

Abstract

The incorporation of aromatic difluoromethyl motifs has proven to be a fruitful strategy for enhancing the therapeutic profiles of modern pharmaceutical candidates. While the defluorofunctionalization of trifluoromethylarenes offers a promising pathway toward diverse aromatic difluoromethyl compounds, current methods are predominantly limited to two-component reactions. Multicomponent cascade reactions (MCRs) involving a transient aromatic difluoromethyl radical are still uncommon and highly sought after, owing to their capacity to rapidly generate challenging molecular structures. In this study, we present a photocatalytic manifold that combines commercially available trifluoromethylarenes, feedstock dienes, and various nucleophiles to achieve a modular defluorinative MCR. This method features mild reaction conditions and a broad substrate scope with excellent functional group compatibility. Furthermore, this protocol enables a previously unreported process of defluorinative editing for the resulting MCR aromatic difluoromethyl adducts. Preliminary mechanistic studies support the proposed photoexcited palladium catalytic cycle.
Keywords: defluorination, photocatalysis, multicomponent reaction, palladium-catalyzed, difluoromethyl motifs
Introduction
Organofluorine compounds are an important class of molecules found extensively across natural products, pharmaceuticals, agrochemicals, and advanced materials.1−8 In particular, aromatic difluoromethyls (ArCF2), which are prevalent in drug molecules and serve as privileged bioisosteres of benzoyl groups,9−17 have garnered increasing attention due to their high lipophilicity, metabolic stability and desirable electronic properties (Figure 1a).18 Consequently, there is a growing demand for the development of direct methodologies for the efficient and selective integration of ArCF2 moieties into various organic frameworks. Over the past few decades, considerable efforts have been devoted to construct this valuable motif through deoxyfluorination,19,20 site-selective C–H fluorination21-23 and fluoroalkylation.24−28 In contrast, the selective functionalization of a single C–F bond in commercially available trifluoromethylated arenes (ArCF3) would generate significant opportunities to readily access the ArCF2 functionality in modern drug discovery.
Figure 1.
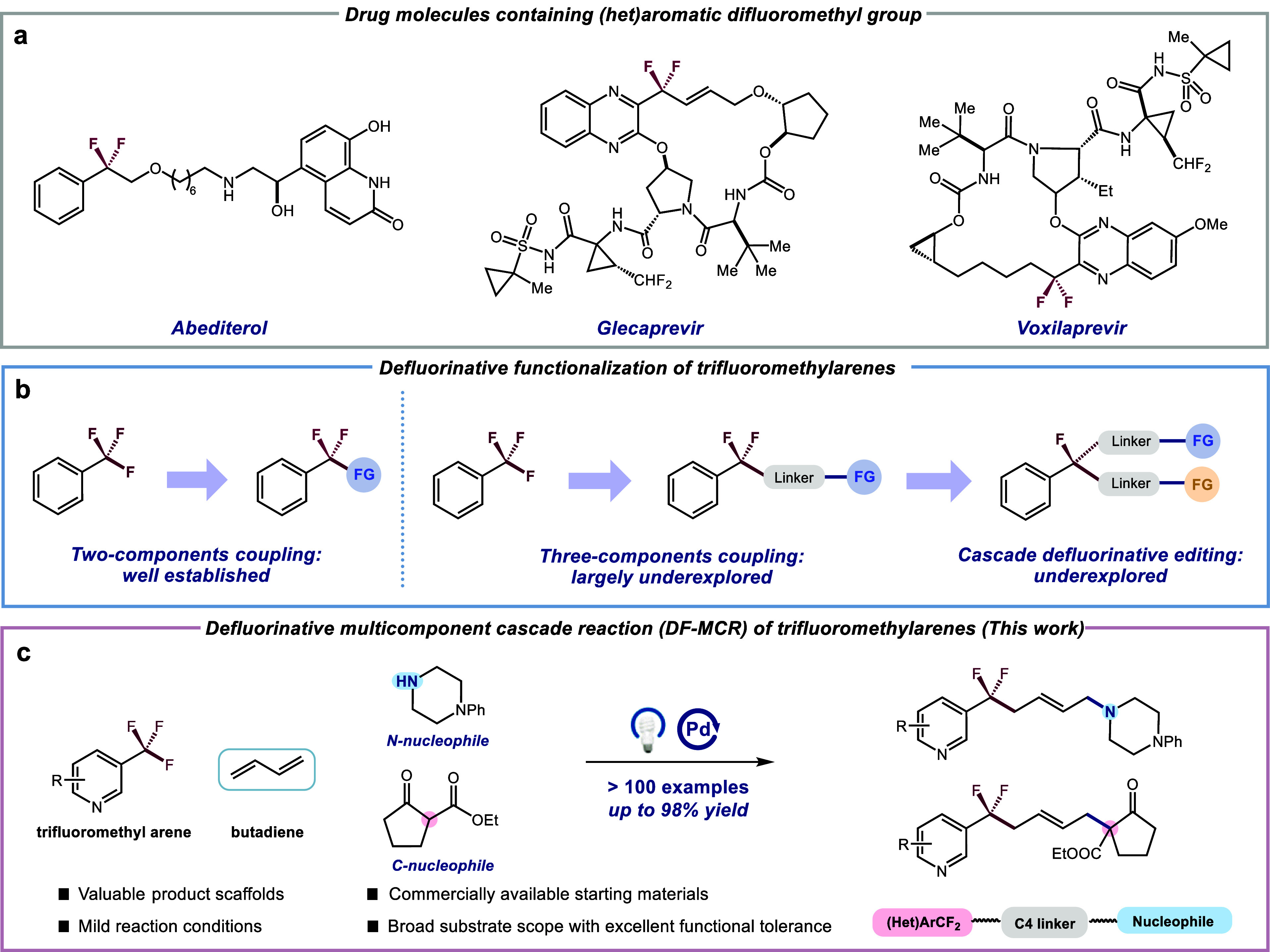
Development and construction of (het)aromatic difluoromethyl-contained motifs. a, Representative drug molecule containing (het)aromatic difluoromethyl group. b, Defluorinative functionalization of trifluoromethylarenes. c, Defluorinative multicomponent cascade reaction (DF-MCR) of trifluoromethylarenes.
Conventional methods for activating C–F bond for this class of substrate typically involve electrochemical reduction,29,30 low-valent metals,31-33 or frustrated Lewis pairs.34,35 However, the reactivity and selectivity of these processes usually suffer from the high energy of C(sp3)–F cleavage (∼481 kJ/mol for PhCF3)36 and the tendency toward exhaustive defluorination.37 Recently, photoredox catalysis has emerged as a potent platform for the C(sp3)–F cleavage of ArCF3. Notable works from research groups led by König,38 Jui,39,40 Gouverneur,41 Molander,42,43 Glorius,44 Zhang,45 and others46−50 have demonstrated the feasibility of single-electron reduction of the C(sp3)–F bond. Nevertheless, these pioneering advancements have primarily focused on two-component coupling, highlighting the inherent limitation of direct addition between ArCF2 radicals and acceptors (H, CO2, alkene, etc.) (Figure 1b, left). Multicomponent cascade reactions, known for their sustainable nature in rapidly synthesizing complex structures, drugs, natural products, and materials in a single step, play an essential role in the synthetic toolkit of organic chemists.51,52 Hence, it is extremely desirable to develop innovative MCRs involving the challenging selective/cascade defluorination of ArCF3, thereby inspiring rational reaction design (Figure 1b, right).53,54 1,3-Butadienes, easily accessible feedstocks and valuable building blocks, present an attractive platform for efficient difunctionalization to access complex and high-value molecules.55−57 Despite the demonstrated utility, a three-component coupling of N-/C-based nucleophiles, butadiene, and ArCF3 for the 1,4-difunctioalization of butadiene, which could provide a straightforward and practical strategy for the rapid synthesis of intriguing ArCF2-C4 linker-nucleophile molecules, has remained elusive thus far.58−61
Drawing inspiration from the recent achievements in visible light-induced palladium catalysis,62−69 we envisioned that the aforementioned transformation could be realized through this paradigm with commercially available materials, eliminating the need for an exogeneous photosensitizer. Herein, we report a modular, practical, and general photoexcited palladium-catalyzed 1,4-difunctionalization of butadiene with ArCF3 and N-/C-based nucleophiles (Figure 1c, > 100 examples, up to 98% yield). The mild reaction conditions tolerate a wide range of functional groups and bioactive molecules that are typically incompatible with traditional palladium catalysis, thereby creating new opportunities to expedite drug discovery and the exploration of advanced materials. Furthermore, the adaptability and versatility of this approach are highlighted through a subsequent defluorinative coupling of the resultant products, which would be challenging to achieve using the currently established methods.
Results and Discussion
We began our investigations by selecting 1,3-butadiene 1, 1-phenylpiperazine 2 and 3,5-bis(trifluoromethyl)-1,1’-biphenyl 3 as the model substrates, and we were delighted to find that the desired product 112 was obtained in 85% yield under the standard condition, albeit as a mixture of E/Z isomers (Table 1, entry 1). It is intriguing that only trace amounts of overdefluorinated side products were detected by LC-MS analysis, likely attributed to the overwhelming loading of trifluoromethylarenes, which surpasses the generation of difluoromethylarene products. Other bases, such as K2CO3, and TMG, proved to be less effective, resulting in lower yields (entries 2 and 3). Furthermore, other palladium species (1.5 mol%) led to inferior results (entries 4 and 5, see Supporting Information for details). Switching the solvent to MeCN resulted in a significant decrease in the yield of the DF-MCR product 112, likely due to the poor solubility for the catalyst and base (43% yield) (entry 6, see Supporting Information for details). The inclusion of two types of phosphine ligands proved to be crucial for enhancing the reaction efficiency. A yield of only 28% was observed in the absence of XantPhos, while a slightly reduced yield of 72% was obtained without the assistance of (o-OMe)Ph2PPh (entries 7 and 8). Finally, control experiments showed that the presence of the palladium catalyst, light, and LiOH was essential for achieving useful efficiencies of this transformation (entries 9–11). Notably, a 91% isolated yield was achieved when the reaction was carried out on a 0.3 mmol scale (entry 12).
Table 1. Optimization of Reaction Conditionsa.

| Entry | Variation of standard condition | Yieldb |
|---|---|---|
| 1 | None | 85%(E/Z = 1:2) |
| 2 | K2CO3 instead of LiOH | 29% |
| 3 | TMG instead of LiOH | 72% |
| 4 | Pd(PPh3)2Cl2 instead of Pd(PPh3)4 | 79% |
| 5 | Pd2(dba)3 instead of Pd(PPh3)4 | 19% |
| 6 | MeCN instead of THF | 43% |
| 7 | no XantPhos | 28% |
| 8 | no (o–OMe)Ph2PPh | 72% |
| 9 | no Pd(PPh3)4 | N.D. |
| 10 | in the dark | N.D. |
| 11 | no LiOH | trace |
| 12 | 0.3 mmol scale | 91%c(E/Z = 1:2) |
Reaction conditions: 1 (0.30 mmol), 2 (0.15 mmol), 3 (0.45 mmol), Pd(PPh3)4 (1.5 mol%), XantPhos (8 mol%), [(o-OMe)Ph]2PPh (8 mol%), LiOH (0.15 mmol), THF (0.1 M), λmax = 440 nm Kessil (40 W), N2, RT – 40 °C, 6 h.
GC yield with 1,3,5-trimethylbenzene as internal standard.
12 h, isolated yield. N.D., not detected.
With the optimized conditions in hand, we first evaluated the generality of the protocol with respect to trifluoromethylarenes. To simplify purification, the products were isolated as the corresponding hydrogenated DF-MCR adducts after reduction with Pd(OH)2/C. As shown in Scheme 1, a wide array of aromatic bis(trifluoromethyl) compounds bearing electron-donating and electron-withdrawing substituents were found to be competent substrates (3-17, 54–95% yield). Notably, various functional groups such as (het)arenes, ethers, unprotected amines, and nitriles were all compatible, providing practical handles for further derivatization. Additionally, aromatic mono(trifluoromethyl) compounds (18–21) underwent smooth reactions to yield the desired products in 39–60% yield. It is worth mentioning that the benzylic alcohol 22 was cleanly preserved, providing MCR product in 51% yield.
Scheme 1. Scope of Trifluoromethylarenes.

Reaction conditions:(Het)ArCF3 (0.9 mmol), 1 (0.6 mmol), 2 (0.3 mmol), Pd(PPh3)4 (1.5 mol%), XantPhos (8 mol%), [(o-OMe)Ph]2PPh (8 mol%), LiOH (0.3 mmol), THF (0.1 M), λmax = 440 nm Kessil (40 W), N2, RT – 40°C, 12 h. Hydrogenation yield is shown in square brackets. See Supporting Information for hydrogenation procedures.
w/o [(o-OMe)Ph]2PPh, with Mg(OTf)2 (20 mol%), 12–24 h.
TMG (0.3 mmol) instead of LiOH as base.
K3PO4 (0.3 mmol) instead of LiOH as base.
Considering the significance of nitrogen-containing heterocycles in bioactive molecules synthesis, we were pleased to find that a broad range of pyridine substrates with trifluoromethyl groups at 2-, 3-, and 4- positions readily engage in this DF-MCR (23–38, 40–81% yield). Interestingly, the addition of 20% Mg(OTf)2 significantly enhance the conversion of the ArCF3 and improved the MCR yields. We assumed that Mg2+ could facilitate C-F bond cleavage or promote single electron transfer (SET) from Pd(0) to (het)aromatic trifluoromethyls by anchoring to the nitrogen atom.70,71 Furthermore, this reactivity could be extended to the quinoline substrates, albeit with slightly lower yields (39 and 40, 41% and 48% yield, respectively).
Next, we explored the range of nucleophiles in this new protocol (Scheme 2). It was gratifying to discover that a diverse array of medicinally relevant nitrogen-based nucleophiles could engage in this transformation. Commercially available morpholine derivatives (41–43), piperidine derivatives (44–52), piperazine derivatives (53–63) and pyrrolidine derivatives (64–68) were all tolerated in this DF-MCRs with moderate to excellent yields (39–95% yield). Pleasingly, the presence of other polar functional groups such as amides and alcohols did not impede the reaction efficiency (52, 56–58, and 65). Moreover, the protocol could employ acyclic amine such as benzylamines (69–74), alkyl amines (75–77) as nucleophiles, affording the desired products in good yields (42–90% yield). Additionally, primary amines (78) were reactive under the optimized conditions, albeit with slightly lower yield (36% yield). Amides were also applicable substrates; however, only 28% yield of the desired product was obtained, likely due to the less nucleophilic nature of succinimide (80). Remarkably, this transformation could also accommodate aromatic amines (81–86) delivering the target products with useful levels of efficiency (45–62% yield). Encouraged by these promising results, we next sought to extend the protocol to carbon-based nucleophiles. Gratifyingly, various 1,3-dicarbonyl cyclic ester or ketones (87–91, 51–73% yield) could serve as coupling partners. Additionally, a range of acyclic 1,3-dicarbonyl compounds (92–95) was examined in the DF-MCRs, yielding the desired products at useful levels (31–75% yield). Notably, this protocol demonstrates good to excellent E/Z selectivity with the C-nucleophiles, in stark contrast to previous instances with N-nucleophiles that usually resulted in low E/Z selectivity. Finally, other 1,3-conjugated dienes were examined. Under standard conditions, 2-methyl-1,3-butadiene exhibited good reactivity with a regioselectivity of 1:3 (96, 66% yield). However, when 1-phenyl-1,3-butadiene was used in the reaction, 1,2-addition was superior to 1,4-addition with 2:1 regioselectivity in 29% yield (97).
Scheme 2. Scope of Nucleophiles.

Reaction conditions: 3 (0.9 mmol), 1 (0.6 mmol), amine (0.3 mmol), Pd(PPh3)4 (1.5 mol%), XantPhos (8 mol%), [(o-OMe)Ph]2PPh (8 mol%), LiOH (0.3–0.9 mmol, when amine hydrochloride was used, 1.0 equiv. of hydrochloride consumed additional 1.0 equiv. of LiOH), THF (0.1 M), λmax = 440 nm Kessil (40 W), N2, RT–40 °C, 12 h. Hydrogenation yield is shown in square brackets. See Supporting Information for hydrogenation procedures.
w/o [(o-OMe)Ph]2PPh, with Mg(OTf)2 (20 mol%).
11 (0.9 mmol), 1 (0.6 mmol), 1,3-dicarbonyl compound (0.3 mmol), Pd(PPh3)4 (1.5 mol%), XantPhos (8 mol%), [(o-OMe)Ph]2PPh (8 mol%), LiOH (0.3 mmol), THF (0.1 M), λmax = 440 nm Kessil (40 W), N2, RT– 40 °C, 12 h. Hydrogenation yield is shown in square brackets. See Supporting Information for hydrogenation procedures.
Having demonstrated the success of these DF-MCRs, we were further motivated to explore late-stage modification of natural products and pharmaceuticals (scheme 3). Trifluoromethylarenes derived from estrone, galactose, and azelnidipine were proved to be effective substrates for late-stage defluorinative coupling with 1,3-butadiene 1 and 1-phenylpiperazine 2 under standard conditions, resulting in the corresponding products 98–100 in 60–82% yields. Furthermore, a series of amine-containing drug molecules could also be employed to furnish the target ArCF2-C4 linker-amines in synthetically useful yields. Methyl amines such as fluoxetine (101), maprotiline (103), doluxetine (104), betahistine (105) and nortriptyline (110) were reacted smoothly to give the desired products in moderate to excellent yields (54–96% yield). Cyclic amines such as troxipide (102), desbenzyl donepezil (106), D-proline (107), sitagliptin (108), nortropine (109), and aprepitant (111) were also competent to deliver satisfying results (65–94% yield), showcasing the broad compatibility of our protocol with complex structural scaffolds.
Scheme 3. Late-stage Modification of Natural Products and Drug Derivatives.

Reaction conditions: ArCF3 (0.9 mmol), 1,3-butadiene (0.6 mmol), amine (0.3 mmol), Pd(PPh3)4 (1.5 mol%), XantPhos (8 mol%), [(o-OMe)Ph]2PPh (8 mol%), LiOH (0.3–0.9 mmol, when amine hydrochloride was used, 1.0 equiv. of hydrochloride consumed additional 1.0 equiv. LiOH), THF (0.1 M), λmax = 440 nm Kessil (40 W), N2, RT – 40°C, 12 h. Hydrogenation yield is shown in square brackets. See Supporting Information for hydrogenation procedures.
To highlight the potential utility of the DF-MCRs in organic synthesis, several applications were showcased. First, a cascade defluorinative coupling of 3 was demonstrated, providing benzylic monofluoromethyl compound 113 under standard condition (scheme 4a). To our knowledge, this represents the first instance of DF-MCRs adducts engaging in a metallaphotoredox-catalyzed cascade defluorinative editing process. It is noteworthy that perfluoroalkylarene 114 could also be utilized for selectively defluorinative coupling, producing the corresponding product 115 in 55% yield (Scheme 4b). Additionally, a gram-scale reaction was performed under the standard conditions, giving the target products 116 in 88% yield (Scheme 4c). Moreover, when piperazine was employed as the nucleophile for iteratively defluorinative coupling, two C–N bonds were formed in a single step to afford 118 in 75% yield (Scheme 4d, right). Similar result was also observed with malonic acid cyclic isopropylidene ester as a carbon-based nucleophile (117, 85% yield) (Scheme 4d, left). Furthermore, a cascade C–N bond formation was carried out to provide 119 and 120 in moderate yield by using t-butyloxycarboryl protected amine 68 as a nucleophile (Scheme 4e). Finally, two medicinally significant drugs (netupitant and betahistine) could be employed in this newly developed three-component cascade coupling to generate the C4-linked drug derivative 121 in 59% yield, a synthesis that would be challenging if using existing synthetic methodologies (Scheme 4f).
Scheme 4. Synthetic Applications.
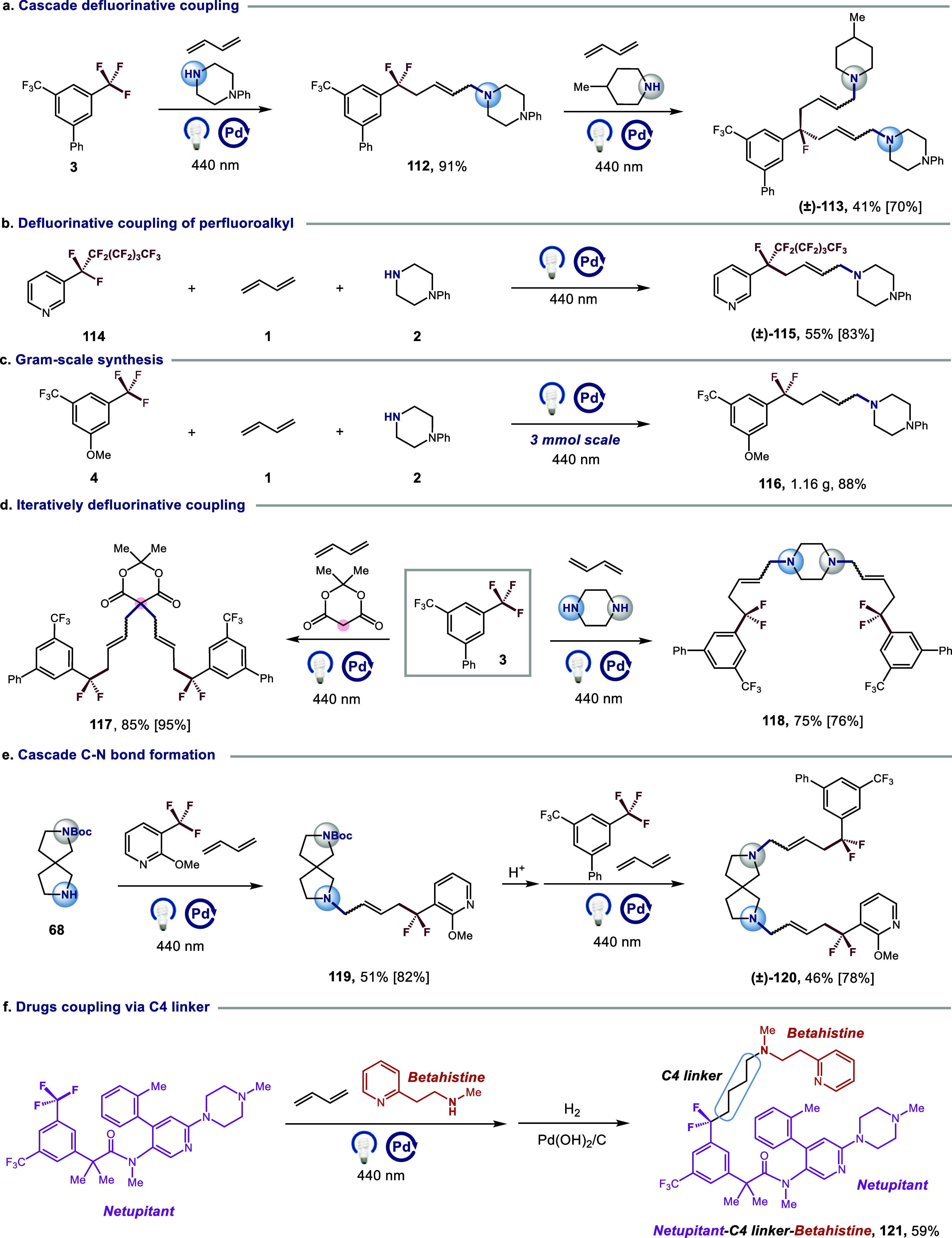
Reaction conditions: ArCF2R (0.9 mmol), 1 (0.6 mmol), amine (0.3 mmol), Pd(PPh3)4 (1.5 mol%), XantPhos (8 mol%), [(o-OMe)Ph]2PPh (8 mol%), LiOH (0.3 mmol), THF (0.1 M), λmax = 440 nm Kessil (40 W), N2, RT – 40 °C, 12 h. Hydrogenation yield is shown in square brackets. See SI for hydrogenation procedures.
To gain insight into this transformation, several mechanistic studies were conducted. First, when the reaction was carried out in the presence of TEMPO, only a trace amount of product 116 was detected, along with the identification of the difluoromethyl-TEMPO adduct 124 (detected by GCMS). This observation suggests the involvement of difluoromethyl radical intermediates in the process (Figure 2a). In contrast, when BHT was used as an inhibitor, we found that the yield of 116 remained largely unaffected, and no BHT adduct 125 was found in the reaction system, likely due to the rapid recombination of the difluoromethyl radical with Pd (I) species to form the difluoromethyl Pd(II) complex (Figure 2a). Furthermore, an α-cyclopropylstyrene 126 was employed in a radical clock experiment, where ring-opening followed by intramolecular cyclization resulted in the formation of the corresponding product 127 in 30% yield, supporting the radical mechanism in this process (Figure 2b). A tracking experiment revealed that the E/Z ratio of 14a did not change over time, indicating that irradiation did not alter the configuration of the double bond during the photocatalysis process (Figure 2c). The slightly favorable Z-configuration suggested by the E/Z ratio led to speculation that the Z-configuration in the metal complex intermediates is more stable than the E-configuration. Thus, density functional theory (DFT) calculations were performed to investigate some details of these two configurations in the Pd (II) complex before product releases. The geometries were optimized in a vacuum at the B3LYP-D3 level. The mixed basis set was adopted (LanL2DZ for Pd and 6–31G* for nonmetallic atoms) and it was improved for single-point calculations (LanL2DZ for Pd and 6–311G** for nonmetallic atoms) with the inclusion of solvent effects of THF (SMD model). As shown in Figure 2d, the π-center of the substrate faces inward to XantPhos in Z- IM, while it faces outward to the ligand in E- IM. This suggests that the substrate may be stabilized by XantPhos in the former, a viewpoint supported by the Gibbs free energy calculations. Specifically, the ΔG of Z-IM is 2.3 kcal mol–1 lower than that of E-IM. This observation may explain the higher proportion of Z-products compared to E-products in the experimental results compared with those reported in the literature. We also conducted UV–vis experiments and found that Pd(PPh3)4 can be excited at approximately 340 nm with moderate absorbance. A similar result was observed upon the addition of XantPhos, resulting in a red shift to around 360 nm and decreased absorbance. It was unexpected that the addition of [(2-OMe)Ph]2PPh led to a significant increase in absorbance (Figure S6). Furthermore, 31P NMR experiments revealed that either XantPhos or [(2-OMe)Ph]2PPh can facilitate the ligand exchange process of Pd(PPh3)4 (Figures S12 and S13). These results suggest the generation of a Pd(0)-XantPhos-[(2-OMe)Ph]2PPh complex in the reaction system, which likely serves as the active catalyst for this reaction.
Figure 2.
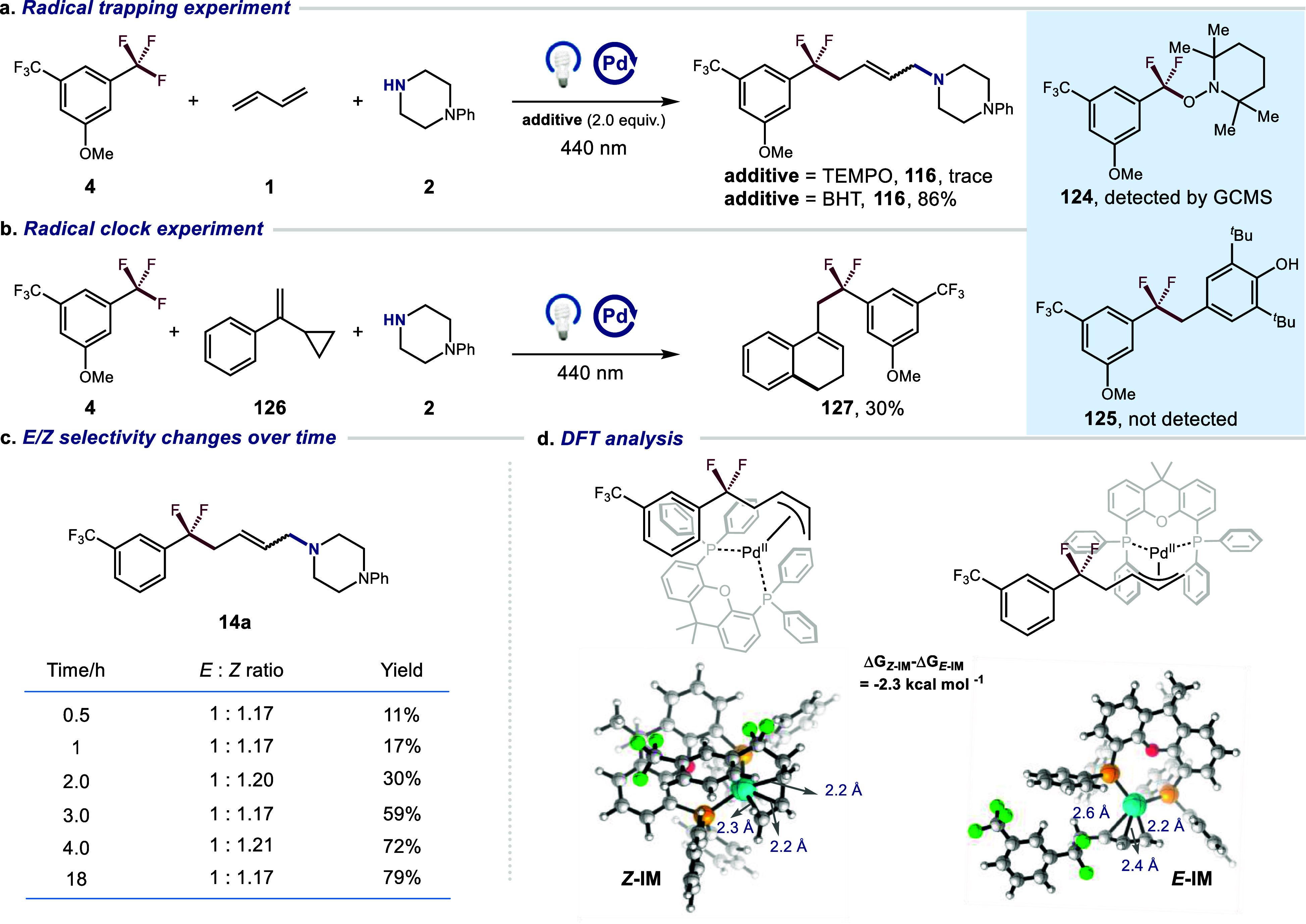
Preliminary mechanistic experiments. a, Radical trapping experiment. b, Radical clock experiment. c, E/Z Selectivity changes over time. d, DFT analysis.
On the basis of mechanistic experiments and earlier precedents,58−69 a putative mechanistic pathway was proposed (Figure 3). Initially, under visible light conditions (blue LED), a direct single electron transfer from excited Pd(0) complex A to trifluoromethylarenes generates intermediate B (Tables S6-S11, Figures S12 and S13), which then produces a hybrid difluoromethyl Pd(I) radical intermediate C via Li+ assisted C-F bond cleavage (Table S2).70,71 Subsequently, C may undergo a radical addition into 1,3-dienes to form hybrid allylic Pd(I) radical species D (path a), or it may proceed through the recombination of the resulting difluoromethyl radical with Pd(I) leading to the formation of the difluoromethyl Pd(II) complex C’(45) (path b). This is followed by radical recombination of D or the migration insertion of C’, resulting in the generation of π-allylpalladium complexes E. Meanwhile, OH– can serve as a suitable base in THF conditions to enhance the nucleophilicity of the amine, which then attacks the π-allylpalladium complexes E to yield the desired DF-MCR product F and regenerate the palladium catalyst in a redox-neutral process.
Figure 3.

Proposed mechanism.
Conclusions
In summary, we have demonstrated a general catalytic protocol for the defluorinative multicomponent coupling of trifluoromethylarenes, dienes, and N-/C-based nucleophiles enabled by the photoexcited palladium catalytic system. These transformations were characterized by their wide applicability, mild redox-neutral conditions, and capacity for the late-stage modification of natural products or drugs. Furthermore, the successful cascade editing of difluoromethyl groups within this protocol underscores its potential for efficiently accessing complex molecular diversity. Mechanistic investigations are presented and discussed.
Methods
General Procedure for Three Components Coupling
To an 8 mL vial equipped with a stir bar was added Pd(PPh3)4 (5.2 mg, 4.6 μmol, 0.015 equiv.), XantPhos (14.0 mg, 0.024 mmol, 0.08 equiv.), bis(2-methoxyphenyl)phenylphosphine (7.8 mg, 0.024 mmol, 0.08 equiv.), substituted trifluoromethylbenzene (0.90 mmol, 3.0 equiv.), amine or 1,3-dicarbonyl compound (if solid or high boiling point liquid, 0.30 mmol, 1.0 equiv.), LiOH (7.2 mg, 0.30 mmol, 1.0 equiv.) and THF (3.0 mL). The solution was degassed by bubbling with nitrogen for 8 min, and 1,3-butadiene (2.0 mol/L in THF, 300 μL, 0.60 mmol, 2.0 equiv) was syringed into the reaction vessel before sealing with parafilm. The reaction was carried out in a steel chamber under the irradiation at room temperature via blue LEDs (40 W, λmax = 440 nm) for 12 h. The reaction mixture was removed from the light, cooled to ambient temperature, and quenched by exposure to air. After the removal of the solvent, the residue was purified by flash chromatography on silica gel to afford the desired product.
Acknowledgments
The authors thank Mingshuo Chen for help discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.4c00899.
General information, synthesis of starting materials, details of reaction optimization, synthetic applications and preliminary mechanistic experiments, computation data, characterization of all compounds, references, and NMR Spectra (PDF)
Author Contributions
X.Z. conceptualized the project. Z.L., K.W., B.Z., P.L., and X.Z. designed, performed, and analyzed experiments. L.B. conducted the DFT calculations. Z.L., B.Z., and X.Z. prepared the manuscript, which was revised by all authors. All authors have given approval to the final version of the manuscript. CRediT: Zhibin Li data curation, methodology, writing - original draft; Lei Bao formal analysis, investigation; Kaihang Wei investigation, methodology; Bei-Bei Zhan investigation, writing - original draft; Ping Lu validation; Xiaheng Zhang conceptualization, funding acquisition, project administration, writing - original draft, writing - review & editing.
We are grateful for financial support from the National Natural Science Foundation of China (22171049, 22301054), the Natural Science Foundation of Zhejiang Province (LDQ23B020001), the Hangzhou leading innovation and entrepreneurship team project (TD2022002) and Research Funds of Hangzhou Institute for Advanced Study, UCAS.
The authors declare the following competing financial interest(s): X. Zhang, Z. Li and K. Wei are inventors on a Chinese patent application (Application No. CN202410093803X).
Supplementary Material
References
- Kirsch P.Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, 2004. [Google Scholar]
- Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- Berger R.; Resnati G.; Metrangolo P.; Weberd E.; Hulliger J. Organic Fluorine Compounds: a Great Opportunity for Enhanced Materials Properties. Chem. Soc. Rev. 2011, 40, 3496–3508. 10.1039/c0cs00221f. [DOI] [PubMed] [Google Scholar]
- Wang J.; Sánchez-Roselló M.; Aceña J. L.; Del Pozo C.; Sorochinsky A. E.; Fustero S.; Soloshonok V. A.; Liu H. Fluorine in Pharmaceutical Industry: Fluorine-containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- Muller K.; Faeh C.; Diederich F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Clayden J. Fluorinated Compounds Present Opportunities for Drug Discovery. Nature 2019, 573, 37–38. 10.1038/d41586-019-02611-7. [DOI] [PubMed] [Google Scholar]
- Fujiwara T.; O’Hagan D. Successful Fluorine-containing Herbicide Agrochemicals. J. Fluor. Chem. 2014, 167, 16–29. 10.1016/j.jfluchem.2014.06.014. [DOI] [Google Scholar]
- Hagmann W. K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- Brahms D. L. S.; Dailey W. P. Fluorinated Carbenes. Chem. Rev. 1996, 96, 1585–1632. 10.1021/cr941141k. [DOI] [PubMed] [Google Scholar]
- Feng Z.; Xiao Y.; Zhang X. Transition-metal (Cu, Pd, Ni)-Catalyzed Difluoroalkylation via Cross-coupling with Difluoroalkyl Halides. Acc. Chem. Res. 2018, 51, 2264–2278. 10.1021/acs.accounts.8b00230. [DOI] [PubMed] [Google Scholar]
- Ma X.; Song Q. Recent Progress on Selective Deconstructive Modes of Halodifluoromethyl and Trifluoromethylcontaining Reagents. Chem. Soc. Rev. 2020, 49, 9197–9219. 10.1039/D0CS00604A. [DOI] [PubMed] [Google Scholar]
- Ai H.; Ma X.; Song Q.; Wu X. C–F Bond Activation under Transition-metal-free Conditions. Sci. China: chem 2021, 64, 1630–1659. 10.1007/s11426-021-1040-2. [DOI] [Google Scholar]
- Li S.; Shu W. Recent Advances in Radical Enabled Selective Csp3–F Bond Activation of Multifluorinated Compounds. Chem. Commun. 2022, 58, 1066–1077. 10.1039/D1CC06446K. [DOI] [PubMed] [Google Scholar]
- Zhao F.; Zhou W.; Zuo Z. Recent Advances in the Synthesis of Difluorinated Architectures from Trifluoromethyl Groups. Adv. Synth. Catal. 2022, 364, 234–267. 10.1002/adsc.202101234. [DOI] [Google Scholar]
- Wang Z.; Sun Y.; Shen L.; Yang W.; Meng F.; Li P. Photochemical and Electrochemical Strategies in C–F Bond Activation and Functionalization. Org. Chem. Front. 2022, 9, 853–873. 10.1039/D1QO01512E. [DOI] [Google Scholar]
- Simur T. T.; Ye T.; Yu Y.; Zhang F.; Wang Y. C–F Bond Functionalizations of Trifluoromethyl Groups via Radical Intermediates. Chin. Chem. Lett. 2022, 33, 1193–1198. 10.1016/j.cclet.2021.08.043. [DOI] [Google Scholar]
- Ni C.; Hu J. The Unique Fluorine Effects in Organic Reactions: Recent Facts and Insights into Fluoroalkylations. Chem. Soc. Rev. 2016, 45, 5441–5454. 10.1039/C6CS00351F. [DOI] [PubMed] [Google Scholar]
- Middleton W. J.; Bingham E. M. α,α-Difluoroarylacetic Acids: Preparation from (Diethylamino)sulfur Trifluoride and α-Oxoarylacetates. J. Org. Chem. 1980, 45, 2883–2887. 10.1021/jo01302a025. [DOI] [Google Scholar]
- Ni C.; Hu M.; Hu J. Good Partnership between Sulfur and Fluorine: Sulfur-based Fluorination and Fluoroalkylation Reagents for Organic Synthesis. Chem. Rev. 2015, 115, 765–825. 10.1021/cr5002386. [DOI] [PubMed] [Google Scholar]
- Xia J.; Zhu C.; Chen C. Visible Light-promoted Metal-free C–H Activation: Diarylketone-catalyzed Selective Benzylic Mono- and Difluorination. J. Am. Chem. Soc. 2013, 135, 17494–17500. 10.1021/ja410815u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P.; Guo S.; Wang L.; Tang P. Silver-catalyzed Oxidative Activation of Benzylic C-H Bonds for the Synthesis of Difluoromethylated Arenes. Angew. Chem., Int. Ed. 2014, 53, 5955–5958. 10.1002/anie.201400225. [DOI] [PubMed] [Google Scholar]
- Ning Y.; Sivaguru P.; Zanoni G.; Anderson E. A.; Bi X. Synthesis of β-Difluoroalkyl Azides via Elusive 1,2-Azide. Chem 2020, 6, 486–496. 10.1016/j.chempr.2019.12.004. [DOI] [Google Scholar]
- Xie J.; Zhang T.; Chen F.; Mehrkens N.; Rominger F.; Rudolph M.; Hashmi A. S. K. Gold-catalyzed Highly Selective Photoredox C(sp2)-H Difluoroalkylation and Perfluoroalkylation of Hydrazones. Angew. Chem., Int. Ed. 2016, 55, 2934–2938. 10.1002/anie.201508622. [DOI] [PubMed] [Google Scholar]
- Fu X.; Xue X.; Zhang X.; Xiao Y.; Zhang S.; Guo Y.; Leng X.; Houk K. N.; Zhang X. Controllable Catalytic Difluorocarbene Transfer Enables Access to Diversified Fluoroalkylated Arenes. Nat. Chem. 2019, 11, 948–956. 10.1038/s41557-019-0331-9. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Nottingham K. G.; Patel C.; Alegre-Requena J. V.; Levy J. N.; Paton R. S.; McNally A. Phosphorus-mediated sp2–sp3 Couplings for C–H Fluoroalkylation of Azines. Nature 2021, 594, 217–222. 10.1038/s41586-021-03567-3. [DOI] [PubMed] [Google Scholar]
- Feng X.; Ren J.; Gao X.; Min Q.; Zhang X. 3,3-Difluoroallyl Sulfonium Salts: Practical and Bench-Stable Reagents for Highly Regioselective gem -Difluoroallylations. Angew. Chem., Int. Ed. 2022, 61, e202210103 10.1002/anie.202210103. [DOI] [PubMed] [Google Scholar]
- Deng D.; Tang S.; Yuan Y.; Xie D.; Zhu Z.; Huang Y.; Liu Y. C-F Insertion Reaction Sheds New Light on the Construction of Fluorinated Compounds. Chin. Chem. Lett. 2024, 35, 109417. 10.1016/j.cclet.2023.109417. [DOI] [Google Scholar]
- Lund H.; Jensen N. J.; Almqvist S.O.; Enzell C. R.; Taticchi A.; Mannervik B. Electroörganic Preparations. XXXVI. Stepwise Reduction of Benzotrifluoride. Acta. Chem. Scand. 1974, 28b, 263–265. 10.3891/acta.chem.scand.28b-0263. [DOI] [Google Scholar]
- Yamauchi Y.; Fukuhara T.; Hara S.; Senboku H. Electrochemical Carboxylation of α,α-Difluorotoluene Derivatives and its Application to the Synthesis of α-Fluorinated Nonsteroidal Anti-inflammatory Drugs. Synlett 2008, 2008, 438–442. 10.1055/s-2008-1032069. [DOI] [Google Scholar]
- Munoz S. B.; Ni C.; Zhang Z.; Wang F.; Shao N.; Mathew T.; Olah G. A.; Prakash G. K. S. Selective Late-stage Hydrodefluorination of Trifluoromethylarenes: a Facile Access to Difluoromethylarenes. Eur. J. Org. Chem. 2017, 2017, 2322–2326. 10.1002/ejoc.201700396. [DOI] [Google Scholar]
- Fuchibe K.; Ohshima Y.; Mitomi K.; Alkiyama T. Low-Valent Niobium-Catalyzed Reduction of α,α,α-Trifluorotoluenes. Org. Lett. 2007, 9, 1497–1499. 10.1021/ol070249m. [DOI] [PubMed] [Google Scholar]
- Amii H.; Hatamoto Y.; Seo M.; Uneyama K. A New C-F Bond-cleavage Route for the Synthesis of Octafluoro[2.2]paracyclophane. J. Org. Chem. 2001, 66, 7216–7218. 10.1021/jo015720i. [DOI] [PubMed] [Google Scholar]
- Stahl T.; Klare H. F. T.; Oestreich M. Main-group Lewis Acids for C–F Bond Activation. ACS Catal. 2013, 3, 1578–1587. 10.1021/cs4003244. [DOI] [Google Scholar]
- Forster F.; Metsänen T. T.; Irran E.; Hrobarik P.; Oestreich M. Cooperative Al–H Bond Activation in DIBAL-H: Catalytic Generation of an Alumenium-ion-like Lewis Acid for Hydrodefluorinative Friedel–Crafts Alkylation. J. Am. Chem. Soc. 2017, 139, 16334–16342. 10.1021/jacs.7b09444. [DOI] [PubMed] [Google Scholar]
- O’Hagan D. Understanding Organofluorine Chemistry. An Introduction to the C–F Bond. Chem. Soc. Rev. 2008, 37, 308–319. 10.1039/B711844A. [DOI] [PubMed] [Google Scholar]
- Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton: 2007. [Google Scholar]
- Chen K.; Berg N.; Gschwind R.; König B. Selective Single C(sp3)–F Bond Cleavage in Trifluoromethylarenes: Merging Visible-Light Catalysis with Lewis Acid Activation. J. Am. Chem. Soc. 2017, 139, 18444–18447. 10.1021/jacs.7b10755. [DOI] [PubMed] [Google Scholar]
- Wang H.; Jui N. T. Catalytic Defluoroalkylation of Trifluoromethylaromatics with Unactivated Alkenes. J. Am. Chem. Soc. 2018, 140, 163–166. 10.1021/jacs.7b12590. [DOI] [PubMed] [Google Scholar]
- Vogt D. B.; Seath C. P.; Wang H.; Jui N. T. Selective C–F Functionalization of Unactivated Trifluoromethylarenes. J. Am. Chem. Soc. 2019, 141, 13203–13211. 10.1021/jacs.9b06004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sap J. B. I.; Straathof N. J. W.; Knauber T.; Meyer C. F.; Médebielle C.; Buglioni L.; Genicot C.; Trabanco A. A.; Noël T.; Am Ende C. W.; Gouverneur V. Organophotoredox Hydrodefluorination of Trifluoromethylarenes with Translational Applicability to Drug Discovery. J. Am. Chem. Soc. 2020, 142, 9181–9187. 10.1021/jacs.0c03881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M. W.; Polites V. C.; Patel S.; Lipson J. E.; Majhi J.; Molander G. A. Photochemical C–F Activation Enables Defluorinative Alkylation of Trifluoroacetates and Acetamides. J. Am. Chem. Soc. 2021, 143, 19648–19654. 10.1021/jacs.1c11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shreiber S. T.; Granados A.; Matsuo B.; Majhi J.; Campbell M. W.; Patel S.; Molander G. A. Visible-light-induced C–F Bond Activation for the Difluoroalkylation of Indoles. Org. Lett. 2022, 24, 8542–8546. 10.1021/acs.orglett.2c03549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J.; Bellotti P.; Heusel C.; Glorius F. Photoredox-Catalyzed Defluorinative Functionalizations of Polyfluorinated Aliphatic Amides and Esters. Angew. Chem., Int. Ed. 2022, 61, e202115456 10.1002/anie.202115456. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Tong F.; Zhang Y.; He C.; Zhang X. Visible-lightinduced Palladium-catalyzed Selective Defluoroarylation of Trifluoromethylarenes with Arylboronic Acids. J. Am. Chem. Soc. 2021, 143, 13971–13979. 10.1021/jacs.1c07459. [DOI] [PubMed] [Google Scholar]
- Sugihara N.; Suzuki K.; Nishimoto Y.; Yasuda M. Photoredoxcatalyzed C–F Bond Allylation of Perfluoroalkylarenes at the Benzylic Position. J. Am. Chem. Soc. 2021, 143, 9308–9313. 10.1021/jacs.1c03760. [DOI] [PubMed] [Google Scholar]
- Liu C.; Shen N.; Shang R. Photocatalytic Defluoroalkylation and Hydrodefluorination of Trifluoromethyls Using o-Phosphinophenolate. Nat. Commun. 2022, 13, 354. 10.1038/s41467-022-28007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S.; Liu S.; Chen L.; Bo Z.; Jing K.; Gao T.; Yu B.; Lan Y.; Luo S.; Yu D. Visible-light Photoredox-catalyzed Selective Carboxylation of C(sp3)–F Bonds with CO2. Chem 2021, 7, 3099–3113. 10.1016/j.chempr.2021.08.004. [DOI] [Google Scholar]
- Huang J.; Gao Q.; Zhong T.; Chen S.; Lin W.; Han J.; Xie J. Photoinduced Copper-catalyzed C–N Coupling with Trifluoromethylated Arenes. Nat. Commun. 2023, 14, 8292. 10.1038/s41467-023-44097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Zhang G.; Zhou Q.; Bian T.; Zhou L.; Zhang Z. Hybrid Palladium-Catalyzed Intramolecular Carboamination of Conjugated Dienes: Synthesis of Functionalized Pyrrolidines via Selective Trifluoromethylarene Defluorination. J. Org. Chem. 2024, 89, 7790–7794. 10.1021/acs.joc.4c00447. [DOI] [PubMed] [Google Scholar]
- Rotstein B. H.; Zaretsky S.; Rai V.; Yudin A. K. Small Heterocycles in Multicomponent Reactions. Chem. Rev. 2014, 114, 8323–8359. 10.1021/cr400615v. [DOI] [PubMed] [Google Scholar]
- Touré B. B.; Hall D. G. Natural Product Synthesis Using Multicomponent Reaction Strategies. Chem. Rev. 2009, 109, 4439–4486. 10.1021/cr800296p. [DOI] [PubMed] [Google Scholar]
- Fan Y.; Huang Z.; Lu Y.; Zhu S.; Chu L. Defluorinative Alkylboration of Alkenes Enabled by Dual Photoredox and Copper Catalysis. Angew. Chem., Int. Ed. 2024, 63, e202315974 10.1002/anie.202315974. [DOI] [PubMed] [Google Scholar]
- Chen M.; Cui Y.; Chen X.; Shang R.; Zhang X. C–F Bond Activation Enables Synthesis of Aryl Difluoromethyl Bicyclopentanes as Benzophenone-type Bioisosteres. Nat. Commun. 2024, 15, 419. 10.1038/s41467-023-44653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes M.; Schwartz L. A.; Krische M. J. Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev. 2018, 118, 6026–6052. 10.1021/acs.chemrev.8b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Huo X.; Jiang X.; Zhang W. Asymmetric Synthesis of Allylic Compounds via Hydrofunctionalisation and Difunctionalisation of Dienes, Allenes, and Alkynes. Chem. Soc. Rev. 2020, 49, 2060–2118. 10.1039/C9CS00400A. [DOI] [PubMed] [Google Scholar]
- Canfield A. M.; Rodina D.; Paradine S. M. Dienes as Versatile Substrates for Transition Metal-Catalyzed Reactions. Angew. Chem., Int. Ed. 2024, 63, e202401550 10.1002/anie.202401550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Bellotti P.; Pflüger P. M.; Schwarz J. L.; Heidrich B.; Glorius F. Three-component, Interrupted Radical Heck/allylic Substitution Cascade Involving Unactivated Alkyl Bromides. J. Am. Chem. Soc. 2020, 142, 10173–10183. 10.1021/jacs.0c03239. [DOI] [PubMed] [Google Scholar]
- Cheung K. P. S.; Kurandina D.; Yata T.; Gevorgyan V. Photoinduced Palladium-catalyzed Carbofunctionalization of Conjugated Dienes Proceeding via Radical-polar Crossover Scenario: 1,2-Aminoalkylation and Beyond. J. Am. Chem. Soc. 2020, 142, 9932–9937. 10.1021/jacs.0c03993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Bellotti P.; Kim S.; Zhang X.; Glorius F. Catalytic Multicomponent Reaction Involving a Ketyl-type Radical. Nat. Synth. 2022, 1, 464–474. 10.1038/s44160-022-00085-6. [DOI] [Google Scholar]
- Cai Y.; Gaurav G.; Ritter T. 1,4-Aminoarylation of Butadienes via Photoinduced Palladium Catalysis. Angew. Chem., Int. Ed. 2024, 63, e202311250 10.1002/anie.202311250. [DOI] [PubMed] [Google Scholar]
- Sarkar S.; Cheung K. P. S.; Gevorgyan V. Recent Advances in Visible Light Induced Palladium Catalysis. Angew. Chem., Int. Ed. 2024, 63, e202311972 10.1002/anie.202311972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Shang R.; Cheng W.; Fu Y. Irradiation-Induced Heck Reaction of Unactivated Alkyl Halides at Room Temperature. J. Am. Chem. Soc. 2017, 139, 18307–18312. 10.1021/jacs.7b10009. [DOI] [PubMed] [Google Scholar]
- Cheng W.; Shang R.; Fu Y. Irradiation-induced Palladium-Catalyzed Decarboxylative Desaturation Enabled by a Dual Ligand System. Nat. Commun. 2018, 9, 5215. 10.1038/s41467-018-07694-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasram M.; Chuentragool P.; Sarkar D.; Gevorgyan V. Photoinduced Formation of Hybrid Aryl Pd-Radical Species Capable of 1,5-HAT: Selective Catalytic Oxidation of Silyl Ethers into Silyl Enol Ethers. J. Am. Chem. Soc. 2016, 138, 6340–6343. 10.1021/jacs.6b01628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuen-Tragool P.; Kurandina D.; Gevorgyan V. Catalysis with Palladium Complexes Photoexcited by Visible Light. Angew. Chem., Int. Ed. 2019, 58, 11586–11598. 10.1002/anie.201813523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kancherla R.; Muralirajan K.; Maity B.; Zhu C.; Krach P. E.; Cavallo L.; Rueping M. Oxidative Addition to Palladium (0) Made Easy through Photoexcited-State Metal Catalysis: Experiment and Computation. Angew. Chem., Int. Ed. 2019, 58, 3412–3416. 10.1002/anie.201811439. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Cao G.; Zhang Z.; Yu D. Visible Light-induced Palladium-catalysis in Organic Synthesis. Chem. Lett. 2019, 48, 181–191. 10.1246/cl.190006. [DOI] [Google Scholar]
- Torres G. M.; Liu Y.; Arndtsen B. A. A Dual Light-Driven Palladium Catalyst: Breaking the Barriers in Carbonylation Reactions. Science 2020, 368, 318–323. 10.1126/science.aba5901. [DOI] [PubMed] [Google Scholar]
- Sheng J.; Cheng X. Electrochemical Mono-Deuterodefluorination of Trifluoromethyl Aromatic Compounds with Deuterium Oxide. CCS Chem. 2024, 6, 230–240. 10.31635/ccschem.023.202302835. [DOI] [Google Scholar]
- Luo Y.; Ma Y.; Li G.; Huo X.; Zhang W. Desymmetrization of Geminal Difluoromethylenes using a Palladium/Copper/Lithium Ternary System for the Stereodivergent Synthesis of Fluorinated Amino Acids. Angew. Chem., Int. Ed. 2023, 62, e202313838 10.1002/anie.202313838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.