

Abstract
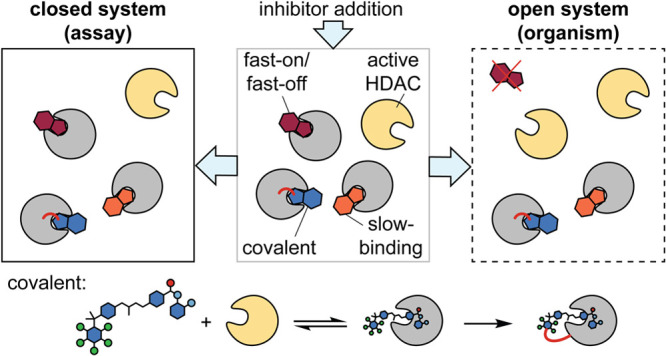
The dysregulated post-translational modification of proteins is an established hallmark of human disease. Through Zn2+-dependent hydrolysis of acyl-lysine modifications, histone deacetylases (HDACs) are key regulators of disease-implicated signaling pathways and tractable drug targets in the clinic. Early targeting of this family of 11 enzymes (HDAC1–11) afforded a first generation of broadly acting inhibitors with medicinal applications in oncology, specifically in cutaneous and peripheral T-cell lymphomas and in multiple myeloma. However, first-generation HDAC inhibitors are often associated with weak-to-modest patient benefits, dose-limited efficacies, pharmacokinetic liabilities, and recurring clinical toxicities. Alternative inhibitor design to target single enzymes and avoid toxic Zn2+-binding moieties have not overcome these limitations. Instead, recent literature has seen a shift toward noncanonical mechanistic approaches focused on slow-binding and covalent inhibition. Such compounds hold the potential of improving the pharmacokinetic and pharmacodynamic profiles of HDAC inhibitors through the extension of the drug–target residence time. This perspective aims to capture this emerging paradigm and discuss its potential to improve the preclinical/clinical outlook of HDAC inhibitors in the coming years.
Keywords: histone deacetylase, covalent inhibitor, slow-binding, inhibitor kinetics, cancer chemotherapy
1. Introduction – HDAC Structure and Function
Histone deacetylases (HDACs) are Zn2+-dependent hydrolases of protein ε-N-acetyllysine (Kac) modifications and of other acyl-amine groups. HDACs were first discovered to erase Kac post-translational modifications (PTMs) from histones,1,2 which determined their name and fundamental role in regulating DNA packing and gene expression. However, research over the past 30 years has uncovered a much wider network of modifications and biological processes regulated by HDACs, as well as deacylation-independent roles in gene regulation and cell signaling.3−5
The structure of the HDAC catalytic domain belongs to the arginase-deacetylase superfamily, which spans multiple enzyme families across the tree of life.4 The α/β arginase-deacetylase fold in HDACs accommodates a Zn2+ ion within a hydrophobic tunnel, which coordinates to the acyl-amine substrate and catalyzes its hydrolysis (Figure 1A).4,6 After initial nucleophilic attack by a water molecule, the resulting tetrahedral intermediate is further stabilized by a Tyr residue within the catalytic tunnel, and two Asp-His pairs help catalysis through proton exchange (Figure 1B).6 The cavity where the Zn2+ ion sits varies in size and structure, as highlighted by the variety of acyl-lysine PTMs that different HDAC isozymes can accommodate (Figure 1C).4 In addition, certain HDACs feature a second cavity or “foot pocket” beyond the Zn2+ ion that has been employed for inhibitor development.7,8 The rim of the hydrophobic tunnel also displays isozyme-specific residues that are critical for their substrate preferences.9 Beyond this rim, HDACs present a variety of topologies and functionalities, which are often linked to their substrate recognition mechanisms and regulatory multiprotein complexes.10,11
Figure 1.
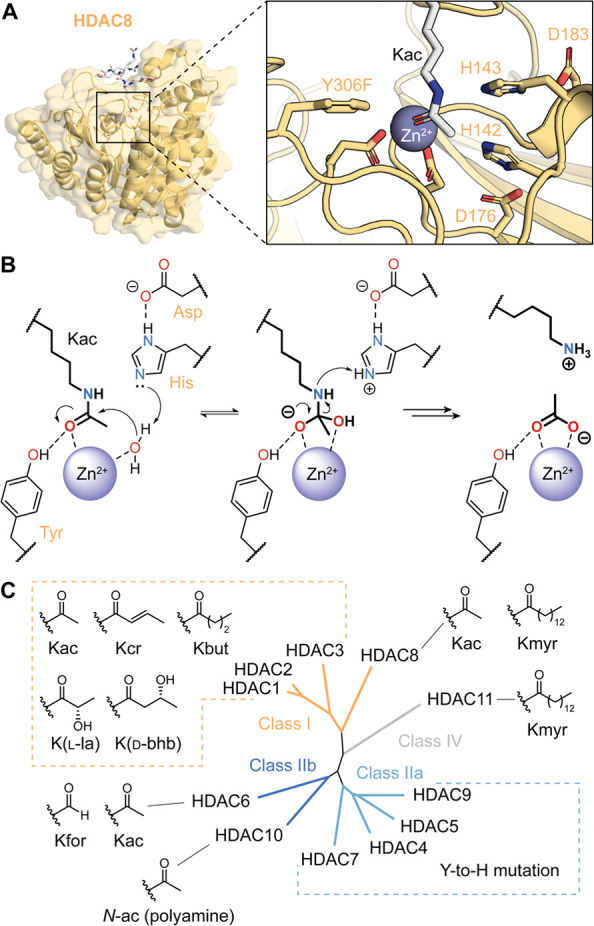
(a) Crystal structure of HDAC8 bound to a Kac substrate, with highlighted residues involved in catalysis (PDB 2V5W, HDAC8 has an inactivating Y306F mutation). (b) Summary of the mechanism of deacetylation by HDACs. (c) Phylogenetic relationship of the 11 human HDACs, and representative acyl substrates.
There are 11 mammalian HDACs divided into 4 classes: class I, class IIa, class IIb, and class IV (Figure 1C). HDACs 1, 2, 3, and 8 (class I) are nuclear enzymes that fulfill the canonical role of histone deacetylation.3 In particular, HDACs 1–3 are the most active deacetylases in the nucleus and regulate chromatin structure and gene expression through modification of histones, chromatin remodelers and transcription factors.12 These three enzymes serve as the deacetylase module of a variety of multiprotein complexes with additional chromatin readers and modifiers.11,13,14 Thus, they are deeply involved in the epigenetic network of gene regulation. HDACs 1–3 also recognize less abundant histone PTMs such as crotonyl-lysine (Kcr)15,16 β-hydroxybutyryl-lysine (Kbhb)17 and lactyl-lysine (Kla),18 which have arisen as key modifications in disease due to their connection to metabolic imbalance (Figure 1C).19,20 In contrast with the other class I HDACs, HDAC8 is a less active histone deacetylase (kcat/KM ∼ 50 M–1 s–1 on H3K9ac peptides,21 compared to ∼500 for HDAC1 and ∼150,000 for HDAC3/NCoR2)18 and has not been found to interact with nuclear multiprotein complexes. Instead, HDAC8 is a more specific deacetylase of histone complexes, chromatin-associated structural proteins and transcription factors.12,22,23 HDAC8 can also hydrolyze longer aliphatic and fatty acid-derived lysine PTMs.24
The class IIa HDACs 4, 5, 7, and 9 are nuclear proteins with marginal catalytic activity, due to a shared mutation of the catalytic Tyr residue to a His (Figure 1B).25 As a result, these proteins are considered to be readers of Kac marks that recruit other factors to chromatin.26 Class IIa HDACs feature large N-terminal extensions that include transcription factor-interacting domains, and phosphorylation sites that lead to nuclear export.27,28 Thus, they serve as regulatory switches in gene activation.29
HDAC6 and HDAC10 belong to class IIb HDACs, and they are highly efficient deacetylases of cytosolic targets (Figure 1C). HDAC6 has two catalytic domains: CD1 that targets C-terminal Kac modifications,30 and CD2 that serves as the main regulator of α-tubulin acetylation.31,32 HDAC6 also targets microtubule-associated proteins33 and can bind ubiquitylated targets through a C-terminal zinc-finger domain.34 HDAC10, on the contrary, does not target acyl-lysine PTMs (kcat/KM > 200-fold lower on Kac substrates compared to HDAC1–3 and HDAC6)35 but rather functions as polyamine deacetylase.36 The respective substrate selectivity of HDAC6_CD1 and HDAC10 are dictated by specific mutations within the active side rim.
A conserved Leu residue is replaced by a “gatekeeper” K353 in HDAC6_CD1, which confers selectivity toward the C-terminal carboxylate, and by E272 in HDAC10, which interacts with the positively charged polyamine scaffold.30,36
Finally, the smallest isozyme and sole member of class IV in mammals is HDAC11.37 Even though HDAC11 is present in the nucleus and involved in gene regulation, its histone deacetylase activity is elusive. Instead, HDAC11 was found to remove fatty acid-derived PTMs such as myristoyl-lysine (Kmyr, kcat/KM ∼ 15,000 M–1 s–1)38 for protein trafficking and cell signaling.38−41
As primary regulators of the cellular acetylome,42 HDACs play important roles at every stage of cellular function, including division, movement, adhesion, metabolism, and signaling.43,44 The deacetylation of histones and other chromatin-associated nuclear targets (e.g., BRCA1, NF-κB, STAT3) renders HDACs crucial regulators of chromatin structure and gene regulation. Importantly, HDACs regulate pro-apoptotic signals (e.g., p21, BAX), tumor suppressor and angiogenesis factors (e.g., p53., HIF1α), as well as biomarkers of cellular adhesion (e.g., CDH1). Thus, HDACs control neoplastic processes45 and their dysregulated activity is strongly liked to tumorigenesis and a variety of human cancers. Despite their general role in oncology, these enzymes have been mostly studied within the context of blood cancers revealing important roles in hematopoiesis, hematopoietic stem cell fate determination, and early stage differentiation.46 This historical link to hematology likely emanated out of the initial studies on erythroid leukemia cell differentiation by Friend, Lapeyre, and Bekhor,47,48 which ultimately led to the regulatory approval of SAHA (vorinostat) in cutaneous T-cell lymphoma (CTCL). However, HDAC targeting keeps holding promise in other types of cancer.49
Outside of oncology, the last 10 years have seen extensive research linking HDAC activity to alternative indications including neurodegenerative disease, metabolic disorders, cardiovascular disease, and viral infections.50 Dysregulated acetylation is a hallmark of neurological disease and by affecting synaptic plasticity, tau phosphorylation, and the expression of cognition-related proteins, HDACs have been widely linked with Alzheimer’s disease (AD).51 In addition, action on neuronal demyelination and the induction of cytosolic protein aggregates has specifically linked class IIb isozyme HDAC6 in Charcot-Marie-Tooth disease (CMT)—a severe neuropathy of the peripheral nervous system.52 The roles of HDACs in the transcription of muscle-specific proteins, autophagy, and microtubule stability, provided a link to Duchenne muscular dystrophy (DMD).53 Moreover, dysregulated HDAC activity related to glucose homeostasis, hepatic fibrogenesis, and adipogenesis has also linked these enzymes to metabolic disorders. Here, HDACs are primarily linked to diabetes mellitus, since multiple HDAC isotypes (HDAC1–3, HDAC6, HDAC11) regulate insulin signaling, metabolic switching, hepatic gluconeogenesis, β-cell apoptosis, and renal inflammation.54 In cardiac disease, HDAC9 has emerged as an important regulator of vascular health and atherosclerosis. This class IIa isozyme was found to play key roles in cholesterol transport, and the development of atherosclerotic lesions.55
Demonstrating the complex nature of epigenetic signatures within these multifactor chronic conditions, in cardiac hypertrophy, while specific isozymes can cause disease (e.g., HDAC2), other HDACs promote protective mechanisms (e.g., HDAC4).56 Finally, viral infections have been linked to deacetylase function with roles across viral entry, fusion, replication, latency, and release.57 A notable example is within human immunodeficiency virus (HIV), which is in dire need of novel and improved therapeutic modalities. In this regard, HDAC6 controls HIV-1 transcription and virion entry and release by affecting microtubule dynamics and cytoskeletal structural integrity. Ultimately, through control of cell-wide acetylation (and downstream signaling cascades), these enzymes are involved in a myriad of human diseases, and targeted epigenetic discovery efforts can provide immense benefit to millions of patients around the world.
2. Non-selective and Selective HDAC Inhibition
As an approach to drug discovery, the targeted inhibition of HDACs to treat human disease is a relatively recent undertaking. While the initial link between histone PTMs and mRNA synthesis was reported in 1964 by Allfrey et al.,58 followed by studies using n-butyrate to affect histone acetylation and cell differentiation (Figure 2A), it is only in the last 30 years that pharmacological modulation of deacetylase enzymes has advanced into a viable clinical strategy.59 At the time of writing this, the field has reached over 1,250 articles per year on www.pubmed.gov (2023–2024), compared to 5 hits in 1990, with growing applications against various therapeutic indications (e.g., cancer, neurology, inflammation) and, more importantly, 6 drug approvals.
Figure 2.
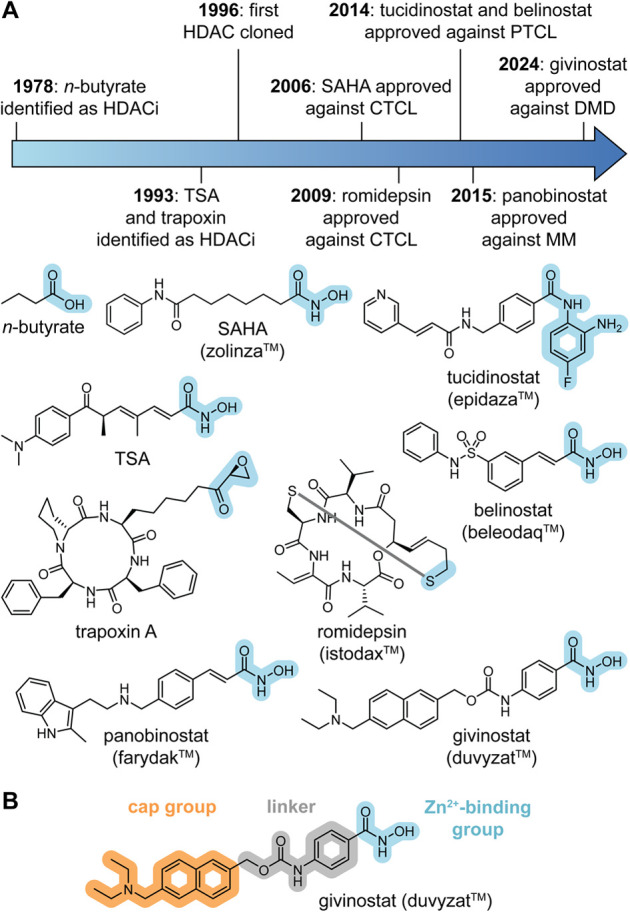
(a) Timeline and chemical structures of early HDACi identification and drug approvals. (b) Common pharmacophore of HDAC inhibitors.
The discovery of early HDAC inhibitors (HDACi) was quite serendipitous, emerging from target-agnostic phenotypic screens of simple polar molecules and entirely unrelated research on antifungal antibiotics such as trichostatin and trapoxin (Figure 2A).60 While studying murine erythroleukemia cells, Friend et al.47 reported that the exposure to mM concentrations of dimethyl sulfoxide induced terminal differentiation and cell growth arrest. Marks et al.61,62 extended this observation to other carbonyl compounds including N-methylacetamide and N-dimethylformamide. Pursuing a predicted metal interaction, a series of bishydroxamic acids were found to be several times more potent than their amide counterparts. Subsequent structure–activity analyses exploring aromatic tail groups and varied methylene spacers ultimately led to the discovery of suberoylanilide hydroxamic acid (SAHA, Sloan-Kettering Institute for Cancer Research, Columbia University, Figure 2A).63−65 Due to its structural similarity to the bacterial metabolite trichostatin A (TSA)—a natural antibiotic and bona fide HDACi1—SAHA was confirmed as an inhibitor of HDACs. These findings, together with the isolation and cloning of the first HDAC in 19962 and the first cocrystal structure of a bacterial deacetylase by Finnin et al.,66 spurred an outburst of interest in deacetylases as ligandable drug targets.
SAHA was found to cause up-regulation in acetylated substrates, influencing gene transcription and ultimately inducing cell cycle arrest and apoptotic cell death. This trend was observed across a wide panel of cancer cell lines.
Target engagement was also confirmed in follow-up crystallographic studies, clearly highlighting a bidentate coordination mode between the hydroxamic acid and the active site Zn2+ ion.67−69 In 2006, SAHA (zolinza, Aton Pharma/Merck & Co.) was approved for the treatment of CTCL. As the first targeted deacetylase inhibitor to enter the clinic, the success of SAHA spurred other clinical oncology studies, which led to several approvals primarily within the context of blood cancers. These included prodrug and natural product depsipeptide romidepsin (istodax, 2009) against CTCL, cinnamoyl hydroxamic acid belinostat (beleodaq, 2014) and 4-fluoro-2-aminobenzanilide tucidinostat (epidaza, China 2014), both against peripheral T-cell lymphoma (PTCL) and cinnamoyl hydroxamic acid panobinostat (farydak, 2015) in combination with bortezomib and dexamethasone against multiple myeloma (MM). Beyond cancer, the phenylhydroxamic acid givinostat (duvyzat, 2024) has recently received approval for the treatment of DMD as the first nononcology HDACi on the market (Figure 2A).50 If we consider the past decade, these breakthroughs have had a strong influence on all subsequent HDACi research - perhaps in a manner greater than other drug classes. Ultimately, these first generation molecules defined a lasting pharmacophore with repeated sets of privileged motifs and specific design strategies.
Standard HDACi are generally competitive acetyl lysine mimetics with 3 defining features: A Zn2+-binding group (ZBG), an elongated linker region, and a surface cap group (Figure 2B). The critical ZBG aims to coordinate the active site Zn2+ through mono- or bidentate modes. In most cases, this interaction represents the central driving force behind the observed potency. While ZBGs have advanced since the early days of sulfoxides, thiols, and simple carboxylates, the N-hydroxamic acid remains the most commonly used motif in HDAC literature. Despite known metabolic issues and a potential for idiosyncratic toxicities, this group persists for several reasons: a particularly superior affinity for Zn2+, simple synthetic routes, high polarity, improved aqueous solubility (as the rest of the molecule tends to be quite lipophilic), as well as precedence in literature (e.g., SAHA), and nature (e.g., TSA).70 Next in line is the o-aminoanilide group (often referred to as benzamide) seen in tucidinostat. While weaker as a Zn2+ chelator, this motif is uniquely selective for class I HDAC1–3 (owing to their larger active sites) and offers several additional benefits: improved systemic stability, slow-binding kinetics, and the capacity for synthetic derivatization. Outside of these two motifs, several novel ZBGs have also been recently reported. Notable examples include trifluoromethylketones, α-ketoamides, 2-(difluoromethyl)-1,3,4-oxadiazoles (DFMOs), and mercaptoacetamides.71
For the linker region, its extended structure spans the lysine tunnel (d = 6–10 Å) and has been largely explored using aliphatic hydrocarbons, vinyl extensions, and aromatic ring systems. Interestingly, recent studies have highlighted the value of linker choice, and heteroatom insertions to capture favorable intratunnel interactions.72 Lastly, the terminal surface cap sees the highest degree of structural variance and is generally the first design element optimized in structure–activity relationship (SAR) programs. This mainly involves the use of varied aromatics and heterocycles, or larger moieties inspired by trapoxin or apicidin. The surface cap is also commonly used to optimize physicochemical properties such as lipophilicity and solubility.
While this first wave of clinical HDACi set an important precedent, their widespread and long-term use was quickly restricted by recurring dose-limiting toxicities (e.g., thrombocytopenia, pyrexia, cardiac arrythmia, diarrhea, fatigue).73 These symptoms were mainly attributed to the indiscriminate inhibition of HDAC enzymes and cell-wide disruption of the acetylome. Without a selectivity scaffold, hydroxamic acids such as SAHA or panobinostat inhibit about half of the 11 HDACs. Moreover, with >300 Zn2+ metalloenzymes in the cell, hydroxamic acids often suffer from side effects due to off-targets. Strikingly, in a recent study, > 20 established hydroxamate-based HDACi were found to also inhibit metallo-β-lactamase domain-containing protein 2 (MBLAC2) - palmitoyl-CoA hydrolase with nM potencies.74 Furthermore, hydroxamic acids are also notoriously susceptible to phase I (reduction, hydrolysis) and phase II (glucuronidation) metabolic processes, with established links to mutagenicity and genotoxicity effects,75 and may also form reactive isocyanates through Lossen rearrangement in vivo.75,76 The acidity of the N-hydroxy group (pKa 8.0–9.5, 25 °C) can also compromise oral bioavailability, intestinal permeability and effective distribution.77,78
Ultimately, poor safety profiles with unpredictable pharmacology and dose-limited clinical use sparked paradigm shifts into alternative design strategies to overcome these issues. These included the aim for more selective inhibitors, the use of alternative ZBGs, combination dosing and, later on, noncanonical inhibition mechanisms (discussed in the next section).10,79
Selective HDACi exploit subtle differences in protein structure to achieve discriminative target engagement. Strategies focus on either a single isozyme (e.g., HDAC6) or a subset of deacetylase enzymes (e.g., HDAC1–3). While HDACs are known to share high catalytic domain sequence identities (35–94%), the active site differences revealed by crystal structures have been targeted as selectivity filters via structure-based approaches.80,81 HDAC1–3 have a slightly larger active site that can accommodate o-aminoanilides, with HDACs 1 and 2 showing an additional L-shaped “foot-pocket” (d = 3–14 Å).8 Studies into HDAC6 revealed a rigid pair of Phe residues flanking the lysine tunnel,80 while a buried catalytic Tyr in HDAC8 forms a surface exposed L1-L6 cavity. The atypical “gatekeeper” residues of HDAC6 and HDAC10 (Lys in HDAC6_CD1 and Glu in HDAC10) have also been targeted for selective inhibition.82 In general, these strategies have been successful and provided class- and subclass-selective molecules against HDAC1–3 (entinostat), HDAC4, 5, 7, 9 (TMP269), or HDAC6, 10 (tubastatin A, Figure 3A) and compounds with more exquisite selectivity against HDAC3 (compound 16),83 HDAC6 (ACY-738, NN-390, TO-317), HDAC8 (PCI-34051, MMH410), HDAC10 (DKFZ-748), and HDAC11 (FT895, Figure 3B).84−92 While useful as chemical probes within biological studies and preclinical animal models, these selective molecules have unfortunately not yet resulted in any significant clinical benefit. This highlights potential disconnects between in vitro selectivity profiles (derived from isolated inhibition activity assays) and subsequent in vivo pharmacology.
Figure 3.
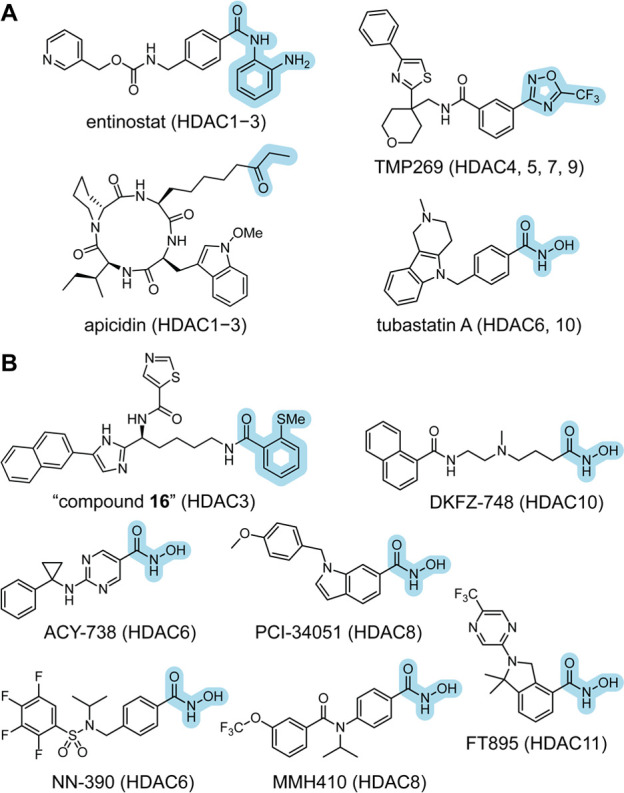
(a) Structure of selected class- and subclass-selective HDACi. (b) Structure of selected isozyme-selective HDACi.
While targeting a single HDAC can be regarded as a safer strategy due to reduced toxicity, it can also lead to lower pharmacological potency. In fact, it has been posited that the clinical efficacy of the first generation of HDACi stems from the inhibition of multiple HDAC isozymes. Thus, it appears that a delicate balance between selectivity, toxicity, and potency must be struck to achieve full clinical potential, within a specific optimal target profile for each indication.
A second strategy to improve upon the first generation of HDACi was the exploration of novel ZBGs with improved pharmacokinetics and acceptable affinities for Zn2+ (Figure 3). While approved HDACi still rely on the hydroxamic acid, it is worth noting that these molecules have been long in development and reflect a time where this motif was in fact the best option. In hindsight, it appears that for the longest time, the field operated with a sense of complacency with respect to the Zn2+ ligand. It was common to install a hydroxamic acid at the start and focus on the optimization of the other structural motifs, without probing alternative ZBGs. This matter has been actively reconsidered in the past decade, with several useful strides to begin departing from the hydroxamic acid. o-Aminoanilides have been investigated extensively due to their added selectivity toward HDAC1–3 and potential for brain permeability,93 with the goal of addressing neurological diseases.
Similarly, trifluoromethyl heterocycles drew attention for targeting class IIa HDACs, which are often more difficult to target with hydroxamic acids (Figure 3A). A more recent example is the emergence of fluorinated oxadiazoles for the selective targeting of HDAC6.94 While this group has been known for several years, it was only recently that Christianson and Hansen showed DFMOs act as mechanism-based slow-binding inhibitors.94,95 Preliminary in vitro studies have shown successful brain penetrance, oral bioavailability, low metabolic clearance and an acceptable safety profile with no genotoxicity, or cardiotoxicity. Despite these benefits, DFMOs also present their own set of challenges, primarily concerning the chemical stability of the oxadiazole ring in the blood and non-neutral pH environments.
A third approach was the use of deacetylase inhibitors as synergizing therapeutics combined with standard-of-care or as adjuvant drugs following a primary treatment course.96 While HDACi have long been studied as single agents, they rarely provided sustained clinical benefits, in part due to the dose-limited efficacies and recurring toxicities. Alternatively, it was postulated that HDACi could help sensitize the epigenetic framework of a diseased cell (e.g., in cancer), allowing the standard-of-care to achieve better clinical benefits. These combinations have been shown to improve efficacy and reduce tumor resistance mechanisms, in addition to allowing smaller and less frequent doses of either medicine resulting in an overall safer dosing regimen.97,98 At the same time, combination strategies enable researchers to “rescue” current inhibitors instead of initiating new drug discovery programs.
On this front, HDACi have been explored across various human cancers, in combination with topoisomerase inhibitors (e.g., doxorubicin), kinase inhibitors (e.g., pazopanib), glucocorticoid antagonists (e.g., dexamethasone), immunotherapies (e.g., anti-PD1 Ab), radiotherapies, as well as other epigenetic drugs (e.g., 5-azacytidine). These studies are summarized in an excellent review by Hontecillas-Prieto et al.99 Furthermore, as epigenetic drug hunters turn their attention beyond oncology, HDACi have also been combined with antiretroviral therapeutics for HIV-1 treatment.100,101 These molecules reactivate latent viral reservoirs, allowing antiretrovirals to eliminate traditionally inaccessible virion populations and improve patient prognoses.57 Through this dual shock-and-kill approach, HDACi provide a promising avenue toward a potential curative therapy against HIV1, with several clinical trials reported in recent years (e.g., NCT03198559, NCT05700630, NCT03525730).57
In the clinic, after a decade without any new drug applications, givinostat was recently approved for the treatment of DMD—an incurable neuromuscular disorder, which primarily affects pediatric males (<6 years old).53,102 Despite its poor selectivity profile as an unselective HDACi, this approval represents an important milestone outside of cancer. Another great example is the CKD series out of Chong Kun Dang Pharmaceutical Corp. (CKD-504, CKD-510, etc.), currently under investigation for the treatment chronic neuropathies (e.g., CMT1A).103 While its structure is not disclosed, clinical candidate CKD-510 is a nonhydroxamate HDACi, highlighting this more recent move away from this suboptimal ZBG. Overall, with the increasingly growing list of HDACi under clinical investigation, it appears that the clinical landscape of these molecules may be on the cusp of a new paradigm outside of oncology, away from traditional structural designs and open to new pharmacological approaches.
3. Non-canonical Inhibitors: A New Paradigm?
The current limitations of HDAC inhibitors in oncology, as well as the search for therapeutic avenues in other diseases, has motivated the investigation of alternative strategies in HDAC inhibition. On the one hand, compounds with long target engagement, i.e., slow kinetics, have arisen as more efficient tools than the first generation of approved drugs. As a result, research is focusing on the kinetic optimization of reversible inhibitors and the development of irreversible covalent strategies. On the other hand, degradation with proteolysis-targeting chimeras (PROTACs) is regarded as a complementary strategy that may expand the scope of HDAC-targeting therapeutics.104−107 Driven by preclinical successes and first-in-human validation against other targets (e.g., ARV-110, ARV-471),108 the HDAC field has rapidly adopted degradation as an alternative. This endeavor has provided reports of >100 HDAC-PROTACs in literature, as it has been reviewed elsewhere.109−111
Similar to the interaction of membrane receptors with their ligands,112 HDACi interact with their targets to form both short- and long-lived complexes.113
The combined rate of dissociation of each binding pose confers an HDAC inhibitor with a characteristic residence time (τ), which determines the overall duration of the inhibition event.114,115 This dynamic aspect of inhibition is often overlooked during compound development, even though high τ values are often indicative of success in the clinic.114,116
In standard end-point assays, inhibitors are assumed to display fast-on/fast-off kinetics (Figure 4A), where τ would typically be on the order of seconds or shorter. Under these circumstances, inhibitors instantly reach equilibrium with their targets, and any alteration of free-compound concentration is rapidly reflected on target occupancy (Figure 4A).115 This is the case for SAHA,113 and initially assumed for most small-molecule HDACi. On the contrary, early work from Gottesfeld and co-workers revealed that an o-aminoanilide SAHA derivative was a slow-binding inhibitor with τ of hours against HDACs 1–3, significantly extending the effect of this compound in cells.113 Subsequent kinetic studies revealed that most o-aminoanilides are slow-binding inhibitors (Figure 4B),117 including entinostat3,4 and compounds targeting the “foot pocket”.118 Since slow-binding inhibitors do not reach equilibrium instantly, their observed potency increases gradually during incubation. More importantly, concentration changes are not immediately reflected on target occupancy, due to their slow dissociation rates from the enzyme, which is highly attractive in an in vivo context (Figure 4B).115 Thus, continuous monitoring of activity and τ determination is essential to determine the potency and selectivity of slow-binding inhibitors, as reflected by the recent o-aminoanilide literature.118−120 Importantly, these compounds can display kinetic selectivity even between a free HDAC enzyme, the same HDAC in a multiprotein complex,120 and between different complexes.121
Figure 4.

Schematic representation of target engagement after addition, incubation and wash of (a) fast-on/fast-off inhibitors, (b) slow-binding inhibitors, (c) reversible covalent inhibitors, and (d) irreversible covalent inhibitors, with examples of HDACi following each kinetic mechanism.
After the identification of o-aminoanilides as slow-binding inhibitors, many other chemotypes have been shown to display slow kinetics. A trifluoromethyl ketone version of SAHA,122 various alkyl hydrazides,119,123 and o-substituted benzamides,124 are all slow-binding inhibitors of HDACs 1–3. Slow inhibition of HDAC8 was achieved with trifluoromethyl ketone inhibitors,125 and of HDAC6 with DFMOs (compound 6, Figure 4B).95,126
The natural products romidepsin and trapoxin A were found to be slow-binding inhibitors with τ of up to several hours, which serves to rationalize their excellent potency in vivo.127−129 Finally, the hydroxamic acid derivatives of apicidin and trapoxin, as well as the archetypal inhibitors panobinostat and TSA, are slow-binding inhibitors of subsets of class I, class IIb and class IV enzymes with different kinetic profiles of selectivity among them (Figure 4B).128,129
Even though most approved HDACi are now known to be slow-binding inhibitors, which highlights the potential of slow kinetics in the clinic, the lack of clear structure-kinetic relationships (SKR) hampers slow-binder design. Most slow-binding inhibitors follow the so-called “mechanism B” of target inhibition, where a first, fast-binding event is detected, followed by a transition to a more stable and long-lived enzyme–inhibitor complex (Figure 5A).114,130 The basis of this transition, often referred to as “induced-fit”, is unknown for most chemotypes, and thus difficult to exploit for SKR studies. Interestingly, recent studies on DFMOs have provided a first rational mechanism B transition, which does not entail an induced fit but rather a mechanism-based transformation. These compounds inhibit HDAC6 with high selectivity and τ of several hours, as the catalytic water molecule in the HDAC6 active site performs a nucleophilic attack on the outermost C=N bond of the oxadiazole and forms a tetrahedral intermediate coordinated to the Zn2+ ion (Figure 5B). The oxadiazole ring then opens to afford a hydrolyzed acyl-hydrazide, and this transition is rate-limiting and likely responsible for the slow-binding behavior.131,132
Figure 5.

(a) Standard mechanisms of slow-binding inhibition, and calculation of inhibitor potency (Ki) and residence time (τ).1,2 (b) Mechanism-based inhibition (as judged by the nucleophilic H2O attack) of HDAC6 by difluoro-1,3,4-oxadiazoles (DFMOs).131−133 (c) Standard mechanisms of covalent inhibition and calculation of inhibitor potency (Ki) and residence time (τ) for reversible inhibitors or inactivation constant (KI, or KInact, not to be confused with Ki) and efficiency for irreversible inhibitors. (d) Cysteine residues nearby the HDAC8 (PDB 2V5W) and HDAC2 (PDB 4LXZ) active sites.
After that, the acyl-hydrazide dissociates or gets further deacylated to the free hydrazide, but none of these species show as high potency, selectivity, or τ as the original DFMO.95,126,131−133 This mechanism has thus served to rationalize the oxadiazole structural and electronic requirements to achieve slow kinetics.131−133 Unfortunately, such knowledge is still missing for other slow-binding HDAC inhibitor scaffolds.
A second strategy to extend the τ of a compound is to include a reactive handle for covalent bond formation. This approach is often more intuitive and easier to design, although it requires finding accessible reactive residues within the HDAC structure. Since the covalent interaction provides a large gain of affinity, this approach can also potentially remove the need for a strong Zn2+ chelator such as the hydroxamic acid. There are two possible mechanisms of covalent inhibition: (1) the formation of a reversible covalent interaction, where the inhibitor can be regenerated and dissociate from the enzyme (Figure 4C), and (2) an irreversible bond formation, where the fate of the complex would be the degradation of the HDAC and/or the inhibitor (Figure 4D).134 In both cases, the system can be modeled as a two-step process with a first fast binding step and a second slow covalent transition. In reversible covalent inhibition, the kinetic profile is similar to the mechanism B of slow kinetics, whereas irreversible covalent inhibition affords a system with theoretical infinite τ values (Figure 5C).
The first two reports of covalent HDAC inhibition were in 2015, on HDAC-targeted compounds that released Cys-reactive species. The laboratories of Yingjie Zhang and Wengfang Chu developed SAHA-based compounds with a phenylsulfonylfuroxan as a NO-donating functionality,135 which was also later incorporated into an HDAC6-targeting scaffold.136 The NO generated by these inhibitors is transferred to neighboring sulfides such as those of Cys residues on the surface and the catalytic pocket of HDACs.135,136 The laboratories of Seth Cohen and Carol Fierke found a similar compound class serendipitously, as their quinonyl-SAHA (SAHA-TAP) prodrug led to modification of multiple Cys residues of HDAC8 by the released quinone cap.137
Class I HDACs and, in particular, HDAC8 have reactive Cys residues on the surface surrounding the active site and within the substrate pocket. As the latter residues are relatively close to the Zn2+ ion (Figure 5D), they are thought to be involved in the sensing of electrophilic Lys modifications.138 Promiscuous reactive fragments can label both internal and surface residues,125,139 and they can be further developed into potent irreversible inhibitors. For instance, 3-ethynylmethylpyridiniums were developed as covalent HDAC8 inhibitors targeting Cys-containing allosteric sites (compound 3, Figure 4D).139 This new approach permits targeting potentially unique pockets for isozyme- or complex-selective inhibition, and the development of potent HDAC inactivators without a Zn2+-binding group.139
HDAC1–3 display less surface Cys residues than HDAC8, but their distribution is similar around the active site (Figure 5D). These four enzymes share a reactive Cys residue 10 Å away from the entrance of the active site with a slight change in position in HDAC8 (HDAC8 C275 vs HDAC2 C274, Figure 5D).140 The addition of a Cys-reactive handle to the cap group of a class I inhibitor thus improves potency by further extending τ on these enzymes.140 This effect was recently shown by modifying the scaffold of the HDAC1–3 inhibitor entinostat with the irreversible pentafluorobenzenesulfonamide electrophyle (YSR734, Figure 4D), which adds covalent targeting to the three enzymes and provides longer lasting effects in cells.140 This proof-of-concept study opens up the possibility of maximizing τ with the addition of Cys-targeting moieties, and provides a framework for identifying enzyme-specific residues toward selective HDAC inactivation. Moreover, recent developments in protein covalent chemistry have expanded the possibilities beyond Cys targeting,141−143 which will facilitate the rational design of future covalent HDAC inhibitors.
4. THE BENEFITS OF IMPROVING INHIBITOR KINETICS
Traditionally, most (if not all) drug discovery programs quantify inhibition events by deriving dissociation constants (Kd), inhibition constants (Ki), or half-maximal inhibitory concentrations (IC50). Governed by thermodynamic equilibria, these macroscopic potency metrics operate within a simple closed system, which assumes invariable receptor–ligand concentrations. While useful for controlled in vitro assays, such a simplistic view of pharmacology breaks down in the complex milieu of a living cell (Figure 6A). In our pursuit of bioactive molecules with clinical benefits in human patients, medicinal chemists must try to adopt experimental models that most closely represent their target environment. In this regard, Copeland et al.115 promoted the use of the drug-target residence time as a central parameter in lead optimization programs, providing a measure of the duration of a drug’s action, and thus a more accurate insight into in vivo drug pharmacology.115
Figure 6.
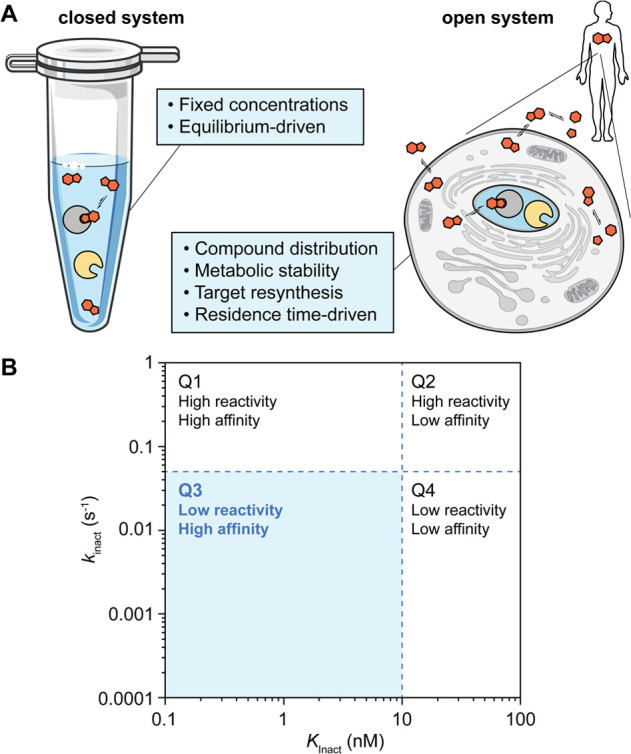
(a) Comparison of closed (laboratory experiment) and open (human body) systems, inspired by Copeland et al.112 Tube icon provided by bioicons.com. (b) Quadrant distribution of irreversible covalent inhibitor properties.
Optimizing residence time (τ), or in the case of irreversible inhibitors their kinact/KI, is an overall more promising strategy (Figure 5A, C), as [drug]free in a cellular environment is in fact in constant flux due to competing mechanisms of absorption, permeation, metabolism, and excretion. Concurrently, [target]free also varies due to competing substrates, protein resynthesis, and degradation pathways (Figure 6A).114 Achieving extended residence times ultimately results in the decoupling of pharmacodynamic (PD) profiles, as a slowly dissociating molecule can outlast its systemic stability (i.e., half-life) or the protein resynthesis rates.130 Thus, slow-binding and covalent inhibitors hold better promise toward achieving smaller and less frequent dosing regimens, as required for most HDACi. Taken to their clinical extreme, these benefits could offer better safety profiles, reduced costs, and improved compliance.
For slow-binding and reversible covalent inhibitors, optimizing τ entails primarily the optimization of their off-rate (Figure 5A, C). Interestingly, Copeland also highlights the importance of on-rates, which can drive target selectivity as well as the subtle phenomenon of drug rebinding. As ligands dissociate from their target, a high local concentration is generated at the binding site interface, which without any rapid clearing mechanism enables rebinding and the extended pharmacological effect. For irreversible covalent inhibitors, there is no off-rate and thus τ cannot be calculated. Instead, the ratio of the covalent bond formation kinetics (kinact) and the inactivation constant (KI, or KInact, not to be confused with Ki) serves as a dynamic measure of the compound’s efficiency (Figure 5C). Here, typical thresholds of kinact = 0.05 s–1 and KInact = 10 nM would define desirable kinetics (Figure 6B), with lead compounds laying under the kinact threshold (Q3). These insights are lost when measuring broad thermodynamic metrics. While previously challenging to ascertain, dynamic parameters can now be regularly measured via kinetic binding experiments through, for example, NMR or surface plasmon resonance, or with continuous enzyme activity assays using fluorogenic substrates.144 In addition, studies are proposing new expedited approaches to estimate these important metrics.145,146 When supported with pharmacokinetic AUCfree values, these data would allow the estimation of in vivo target occupancy, which can help guide covalent dosing regimens.
The true extent of sustained residence times and decoupled PK–PD has been regularly exemplified in the rich field of covalent drugs.147−150 With >50 covalent drugs currently on the market, >15 FDA-approvals in the past 10 years alone and an annual market cap estimated at >US$50 billion, covalent molecules have firmly positioned themselves as arguably one of the most successful clinical strategies to date.151 Covalent inhibitors are known for their powerful potencies (pM–nM range), sustained target engagement (even in the presence of endogenous substrates), and a unique ability to engage intractable drug targets (e.g., flat, solvated, allosteric pockets).152 Previous concerns of nonspecific hyperreactivity have been largely resolved with recent advances in chemoproteomic protein profiling, soft electrophiles with tunable reactivities, and an overall better understanding of the required benchmarks for a successful preclinical/clinical covalent drug candidate.134,149,150,153,154
Regarding targeted HDAC binders, researchers have started to explore the potential benefits of slow-binding or covalent kinetic mechanisms. Shifting to a long-lived (or permanent) binding strategy would disengage inhibitor PD from any inherent PK liabilities and provide more sustained pharmacology.155 While several slow-binding HDACi have been reported in recent literature, we still lack an explicit and predictable framework of the structure-kinetic forces underlying these mechanisms. Thus, it is clear that these are complex and multifactorial processes with important contributions from both the ligand and target protein. For instance, simple hydroxamic acids were long presumed to represent a classic case of potent ligands with fast-on/fast-off kinetics, while bulkier ZBGs such as o-aminoanilides were assumed to be needed for slow-on/slow-off kinetics, due to structural rearrangements in the protein active site (i.e., break an internal H-bond).117 Instead, recent reports have demonstrated that small hydroxamic acids and alkyl-hydrazides can also be slow-binding inhibitors.129,156 On the other hand, covalent inhibitors represent a more mature field of study, with defined structure–activity trends, established design principles and relatively predictable molecular recognition. Several HDAC isozymes have also been shown to have nucleophilic residues within targetable distances of their catalytic domains, as seen in studies on HDAC2 (YSR734), HDAC6 (SAHA-TAP), and HDAC8 (Compound 3).137,140,157 At this early stage, these molecules remain as useful chemical starting points and further optimization efforts are needed to effectively probe the potential benefits of irreversible HDACi binders. As the field compares the preclinical tractability of reversible, slow-binding, or covalent molecules, detailed research into current structure-kinetic trends would be a great step toward improving HDAC targeting.
5. Conclusion
Dysregulated acetylation mechanisms are an established hallmark of human disease. Intensive research into these signatures ultimately led to the discovery and classification of a family of deacetylases with Zn2+-dependent activity (HDACs 1–11). With cell-wide influence on post-translational acylation of proteins and acetylation of polyamines, these hydrolase enzymes have established themselves as master regulators of cell homeostasis.50 At the same time, uncontrolled HDAC activity has broad implications in cellular dysfunction and disease progression, including cancer, neurodegeneration, inflammation and infection. Thanks to early phenotypic results and extensive crystallographic work, HDACs are now useful drug targets with a wealth of inhibitors reported, generally acting as competitive acetyl-lysine mimetics.4 However, besides a handful regulatory approvals, the clinical adoption of first gen. HDACi (e.g., SAHA, belinostat, panobinostat) is far from widespread, and it is largely confined to small patient populations with specific disease states. Arguably, the causes of these limitations are the nonspecific inhibitory profiles and the use of warheads with harmful metabolites, which afford dose-limited efficacies and recurring clinical toxicities.
To address limitations in HDAC targeting, several paradigms shifts have occurred toward the next generation of therapeutics, namely isozyme-specific inhibition, non-hydroxamate ZBGs, combination therapies and targeted protein degradation.10 Along with these strategies, recent literature has seen a notable rise of the novel kinetic mechanisms of inhibition of slow-binding kinetics and covalent targeting. These mechanisms hold promise in achieving more sustained efficacies in vivo and better pharmacokinetic profiles, through extended drug-target residence times. To accomplish this, studies have explored substituted o-aminoanilides,8 alkyl hydrazides,158 mechanism-based difluoromethyl-1,3,4-oxadiazoles,95 quinonyl pro-drugs,137 ethynylmethylpyridiniums,139 and irreversible perfluorinated arylsulfonamides.140 Interestingly, recent reevaluations revealed the slow-binding properties of successful first gen. HDACi, further supporting this approach.
While great strides have been made and the field is in the midst of a transformation, these studies remain in their early stages.159−161 Future efforts on this front will likely focus on establishing reproducible structure-kinetic relationships, delineating isozyme-specific slow-binding and covalent strategies, and establishing tractable preclinical and clinical guidelines for better slow-binding and covalent inhibitor design. All in all, we envision that HDACi with improved kinetic profiles hold promise toward extending the clinical benefits of targeting HDACs in human disease.
Acknowledgments
This work was supported by the Swiss National Science Foundation (SNSF Swiss Postdoctoral Fellowship no. TMPFP2_217187 to C.M.-Y.) and the United Arab Emirates University (research start-up grant #12S156 to Y.S.R.).
Glossary
ABBREVIATIONS
- AD
Alzheimer’s disease
- CMT
Charcot-Marie-Tooth
- CTCL
cutaneous T-cell lymphoma
- DFMO
2-(difluoromethyl)-1,3,4-oxadiazoles
- DMD
Duchenne muscular dystrophy
- HDAC
histone deacetylase
- HDACi
HDAC inhibitor
- HIV
human immunodeficiency virus
- MM
multiple myeloma
- PD
pharmacodynamics
- PK
pharmacokinetics
- PROTAC
protein-targeting chimera
- PTCL
peripheral T-cell lymphoma
- PTM
post-translational modification
- ZBG
Zn2+-binding group
Author Contributions
Y.S.R and C.M-Y. contributed equally. The manuscript was written through contributions of all authors. All of the authors approved the final version of the manuscript.
The authors declare no competing financial interest.
References
- Yoshida M.; Kijima M.; Akita M.; Beppu T. Potent and Specific Inhibition of Mammalian Histone Deacetylase Both in Vivo and in Vitro by Trichostatin A. J. Biol. Chem. 1990, 265 (28), 17174–17179. 10.1016/S0021-9258(17)44885-X. [DOI] [PubMed] [Google Scholar]
- Taunton J.; Hassig C. A.; Schreiber S. L. A Mammalian Histone Deacetylase Related to the Yeast Transcriptional Regulator Rpd3p. Science (1979) 1996, 272 (5260), 408–411. 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- Yang X. J.; Seto E. The Rpd3/Hda1 Family of Lysine Deacetylases: From Bacteria and Yeast to Mice and Men. Nat. Rev. Mol. Cell Biol. 2008, 9 (3), 206–218. 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter N. J.; Christianson D. W. Structure, Mechanism, and Inhibition of the Zinc-Dependent Histone Deacetylases. Curr. Opin Struct Biol. 2019, 59, 9–18. 10.1016/j.sbi.2019.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T. C. S.; Chan A. H. Y.; Ganesan A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63 (21), 12460–12484. 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- Gantt S. M. L.; Decroos C.; Lee M. S.; Gullett L. E.; Bowman C. M.; Christianson D. W.; Fierke C. A. General Base-General Acid Catalysis in Human Histone Deacetylase 8. Biochemistry 2016, 55 (5), 820–832. 10.1021/acs.biochem.5b01327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. F.; Wiest O.; Helquist P.; Lan-Hargest H. Y.; Wiech N. L. On the Function of the 14 A Long Internal Cavity of Histone Deacetylase-like Protein: Implications for the Design of Histone Deacetylase Inhibitors. J. Med. Chem. 2004, 47 (13), 3409–3417. 10.1021/jm0498497. [DOI] [PubMed] [Google Scholar]
- Bressi J. C.; Jennings A. J.; Skene R.; Wu Y.; Melkus R.; De Jong R.; O’Connell S.; Grimshaw C. E.; Navre M.; Gangloff A. R. Exploration of the HDAC2 Foot Pocket: Synthesis and SAR of Substituted N-(2-Aminophenyl)Benzamides. Bioorg. Med. Chem. Lett. 2010, 20 (10), 3142–3145. 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- Christianson D. W. Chemical Versatility in Catalysis and Inhibition of the Class IIb Histone Deacetylases. Acc. Chem. Res. 2024, 57 (8), 1135–1148. 10.1021/acs.accounts.3c00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maolanon A. R.; Madsen A. S.; Olsen C. A. Innovative Strategies for Selective Inhibition of Histone Deacetylases. Cell Chemical Biology 2016, 23, 759–768. 10.1016/j.chembiol.2016.06.011. [DOI] [PubMed] [Google Scholar]
- Millard C. J.; Watson P. J.; Fairall L.; Schwabe J. W. R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38 (4), 363–377. 10.1016/j.tips.2016.12.006. [DOI] [PubMed] [Google Scholar]
- Scholz C.; Weinert B. T.; Wagner S. A.; Beli P.; Miyake Y.; Qi J.; Jensen L. J.; Streicher W.; McCarthy A. R.; Westwood N. J.; Lain S.; Cox J.; Matthias P.; Mann M.; Bradner J. E.; Choudhary C. Acetylation Site Specificities of Lysine Deacetylase Inhibitors in Human Cells. Nat. Biotechnol. 2015, 33 (4), 415–423. 10.1038/nbt.3130. [DOI] [PubMed] [Google Scholar]
- Guenther M. G.; Barak O.; Lazar M. A. The SMRT and N-CoR Corepressors Are Activating Cofactors for Histone Deacetylase 3. Mol. Cell. Biol. 2001, 21 (18), 6091–6101. 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M.; Hopf C.; Savitski M. M.; Dittmann A.; Grandi P.; Michon A. M.; Schlegl J.; Abraham Y.; Becher I.; Bergamini G.; Boesche M.; Delling M.; Dumpelfeld B.; Eberhard D.; Huthmacher C.; Mathieson T.; Poeckel D.; Reader V.; Strunk K.; Sweetman G.; Kruse U.; Neubauer G.; Ramsden N. G.; Drewes G. Chemoproteomics Profiling of HDAC Inhibitors Reveals Selective Targeting of HDAC Complexes. Nat. Biotechnol. 2011, 29 (3), 255–265. 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Madsen A. S.; Olsen C. A. Profiling of Substrates for Zinc-Dependent Lysine Deacylase Enzymes: HDAC3 Exhibits Decrotonylase Activity In Vitro. Angew. Chem. 2012, 124 (36), 9217–9221. 10.1002/ange.201203754. [DOI] [PubMed] [Google Scholar]
- Wei W.; Liu X.; Chen J.; Gao S.; Lu L.; Zhang H.; Ding G.; Wang Z.; Chen Z.; Shi T.; Li J.; Yu J.; Wong J. Class I Histone Deacetylases Are Major Histone Decrotonylases: Evidence for Critical and Broad Function of Histone Crotonylation in Transcription. Cell Res. 2017, 27 (7), 898–915. 10.1038/cr.2017.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Zhang D.; Weng Y.; Delaney K.; Tang Z.; Yan C.; Qi S.; Peng C.; Cole P. A.; Roeder R. G.; Zhao Y. The Regulatory Enzymes and Protein Substrates for the Lysine Beta-Hydroxybutyrylation Pathway. Sci. Adv. 2021, 7 (9), eabe2771. 10.1126/sciadv.abe2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Yruela C.; Zhang D.; Wei W.; Baek M.; Liu W.; Gao J.; Dankova D.; Nielsen A. L.; Bolding J. E.; Yang L.; Jameson S. T.; Wong J.; Olsen C. A.; Zhao Y. Class I Histone Deacetylases (HDAC1–3) Are Histone Lysine Delactylases. Sci. Adv. 2022, 8 (3), eabi6696. 10.1126/sciadv.abi6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari B. R.; Zhang D.; Allis C. D.; Zhao Y. Metabolic Regulation of Gene Expression through Histone Acylations. Nat. Rev. Mol. Cell Biol. 2017, 18 (2), 90–101. 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.; Tang Z.; Huang H.; Zhou G.; Cui C.; Weng Y.; Liu W.; Kim S.; Lee S.; Perez-Neut M.; Ding J.; Czyz D.; Hu R.; Ye Z.; He M.; Zheng Y. G.; Shuman H. A.; Dai L.; Ren B.; Roeder R. G.; Becker L.; Zhao Y. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature 2019, 574 (7779), 575–580. 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda C. A.; Wolfson N. A.; Leng K. R.; Kuo Y. M.; Andrews A. J.; Fierke C. A. HDAC8 Substrate Selectivity Is Determined by Long- and Short-Range Interactions Leading to Enhanced Reactivity for Full-Length Histone Substrates Compared with Peptides. J. Biol. Chem. 2017, 292 (52), 21568–21577. 10.1074/jbc.M117.811026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson N. A.; Pitcairn C. A.; Fierke C. A. HDAC8 Substrates: Histones and Beyond. Biopolymers 2013, 99, 112–126. 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti A.; Oehme I.; Witt O.; Oliveira G.; Sippl W.; Romier C.; Pierce R. J.; Jung M. HDAC8: A Multifaceted Target for Therapeutic Interventions. Trends Pharmacol. Sci. 2015, 36 (7), 481–492. 10.1016/j.tips.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Aramsangtienchai P.; Spiegelman N. A.; He B.; Miller S. P.; Dai L.; Zhao Y.; Lin H. HDAC8 Catalyzes the Hydrolysis of Long Chain Fatty Acyl Lysine. ACS Chem. Biol. 2016, 11 (10), 2685–2692. 10.1021/acschembio.6b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahm A.; Paolini C.; Pallaoro M.; Nardi M. C.; Jones P.; Neddermann P.; Sambucini S.; Bottomley M. J.; Lo Surdo P.; Carfí A.; Koch U.; De Francesco R.; Steinkühler C.; Gallinari P. Unraveling the Hidden Catalytic Activity of Vertebrate Class IIa Histone Deacetylases. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (44), 17335–17340. 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Andrade R.; Hanna A. A.; Pflum M. K. H. Evidence That HDAC7 Acts as an Epigenetic “Reader” of AR Acetylation through NCoR-HDAC3 Dissociation. Cell Chem. Biol. 2022, 29 (7), 1162–1173e5. 10.1016/j.chembiol.2022.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey T. A.; Zhang C. L.; Olson E. N. Activation of the Myocyte Enhancer Factor-2 Transcription Factor by Calcium/Calmodulin-Dependent Protein Kinase-Stimulated Binding of 14–3-3 to Histone Deacetylase 5. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (26), 14400–14405. 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinger C. M.; Schreiber S. L. Regulation of Histone Deacetylase 4 and 5 and Transcriptional Activity by 14–3-3-Dependent Cellular Localization. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (14), 7835–7840. 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra M.; Verdin E. Regulatory Signal Transduction Pathways for Class IIa Histone Deacetylases. Curr. Opin Pharmacol 2010, 10 (4), 454–460. 10.1016/j.coph.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Hai Y.; Christianson D. W. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol. 2016, 12 (9), 741–747. 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbert C.; Guardiola A.; Shao R.; Kawaguchi Y.; Ito A.; Nixon A.; Yoshida M.; Wang X. F.; Yao T. P. HDAC6 Is a Microtubule-Associated Deacetylase. Nature 2002, 417 (6887), 455–458. 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Haggarty S. J.; Koeller K. M.; Wong J. C.; Grozinger C. M.; Schreiber S. L. Domain-Selective Small-Molecule Inhibitor of Histone Deacetylase 6 (HDAC6)-Mediated Tubulin Deacetylation. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (8), 4389–4394. 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen T. J.; Guo J. L.; Hurtado D. E.; Kwong L. K.; Mills I. P.; Trojanowski J. Q.; Lee V. M. The Acetylation of Tau Inhibits Its Function and Promotes Pathological Tau Aggregation. Nat. Commun. 2011, 2, 252. 10.1038/ncomms1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook S. S.; Orian A.; Cowley S. M.; Eisenman R. N. Histone Deacetylase 6 Binds Polyubiquitin through Its Zinc Finger (PAZ. Domain) and Copurifies with Deubiquitinating Enzymes. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (21), 13425–13430. 10.1073/pnas.172511699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz B. E.; Misialek S.; Wu J.; Tang J.; Conn M. T.; Tahilramani R.; Wong L. Kinetics and Comparative Reactivity of Human Class I and Class IIb Histone Deacetylases. Biochemistry 2004, 43 (34), 11083–11091. 10.1021/bi0494471. [DOI] [PubMed] [Google Scholar]
- Hai Y.; Shinsky S. A.; Porter N. J.; Christianson D. W. ARTICLE Histone Deacetylase 10 Structure and Molecular Function as a Polyamine Deacetylase. Nat. Commun. 2017, 8, 15368. 10.1038/ncomms15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoretti I. V.; Lee Y. M.; Goodson H. V. Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. J. Mol. Biol. 2004, 338 (1), 17–31. 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Moreno-Yruela C.; Galleano I.; Madsen A. S.; Olsen C. A. Histone Deacetylase 11 Is an ε-N-Myristoyllysine Hydrolase. Cell Chem. Biol. 2018, 25 (7), 849–856. 10.1016/j.chembiol.2018.04.007. [DOI] [PubMed] [Google Scholar]
- Kutil Z.; Novakova Z.; Meleshin M.; Mikesova J.; Schutkowski M.; Barinka C. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem. Biol. 2018, 13 (3), 685–693. 10.1021/acschembio.7b00942. [DOI] [PubMed] [Google Scholar]
- Cao J.; Sun L.; Aramsangtienchai P.; Spiegelman N. A.; Zhang X.; Huang W.; Seto E.; Lin H. HDAC11 Regulates Type I Interferon Signaling through Defatty-Acylation of SHMT2. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (12), 5487–5492. 10.1073/pnas.1815365116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi R. A.; Robinson E. L.; Hu T.; Cao J.; Hong J. Y.; Tharp C. A.; Qasim H.; Gavin K. M.; Pires da Silva J.; Major J. L.; McConnell B. K.; Seto E.; Lin H.; McKinsey T. A. Reversible Lysine Fatty Acylation of an Anchoring Protein Mediates Adipocyte Adrenergic Signaling. Proc. Natl. Acad. Sci. U. S. A. 2022, 119 (7), e2119678119 10.1073/pnas.2119678119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C.; Weinert B. T.; Nishida Y.; Verdin E.; Mann M. The Growing Landscape of Lysine Acetylation Links Metabolism and Cell Signalling. Nature Reviews Molecular Cell Biology 2014 15:8 2014, 15 (8), 536–550. 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- Seto E.; Yoshida M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb Perspect Biol. 2014, 6 (4), a018713. 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi P. M.; Cole K. E.; Dowling D. P.; Christianson D. W. Structure, Mechanism, and Inhibition of Histone Deacetylases and Related Metalloenzymes. Curr. Opin Struct Biol. 2011, 21 (6), 735. 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. 2016, 6 (10), a026831. 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrarelli L. K. HDAC Inhibitors in Solid Tumors and Blood Cancers. Science Signaling 2016, 9 (446), ec216. 10.1126/scisignal.aaj2316. [DOI] [Google Scholar]
- Friend C.; Scher W.; Holland J. G.; Sato T. Hemoglobin Synthesis in Murine Virus-Induced Leukemic Cells in Vitro: Stimulation of Erythroid Differentiation by Dimethyl Sulfoxide. Proc. Natl. Acad. Sci. U. S. A. 1971, 68 (2), 378–382. 10.1073/pnas.68.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapeyre J. N.; Bekhor I. Effects of 5-Bromo-2′-Deoxyuridine and Dimethyl Sulfoxide on Properties and Structure of Chromatin. J. Mol. Biol. 1974, 89 (1), 137–162. 10.1016/0022-2836(74)90167-3. [DOI] [PubMed] [Google Scholar]
- Hogg S. J.; Beavis P. A.; Dawson M. A.; Johnstone R. W. Targeting the Epigenetic Regulation of Antitumour Immunity. Nature Reviews Drug Discovery 2020 19:11 2020, 19 (11), 776–800. 10.1038/s41573-020-0077-5. [DOI] [PubMed] [Google Scholar]
- Raouf Y. S. Targeting Histone Deacetylases: Emerging Applications beyond Cancer. Drug Discov Today 2024, 29 (9), 104094. 10.1016/j.drudis.2024.104094. [DOI] [PubMed] [Google Scholar]
- Onishi T.; Maeda R.; Terada M.; Sato S.; Fujii T.; Ito M.; Hashikami K.; Kawamoto T.; Tanaka M. A Novel Orally Active HDAC6 Inhibitor T-518 Shows a Therapeutic Potential for Alzheimer’s Disease and Tauopathy in Mice. Scientific Reports 2021 11:1 2021, 11 (1), 1–11. 10.1038/s41598-021-94923-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Ydewalle C.; Krishnan J.; Chiheb D. M; Van Damme P.; Irobi J.; Kozikowski A. P; Berghe P. V.; Timmerman V.; Robberecht W.; Van Den Bosch L. HDAC6 Inhibitors Reverse Axonal Loss in a Mouse Model of Mutant HSPB1-Induced Charcot-Marie-Tooth Disease. Nature Medicine 2011 17:8 2011, 17 (8), 968–974. 10.1038/nm.2396. [DOI] [PubMed] [Google Scholar]
- Mullard A. FDA Approves an HDAC Inhibitor for Duchenne Muscular Dystrophy. Nat. Rev. Drug Discov 2024, 23, 329. 10.1038/d41573-024-00066-8. [DOI] [PubMed] [Google Scholar]
- Hou Q.; Kan S.; Wang Z.; Shi J.; Zeng C.; Yang D.; Jiang S.; Liu Z. Inhibition of HDAC6 With CAY10603 Ameliorates Diabetic Kidney Disease by Suppressing NLRP3 Inflammasome. Front Pharmacol 2022, 13, 1. 10.3389/fphar.2022.938391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.; Natarajan R. HDAC9: An Inflammatory Link in Atherosclerosis. Circ. Res. 2020, 127 (6), 824. 10.1161/CIRCRESAHA.120.317723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee H. J.; Kim S. H.; Jeong S. M.; Sim D. S.; Hong Y. J.; Jeong M. H. The Role of Class I and IIa Histone Deacetylases in Atherosclerosis and Vascular Calcification. Journal of Cardiovascular Intervention 2024, 3 (3), 158–176. 10.54912/jci.2024.0011. [DOI] [Google Scholar]
- Shirakawa K.; Chavez L.; Hakre S.; Calvanese V.; Verdin E. Reactivation of Latent HIV by Histone Deacetylase Inhibitors. Trends Microbiol 2013, 21 (6), 277–285. 10.1016/j.tim.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALLFREY V. G.; FAULKNER R.; MIRSKY A. E. ACETYLATION AND METHYLATION OF HISTONES AND THEIR POSSIBLE ROLE IN THE. Proceedings of the National Academy of Sciences of the United States of 1964, 51 (5), 786–794. 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince H. M.; Bishton M. J.; Harrison S. J. Clinical Studies of Histone Deacetylase Inhibitors. Clin. Cancer Res. 2009, 15, 3958–3969. 10.1158/1078-0432.CCR-08-2785. [DOI] [PubMed] [Google Scholar]
- Marks P. A.; Breslow R. Dimethyl Sulfoxide to Vorinostat: Development of This Histone Deacetylase Inhibitor as an Anticancer Drug. Nature Biotechnology 2007 25:1 2007, 25 (1), 84–90. 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- Reuben R. C.; Wife R. I.; Breslow R.; Rifkind R. A.; Marks P. A. A New Group of Potent Inducers of Differentiation in Murine Erythroleukemia Cells. Proc. Natl. Acad. Sci. U. S. A. 1976, 73 (3), 862–866. 10.1073/pnas.73.3.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M.; Levy J.; Terada M.; Breslow R.; Rifkind R. A.; Marks P. A. Induction of Erythroid Differentiation in Murine Virus Infected Erythroleukemia Cells by Highly Polar Compounds. Proc. Natl. Acad. Sci. U. S. A. 1975, 72 (3), 1003–1006. 10.1073/pnas.72.3.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon V. M.; Webb Y.; Merger R.; Sheppard T.; Jursic B.; Ngo L.; Civoli F.; Breslow R.; Rifkind R. A.; Marks P. A. Second Generation Hybrid Polar Compounds Are Potent Inducers of Transformed Cell Differentiation. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (12), 5705–5708. 10.1073/pnas.93.12.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow R; Jursic B; Yan Z F; Friedman E; Leng L; Ngo L; Rifkind R A; Marks P A Potent Cytodifferentiating Agents Related to Hexamethylenebisacetamide. Proc. Natl. Acad. Sci. U. S. A. 1991, 88 (13), 5542–5546. 10.1073/pnas.88.13.5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon V. M.; Emiliani S.; Verdin E.; Webb Y.; Breslow R.; Rifkind R. A.; Marks P. A. A Class of Hybrid Polar Inducers of Transformed Cell Differentiation Inhibits Histone Deacetylases. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (6), 3003–3007. 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnin M. S.; Donigian J. R.; Cohen A.; Richon V. M.; Rifkind R. A.; Marks P. A.; Breslow R.; Pavletich N. P. Structures of a Histone Deacetylase Homologue Bound to the TSA and SAHA Inhibitors. Nature 1999, 401 (6749), 188–193. 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- Marks P. A. Discovery and Development of SAHA as an Anticancer Agent. Oncogene 2007, 1351–1356. 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- Yang F.; Zhao N.; Hu Y.; Jiang C.; Zhang H. The Development Process: From SAHA to Hydroxamate HDAC Inhibitors with Branched CAP Region and Linear Linker. Chem. Biodivers 2020, 17 (1), e1900427 10.1002/cbdv.201900427. [DOI] [PubMed] [Google Scholar]
- Mann B. S.; Johnson J. R.; Cohen M. H.; Justice R.; Pazdur R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T Cell Lymphoma. Oncologist 2007, 12 (10), 1247–1252. 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- Yang X. J.; Seto E. HATs and HDACs: From Structure, Function and Regulation to Novel Strategies for Therapy and Prevention. Oncogene 2007, 26, 5310–5318. 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Zhang J.; Jiang Q.; Zhang L.; Song W. Zinc Binding Groups for Histone Deacetylase Inhibitors. J. Enzyme Inhib Med. Chem. 2018, 33 (1), 714. 10.1080/14756366.2017.1417274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steimbach R. R.; Herbst-Gervasoni C. J.; Lechner S.; Stewart T. M.; Klinke G.; Ridinger J.; Géraldy M. N. E.; Tihanyi G.; Foley J. R.; Uhrig U.; Kuster B.; Poschet G.; Casero R. A.; Médard G.; Oehme I.; Christianson D. W.; Gunkel N.; Miller A. K. Aza-SAHA Derivatives Are Selective Histone Deacetylase 10 Chemical Probes That Inhibit Polyamine Deacetylation and Phenocopy HDAC10 Knockout. J. Am. Chem. Soc. 2022, 144 (41), 18861–18875. 10.1021/jacs.2c05030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S.; Bates S. E.; Wright J. J.; Espinoza-Delgado I.; Piekarz R. L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. 10.3390/ph3092751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner S.; Malgapo M. I. P.; Grätz C.; Steimbach R. R.; Baron A.; Rüther P.; Nadal S.; Stumpf C.; Loos C.; Ku X.; Prokofeva P.; Lautenbacher L.; Heimburg T.; Würf V.; Meng C.; Wilhelm M.; Sippl W.; Kleigrewe K.; Pauling J. K.; Kramer K.; Miller A. K.; Pfaffl M. W.; Linder M. E.; Kuster B.; Médard G. Target Deconvolution of HDAC Pharmacopoeia Reveals MBLAC2 as Common Off-Target. Nature Chemical Biology 2022 18:8 2022, 18 (8), 812–820. 10.1038/s41589-022-01015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S.; Kozikowski A. P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors—What Some May Have Forgotten or Would Rather Forget?. ChemMedChem. 2016, 11 (1), 15. 10.1002/cmdc.201500486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M.; Alsarraf J.; Araji N.; Tranoy-Opalinski I.; Renoux B.; Papot S. The Lossen Rearrangement from Free Hydroxamic Acids. Organic and Biomolecular Chemistry 2019, 17, 5420–5427. 10.1039/C9OB00789J. [DOI] [PubMed] [Google Scholar]
- Ching Yung Wang Mutagenicity of Hydroxamic Acids for Salmonella Typhimurium. Mutation Research - Fundamental and Molecular Mechanisms of Mutagenesis 1977, 56 (1), 7–12. 10.1016/0027-5107(77)90235-4. [DOI] [PubMed] [Google Scholar]
- Hermant P.; Bosc D.; Piveteau C.; Gealageas R.; Lam B.; Ronco C.; Roignant M.; Tolojanahary H.; Jean L.; Renard P. Y.; Lemdani M.; Bourotte M.; Herledan A.; Bedart C.; Biela A.; Leroux F.; Deprez B.; Deprez-Poulain R. Controlling Plasma Stability of Hydroxamic Acids: A MedChem Toolbox. J. Med. Chem. 2017, 60 (21), 9067–9089. 10.1021/acs.jmedchem.7b01444. [DOI] [PubMed] [Google Scholar]
- Yang F.; Zhao N.; Ge D.; Chen Y. Next-Generation of Selective Histone Deacetylase Inhibitors. RSC Adv. 2019, 9 (34), 19571–19583. 10.1039/C9RA02985K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter N. J.; Wagner F. F.; Christianson D. W. Entropy as a Driver of Selectivity for Inhibitor Binding to Histone Deacetylase 6. Biochemistry 2018, 57 (26), 3916–3924. 10.1021/acs.biochem.8b00367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter N. J.; Mahendran A.; Breslow R.; Christianson D. W. Unusual Zinc-Binding Mode of HDAC6-Selective Hydroxamate Inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (51), 13459–13464. 10.1073/pnas.1718823114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst-Gervasoni C. J.; Steimbach R. R.; Morgen M.; Miller A. K.; Christianson D. W. Structural Basis for the Selective Inhibition of HDAC10, the Cytosolic Polyamine Deacetylase. ACS Chem. Biol. 2020, 15 (8), 2154–2163. 10.1021/acschembio.0c00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Yu Y.; Kelly J.; Sha D.; Alhassan A. B.; Yu W.; Maletic M. M.; Duffy J. L.; Klein D. J.; Holloway M. K.; Carroll S.; Howell B. J.; Barnard R. J. O.; Wolkenberg S.; Kozlowski J. A. Discovery of Highly Selective and Potent HDAC3 Inhibitors Based on a 2-Substituted Benzamide Zinc Binding Group. ACS Med. Chem. Lett. 2020, 11 (12), 2476–2483. 10.1021/acsmedchemlett.0c00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan M. M.; Israelian J.; Nawar N.; Ganda G.; Manaswiyoungkul P.; Raouf Y. S.; Armstrong D.; Sedighi A.; Olaoye O. O.; Erdogan F.; Cabral A. D.; Angeles F.; Altintas R.; De Araujo E. D.; Gunning P. T. Characterization of Conformationally Constrained Benzanilide Scaffolds for Potent and Selective HDAC8 Targeting. J. Med. Chem. 2020, 63 (15), 8634–8648. 10.1021/acs.jmedchem.0c01025. [DOI] [PubMed] [Google Scholar]
- Shouksmith A. E.; Gawel J. M.; Nawar N.; Sina D.; Raouf Y. S.; Bukhari S.; He L.; Johns A. E.; Manaswiyoungkul P.; Olaoye O. O.; Cabral A. D.; Sedighi A.; de Araujo E. D.; Gunning P. T. Class I/IIb-Selective HDAC Inhibitor Exhibits Oral Bioavailability and Therapeutic Efficacy in Acute Myeloid Leukemia. ACS Med. Chem. Lett. 2020, 11 (1), 56–64. 10.1021/acsmedchemlett.9b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawel J. M.; Shouksmith A. E.; Raouf Y. S.; Nawar N.; Toutah K.; Bukhari S.; Manaswiyoungkul P.; Olaoye O. O.; Israelian J.; Radu T. B.; Cabral A. D.; Sina D.; Sedighi A.; de Araujo E. D.; Gunning P. T. PTG-0861: A Novel HDAC6-Selective Inhibitor as a Therapeutic Strategy in Acute Myeloid Leukaemia. Eur. J. Med. Chem. 2020, 201, 112411. 10.1016/j.ejmech.2020.112411. [DOI] [PubMed] [Google Scholar]
- Toutah K.; Nawar N.; Timonen S.; Sorger H.; Raouf Y. S.; Bukhari S.; von Jan J.; Ianevski A.; Gawel J. M.; Olaoye O. O.; Geletu M.; Abdeldayem A.; Israelian J.; Radu T. B.; Sedighi A.; Bhatti M. N.; Hassan M. M.; Manaswiyoungkul P.; Shouksmith A. E.; Neubauer H. A.; de Araujo E. D.; Aittokallio T.; Krämer O. H.; Moriggl R.; Mustjoki S.; Herling M.; Gunning P. T. Development of HDAC Inhibitors Exhibiting Therapeutic Potential in T-Cell Prolymphocytic Leukemia. J. Med. Chem. 2021, 64 (12), 8486–8509. 10.1021/acs.jmedchem.1c00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari N.; Amin S. A.; Jha T. Selective and Nonselective HDAC8 Inhibitors: A Therapeutic Patent Review. Pharm. Pat Anal 2018, 7 (6), 259–276. 10.4155/ppa-2018-0019. [DOI] [PubMed] [Google Scholar]
- Khabele D. The Therapeutic Potential of Class I Selective Histone Deacetylase Inhibitors in Ovarian Cancer. Front Oncol 2014, 4, 111. 10.3389/fonc.2014.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géraldy M.; Morgen M.; Sehr P.; Steimbach R. R.; Moi D.; Ridinger J.; Oehme I.; Witt O.; Malz M.; Nogueira M. S.; Koch O.; Gunkel N.; Miller A. K. Selective Inhibition of Histone Deacetylase 10: Hydrogen Bonding to the Gatekeeper Residue Is Implicated. J. Med. Chem. 2019, 62 (9), 4426–4443. 10.1021/acs.jmedchem.8b01936. [DOI] [PubMed] [Google Scholar]
- Huang P.; Almeciga-Pinto I.; Jarpe M.; van Duzer J. H.; Mazitschek R.; Yang M.; Jones S. S.; Quayle S. N. Selective HDAC Inhibition by ACY-241 Enhances the Activity of Paclitaxel in Solid Tumor Models. Oncotarget 2017, 8 (2), 2694–2707. 10.18632/oncotarget.13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl D. T.; Raje N.; Jagannath S.; Richardson P.; Hari P.; Orlowski R.; Supko J. G.; Tamang D.; Yang M.; Jones S. S.; Wheeler C.; Markelewicz R. J.; Lonial S. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23 (13), 3307–3315. 10.1158/1078-0432.CCR-16-2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonini M. V.; Camargo L. M.; Dong E.; Maloku E.; Veldic M.; Costa E.; Guidotti A. The Benzamide MS-275 Is a Potent, Long-Lasting Brain Region-Selective Inhibitor of Histone Deacetylases. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (5), 1587–1592. 10.1073/pnas.0510341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keuler T.; König B.; Bückreiß N.; Kraft F. B.; König P.; Schäker-Hübner L.; Steinebach C.; Bendas G.; Gütschow M.; Hansen F. K. Development of the First Non-Hydroxamate Selective HDAC6 Degraders. Chem. Commun. 2022, 58 (79), 11087–11090. 10.1039/D2CC03712B. [DOI] [PubMed] [Google Scholar]
- König B.; Watson P. R.; Reßing N.; Cragin A. D.; Schäker-Hübner L.; Christianson D. W.; Hansen F. K. Difluoromethyl-1,3,4-Oxadiazoles Are Selective, Mechanism-Based, and Essentially Irreversible Inhibitors of Histone Deacetylase 6. J. Med. Chem. 2023, 66 (19), 13821–13837. 10.1021/acs.jmedchem.3c01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurn K. T.; Thomas S.; Moore A.; Munster P. N. Rational Therapeutic Combinations with Histone Deacetylase Inhibitors for the Treatment of Cancer. Future Oncology 2011, 7, 263–283. 10.2217/fon.11.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraweera A.; O’Byrne K. J.; Richard D. J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Frontiers in Oncology 2018, 8, 92. 10.3389/fonc.2018.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao M.-W.; Chang L.-H.; Tu H.-J.; Chang C.-D.; Lai M.-J.; Chen Y.-Y.; Liou J.-P.; Teng C.-M.; Pan S.-L. Combination Treatment Strategy for Pancreatic Cancer Involving the Novel HDAC Inhibitor MPT0E028 with a MEK Inhibitor beyond K-Ras Status. Clin Epigenetics 2019, 11 (1), 85. 10.1186/s13148-019-0681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hontecillas-Prieto L.; Flores-Campos R.; Silver A.; de Álava E.; Hajji N.; García-Domínguez D. J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front Genet 2020, 11, 578011. 10.3389/fgene.2020.578011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal A.; Bauer J.; Hey J.; Papageorgiou D. N.; Stepanova E.; Daskalakis M.; Scheid J.; Dubbelaar M.; Klimovich B.; Schwarz D.; Märklin M.; Roerden M.; Lin Y. Y.; Ma T.; Mücke O.; Rammensee H. G.; Lübbert M.; Loayza-Puch F.; Krijgsveld J.; Walz J. S.; Plass C. DNMT and HDAC Inhibition Induces Immunogenic Neoantigens from Human Endogenous Retroviral Element-Derived Transcripts. Nature Communications 2023 14:1 2023, 14 (1), 1–19. 10.1038/s41467-023-42417-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbein G.; Wendling D. Histone Deacetylases in Viral Infections. Clin Epigenetics 2010, 1 (1–2), 13. 10.1007/s13148-010-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licandro S. A.; Crippa L.; Pomarico R.; Perego R.; Fossati G.; Leoni F.; Steinkühler C. The Pan HDAC Inhibitor Givinostat Improves Muscle Function and Histological Parameters in Two Duchenne Muscular Dystrophy Murine Models Expressing Different Haplotypes of the LTBP4 Gene. Skelet Muscle 2021, 11 (1), 1–22. 10.1186/s13395-021-00273-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae D.; Lee J. Y.; Ha N.; Park J.; Baek J.; Suh D.; Lim H. S.; Ko S. M.; Kim T.; Som Jeong D.; Son W. c. CKD-506: A Novel HDAC6-Selective Inhibitor That Exerts Therapeutic Effects in a Rodent Model of Multiple Sclerosis. Scientific Reports 2021, 11 (1), 1–16. 10.1038/s41598-021-93232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley J. P.; Baker I. M.; Pytel W. A.; Lin L. Y.; Bowman K. J.; Schwabe J. W. R.; Cowley S. M.; Hodgkinson J. T. Optimization of Class i Histone Deacetylase PROTACs Reveals That HDAC1/2 Degradation Is Critical to Induce Apoptosis and Cell Arrest in Cancer Cells. J. Med. Chem. 2022, 65 (7), 5642–5659. 10.1021/acs.jmedchem.1c02179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Yang K.; Zhang Z.; Leisten E. D.; Li Z.; Xie H.; Liu J.; Smith K. A.; Novakova Z.; Barinka C.; Tang W. Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem. 2019, 62 (15), 7042–7057. 10.1021/acs.jmedchem.9b00516. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Hale S.; Awasthee N.; Meng C.; Zhang X.; Liu Y.; Ding H.; Huo Z.; Lv D.; Zhang W.; He M.; Zheng G.; Liao D. HDAC3 and HDAC8 PROTAC Dual Degrader Reveals Roles of Histone Acetylation in Gene Regulation. Cell Chem. Biol. 2023, 30 (11), 1421–1435e12. 10.1016/j.chembiol.2023.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macabuag N.; Esmieu W.; Breccia P.; Jarvis R.; Blackaby W.; Lazari O.; Urbonas L.; Eznarriaga M.; Williams R.; Strijbosch A.; Van de Bospoort R.; Matthews K.; Clissold C.; Ladduwahetty T.; Vater H.; Heaphy P.; Stafford D. G.; Wang H. J.; Mangette J. E.; McAllister G.; Beaumont V.; Vogt T. F.; Wilkinson H. A.; Doherty E. M.; Dominguez C. Developing HDAC4-Selective Protein Degraders To Investigate the Role of HDAC4 in Huntington’s Disease Pathology. J. Med. Chem. 2022, 65 (18), 12445–12459. 10.1021/acs.jmedchem.2c01149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Gao H.; Yang Y.; He M.; Wu Y.; Song Y.; Tong Y.; Rao Y. Protacs: Great Opportunities for Academia and Industry. Signal Transduction and Targeted Therapy 2019, 4, 1–33. 10.1038/s41392-019-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues D. A.; Roe A.; Griffith D.; Chonghaile T. N. Advances in the Design and Development of PROTAC-Mediated HDAC Degradation. Curr. Top Med. Chem. 2022, 22 (5), 408–424. 10.2174/1568026621666211015092047. [DOI] [PubMed] [Google Scholar]
- Fischer F.; Alves Avelar L. A.; Murray L.; Kurz T. Designing HDAC-PROTACs: Lessons Learned so Far. Future Med. Chem. 2022, 14 (3), 143–166. 10.4155/fmc-2021-0206. [DOI] [PubMed] [Google Scholar]
- Chen S.; Zheng Y.; Liang B.; Yin Y.; Yao J.; Wang Q.; Liu Y.; Neamati N. The Application of PROTAC in HDAC. Eur. J. Med. Chem. 2023, 260, 115746. 10.1016/j.ejmech.2023.115746. [DOI] [PubMed] [Google Scholar]
- Tummino P. J.; Copeland R. A. Residence Time of Receptor- Ligand Complexes and Its Effect on Biological Function. Biochemistry 2008, 47 (20), 5481–5492. 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Chou C. J.; Herman D.; Gottesfeld J. M. Pimelic Diphenylamide 106 Is a Slow, Tight-Binding Inhibitor of Class I Histone Deacetylases. J. Biol. Chem. 2008, 283 (51), 35402–35409. 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. The Drug-Target Residence Time Model: A 10-Year Retrospective. Nat. Rev. Drug Discov 2016, 15 (2), 87–95. 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Pompliano D. L.; Meek T. D. Drug-Target Residence Time and Its Implications for Lead Optimization. Nat. Rev. Drug Discovery 2006, 5 (9), 730–739. 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Georgi V.; Schiele F.; Berger B. T.; Steffen A.; Marin Zapata P. A.; Briem H.; Menz S.; Preusse C.; Vasta J. D.; Robers M. B.; Brands M.; Knapp S.; Fernandez-Montalvan A. Binding Kinetics Survey of the Drugged Kinome. J. Am. Chem. Soc. 2018, 140 (46), 15774–15782. 10.1021/jacs.8b08048. [DOI] [PubMed] [Google Scholar]
- Lauffer B. E. L.; Mintzer R.; Fong R.; Mukund S.; Tam C.; Zilberleyb I.; Flicke B.; Ritscher A.; Fedorowicz G.; Vallero R.; Ortwine D. F.; Gunzner J.; Modrusan Z.; Neumann L.; Koth C. M.; Lupardus P. J.; Kaminker J. S.; Heise C. E.; Steiner P. Histone Deacetylase (HDAC) Inhibitor Kinetic Rate Constants Correlate with Cellular Histone Acetylation but Not Transcription and Cell Viability. J. Biol. Chem. 2013, 288, 26926–26943. 10.1074/jbc.M113.490706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y.; Zhan P.; Tojo T.; Jaikhan P.; Ota Y.; Suzuki M.; Li Y.; Hui Z.; Moriyama Y.; Takada Y.; Yamashita Y.; Oba M.; Uchida S.; Masuda M.; Ito S.; Sowa Y.; Sakai T.; Suzuki T. Discovery of Selective Histone Deacetylase 1 and 2 Inhibitors: Screening of a Focused Library Constructed by Click Chemistry, Kinetic Binding Analysis, and Biological Evaluation. J. Med. Chem. 2023, 66 (22), 15171–15188. 10.1021/acs.jmedchem.3c01095. [DOI] [PubMed] [Google Scholar]
- Moreno-Yruela C.; Olsen C. A. Determination of Slow-Binding HDAC Inhibitor Potency and Subclass Selectivity. ACS Med. Chem. Lett. 2022, 13 (5), 779–785. 10.1021/acsmedchemlett.1c00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazitschek R.; Payne N. Resolving the Deceptive Isoform and Complex Selectivity of HDAC1/2 Inhibitors. Cell Chemical Biology 2021, 29 (7), 1140–1152. 10.2139/ssrn.3960267. [DOI] [PubMed] [Google Scholar]
- Becher I.; Dittmann A.; Savitski M. M.; Hopf C.; Drewes G.; Bantscheff M. Chemoproteomics Reveals Time-Dependent Binding of Histone Deacetylase Inhibitors to Endogenous Repressor Complexes. ACS Chem. Biol. 2014, 9 (8), 1736–1746. 10.1021/cb500235n. [DOI] [PubMed] [Google Scholar]
- Madsen A. S.; Olsen C. A. A Potent Trifluoromethyl Ketone Histone Deacetylase Inhibitor Exhibits Class-Dependent Mechanism of Action. Medchemcomm 2016, 7 (3), 464–470. 10.1039/C5MD00451A. [DOI] [Google Scholar]
- Li X.; Peterson Y. K.; Inks E. S.; Himes R. A.; Li J.; Zhang Y.; Kong X.; Chou C. J. Class I HDAC Inhibitors Display Different Antitumor Mechanism in Leukemia and Prostatic Cancer Cells Depending on Their P53 Status. J. Med. Chem. 2018, 61 (6), 2589–2603. 10.1021/acs.jmedchem.8b00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Yu Y.; Kelly J.; Sha D.; Alhassan A. B.; Yu W.; Maletic M. M.; Duffy J. L.; Klein D. J.; Holloway M. K.; Carroll S.; Howell B. J.; Barnard R. J. O.; Wolkenberg S.; Kozlowski J. A. Discovery of Highly Selective and Potent HDAC3 Inhibitors Based on a 2-Substituted Benzamide Zinc Binding Group. ACS Med. Chem. Lett. 2020, 11 (12), 2476–2483. 10.1021/acsmedchemlett.0c00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopranovic A.; Meyer-Almes F. J. Rapid Determination of Kinetic Constants for Slow-Binding Inhibitors and Inactivators of Human Histone Deacetylase 8. Int. J. Mol. Sci. 2024, 25 (11), 5593. 10.3390/ijms25115593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellupica E.; Caprini G.; Cordella P.; Cukier C.; Fossati G.; Marchini M.; Rocchio I.; Sandrone G.; Vanoni M. A.; Vergani B.; Zrubek K.; Stevenazzi A.; Steinkuhler C. Difluoromethyl-1,3,4-Oxadiazoles Are Slow-Binding Substrate Analog Inhibitors of Histone Deacetylase 6 with Unprecedented Isotype Selectivity. J. Biol. Chem. 2023, 299 (1), 102800. 10.1016/j.jbc.2022.102800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robers M. B.; Dart M. L.; Woodroofe C. C.; Zimprich C. A.; Kirkland T. A.; Machleidt T.; Kupcho K. R.; Levin S.; Hartnett J. R.; Zimmerman K.; Niles A. L.; Ohana R. F.; Daniels D. L.; Slater M.; Wood M. G.; Cong M.; Cheng Y. Q.; Wood K. V. Target Engagement and Drug Residence Time Can Be Observed in Living Cells with BRET. Nat. Commun. 2015, 6, 10091. 10.1038/ncomms10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitir B.; Maolanon A. R.; Ohm R. G.; Colaço A. R.; Fristrup P.; Madsen A. S.; Olsen C. A. Chemical Editing of Macrocyclic Natural Products and Kinetic Profiling Reveal Slow, Tight-Binding Histone Deacetylase Inhibitors with Picomolar Affinities. Biochemistry 2017, 56 (38), 5134–5146. 10.1021/acs.biochem.7b00725. [DOI] [PubMed] [Google Scholar]
- Moreno-Yruela C.; Fass D. M.; Cheng C.; Herz J.; Olsen C. A.; Haggarty S. J. Kinetic Tuning of HDAC Inhibitors Affords Potent Inducers of Progranulin Expression. ACS Chem. Neurosci. 2019, 10 (8), 3769–3777. 10.1021/acschemneuro.9b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J. F.; Walsh C. T. The Behavior and Significance of Slow-Binding Enzyme Inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988, 61, 201–301. 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- Ripa L.; Sandmark J.; Hughes G.; Shamovsky I.; Gunnarsson A.; Johansson J.; Llinas A.; Collins M.; Jung B.; Noven A.; Pemberton N.; Mogemark M.; Xiong Y.; Li Q.; Tangefjord S.; Ek M.; Astrand A. Selective and Bioavailable HDAC6 2-(Difluoromethyl)-1,3,4-Oxadiazole Substrate Inhibitors and Modeling of Their Bioactivation Mechanism. J. Med. Chem. 2023, 66 (20), 14188–14207. 10.1021/acs.jmedchem.3c01269. [DOI] [PubMed] [Google Scholar]
- Motlova L.; Snajdr I.; Kutil Z.; Andris E.; Ptacek J.; Novotna A.; Novakova Z.; Havlinova B.; Tueckmantel W.; Draberova H.; Majer P.; Schutkowski M.; Kozikowski A.; Rulisek L.; Barinka C. Comprehensive Mechanistic View of the Hydrolysis of Oxadiazole-Based Inhibitors by Histone Deacetylase 6 (HDAC6). ACS Chem. Biol. 2023, 18 (7), 1594–1610. 10.1021/acschembio.3c00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellupica E.; Gaiassi A.; Rocchio I.; Rovelli G.; Pomarico R.; Sandrone G.; Caprini G.; Cordella P.; Cukier C.; Fossati G.; Marchini M.; Bebel A.; Airoldi C.; Palmioli A.; Stevenazzi A.; Steinkuhler C.; Vergani B. Mechanistic and Structural Insights on Difluoromethyl-1,3,4-Oxadiazole Inhibitors of HDAC6. Int. J. Mol. Sci. 2024, 25 (11), 5885. 10.3390/ijms25115885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie T. A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem., Int. Ed. 2016, 55 (43), 13408–13421. 10.1002/anie.201601091. [DOI] [PubMed] [Google Scholar]
- Duan W.; Li J.; Inks E. S.; Chou C. J.; Jia Y.; Chu X.; Li X.; Xu W.; Zhang Y. Design, Synthesis, and Antitumor Evaluation of Novel Histone Deacetylase Inhibitors Equipped with a Phenylsulfonylfuroxan Module as a Nitric Oxide Donor. J. Med. Chem. 2015, 58 (10), 4325–4338. 10.1021/acs.jmedchem.5b00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Liu C.; Chai Q.; Zhao C.; Meng H.; Xue X.; Yao T. P.; Zhang Y. Discovery of the First Irreversible HDAC6 Isoform Selective Inhibitor with Potent Anti-Multiple Myeloma Activity. J. Med. Chem. 2023, 66 (14), 10080–10091. 10.1021/acs.jmedchem.3c00977. [DOI] [PubMed] [Google Scholar]
- Daniel K. B.; Sullivan E. D.; Chen Y.; Chan J. C.; Jennings P. A.; Fierke C. A.; Cohen S. M. Dual-Mode HDAC Prodrug for Covalent Modification and Subsequent Inhibitor Release. J. Med. Chem. 2015, 58 (11), 4812–4821. 10.1021/acs.jmedchem.5b00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto M. P.; Greenberg M. M. Histone Deacetylase 1 Inhibition by Peptides Containing a DNA Damage-Induced, Nonenzymatic, Histone Covalent Modification. Biochemistry 2023, 62 (8), 1388–1393. 10.1021/acs.biochem.3c00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muth M.; Jansch N.; Kopranovic A.; Kramer A.; Wossner N.; Jung M.; Kirschhofer F.; Brenner-Weiss G.; Meyer-Almes F. J. Covalent Inhibition of Histone Deacetylase 8 by 3,4-Dihydro-2H-Pyrimido[1,2-c][1,3]Benzothiazin-6-Imine. Biochim Biophys Acta Gen Subj 2019, 1863 (3), 577–585. 10.1016/j.bbagen.2019.01.001. [DOI] [PubMed] [Google Scholar]
- Raouf Y. S.; Sedighi A.; Geletu M.; Frere G. A.; Allan R. G.; Nawar N.; de Araujo E. D.; Gunning P. T. Discovery of YSR734: A Covalent HDAC Inhibitor with Cellular Activity in Acute Myeloid Leukemia and Duchenne Muscular Dystrophy. J. Med. Chem. 2023, 66 (24), 16658–16679. 10.1021/acs.jmedchem.3c01236. [DOI] [PubMed] [Google Scholar]
- Kim H.; Hwang Y. S.; Kim M.; Park S. B. Recent Advances in the Development of Covalent Inhibitors. RSC Med. Chem. 2021, 12 (7), 1037–1045. 10.1039/D1MD00068C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulussen F. M.; Grossmann T. N. Peptide-Based Covalent Inhibitors of Protein-Protein Interactions. J. Pept Sci. 2023, 29 (1), e3457. 10.1002/psc.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faridoon; Ng R.; Zhang G.; Li J. J. An Update on the Discovery and Development of Reversible Covalent Inhibitors. Med. Chem. Res. 2023, 32 (6), 1039–1062. 10.1007/s00044-023-03065-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B.; Xu J.; Liu S.; Li Q. X. Characterization of Small Molecule-Protein Interactions Using SPR Method. Methods Mol. Biol. 2023, 2690, 149–159. 10.1007/978-1-0716-3327-4_15. [DOI] [PubMed] [Google Scholar]
- Miyahisa I.; Sameshima T.; Hixon M. S. Rapid Determination of the Specificity Constant of Irreversible Inhibitors (Kinact/KI) by Means of an Endpoint Competition Assay. Angew. Chem., Int. Ed. Engl. 2015, 54 (47), 14099–14102. 10.1002/anie.201505800. [DOI] [PubMed] [Google Scholar]
- Strelow J. M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. J. Biomol Screen 2017, 22 (1), 3–20. 10.1177/1087057116671509. [DOI] [PubMed] [Google Scholar]
- Boike L.; Henning N. J.; Nomura D. K. Advances in Covalent Drug Discovery. Nature Reviews Drug Discovery 2022, 21, 881–898. 10.1038/s41573-022-00542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cesco S.; Kurian J.; Dufresne C.; Mittermaier A. K.; Moitessier N. Covalent Inhibitors Design and Discovery. Eur. J. Med. Chem. 2017, 138, 96–114. 10.1016/j.ejmech.2017.06.019. [DOI] [PubMed] [Google Scholar]
- Abdeldayem A.; Raouf Y. S.; Constantinescu S. N.; Moriggl R.; Gunning P. T. Advances in Covalent Kinase Inhibitors. Chem. Soc. Rev. 2020, 49 (9), 2617–2687. 10.1039/C9CS00720B. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The Resurgence of Covalent Drugs. Nature Reviews Drug Discovery 2011 10:4 2011, 10 (4), 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Gehringer M.; Laufer S. A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62 (12), 5673–5724. 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- Hillebrand L.; Liang X. J.; Serafim R. A. M.; Gehringer M. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: An Update. J. Med. Chem. 2024, 67 (10), 7668–7758. 10.1021/acs.jmedchem.3c01825. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Bourne P. E. Progress with Covalent Small-Molecule Kinase Inhibitors. Drug Discov Today 2018, 23 (3), 727–735. 10.1016/j.drudis.2018.01.035. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Burgess J.; Colclough N.; Davies N. L.; Lenz E. M.; Orton A. L.; Ward R. A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf Model 2017, 57 (12), 3124–3137. 10.1021/acs.jcim.7b00553. [DOI] [PubMed] [Google Scholar]
- Lee K. S. S.; Yang J.; Niu J.; Ng C. J.; Wagner K. M.; Dong H.; Kodani S. D.; Wan D.; Morisseau C.; Hammock B. D. Drug-Target Residence Time Affects in Vivo Target Occupancy through Multiple Pathways. ACS Cent Sci. 2019, 5 (9), 1614–1624. 10.1021/acscentsci.9b00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Jiang Y.; Peterson Y. K.; Xu T.; Himes R. A.; Luo X.; Yin G.; Inks E. S.; Dolloff N.; Halene S.; Chan S. S. L.; Chou C. J. Design of Hydrazide-Bearing HDACIs Based on Panobinostat and Their P53 and FLT3-ITD Dependency in Antileukemia Activity. J. Med. Chem. 2020, 63 (10), 5501–5525. 10.1021/acs.jmedchem.0c00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeley A. B.; Kopranovic A.; Di Lorenzo V.; Abranyi-Balogh P.; Jansch N.; Lai L. N.; Petri L.; Orgovan Z.; Poloske D.; Orlova A.; Nemeth A. G.; Desczyk C.; Imre T.; Bajusz D.; Moriggl R.; Meyer-Almes F. J.; Keseru G. M. Electrophilic MiniFrags Revealed Unprecedented Binding Sites for Covalent HDAC8 Inhibitors. J. Med. Chem. 2024, 67 (1), 572–585. 10.1021/acs.jmedchem.3c01779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure J. J.; Zhang C.; Inks E. S.; Peterson Y. K.; Li J.; Chou C. J. Development of Allosteric Hydrazide-Containing Class i Histone Deacetylase Inhibitors for Use in Acute Myeloid Leukemia. J. Med. Chem. 2016, 59 (21), 9942–9959. 10.1021/acs.jmedchem.6b01385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões-Pires C.; Zwick V.; Nurisso A.; Schenker E.; Carrupt P. A.; Cuendet M. HDAC6 as a Target for Neurodegenerative Diseases: What Makes It Different from the Other HDACs?. Mol. Neurodegener 2013, 8 (1), 1–16. 10.1186/1750-1326-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Qin G.; Zhao T. C. HDAC4: Mechanism of Regulation and Biological Functions. Epigenomics 2014, 6, 139–150. 10.2217/epi.13.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewanjee S.; Vallamkondu J.; Kalra R. S.; Chakraborty P.; Gangopadhyay M.; Sahu R.; Medala V.; John A.; Reddy P. H.; De Feo V.; Kandimalla R. The Emerging Role of HDACs: Pathology and Therapeutic Targets in Diabetes Mellitus. Cells 2021, 10 (6), 1340. 10.3390/cells10061340. [DOI] [PMC free article] [PubMed] [Google Scholar]