium containing recombinant mouse (rm) IL-7 (12.5 ng/ml; catalog #217-17, PeproTech) and rmIL-15 (25 ng/ml; catalog #210-15, PeproTech) for 4 days. Cells were then sorted using a Reflection (iCyt), MoFlo (BD Biosciences), or BigFoot (Thermo Fisher Scientific) cell sorter based on fluorescent protein expression (Ametrine, GFP, or mCherry as indicated in methods and/or figure legends) and adoptively transferred into recipient mice.
In vivo adoptive transfer, infection, and recall assay
For adoptive transfer of naive OT-I or P14 cells, a total of 1–2×104 cells were transferred intravenously (i.v.) into naïve C57BL/6 mice. For adoptive transfer of retrovirus-transduced OT-I cells, a total of 1-2×104 [for experiments at day >5 post-infection (p.i.)] or 2×105 (for experiments at day <5 p.i.) cells were transferred i.v. into naïve mice. 1×105 retrovirus-transduced YopE-I cells were transferred i.v. into naive mice. We used single-color transfer experiments to examine YopE-I recall responses to WT Yersinia pseudotuberculosis (Yptb) (32777 strain) secondary infection, and dual-color transfer systems for all other experiments to examine cell-intrinsic effects of the applied genetic perturbations. For the single-color transfer system, YopE-I cells were transduced with sgNTC or sgFlcn expressing GFP and transferred into separate Tcra−/− mice. For the dual-color transfer system, OT-I or P14 cells were transduced with sgNTC (called "spike") or sgRNA expressing different fluorescent proteins (Ametrine, GFP, or mCherry), and then co-transferred at a 1:1 ratio into the same host, followed by infection with LM-OVA or LCMV-Armstrong 1–3 hours later.
For infection, 3×104 colony forming units (CFU) of Listeria monocytogenes expressing ovalbumin (LM-OVA) were injected i.v., and 2×105 plaque forming units (PFU) of LCMV-Armstrong strain virus were injected intraperitoneally (i.p.). Mice were infected with 1×107 CFU of mutant Yptb (YptbΔyopM) by oral gavage.29 For secondary Yptb infection at day 30 p.i., mice were challenged by oral gavage with 5×109 CFU wild-type (WT) Yptb, and bacterial burden was assessed in the spleen after 3 days, as previously described.29,34 Bacterial burden was determined by serial plating on MacConkey plates and incubating at room temperature for 48 h. Colonies were counted to calculate the CFU per gram (CFU/g) in the spleen.
In vivo treatments and cell trafficking assay
To distinguish vascular-associated CD8+ T cells, 3 μg of anti-CD8α antibody conjugated to biotin (53-6.7, Thermo Fisher Scientific) was injected intravenously into mice 3 minutes prior to euthanasia, as previously described.33 For flow cytometric detection of i.v. injected anti-CD8α antibody, fluorochrome-conjugated streptavidin (Thermo Fisher Scientific) was used.
For low-protein dietary treatments, mice were fed irradiated control diet (5CC7 Baker amino acid with 16% total protein, TestDiet) or irradiated isocaloric low-protein diet (5BT9 Mod TestDiet 5CC7 with 2% total protein, TestDiet), as previously described.97 Briefly, 8-week-old C57BL/6 mice were pre-treated with the diets for 2.5 weeks, followed by transfer of WT CD45.1+ OT-I cells one day prior to LM-OVA infection. Mice were maintained on control and low-protein dietary treatment conditions throughout the course of the experiment.
In vivo cell trafficking was performed as previously described.7 Briefly, P14 cells were isolated from the spleen and pLN of LCMV-Armstrong-infected mice at day 6 p.i., sorted according to congenic markers, and WT and Flcn-deficient P14 cells were adoptively transferred at a 1:1 ratio into recipient mice that were infected with LCMV-Armstrong 5 days prior. At 20 h after secondary transfer, cells were isolated from the spleen, siIEL, liver, and lung, and P14 cells were analyzed by flow cytometry.
Cell culture assays
In vitro TGF-β and retinoic acid (RA) cultures were done as previously described.98 Briefly, naive CD8+ T cells that were isolated from the spleen were cultured in Click’s medium (catalog #9195, FujiFilm Irvine Scientific; supplemented with 10% FBS, β-mercaptoethanol and 1× penicillin–streptomycin-L-glutamine) with irradiated T cell-depleted splenocytes (for antigen presenting cells), 5 μg/ml soluble anti-CD3, 5 μg/ml soluble anti-CD28, and 100 IU/ml rhIL-2 (catalog #23-6019, Sigma-Aldrich) together with 5 ng/ml rhTGF-β1 (catalog #240-B, R&D) or 30 nM RA (catalog #R2625, Sigma-Aldrich) for 5 days at 37°C. At day 5, cell surface expression of CD103 (for cultures with TGF-β) or CCR9 (for cultures with RA) was assessed by flow cytometry using the antibodies indicated above (see Flow Cytometry subsection).
For in vitro cultures using amino acid-deficient medium, naive CD8+ T cells were isolated from the spleen and activated with 5 μg/ml plate-bound anti-CD3 and 5 μg/ml plate-bound anti-CD28 for 48 h, and then expanded in cytokines (5 ng/ml rmIL-7, 25 ng/ml rmIL-15, and 100 IU/ml rhIL-2) for 24 h at 37 °C; this system was chosen to bypass the requirements of amino acids to promote the initial activation of T cells,13 and also to mimic our conditions of gene perturbation in pre-activated CD8+ T cells. Cells were then transferred into control RPMI medium (prepared from powder; catalog #R8999-04A, US Biological) or single amino acid free RPMI medium (prepared from powder as above and supplemented with all essential and non-essential amino acids78 except for the one indicated in figures) containing 10% dialyzed FBS (A3382001, Thermo Fisher Scientific), 5 ng/ml rmIL-7, 25 ng/ml rmIL-15, and 100 IU/ml rhIL-2, with or without 5 ng/ml rhTGF-β1 for 20 h40 [for imaging analysis of nuclear Tfeb (see Immunofluorescence subsection)] to 2.5 days53 (for flow cytometry analysis of LAMP-1 expression and TRM-like differentiation based on CD69 and CD103 cell surface expression and transcriptome profiling) at 37°C.
Immunofluorescence
sgNTC (Ametrine+ or GFP+)-, sgFlcn (GFP+)-, sgLamtor4, sgRraga, or sgRragc (all Ametrine+)-transduced OT-I cells were sort-purified from the siIEL or spleen of mice at day 7.5 p.i., followed by nuclear Tfeb quantification. To examine the effects of amino acid deprivation on Tfeb nuclear localization, WT CD8+ T cells cultured in control or amino acid-deficient (aa−) medium for 20 h prior to imaging, as described above. For nuclear Tfeb quantification, cells were allowed to settle onto poly-D-lysine-coated coverslips for 10 min at 37 °C prior to fixation with 4% paraformaldehyde for 10 min. Cells were permeabilized with 0.1% Triton X100 for 3 min prior to blocking in PBS containing 2% BSA, 5% normal donkey serum and 0.05% Tween-20. Cells were incubated with anti-Tfeb (rabbit polyclonal, catalog #13372-1-AP, ProteinTech) and detected with Alexa Fluor Plus 647-labeled donkey anti-rabbit secondary antibody (catalog #A32795, Thermo Fisher Scientific), while Alexa Fluor 568-labeled phalloidin (catalog #A12380; Thermo Fisher Scientific) was utilized to detect F-actin. Coverslips were mounted in Vectashield Vibrance mounting media with DAPI (catalog #H-1800, Vector Laboratories) and were imaged using a Marianas spinning disk confocal (3i; Intelligent Imaging Innovations) equipped with Sora (Yokagawa), Prime 95B sCMOS camera (Photometrics) and a 1.45 NA 100× oil objective. Images were acquired and analyzed using Slidebook software (version 6.0.24; 3i).
For imaging of OT-I cells in the small intestine, sgNTC (mCherry+)- and sgFlcn (GFP+)-transduced OT-I cells were co-adoptively transferred into mice, followed by LM-OVA infection. At day 7 p.i., tissues were fixed in 4% paraformaldehyde prior to embedding in tissue freezing medium. 10 μm thick cryosections were blocked in PBS containing 2% bovine serum albumin, 5% normal donkey serum and 0.05% Tween-20 followed by incubation with the following reagents: GFP booster (catalog #gba488, Chromotek/ ProteinTech), anti-mCherry (catalog #orb11618, Biorbyt), and biotin-labeled anti-Epcam (clone G8.8, Biolegend). Sections were washed in PBS followed by incubation with Alexa Fluor Plus 555-labeled secondary antibody (catalog #A32816, Thermo Fisher Scientific) and Alexa Fluor 647-labeled streptavidin (catalog #S21374, Thermo Fisher Scientific). Sections were mounted with Vectashield Vibrance mounting media with DAPI (Vector Laboratories; Catalog H-1800) and imaged using an inverted Ti2 eclipse microscope (Nikon Instruments) equipped with a 20× 0.75 NA Plan Apo objective, SOLA light engine LED light source (Lumencorp) and Orca Fusion digital CMOS camera (Hamamatsu). Images were acquired and analyzed using NIS Elements software (version 5.30.05, Nikon Instruments).
RNA isolation and real-time PCR
Real-time quantitative PCR analysis was performed using primers and Sybr Green PCR Master Mix (catalog #4309155, Thermo Fisher Scientific), as previously described.78 Briefly, all RNA was isolated from 1×105 sort-purified splenic or siIEL OT-I cells using the RNeasy Micro Kit according to the manufacturer’s instructions (catalog #74004, Qiagen), and mRNA was reverse transcribed to cDNA for subsequent real-time PCR analysis using High-Capacity cDNA Reverse Transcription kit (catalog #4374966, Thermo Fisher Scientific). Gene amplification was assessed using an Applied Biosystems QuantStudio 7 Flex quantitative PCR machine (Thermo Fisher Scientific). Primer sequences used were: Tgfbr1 (forward 5’- TCTGCATTGCACTTATGCTGA; reverse 5’- AAAGGGCGATCTAGTGATGGA) and Tgfbr2 (forward 5’- GACTGTCCACTTGCGACAAC; reverse 5’- GGCAAACCGTCTCCAGAGTAA).
In vivo CRISPR–Cas9 screening
Lentiviral sgRNA mitochondria–lysosome library construction
A custom mouse mitochondria-lysosome library containing sgRNA targeting 1,589 genes was synthesized based on the gene lists in the MitoCarta 2.0 (refs.Calvo et al.99 and Pagliarini et al.100) and GO: 0005764 Lysosome (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) databases. A total of four sgRNAs were designed for each gene, and 500 non-targeting controls were also included (Table S6). The synthesis, purification, and quality control of the library was performed by the Center for Advanced Genome Engineering at St. Jude Children’s Research Hospital as previously described.74
In vivo screening
The in vivo screening approach was modified based on previous studies.25,74 Naive Cas9+ OT-I cells were isolated and pooled from 12 Cas9+ OT-I mice and activated overnight with plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) antibodies. Cells were then transduced with the lentiviral mitochondria–lysosome library at a MOI of 0.3 to achieve 20% transduction efficiency and cultured for 4 days in Click’s medium containing rmIL-7 (12.5 ng/ml) and rmIL-15 (25 ng/ml) to allow for gene editing. Transduced OT-I cells were sorted based on Ametrine expression and 6×105 cells were then transferred i.v. into naive C57BL/6 mice, followed by LM-OVA infection 2 h later. Triplicate aliquots of 1×106 transduced OT-I cells were saved as ‘input’ (approximately 145× coverage per sgRNA). A total of 120 recipient mice were randomly divided into 6 groups (3 groups at day 7.5 p.i. and 3 groups at day 30 p.i.) as biological replicates. At days 7.5 and 30 p.i., donor-derived OT-I cells were sorted from the spleen [bulk OT-I (days 7.5 and 30), CD44hiCD62Llo TEM OT-I (day 30), CD44hiCD62Lhi TCM OT-I (day 30)] and the siIEL [bulk OT-I (days 7.5 and 30), CD103+ OT-I (day 7.5), and CD103− OT-I (day 7.5)], and genomic DNA was extracted. A minimum of 3×105 OT-I cells per sample (approximately 40× coverage per sgRNA) was recovered for sgRNA sequencing.
Sequencing library preparation
The library was prepared as previously described.25,74 Briefly, genomic DNA was extracted by using the DNeasy Blood and Tissue Kit (catalog #69504, Qiagen) according to the manufacturer’s instructions. Primary PCR was performed by using the KOD Hot Start DNA Polymerase (catalog #71086, Millipore) and the following pair of Nextera NGS primers: Nextera NGS-F: TCGTCGGCAGCGTCAGA TGTGTATAAGAGACAGTTGTGGAAAGGACGAAACACCG; Nextera NGS-R:
GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCCACTTTTTCAAGTTGATAACGG. Primary PCR products were purified using the AMPure XP beads (catalog #A63881, Beckman Coulter), followed by a second PCR to add adaptors and indexes to barcode each sample. Hi-Seq 50-bp single-end sequencing (Illumina) was performed.
Data processing and analysis of in vivo CRISPR screening
For data analysis, raw FASTQ files obtained after sequencing were demultiplexed using the HiSeq Analysis software (Illumina), as described.74 Single-end reads were trimmed, matched against sgRNA sequences from the sgRNA library, and counted by mageck software (version 0.5.9.4). Read counts for sgRNAs were normalized against median read counts across all samples. For each sgRNA and corresponding gene, the fold change (FC; log2-transformed ratio), P value and false discovery rate (FDR) for enrichment were calculated between indicated groups by mageck test command. The read counts of each sgRNA were also analyzed by DrugZ software80 to calculate the normZ (Normalized z-score) score for enrichment at gene level between indicated groups. From the following comparisons, the normZ scores at gene level were hierarchically clustered into four clusters (C1, C2, C3 and C4) by heatmap.2 function in gplots R package (version 3.1.1): 1. siIEL TEFF versus splenic TEFF (day 7.5); 2. siIEL TRM versus splenic TCIRC (day 30); 3. siIEL TRM versus splenic TCM (day 30); 4. siIEL TRM versus splenic TEM (day 30). This clustering analysis identified candidate genes with perturbation effects (∣z-score∣ > 1.69) at effector (comparison 1) and memory (comparisons 2–4) phases of T cell responses. Negative z-score values indicate putative positive regulators and positive z-score values indicate putative negative regulators. Functional enrichment of the genes in the four clusters was performed using funcEnrich.Fisher function in NetBID2 R package (version 2.0.2)81 using pooled HALLMARK, KEGG, and GO gene sets from the Molecular Signatures Database (MsigDB).
Protein-protein interaction network analysis
PPI network analysis was performed using our in-house JUMPn software (version 0.19.006) as previously described.19,20,82,101 Briefly, the genes in clusters 1–4 (C1–C4) were superimposed onto a composite PPI database (includes the STRING (v10),102 BioPlex,103 and InWeb_IM104 databases) with edge confidence scores filtered by best fitting the scale-free network property.105 The PPI network was then visualized by Cytoscape (version 3.7.256).83 The primary protein modules were then identified by MCODE algorithm84 in Cytoscape app clusterMaker106 and annotated by core enriched categories.
Measurement of genome editing efficiency
Assessment of genome editing efficiency by sgRNAs was performed as previously described.25 Briefly, targeted amplicons were generated using gene-specific primers with partial Illumina adaptor overhangs and sequenced as previously described.107 Cell pellets of approximately 1×105 cells were lysed and used to generate gene-specific amplicons by first round PCR, followed by a second round of PCR to index the samples. Indexed amplicons were pooled with other targeted amplicons for other loci to create sequence diversity. Additionally, 10% PhiX Sequencing Control V3 (Illumina) was added to the pooled amplicon library prior to running the sample on a Miseq Sequencer System (Illumina) to generate paired 2× 250bp reads. Samples were demultiplexed using the index sequences, fastq files were generated, and insertion and deletion (indel) mutation analyses were performed using CRIS.py.85 We achieved high efficiency of genome editing (56%–96%) for all target genes tested (Table S6).
Genetic interaction screening
For the in vivo genetic interaction screen, naive Cas9+ OT-I cells were isolated and activated overnight with plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) antibodies. Cells were then co-transduced retrovirus expressing sgFlcn (GFP+) and with the lentiviral mitochondria-lysosome library (Ametrine+) at a MOI of 0.3 to achieve 20% transduction efficiency and cultured for 4 days in Click’s medium containing rmIL-7 (12.5 ng/ml) and rmIL-15 (25 ng/ml) to allow for gene editing. Transduced OT-I cells were sorted based on GFP and Ametrine co-expression and 6×105 cells were then transferred i.v. into naive C57BL/6 mice, followed by LM-OVA infection 2 h later. Triplicate aliquots of 1×106 transduced OT-I cells were saved as ‘input’ (approximately 145× coverage per sgRNA). A total of 60 recipient mice were randomly divided into 3 groups as biological replicates. At day 7.5 p.i., donor-derived OT-I were sorted from the spleen (bulk OT-I) and the siIEL [bulk OT-I (day 7.5), CD103+ OT-I (day 7.5), CD103− OT-I (day 7.5)], and genomic DNA was extracted. A minimum of 3×105 OT-I cells per sample (approximately 40× coverage per sgRNA) was recovered for sgRNA sequencing, and library preparation was performed. The sequencing library preparation and sample sequencing was performed as described above.
Data processing and analysis of the genetic interaction screening
For CRISPR screens with WT or sgFlcn-transduced OT-I cells, FASTQ read files obtained after sequencing were demultiplexed using the Hi-Seq analysis software (Illumina) and processed using mageck software (version 0.5.9.4).79 Raw count tables were generated by mageck count command by matching sequence of the aforementioned mitochondria–lysosome library. Read counts for sgRNAs were normalized against median read counts across all samples for each screening.
For each gene or sgRNA in the mitochondria–lysosome library, the log2FC for enrichment or depletion was calculated with mageck test command, with gene-lfc-method parameter as mean and control-sgrna parameter using the list of non-targeting control sgRNAs. The log2FC values of each genetic perturbation from WT and sgFlcn-transduced OT-I cell screens were then compared, with the following cut-offs applied. For Figure S6A (upper panel): Genetic perturbations that alleviated the increased accumulation of Flcn-deficient siIEL cells relative to spleen cells were identified based on log2FC (sgFlcn-transduced siIEL OT-I cells versus sgFlcn-transduced spleen OT-I cells) < −0.5, and then we filtered out those genetic perturbations that lowered siIEL (versus spleen) accumulation in the WT (Flcn-sufficient) background from the aforementioned in vivo CRISPR screen based on log2FC (WT siIEL OT-I cells versus WT spleen OT-I cells) < −0.5), to nominate a total of 167 putative targets. For Figure S6A (lower panel): Genetic perturbations that mitigated the increased ratio of Flcn-deficient CD103+ relative to Flcn-deficient CD103− siIEL cells were identified based on log2FC (sgFlcn-transduced CD103+ siIEL versus sgFlcn-transduced CD103− siIEL OT-I cells) < −0.5), and then we removed those genetic perturbations that also reduced CD103+ siIEL percentage in WT cells based on log2FC (WT CD103+ siIEL versus WT CD103− siIEL OT-I cells) < −0.5), to identify another set of 167 putative targets. For Figure 6B, two Flcn-dependent parameters were used for comparison to nominate the candidates: (1) siIEL OT-I versus splenic OT-I cells to identify factors with selective accumulation of siIEL cells; (2) CD103+ versus CD103− siIEL OT-I cells to uncover factors mediating early TRM programming. Within each parameter, cutoffs were applied in the fold-change/fold-change (FC/FC) plot to identify those factors that had rescue effects on the above parameters in sgFlcn-transduced OT-I cells (log2FC < −0.5), leading to the identification of 57 candidates. To further uncover candidate genes that had selective rescue effects of these two phenotypes induced by Flcn deletion, out of these 57 genes, we excluded those that also had perturbation effects in the WT (Flcn-sufficient) background from the aforementioned in vivo CRISPR screen based on ∣ log2FC∣ >0.5, which identified 12 candidate genes as shown in Figure 6B. For Figure 6F, two cut-offs were applied to the FC/FC plot of the 57 genes identified in Figure 6B to identify those factors that had rescue effects in the above parameters in both sgFlcn-transduced OT-I cells and WT OT-I cells: (1) WT siIEL OT-I versus WT splenic OT-I cells (log2FC < −0.5) and (2) WT CD103+ versus WT CD103− siIEL OT-I cells (log2FC < −0.5).
Transcriptome profiling
Microarray analysis was performed on the following samples in different batches: 1) sgNTC (Ametrine+)- and sgFlcn (GFP+)-transduced OT-I cells were sort-purified from spleen and siIEL of the same LM-OVA-infected mice at day 7.5 p.i.; 2) sgNTC (GFP+)- and sgLamtor4, sgRraga, or sgRragc (all Ametrine+)-transduced OT-I cells were sort-purified from the siIEL of the same LM-OVA-infected mice (with their respective sgNTC controls) at day 7.5 p.i.; 3) OT-I cells from WT (Flcnfl/flCas9+) or Flcn-deficient (Cd4Cre Flcnfl/flCas9+) mice were transduced with sgNTC, sgTfeb, or sgTfe3 in various combinations as indicated in figures and transferred into WT mice, followed by LM-OVA infection. At day 7.5 p.i., siIEL OT-I cells were sort-purified based on GFP and Ametrine expression; 4) OT-I cells were activated in control or arginine-deficient medium with or without stimulation with 5 ng/ml rhTGF-β1 for 2.5 days as described above (see Cell culture assays subsection), and live cells were sort-purified using the fixable viability dye. For all microarray analyses, RNA was isolated from 1×104−1×105 sort-purified cells using the RNeasy Micro Kit according to the manufacturer instructions (catalog #74004, Qiagen). RNA samples were analyzed by microarray analysis using the Clariom S Mouse array (Thermo Fisher Scientific).
Microarray data analysis
For microarray analyses, the gene expression probe signals were quantile normalized and summarized by the RMA algorithm by Affymetrix Expression Console (version 1.4.1), then the differential gene expression analysis was performed by R package limma (version 3.46.0).86 False discovery rate (FDR) was estimated by Benjamini–Hochberg method. Heatmaps were generated using ComplexHeatmap (version 2.6.2) to show the average expression of genes from biological replicates of the same genotype. For co-expression network analysis of OT-I cells transduced or co-transduced with sgRNAs targeting Flcn, Tfeb, Tfe3 or their combination (or sgNTC; related to Figures 3 and S3), weighted gene correlation network analysis (WGCNA)45,89 was performed using WGCNA R package (version 1.66). We defined co-expression clusters using differentially expressed genes (∣log2 fold-change∣ > 0.5 and FDR < 0.05) in at least one of the comparisons. Pearson correlation matrix was calculated using the samples from the above mice, followed by an adjacency matrix calculation, with correlation matrix raised to a power of 9 using scale-free topology criterion.45 Co-expression clusters were defined by hybrid, dynamic tree-cutting method, with minimum height for merging module set at 0.2. A consensus trend for each co-expression cluster was defined based on the first principal component (eigengene) and cluster membership was defined as Pearson correlation between individual genes and the consensus trend of the co-expression clusters. Genes were assigned to most correlated co-expression cluster with cutoff of correlation coefficient r > 0.7. Principal component analysis was performed using function prcomp () in R. Microarray data have been deposited into the GEO series database GEO: GSE231502.
GSEA, functional enrichment, and signature curation
Genes were ranked by the fold change generated by differential expression analysis. The pre-ranked gene set enrichment analysis (GSEA)87 was performed as previously described108 against gene sets from the Hallmark collection combined with the various curated gene signatures as indicated in figure legends (see more details below), GO collection, or KEGG collection from the Molecular Signatures Database (https://www.broadinstitute.org/gsea/msigdb/, v7.4). Functional enrichment was performed using funcEnrich.Fisher function in NetBID2 R package (version 2.0.2)81 using the pathway collection containing the various curated gene signatures as indicated in figure legends (see more details below), including the Hallmark collection, GO collection, or KEGG collection from the abovementioned MSigDB. For signature curation, the comparisons were performed as indicated. The Flcn-suppressed, Flcn-activated, and Tfeb-activated signatures were defined based on the differential expression of genes from in-house generated microarray datasets as follows:
-
Flcn-suppressed signature included the shared elevated genes from the following pairwise comparisons:
a day 7.5 p.i. sgFlcn-transduced siIEL OT-I cells versus sgNTC-transduced siIEL OT-I cells (log2FC > 0.5, FDR < 0.05).
b day 7.5 p.i. sgFlcn-transduced spleen OT-I cells versus sgNTC-transduced spleen OT-I cells (log2FC > 0.5, FDR < 0.05).
-
Flcn-activated signature included the shared reduced genes from the following pairwise comparisons:
a day 7.5 p.i. sgFlcn-transduced siIEL OT-I cells versus sgNTC-transduced siIEL OT-I cells (log2FC < −0.5, FDR < 0.05).
b day 7.5 p.i. sgFlcn-transduced spleen OT-I cells versus sgNTC-transduced spleen OT-I cells (log2FC < −0.5, FDR < 0.05).
Tfeb-activated signature: day 7.5 p.i. sgTfeb-transduced siIEL OT-I cells versus sgNTC-transduced siIEL OT-I cells reduced genes (log2FC < −0.5, FDR < 0.05).
The TGF-β-regulated signatures were defined by the following comparisons using transcriptome data from a public dataset53:
TGF-β-activated signature (GSE125471): TGF-β stimulated CD8+ T cells versus unstimulated CD8+ T cells elevated genes (log2FC > 0.5, FDR < 0.05).
TGF-β-suppressed signature (GSE125471): TGF-β stimulated CD8+ T cells versus unstimulated CD8+ T cells reduced genes (log2FC < −0.5, FDR < 0.05).
To generate the curated siIEL TRM, TCM, and TEM signatures, we used public datasets7,8,46 to define the signatures using the following comparisons:
siIEL TRM (day 35) signature (GSE107278): Day 35 p.i. siIEL TRM versus spleen TCM elevated genes (log2FC > 0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
TCM (day 35) signature (GSE107278): Day 35 p.i. siIEL TRM versus spleen TCM reduced genes (log2FC < −0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
siIEL TRM (day 35) signature #2 (GSE107278): Day 35 p.i. siIEL TRM versus spleen TEM elevated genes (log2FC > 0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
TEM (day 35) signature (GSE107278): Day 35 p.i. siIEL TRM versus spleen TEM reduced genes (log2FC < −0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
siIEL TRM (day 55) signature (GSE157072): Day 55 p.i. siIEL TRM versus spleen TCM elevated genes (log2FC > 0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
TCM (day 55) signature (GSE157072): Day 55 p.i. siIEL TRM versus spleen TCM reduced genes (log2FC < −0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
siIEL TRM (day 55) signature #2 (GSE157072): Day 55 p.i. siIEL TRM versus spleen TEM elevated genes (log2FC > 0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
TEM (day 55) signature (GSE157072): Day 55 p.i. siIEL TRM versus spleen TEM reduced genes (log2FC < −0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
siIEL TRM signature (GSE47045): siIEL TRM versus spleen TCM elevated genes (log2FC > 0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
TCM signature (GSE47045): siIEL TRM versus spleen TCM reduced genes (log2FC < −0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
siIEL TRM signature #2 (GSE47045): siIEL TRM versus spleen TEM elevated genes (log2FC > 0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
TEM signature (GSE47045): siIEL TRM versus spleen TEM reduced genes (log2FC < −0.5, FDR < 0.05), in which top 200 genes (ranked by log2FC) were selected.
The core TRM and core TCIRC signatures were defined previously.7 The set of ‘putative Tfeb target genes’ signature was derived from a public dataset, which identified Tfeb targets by integrating Tfeb ChIP-seq and Tfeb overexpression analyses.39
Single-cell RNA-sequencing
Library preparation
sgNTC- or sgFlcn-transduced OT-I cells were transferred at a 1:1 ratio to naive mice that were subsequently infected with LM-OVA. OT-I cells were sort-purified from the spleen and siIEL of the same host on days 4.5 and 7.5 post-LM-OVA infection (n = 2 biological replicates per group, pooled from two mice). Preparation of the single-cell libraries were performed as previously described.25 Briefly, sort-purified cells were pelleted by centrifugation at 2,000 rpm for 5 min. The supernatant was discarded, and cells were resuspended in 1×PBS (Thermo Fisher Scientific) with 0.04% BSA (Amresco) at a concentration of 1×106 cells/ml. Single-cell suspensions were loaded onto the Chromium Controller according to their respective cell counts to generate 9,000 single-cell gel beads in emulsion per sample. Each sample was loaded into a separate channel and single cell libraries were prepared using the Chromium Single Cell 3’ (version 3.1) Library and Gel Bead Kit (10X Genomics) according to the manufacturer’s instructions. The complementary DNA (cDNA) content of each sample was quantified and quality-checked using a high-sensitivity D5000 ScreenTape with a TapeStation (Agilent Technologies) after cDNA amplification of 12 cycles to determine the number of PCR amplification cycles for preparing a sufficient sequencing library. After library quantification and quality-checking, samples were diluted for loading onto the NovaSeq (Illumina) with a 2×100-bp paired-end kit using the following read length: 28-bp read 1, 10-bp i7 index, 10-bp i5 index, and 90-bp read 2. An average sequencing depth of 300 million reads per sample was obtained (approximately 30,000 reads per cell). scRNA-seq data have been deposited into the GEO series database GEO: GSE231502.
Data analysis
For gene expression sequencing, the filtered count matrices were read into the R package Seurat (version 4.0).88 Within each dataset, the processing pipeline was as follows: Samples were merged into a single Seurat object for consistent filtering, and features detected in fewer than 5 cells were removed from the dataset. Feature count and unique molecular identifier (UMI) count distributions were then visually inspected to determine appropriate cutoffs for each dataset. Cells with abnormally low features or UMI counts or high mitochondrial read percentages (potentially dead or damaged cells) were removed. Cells with abnormally high UMI counts (potentially multiple cells in a single droplet) were also removed. For profiling adoptively transferred CD8+ T cells from the siIEL, a total of 34,095 cells were retained with an average of 2,715 genes per cell (UMI, median: 8,554; range: 1,908–39,972). After quality control, libraries were normalized by Seurat function NormalizeData with scale.factor = 106).
For analysis of the siIEL CD8+ T cells, days 4.5 and 7.5 sgNTC- and sgFlcn-transduced OT-I cells (n = 2 replicates for each genotype per time point, pooled from two mice) were isolated and re-clustered using the Seurat workflow described above. To remove batch effect between datasets, the package Harmony was used to integrate CD8+ T cells from the eight samples. After the RunHarmony step with default parameters, the Seurat functions RunUMAP (dims = 1:30), FindNeighbors (dims = 1:30), and FindClusters (resolution = 0.1) were performed on the ‘Harmony’ reduction to achieve 3 subclusters, which was labeled as Itgae+ (encoding for CD103), Itgae−, and cycling cell clusters. The activity scores of curated signatures were calculated by Seurat function AddModuleScore. The pseudotime trajectory analysis of the 3 clusters (including day 4.5 and day 7.5 sgNTC- and sgFlcn-transduced cells) was performed by Slingshot R52 package using default parameters.
ATAC-sequencing
Library preparation
sgNTC- or sgFlcn-transduced OT-I cells were sort-purified from the spleen and siIEL from the same host on day 7.5 post-LM-OVA infection (n = 5 biological replicates per group). To prepare the ATAC-seq library, purified OT-I cells were incubated in 50 μl of ATAC-seq lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630) on ice for 10 min. The nuclei were then pelleted at 500g for 10 min at 4 °C. The supernatant was removed and the pellet was resuspended in 50 μl of transposase reaction mix (25 μl 2× TD buffer, 22.5 μl nuclease-free water and 2.5 μl transposase) and incubated for 30 min at 37 °C. The DNA was then cleaned up using the Qiagen MinElute kit (catalog #28004), and barcoded using the NEBNext HiFi kit according to the manufacturer’s instructions and amplified as previously described.25,74 A test amplification curve was performed using 5 μl of the PCR product to determine the optimal cycle number (i.e., linear part of amplification curve), and the remaining 45 μl sample was amplified using the optimal cycle number on an Applied Biosystems 7900HT quantitative PCR machine. The PCR products were purified using AMPure XP beads followed by two 70% (v/v) ethanol washes, and elution of DNA I buffer EB (Qiagen). Each sample was quantified using a TapeStation (Agilent) and then sequenced on an Illumina NovaSeq to a sequencing depth of 200 million reads per sample. ATAC-seq data have been deposited into the GEO series database GSE231502.
ATAC-seq data processing
ATAC-seq analysis was performed by following pipelines previously described.74,109 In brief, 2×50-bp paired-end reads obtained from NovaSeq were trimming for Nextera adaptor by trimmomatic (v0.36, paired-end mode, with parameter LEADING:10 TRAILING:10 SLIDINGWINDOW:4:18 MINLEN:25) and aligned to mouse genome mm9 downloaded from gencode release M1 (https://www.gencodegenes.org/mouse/releases.html) by BWA (version 0.7.16, default parameters).110 Duplicated reads were then marked with Picard (v2.9.4) and only non-duplicated proper paired reads have been kept by samtools (parameter ‘-q 1 -F 1804’ v1.9).111 After adjustment of Tn5 shift (reads were offset by +4 bp for the sense strand and −5 bp for the antisense strand), we separated reads into nucleosome-free, mononucleosome, dinucleosome and trinucleosome [as previously described37] by fragment size and generated.bigwig files by using the centre 80-bp of fragments and scaled to 30×106 nucleosome-free reads. We observed reasonable nucleosome-free peaks and a pattern of mono-, di- and tri-nucleosomes on IGV (v2.4.13).112 All samples in this study had approximately 20×106 nucleosome-free reads, indicative of good data quality. Next, peaks were called on nucleosome-free reads by MACS2 (v2.1.1.20160309, with default parameters with ‘–extsize 200–nomodel’).113 To assure replicability, we first finalized nucleosome-free regions for each sample and retained a peak only if it called with a higher cut-off (MACS2 -q 0.05). We further generated consensus peaks for each group by keeping peaks that were present in at least 50% of the replicates and discarding the remaining, non-reproducible peaks. The reproducible peaks were further merged between sgNTC- and sgFlcn-transduced samples if they overlapped by 100-bp and then nucleosome-free reads from each of the eight samples was counted using bedtools (version 2.25.0).114 To identify the differentially accessible open chromatin regions (OCRs), we first normalized raw nucleosome-free read counts per million (CPM) followed by differential accessibility analysis by implementation of the negative binomial model in the DESeq2 R package.115 FDR-corrected p value < 0.05 and ∣log2 FC∣ >0.5 were used as cut-offs for more- or less-accessible regions in sgFlcn-transduced samples compared to their sgNTC-transduced spike cells. We then assigned the differentially accessible OCRs in the ATAC-seq data for the nearest genes to generate a list of DA genes using HOMER.116
Motif and footprinting analysis
For motif analysis,109 we further selected 1,000 unchanged regions log2FC < 0.05 and FDR-corrected P value > 0.5 as control regions. FIMO from MEME suite (v4.11.3, ‘–thresh 1e-4–motif-pseudo 0.0001’)90 was used for scanning motifs (TRANSFAC database release 2019, only included Vertebrata and not 3D structure-based) matches in the nucleosome-free regions and two-tailed Fisher’s exact test was used to determine whether a motif was significantly enriched in differentially accessible compared to the control regions. To perform footprinting analysis of transcription-factor binding site analysis, the RGT HINT application was used to infer transcription factor activity and to plot the results.38
QUANTIFICATION AND STATISTICAL ANALYSIS
For biological experiment (non-omics) analyses, data were analyzed using Prism 10 software (GraphPad) by two-tailed paired Student’s t-test, two-tailed unpaired Student’s t-test, two-tailed Mann-Whitney test, one-way ANOVA, or two-way ANOVA as indicated in the figure legends. p < 0.05 was considered significant, with *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; NS, not significant. Data are presented as mean ± SEM. Statistical parameters are reported in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-TCRβ (H57-597), Brilliant Violet 711 | Biolegend | Cat# 109243; RRID:AB_2629564 |
| anti-CD8α (53-6.7), PerCP-Cyanine5.5 | Cytek/Tonbo | Cat# 65-0081-U100; RRID:AB_2621882 |
| anti-CD8α (53-6.7), Biotin | Thermo Fisher Scientific | Cat# 13-0081-82; RRID: AB_466346 |
| anti-CD103 (2E7), APC | Thermo Fisher Scientific | Cat# 17-1031-82; RRID: AB_1106992 |
| anti-CD103 (2E7), Brilliant Violet 711 | Thermo Fisher Scientific | Cat# 407-1031-82; RRID: AB_2942156 |
| anti-CD69 (H1.2F3), PE/Cyanine 7 | Biolegend | Cat# 104512; RRID: AB_493564 |
| anti-CCR9 (CW-1.2), Brilliant Violet 421 | BD Bioscience | Cat# 565412; RRID: AB_2739223 |
| anti-CD45.1 (A20), Brilliant Violet 711 | Biolegend | Cat# 110739; RRID: AB_2562605 |
| anti-CD45.2 (104), PE/Cyanine 7 | Biolegend | Cat# 109830; RRID: AB_1186098 |
| anti-Thy1.1 (OX-7), PE | Biolegend | Cat# 202524; RRID: AB_1595524 |
| anti-Thy1.2 (53-2.1), Brilliant Violet 421 | Biolegend | Cat# 140327; RRID: AB_2686992 |
| anti-CD62L (MEL-14), Brilliant Violet 711 | Biolegend | Cat# 104445; RRID: AB_2564215 |
| anti-α4β7 (DATK32), PE | Thermo Fisher Scientific | Cat# 12-5887-82; RRID: AB_657803 |
| anti-KLRG1 (2F1), Brilliant Violet 605 | Biolegend | Cat# 138419; RRID: AB_2563357 |
| anti-LAMP-1 (1D4B), PE | Thermo Fisher Scientific | Cat# 12-1071-82; RRID: AB_657554 |
| anti-Ki67 (SolA15), APC | Thermo Fisher Scientific | Cat# 17-5698-82; RRID: AB_2688057 |
| anti-IFN-γ (XMG1.2), PE/Cyanine 7 | Biolegend | Cat# 505826; RRID: AB_2295770 |
| anti-TNF-α (MP6-XT22), APC | Biolegend | Cat# 506308; RRID: AB_315429 |
| anti-phosphorylated-S6 (Ser235–Ser236) (D57.2.2E), Pacific Blue | Cell Signaling Technology | Cat# 8520; RRID: AB_2797646 |
| anti-phosphorylated-4EBP1 (Thr37–Thr46) (236B4), Alexa Fluor 647 | Cell Signaling Technology | Cat# 5123; RRID: AB_2097838 |
| anti-phosphorylated-Smad2-Smad3 (072-670), PE | BD Biosciences | Cat# 562586; RRID: AB_11151915 |
| anti-active caspase-3 (C92-605), PE | BD Biosciences | Cat# 561011; RRID: AB_2033931 |
| anti-BrdU (Bu20a), APC | Biolegend | Cat# 339808; RRID: AB_10895898 |
| anti-Epcam (G8.8), biotin | Biolegend | Cat# 118203; RRID: AB_1134174 |
| anti-Goat IgG (H+L) secondary antibody, Alexa Fluor Plus 555 | Thermo Fisher Scientific | Cat# A32816; RRID: AB_2762839 |
| Streptavidin, PE | Thermo Fisher Scientific | Cat# SA10041 |
| Streptavidin, APC-eFluor 780 | Thermo Fisher Scientific | Cat# 47-4317-82; RRID: AB_10366688 |
| Phalloidin, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A12380 |
| anti-Tfeb | ProteinTech | Cat# 13372-1-AP; RRID: AB_2199611 |
| anti-rabbit IgG (H+L), Alexa Fluor Plus 647 | Thermo Fisher Scientific | Cat# A32795; RRID: AB_2762835 |
| anti-mCherry | Biorbyt | Cat# orb11618; RRID: AB_2687829 |
| Purified anti-mouse CD3 | Bio-X-Cell | Cat# BE0001-1; RRID: AB_1107634 |
| Purified anti-mouse CD28 | Bio-X-Cell | Cat # BE0015-1; RRID: AB_1107624 |
| Bacterial and virus strains | ||
| Listeria monocytogenes expressing ovalbumin (LM-OVA) | In house | N/A |
| Yersinia pseudotuberculosis mutant (Yptb ΔyopM) | Laboratory of Dr. Yasmine Belkaid | N/A |
| Yersinia pseudotuberculosis (WT Yptb) (32777 strain) | Laboratory of Dr. Yasmine Belkaid | N/A |
| LCMV-Armstrong | In house | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| OVA peptide (257–264) | Macromolecular Synthesis Core Facility, St. Jude Children’s Research Hospital | N/A |
| Collagenase, type IV | Worthington Biochemicals | Cat# LS004188 |
| Collagenase, type I | Worthington Biochemicals | Cat# LS004194 |
| Bovine pancreatic deoxyribonuclease I (DNase I) | Sigma-Aldrich | Cat# DN25-1G |
| Percoll | GE Healthcare | Cat#1 7089101 |
| MgCl2 | Ambion | Cat# AM9530G |
| CaCl2 | Thermo Fisher Scientific | Cat# J63122 |
| ACK buffer | Thermo Fisher Scientific | Cat# A1049201 |
| HEPES | Gibco | Cat# 15630-080 |
| β-mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| EDTA | Thermo Fisher Scientific | Cat# 15575020 |
| Dithiothreitol | Sigma-Aldrich | Cat# D9779 |
| DMEM | Thermo Fisher Scientific | Cat# 11965118 |
| RPMI 1640 | Thermo Fisher Scientific | Cat# 11875085 |
| Click’s medium | FujiFilm Irvine Scientific | Cat# 9195 |
| Penicillin–streptomycin–L-glutamine | Thermo Fisher Scientific | Cat# 15140122 |
| rmIL-7 | PeproTech | Cat# 217-17 |
| rmIL-15 | PeproTech | Cat# 210-15 |
| rhIL-2 | Sigma-Aldrich | Cat# 23-6019 |
| rhTGF-β | R&D | Cat# 240-B |
| Retinoic acid | Sigma-Aldrich | Cat# R2625 |
| Dialyzed FBS | Thermo Fisher Scientific | Cat# A3382001 |
| RPMI 1640 medium without amino acids | US Biological | Cat# R8999-04A |
| MEM amino acids solution | Thermo Fisher Scientific | Cat# 11130051 |
| MEM non-essential amino acids solution | Thermo Fisher Scientific | Cat# 11140050 |
| L-Alanine | Sigma-Aldrich | Cat# A7469 |
| Glycine | Sigma-Aldrich | Cat# 50046 |
| L-Asparagine | Sigma-Aldrich | Cat# A4159 |
| L-Aspartic acid | Sigma-Aldrich | Cat# A8949 |
| L-Glutamic acid | Sigma-Aldrich | Cat# 49449 |
| L-Glutamine | Thermo Fisher Scientific | Cat# A2916801 |
| L-Proline | Sigma-Aldrich | Cat# 81709 |
| L-Serine | Sigma-Aldrich | Cat# 84959 |
| L-Arginine | Sigma-Aldrich | Cat# A8094 |
| L-Cystine dihydrochloride | Sigma-Aldrich | Cat# C2526 |
| L-Histidine | Sigma-Aldrich | Cat# 53319 |
| L-Isoleucine | Sigma-Aldrich | Cat# 58879 |
| L-Leucine | Sigma-Aldrich | Cat# L8912 |
| L-Lysine monohydrochloride | Sigma-Aldrich | Cat# 62929 |
| L-Methionine | Sigma-Aldrich | Cat# 64319 |
| L-Phenylalanine | Sigma-Aldrich | Cat# P5482 |
| L-Threonine | Sigma-Aldrich | Cat# T8441 |
| L-Tryptophan | Sigma-Aldrich | Cat# 93659 |
| L-Tyrosine | Sigma-Aldrich | Cat# 93829 |
| L-Valine | Sigma-Aldrich | Cat# V0513 |
| Triton X-100 | Sigma-Aldrich | Cat# 93443 |
| Tween-20 | Fisher Scientific | Cat# BP337-500 |
| Formaldehyde | Polysciences | Cat# 18814-20 |
| Paraformaldehyde | Thermo Fisher Scientific | Cat# J19943-K2 |
| Normal donkey serum | Jackson ImmunoResearch | Cat# 017-000-121 |
| Poly-D-lysine coated coverslips | Electron Microscopy Sciences | Cat# 72294-04 |
| GolgiStop | BD Biosciences | Cat# 554724 |
| GolgiPlug | BD Biosciences | Cat# 555029 |
| Fixable Viability Dye eFluor 780 | Thermo Fisher Scientific | Cat# 65-0865-14 |
| Polybrene | Sigma-Aldrich | Cat# TR-1003 |
| KOD Hot Start DNA Polymerase | Sigma-Aldrich | Cat# 71086 |
| AMPure XP beads | Beckman Coulter | Cat# A63881 |
| Baker amino acid with 16% total protein (control diet) | TestDiet | Cat# 5CC7 |
| Modified TestDiet 5CC7 with 2% total protein | TestDiet | Cat# 5BT9 |
| Critical commercial assays | ||
| APC BrdU flow kit | BD Biosciences | Cat# 552598 |
| CytoFix/CytoPerm fixation/permeabilization kit | BD Biosciences | Cat# 554714 |
| Phosflow lyse/fix buffer | BD Biosciences | Cat# 558049 |
| Phosflow perm buffer III | BD Biosciences | Cat# 558050 |
| GFP booster | Chromotek/ProteinTech | Cat# gba488 |
| Vectashield Vibrance mounting media with DAPI | Vector Laboratories | Cat# H-1800 |
| Naïve CD8+ T cell isolation kit | Miltenyi Biotec | Cat# 130-096-543 |
| RNeasy Micro Kit | QIAGEN | Cat# 74004 |
| DNeasy Blood & Tissue Kits | QIAGEN | Cat# 69504 |
| Transfection reagent | Mirus | Cat# MIR2706 |
| Clariom S mouse array | Thermo Fisher Scientific | Cat# 902930 |
| Nextera DNA sample preparation kit | Illumina | Cat# FC-121-1031 |
| NEBNext HiFi 2 × PCR master mix | NEB | Cat# M0541S |
| MinElute kit | Qiagen | Cat# 28004 |
| High Sensitivity D5000 ScreenTape | Agilent | Cat# 5067-5592 |
| High Sensitivity D5000 Reagents | Agilent | Cat# 5067-5593 |
| Chromium Next GEM Single Cell 3' GEM, Library & Gel Bead Kit v3.1 | 10X Genomics | Cat# PN-1000128 |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | Cat# PN-1000127 |
| Chromium i7 Sample Index Plate | 10X Genomics | Cat# PN-220103 |
| High-Capacity cDNA Reverse Transcription kit | Thermo Fisher Scientific | Cat# 4374966 |
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat# 4309155 |
| Deposited data | ||
| Data files for microarray | This paper | GEO: GSE231502 |
| Processed single-cell RNA sequencing data | This paper | GEO: GSE231502 |
| Data files for ATAC-seq | This paper | GEO: GSE231502 |
| Publicly available microarray and RNA sequencing data | Nath et al.53; Milner et al.7; Milner et al.46; Mackay et al.8 | GEO: GSE125471, GSE107278, GSE157072, GSE47045 |
| Publicly available single-cell RNA sequencing data | Kurd et al.42; Crowl et al.27; Boland et al.28 | GEO: GSE131847, GSE182276, GSE125527 |
| Publicly available putative Tfeb target genes | Palmieri et al.39 | N/A |
| Experimental models: Cell lines | ||
| Plat-E | Laboratory of Dr. Yun-Cai Liu, La Jolla Institute of Immunology | N/A |
| HEK293T | ATCC | Cat# CRL-3216 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# JAX: 000664; RRID: IMSR_JAX:000664 |
| Mouse: OT-I | The Jackson Laboratory | Cat# JAX: 003831; RRID: IMSR_JAX:003831 |
| Mouse: P14 | Laboratory of Dr. Benjamin A. Youngblood, St. Jude Children’s Research Hospital | N/A |
| Mouse: YopE-I | Laboratory of Dr. Yasmine Belkaid | N/A |
| Mouse: Cd4Cre: Tg(Cd4-cre)1Cwi/BfluJ | The Jackson Laboratory | Cat# JAX: 017336; RRID: IMSR_JAX:017336 |
| Mouse: Flcnfl/fl | Laboratory of Dr. Laura S. Schmidt, National Cancer Institute-Frederick | N/A |
| Mouse: Tfebfl/fl | Laboratory of Dr. Andrea Ballabio, Telethon Institute of Genetics and Medicine | N/A |
| Mouse: Tfe3−/− | The Jackson Laboratory | Cat# JAX: 042292; RRID: MMRRC_042292-JAX |
| Mouse: Tcra−/− | The Jackson Laboratory | Cat# JAX: 002116; RRID: IMSR_JAX:002116 |
| Mouse: Rosa26-Cas9 knock-in mice | The Jackson Laboratory | Cat# JAX: 026179; RRID: IMSR_JAX:026179 |
| Oligonucleotides | ||
| Nextera NGS-F: TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTTGTGGAAAGGACGAAACACCG | This paper | N/A |
| Nextera NGS-R: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCCACTTTTTCAAGTTGATAACGG | This paper | N/A |
| Tgfbr1-F: TCTGCATTGCACTTATGCTGA | This paper | N/A |
| Tgfbr1-R: AAAGGGCGATCTAGTGATGGA | This paper | N/A |
| Tgfbr2-F: GACTGTCCACTTGCGACAAC | This paper | N/A |
| Tgfbr2-R: GGCAAACCGTCTCCAGAGTAA | This paper | N/A |
| Actb-F: GGCTGTATTCCCCTCCATCG | This paper | N/A |
| Actb-R: CCAGTTGGTAACAATGCCATGT | This paper | N/A |
| sgRNA targeting sequences | This paper | N/A |
| Recombinant DNA | ||
| psPAX2 | N/A | Addgene plasmid # 12260 |
| pCAG4-Eco | N/A | Addgene plasmid # 35617 |
| pMIG-II-retroviral vector | N/A | Addgene #52107 |
| pCL-Eco | N/A | Addgene #12371 |
| Constitutively active Tfeb sequence | N/A | Addgene #79014 |
| Lentiviral mitochondria–lysosome library | This paper | N/A |
| Software and algorithms | ||
| FACSDiva software (version 8) | BD Biosciences | https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsdiva-software |
| FlowJo (version 10.10.0) | BD Biosciences | https://www.flowjo.com/ |
| Prism (version 10.2.2) | GraphPad | https://www.graphpad.com/features |
| NIS Elements software (version 5.30.05) | Nikon Instruments | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| HiSeq analysis software | Illumina | https://support.illumina.com/sequencing/sequencing_software/hiseq-analysis-software-v2-1.html |
| MAGeCK software (version 0.5.9.4) | Li et al.79 | https://www.encodeproject.org/software/mageck/ |
| DrugZ software | Colic et al.80 | https://github.com/hart-lab/drugz |
| NetBID2 R package (version 2.0.2) | Dong et al.81 | https://github.com/jyyulab/NetBID |
| JUMPn software (version 0.19.006) | Vanderwall et al..82 | N/A |
| Cytoscape (version 3.7.256) | Shannon et al.83 | https://cytoscape.org/index.html |
| MCODE algorithm | Bader and Hogue84 | N/A |
| CRIS.py | Connelly and Pruett-Miller85 | https://github.com/patrickc01/CRIS.py |
| Limma R package (version 3.46.0) | Ritchie et al.86 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Gene set enrichment analysis (GSEA) | Subramanian et al.87 | https://www.gsea-msigdb.org/gsea/msigdb/ |
| Seurat R package (version 4.0) | Butler et al.88 | https://satijalab.org/seurat/ |
| Slingshot R package (version 2.12.0) | Street et al.52 | https://www.bioconductor.org/packages/release/bioc/html/slingshot.html |
| gplots R package (version 3.1.1) | N/A | https://cran.r-project.org/web/packages/gplots/index.html |
| Harmony R package (version 1.0) | N/A | https://portals.broadinstitute.org/harmony/ |
| DESeq2 R package (version1.43.5) | N/A | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| limma R package (version 3.46.0) | N/A | https://bioconductor.org/packages/release/bioc/html/limma.html |
| WGCNA R package (version 1.66) | Langfelder and Horvath89 | https://cran.r-project.org/web/packages/WGCNA/index.html |
| FIMO from MEME suite (version 4.11.3) | Bailey et al.90 | https://meme-suite.org/meme/ |
| RGT HINT software | Li et al.38 | https://reg-gen.readthedocs.io/en/latest/hint/introduction.html |
| Picard (version 2.9.4) | N/A | https://broadinstitute.github.io/picard/ |
| Samtools (version 1.9) | N/A | https://www.htslib.org/ |
| IGV (version 2.4.13) | N/A | https://igv.org/ |
| MACS2 | N/A | https://github.com/macs3-project/MACS |
| bedtools (version 2.25.0) | N/A | https://bedtools.readthedocs.io/en/latest/ |
| HOMER software | N/A | http://homer.ucsd.edu/homer/ |
| Other | ||
| LSR Fortessa flow cytometer | BD Biosciences | N/A |
| LSRII flow cytometer | BD Biosciences | N/A |
| Symphony A3 flow cytometer | BD Biosciences | N/A |
| Reflection cell sorter | iCyt | N/A |
| MoFlo cell sorter | BD Biosciences | N/A |
| BigFoot cell sorter | Thermo Fisher Scientific | N/A |
| Miseq and NovaSeq | Illumina | N/A |
| Inverted Ti2 eclipse microscope | Nikon Instruments | N/A |
| Applied Biosystems QuantStudio 7 Flex quantitative PCR machine | Thermo Fisher Scientific | N/A |
| CRISPick | Broad Institute | https://portals.broadinstitute.org/gppx/crispick/public |
Highlights.
Systematic discovery of mitochondrial and lysosomal pathways in CD8+ TRM formation
Lysosomal signaling and amino acids shape Tfeb-driven TRM development
Small intestine-specific TRM programming is impeded by the Flcn-Tfeb signaling axis
Acss1 and Mrpl52 empower early siIEL TRM formation in distinct contexts
ACKNOWLEDGMENTS
The authors acknowledge M. Hendren and R. Walton for animal colony management; S. Rankin for technical support and help with imaging data analyses; X. Meng for help with generating bioinformatics plots; C. Guo, X. Sun, and W. Su for critical reading of the manuscript; St. Jude’s Immunology flow cytometry core facility for cell sorting; and St. Jude’s Hartwell Center for microarray, ATAC-seq, and scRNA-seq profiling. Portions of the schematics were created using BioRender (BioRender.com). This work was supported by ALSAC and National Institutes of Health (NIH) grants CA253188, AI131703, Al105887, Al140761, Al150241, and Al150514 (to H.C.). The Hartwell Center and Center for Advanced Genome Engineering at St. Jude are funded by the Cancer Center Support Grant (P30 CA021765). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
DECLARATION OF INTERESTS
H. C. consults or consulted for Kumquat Biosciences, Inc.; TCura Bioscience; Chugai Pharmaceuticals; and ONO Pharmaceutical Co and is a co-inventor on patents/patent applications in the fields of immunotherapy. A.W.G. serves on the scientific advisory boards of ArsenalBio and Foundery Innovations.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2024.09.013.
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hongbo Chi (hongbo.chi@stjude.org).
Materials availability
Materials generated in the study will be available upon request to the lead contact with a completed Materials Transfer Agreement.
Data and code availability
All microarray, scRNA-seq, and ATAC-seq data generated for this paper have been deposited to the NCBI Gene Expression Omnibus (GEO) database and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
REFERENCES
- 1.Chi H, Pepper M, and Thomas PG (2024). Principles and therapeutic applications of adaptive immunity. Cell 187, 2052–2078. 10.1016/j.cell.2024.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sasson SC, Gordon CL, Christo SN, Klenerman P, and Mackay LK (2020). Local heroes or villains: tissue-resident memory T cells in human health and disease. Cell. Mol. Immunol 17, 113–122. 10.1038/s41423-019-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jameson SC, and Masopust D (2018). Understanding Subset Diversity in T Cell Memory. Immunity 48, 214–226. 10.1016/j.immuni.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masopust D, and Soerens AG (2019). Tissue-Resident T Cells and Other Resident Leukocytes. Annu. Rev. Immunol 37, 521–546. 10.1146/annurev-immunol-042617-053214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okła K, Farber DL, and Zou W (2021). Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med 218, e20201605. 10.1084/jem.20201605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kok L, Masopust D, and Schumacher TN (2022). The precursors of CD8+ tissue resident memory T cells: from lymphoid organs to infected tissues. Nat. Rev. Immunol 22, 283–293. 10.1038/s41577-021-00590-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, et al. (2017). Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552, 253–257. 10.1038/nature24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. (2013). The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol 14, 1294–1301. 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 9.Kok L, Dijkgraaf FE, Urbanus J, Bresser K, Vredevoogd DW, Cardoso RF, Perié L, Beltman JB, and Schumacher TN (2020). A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J. Exp. Med 217, e20191711. 10.1084/jem.20191711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, et al. (2012). Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol 188, 4866–4875. 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang N, and Bevan MJ (2013). Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696. 10.1016/j.immuni.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chapman NM, and Chi H (2022). Metabolic adaptation of lymphocytes in immunity and disease. Immunity 55, 14–30. 10.1016/j.immuni.2021.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raynor JL, and Chi H (2024). Nutrients: Signal 4 in T cell immunity. J. Exp. Med 221, e20221839. 10.1084/jem.20221839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reina-Campos M, Heeg M, Kennewick K, Mathews IT, Galletti G, Luna V, Nguyen Q, Huang H, Milner JJ, Hu KH, et al. (2023). Metabolic programs of T cell tissue residency empower tumour immunity. Nature 621, 179–187. 10.1038/s41586-023-06483-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reina-Campos M, Scharping NE, and Goldrath AW (2021). CD8+ T cell metabolism in infection and cancer. Nat. Rev. Immunol 21, 718–738. 10.1038/s41577-021-00537-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konjar Š, Frising UC, Ferreira C, Hinterleitner R, Mayassi T, Zhang Q, Blankenhaus B, Haberman N, Loo Y, Guedes J, et al. (2018). Mitochondria maintain controlled activation state of epithelial-resident T lymphocytes. Sci. Immunol 3, eaan2543. 10.1126/sciimmunol.aan2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, et al. (2017). Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256. 10.1038/nature21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konjar Š, Ferreira C, Carvalho FS, Figueiredo-Campos P, Fanczal J, Ribeiro S, Morais VA, and Veldhoen M (2022). Intestinal tissue-resident T cell activation depends on metabolite availability. Proc. Natl. Acad. Sci. USA 119, e2202144119. 10.1073/pnas.2202144119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan H, Yang K, Li Y, Shaw TI, Wang Y, Blanco DB, Wang X, Cho JH, Wang H, Rankin S, et al. (2017). Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 46, 488–503. 10.1016/j.immuni.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, Yarbro JM, Han X, Lutz BM, Rao S, et al. (2020). Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron 105, 975–991.e7. 10.1016/j.neuron.2019.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voisinne G, Kersse K, Chaoui K, Lu L, Chaix J, Zhang L, Goncalves Menoita M, Girard L, Ounoughene Y, Wang H, et al. (2019). Quantitative interactomics in primary T cells unveils TCR signal diversification extent and dynamics. Nat. Immunol 20, 1530–1541. 10.1038/s41590-019-0489-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long L, Wei J, Lim SA, Raynor JL, Shi H, Connelly JP, Wang H, Guy C, Xie B, Chapman NM, et al. (2021). CRISPR screens unveil signal hubs for nutrient licensing of T cell immunity. Nature 600, 308–313. 10.1038/s41586-021-04109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, and Guan KL (2019). mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol 21, 63–71. 10.1038/s41556-018-0205-1. [DOI] [PubMed] [Google Scholar]
- 24.Liu GY, and Sabatini DM (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol 21, 183–203. 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang H, Zhou P, Wei J, Long L, Shi H, Dhungana Y, Chapman NM, Fu G, Saravia J, Raynor JL, et al. (2021). In vivo CRISPR screening reveals nutrient signaling processes underpinning CD8+ T cell fate decisions. Cell 184, 1245–1261.e21. 10.1016/j.cell.2021.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasumi H, Baba M, Hong SB, Hasumi Y, Huang Y, Yao M, Valera VA, Linehan WM, and Schmidt LS (2008). Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene 415, 60–67. 10.1016/j.gene.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crowl JT, Heeg M, Ferry A, Milner JJ, Omilusik KD, Toma C, He Z, Chang JT, and Goldrath AW (2022). Tissue-resident memory CD8+ T cells possess unique transcriptional, epigenetic and functional adaptations to different tissue environments. Nat. Immunol 23, 1121–1131. 10.1038/s41590-022-01229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boland BS, He Z, Tsai MS, Olvera JG, Omilusik KD, Duong HG, Kim ES, Limary AE, Jin W, Milner JJ, et al. (2020). Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci. Immunol 5, eabb4432. 10.1126/sciimmunol.abb4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collins N, Han SJ, Enamorado M, Link VM, Huang B, Moseman EA, Kishton RJ, Shannon JP, Dixit D, Schwab SR, et al. (2019). The Bone Marrow Protects and Optimizes Immunological Memoryduring Dietary Restriction. Cell 178, 1088–1101.e15. 10.1016/j.cell.2019.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fung HY, Teryek M, Lemenze AD, and Bergsbaken T (2022). CD103 fate mapping reveals that intestinal CD103− tissue-resident memory T cells are the primary responders to secondary infection. Sci. Immunol 7, eabl9925. 10.1126/sciimmunol.abl9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Hoesslin M, Kuhlmann M, de Almeida GP, Kanev K, Wurmser C, Gerullis AK, Roelli P, Berner J, and Zehn D (2022). Secondary infections rejuvenate the intestinal CD103+ tissue-resident memory T cell pool. Sci. Immunol 7, eabp9553. 10.1126/sciimmunol.abp9553. [DOI] [PubMed] [Google Scholar]
- 32.Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, Schenkel JM, Mitchell JS, Vezys V, Fife BT, et al. (2018). T Cells in Nonlymphoid Tissues Give Rise to Lymph-Node-Resident Memory T Cells. Immunity 48, 327–338.e5. 10.1016/j.immuni.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, Southern PJ, and Masopust D (2015). Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161, 737–749. 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han SJ, Glatman Zaretsky A, Andrade-Oliveira V, Collins N, Dzutsev A, Shaik J, Morais da Fonseca D, Harrison OJ, Tamoutounour S, Byrd AL, et al. (2017). White Adipose Tissue Is a Reservoir for Memory T Cells and Promotes Protective Memory Responses to Infection. Immunity 47, 1154–1168.e6. 10.1016/j.immuni.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fonseca DM, Hand TW, Han SJ, Gerner MY, Zaretsky A, Byrd AL, Harrison OJ, Ortiz AM, Quinones M, Trinchieri G, et al. (2015). Microbiota-Dependent Sequelae of Acute Infection Compromise Tissue-Specific Immunity. Cell 163, 354–366. 10.1016/j.cell.2015.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Z, Schulz MH, Look T, Begemann M, Zenke M, and Costa IG (2019). Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 20, 45. 10.1186/s13059-019-1642-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, and Ballabio A (2011). Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet 20, 3852–3866. 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 40.Martina JA, Diab HI, Lishu L, Jeong-A L, Patange S, Raben N, and Puertollano R (2014). The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal 7, ra9. 10.1126/scisignal.2004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 42.Kurd NS, He Z, Louis TL, Milner JJ, Omilusik KD, Jin W, Tsai MS, Widjaja CE, Kanbar JN, Olvera JG, et al. (2020). Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol 5, eaaz6894. 10.1126/sciimmunol.aaz6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108. 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young NP, Kamireddy A, Van Nostrand JL, Eichner LJ, Shokhirev MN, Dayn Y, and Shaw RJ (2016). AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. 30, 535–552. 10.1101/gad.274142.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol 4, Article17. 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 46.Milner JJ, Nguyen H, Omilusik K, Reina-Campos M, Tsai M, Toma C, Delpoux A, Boland BS, Hedrick SM, Chang JT, et al. (2020). Delineation of a molecularly distinct terminally differentiated memory CD8 T cell population. Proc. Natl. Acad. Sci. USA 117, 25667–25678. 10.1073/pnas.2008571117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frizzell H, Fonseca R, Christo SN, Evrard M, Cruz-Gomez S, Zanluqui NG, von Scheidt B, Freestone D, Park SL, McWilliam HEG, et al. (2020). Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Sci. Immunol 5, eaay9283. 10.1126/sciimmunol.aay9283. [DOI] [PubMed] [Google Scholar]
- 48.Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, et al. (2010). Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med 207, 553–564. 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu Z, Khairallah C, Chu TH, Imperato JN, Lei X, Romanov G, Atakilit A, Puddington L, and Sheridan BS (2023). Retinoic acid signaling during priming licenses intestinal CD103+ CD8 TRM cell differentiation. J. Exp. Med 220, e20210923. 10.1084/jem.20210923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farsakoglu Y, McDonald B, and Kaech SM (2021). Motility Matters: How CD8+ T-Cell Trafficking Influences Effector and Memory Cell Differentiation. Cold Spring Harb. Perspect. Biol 13, a038075. 10.1101/cshperspect.a038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, and Song SY (2004). Retinoic acid imprints gut-homing specificity on T cells. Immunity 21, 527–538. 10.1016/j.immuni.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 52.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477. 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nath AP, Braun A, Ritchie SC, Carbone FR, Mackay LK, Gebhardt T, and Inouye M (2019). Comparative analysis reveals a role for TGF-beta in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS One 14, e0210495. 10.1371/journal.pone.0210495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen W. (2023). TGF-beta Regulation of T Cells. Annu. Rev. Immunol 41, 483–512. 10.1146/annurev-immunol-101921-045939. [DOI] [PubMed] [Google Scholar]
- 55.Tu E, Chia CPZ, Chen W, Zhang D, Park SA, Jin W, Wang D, Alegre ML, Zhang YE, Sun L, et al. (2018). T Cell Receptor-Regulated TGF-beta Type I Receptor Expression Determines T Cell Quiescence and Activation. Immunity 48, 745–759.e6. 10.1016/j.immuni.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borges da Silva H, Peng C, Wang H, Wanhainen KM, Ma C, Lopez S, Khoruts A, Zhang N, and Jameson SC (2020). Sensing of ATP via the Purinergic Receptor P2RX7 Promotes CD8+ Trm Cell Generation by Enhancing Their Sensitivity to the Cytokine TGF-β. Immunity 53, 158–171.e6. 10.1016/j.immuni.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fujino T, Kondo J, Ishikawa M, Morikawa K, and Yamamoto TT (2001). Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J. Biol. Chem 276, 11420–11426. 10.1074/jbc.M008782200. [DOI] [PubMed] [Google Scholar]
- 58.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, and Verdin E (2006). Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA 103, 10224–10229. 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milner JJ, Toma C, He Z, Kurd NS, Nguyen QP, McDonald B, Quezada L, Widjaja CE, Witherden DA, Crowl JT, et al. (2020). Heterogenous Populations of Tissue-Resident CD8+ T Cells Are Generated in Response to Infection and Malignancy. Immunity 52, 808–824.e7. 10.1016/j.immuni.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lawrence RE, and Zoncu R (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol 21, 133–142. 10.1038/s41556-018-0244-7. [DOI] [PubMed] [Google Scholar]
- 61.Chakrabarty RP, and Chandel NS (2021). Mitochondria as Signaling Organelles Control Mammalian Stem Cell Fate. Cell Stem Cell 28, 394–408. 10.1016/j.stem.2021.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Borges da Silva H, Beura LK, Wang H, Hanse EA, Gore R, Scott MC, Walsh DA, Block KE, Fonseca R, Yan Y, et al. (2018). The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8+ T cells. Nature 559, 264–268. 10.1038/s41586-018-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M, et al. (2016). L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167, 829–842.e13. 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, and Sabatini DM (2013). Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493, 679–683. 10.1038/nature11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin HR, Citron YR, Wang L, Tribouillard L, Goul CS, Stipp R, Sugasawa Y, Jain A, Samson N, Lim CY, et al. (2022). Lysosomal GPCR-like protein LYCHOS signals cholesterol sufficiency to mTORC1. Science 377, 1290–1298. 10.1126/science.abg6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Malik N, Ferreira BI, Hollstein PE, Curtis SD, Trefts E, Weiser Novak S, Yu J, Gilson R, Hellberg K, Fang L, et al. (2023). Induction of lysosomal and mitochondrial biogenesis by AMPK phosphorylation of FNIP1. Science 380, eabj5559. 10.1126/science.abj5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, Lee PH, Shin M, Patel SJ, Yu Z, et al. (2019). T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363, eaau0135. 10.1126/science.aau0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. (2019). Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021. 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qiu J, Villa M, Sanin DE, Buck MD, O’Sullivan D, Ching R, Matsushita M, Grzes KM, Winkler F, Chang CH, et al. (2019). Acetate Promotes T Cell Effector Function during Glucose Restriction. Cell Rep. 27, 2063–2074.e5. 10.1016/j.celrep.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balmer ML, Ma EH, Bantug GR, Grählert J, Pfister S, Glatter T, Jauch A, Dimeloe S, Slack E, Dehio P, et al. (2016). Memory CD8+ T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity 44, 1312–1324. 10.1016/j.immuni.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 71.Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, et al. (2014). Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat. Immunol 15, 1152–1161. 10.1038/ni.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Swadling L, Pallett LJ, Diniz MO, Baker JM, Amin OE, Stegmann KA, Burton AR, Schmidt NM, Jeffery-Smith A, Zakeri N, et al. (2020). Human Liver Memory CD8+ T Cells Use Autophagy for Tissue Residence. Cell Rep 30, 687–698.e6. 10.1016/j.celrep.2019.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, Wang Y, Wang YD, Qian C, Xu B, et al. (2019). Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476. 10.1038/s41586-019-1821-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang K, and Kallies A (2021). Tissue-specific differentiation of CD8+ resident memory T cells. Trends Immunol. 42, 876–890. 10.1016/j.it.2021.08.002. [DOI] [PubMed] [Google Scholar]
- 76.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, and Ahmed R (2009). mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112. 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chapman NM, Boothby MR, and Chi H (2020). Metabolic coordination of T cell quiescence and activation. Nat. Rev. Immunol 20, 55–70. 10.1038/s41577-019-0203-y. [DOI] [PubMed] [Google Scholar]
- 78.Shi H, Chapman NM, Wen J, Guy C, Long L, Dhungana Y, Rankin S, Pelletier S, Vogel P, Wang H, et al. (2019). Amino Acids License Kinase mTORC1 Activity and Treg Cell Function via Small G Proteins Rag and Rheb. Immunity 51, 1012–1027.e7. 10.1016/j.immuni.2019.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Colic M, Wang G, Zimmermann M, Mascall K, McLaughlin M, Bertolet L, Lenoir WF, Moffat J, Angers S, Durocher D, et al. (2019). Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Med. 11, 52. 10.1186/s13073-019-0665-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dong X, Ding L, Thrasher A, Wang X, Liu J, Pan Q, Rash J, Dhungana Y, Yang X, Risch I, et al. (2023). NetBID2 provides comprehensive hidden driver analysis. Nat. Commun 14, 2581. 10.1038/s41467-023-38335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vanderwall D, Suresh P, Fu Y, Cho JH, Shaw TI, Mishra A, High AA, Peng J, and Li Y (2021). JUMPn: A Streamlined Application for Protein Co-Expression Clustering and Network Analysis in Proteomics. J. Vis. Exp 176, e62796. 10.3791/62796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bader GD, and Hogue CW (2003). An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4, 2. 10.1186/1471-2105-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Connelly JP, and Pruett-Miller SM (2019). CRIS.py: A Versatile and High-throughput Analysis Program for CRISPR-based Genome Editing. Sci. Rep 9, 4194. 10.1038/s41598-019-40896-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, and Carbone FR (1994). T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27. 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 92.Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang K, Neale G, Green DR, He W, and Chi H (2011). The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat. Immunol 12, 888–897. 10.1038/ni.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baba M, Furihata M, Hong SB, Tessarollo L, Haines DC, Southon E, Patel V, Igarashi P, Alvord WG, Leighty R, et al. (2008). Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Natl. Cancer Inst 100, 140–154. 10.1093/jnci/djm288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pircher H, Bürki K, Lang R, Hengartner H, and Zinkernagel RM (1989). Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature 342, 559–561. 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 96.Lim AI, McFadden T, Link VM, Han SJ, Karlsson RM, Stacy A, Farley TK, Lima-Junior DS, Harrison OJ, Desai JV, et al. (2021). Prenatal maternal infection promotes tissue-specific immunity and inflammation in offspring. Science 373, eabf3002. 10.1126/science.abf3002. [DOI] [PubMed] [Google Scholar]
- 97.Zhang X, Li S, Malik I, Do MH, Ji L, Chou C, Shi W, Capistrano KJ, Zhang J, Hsu TW, et al. (2023). Reprogramming tumour-associated macrophages to outcompete cancer cells. Nature 619, 616–623. 10.1038/s41586-023-06256-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang C, Thangamani S, Kim M, Gu BH, Lee JH, Taparowsky EJ, and Kim CH (2013). BATF is required for normal expression of gut-homing receptors by T helper cells in response to retinoic acid. J. Exp. Med 210, 475–489. 10.1084/jem.20121088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Calvo SE, Clauser KR, and Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257. 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123. 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang H, Diaz AK, Shaw TI, Li Y, Niu M, Cho JH, Paugh BS, Zhang Y, Sifford J, Bai B, et al. (2019). Deep multiomics profiling of brain tumors identifies signaling networks downstream of cancer driver genes. Nat. Commun 10, 3718. 10.1038/s41467-019-11661-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162, 425–440. 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Li T, Wernersson R, Hansen RB, Horn H, Mercer J, Slodkowicz G, Workman CT, Rigina O, Rapacki K, Stærfeldt HH, et al. (2017). A scored human protein-protein interaction network to catalyze genomic interpretation. Nat. Methods 14, 61–64. 10.1038/nmeth.4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barabási AL, and Oltvai ZN (2004). Network biology: understanding the cell’s functional organization. Nat. Rev. Genet 5, 101–113. 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 106.Morris JH, Apeltsin L, Newman AM, Baumbach J, Wittkop T, Su G, Bader GD, and Ferrin TE (2011). clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics 12, 436. 10.1186/1471-2105-12-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sentmanat MF, Peters ST, Florian CP, Connelly JP, and Pruett-Miller SM (2018). A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep 8, 888. 10.1038/s41598-018-19441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, Wadi L, Meyer M, Wong J, Xu C, et al. (2019). Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc 14, 482–517. 10.1038/s41596-018-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, Dhungana Y, Rankin S, Chen W, Rosencrance C, et al. (2019). Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature 565, 101–105. 10.1038/s41586-018-0806-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.