

Abstract
Background
Accurate and comprehensive genomic annotation, including the full list of protein-coding genes, is vital for understanding the molecular mechanisms of human biology. We have previously shown that the genome contains a multitude of yet hidden functional exons and transcripts, some of which might represent novel mRNAs. These results resonate with those from other groups and strongly argue that two decades after the completion of the first draft of the human genome sequence, the current annotation of human genes and transcripts remains far from being complete.
Results
Using a targeted RNA enrichment technique, we showed that one of the novel functional exons previously discovered by us and currently annotated as part of a long non-coding RNA, is actually a part of a novel protein-coding gene, InSETG-4, which encodes a novel human protein with no known homologs or motifs. We found that InSETG-4 is induced by various DNA-damaging agents across multiple cell types and therefore might represent a novel component of DNA damage response. Despite its low abundance in bulk cell populations, InSETG-4 exhibited expression restricted to a small fraction of cells, as demonstrated by the amplification-based single-molecule fluorescence in situ hybridization (asmFISH) analysis.
Conclusions
This study argues that yet undiscovered human protein-coding genes exist and provides an example of how targeted RNA enrichment techniques can help to fill this major gap in our knowledge of the information encoded in the human genome.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12915-024-02069-8.
Keywords: Targeted RNA enrichment, Genomic “dark matter”, Novel gene, Novel protein, DNA damage response, Rapid amplification of cDNA ends, Nanopore sequencing, Mass spectrometry, Single-cell analysis, Single-molecule fluorescence in situ hybridization
Background
Since the release of the first draft of the human genome, ongoing efforts have addressed gaps and corrected errors [1, 2]. Despite these advancements, human gene annotation remains far from complete. This is particularly evident from the numerous novel transcripts, many of which are located in the genomic “dark matter” regions, discovered through both bulk and single-cell transcriptomic analyses [3–11]. The increasing recognition of the transcriptome’s complexity has further highlighted the limitations of current annotations [12–16]. This hinders our understanding of human biology and presents significant challenges to understanding the basic mechanisms of development and disease [17, 18], underscoring the urgent need for improvement of our understanding of genes and transcripts encoded in the human genome.
In our recent study, we provided the evidence for the widespread existence of functional novel and non-canonical human transcripts in the human genome through a genome-wide forward-genetics survey of functional elements using lentivirus-based insertional mutagenesis [19]. Inactivation of the exons of these transcripts, found in both intragenic and intergenic regions, significantly affected cellular survival in response to stress. Although typically present in low abundance, their expression is markedly elevated under stress caused by anticancer drug treatments, suggesting that these RNAs represent a hidden layer of responses to cellular stress.
As a part of our previous study, we identified a novel functional human exon InSETe-60, previously predicted by the GENSCAN algorithm [20]. Disruption of InSETe-60 exon by multiple independent lentivirus integration events consistently resulted in reduced cell survival following treatment with etoposide [19], an anticancer drug that inhibits DNA topoisomerase II and induces DNA breaks in cells [21]. We found that InSETe-60 exon was part of a novel gene, named by us InSETG-4 [19].
In this study, we show that InSETG-4 represents a novel human protein-coding locus that was previously annotated only as a long non-coding RNA (lncRNA). We found that InSETG-4 could be involved in DNA damage response. The gene exhibits a highly specific pattern of expression restricted to a sub-population of cells even in a cultured cell line, which might be a reason why it was not previously identified. Our findings underscore the need for detailed, locus-specific annotations of the human genome using sensitive targeted enrichment assays in order to refine our understanding of specific genomic regions and to discover and annotate novel genes.
Results
Transcript complexity in the InSETG-4 locus
To elucidate the complexity of transcript isoforms produced by the InSETG-4 locus, we employed rapid amplification of cDNA ends (RACE) coupled with nanopore sequencing (RACE-Nano-Seq), as illustrated in Fig. 1. The RACE-Nano-Seq assay from our initial study was conducted using only a single anchor exon, the original InSETe-60, and a limited sequencing depth [19]. This led to the identification of the InSETG-4A transcript. In the current study, we performed a RACE-Nano-Seq assay on polyA + RNA isolated from the human leukemia K562 cells treated with etoposide for 36 h, utilizing a significantly higher (~ 300-fold) sequencing depth and using two additional exons of InSETG-4A as the anchor points for RACE (Fig. 2, Additional file 1: Table S1). This approach significantly enhanced our ability to capture the full complexity of transcript isoforms from the InSETG-4 locus and enabled a comprehensive characterization of the relative abundance of various transcription start sites (TSSs) and transcription termination sites (TTSs) within the locus. The major TSS in the InSETG-4 locus detected by 5′ RACE-Nano-Seq corresponded to the predominant TSS detected by the FANTOM 5 consortium using the cap analysis of gene expression (CAGE) technology [22–24] (Fig. 2) and obtained from a comprehensive dataset of 1816 human primary cells, cell lines, and tissue samples. A total of 407 CAGE samples, including 43 from normal human tissues and 142 from primary cell types, had at least one CAGE tag within a ± 10 bp window of the major TSS in InSETG-4, with K562 being one of the top expressing cell types (Additional file 1: Table S2). This result further confirmed the validity of the RACE-Nano-Seq assay. Besides K562, the highest expression of InSETG-4 was observed in the primary CD14 + monocytes (Additional file 1: Table S2). Overall, these results suggested that the expression of this gene is not restricted to malignant cells.
Fig. 1.
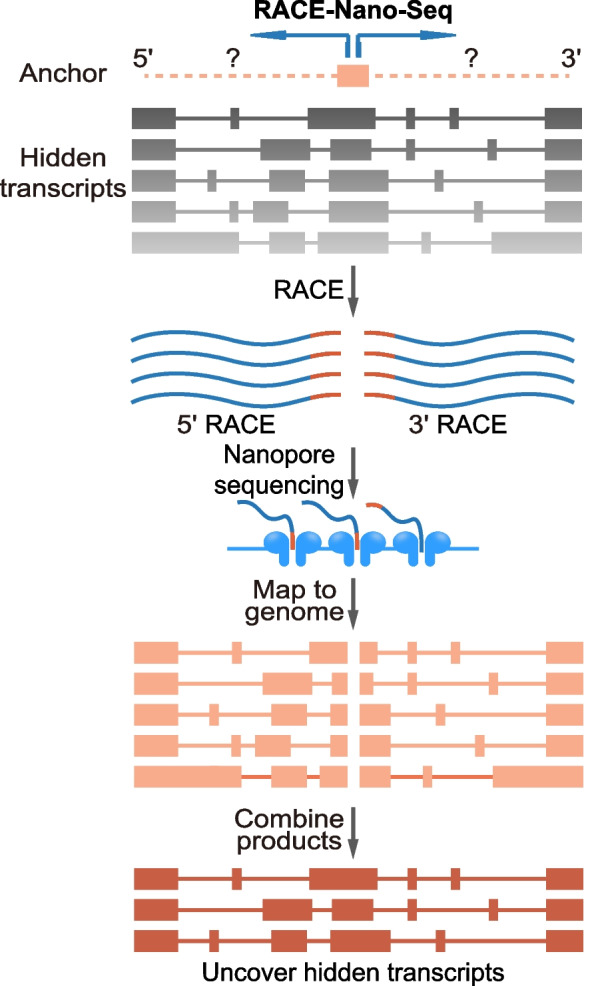
Schematic diagram of rapid amplification of cDNA ends (RACE) coupledwith nanopore sequencing (RACE-Nano-Seq). Primers for 5′ and 3′ RACE-Nano-Seq shown by the blue divergent arrows are designed against a selected genomic anchor region. The 5′ and 3′ ends of the hidden transcripts are enriched via RACE, followed by nanopore sequencing to obtain the full-length sequences. The alignments of the 5′ and 3′ RACE-Nano-Seq sequences are merged to generate complete predicted transcripts
Fig. 2.

Structures of InSETG-4 transcripts identified by RACE-Nano-Seq. A In our previous work [19], an unannotated GENSCAN-predicted exon InSETe-60 was found to harbor both individual lentiviral insertions affecting cellular fitness (IACFs) or their clusters (CACFs). The InSETe-60 exon was then used as the anchor to identify the InSETG-4A transcript. In this study, the exons 2 and exon 3 of InSETG-4A were used as anchors for RACE-Nano-Seq and the most abundant RACE-Nano-Seq products derived from these exons are shown in purple and blue, respectively. Also, the merged TSS and TTS tracks from the 5′ and 3′ RACE-Nano-Seq for each anchor exons are shown. The positions of the RT-qPCR amplicons either common or specific to all three InSETG-4A/B/C transcripts are illustrated. The predicted major open reading frame (ORF) of the InSETG-4 transcripts is indicated in cyan. The 3 ORFs have the same sequences, with the InSETG-4B ORF borrowing the four terminal bases from a different exon. The FANTOM 5 CAGE track represents the total count of CAGE reads from all the CAGE samples. B The relative expression levels (log2) of InSETG-4A/B/C transcripts in K562 cells treated with etoposide for 24 h. The expression levels were normalized to InSETG-4A. Error bars represent standard deviations (SD) from three biological replicates. Asterisks denote significant differences (*p < 0.05, two-sided homoscedastic t-test)
Using 5′ and 3′ RACE-Nano-Seq, we identified a total of 168 exon-exon junctions (EEJs), of which 126 were canonical, displaying the consensus GT-AG splice site. Interestingly, the predicted 5′ boundary of InSETe-60 aligns well with the 5′ exon boundaries detected by RACE-Nano-Seq (Fig. 2A), highlighting the precision of the GENSCAN prediction. In addition, we identified 17 TSS and 110 TTS islands defined by merging individual nucleotide-level TSSs or TTSs found within ± 10 nt from each other. When applying a more stringent threshold of ≥ 5 reads, the numbers of detected EEJs, TSS islands, and TTS islands were 56 (55 with the canonical splice sites), 17, and 46, respectively (Fig. 2A). Among those, 44 EEJs, 17 TTS islands, and 45 TSS islands were not annotated in the GENCODE database. The marked drop in the fraction of the non-canonical EEJs suggested that the corresponding transcripts have lower abundance than the ones with the canonical splice sites. Transcripts utilizing non-canonical splice sites have been detected before in different species [25, 26]. Even though they were also found to have lower abundance, the conservation of the splice sites across species suggested that at least a fraction of them have functional relevance [25, 26]. Based on the number of sequences with unique splicing patterns derived from the two anchor exons (Additional file 1: Table S3), we estimated that this locus in K562 cells produced at least 48 novel transcripts, using a stringent threshold of ≥ 5 reads.
To further elucidate the potential biological functions of the different transcripts in this locus, we combined the common and predominant 5′ RACE sequence with three representative 3′ RACE sequences. This approach revealed three different transcript isoforms originating from the predominant TSS. Among these, InSETG-4A, predicted from the initial RACE-Nano-Seq assay [19], was the longest isoform (Fig. 2A, Additional file 1: Table S4). InSETG-4B was identified as the isoform containing the largest number of exons, while InSETG-4C emerged as the isoform with the most abundant TTS (Fig. 2A, Additional file 1: Table S4). Since InSETG-4C had the most abundant TSS and TTS, it is likely to be the predominant transcript from this locus. All three transcripts shared a common major predicted open reading frame (ORF), defined as the longest ORF starting with an ATG (Fig. 2A, Additional file 1: Table S4). Notably, the 3′ untranslated region (UTR) of InSETG-4C consist of a single exon, while those of InSETG-4A and InSETG-4B are composed of multiple exons (Fig. 2A). The sequences of InSETG-4A and InSETG-4B contain EEJs downstream of the stop codon, making them potentially susceptible to degradation by the nonsense-mediated mRNA decay (NMD) pathway [27–29]. The relative expression levels of the three InSETG-4 transcripts were further confirmed by RT-qPCR using primers specific to each of the three InSETG-4 transcripts (Fig. 2A). While it would be very hard or impossible to design primers absolutely specific to a particular isoform given the transcript complexity in the locus, consistent with the evidence above, InSETG-4C exhibited the highest abundance (Fig. 2B, Additional file 1: Table S5). These results further support the notion that the InSETG-4A and InSETG-4B transcripts might be degraded through NMD. Nonetheless, some mammalian transcripts with EEJ in their 3′ UTRs evade NMD [29]. Therefore, the InSETG-4A and InSETG-4B isoforms might still have function and not merely represent NMD substrates.
The InSETG-4 locus is located far from annotated protein-coding genes—the nearest up- or down-stream protein-coding genes are > 60 kb and > 150 kb away, respectively (Fig. 3A). The InSETG-4A/B/C transcripts overlap to varying degree with several annotated transcripts corresponding to a lncRNA ENSG00000230387 (Fig. 3A, B). The most abundant transcripts of the locus, InSETG-4C, overlap the lncRNA, but have a different splicing pattern, while the InSETG-4A and InSETG-4B transcripts that share multiple exons with the lncRNA represent relatively minor isoforms (Fig. 3B). The TSSs of both the InSETG-4A/B/C transcripts and the lncRNA align with promoter-like signatures in the ENCODE candidate cis-regulatory elements (cCREs) database [30–32]. However, the lncRNA TSS has only a background level of the CAGE signal from multiple human samples (Fig. 3B), suggesting that the lncRNA represents either a minor transcriptional output from the locus, or it has a highly specialized expression pattern. Consistent with this, the ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) data for the H3K4Me3 promoter-associated chromatin mark reveal a signal only near the TSS of InSETG-4 [31, 33, 34]. The presence of the longer transcripts extending beyond the lncRNA is further supported by the human spliced expressed sequence tag (EST) HY086882 (Fig. 3B). However, 5′ RACE-Nano-Seq extended the transcript boundary beyond the 5′ end of the EST (Fig. 3B). Both the absence of the cCREs and background levels of the H3K4Me3 ChIP-seq and CAGE signals at the 5′ end of the EST (Fig. 3B) suggested that the EST was incomplete and did not identify the correct TSS.
Fig. 3.

Existing annotations in the InSETG-4 locus. A Zoom-out view of the genomic context of the InSETG-4 locus. Non-coding RNAs and protein-coding genes are shown in green and blue, respectively. Dash arrows indicate distances to nearby protein-coding genes. B Zoom-in view and details of the existing annotations in the InSETG-4 locus. The original GENSCAN-predicted exon, InSETe-60, is highlighted in yellow. The lncRNAs annotated by GENCODE are shown in green. The FANTOM 5 CAGE track represents the total count of CAGE reads from all the CAGE samples
Taken together, these results showed that the most abundant TSS of the InSETG-4 locus is located ~ 32 kb from the 5′-end of the current annotation, which might represent a minor transcript from the locus. Furthermore, the sequence analysis of the transcripts originating from that TSS suggested that InSETG-4 might be a protein-coding gene.
InSETG-4 represents a novel protein-coding gene
Sequence analysis revealed that all InSETG-4A/B/C transcripts shared a common major ORF of 151 amino acids and almost identical 5′ UTRs—the 5′ UTR of InSETG-4C was 4 nucleotides (nt) longer due to a slightly different TSS (Fig. 4A). All 5′ UTRs harbored upstream ORFs (uORFs) with “ATG” as the initiation codon, spanning 45–57 nt. Notably, the uORFs may also potentially destabilize these transcripts through NMD [27, 28]. In contrast, the sequences of the 3′ UTRs of these transcripts, ranging from 1181 to 1231 nt, differed significantly: InSETG-4B and InSETG-4C had no common 3′ UTR sequences and InSETG-4A and InSETG-4B shared 53.2% bases (Fig. 2, Fig. 3A, and Fig. 4A).
Fig. 4.

Confirmation of the protein-coding potential of InSETG-4 using GFP fusions. A Schematic diagram of the predicted major ORFs (green) and the untranslated regions (UTRs, orange) in the InSETG-4A/B/C transcripts. B The log2 of the cytosol/nucleus ratios of GAPDH mRNA and the spliced (“exon”) and primary (“intron”) InSETG-4 transcripts relative to the ratio of a nuclear-localized very long intergenic non-coding RNA (vlincRNA), ID-838, which is set as zero. C Expression of the ORF-GFP fusions of the InSETG-4 transcripts in 293FT cells. Scale bar, 100 μm. D Flow cytometry analysis of the 293FT cells transfected with the vectors containing ORF-GFP fusions of the InSETG-4 transcripts. For panels C and D, data from one representative biological replicate were shown. For results from all three biological replicates, see Additional file 2: Fig. S1. E, F Ratio of GFP-positive cells (E) and mean fluorescence intensity (MFI) (F) in the ORF-GFP transfected cells versus the untransfected control. Error bars in panels B, E, and F represent SD of three biological replicates. Statistical significance in panels E and F was determined using two-sided Wilcoxon rank sum test, with the p values indicated in the figure. G Confocal microscopy assay of the subcellular localization of the ORF-GFP fusion of InSETG-4C in 293FT cells. Scale bar, 10 μm
To investigate whether the InSETG-4A/B/C transcripts represent real mRNAs in the cell, we first determined their cytosolic vs. nuclear abundance using the GAPDH mRNA and a nuclear-localized very long intergenic non-coding RNA (vlincRNA) [35] as the controls for respectively cytosol- and nucleus-enriched transcripts. We found that the cytosol/nucleus ratio of the spliced InSETG-4 transcripts was similar to that of GAPDH mRNA (Fig. 4B, Additional file 1: Table S6). On the other hand, the cytosol/nucleus ratio of the unspliced version of the InSETG-4A/B/C transcript using primers in the intron common to all three transcripts was comparable to the vlincRNA (Fig. 4B, Additional file 1: Table S6). These results proved that the spliced InSETG-4A/B/C transcripts are exported into the cytosol.
However, some lncRNAs have been shown to function in cytosol [36, 37]. Therefore, as the next step, we directly tested the protein-coding capacity of the predicted InSETG-4A/B/C ORF. We constructed vectors containing the fusions between the ORF green fluorescent protein (GFP) flanked by the cognate 3′ and 5′ UTRs of each of the three InSETG-4 transcripts (Fig. 4C) and transfected these vectors into 293FT cells to test the in vivo translation of the InSETG-4 ORF. Microscopy analysis indicated that cells transfected with each of the three vectors had GFP fluorescence (Fig. 4C). Flow cytometry assays confirmed these findings, showing a significant increase in GFP signals in cells transfected with the vectors compared to the untransfected control, as reflected by both the proportion of GFP-positive cells and the mean fluorescence intensity (MFI) (Fig. 4D–F). Notably, both microscopy and flow cytometry assays revealed that cells transfected with the InSETG-4C ORF vector exhibited the most intense GFP signal (Fig. 4C–F), suggesting that this isoform might be more highly translated. A confocal microscopy analysis using the ORF-GFP fusion revealed that the protein is predominantly localized in the cytosol (Fig. 4G).
However, GFP could be produced from initiation codons within its own ORF or from other initiation codons not part of the predicted ORF. To address this, in addition to the ORF-GFP assay, we constructed vectors without fusing the ORF with the GFP (Fig. 5A) and performed mass spectrometry analysis on cells transfected with these vectors. GFP was placed in the opposite orientation on the vector under the control of its own promoter and served as the positive control (Fig. 5A). We identified a total of four tryptic peptides—HLASQGLAVK, DCWTSVFGAGK, ADMVSAVIPGAPLMMLK, and HLCSCCPQSPLPGYCQLPGPTFPK—predicted to be encoded by the major InSETG-4A/B/C ORF, achieving a total sequence coverage of 41.1% (Fig. 5A, B). The number of peptides detected per sample ranged from one to three, with the sequence coverage varying from 11 to 27% (Fig. 5B). No additional peptides were found in other predicted ORFs within the InSETG-4A/B/C transcript sequences, including the ones utilizing alternative initiation codons, suggesting that the major ORF is the only one that is translated. However, we cannot totally exclude the translation of other ORFs products, which might not have been detected for technical reasons. These results conclusively demonstrate that all three tested InSETG-4 transcripts could function as mRNAs in the cell.
Fig. 5.

Confirmation of the protein-coding potential of InSETG-4 using mass spectrometry (MS). A The diagram of the vector transfected into 293FT cells for LC–MS/MS analysis is shown above the representative mass spectra of the four detected tryptic peptides, with y ions indicated in blue and b ions in red. B Left, the major ORF sequence of InSETG-4 with the four peptides detected by MS underlined and marked in purple. Right, summary of the InSETG-4 peptides detected in different InSETG-4 transcripts and biological replicates. The peptides detected for the GFP expressed from an independent promoter are shown as positive controls
The InSETG-4 encodes a novel protein that has no significant hits to any known proteins in any species and no known motifs based on the homology analysis with BLASTP [38] and motif analysis with PROSITE [39] and Motif Scan [40]. The protein has a molecular weight of 16,230.9 Da and a theoretical isoelectric point (pI) of 8.77. The grand average of hydropathicity (GRAVY) is − 0.130, suggesting the protein is hydrophilic and is likely functional in aqueous environments such as the cytoplasm or extracellular fluid.
InSETG-4 is induced by multiple DNA-damaging agents
The expression of InSETG-4 was found to be induced by etoposide in our previous study [19], suggesting the involvement of InSETG-4 in the DNA damage response. Here, we further investigated the response of InSETG-4 expression to various DNA-damaging conditions using RT-qPCR with primers common to the three InSETG-4A/B/C transcripts (Fig. 3B).
To investigate the kinetics of InSETG-4 induction by etoposide in K562 cells, we performed a time course treatment and found that the peak induction was observed at 24 h of the treatment (Fig. 6A, Additional file 1: Table S7). Subsequently, we examined InSETG-4 expression in response to 24 h of etoposide treatment across six human cell lines (K562, 293FT, UT-7, LAMA-84, HepG2, and HeLa) and found that the gene was induced in all the cell types (Fig. 6B, Additional file 1: Table S7). Notably, K562 cells exhibited the highest levels of InSETG-4 expression both before and after the etoposide exposure (Fig. 6B, Additional file 1: Table S7). In addition to etoposide, InSETG-4 was induced by three other DNA-damaging agents that are known to cause various types of DNA lesions [41–43]: methyl methanesulfonate (MMS), hydrogen peroxide (H2O2), and X-ray irradiation (Fig. 6C, D, Additional file 1: Table S7). Both MMS and H2O2 significantly induced InSETG-4 expression 24 h after treatment (Fig. 6C), suggesting a generalized response of InSETG-4 to DNA damage. InSETG-4 was also significantly induced by X-ray irradiation, with the peak induction occurring 6 h post-irradiation (Fig. 6D, Additional file 1: Table S7). Altogether, these findings indicated that InSETG-4 consistently responds to various forms of DNA damage across different cell types.
Fig. 6.

Induction of the InSETG-4 expression in response to DNA damage. A A time course of etoposide induction of InSETG-4 relative to DMSO-treated K562 cells. B Left and middle: expression levels of InSETG-4 normalized to HeLa cells after 24 h of exposure to DMSO control (left) or etoposide (middle) across different cell lines. Right: induction of InSETG-4 after 24 h of exposure to etoposide relative to the DMSO controls. C Induction of InSETG-4 by 1 mM methyl methanesulfonate (MMS) or 1.5 mM hydrogen peroxide (H2O2) for 24 h using DMSO or H2O as the controls, respectively, in K562 cell line. D Time course of InSETG-4 induction by 50 Gy X-ray irradiation in K562 cell line relative to untreated cells. E Effect of ATM, ATR, and DNA-PKcs inhibitors on the InSETG-4 induction by etoposide. Error bars represent SD. Asterisks denote significant differences (*p < 0.05, two-sided homoscedastic t-test)
All of the four DNA-damaging agents tested above can cause single- and double-strand DNA breaks among other types of DNA damage [21, 41–45]. To test whether the known signal transduction pathways that mediate cellular response to DNA breaks are involved in the induction of InSETG-4, we employed inhibitors targeting three key protein kinases: ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3 related (ATR), and DNA-dependent protein kinase catalytic subunit (DNA-PKcs). These kinases are the most upstream kinases of DNA damage response pathway [46]. They belong to the phosphoinositide 3-kinase (PI3K)-related kinases (PIKKs) family, with ATM and DNA-PKcs primarily induced by DNA double-stranded breaks (DSBs), while ATR mainly responds to single-stranded DNA (ssDNA) regions arise during DNA replication [47]. Recruited to DNA damage sites, these kinases activate diverse downstream processes of DNA damage response, including DNA repair, cell cycle control, senescence, and apoptosis [47]. The induction of InSETG-4 was suppressed, albeit not significantly, by the individual treatments with the ATR or DNA-PKcs, but not by the ATM inhibitors (Fig. 6E, Additional file 1: Table S8). However, the induction was totally abolished by the DNA-PKcs inhibitor in combination with either the ATR or ATM inhibitor (Fig. 6E, Additional file 1: Table S8). On the other hand, the combination of the ATR and ATM inhibitors did not suppress the induction more than the ATR inhibitor by itself. These results suggested that full induction of InSETG-4 by the etoposide treatment is mediated by the combined activities of DNA-PKcs and either ATR or ATM. Consistent with this, the treatment with all three inhibitors together completely abolished the induction of InSETG-4 by etoposide (Fig. 6E, Additional file 1: Table S8). In summary, all three canonical DNA break signaling pathways are involved in the regulation of InSETG-4’s expression in a redundant fashion.
Cell-specific expression of InSETG-4
InSETG-4, located in the genomic “dark matter” region, exhibits relatively lower abundance compared to a typical protein-coding gene. Based on the bulk-cell RT-qPCR analysis, the highest expression level was still ~ 10,000-fold lower than that of GAPDH mRNA estimated to be present at ~ 1000 copies per cell in K562. These results implied that InSETG-4 might be expressed in a sub-population of K562 cells. To further explore the expression and sub-cellular localization of the InSETG-4 transcripts, we employed amplification-based single-molecule fluorescence in situ hybridization (asmFISH) method that can detect single RNA molecules in individual cells [48]. For this experiment, we used DNA ligation probes (DLPs) designed against the shared sequence of InSETG-4A and InSETG-4C, as well as for InSETG-4B (Fig. 7A), using DLPs targeting the bacterial DapB gene as the control.
Fig. 7.

Single-cell analysis of the InSETG-4 expression using the asmFISH assay. A Schematic diagram of the locations of probes for the amplification-based single-molecule fluorescence in situ hybridization (asmFISH) assay. Two pairs of the DNA ligation probes (DLPs) for asmFISH were designed for InSETG-4A/C and InSETG-4B as indicated by the purple and red arrows, respectively. B–D Statistical analysis of the asmFISH assay results based on the ratio of InSETG-4 transcripts positive cells (B), average transcript copies per cell (C), and transcript copies per positive cell in etoposide-treated cells (D). In panels B and C, statistical significances were calculated using two-sided Wilcoxon rank sum test, with the p values indicated in the figures. E, F Microscopy images showing the expression of InSETG-4A/C (magenta) (E) and InSETG-4B (red) (F) in K562 cells exposed to etoposide (180 μM) for 24 h. The blue DAPI stain marks nuclei. The bacterial DapB gene was used as the negative control. InSETG-4 loci are indicated by white arrows. Scale bar: 5 μm
To validate the specificity of the asmFISH method for InSETG-4 detection, we applied it to cells treated with etoposide or DMSO. Since the InSETG-4 transcripts were induced by etoposide as detected by RT-qPCR, we expected to observe the same by asmFISH. The etoposide treatment significantly increased the median fractions of cells containing InSETG-4 transcripts from 3.1% for InSETG-4A/C and 0.5% for InSETG-4B to 9.4% and 2.6%, respectively (Fig. 7B, Additional file 1: Table S9). The treatment also increased the median average copy number of InSETG-4 transcripts per cell from around 0.04 for InSETG-4A/C and 0.008 for InSETG-4B to 0.16 and 0.03 by 4.1- and 4.4-folds, respectively (Fig. 7C, Additional file 1: Table S9).
While the majority of the InSETG-4-positive cells contained only one copy of the InSETG-4 transcript (63.7% for InSETG-4A/C and 80.3% for InSETG-4B), a subset of these cells had multiple copies, with as many as 11 copies of InSETG-4A/C observed in etoposide-treated cells (Fig. 7D, Additional file 1: Table S10). Figure 7E and F illustrates representative cells with relatively high copies of InSETG-4 transcripts, which exhibited punctate patterns and were predominantly located in the cytosol. No signal was observed with the control bacterial DapB probes (Fig. 7E and F). Overall, the results of asmFISH were consistent with those of RT-qPCR and showed that the expression of InSETG-4 is limited to a sub-population of K562 cells. This analysis also revealed heterogeneity of the copy number of InSETG-4 per cell, with some cells having relatively high levels of these transcripts.
Discussion
This study showcases a path from the initial identification of a novel-predicted functional exon InSETe-60 in the human genome to the discovery and initial characterization of a novel gene to which this exon belongs using a targeted RNA enrichment technique RACE-Nano-Seq. We have shown that the novel gene, named by us InSETG-4, encodes a novel protein that has no homologs among known proteins or known amino acid motifs. By virtue of being induced by several DNA damage treatments, in multiple cell types, and more importantly by showing that the induction is mediated by some of the canonical DNA damage response pathways, it appears that the gene might represent a novel component of DNA damage response. This aligns with our previous findings from the phenotypic screen in which inactivation of the novel exon InSETe-60 led to a reduced cell survival in response to the etoposide treatment which causes DNA breaks [19]. However, additional studies are required to pinpoint the exact function of InSETG-4 transcripts within the DNA damage response.
The analysis of the CAGE dataset from the FANTOM 5 consortium generated from hundreds of diverse human cell types [22, 23] did not reveal a cell type where the expression of this gene is much higher. The highest level of InSETG-4 expression was found in primary CD14 + monocytes and it was similar to that in K562 cells (Additional file 1: Table S2). Furthermore, even after induction by DNA damage, the expression of InSETG-4 is confined to a sub-population of K562 cells. Therefore, this gene does appear to have a very restricted pattern of expression. Previous single-cell analyses have revealed that transcripts with low or undetectable expression levels in bulk samples can be highly abundant in specific cell types [6–8]. The uneven distribution holds true even in cultured cell lines previously considered to be more homogenous [6], consistent with our asmFISH results. One possible explanation for the uneven expression level of InSETG-4 among sub-populations of cells is a higher level of DNA damage in these cells. However, other factors such as cell-to-cell variation in cell-cycle stage, differentiation state, metabolic activity, and epigenetic modifications could also cause the observed variability. These observations suggest that low-abundant transcripts, often considered transcriptional noise, may instead have specialized function and/or function only in specific cell types or sub-population of cells. Such transcripts can be easily missed in genome-wide transcriptome surveys and instead, require targeted RNA enrichment techniques for detection [15].
Our results highlight the complexity of the information encoded in the human genome. Overall, our data suggested that at the very least, 48 novel transcripts are produced by the InSETG-4 locus in K562 cells. However, the combinations of the transcript diversity revealed by 5′ and 3′ RACE-Nano-Seq suggested that this number could be at least an order of magnitude more. Furthermore, some EEJs, TSSs and TTSs were found only using specific anchor exons, suggesting that increasing the number of anchor exons might significantly increase the number of novel transcripts just in this one cell type. Expansion of this analysis to other cell types could reveal even more transcripts emanating from this locus. Interestingly, all three transcripts from this locus tested in this study appear to function as mRNAs, albeit with varying efficiency that is most likely due to the differences in their 3′ UTR sequences. However, as shown in Fig. 2, we found alternative splicing events in the exons that encode the ORF, therefore it is likely that the InSETG-4 locus produces more than one amino-acid sequence.
The findings from this study also highlight the fact that the existing annotations are far from being complete. These results are consistent with the estimates from other researchers that, despite the completion of the initial human genome sequencing over two decades ago, the exact number of protein-coding genes is still unknown [18, 49]. Current tools for gene prediction and annotation have limitations, as demonstrated by the identification of novel peptides or proteins derived from non-coding RNAs or non-canonical ORFs that are not included in existing annotation databases [19, 50–53]. The case of InSETG-4 exemplifies some of the problems and challenges faced by the discovery of protein-coding genes. First, both the lack of homologs among other known proteins and the absence of the known domains would hamper the bioinformatic predictions of such sequences. Second, the above problem would be exacerbated by the relatively short length of the amino acid sequence. Third, the restricted pattern of expression would complicate the detection of such transcripts using traditional whole-genome transcriptome surveys. Fourth, the complexity of the transcriptional output from the locus would limit the utility of the short-read next-generation sequencing techniques commonly used for the whole-genome transcriptome analyses. Overall, this study shows that the current annotations of the human genome and human proteome are far from completion.
Conclusions
Complete and accurate annotation of the genome is obviously the basic foundation for understanding the biological processes happening in the cell and interpretation of information encoded in the sequence. The case of the InSETe-60—an in silico predicted exon later shown to have functional relevance—serves as another illustration of this basic premise. The exon was originally predicted to be a part of a lncRNA, which appears to be a minor product from the locus, particularly in K562 cells where InSETe-60 was shown to be functional. Instead, the more extensive annotation efforts performed here suggest that InSETe-60 appears to function via being a part of a novel protein-coding gene. This realization in turn would lead to very different experimental strategies to fully understand the functionality of the DNA sequence encoding this exon. We believe that the lessons learnt here with InSETe-60 are applicable to any genomic sequence and emphasize the need for targeted RNA enrichment methods and advanced sequencing techniques to refine our understanding of the human genome. Targeted RNA sequencing strategies such as RACE-Nano-Seq or CaptureSeq [13, 14] should be routinely used to uncover the true complexity of genomic regions of interest, especially those that are currently poorly understood. Finally, our results show that the current list of the human protein-coding genes is incomplete and raise a question of how many such genes still remain to be discovered.
Methods
Biological resources
The human chronic myeloid leukemia cell line K562 and human hepatoma/hepatoblastoma cell line HepG2 were obtained from Cell Bank of Chinese Academy of Sciences (Shanghai, China). The human cervix carcinoma cell line HeLa, acute myeloid leukemia cell line UT-7, chronic myeloid leukemia in blast crisis cell line LAMA-84, and human embryonal kidney cell line 293FT were obtained from the Cell Resource Center, Peking Union Medical College (which is part of the National Science and Technology Infrastructure, the National Biomedical Cell-Line Resource, NSTI-BMCR. http://cellresource.cn/) (Beijing, China). All cell lines were authenticated by short tandem repeat (STR) profiling and not tested for mycoplasma contamination.
The K562, HepG2, HeLa, UT-7, and LAMA-84 cell lines were grown in RPMI 1640 medium (Gibco) and 293FT were cultured in DMEM high glucose medium (Gibco). All media were supplemented with 10% fetal bovine serum (FBS) (ExCell Bio, Uruguay) and 1% penicillin–streptomycin (PS) (Gibco), except for the media used for LAMA-84, which contained 20% FBS. The media for UT-7 also contained 5–7 U/mL erythropoietin (Proteintech). All cell lines were maintained at 37℃ in 5% CO2.
RACE-Nano-Seq
PolyA + RNA isolation
K562 cells were seeded at a density of 0.5 × 106 cells /mL and cultured for 16 h prior to etoposide treatment. The cells were then treated with 90 μM etoposide (Abmole) for 36 h. Total RNA was extracted using TRNzol Universal (Tiangen, DP424) and the Total RNA Kit I (Omega, R6438-02) according to the manufacturer’s instructions. PolyA + RNA was isolated from total RNA using mRNA capture beads (VAHTS, N401).
RACE and nanopore sequencing
Separate reactions were conducted for the 5′ RACE and 3′ RACE for each anchor exon essentially as described in our previous publication [19]. All RACE primers are listed in Additional file 1: Table S11. The 5′ RACE and 3′ RACE products from each reaction were mixed in equal volumes and purified with two volumes of VAHTS DNA Clean Beads (Vazyme) to a final volume of 50 μL. The concentrations were measured using Merinton SMA6000 spectrophotometer. The pooled products of 5′ RACE and 3′ RACE were sequenced using Oxford Nanopore Technologies platform by BENAGEN Corporation (Wuhan, China). The sequencing library was prepared with the Ligation Sequencing Kit (SQK-PCS109) from Oxford Nanopore Technologies Inc. (Oxford, UK). Sequencing was performed on a FLO-PRO002 R10.4 flow cell using Oxford Nanopore PromethION (Oxford Nanopore Technologies Inc., Oxford, UK). A total of 23.2 GB of raw data were obtained after 48 h of sequencing. The reads were based-called in real-time using MinKNOW (v20.10.6) and integrated with Guppy (v4.2.3).
Data analysis
Raw nanopore sequencing data were initially filtered to retain sequences with an average quality score of 7 or higher, resulting in 18.0 GB of clean data. Subsequently, the filtered reads were aligned to the human GRCh38/hg38 reference genome using Minimap2 (v2.24-r1122) in spliced alignment mode with the command: minimap2 -ax splice -ub -G400000 --end-seed-pen 30 [54]. The “-ub” option was used to find “GT-AG” splice junctions on both strands, the “-G400000” option allowed a maximum intron length of 400,000 nucleotides, and the “--end-seed-pen 30” option helped to avoid tiny terminal sequences. Supplementary and low-quality alignments were filtered out using SAMTools (v1.10) with the command: samtools view -F0 × 900 -q 60 [55]. High-quality alignments were then aligned to anchor exons using the “intersect” function of the BEDTools suite (v2.30.0) [56].
To determine the TSSs or TTSs of InSETG-4, we first ensured that the reads mapped to the appropriate strand of the gene. Subsequently, the TSSs or TTSs were defined as the first or last aligned bases of the respectively 5′ or 3′ RACE-Nano-Seq reads. The normalized abundance of TSSs or TTSs was calculated using the following formula:
where C represents the counts of each TSS or TTS from respectably 5′ or 3′ RACE-Nano-Seq assays, and T denotes the total number of reads in the corresponding assay.
The overlap analysis of the TSSs of InSETG-4A/B/C transcripts with FANTOM 5 CAGE tags was conducted using the “window” function in the BEDTools suite [56]. The parameters used were “-l 10 -r 10 -sm” in order to include 10 bases upstream and downstream of the TSSs and to ensure strand-specific matches.
Protein-coding potential assessment using in vivo fluorescence marker analysis
The ORF prediction of the InSETG-4A, InSETG-4B, and InSETG-4C transcripts were predicted using ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/). The sequence encoding GFP was inserted in-frame after the major predicted 151 aa ORF in each InSETG-4A/B/C sequence and cloned into a CMV promoter-driven expression vector. The CMV-ORF-GFP vectors were constructed by SyngenTech Corporation (Beijing, China). Plasmids were isolated using the PureLink™ HiPure Plasmid Midiprep Kit (Invitrogen, K21005). The vectors were transfected into 293FT cells using the EpFed™ transfection reagent (SyngenTech). As controls, 293FT cells were either transfected with GFP-overexpressing vectors (positive control) or left untransfected (negative control). Fluorescence was observed 48 h post-transfection using a fluorescence microscope (Observer.D1, Zeiss, US). GFP fluorescence was quantified by flow cytometry (Cytoflex LX, Beckman Coulter, US), and data were analyzed with FlowJo software. The experiment included three biological replicates, each with three technical replicates for flow cytometry analysis. To determine the subcellular localization of the protein encoded by InSETG-4, 293FT cells were transfected with the ORF-GFP fusion of InSETG-4C as described above. After the transfection, the cells were stained with 1 × Hoechst 33,342 Staining Solution for Live Cells (Beyotime, C1028, China) for 10 min to visualize the nuclei. The stained cells were then examined using a Leica TCS SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany).
LC–MS/MS analysis
Sequences of the InSETG-4A, InSETG-4B, or InSETG-4C transcripts were cloned into a CMV promoter-driven expression vector containing GFP driven by the EF1α promoter in the opposite orientation. The vectors were transfected into 293FT cells using the EpFed™ transfection reagent (SyngenTech). GFP + cells were sorted using a CytoFLEX SRT flow cytometer (Beckman Coulter, US) and subsequently subjected to total protein extraction. Cells were lysed using 1% sodium deoxycholate (SDC) solution (1% SDC in pH 8 Tris–HCl buffer, containing 1 × protease inhibitor cocktail (APExBIO, K4001)) and sonicated with a BIORUPTOR PLUS (DIAGENODE) at high power for 30 s on and 30 s off for 5 cycles. Protein concentration was measured using a BCA Protein Assay Kit (GLPBIO, GK10009) on a Tecan Spark multimode microplate reader.
Protein solutions were subjected to in-solution trypsin digestion (SignalChem, T575-31N-100) and subsequently dried. Samples were then analyzed on an EASY-nLC 1200 (Thermo Scientific) coupled to an Orbitrap Fusion Lumos (Thermo Scientific) equipped with an EASY-IC ion source. The peptides were dissolved in 10 μl 0.1% formic acid and auto-sampled directly onto a homemade C18 column (35 cm × 75 μm i.d., 2.5 μm 100 Å). The samples were eluted over 120 min with linear gradients of 3–35% acetonitrile in 0.1% formic acid at a flow rate of 300 nL/min. The raw files were analyzed using Proteome Discoverer 2.5 software against all InSETG-4 predicted ORFs.
InSETG-4 ORFs were predicted using ORFfinder (v0.4.3, https://ftp.ncbi.nlm.nih.gov/genomes/TOOLS/ORFfinder/linux-i64/). The following parameters were applied: “-ml 30 -s 0 -strand plus”, “-ml 30 -s 0 -n true -strand plus”, “-ml 30 -s 1 -strand plus”, and “-ml 30 -s 1 -n true -strand plus”. Here, “-ml 30” specifies a minimum ORF length of 30 nucleotides. The “-s 0” uses the initiation codon “ATG” only, while “-s 1” includes both “ATG” and alternative initiation codons. The “-n true” option ignores nested ORFs (default is false), and “-strand plus” outputs ORFs on the plus strand only. All predicted ORFs obtained using these parameters were combined, and duplicates were removed using SeqKit (v2.5.0) [57] with the command “seqkit rmdup -s -i”.
The homology analysis was conducted using BLASTP [38] against the non-redundant protein sequences (nr) database. The motif analysis was performed using two online resources: PROSITE (https://prosite.expasy.org/scanprosite/) [39] with default settings and Motif Scan (https://myhits.sib.swiss/cgi-bin/motif_scan/) [40] against all available motif collection. The physicochemical properties of the protein were predicted using ProtParm web server (https://web.expasy.org/protparam/) [58].
Treatment of cells with DNA-damaging reagents
For the chemical treatments, all cell lines were seeded at the density of 0.5 × 106 cells /mL in 2 mL of medium without PS for 16 h before the treatment. For etoposide time-course treatment, K562 cells were treated with 90 μM etoposide (Abmole Bioscience Inc.) or 0.1% (v/v) DMSO as a control at various time points: 0, 3, 6, 12, 24, and 36 h. Each time point included two biological replicates. For the treatment of etoposide across different cell lines, K562, HepG2, Hela, UT-7, LAMA-84, and 293FT cells were treated with 90 μM etoposide or 0.1% (v/v) DMSO as a control for 24 h, with three biological replicates per condition.
For the treatments with MMS or H2O2, K562 cells were exposed to 1 mM MMS (Sigma, 129,925) or 1.5 mM H2O2 (Caoshanhu, China) for 24 h. Controls were 0.1% (v/v) DMSO for MMS treatment and 0.1% (v/v) H2O for H2O2 treatment. Three biological replicates were performed.
For the treatment with the inhibitors of DNA-PKcs, ATM, and ATR, K562 cells were treated with 90 μM etoposide in combination with one or more of DNA damage sensor inhibitors for 24 h. The inhibitors used were ATM inhibitor KU-60019 (17 μM), ATR inhibitor AZ20 (7 μM), and DNA-PKcs inhibitor NU7441 (5 μM) (all from Abmole Bioscience Inc.). The treatment groups were: KU60019 + etoposide, AZ20 + etoposide, NU7441 + etoposide, KU-60019 + AZ20 + etoposide, KU-60019 + NU7441 + etoposide, AZ20 + NU7441 + etoposide, and KU-60019 + AZ20 + NU7441 + etoposide. Each combination was tested with three biological replicates.
For the X-ray treatments, K562 cells were seeded at the density of 0.25 × 106 cells/mL in 30 mL of medium without PS for 16 h before X-irradiation. The cells were then exposed to 50 Gy using an RS2000 X-ray irradiator (Radsource Technologies Asia Limited, 160 kV, 25 mA), with unexposed cells serving as the control. After irradiation, 2 mL of cells were seeded into 6-well plates and further incubated of 3, 6, 12, 24, and 48 h. Three biological replicates were performed.
Total RNA from all the samples was extracted using TRNzol Universal (Tiangen, DP424) and total RNA kit I (Omega, R6438-02) according to the manufacturer’s instructions. Total RNA quantification was performed using a Merinton SMA6000 spectrophotometer for subsequent experiments.
Isolation of cytosolic and nuclear RNA
K562 cells treated with 90 μM etoposide for 24 h were lysed in 175 μL of pre-chilled lysis buffer (50 mM Tris–HCl (Thermo Scientific), 140 mM NaCl (Thermo Scientific), 1.5 mM MgCl2 (Thermo Scientific), 0.5% Nonidet P-40 (VWR), 1000 U/mL RNase inhibitor (Takara), and 1 mM DTT (Thermo Scientific)) and incubated on ice for 5 min. The lysate was centrifuged at 300 g for 2 min at 4 °C, and the supernatant containing the cytosol was transferred to a new tube. The nuclei and cell debris were washed twice with 500 μL of cold 1 × PBS, followed by centrifugation at 1500 rpm for 5 min at 4 °C, after which the supernatant was discarded. Cytosolic and nuclear RNA were then extracted using TRNzol Universal (Tiangen, DP424) and Total RNA kit I (Omega, R6438-02) according to the manufacturer’s instructions. RNA quantification was performed using a Merinton SMA6000 spectrophotometer for subsequent experiments.
RT-qPCR
cDNA synthesis was carried out with the PrimeScript™ II 1st strand cDNA Synthesis Kit (Takara, 6210A). RT-qPCR was conducted using PowerUp SYBR Green Mast Mix (Life Technologies) on an Mx3005P cycler (Agilent Technologies). For samples without a Ct value, a Ct value of 40 was assigned, as 40 cycles of amplification were performed. The RT-qPCR primers are listed in Additional file 1: Table S12.
AsmFISH analysis
AsmFISH was performed following the procedure reported previously [48] with K562 cells treated with etoposide (180 μM) or DMSO as a control for 24 h. The images were acquired with a Leica DM6B fluorescence microscope (Leica Microsystems, Wetzlar, Germany). For each sample, five to seven fields of view were analyzed. Quantitative analysis of the images was carried out using CellProfiler (v4.2.6) software [59]. The asmFISH probes are listed in the Additional file 1: Table S13.
Supplementary Information
Additional file 1: Fig.S1. Confirmation of the protein-coding potential of InSETG-4 using the ORF-GFP fusions.
Additional file 2: Tables S1-S12. Table S1. InSETG-4 read count of two RACE-Nano-Seq assays. Table S2. Detection of the InSETG-4A/B/C transcripts in different FANTOM5 CAGE samples. Table S3. Number of sequences with unique splicing patterns derived from the two anchor exons. Table S4. Genomic annotation of InSETG-4A/B/C transcripts in the GTF format. Table S5. The relative expression abundance of InSETG-4A/B/C. Table S6. The log2 cytosol/nucleus ratios of InSETG-4 and GAPDH . Table S7. Expression profile of InSETG-4 in response to DNA-damaging agents. Table S8. Induction of InSETG-4 in response to etoposide in presence of inhibitors of DNA break signaling pathways. Table S9. Single-cell expression profile of InSETG-4 measured by asmFISH. Table S10. Distribution of InSETG-4 copies per positive cell in positive cells as detected by asmFISH. Table S11. RACE primers. Table S12. RT-qPCR primers. Table S13. AsmFISH probes.
Acknowledgements
We thank Yaying Wu, Zheni Xu, and Dr. Changchuan Xie from the School of Life Sciences, Xiamen University for mass spectrometry experiments and data analysis.
Abbreviations
- AsmFISH
Amplification-based single-molecule fluorescence in situ hybridization
- ATM
Ataxia telangiectasia mutated
- ATR
Ataxia telangiectasia and Rad3 related
- CACF
Cluster affecting cellular fitness
- CAGE
Cap analysis of gene expression
- cCRE
Candidate cis-regulatory element
- ChIP-seq
Chromatin immunoprecipitation sequencing
- DLP
DNA ligation probe
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- DSB
Double-stranded break
- EEJ
Exon–exon junction
- EST
Expressed sequence tag
- FBS
Fetal bovine serum
- GFP
Green fluorescent protein
- GRAVY
Grand average of hydropathicity
- H2O2
Hydrogen peroxide
- IACF
Insertion affecting cellular fitness
- LncRNA
Long non-coding RNA
- MFI
Mean fluorescence intensity
- MMS
Methyl methanesulfonate
- MS
Mass spectrometry
- NMD
Nonsense-mediated mRNA decay
- nr
Non-redundant protein sequences
- nt
Nucleotide
- ORF
Open reading frame
- pI
Isoelectric point
- PI3K
Phosphoinositide 3-kinase
- PIKK
PI3K-related kinase
- PS
Penicillin–streptomycin
- RACE
Rapid amplification of cDNA ends
- RACE-Nano-Seq
RACE coupled with nanopore sequencing
- SD
Standard deviation
- SDC
Sodium deoxycholate
- ssDNA
Single-stranded DNA
- STR
Short tandem repeat
- TSS
Transcription start site
- TTS
Transcription termination site
- uORF
Upstream ORF
- UTR
Untranslated region
- VlincRNA
Very long intergenic non-coding RNA
Authors’ contributions
P.K. conceived the project with contributions from D.X. and R.K. P.K. and D.X. supervised the project and wrote the manuscript. R.K. supervised the asmFISH experiment. L.T. performed the experiments and contributed to the manuscript writing. L.L., W.M., and X.H. assisted with the experiments. Y.D. assisted with the writing of the manuscript. All authors approved the manuscript.
Funding
D.X. is supported by the National Natural Science Foundation of China (32000441), the Natural Science Foundation of Fujian Province, China (2023J01130), the Fundamental Research Funds for the Central Universities of Huaqiao University (ZQN-924), and the Scientific Research Funds of Huaqiao University (18BS205). P.K. is supported by the National Natural Science Foundation of China (32170619) and the Research Fund for International Senior Scientists from the National Natural Science Foundation of China (32150710525).
Data availability
The raw RACE-Nano-Seq data have been deposited in the SRA under accession number SRR30285700. The processed data are available in GEO under accession number GSE275424. The nucleotide sequences of InSETG-4A, InSETG-4B, and InSETG-4C, as well as the major CDS are available in GenBank under accession numbers PQ197210, PQ197211, PQ197212, and PQ197213, respectively. Materials used in this study can be requested from the corresponding authors upon reasonable request.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dongyang Xu, Email: xudongyang@hqu.edu.cn.
Rongqin Ke, Email: rke@hqu.edu.cn.
Philipp Kapranov, Email: philippk@xmu.edu.cn.
References
- 1.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291:1304–51. [DOI] [PubMed] [Google Scholar]
- 2.Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, et al. The complete sequence of a human genome. Science. 2022;376:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeo G, Holste D, Kreiman G, Burge CB. Variation in alternative splicing across human tissues. Genome Biol. 2004;5:R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang YE, Landback P, Vibranovski M, Long M. New genes expressed in human brains: implications for annotating evolving genomes. BioEssays. 2012;34:982–91. [DOI] [PubMed] [Google Scholar]
- 5.Yan L, Yang M, Guo H, Yang L, Wu J, Li R, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20:1131–9. [DOI] [PubMed] [Google Scholar]
- 6.Liu SJ, Nowakowski TJ, Pollen AA, Lui JH, Horlbeck MA, Attenello FJ, et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016;17:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nilsson F, Storm P, Sozzi E, Hidalgo Gil D, Birtele M, Sharma Y, et al. Single-cell profiling of coding and noncoding genes in human dopamine neuron differentiation. Cells. 2021;10:137. [DOI] [PMC free article] [PubMed]
- 8.Bocchi VD, Conforti P, Vezzoli E, Besusso D, Cappadona C, Lischetti T, et al. The coding and long noncoding single-cell atlas of the developing human fetal striatum. Science. 2021;372:eabf5759. [DOI] [PubMed]
- 9.Djebali S, Lagarde J, Kapranov P, Lacroix V, Borel C, Mudge JM, et al. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS ONE. 2012;7: e28213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagarde J, Uszczynska-Ratajczak B, Santoyo-Lopez J, Gonzalez JM, Tapanari E, Mudge JM, et al. Extension of human lncRNA transcripts by RACE coupled with long-read high-throughput sequencing (RACE-Seq). Nat Commun. 2016;7:12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lagarde J, Uszczynska-Ratajczak B, Carbonell S, Pérez-Lluch S, Abad A, Davis C, et al. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat Genet. 2017;49:1731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapranov P, Drenkow J, Cheng J, Long J, Helt G, Dike S, et al. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 2005;15:987–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercer TR, Gerhardt DJ, Dinger ME, Crawford J, Trapnell C, Jeddeloh JA, et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol. 2011;30:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deveson IW, Brunck ME, Blackburn J, Tseng E, Hon T, Clark TA, et al. Universal Alternative Splicing of Noncoding Exons. Cell Syst. 2018;6:245–255.e5. [DOI] [PubMed] [Google Scholar]
- 15.Xu D, Tang L, Kapranov P. Complexities of mammalian transcriptome revealed by targeted RNA enrichment techniques. Trends Genet. 2023;39:320–33. [DOI] [PubMed] [Google Scholar]
- 16.Mattick JS, Amaral PP, Carninci P, Carpenter S, Chang HY, Chen L-L, et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol. 2023;24:430–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang D, Guelfi S, Garcia-Ruiz S, Costa B, Reynolds RH, D’Sa K, et al. Incomplete annotation has a disproportionate impact on our understanding of Mendelian and complex neurogenetic disorders. Sci Adv. 2020;6:eaay8299. [DOI] [PMC free article] [PubMed]
- 18.Amaral P, Carbonell-Sala S, De La Vega FM, Faial T, Frankish A, Gingeras T, et al. The status of the human gene catalogue. Nature. 2023;622:41–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu D, Tang L, Zhou J, Wang F, Cao H, Huang Y, et al. Evidence for widespread existence of functional novel and non-canonical human transcripts. BMC Biol. 2023;21:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. [DOI] [PubMed] [Google Scholar]
- 21.Wu C-C, Li T-K, Farh L, Lin L-Y, Lin T-S, Yu Y-J, et al. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science. 2011;333:459–62. [DOI] [PubMed] [Google Scholar]
- 22.FANTOM Consortium and the RIKEN PMI and CLST (DGT), Forrest ARR, Kawaji H, Rehli M, Baillie JK, de Hoon MJL, et al. A promoter-level mammalian expression atlas. Nature. 2014;507:462–70. [DOI] [PMC free article] [PubMed]
- 23.Kanamori-Katayama M, Itoh M, Kawaji H, Lassmann T, Katayama S, Kojima M, et al. Unamplified cap analysis of gene expression on a single-molecule sequencer. Genome Res. 2011;21:1150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lizio M, Harshbarger J, Shimoji H, Severin J, Kasukawa T, Sahin S, et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015;16:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parada GE, Munita R, Cerda CA, Gysling K. A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res. 2014;42:10564–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pucker B, Brockington SF. Genome-wide analyses supported by RNA-Seq reveal non-canonical splice sites in plant genomes. BMC Genomics. 2018;19:980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–8. [DOI] [PubMed] [Google Scholar]
- 28.Yi Z, Sanjeev M, Singh G. The branched nature of the nonsense-mediated mRNA decay pathway. Trends Genet. 2021;37:143–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karousis ED, Gypas F, Zavolan M, Mühlemann O. Nanopore sequencing reveals endogenous NMD-targeted isoforms in human cells. Genome Biol. 2021;22:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ENCODE Project Consortium, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. [DOI] [PMC free article] [PubMed]
- 31.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.ENCODE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011;9: e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo Y, Hitz BC, Gabdank I, Hilton JA, Kagda MS, Lam B, et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 2020;48:D882–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hitz BC, Jin-Wook L, Jolanki O, Kagda MS, Graham K, Sud P, et al. The ENCODE uniform analysis pipelines bioRxiv. 2023. 10.1101/2023.04.04.535623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.St Laurent G, Shtokalo D, Dong B, Tackett MR, Fan X, Lazorthes S, et al. VlincRNAs controlled by retroviral elements are a hallmark of pluripotency and cancer. Genome Biol. 2013;14:R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Heesch S, van Iterson M, Jacobi J, Boymans S, Essers PB, de Bruijn E, et al. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014;15:R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noh JH, Kim KM, McClusky WG, Abdelmohsen K, Gorospe M. Cytoplasmic functions of long noncoding RNAs. Wiley Interdiscip Rev RNA. 2018;9: e1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. [DOI] [PubMed] [Google Scholar]
- 39.Sigrist CJA, de Castro E, Cerutti L, Cuche BA, Hulo N, Bridge A, et al. New and continuing developments at PROSITE. Nucleic Acids Res. 2013;41 Database issue:D344–7. [DOI] [PMC free article] [PubMed]
- 40.Pagni M, Ioannidis V, Cerutti L, Zahn-Zabal M, Jongeneel CV, Hau J, et al. MyHits: improvements to an interactive resource for analyzing protein sequences. Nucleic Acids Res. 2007;35 Web Server issue:W433–7. [DOI] [PMC free article] [PubMed]
- 41.Pascucci B, Russo MT, Crescenzi M, Bignami M, Dogliotti E. The accumulation of MMS-induced single strand breaks in G1 phase is recombinogenic in DNA polymerase beta defective mammalian cells. Nucleic Acids Res. 2005;33:280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Driessens N, Versteyhe S, Ghaddhab C, Burniat A, De Deken X, Van Sande J, et al. Hydrogen peroxide induces DNA single- and double-strand breaks in thyroid cells and is therefore a potential mutagen for this organ. Endocr Relat Cancer. 2009;16:845–56. [DOI] [PubMed] [Google Scholar]
- 43.Zhao H, Zhuang Y, Li R, Liu Y, Mei Z, He Z, et al. Effects of different doses of X-ray irradiation on cell apoptosis, cell cycle, DNA damage repair and glycolysis in HeLa cells. Oncol Lett. 2019;17:42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58:235–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Almeida LC, Calil FA, Machado-Neto JA, Costa-Lotufo LV. DNA damaging agents and DNA repair: From carcinogenesis to cancer therapy. Cancer Genet. 2021;252–253:6–24. [DOI] [PubMed] [Google Scholar]
- 46.Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5:a012716. [DOI] [PMC free article] [PubMed]
- 47.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell. 2017;66:801–17. [DOI] [PubMed] [Google Scholar]
- 48.Lin C, Jiang M, Liu L, Chen X, Zhao Y, Chen L, et al. Imaging of individual transcripts by amplification-based single-molecule fluorescence in situ hybridization. N Biotechnol. 2021;61:116–23. [DOI] [PubMed] [Google Scholar]
- 49.Frankish A, Carbonell-Sala S, Diekhans M, Jungreis I, Loveland JE, Mudge JM, et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 2023;51:D942–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prabakaran S, Hemberg M, Chauhan R, Winter D, Tweedie-Cullen RY, Dittrich C, et al. Quantitative profiling of peptides from RNAs classified as noncoding. Nat Commun. 2014;5:5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fields AP, Rodriguez EH, Jovanovic M, Stern-Ginossar N, Haas BJ, Mertins P, et al. A regression-based analysis of ribosome-profiling data reveals a conserved complexity to mammalian translation. Mol Cell. 2015;60:816–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Brunner A-D, Cogan JZ, Nuñez JK, Fields AP, Adamson B, et al. Pervasive functional translation of noncanonical human open reading frames. Science. 2020;367:1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE. 2016;11: e0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–52. [DOI] [PubMed] [Google Scholar]
- 59.Stirling DR, Swain-Bowden MJ, Lucas AM, Carpenter AE, Cimini BA, Goodman A. Cell Profiler 4: improvements in speed, utility and usability. BMC Bioinformatics. 2021;22:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Fig.S1. Confirmation of the protein-coding potential of InSETG-4 using the ORF-GFP fusions.
Additional file 2: Tables S1-S12. Table S1. InSETG-4 read count of two RACE-Nano-Seq assays. Table S2. Detection of the InSETG-4A/B/C transcripts in different FANTOM5 CAGE samples. Table S3. Number of sequences with unique splicing patterns derived from the two anchor exons. Table S4. Genomic annotation of InSETG-4A/B/C transcripts in the GTF format. Table S5. The relative expression abundance of InSETG-4A/B/C. Table S6. The log2 cytosol/nucleus ratios of InSETG-4 and GAPDH . Table S7. Expression profile of InSETG-4 in response to DNA-damaging agents. Table S8. Induction of InSETG-4 in response to etoposide in presence of inhibitors of DNA break signaling pathways. Table S9. Single-cell expression profile of InSETG-4 measured by asmFISH. Table S10. Distribution of InSETG-4 copies per positive cell in positive cells as detected by asmFISH. Table S11. RACE primers. Table S12. RT-qPCR primers. Table S13. AsmFISH probes.
Data Availability Statement
The raw RACE-Nano-Seq data have been deposited in the SRA under accession number SRR30285700. The processed data are available in GEO under accession number GSE275424. The nucleotide sequences of InSETG-4A, InSETG-4B, and InSETG-4C, as well as the major CDS are available in GenBank under accession numbers PQ197210, PQ197211, PQ197212, and PQ197213, respectively. Materials used in this study can be requested from the corresponding authors upon reasonable request.