

ABSTRACT
Mitochondria are essential and highly dynamic organelles whose morphology is determined by a steady-state balance between fusion and fission. Mitochondrial morphology and function are tightly connected. Because they are involved in many important cellular processes, including energy production, cell-autonomous immunity, and apoptosis, mitochondria present an attractive target for pathogens. Here, we explore the relationship between host cell mitochondria and intracellular bacteria, with a focus on mitochondrial morphology and function, as well as apoptosis. Modulation of apoptosis can allow bacteria to establish their replicative niche or support bacterial dissemination. Furthermore, bacteria can manipulate mitochondrial morphology and function through secreted effector proteins and can also contribute to the establishment of a successful infection, e.g., by favoring access to nutrients and/or evasion of the immune system.
INTRODUCTION
Mitochondria are dynamic organelles, which are fundamental to eukaryotic cell function. They originated from an endosymbiotic alphaproteobacterium of the genus Rickettsia, which was internalized by the ancestor of all eukaryotes (1). Consistent with this endosymbiotic event, mitochondria are surrounded by a double membrane and still share molecular and morphological features with prokaryotic cells, such as the ability to create energy in the form of ATP through aerobic respiration. To do so, mitochondria oxidize nutrients in a process termed oxidative phosphorylation, which involves the creation and harnessing of a membrane potential across the inner mitochondrial membrane, resulting in ATP synthesis.
Apart from energy production, mitochondria carry out essential steps of heme, iron-sulfur cluster, and amino acid biosynthesis as well as fatty acid oxidation and play an important role in calcium homeostasis and cell-autonomous innate immunity (2). In this context, mitochondria display antimicrobial activity through reactive oxygen species (ROS) production and through signaling. Mitochondrial innate immune signaling is mediated by the mitochondrial antiviral signaling protein (MAVS) and results in an interferon response (2, 3). Importantly, mitochondria also play a key role in apoptosis, as the intrinsic apoptosis pathway converges on mitochondrial outer membrane permeabilization (MOMP), which represents a point of no return. Mitochondrion-mediated apoptosis is highly regulated by members of the B cell lymphoma 2 (Bcl-2) protein family; proapoptotic BH3-only proteins are activated by intracellular stress signals, overcome the inhibitory effect of antiapoptotic Bcl-2 proteins, and enhance recruitment of Bcl2-associated X protein (Bax) and Bcl-2 antagonist or killer (Bak) to the mitochondrial outer membrane. There, Bax and Bak oligomerization results in MOMP and allows the release of cytochrome c, second mitochondrion-derived activator of caspases (SMAC), and Omi, promoting caspase activation and apoptosis (4).
Along with innate immune signaling and apoptosis, the highly dynamic morphology of mitochondria is one of the characteristics of the organelle that clearly differentiate it from most bacteria. Mitochondrial morphology is determined by a steady-state balance between the opposing events of fusion and fission, which are mediated by a set of dynamin-related GTPases. Mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) coordinate outer membrane fusion by homo- and heterotypic interactions, while optic atrophy 1 (Opa1) mediates fusion of the inner membrane. The current model for mitochondrial fission involves initial mitochondrial constriction through the endoplasmic reticulum (ER) and actin, followed by recruitment of dynamin-related protein 1 (Drp1) to its receptors on the mitochondrial outer membrane. There, Drp1 oligomerizes to form ring-like structures and mediates GTP-dependent mitochondrial fission in conjunction with dynamin 2 (5). Interestingly, Drp1-dependent mitochondrial fission is observed during apoptosis but is not strictly required for its progression (4). Depending on the cell type and functional status of mitochondria, they can adapt their morphology according to cellular energy demands (6) and move along cytoskeletal tracks with the help of molecular motors (7).
Owing to their central role in multiple cellular processes, mitochondria are an attractive target for pathogens. Modulation of mitochondrial functions can be advantageous for bacteria in terms of access to nutrients and/or evasion of the humoral immune system. Here, we explore the relationship between intracellular bacteria and host cell mitochondria, primarily focusing on the effect of the bacteria on mitochondrial morphology and manipulation of host cell death. Manipulation of cell death allows the bacteria to either preserve their intracellular niche by enhancing survival of the host cell or favor dissemination by inducing host cell death. We present examples of both cytosolic and intravacuolar pathogenic bacteria, including Listeria monocytogenes, Shigella flexneri, Rickettsia spp., Legionella pneumophila, Mycobacterium tuberculosis, Salmonella enterica, Chlamydia spp., and Ehrlichia chaffeensis. While cytosolic bacteria are able to directly interact with mitochondria and other organelles, intravacuolar pathogens are confined within a membrane-enclosed vacuole and employ specialized secretion systems to introduce effector proteins into the host cell cytoplasm that target mitochondria.
CYTOSOLIC BACTERIA
Listeria monocytogenes
The Gram-positive bacterium L. monocytogenes is a facultative intracellular pathogen causing the foodborne disease listeriosis, which mainly and most severely affects immunocompromised individuals. L. monocytogenes is capable of invading both phagocytic and nonphagocytic cells and employs the phospholipases PlcA and PlcB and the pore-forming toxin listeriolysin O (LLO) to escape the phagosome (8). Inside the cytosol, L. monocytogenes replicates and hijacks the host actin polymerization machinery in order to spread nonlytically to neighboring cells (9). Infection of epithelial cells with L. monocytogenes interferes with mitochondrial dynamics and induces a strong and rapid but transient fragmentation of the mitochondrial network at early time points of infection. The fragmentation is specific to virulent L. monocytogenes, and the secreted toxin LLO has been identified as the causative factor, but the exact mechanism remains to be elucidated. LLO appears not to localize to mitochondria, but rather oligomerizes and forms pores in the plasma membrane, causing a calcium influx, which is crucial for the induction of mitochondrial fission (10) (Fig. 1). Moreover, the L. monocytogenes-induced mitochondrial fragmentation is atypical, as it is independent of Opa1 and Drp1. Indeed, Drp1 dissociates from mitochondria upon infection. On the other hand, the ER and actin, which both have been suggested as regulators of canonical mitochondrial fragmentation, play a role in this type of mitochondrial fission (11).
FIGURE 1.
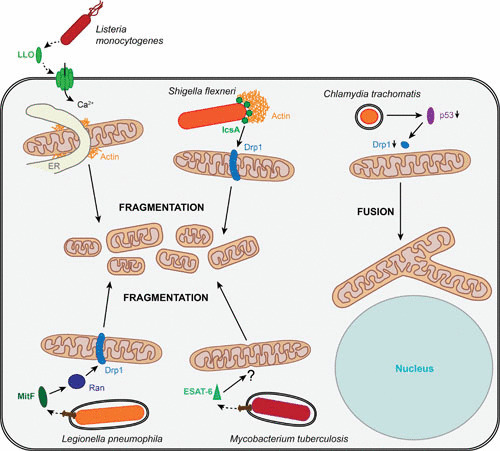
Strategies of intracellular bacteria to interfere with mitochondrial morphology. In epithelial cells, the secreted L. monocytogenes toxin LLO induces rapid mitochondrial fragmentation by pore formation in the plasma membrane, enabling calcium influx. Independent of Drp1 and Opa1, L. monocytogenes-induced mitochondrial fragmentation is of an atypical type; however, the ER and actin appear to play a regulatory role. The S. flexneri surface protein IcsA leads to Drp1-dependent mitochondrial fission in epithelial cells. Infecting macrophages, L. pneumophila induces mitochondrial fragmentation in macrophages by the secreted MitF, which activates Ran GTPase and triggers Drp1 recruitment. Infection of epithelial cells with M. tuberculosis leads to mitochondrial fission, induced by the bacterial pore-forming toxin ESAT-6. In contrast to the other bacteria shown here, C. trachomatis stabilizes the mitochondrial network; downregulating p53, the bacterium inhibits Drp1 expression and recruitment and prevents mitochondrial fragmentation.
L. monocytogenes-induced mitochondrial fragmentation is not associated with apoptosis, as classical apoptotic markers such as cytochrome c release and Bax recruitment to mitochondria are absent. Nevertheless, LLO impacts mitochondrial function, since it causes a dissipation of the mitochondrial membrane potential as well as a drop in respiration activity and cellular ATP levels (10). Whether mitochondrial fragmentation directly impacts the host cell metabolic switch to glycolysis (12) remains speculative. As both the mitochondrial network morphology and ATP level recover within a few hours, mitochondria seem not to be terminally damaged upon infection. Interestingly, mitochondrial dynamics plays an important role in L. monocytogenes infection. It was shown that treatment with small interfering RNA favoring mitochondrial fusion augments the infection efficiency, whereas cells with fragmented mitochondria are less susceptible to L. monocytogenes infection. Based on these observations, it was proposed that L. monocytogenes targets mitochondria to temporarily impair mitochondrial functions in order to establish its replication niche (10). Subsequent studies showed that one of the mitochondrial functions, i.e., cell-autonomous innate immune signaling through MAVS, is not active during L. monocytogenes infection, and innate immune signaling is rather mediated by peroxisome-localized MAVS (13).
Shigella flexneri
S. flexneri is a Gram-negative bacterium which causes shigellosis, an inflammatory disease of the colon leading to tissue destruction, and a leading cause of diarrhea in the developing world. After crossing the colonic epithelium, the facultative intracellular pathogen infects both myeloid immune cells and intestinal epithelial cells. S. flexneri injects secreted effectors into the host cell by its type III secretion system (T3SS) to induce membrane ruffling, resulting in enterocyte invasion. The bacterium then rapidly escapes from the phagosome and proliferates in the cytosol, where it employs the host cell actin machinery for intracellular motility as well as for cell-to-cell spread (14). Interestingly, mitochondria were observed at bacterial invasion sites and appear to be entrapped in an actin meshwork induced by the bacterium (15). The authors of the study proposed a model in which an increase in mitochondrial calcium concentration would activate mitochondrial ATP production to locally provide ATP for further actin polymerization (15).
At later time points of infection (3.5 h), S. flexneri infection induced mitochondrial fission, which was dependent on Drp1 (16). Another study reported local mitochondrial fragmentation in the context of counteracting host cell defense through septin cages. Septin cages have been described to reduce infection by actin-polymerizing bacteria, targeting them to autophagosomes, thus limiting both their motility and dissemination (17). Whereas mitochondrial recruitment to S. flexneri contributes to the formation of septin cages, local mitochondrial fission induced by the bacterial surface protein IcsA (Fig. 1) has been shown to prevent septin cage formation (18).
At the functional level, S. flexneri has been found to cause a dissipation of the mitochondrial membrane potential and a decrease in the cellular ATP levels by around 50%, correlating with S. flexneri-induced necrotic cell death in both epithelial cells (19) and macrophages (20). In epithelial cells, necrosis is counterbalanced by the bacterium, inducing Nod1 signaling. This signaling activates the prosurvival nuclear factor κB (NF-κB) signaling pathway, which in turn inhibits the BNIP3-CypD-dependent opening of the mitochondrial permeability transition pore, a crucial player in the induction of necrosis (19).
Rickettsia spp.
Rickettsia species are Gram-negative obligate intracellular bacteria and are further classified into two major antigenically defined groups, the typhus group and the spotted fever group. Rickettsia prowazekii represents the prototype of the typhus group and is transmitted by lice to humans, causing epidemic typhus. Rickettsia rickettsii and Rickettsia conorii belong to the spotted fever group and are the causative agents of Rocky Mountain spotted fever and Mediterranean spotted fever, respectively. R. prowazekii, R. rickettsii, and R. conorii preferentially infect vascular endothelial cells lining small and medium-sized blood vessels. After entering the host cell via induced phagocytosis, the bacteria rapidly escape the phagosome and replicate in the cytoplasm (21, 22). It has been shown that R. prowazekii and several spotted fever group species import the host mitochondrial protein VDAC1 (voltage-dependent anion-selective channel 1), which localizes to contact sites between inner and outer bacterial membranes and appears to be functional. The authors suggested that a primitive protein import mechanism hijacking mitochondrial proteins underlies the obligate endosymbiotic lifestyle of rickettsiae (23, 24). In order to maintain the endothelial cell as a bacterial replication niche, R. rickettsii suppresses apoptosis by activation of the prosurvival protein NF-κB (25). By regulating levels and localization of pro- and antiapoptotic Bcl2-family proteins, R. rickettsii furthermore maintains mitochondrial integrity and inhibits the activation of caspases 8 and 9 (26, 27).
VACUOLAR BACTERIA
Legionella pneumophila
L. pneumophila is a facultative intracellular bacterium infecting a wide range of hosts, ranging from amoebae to humans. In humans, L. pneumophila replicates in alveolar macrophages and causes Legionnaires’ disease, a serious pulmonary infection. Inside the host cell, the Gram-negative bacterium resides inside an L. pneumophila-containing vacuole (LCV) and is able to evade fusion with the endosome. To manipulate host functions and allow bacterial replication, L. pneumophila injects more than 200 bacterial proteins into host cells via a type IV secretion system (T4SS) (28). It has been reported that 30% of LCVs associate with mitochondria as early as 15 min after infection, and the proportion increases to up to 65% after 1 h of infection (29). A close proximity of mitochondria to LCVs was also shown in L. pneumophila-infected amoebae (30).
In contrast to these data, more recent time-lapse imaging analyses failed to highlight a stable association of mitochondria with LCVs in Drosophila S2 cells (31) or in human primary macrophages (32). The latter study identified transient and highly dynamic mitochondrion-LCV contacts with virulent or avirulent (T4SS-deficient) strains (32). Escoll and colleagues further demonstrated that L. pneumophila induces mitochondrial fragmentation without induction of host cell apoptosis at 6 h postinfection (32). Indeed, previous studies had already proposed that L. pneumophila inhibits apoptosis, as the number of apoptotic cells remains stable despite effective bacterial replication (33). The secreted bacterial effectors SdhA (34) and SidF were later shown to prevent apoptosis. While the mode of SdhA action remains elusive, SidF mediates apoptotic resistance by specifically interacting with the proapoptotic Bcl-2 proteins BNIP3 and Bcl-rambo (35).
The bacterial factor inducing mitochondrial fragmentation upon L. pneumophila infection has been identified and termed mitochondrial fragmentation factor (MitF). MitF is a T4SS-secreted effector that was shown to promote Drp1-dependent mitochondrial fragmentation through a yet-to-be-discovered mechanism involving the nuclear transport factors Ran and Ran-binding protein B2 (RanBP2) as host targets of MitF (Fig. 1). Furthermore, L. pneumophila-induced mitochondrial fragmentation correlates with an alteration of the host cell energy metabolism by impairing mitochondrial respiration, leading to reduced mitochondrial ATP production and a decrease in ATP levels. Simultaneously, host cell glycolysis is upregulated and was shown to favor L. pneumophila intracellular replication, while mitochondrial respiration appears to be dispensable (32). In line with these findings, it was reported that the secreted L. pneumophila mitochondrial carrier protein LncP is not crucial for bacterial proliferation, even though it is targeted to the mitochondrial inner membrane, where it transports ATP from the matrix to the intermembrane space (36).
Mycobacterium tuberculosis
M. tuberculosis is the causative agent of tuberculosis, an infectious disease affecting approximately one-third of the world’s population asymptomatically and leading to 1.8 million deaths annually (37). A facultative intracellular bacterium, M. tuberculosis colonizes primarily human monocytes and macrophages, where it replicates in a specialized phagosomal compartment. By preventing the fusion of the phagosome with lysosomes and inhibiting phagosomal acidification, M. tuberculosis preserves its intracellular niche and replicates. Eventually, M. tuberculosis induces necrosis of the host cell in order to spread to neighboring cells. The initiation of apoptosis is therefore considered a defense strategy of host cells to restrict M. tuberculosis spreading, as apoptotic cells maintain their contents inside and are cleared by phagocytes (38). In agreement with this hypothesis, infection of macrophages with an avirulent M. tuberculosis strain induces apoptosis at a higher level than infection with a virulent strain (39). Consistently, infection with virulent M. tuberculosis has been shown to upregulate antiapoptotic proteins such as Bcl-2 (40) and myeloid cell leukemia 1 (Mcl-1) (41). Regarding the interaction of M. tuberculosis and mitochondria, recent studies focused on mitochondrial implication in cell death modulation. Chen and colleagues correlated the primarily induced cell death in macrophages with mitochondrial membrane perturbations. While both virulent and avirulent strains lead to transient MOMP and cytochrome c release, only the virulent strain causes significantly more mitochondrial permeability transition at early infection time points (6 h). As a consequence, the virulent strain rapidly triggers necrosis, whereas the avirulent strain induces apoptosis only 48 h after infection (42).
Metabolomic profiling on aqueous tissue extracts suggested that M. tuberculosis infection in mice leads to an upregulation of host cell glycolysis (80), consistent with previous findings reporting mitochondrial damage (42, 43). In contrast, Jamwal and colleagues observed an increased mitochondrial membrane potential in cells infected with the virulent strain (44). Furthermore, the authors analyzed the mitochondrial response to infection at the proteomic level, revealing infection-induced upregulation of the mitochondrial protein VDAC2 (44), which appears to prevent apoptosis by keeping the proapoptotic protein Bak inactive (45).
Studies on the effect of M. tuberculosis infection on mitochondrial morphology are controversial. While one study described mitochondrial swelling and a reduction of the mitochondrial matrix density in J744 macrophages infected with virulent and avirulent M. tuberculosis (43), another study reported different mitochondrial phenotypes and activities depending on the virulence of the strain. Upon infection with virulent H37Rv, monocytic THP-1 cells display elongated mitochondria with an increased electron density and augmented activity; in contrast, cells infected with the avirulent H37Ra strain appear to have less electron-dense mitochondria, which are considered nonfunctional (44). Infection of alveolar epithelial cells with a virulent strain caused mitochondrial fragmentation and aggregation in the perinuclear region at late time points of infection (48 h). The pore-forming toxin ESAT-6 has been proposed as the virulence factor responsible for this effect, as its absence prevents mitochondrial fragmentation and aggregation (46) (Fig. 1).
Salmonella enterica
S. enterica is a foodborne Gram-negative facultative intracellular bacterium causing gastroenteritis. To establish infection, S. enterica manipulates host cell functions through a plethora of secreted effector proteins. These effectors are secreted through two T3SSs and contribute to the very early steps of infection by inducing membrane ruffling, which mediates pathogen uptake into nonphagocytic host cells, where S. enterica then persists in a vacuole (47). In order to ensure bacterial survival and replication within epithelial cells, S. enterica remodels the vacuole to prevent its fusion with lysosomes (48) and inhibits host cell apoptosis. In recent years, Salmonella outer protein B (SopB) and fimbrial protein subunit A (FimA) have been identified as two secreted bacterial effector proteins which interfere with mitochondrial functions such as ROS production and apoptosis. SopB binds to cytosolic tumor necrosis factor receptor-associated factor 6 (TRAF6) and delays its mitochondrial recruitment, which causes decreased generation of mitochondrial ROS. In addition to this, Bax translocation to mitochondria and pore formation are inhibited, and induction of apoptosis by cytochrome c release is prevented (49). On the other hand, the soluble form of the pilus protein FimA targets mitochondria, where it binds to the outer mitochondrial membrane protein VDAC1. This results in a tight association between VDAC1 and the mitochondrial hexokinases, suppressing the integration of the pore-forming protein Bax into the outer mitochondrial membrane and the release of cytochrome c (50). Two mechanisms thus converge to prevent cytochrome c release from mitochondria in S. enterica-infected cells. Another effector, called Salmonella outer protein A (SopA), has been reported to localize to mitochondria (51). SopA was lately described as interacting with two host E3 ubiquitin ligases, TRIM56 and TRIM65, inducing an innate immune response involving MAVS activation and characterized by enhanced interferon beta signaling (52).
In contrast to S. enterica infection in epithelial cells, S. enterica infection induces programmed cell death in macrophages. In this context, Hernandez and colleagues proposed that the effector SipB localizes to mitochondria and disrupts mitochondrial morphology, causing swelling and loss of cristae integrity, triggering mitochondrial disruption and resulting in a caspase 1-independent and autophagy-mediated cell death (53).
Chlamydia spp.
The genus Chlamydia comprises three Gram-negative bacterial species, which are pathogenic to humans: Chlamydia trachomatis, C. pneumoniae, and C. psittaci. C. trachomatis is one of the most common sexually transmitted bacteria and causes trachoma, a severe eye infection that can lead to blindness. C. pneumoniae and C. psittaci are associated with respiratory infections such as pneumonia. The obligate intracellular Chlamydia species exhibit a characteristic biphasic life cycle with two distinct developmental forms. The infectious elementary bodies (EBs) attach to epithelial cells and are taken up by phagocytosis. Inside host cells, EBs reside inside membrane-bound inclusions, where they differentiate into metabolically active reticulate bodies (RBs). RBs proliferate by binary fission and undergo maturation to again form infectious EBs, which are released upon host cell lysis in order to infect new cells (54). Several Chlamydia species have been shown to prevent host cell apoptosis by acting on mitochondria (55). Upon C. trachomatis and C. pneumoniae infection, the chlamydial protease-like activity factor induces the degradation of proapoptotic BH3-only proteins, such as Bim, Puma, and Bad, thereby suppressing cytochrome c release from mitochondria and mediating cellular resistance to apoptosis (56, 57).
In terms of energy supply, Chlamydia species have been described as “energy parasites,” as they depend on the import of host cell ATP and metabolites (58, 59). In agreement with this hypothesis, chlamydial infections influence localization and morphology of mitochondria, presumably to obtain ATP and metabolites. By employing electron microscopy, Masumoto identified a tight association of C. psittaci with mitochondria, supporting the former biochemical observations. Mitochondrial association occurred approximately 12 h postinfection, at the time when RBs start to replicate (60).
Interestingly, although all Chlamydia species possess genes encoding ATP transporters, the recruitment of mitochondria to the inclusion is unique to C. psittaci and was not observed for C. trachomatis or C. pneumoniae (61). Instead, C. trachomatis stabilizes the mitochondrial network upon infection-induced stress in order to preserve the mitochondrial ATP production capacity. To do so, C. trachomatis prevents Drp1-mediated mitochondrial fragmentation by downregulating p53 (62), a known regulator of Drp1 expression (63) (Fig. 1), and apoptosis. Mitochondrial morphology affects C. trachomatis infection, and cells with fragmented mitochondria display decreased infection levels (62). Liang and colleagues demonstrated a dependency of C. trachomatis EBs on mitochondrial energy production in epithelial cells; however, the authors also showed that later during infection, C. trachomatis RBs rely only to a limited extent on mitochondrial ATP and employ a sodium gradient to produce energy (64). In contrast to C. trachomatis infection, C. pneumoniae infection causes mitochondrial dysfunction characterized by mitochondrial hyperpolarization, increased ROS generation, and the induction of a metabolic switch to host cell glycolysis. Consistently, impairment of mitochondrial function enhances growth of C. pneumoniae inclusions (65).
Ehrlichia chaffeensis
E. chaffeensis is an obligate intracellular bacterium which causes the tick-borne disease human monocytic ehrlichiosis. This Gram-negative pathogen infects and proliferates inside monocytes and macrophages, where it resides inside vacuoles and forms characteristic mulberry-like bacterial aggregates, which are referred to as morulae. The first evidence that mitochondria play an important role for E. chaffeensis infection was reported by Popov and colleagues; they observed mitochondria closely associated with bacteria-containing morulae in infected macrophages (67). Functional studies of the interaction revealed that E. chaffeensis infection does not cause a change in the mitochondrial membrane potential; however, it reduces mitochondrial DNA synthesis and transcription of mitochondrial genes (68). These observations suggest that E. chaffeensis inhibits mitochondrial activity. Surprisingly, a screen in Drosophila melanogaster identified seven mitochondrion-associated genes whose mutation results in increased resistance to infection (69). A key process in E. chaffeensis pathogenicity is the secretion of effector proteins, which allow evasion of bacterial killing by preventing lysosomal degradation and inhibiting apoptosis, thereby preserving the bacterial replication niche (66). For example, E. chaffeensis secretes the bacterial effector ECH0825, which localizes to mitochondria and inhibits Bax-induced apoptosis. It has been furthermore proposed that ECH0825 prevents ROS-induced cellular stress and apoptosis by upregulating mitochondrial manganese superoxide dismutase (70). Transcriptional profiling of cells infected with E. chaffeensis revealed the induction of the antiapoptotic protein NF-κB and of the Bcl2 proteins Bcl2A1 and Mcl-1 as well as repression of the proapoptotic proteins Bik and BNIP3 (71).
CONCLUSIONS
In this article, we summarize several findings illustrating the importance of mitochondria during bacterial infection, involving the manipulation of mitochondrial morphology and function or the recruitment of mitochondria to the infection site. While viruses induce either mitochondrial fission or fusion (72), to date, bacterial infections seem to mainly induce mitochondrial fission. Although such fission appears to proceed either via the classical, Drp1-dependent fission pathway (S. flexneri and L. pneumophila) or through an atypical, Drp1-independent pathway (L. monocytogenes), fragmentation of the mitochondrial network may represent a common bacterial strategy to impact different mitochondrial functions, or a cellular stress response. Strikingly, mitochondrial morphology can impact the success of infection; cells with fragmented mitochondria display a reduced rate of infection by both L. monocytogenes and C. trachomatis.
Several bacteria use a similar mechanism to interfere with mitochondrial morphology and function, relying on secreted bacterial effectors. These effectors induce changes in mitochondrial structure, dynamics, and functionality which allow bacteria to preserve their replicative niche. Many of the bacterial effectors target Bcl-2 family members by modulating their expression and activity in order to suppress apoptosis. Several secreted effectors that affect mitochondrial morphology and function are pore-forming toxins, and pore formation is essential for their effects. While L. monocytogenes enables calcium influx through pores in the plasma membrane formed by the secreted toxin LLO (Fig. 1), vacuolating cytotoxin A (VacA) injected by extracellular Helicobacter pylori localizes to mitochondria and has been shown to form pores in the inner mitochondrial membrane, inducing a loss of the mitochondrial membrane potential and apoptosis (73, 74) (Fig. 2). In contrast, the exact mechanism by which the pore-forming toxin ESAT-6 from M. tuberculosis affects mitochondria remains speculative (Fig. 1). Other secreted bacterial effectors rely on different mechanisms to induce mitochondrial fission: for example, L. pneumophila MitF indirectly triggers Drp1 oligomerization to cause mitochondrial fragmentation (Fig. 1).
FIGURE 2.
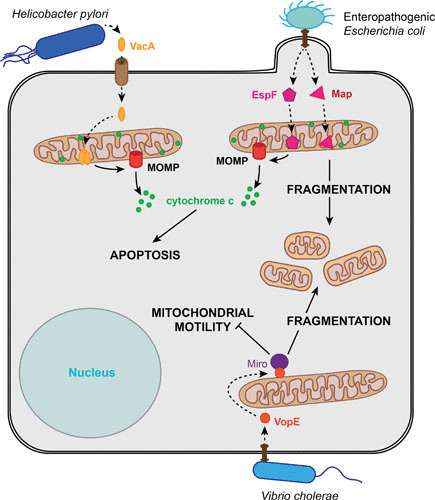
Relationship between extracellular bacteria and host cell mitochondria. The extracellular bacterium H. pylori secretes VacA, a pore-forming toxin, which localizes to the mitochondrial inner membrane and induces MOMP, resulting in apoptosis. EPEC interferes with mitochondrial morphology and function by the secretion of the effector proteins Map and EspF. Both proteins localize to the mitochondrial matrix, where Map leads to mitochondrial fragmentation and EspF induces MOMP and subsequent apoptosis. V. cholerae secreted VopE is a GTPase-activating protein that inactivates Miro at mitochondria, causing mitochondrial fragmentation and inhibiting kinesin-dependent mitochondrial motility. Movement is represented by dashed arrows, while solid arrows indicate induction.
Several extracellular bacteria also secrete effectors that target mitochondrial dynamics and function through diverse mechanisms. The extracellular bacterium Vibrio cholerae secretes a GTPase-activating protein (VopE), which promotes mitochondrial fragmentation and prevents kinesin-dependent mitochondrial motility (Fig. 2). Thereby, VopE inhibits mitochondrial perinuclear clustering and MAVS-dependent innate immune responses (75). Enteropathogenic Escherichia coli (EPEC) effector proteins mitochondria-associated protein (Map) and EPEC-secreted protein F (EspF) display yet another way of targeting mitochondria, as they localize to the mitochondrial matrix and act from within (Fig. 2). While both proteins disrupt the mitochondrial membrane potential, EspF has been shown to trigger apoptosis (76), while Map causes mitochondrial fragmentation and might work in an antiapoptotic fashion and control other mitochondrion-regulated cellular processes (77, 78).
Another way by which bacteria affect mitochondrial morphology is through the recruitment of mitochondria. This has been observed for several cytosolic and intravacuolar bacteria, which thereby presumably benefit from mitochondrion-derived ATP or other metabolites. An extreme example of a bacterium that may benefit from mitochondrial functions or metabolites is the alphaproteobacterium “Candidatus Midichloria mitochondrii,” which invades mitochondria of tick ovarian cells (79).
Bacterial infection can affect not only mitochondrial morphology but also the host cell energy metabolism. In L. pneumophila infection, mitochondrial fragmentation correlates with a decrease in mitochondrial respiration. A similar scenario might apply to L. monocytogenes infection. Interestingly, although C. pneumoniae infection causes mitochondrial hyperpolarization and increased ROS production, it also triggers host cell glycolysis. Reprogramming of the host cell energy metabolism has also been reported for other intracellular bacteria, such as L. monocytogenes and M. tuberculosis. In contrast, Francisella tularensis infection was found to inhibit glycolysis in macrophages (81). However, the link between mitochondria and these infection-induced metabolic changes remains largely unknown.
The complex interactions between bacteria and mitochondria summarized here highlight the importance of this organelle in infection. Future studies will shed more light on the mechanisms by which bacteria affect mitochondria and afford a better understanding of the specific roles that mitochondria play during different stages of bacterial infection.
Contributor Information
Anna Spier, Institut Pasteur, Unité des Interactions Bactéries-Cellules, Paris, France; Institut National de la Santé et de la Recherche Médicale, U604, Paris, France; Institut National de la Recherche Agronomique, USC2020, Paris, France; Bio Sorbonne Paris Cité, Université Paris Diderot, Paris, France.
Fabrizia Stavru, Institut Pasteur, Unité des Interactions Bactéries-Cellules, Paris, France; Institut National de la Santé et de la Recherche Médicale, U604, Paris, France; Institut National de la Recherche Agronomique, USC2020, Paris, France; Centre National de la Recherche Scientifique, SNC 5101, France.
Pascale Cossart, Institut Pasteur, Unité des Interactions Bactéries-Cellules, Paris, France; Institut National de la Santé et de la Recherche Médicale, U604, Paris, France; Institut National de la Recherche Agronomique, USC2020, Paris, France.
Pascale Cossart, Institut Pasteur, Paris, France.
Craig R. Roy, Yale University School of Medicine, New Haven, Connecticut
Philippe Sansonetti, Institut Pasteur, Paris, France.
REFERENCES
- 1.Roger AJ, Muñoz-Gómez SA, Kamikawa R. 2017. The origin and diversification of Mitochondria. Curr Biol 27:R1177–R1192 10.1016/j.cub.2017.09.015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Nunnari J, Suomalainen A. 2012. Mitochondria: in sickness and in health. Cell 148:1145–1159 10.1016/j.cell.2012.02.035. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seth RB, Sun L, Ea C-K, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682 10.1016/j.cell.2005.08.012. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Tait SWG, Green DR. 2010. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11:621–632 10.1038/nrm2952. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Pagliuso A, Cossart P, Stavru F. 2018. The ever-growing complexity of the mitochondrial fission machinery. Cell Mol Life Sci 75:355–374 10.1007/s00018-017-2603-0. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wai T, Langer T. 2016. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab 27:105–117 10.1016/j.tem.2015.12.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Mishra P, Chan DC. 2014. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15:634–646 10.1038/nrm3877. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamon MA, Ribet D, Stavru F, Cossart P. 2012. Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol 20:360–368 10.1016/j.tim.2012.04.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 9.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. 1992. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68:521–531 10.1016/0092-8674(92)90188-I. [DOI] [PubMed] [Google Scholar]
- 10.Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P. 2011. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc Natl Acad Sci USA 108:3612–3617 10.1073/pnas.1100126108. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stavru F, Palmer AE, Wang C, Youle RJ, Cossart P. 2013. Atypical mitochondrial fission upon bacterial infection. Proc Natl Acad Sci USA 110:16003–16008 10.1073/pnas.1315784110. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillmaier N, Götz A, Schulz A, Eisenreich W, Goebel W. 2012. Metabolic responses of primary and transformed cells to intracellular Listeria monocytogenes. PLoS One 7:e52378 10.1371/journal.pone.0052378. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, Durbin AF, Boulant S, Gehrke L, Cossart P, Kagan JC. 2014. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol 15:717–726 10.1038/ni.2915. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Killackey SA, Sorbara MT, Girardin SE. 2016. Cellular aspects of Shigella pathogenesis: focus on the manipulation of host cell processes. Front Cell Infect Microbiol 6:38 10.3389/fcimb.2016.00038. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tran Van Nhieu G, Kai Liu B, Zhang J, Pierre F, Prigent S, Sansonetti P, Erneux C, Kuk Kim J, Suh PG, Dupont G, Combettes L. 2013. Actin-based confinement of calcium responses during Shigella invasion. Nat Commun 4:1567 10.1038/ncomms2561. [PubMed] [DOI] [PubMed] [Google Scholar]
- 16.Lum M, Morona R. 2014. Dynamin-related protein Drp1 and mitochondria are important for Shigella flexneri infection. Int J Med Microbiol 304:530–541 10.1016/j.ijmm.2014.03.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 17.Mostowy S, Bonazzi M, Hamon MA, Tham TN, Mallet A, Lelek M, Gouin E, Demangel C, Brosch R, Zimmer C, Sartori A, Kinoshita M, Lecuit M, Cossart P. 2010. Entrapment of intracytosolic bacteria by septin cage-like structures. Cell Host Microbe 8:433–444 10.1016/j.chom.2010.10.009. [PubMed] [DOI] [PubMed] [Google Scholar]
- 18.Sirianni A, Krokowski S, Lobato-Márquez D, Buranyi S, Pfanzelter J, Galea D, Willis A, Culley S, Henriques R, Larrouy-Maumus G, Hollinshead M, Sancho-Shimizu V, Way M, Mostowy S. 2016. Mitochondria mediate septin cage assembly to promote autophagy of Shigella. EMBO Rep 17:1029–1043 10.15252/embr.201541832. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carneiro LAM, Travassos LH, Soares F, Tattoli I, Magalhaes JG, Bozza MT, Plotkowski MC, Sansonetti PJ, Molkentin JD, Philpott DJ, Girardin SE. 2009. Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe 5:123–136 10.1016/j.chom.2008.12.011. [PubMed] [DOI] [PubMed] [Google Scholar]
- 20.Koterski JF, Nahvi M, Venkatesan MM, Haimovich B. 2005. Virulent Shigella flexneri causes damage to mitochondria and triggers necrosis in infected human monocyte-derived macrophages. Infect Immun 73:504–513 10.1128/IAI.73.1.504-513.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahni SK, Rydkina E. 2009. Host-cell interactions with pathogenic Rickettsia species. Future Microbiol 4:323–339 10.2217/fmb.09.6. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez JJ, Cossart P. 2004. Early signaling events involved in the entry of Rickettsia conorii into mammalian cells. J Cell Sci 117:5097–5106 10.1242/jcs.01382. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Emelyanov VV, Vyssokikh MY. 2006. On the nature of obligate intracellular symbiosis of rickettsiae—Rickettsia prowazekii cells import mitochondrial porin. Biochemistry (Mosc) 71:730–735 10.1134/S0006297906070054. [DOI] [PubMed] [Google Scholar]
- 24.Emelyanov VV. 2009. Mitochondrial porin VDAC 1 seems to be functional in rickettsial cells. Ann N Y Acad Sci 1166:38–48 10.1111/j.1749-6632.2009.04513.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 25.Clifton DR, Goss RA, Sahni SK, van Antwerp D, Baggs RB, Marder VJ, Silverman DJ, Sporn LA. 1998. NF-κB-dependent inhibition of apoptosis is essential for host cellsurvival during Rickettsia rickettsii infection. Proc Natl Acad Sci USA 95:4646–4651 10.1073/pnas.95.8.4646. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joshi SG, Francis CW, Silverman DJ, Sahni SK. 2003. Nuclear factor κB protects against host cell apoptosis during Rickettsia rickettsii infection by inhibiting activation of apical and effector caspases and maintaining mitochondrial integrity. Infect Immun 71:4127–4136 10.1128/IAI.71.7.4127-4136.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joshi SG, Francis CW, Silverman DJ, Sahni SK. 2004. NF-kappaB activation suppresses host cell apoptosis during Rickettsia rickettsii infection via regulatory effects on intracellular localization or levels of apoptogenic and anti-apoptotic proteins. FEMS Microbiol Lett 234:333–341. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Newton HJ, Ang DKY, van Driel IR, Hartland EL. 2010. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin Microbiol Rev 23:274–298 10.1128/CMR.00052-09. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horwitz MA. 1983. Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J Exp Med 158:1319–1331 10.1084/jem.158.4.1319. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newsome AL, Baker RL, Miller RD, Arnold RR. 1985. Interactions between Naegleria fowleri and Legionella pneumophila. Infect Immun 50:449–452. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun EW, Wagner ML, Maize A, Kemler D, Garland-Kuntz E, Xu L, Luo ZQ, Hollenbeck PJ. 2013. Legionella pneumophila infection of Drosophila S2 cells induces only minor changes in mitochondrial dynamics. PLoS One 8:e62972 10.1371/journal.pone.0062972. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Escoll P, Song OR, Viana F, Steiner B, Lagache T, Olivo-Marin JC, Impens F, Brodin P, Hilbi H, Buchrieser C. 2017. Legionella pneumophila modulates mitochondrial dynamics to trigger metabolic repurposing of infected macrophages. Cell Host Microbe 22:302–316.E7 10.1016/j.chom.2017.07.020. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.Derré I, Isberg RR. 2004. Macrophages from mice with the restrictive Lgn1 allele exhibit multifactorial resistance to Legionella pneumophila. Infect Immun 72:6221–6229 10.1128/IAI.72.11.6221-6229.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. 2006. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc Natl Acad Sci USA 103:18745–18750 10.1073/pnas.0609012103. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banga S, Gao P, Shen X, Fiscus V, Zong W-X, Chen L, Luo Z-Q. 2007. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc Natl Acad Sci USA 104:5121–5126 10.1073/pnas.0611030104. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dolezal P, Aili M, Tong J, Jiang JH, Marobbio CM, Lee SF, Schuelein R, Belluzzo S, Binova E, Mousnier A, Frankel G, Giannuzzi G, Palmieri F, Gabriel K, Naderer T, Hartland EL, Lithgow T. 2012. Legionella pneumophila secretes a mitochondrial carrier protein during infection. PLoS Pathog 8:e1002459. CORRECTION PLoS Pathog 8:10.1371/annotation/5039541e-b48a-4cfc-84b1-21566e311a62. CORRECTION PLoS Pathog 8:10.1371/annotation/ee7c807b-032c-4d1f-b5ac-0f6620a2ef24. 10.1371/journal.ppat.1002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Corbett EL, Watt CJ, Walker N, Maher D, Williams BG, Raviglione MC, Dye C. 2003. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch Intern Med 163:1009–1021 10.1001/archinte.163.9.1009. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Dubey RK. 2016. Assuming the role of mitochondria in mycobacterial infection. Int J Mycobacteriol 5:379–383 10.1016/j.ijmyco.2016.06.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 39.Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, Kornfeld H. 1997. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun 65:298–304. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J, Jiang R, Takayama H, Tanaka Y. 2005. Survival of virulent Mycobacterium tuberculosis involves preventing apoptosis induced by Bcl-2 upregulation and release resulting from necrosis in J774 macrophages. Microbiol Immunol 49:845–852 10.1111/j.1348-0421.2005.tb03673.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 41.Sly LM, Hingley-Wilson SM, Reiner NE, McMaster WR. 2003. Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J Immunol 170:430–437 10.4049/jimmunol.170.1.430. [PubMed] [DOI] [PubMed] [Google Scholar]
- 42.Chen M, Gan H, Remold HG. 2006. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J Immunol 176:3707–3716 10.4049/jimmunol.176.6.3707. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.Abarca-Rojano E, Rosas-Medina P, Zamudio-Cortéz P, Mondragón-Flores R, Sánchez-García FJ. 2003. Mycobacterium tuberculosis virulence correlates with mitochondrial cytochrome c release in infected macrophages. Scand J Immunol 58:419–427 10.1046/j.1365-3083.2003.01318.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 44.Jamwal S, Midha MK, Verma HN, Basu A, Rao KVS, Manivel V. 2013. Characterizing virulence-specific perturbations in the mitochondrial function of macrophages infected with Mycobacterium tuberculosis. Sci Rep 3:1328 10.1038/srep01328. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng EH-Y, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. 2003. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301:513–517. [PubMed] [DOI] [PubMed] [Google Scholar]
- 46.Fine-Coulson K, Giguère S, Quinn FD, Reaves BJ. 2015. Infection of A549 human type II epithelial cells with Mycobacterium tuberculosis induces changes in mitochondrial morphology, distribution and mass that are dependent on the early secreted antigen, ESAT-6. Microbes Infect 17:689–697 10.1016/j.micinf.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 47.Cossart P, Sansonetti PJ. 2004. Bacterial invasion: the paradigm of enteroinvasive pathogens. Science 304:242–248. [PubMed] [DOI] [PubMed] [Google Scholar]
- 48.Eng SK, Pusparajah P, Ab Mutalib NS, Ser HL, Chan KG, Lee LH. 2015. Salmonella: a review on pathogenesis, epidemiology and antibiotic resistance. Front Life Sci 8:284–293 10.1080/21553769.2015.1051243. [DOI] [Google Scholar]
- 49.Ruan H, Zhang Z, Tian L, Wang S, Hu S, Qiao JJ. 2016. The Salmonella effector SopB prevents ROS-induced apoptosis of epithelial cells by retarding TRAF6 recruitment to mitochondria. Biochem Biophys Res Commun 478:618–623 10.1016/j.bbrc.2016.07.116. [PubMed] [DOI] [PubMed] [Google Scholar]
- 50.Sukumaran SK, Fu NY, Tin CB, Wan KF, Lee SS, Yu VC. 2010. A soluble form of the pilus protein FimA targets the VDAC-hexokinase complex at mitochondria to suppress host cell apoptosis. Mol Cell 37:768–783 10.1016/j.molcel.2010.02.015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 51.Layton AN, Brown PJ, Galyov EE. 2005. The Salmonella translocated effector SopA is targeted to the mitochondria of infected cells. J Bacteriol 187:3565–3571 10.1128/JB.187.10.3565-3571.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kamanova J, Sun H, Lara-Tejero M, Galán JE. 2016. The Salmonella effector protein SopA modulates innate immune responses by targeting TRIM E3 ligase family members. PLoS Pathog 12:e1005552 10.1371/journal.ppat.1005552. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hernandez LD, Pypaert M, Flavell RA, Galán JE. 2003. A Salmonella protein causes macrophage cell death by inducing autophagy. J Cell Biol 163:1123–1131 10.1083/jcb.200309161. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elwell C, Mirrashidi K, Engel J. 2016. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 14:385–400 10.1038/nrmicro.2016.30. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fischer SF, Harlander T, Vier J, Häcker G. 2004. Protection against CD95-induced apoptosis by chlamydial infection at a mitochondrial step. Infect Immun 72:1107–1115 10.1128/IAI.72.2.1107-1115.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fischer SF, Vier J, Kirschnek S, Klos A, Hess S, Ying S, Häcker G. 2004. Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. J Exp Med 200:905–916 10.1084/jem.20040402. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med 187:487–496 10.1084/jem.187.4.487. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moulder JW. 1962. Structure and chemical composition of isolated particles. Ann N Y Acad Sci 98:92–99 10.1111/j.1749-6632.1962.tb30535.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 59.Hatch TP, Al-Hossainy E, Silverman JA. 1982. Adenine nucleotide and lysine transport in Chlamydia psittaci. J Bacteriol 150:662–670. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsumoto A. 1981. Isolation and electron microscopic observations of intracytoplasmic inclusions containing Chlamydia psittaci. J Bacteriol 145:605–612. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsumoto A, Bessho H, Uehira K, Suda T. 1991. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. J Electron Microsc (Tokyo) 40:356–363. [PubMed] [Google Scholar]
- 62.Chowdhury SR, Reimer A, Sharan M, Kozjak-Pavlovic V, Eulalio A, Prusty BK, Fraunholz M, Karunakaran K, Rudel T. 2017. Chlamydia preserves the mitochondrial network necessary for replication via microRNA-dependent inhibition of fission. J Cell Biol 216:1071–1089 10.1083/jcb.201608063. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. 2010. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet 6:e1000795. CORRECTION PLoS Genet 6:10.1371/annotation/4050116d-8daa-4b5a-99e9-34cdd13f6a26. 10.1371/journal.pgen.1000795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liang P, Rosas-Lemus M, Patel D, Fang X, Tuz K, Juárez O. 2018. Dynamic energy dependency of Chlamydia trachomatis on host cell metabolism during intracellular growth: role of sodium-based energetics in chlamydial ATP generation. J Biol Chem 293:510–522 10.1074/jbc.M117.797209. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Käding N, Kaufhold I, Müller C, Szaszák M, Shima K, Weinmaier T, Lomas R, Conesa A, Schmitt-Kopplin P, Rattei T, Rupp J. 2017. Growth of Chlamydia pneumoniae is enhanced in cells with impaired mitochondrial function. Front Cell Infect Microbiol 7:499 10.3389/fcimb.2017.00499. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rikihisa Y. 2015. Molecular pathogenesis of Ehrlichia chaffeensis infection. Annu Rev Microbiol 69:283–304 10.1146/annurev-micro-091014-104411. [PubMed] [DOI] [PubMed] [Google Scholar]
- 67.Popov VL, Chen S-M, Feng H-M, Walker DH. 1995. Ultrastructural variation of cultured Ehrlichia chaffeensis. J Med Microbiol 43:411–421 10.1099/00222615-43-6-411. [PubMed] [DOI] [PubMed] [Google Scholar]
- 68.Liu Y, Zhang Z, Jiang Y, Zhang L, Popov VL, Zhang J, Walker DH, Yu XJ. 2011. Obligate intracellular bacterium Ehrlichia inhibiting mitochondrial activity. Microbes Infect 13:232–238 10.1016/j.micinf.2010.10.021. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Von Ohlen T, Luce-Fedrow A, Ortega MT, Ganta RR, Chapes SK. 2012. Identification of critical host mitochondrion-associated genes during Ehrlichia chaffeensis infections. Infect Immun 80:3576–3586 10.1128/IAI.00670-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu H, Bao W, Lin M, Niu H, Rikihisa Y. 2012. Ehrlichia type IV secretion effector ECH0825 is translocated to mitochondria and curbs ROS and apoptosis by upregulating host MnSOD. Cell Microbiol 14:1037–1050 10.1111/j.1462-5822.2012.01775.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang JZ, Sinha M, Luxon BA, Yu XJ. 2004. Survival strategy of obligately intracellular Ehrlichia chaffeensis: novel modulation of immune response and host cell cycles. Infect Immun 72:498–507 10.1128/IAI.72.1.498-507.2004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khan M, Syed GH, Kim SJ, Siddiqui A. 2015. Mitochondrial dynamics and viral infections: a close nexus. Biochim Biophys Acta 1853(10 Pt B):2822–2833 10.1016/j.bbamcr.2014.12.040. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Willhite DC, Blanke SR. 2004. Helicobacter pylori vacuolating cytotoxin enters cells, localizes to the mitochondria, and induces mitochondrial membrane permeability changes correlated to toxin channel activity. Cell Microbiol 6:143–154 10.1046/j.1462-5822.2003.00347.x. [DOI] [PubMed] [Google Scholar]
- 74.Foo JH, Culvenor JG, Ferrero RL, Kwok T, Lithgow T, Gabriel K. 2010. Both the p33 and p55 subunits of the Helicobacter pylori VacA toxin are targeted to mammalian mitochondria. J Mol Biol 401:792–798 10.1016/j.jmb.2010.06.065. [PubMed] [DOI] [PubMed] [Google Scholar]
- 75.Suzuki M, Danilchanka O, Mekalanos JJ. 2014. Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe 16:581–591 10.1016/j.chom.2014.09.015. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nougayrède JP, Donnenberg MS. 2004. Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cell Microbiol 6:1097–1111 10.1111/j.1462-5822.2004.00421.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Kenny B, Jepson M. 2000. Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cell Microbiol 2:579–590 10.1046/j.1462-5822.2000.00082.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 78.Papatheodorou P, Domańska G, Öxle M, Mathieu J, Selchow O, Kenny B, Rassow J. 2006. The enteropathogenic Escherichia coli (EPEC) Map effector is imported into the mitochondrial matrix by the TOM/Hsp70 system and alters organelle morphology. Cell Microbiol 8:677–689 10.1111/j.1462-5822.2005.00660.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Sassera D, Beninati T, Bandi C, Bouman EAP, Sacchi L, Fabbi M, Lo N. 2006. ‘Candidatus Midichloria mitochondrii’, an endosymbiont of the tick Ixodes ricinus with a unique intramitochondrial lifestyle. Int J Syst Evol Microbiol 56:2535–2540 10.1099/ijs.0.64386-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 80.Shin JH, Yang JY, Jeon BY, Yoon YJ, Cho SN, Kang YH, Ryu DH, Hwang GS. 2011. 1H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J Proteome Res 10:2238–2247 10.1021/pr101054m. [PubMed] [DOI] [PubMed] [Google Scholar]
- 81.Wyatt EV, Diaz K, Griffin AJ, Rassmussen JA, Crane DD, Jones BD, Bosio CM. 2016. Metabolic reprogramming of host cells by virulent Francisella tularensis for optimal replication and modulation of inflammation. J Immunol 196:4227–4236. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]