

Summary
Interleukin-2 (IL-2) variants with increased CD25 dependence that selectively expand Foxp3+ regulatory T (TR) cells are in clinical trials for treating inflammatory diseases. Using an Fc-fused IL-2 mutein (Fc.IL-2 mutein) we developed that prevents diabetes in non-obese diabetic (NOD) mice, we show that Fc.IL-2 mutein induced an activated TR population with elevated proliferation, a transcriptional program associated with Stat5- and T cell receptor-dependent gene modules, and high IL-10 and CTLA-4 expression. Increased IL-10 signaling limited surface major histocompatibility complex class II upregulation during conventional dendritic cell (cDC) maturation, while increased CTLA-4-dependent transendocytosis led to the transfer of CD80 and CD86 co-stimulatory ligands from maturing cDCs to TR cells. In NOD mice, Fc.IL-2 mutein treatment promoted the suppression of cDCs in the inflamed pancreas and pancreatic lymph nodes, resulting in T cell anergy. Thus, IL-2 mutein-expanded TR cells have enhanced functional properties and restrict cDC function, offering promise for targeted immunotherapy use in autoimmune disease.
Keywords: regulatory T cells, IL-2, CTLA-4, anergy, tolerance
Graphical abstract
Highlights
-
•
A regulatory T (TR) cell-selective IL-2 mutein activates TR
-
•
CTLA4 and IL-10 function in TR cells is enhanced by IL-2 mutein stimulation
-
•
IL-2 mutein-stimulated TR cells suppress dendritic cell activity
-
•
Anergic T cells are found in the pancreatic lymph node of IL-2 mutein-treated NOD mice
Regulatory T (TR) cell expansion by interleukin-2 (IL-2) muteins is a promising therapy for autoimmune and inflammatory diseases. Jamison et al. show that IL-2 mutein-stimulated TR cells are highly activated and inhibit dendritic cell function via CTLA-4 and IL-10, leading to induction of anergic phenotype T cells in an autoimmune disease model.
Introduction
Regulatory T (TR) cells expressing forkhead box P3 (Foxp3) are required to suppress immune responses against self-antigens and prevent autoimmunity. The cytokine interleukin-2 (IL-2) and downstream signals mediated by the transcription factor Stat5 support the development, maintenance, and function of TR cells.1 IL-2 also promotes effector and memory CD8+ and CD4+ T cell responses in infection and anti-tumor immunity.2,3 The opposing functions of IL-2 are explained by varying IL-2 receptor (IL-2R) expression patterns and responsiveness thresholds among cell types. At low concentrations, IL-2 stimulates TR cells constitutively expressing the high-affinity IL-2R (CD25, CD122, and CD132).4 In contrast, memory T cells and natural killer cells expressing the low-affinity IL-2R (CD122 and CD132) are activated at higher concentrations. Furthermore, effector T cells upregulate CD25 after activation, allowing them to compete with TR cells for limiting amounts of IL-2. Low-dose IL-2 has been explored as a potential treatment for autoimmune diseases due to its ability to boost the expansion of Foxp3+ TR cells.4 However, the effects on both regulatory and effector T cell populations make balancing the safety and efficacy of low-dose IL-2 challenging, and results in clinical trials have been inconsistent,5,6,7 prompting the search for more targeted therapeutic approaches.
Engineered variants of IL-2 (known as IL-2 mutant proteins or muteins) designed to preferentially stimulate TR cells with less off-target specificity have been developed. One approach is to decrease the affinity of IL-2 for CD122, thereby increasing CD25 dependence and improving TR cell selectivity.8 In recent work, we introduced two amino acid substitutions into murine IL-2 that reduce CD122 binding (N103R and V106D, equivalent to the N88 and V91 positions of human IL-2) to create a CD25-biased IL-2 mutein.9 To improve pharmacokinetic properties, this IL-2 mutein (Fc.Mut24) was fused to an immunoglobulin G2a Fc domain mutated to reduce FcR binding and effector function. Although Fc.Mut24 is a weaker agonist of IL-2R signaling than an Fc-fused wild-type IL-2 (Fc.WT) in vitro, it is significantly better at expanding TR cells in vivo, due in part to decreased receptor-mediated clearance that results in an extended biological half-life and sustained IL-2R signaling.9 Furthermore, unlike Fc.WT, Fc.Mut24 can be administered at high doses while maintaining TR cell selectivity and halting disease progression in the non-obese diabetic (NOD) model of type 1 diabetes. Several human Fc-fused IL-2 (Fc.IL-2) muteins with similar mutations to Fc.Mut24 are in early-stage clinical development for treating autoimmune disorders,10 and phase 1 trials have demonstrated substantial TR cell expansion and safety.11
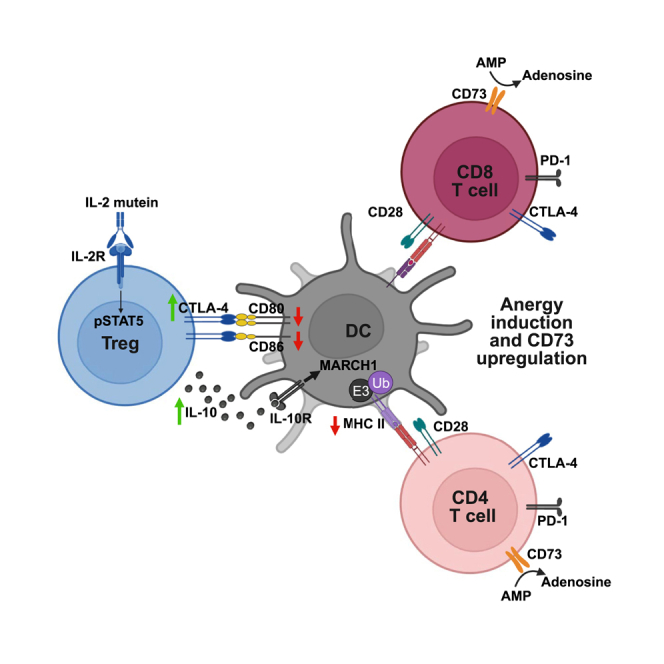
It is critical to better understand the mechanisms of action of TR-selective Fc.IL-2 muteins to optimize therapeutic use. One potential benefit of Fc.IL-2 muteins over other therapies for inflammatory diseases, such as biologics that block pro-inflammatory cytokines, is their ability to induce durable tolerance by increasing the balance between TR cells and conventional T (Tconv) cells at the site of tissue inflammation during critical windows of disease development.9 An increased TR-to-Tconv ratio is associated with induction of anergy,12 a tolerance mechanism resulting from antigen recognition without sufficient co-stimulatory signals,13 and TR cells are required for the in vivo induction of anergy.14,15 A key feature of TR-mediated suppression is the counter-regulation of antigen-presenting cell (APC) function. TR cells are primary producers of the immunosuppressive cytokine IL-10, a potent inhibitor of APCs such as macrophages and dendritic cells (DCs).16,17 Additionally, the checkpoint receptor CTLA-4 is required for TR-suppressive activity and functions in a cell-extrinsic manner by capturing the co-stimulatory ligands CD80 and CD86 that it shares with CD28 via CTLA-4-dependent transendocytosis.18,19,20 In this process, CD80 and CD86 are transferred from the APC membrane to intracellular compartments in TR cells and undergo subsequent degradation. Thus, TR cells can promote anergy in autoreactive Tconv cells by inhibiting APC maturation via IL-10 and restricting CD28 co-stimulation via CTLA-4.
Our development of Fc.Mut24 provided a tool to test whether sustained IL-2R signaling not only increased TR cell numbers but also influenced TR cell activation and function. We found that Fc.Mut24 treatment induced a novel transcriptional, phenotypic, and functional state in TR cells indicative of activation via both IL-2R and T cell receptor (TCR) signaling. Using in vivo models of DC maturation and examining pre-diabetic NOD mice, we found that Fc.Mut24 treatment led to the downregulation of CD80/CD86 and major histocompatibility complex (MHC) class II on DCs via CTLA-4-dependent transendocytosis or IL-10 production by TR cells, respectively. The modulation of DC function was associated with T cell anergy and increased CD73 expression by CD4+ and CD8+ Tconv cells in NOD mice. Our study links IL-2R signaling in TR cells with IL-10-mediated suppression of DCs and CTLA-4-dependent transendocytosis, thereby demonstrating how three molecular pathways known to play essential roles in immune regulation converge on the induction of T cell tolerance.
Results
Induction of Stat5 and TCR-dependent programs in Foxp3+ TR cells by Fc.Mut24 treatment
To investigate how sustained IL-2R signaling with Fc.Mut24 impacted the transcriptional state of TR cells, we treated C57BL/6 (B6) Foxp3-mRFP mice with PBS (vehicle control) or a single 10-μg dose of either Fc.WT or Fc.Mut24. As expected, TR cell percentages in the spleen significantly increased 3 days after Fc.Mut24 treatment compared to mice given PBS or Fc.WT (Figure 1A). We performed single-cell RNA sequencing (scRNA-seq) using the 10× Genomics platform on sorted CD4+ Foxp3-mRFP+ splenic TR cells with four mice per treatment group. After filtering and normalization, 42,698 TR cells were recovered, with similar numbers in each group.
Figure 1.

Fc.Mut24 induces a distinct Foxp3+ TR cell transcriptional state
(A) Percentage of Foxp3+ TR from different treatment groups. Representative flow-cytometry plots from sorting are shown. Gates were set on live, CD4+ cells. Graph shows mean ± SD with individual data points (n = 4); ∗∗∗∗p ≤ 0.0001, two-tailed unpaired t tests.
(B) UMAP-based clustering of merged scRNA-seq profiles of TR from PBS control, Fc.WT-treated, and Fc.Mut24-treated mice. Individual cells are colored by cluster assignments. A complete list of the differentially expressed genes for each cluster is available in Table S1.
(C) UMAP-based clustering of scRNA-seq profiles of TR by treatment group.
(D) Bar graph showing the frequency of TR clusters in each treatment group.
(E) Heatmap showing the mean expression (Z score) of representative signature genes (rows) for each cluster (columns).
(F) Violin plots with gene module expression for Stat5-dependent genes (Chinen et al.21) or TCR-dependent genes (Levine et al.22) in TR cells; ∗∗∗∗p ≤ 0.0001, Wilcoxon signed rank test.
(G–I) Biaxial plots showing expression of Stat5-dependent versus TCR-dependent genes in each treatment group colored by TR cluster (top) or using a high and low cutoff based on co-expression of both modules (bottom).
(J) Violin plots showing normalized expression of select genes associated with TR activation between high and low populations from all treatment groups based on cutoff in (G)–(I) (bottom row); ∗∗∗∗p ≤ 0.0001, Wilcoxon signed rank test.
Uniform manifold approximation and projection (UMAP) analysis and unsupervised graphical clustering based on features of all cells organized the TR cells into six clusters (Figures 1B–1D). Whereas the vast majority of cells from PBS controls or Fc.WT-treated mice were found in clusters 1 and 2, TR cells from Fc.Mut24-treated mice were predominantly found in clusters 2, 3, 4, and 5 (Figures 1C and 1D). A small fraction of TR cells from all treatment groups were found in cluster 6. These clusters were annotated based on differential gene expression (Figure 1E). Genes previously associated with naive-like central TR (cTR) cells such as Ly6c1, Bcl2, Ccr7, and Sell were highly expressed in cluster 1, whereas genes enriched in cluster 2 included Izumo1r, Cd44, Pdcd1, Lag3, Icos, Tigit, and Ctla4 that define effector TR (eTR) cells.23,24 Genes induced by Stat5 activation, including Gzmb, Ifitm2, Ifitm3, Vim, Socs2, Il2ra, and Cish, were enriched in clusters 3, 4, and 5.25,26,27 Based on the expression of genes controlling cell proliferation and DNA replication or repair, such as Hells, Rad51, Cenpf, Mki67, and Pclaf, clusters 4 and 5 represent actively dividing cells. Genes that play a role in chromatin remodeling or microtubule assembly such as Top2a, Hist1h1b, Hist1h2ae, Tuba1b, Tubb4b, and Tubb55 were highly expressed in cluster 5, distinguishing these cells from those in cluster 4. Genes upregulated by T cell activation, such as Zap70, Tnfrsf9, Tnfrsf4, and Orai1, were highly enriched in cluster 6.
The cluster annotations were further supported by examining differential transcription factor (TF) expression (Figure S1A). This showed enrichment of Lef1, Tcf7, and Bach2 associated with T cell stemness in cluster 1,28,29 whereas cluster 2 expressed TFs associated with TR cell effector function or stability such as Hif1a, Batf, Maf, and Ikzf2,30,31,32,33 and cluster 3 had the highest expression of Foxp3, a known target gene of Stat5 in TR cells.34 TFs involved in cell-cycle progression, such as E2f1, E2f2, Mybl2, and Tfdp1, were expressed in clusters 4 and 5, while Tox, Nr4a1, Nr4a3, and Egr2, which are associated with NFAT activation downstream of antigen receptor signaling, were expressed in cluster 6.35,36,37
In addition to IL-2R signaling that supports the survival of cTR cells,23 the abundance and proliferation of eTR cells are controlled by signaling through the TCR.22 Surprisingly, analysis of genes regulated in TR cells by either Stat5 activation21 or TCR signaling22 showed that Fc.Mut24 treatment upregulated both of these transcriptional programs relative to PBS or Fc.WT (Figure 1F). In TR cells from PBS controls, there was an inverse correlation between the expression of Stat5- and TCR-dependent programs, and these gene sets were associated with cTR cells in cluster 1 and eTR cells in cluster 2, respectively (Figure 1G). In contrast, TR cells from Fc.Mut24-treated and, to a lesser extent, Fc.WT-treated mice co-expressed the Stat5- and TCR-dependent programs, and in response to Fc.Mut24 this was associated with clusters 3, 4, and 5 (Figures 1H and 1I). Cells with high expression of both Stat5- and TCR-dependent programs had increased expression of genes associated with highly functional TR cells, including Il2ra, Ctla4, Icos, Tnfrsf18, and Tgfb1 (Figures 1G–1J). Despite the prevalence of a TCR-dependent gene signature in TR cells from Fc.Mut24-treated mice, single-cell TCR sequencing showed that Fc.Mut24-expanded TR cells maintained a diverse TCR repertoire that was comparable to PBS controls or Fc.WT-treated mice and had limited clonal expansion (Figures S1B and S1C). Thus, Fc.WT and Fc.Mut24 have divergent effects on TR cell expansion and gene-expression profiles, and prolonged IL-2R signaling in TR cells by Fc.Mut24 is uniquely characterized by co-expression of Stat5- and TCR-dependent gene signatures associated with activated and highly functional TR cells.
Unique phenotypic features of Foxp3+ TR cells induced by Fc.Mut24 treatment
Given the transcriptional changes induced by Fc.Mut24 treatment, we further analyzed its role in modulating TR phenotype and function. We injected B6 mice with 10 μg of Fc.Mut24 and examined splenic TR cells 3 days later. We observed an ∼8-fold expansion in TR cell number in Fc.Mut24-treated mice compared to mice given PBS (Figure 2A), and nearly all TR cells were Ki-67+ after treatment (Figure 2A). Fc.Mut24 treatment increased the expression of CD44 (Figure 2A), a marker of T cell activation previously associated with proliferating TR cells in the steady state.38 Expression of the hallmark TR identity markers CD25, CTLA-4, and ICOS were also increased (Figure 2A). In contrast, expression of the co-inhibitory receptor programmed cell death 1 (PD-10), which antagonizes TR function and is downregulated by Stat5 activation,39,40 did not increase (Figure 2A).
Figure 2.

Fc.Mut24 activates different Foxp3+ TR cell subsets
(A) Number and proliferation (Ki-67) of CD25+ Foxp3+ TR and the expression of indicated TR activation markers. Gates were set on live, CD4+ cells.
(B) Percentage, number, and proliferation of CD44lo CD62Lhi central TR (cTR), CD44hi CD62Lhi central-effector TR (ceTR), and CD44hi CD62Llo effector TR (eTR) in each treatment group. Gates are indicated on representative flow-cytometry plots.
(C) Expression of indicated TR activation markers on TR subsets.
All graphs show mean ± SD with individual data points (n = 6–11); ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗∗p ≤ 0.0001, two-tailed unpaired t tests, or multiple unpaired t tests. n.s., not significant. Data are representative of 2–3 independent experiments.
We analyzed CD44lo CD62Lhi cTR and CD44hi CD62Llo eTR populations, since cTR cells in secondary lymphoid tissues are more dependent on IL-2R signaling during homeostasis.23,41 There was close to an equal expansion of cTR (6-fold) and eTR (7-fold) in response to Fc.Mut24 treatment (Figure 2B). However, a population of CD44hi CD62Lhi TR cells, which we called central-effector (ceTR) cells, had the largest increase in both percentage and cell number (17-fold) in Fc.Mut24-treated mice (Figure 2B). Fc.Mut24 treatment upregulated CD25 on both cTR and eTR cells but, similar to the expansion of cell number, ceTR cells had the highest overall CD25 expression (Figure 2B). The expression of CTLA-4 and ICOS was increased on all TR cell subsets after Fc.Mut24 treatment, with the highest levels of these markers observed on eTR cells followed by ceTR cells (Figure 2C). All three of these expanded TR cell populations were highly suppressive based on their ability to inhibit Tconv cell proliferation and expression of activation markers in in vitro co-culture assays (Figure S2). We also found that although Fc.Mut24 treatment had no effect on PD-1 when the total number of TR cells were analyzed, PD-1 expression was specifically reduced in eTR cells (Figure 2C), which have the highest baseline PD-1 levels due to strong TCR signaling.42
CTLA-4 cycling by Foxp3+ TR cells is enhanced by Fc.Mut24 treatment
Although CTLA-4 is constitutively expressed by eTR cells, it is largely restricted to intracellular vesicles due to rapid internalization from the plasma membrane via clathrin-mediated endocytosis.43 Following TCR stimulation, the intracellular pool of CTLA-4 is mobilized and rapidly cycles between these vesicles and the cell surface, allowing CTLA-4 to bind and strip CD80 and CD86 from APCs via transendocytosis.19,20 Therefore, to determine whether CTLA-4 function may be increased after Fc.Mut24 treatment, we measured surface CTLA-4 by staining live cells at 4°C for 30 min and stained for the cycling pool of functional CTLA-4 molecules by labeling for 2 h at 37°C. We found that Fc.Mut24 treatment dramatically increased both surface and cycling CTLA-4 in TR cells but not in Tconv cells, with ∼17% of TR cells cycling CTLA-4 after treatment (Figure 3A). Additionally, analysis of cTR, ceTR, and eTR cells showed significantly increased CTLA-4 surface expression and cycling in all TR subsets after Fc.Mut24 treatment (Figure 3B). Consistent with these results, the expression of Trat1 (TRIM), Lax1 (LAX), and Rab8 that form a multimeric complex regulating post-Golgi trafficking of CTLA-4 to the cell surface were upregulated in TR cells from Fc.Mut24-treated mice (Figure S3A).44 Fc.Mut24 treatment also increased the expression of the guanine nucleotide exchange factor Def6 and the small guanosine triphosphatase Rab11 (Rab11a and Rab11b), which interact to control endosomal recycling of CTLA-4 to the cell surface (Figure S3B).45,46
Figure 3.

Fc.Mut24 increases CTLA-4 cycling by different Foxp3+ TR cell subsets
(A) Percentage of CTLA-4+ cells between different treatment groups and staining conditions. Gates were set on live, CD4+, Foxp3+ TR, or Foxp3− Tconv cells.
(B) Percentage of cycling CTLA-4+ cells between CD44lo CD62Lhi central TR (cTR), CD44hi CD62Lhi central-effector TR (ceTR), and CD44hi CD62Llo effector TR (eTR). Representative flow-cytometry plots for cycling CTLA-4 are shown.
All graphs show mean ± SD with individual data points (n = 6–9); ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗∗p ≤ 0.0001, multiple unpaired t tests. Data are representative of two independent experiments.
CTLA-4-dependent capture of co-stimulatory ligands by Foxp3+ TR cells is enhanced by Fc.Mut24 treatment
To determine whether the increased CTLA-4 cycling in TR cells resulted in increased CTLA-4-dependent transendocytosis of co-stimulatory ligands, we isolated CD4+ T cells from PBS- or Fc.Mut24-treated mice, co-cultured them with NIH/3T3 fibroblasts expressing GFP-tagged CD86 for 2 h, and analyzed ligand capture by flow cytometry (Figure 4A). In accordance with the CTLA-4 cycling results (Figure 3A), almost no Tconv cells captured the CD86-GFP ligand, and only a low level of CD86-GFP capture (<5% of cells) was observed in TR cells from PBS controls (Figure 4B). However, this capture significantly increased to ∼17% of TR cells after Fc.Mut24 treatment (Figure 4B). Addition of the anti-CTLA-4 blocking antibody clone UC10-4F10-11 (4F10) reduced CD86-GFP capture, and internalization of captured ligand was confirmed by the accumulation of CD86-GFP in the presence of the lysosomal inhibitor bafilomycin A1 (BafA) (Figure 4C). In line with a previous report,20 we found that eTR cells exhibited the highest level of CTLA-4 transendocytosis in PBS controls (Figures S4A and S4B). However, after Fc.Mut24 treatment, ceTR cells captured CD86-GFP ligand as efficiently as eTR cells, and treatment also significantly increased ligand capture by cTR cells (Figures S4A and S4B).
Figure 4.

Fc.Mut24 increases CTLA-4-dependent transendocytosis of CD80 and CD86 by Foxp3+ TR cells
(A) Experimental design schematic for ex vivo CTLA-4 transendocytosis assay. Gates were set on live, CD4+, Foxp3+ TR, or Foxp3− Tconv cells.
(B) Capture of CD86-GFP by Foxp3+ TR and Foxp3− Tconv between different treatment groups. Representative flow-cytometry plots for TR are shown. Graph shows mean ± SD with individual data points (n = 7); ∗∗∗∗p ≤ 0.0001, multiple unpaired t tests. Data are representative of 2–3 independent experiments.
(C) Capture of CD86-GFP by Foxp3+ TR between different co-culture conditions. Representative flow-cytometry plots from Fc.Mut24-treated mice are shown. Graph shows mean ± SD with individual data points (n = 7); ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, multiple paired t tests. Data are representative of 2–3 independent experiments.
(D) Experimental design schematic for in vivo CTLA-4 transendocytosis assay. Gates were set on CD4+, Foxp3+ TR, or Foxp3− Tconv.
(E) Capture of CD80-mCherry by Foxp3+ TR and Foxp3− Tconv between different treatment groups. Representative flow-cytometry plots are shown. The graph shows Foxp3+ TR data, and mean ± SD with individual data points (n = 5 per group); ∗p ≤ 0.05, ∗∗∗∗p ≤ 0.0001, multiple unpaired t tests. Data are representative of three independent experiments.
To determine whether Fc.Mut24 also enhanced CTLA-4-dependent transendocytosis by TR cells in vivo, we set up a model in which CD4+ T cells were adoptively transferred into CD80-mCherry.Rag2−/− mice followed by Fc.Mut24 treatment (Figure 4D). The CD80-mCherry fusion gene is knocked into the endogenous locus in these recipient animals, allowing CD80 transfer from APCs to T cells to be monitored by flow cytometry (C.J.W., D.M.S., and L.S.K.W., unpublished data). In addition to Fc.Mut24 treatment, recipients received three injections of isotype or anti-CTLA-4 blocking antibody (4F10) to confirm the role of CTLA-4 in ligand capture (Figure 4D). Similar to our ex vivo CTLA-4 transendocytosis assay, <5% of Foxp3+ TR cells from PBS controls captured CD80-mCherry ligand in vivo, while virtually no Tconv cells did (Figure 4E). However, Fc.Mut24 treatment increased the percentage of CD80-mCherry+ TR cells with no changes observed for Tconv cells, and administration of 4F10 almost completely blocked CD80-mCherry ligand capture by TR cells (Figure 4E). Thus, in addition to stimulating TR expansion, Fc.Mut24 enhances TR function by increasing CTLA-4 cycling and transendocytosis of co-stimulatory ligands by multiple subsets of Foxp3+ TR cells.
Upregulation of co-stimulatory ligand and MHC class II protein expression during cDC maturation is suppressed by Fc.Mut24 treatment
We next examined how elevated function of CTLA-4 in TR cells induced by Fc.Mut24 treatment impacted the maturation of different APC populations following inflammatory stimulation. In secondary lymphoid organs, two primary cDC subsets have been described. Type 1 conventional DCs (cDC1s) depend on the transcription factors Batf3 and Irf847 and are essential for cross-presentation and priming of CD8+ T cells as well as T helper 1 (Th1) cells.48 In contrast, type 2 conventional DCs (cDC2s) are Irf4 dependent, and cDC2s prime Th17 and Th2 responses.49 To determine the impact of Fc.Mut24 treatment on cDC1s and cDC2s in vivo, we treated mice with PBS or Fc.Mut24, and 3 days later some mice received a low dose of lipopolysaccharide (LPS) intraperitoneally to promote cDC maturation in the spleen (Figure 5A). Whereas Fc.Mut24 treatment had no impact on 33D1+ cDC2s frequency in the spleen, both the percentage and number of splenic CD8α+ cDC1s were increased after treatment (Figure S5). However, this expansion was lost in the presence of LPS, where Fc.Mut24 treatment was associated with a decreased abundance of cDC1s.
Figure 5.

Fc.Mut24 promotes CTLA-4-dependent transendocytosis of CD80 and CD86 during dendritic cell maturation
(A) Experimental design schematic for the in vivo dendritic cell (DC) maturation model. See Figure S5A for the gating strategy.
(B–E) Expression of indicated DC maturation markers. Representative histograms are shown. Graphs show mean ± SD with individual data points (n = 7–10); ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, multiple unpaired t tests. Ctrl, control. For MHC class II expression, data are shown as fold-change geometric MFI over the mean of PBS control (dotted line). Data are representative of 2–3 independent experiments.
(F) Experimental design schematic for the anti-CTLA-4 study.
(G) Expression of indicated DC maturation markers between isotype- and anti-CTLA-4 antibody-treated mice. Graphs show mean ± SD with individual data points (n = 7); ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, multiple unpaired t tests. n.s., not significant. Data are representative of 2–3 independent experiments.
LPS stimulation in control mice led to classical signs of DC maturation, such as increased surface expression of co-stimulatory ligands (CD80 and CD86), co-stimulatory receptors (CD40), and antigen-presentation molecules (MHC class II) on both cDC1s and cDC2s (Figures 5B–5E). However, in mice previously treated with Fc.Mut24, the LPS-mediated upregulation of CD80 and CD86 on both cDC1s and cDC2s was almost completely blocked (Figures 5B and 5C), whereas there was no effect on upregulation of CD40 (Figure 5D). LPS-mediated upregulation of MHC class II surface expression was also partially reduced in both cDC1s and cDC2s in Fc.Mut24-treated mice (Figure 5E). In the absence of LPS, the effects of Fc.Mut24 treatment were less dramatic but included increased expression of CD40 by cDC1s (Figure 5D), decreased expression of CD80 and CD86 by cDC1s (Figures 5B and 5C), and decreased expression of CD80 by cDC2s (Figure 5B).
To further assess changes in cDCs from Fc.Mut24-treated mice, we performed bulk RNA-seq on sorted cDC1s and cDC2s from each of the four groups of mice outlined in Figure 5A cDC1s expressed canonical genes such as Xcr1, Clec9a, Itgae (CD103), and Irf8, whereas cDC2s expressed Irf4, Cd4, and Sirpa (Figure S6A). Principal component analysis (PCA) revealed that unstimulated (Ctrl) samples separated from LPS-stimulated samples across the PC1 axis for cDC1s and cDC2s, indicating a strong transcriptional response to LPS in both cell types (Figure S7A). Analysis of all differentially expressed genes between unstimulated and LPS-stimulated samples demonstrated that Fc.Mut24 had minimal impact on LPS-induced changes in gene expression in cDC1s and essentially no impact in cDC2s (Figure S7B). For cDC1s in Fc.Mut24-treated mice, a subset of 634 genes did have blunted LPS-mediated upregulation (highlighted in the red rectangle, Figure S7B). Gene set enrichment analysis showed that the hallmark gene set TNFA_Signaling_via_NFKB was enriched in LPS-stimulated cDC1s from control mice compared to Fc.Mut24-treated mice (Figure S7C). Consistent with this, cDC1s from Fc.Mut24-treated mice had slightly reduced upregulation of nuclear factor (NF)-κB-regulated genes associated with cDC maturation such as Cd80, Il15, Il15ra, Ccr7, and Ccl5 (Figure S7D).50,51,52 Although Cd80 expression was reduced in LPS-stimulated cDC1s from Fc.Mut24-treated mice, there were no changes in Cd86 (Figure S6E). Additionally, genes encoding MHC class II (H2-Ab1 and H2-Aa) were increased in Fc.Mut24-treated mice (Figure S7E), in contrast to what was observed for MHC class II protein expression (Figure 5E). Together, these data show that Fc.Mut24 treatment has a minor effect on the transcriptional response of cDC1s but not cDC2s to a strong inflammatory stimulus, and suggest that regulation of CD80, CD86, and MHC class II expression by TR cells primarily occurs post-transcriptionally.
Fc.Mut24-mediated suppression of co-stimulatory ligand protein expression in cDCs is CTLA-4 dependent
Whereas CTLA-4 expression by TR cells can downregulate CD80 and CD86 on APCs in various settings in vitro,18,19,53,54 fewer in vivo studies have shown that this is a CTLA-4-dependent process.20,55 In addition to the CTLA-4 transendocytosis model, an alternative model of trogocytosis has been proposed, in which CD80 and CD86 plus other membrane proteins such as CD40 and MHC class II are concomitantly transferred from APCs to the TR cell surface.56 We therefore wanted to understand to what extent the downregulation of CD80, CD86, and MHC class II on cDCs we observed after Fc.Mut24 treatment was dependent on CTLA-4 expression. For this, we used the same LPS stimulation model with Fc.Mut24 treatment and added the blocking 4F10 anti-CTLA4 antibody (Figure 5F), which does not cause FcR-dependent depletion of TR cells as seen with other anti-CTLA-4 monoclonal antibodies,57 which we confirmed in our studies (Figure S8A). 4F10 treatment did reduce total CTLA-4 expression on Foxp3+ TR cells but slightly increased TR ICOS expression (Figure S8B), likely due to increased CD28 co-stimulation. Importantly, 4F10 treatment in Fc.Mut24-treated mice partially restored CD80 and CD86 expression by cDC1s and cDC2s, with no effect on MHC class II (Figure 5G). Therefore, our data are consistent with the CTLA-4 transendocytosis model in which cognate CD80 and CD86 ligands, but not other membrane proteins such as MHC class II, are stripped from cDCs in a CTLA-4-dependent manner.
CD80 also promotes T cell function by binding in cis to PD-L1 (PD-1 ligand) and preventing it from binding to the checkpoint receptor PD-1.58 Thus, loss of CD80 from the surface of DCs following Fc.Mut24 treatment could further suppress T cell activation by promoting functional PD-L1 expression. To test this, we treated control or Fc.Mut24 pre-treated mice with LPS as above and examined the binding of splenic cDC1s and cDC2s to a fluorescently labeled PD-1-Fc fusion protein. Indeed, we found that downregulation of CD80 in Fc.Mut24-treated mice was associated with significantly increased PD-1 binding to the surface of both cDC1s and cDC2s, and this was partially blocked when mice were also treated with the 4F10 anti-CTLA-4 antibody (Figure S9). These data indicate that the enhanced function of CTLA-4hi TR cells following Fc.Mut24 treatment modulates DC activity through both direct depletion of co-stimulatory ligands and consequent enhancement of PD-1 checkpoint signaling.
Fc.Mut24-mediated suppression of MHC class II protein expression in cDC1s is IL-10 dependent
In addition to CTLA-4 expression, IL-10 production by TR cells may also regulate cDC function in Fc.Mut24-treated mice. We found that IL-10 production was mostly restricted to CD44hiCTLA-4+ TR cells in both PBS controls and Fc.Mut24-treated mice (Figure 6A). Although the percentage of IL-10+ TR cells did not change after Fc.Mut24 treatment, the absolute number of IL-10+ TR cells significantly increased, as well as the amount of IL-10 produced on a per-cell basis (Figure 6B). There was also a slight but significant increase in the percentage of IL-10+ cells among the Foxp3− Tconv cell population, but this was not reflected by an increase in cell number (Figure 6B).
Figure 6.

Fc.Mut24 promotes IL-10-dependent downregulation of MHC class II during dendritic cell maturation
(A) Representative flow-cytometry plots of IL-10 and CTLA-4 expression by TR are shown in PBS- and Fc.Mut24-treated mice.
(B) Graphs show mean ± SD with individual data points for IL-10 percentage, IL-10+ cell number, and mean fluorescence intensity (MFI) of IL-10 staining in Tconv and TR cells (n = 8–9); ∗∗p ≤ 0.01, ∗∗∗∗p ≤ 0.0001, multiple unpaired t tests. The MFI shown is gated on IL-10+ TR. Data are representative of two independent experiments.
(C) Experimental design schematic for IL-10 receptor β knockout (Il10rb−/−) study.
(D and E) Expression of indicated DC maturation markers between wild-type and Il10rb−/− cDC1s (D) and cDC2s (E). Graphs show mean ± SD with individual data points (n = 3); ∗p ≤ 0.05, multiple paired t tests.
To analyze the contribution of IL-10 receptor signaling to the regulation of CD80, CD86, and MHC class II in cDCs, we generated mixed bone marrow (BM) chimeric mice in which lethally irradiated CD45.1+ hosts were reconstituted with 90% wild-type CD45.1+ BM and 10% BM from CD45.2+ IL-10 receptor subunit β knockout (Il10rb−/−) mice (Figure 6C). We used a low frequency of Il10rb−/− cells to prevent recipients from developing chronic colitis or sensitivity to LPS-induced shock that occurs in global ll10rb−/− knockout mice.59,60 This allowed us to directly compare IL-10-receptor-sufficient and -deficient cDCs within the same animals following LPS stimulation in the absence or presence of Fc.Mut24 treatment. We found that MHC class II expression was partially restored on Il10rb−/− compared to wild-type cDC1s from Fc.Mut24-treated mice, without any changes in CD80 or CD86 (Figure 6D). In PBS-treated controls, lack of IL-10 signaling did increase CD80 and CD86 levels by cDC1s, demonstrating that their expression of co-stimulatory ligands in the steady state is regulated by mechanisms other than CTLA-4-dependent transendocytosis (Figure 6D). In contrast to cDC1s, we did not detect any significant changes in MHC class II, CD80, or CD86 expression by Il10rb−/− versus wild-type cDC2s (Figure 6E). IL-10 regulates MHC class II post-transcriptionally in DCs by inducing expression of the E3 ubiquitin ligase March-I that targets MHC class II molecules for degradation.61,62 Consistent with a potential role for March-I in decreasing MHC class II expression in cDC1s, we found that although not statistically significant (false discovery rate = 0.065), there was a trend toward increased March1 expression in LPS-stimulated cDC1s from mice treated with Fc.Mut24 relative to PBS controls, with minimal difference in cDC2s (Figure S10A). Thus, during Fc.Mut24 treatment, IL-10 receptor signaling in cDC1s plays a role in downregulating MHC class II surface expression, likely through a March-1-dependent mechanism.
Co-stimulatory ligand and MHC class II protein expression in cDCs is suppressed by Fc.Mut24 treatment in autoimmune diabetes
Fc.Mut24 treatment in pre-diabetic NOD mice prevents diabetes onset and reduces the severity of islet infiltration.9 However, the mechanisms responsible for durable disease protection have not been explored, and whether distinct types of APCs are suppressed by TR cells during an autoimmune response is undetermined. In NOD mice, pancreatic islet-infiltrating cDC1s and islet-resident macrophages play essential and non-redundant roles in the development of autoimmune diabetes.63,64 In an early event in the inflammatory cascade, migratory cDC1s (XCR1+CD103+) prime islet-reactive CD8+ and CD4+ T cells in the pancreatic lymph node (pancLN), and cDC1s also present autoantigens to T cells in the islets during disease progression,63 whereas resident macrophages (F4/80+) promote the early entrance of CD4+ T cells and cDCs into the islets.64
Treatment of pre-diabetic NOD mice with Fc.Mut24 significantly increased the percentage and number of Foxp3+ TR cells in the spleen, pancLN, and pancreas, with almost all cells being Ki-67+ (Figure S11A), with further upregulation of CD25, Foxp3, CTLA-4, and ICOS, and downregulation of PD-1 by TR cells in all tissue sites (Figure S11B). To establish how TR expansion and activation affect disease-relevant APCs, we analyzed XCR1+CD103+ migratory cDC1s and Sirpα+ cDC2s in the pancLN and pancreas, as well as pancreatic F4/80+ macrophages in PBS controls or Fc.Mut24-treated mice (Figures 7A and S12A). There was an expansion of XCR1+ cDC1 number in the spleen, pancLN, and pancreas after Fc.Mut24 treatment and a slight increase in splenic Sirpα+ cDC2 number, but no changes in pancreatic F4/80+ macrophages (Figure S12B). In agreement with reports showing a pro-inflammatory phenotype,63,64,65 pancreatic F4/80+ macrophages had high expression of CD80, CD86, CD40, and MHC class II, and treatment with Fc.Mut24 did not alter the expression of any of these markers (Figure 7A). In contrast, CD80 expression was significantly reduced in migratory cDC1s from the pancLN and pancreas after treatment (Figure 7A). CD86 expression was also lower in pancreatic cDC1s from Fc.Mut24-treated mice, in addition to lower expression of CD80 and MHC class II in pancreatic cDC2s (Figure 7A). Across all cell types and tissues examined, CD40 was not downregulated by Fc.Mut24 treatment (Figure 7A). Together, these data demonstrate that TR cells in lymphoid tissues and at the site of tissue inflammation are activated with Fc.Mut24, leading to tissue- and cell-type-dependent effects on co-stimulatory ligand expression by disease-promoting cDCs.
Figure 7.

Fc.Mut24 limits dendritic cell maturation and induces anergic T cells during the progression of autoimmune diabetes
(A) Expression of indicated maturation markers on different subsets of CD11c+ MHC II+ antigen-presenting cells isolated from the spleen, pancreatic lymph node (pancLN), and pancreas of 8- to 10-week-old female non-obese diabetic (NOD) mice treated with PBS or Fc.Mut24 4 days previously. See Figure S12A for the gating strategy.
(B) Percentage and number of CD73hi FR4−, CD73hi FR4intermediate, and CD73hi FR4hi conventional T (Tconv) cells isolated from the pancLN and expression of indicated markers on CD73hi FR4− cells. Gates were set on live, CD45+, CD4+, CD44hi, Foxp3− cells.
(C) Percentage and number of CD73hi FR4− Tconv cells isolated from the pancLN and expression of indicated markers. Gates were set on live, CD45+, CD8+, CD44hi, Foxp3− cells.
All graphs show mean ± SD with individual data points (n = 6–9); ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, two-tailed unpaired t tests or multiple unpaired t tests. Data are representative of 2–3 independent experiments.
Induction of anergic T cells by Fc.Mut24 treatment in autoimmune diabetes
Since the upregulation of CTLA-4 by Foxp3+ TR cells in the pancLN of Fc.Mut24-treated NOD mice correlated with decreased CD80 expression by migratory cDC1s, we wanted to determine the impact on anergy in Tconv cells at this site. We first analyzed CD4+ CD44hi Foxp3− cells for co-expression of CD73 and FR4, which defines an anergic population that has the ability to differentiate into Foxp3+ TR cells.66 Differentiation of anergic CD4+ T cells into suppressive Foxp3− Tr1 cells by tolerogenic DCs has also been reported and is associated with the upregulation of CD73, CTLA-4, and PD-1.67,68 We found that the percentage and number of anergic CD73hi FR4hi cells as well as CD73hi FR4intermediate and CD73hi FR4− cells in the pancLN significantly increased after Fc.Mut24 treatment (Figure 7B). The CD73hi FR4− population also had increased expression of CTLA-4 and PD-1 in Fc.Mut24-treated mice (Figure 7B), consistent with what was previously reported for induced Foxp3− Tr1 cells. When analyzing CD8+ CD44hi cells, we observed a similar expansion of CD73hi FR4− cells after Fc.Mut24 treatment in addition to the upregulation of CTLA-4 and PD-1 by this population (Figure 7C). Analysis of CD4+ CD44hi Foxp3− and CD8+ CD44hi cells in the pancLN of NOD mice at different times after IL-2 mutein treatment showed that elevated CD73 expression was transient and largely mirrored the expansion of Foxp3+ TR cells (Figure S13). These findings indicate that Fc.Mut24-expanded Foxp3+ TR cells promote anergic phenotypes in autoreactive Tconv cells, with the potential for generating suppressive CD4+ and CD8+ T cells lacking Foxp3 expression that may further amplify immune regulation as an example of infectious tolerance to promote durable disease protection.69
Discussion
Despite the current testing of CD25-biased Fc.IL-2 muteins with enhanced Foxp3+ TR cell selectivity in clinical trials,10 the molecular mechanisms by which these therapeutics induce immune tolerance and prevent disease progression in models of autoimmune disease have not been well characterized. In addition, although the role of IL-2 in maintaining the homeostasis of Foxp3+ TR cells is well documented23,70 and there is an indispensable role of the IL-2R in TR suppressor activity,21 there is still a lack of complete understanding of how IL-2R signaling influences TR cell differentiation and function.
We used a murine Fc.IL-2 mutein (Fc.Mut24) that we previously developed for pre-clinical studies as a tool to understand how treatment influences Foxp3+ TR-suppressive function in vivo. Fc.Mut24 treatment induced a large expansion of ceTR cells with high expression of the activation marker CD44 and the lymphoid trafficking molecule CD62L, increased the expression of immunoregulatory molecules such as CTLA-4 and IL-10, and decreased the expression of molecules that antagonize TR function, such as PD-1. Similar to our findings, low-dose IL-2 therapy promotes the upregulation of CD44 and downregulation of PD-1 on antigen-specific CD8+ T cells during chronic viral infection and is associated with enhanced effector function.71 Although the origin of the ceTR cell population after Fc.Mut24 treatment will require further investigation, these cells may derive from cTR cells that upregulate CD44 or eTR cells that upregulate CD62L. Neither Cd44 nor Sell (the gene encoding CD62L) appear to be direct targets of Stat5 in TR cells, yet both are modulated by TCR signaling and their altered expression in Fc.Mut24-treated mice could reflect prolonged IL-2R and TCR synergy due to the enhanced half-life and signaling of Fc.Mut24. Consistent with this hypothesis, we found that Fc.Mut24 treatment induced a distinct transcriptional profile indicative of both IL-2R and TCR signaling, and a recent report showed that TR cell expansion with Fc.IL-2 muteins was blunted by the blockade of MHC class II.72 Whether unique phenotypes are observed in human Foxp3+ TR cells after Fc.IL-2 mutein treatment will be interesting to examine, given the heterogeneity in human TR cell populations that we and others have described.73,74
Our data demonstrate that a major mechanism by which Fc.Mut24 treatment alters TR function is through increasing CTLA-4 expression and cycling from intracellular compartments to the cell surface in TR cells but not Foxp3− Tconv cells. Previous work has pointed to TCR signaling as the principal regulator of CTLA-4 function, as T cell activation via anti-CD3 increases CTLA-4 cycling in both TR and Tconv cells, albeit with different kinetics between cell types.20,43 However, these experiments were performed using total CD4+ T cells rather than purified populations, and it cannot be ruled out that IL-2 produced by Tconv cells upon activation synergized with TCR signaling to enhance CTLA-4 cycling in TR cells. A connection between IL-2-induced immune regulation and the CTLA-4 pathway was previously suggested, since IL-2 induces the surface and intracellular expression of CTLA-4 in murine and human T cells in vitro,75,76 and acute IL-2 blockade in mice decreases CTLA-4 expression by TR cells.77 Transgenic expression of CTLA-4 also rescues the lymphoproliferative disease that occurs in IL-2-deficient mice,78 suggesting that impaired CTLA-4 function in the absence of IL-2 contributes to immune pathology. In support of our CTLA-4 cycling results, Fc.Mut24 treatment significantly increased CTLA-4-dependent transendocytosis of CD86 ex vivo and CD80 in vivo by TR but not Tconv cells. We were particularly interested to find that cTR cells, which usually have a naive phenotype and relatively low CTLA-4 expression, gained the ability to capture CD86 in Fc.Mut24-treated mice. As cTR cells are generally found at high density within T cell zones of secondary lymphoid tissues where Tconv-DC interactions and T cell priming occur, this increases their potential to contribute to immune regulation after Fc.Mut24 treatment.
To determine whether TR expansion and activation after Fc.Mut24 treatment resulted in the suppression of cDC function, we established a model of LPS-mediated maturation. In these experiments and others in NOD mice, Fc.Mut24 increased the abundance of cDC1s in multiple tissues. While we did not further explore this mechanistically, IL-2 can induce cDC expansion by increasing T cell production of cytokines such as Fms-like tyrosine kinase 3 ligand (Flt3l).79 Importantly, Fc.Mut24 significantly enhanced CTLA-4-dependent transendocytosis of CD80 and CD86 during splenic cDC1 and cDC2 maturation, in addition to downregulation of surface MHC class II, which was not CTLA-4 dependent but due to IL-10 signaling in cDC1s. Loss of CD80 also increased binding of PD-1 to both cDC1s and cDC2s, likely due to the release of PD-L1 from inhibitory cis interaction with CD80, and thus interaction with TR cells in Fc.Mut24-treated mice significantly alters the balance of co-stimulatory and co-inhibitory ligands presented by DCs. By contrast, Fc.Mut24 treatment slightly blunted the overall transcriptional response of cDC1s but had no effect on cDC2s following LPS stimulation. While the reasons for the differential transcriptional regulation of splenic cDC1s versus cDC2 subsets are not apparent, at steady state cDC1s are found within splenic T cell zones, whereas cDC2s are located at the marginal zone bridging channel between the red pulp and white pulp,80 possibly allowing for more sustained interactions between TR cells and cDC1s. In human monocytes, IL-10 prevents IκB degradation as well as nuclear translocation of NF-κB and binding to target genes.81 Therefore, IL-10-mediated downregulation of the NF-κB pathway may reduce CD80 transcription in cDC1s and blunt their transcriptional response to LPS. We found no evidence of CD40 downregulation during cDC maturation, similar to other studies on TR cells activated via TCR stimulation, which downregulate CD80 and CD86, but not CD40, on splenic DCs.54 These results support a model in which increased CTLA-4-dependent transendocytosis and IL-10 production are important mechanisms used by TR cells to modulate cDC function following Fc.Mut24 treatment and establish that in the presence of a strong inflammatory stimulus, TR cells do not completely inhibit cDC maturation.
In cancer models, TR cells suppress cDC1s in both the tumor-draining lymph node and the tumor,82,83 but it is unclear whether similar findings apply to target organs in autoimmune disease. Using 8- to 10 week-old female NOD mice, an age at which there is already established autoimmunity and infiltration of the pancreatic islets but no overt diabetes,84 we found that cDC2s and migratory cDC1s from the pancreas of Fc.Mut24-treated mice had reduced CD80 expression, and there was reduced CD86 expression by cDC1s. These migratory cDC1s, which are required for the initiation of disease development in NOD mice,63 also had lower CD80 expression in the pancreatic lymph node. Consistent with reduced expression of the CD28 ligands and MHC class II by DCs, the frequency of anergic phenotype CD73hi FR4hi CD4+ T cells in the pancreatic lymph node and CD73hi FR4− T cells that were either CD4+ or CD8+ significantly increased after Fc.Mut24 treatment. In CD8+ T cells, a lack of CD28 co-stimulation during activation leads to CD73 upregulation and the generation of regulatory cells that suppress effector T cell responses through CD73-mediated adenosine production.85 Similarly, CD4+ T cells activated without CD28 co-stimulation have elevated levels of the Nt5e gene encoding CD73,86 suggesting that CD73 upregulation is a common mechanism of T cell tolerance in the absence of sufficient co-stimulation. Adenosine produced by CD73 can further induce IL-10 expression in immune cells such as macrophages and DCs,87,88 and thus a suppressive CTLA-4 → CD73 → IL-10 axis likely contributes to immune regulation and induction of anergic phenotype cells after Fc.Mut24 IL-2 mutein treatment.
High levels of IL-2R signaling promote the terminal differentiation of short-lived but highly suppressive TR cells.89 This is analogous to the ability of strong IL-2R signaling to induce terminally differentiated short-lived effector CD8+ T cells2 and highlights a conserved role for IL-2R signaling driving both TR and Tconv effector function. While the mechanisms by which strong IL-2R signaling potentiates TR-suppressive function are not completely understood, Stat5 activation appears to increase TR cell adherence to DCs in vitro, reducing their expression of CD80 and CD86.21 Exposure of TR cells to IL-2 also increases TR-DC interactions in vivo, leading to suppression of DC function and T cell priming in a contact-dependent but MHC class II-independent manner.90 We postulate that extensive IL-2R signaling in TR cells increases CTLA-4 expression and TR-DC adhesion, allowing TR cells to strip DCs of CD80 and CD86 in an MHC class II-independent manner, which is consistent with the established models of TR cell-mediated bystander suppression.
CD25-biased Fc.IL-2 muteins have unique properties, such as sustained IL-2 signaling, that promote the expansion and differentiation of highly activated Foxp3+ TR cells. We show that these Fc.IL-2 mutein-expanded TR cells have a distinct transcriptional and functional profile and can regulate DCs using multiple molecular mechanisms resulting in reduced co-stimulatory ligand availability that is accompanied by induction of T cell anergy and transient expression of the immunoinhibitory protein CD73 by Tconv cells. The relationship between sustained IL-2R signaling, TR effector function, and anergy induction is highly desirable under conditions of persistent autoreactive T cell activation for establishing lasting immune tolerance. Indeed, the ability of Fc.IL-2 muteins to enhance TR cell-mediated suppression of DCs during an autoimmune response distinguishes this class of therapeutics from other biologics that block pathogenic effector T cell pathways, further warranting their use in the prevention and treatment of chronic inflammatory diseases.
Limitations of the study
All experiments in this study were performed in mice with a corresponding murine IL-2 mutein, and their applicability to human TR cells with different human IL-2 muteins has not been confirmed. Comparing in vivo responses of TR cells to Fc.WT and Fc.Mut24 IL-2 is complicated by the dramatically different half-lives of these proteins as demonstrated in our previous study.9 TR cells function via a large number of different mechanisms, and although we have shown that CTLA-4 and IL-10 function is increased following treatment with Fc.Mut24 to suppress the activity of various DC populations, induction of full tolerance may also require other suppressive pathways not examined in this study. Induction of Tconv cell anergy in the pancLN of NOD mice is inferred based on the phenotypic markers CD73 and FR4 but is not directly demonstrated.
Resource availability
Lead contact
Requests for resources and reagents can be directed to the lead contact, Daniel J. Campbell, dcampbell@benaroyaresearch.org.
Materials availability
Materials are available by request from Daniel J. Campbell.
Data and code availability
-
•
Bulk and single-cell RNA-seq data have been deposited into the Gene Expression Omnibus under series GEO: GSE249482.
-
•
Code used for analyses can be found at http://doi.org/10.5281/zenodo.13315646.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
We thank Shivani Srivastava (Fred Hutchinson Cancer Center) for Plat-E packaging cells, Francesco Marangoni (UC Irvine) for the signal-amplification protocol used for the staining of surface and cycling CTLA-4, the Cell and Tissue Analysis Group at the Benaroya Research Institute (BRI) for flow cytometry and sorting expertise, and the Genomics Core at BRI for bulk and single-cell RNA-seq. Flow-cytometry and genomics equipment used in this study was provided by generous support from the M.J. Murdock Charitable Trust. This work was supported by National Institutes of Health grants (5R01AI154773, 5R01AI136475, and 1R21AI172140) to D.J.C., an MRC Programme grant to L.S.K.W. (MR/N001435/1), and a postdoctoral fellowship from the Washington Research Foundation to B.L.J.
Author contributions
B.L.J. designed and performed the experiments, analyzed and interpreted the data, prepared the figures, and co-wrote the manuscript. L.S.K.W. and D.M.S. generated the CD80-mCherry mice. C.J.W. and L.S.K.W. designed and performed experiments on in vivo CTLA-4 transendocytosis in Figures 4D and 4E. L.J.Z. and D.J.C. performed experiments in Figures S2 and S13. C.J.W., L.S.K.W., and M.A.G. edited the manuscript. M.L. and H.A.D. analyzed the data and assisted with figure generation. D.J.C. and M.A.G. conceptualized the study, and D.J.C. supervised the study and co-wrote the manuscript.
Declaration of interests
D.J.C. is a member of the scientific advisory board and holds stock options in Sonoma Biotherapeutics.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD4 BV650 (RM4-5) | BioLegend | Cat# 100555; RRID: AB_2562529 |
| Anti-mouse CD4 BV421 (RM4-5) | BioLegend | Cat# 100563; RRID:AB_2563052 |
| Anti-mouse CD45 BUV395 (30-F11) | BD Biosciences | Cat# 564279; RRID:AB_2651134 |
| Anti-mouse TCRβ BUV737 (H57-597) | BD Biosciences | Cat# 612821; RRID:AB_2870145 |
| Anti-mouse CD25 BV785 (PC61) | BioLegend | Cat# 102051; RRID: AB_2564131 |
| Anti-mouse Foxp3 PE (FJK-16s) | eBioscience (ThermoFisher) | Cat# 12-5773-82; RRID:AB_465936 |
| Anti-mouse Foxp3 eFluor450 (FJK-16s) | eBioscience (ThermoFisher) | Cat# 48-5773-82; RRID:AB_1518812 |
| Anti-mouse Ki-67 AF488 (11F6) | BioLegend | Cat# 151204; RRID:AB_2566800 |
| Anti-mouse CD44 AF700 (IM7) | BioLegend | Cat# 103026; RRID:AB_493713 |
| Anti-mouse CD44 BV510 (IM7) | BioLegend | Cat# 103043; RRID:AB_2561391 |
| Anti-mouse CD62L BV711 (MEL-14) | BioLegend | Cat# 104445; RRID:AB_2564215 |
| Anti-mouse ICOS BV510 (C398.4A) | BioLegend | Cat# 313525; RRID:AB_2562642 |
| Anti-mouse PD-1 PE-Cy7 (RMP1-30) | BioLegend | Cat# 109110; RRID:AB_572017 |
| Anti-mouse PD-1 BUV805 (29F.1A12) | BD Biosciences | Cat# 568608 |
| Anti-mouse CTLA-4 PE-Dazzle594 (UC10-4B9) | BioLegend | Cat# 106318; RRID:AB_2564496 |
| Anti-mouse CTLA-4 APC (UC10-4F10-11) | BD Biosciences | Cat# 564331; RRID:AB_2738751 |
| Anti-mouse CTLA-4 PE (UC10-4F10-11) | BD Biosciences | Cat# 553720; RRID:AB_395005 |
| Biotin anti-Allophycocyanin (APC003) | BioLegend | Cat# 408004; RRID:AB_345360 |
| Anti-mouse IL-10 BV421 (JES5-16E3) | BioLegend | Cat# 505022; RRID:AB_2563240 |
| Anti-mouse CD16/32 (93) | BioLegend | Cat# 101320; RRID:AB_1574975 |
| Anti-mouse CD5 AF488 (53–7.3) | BioLegend | Cat# 100612; RRID:AB_493165 |
| Anti-mouse CD19 AF488 (6D5) | BioLegend | Cat# 115521; RRID:AB_389307 |
| Anti-mouse GR-1 AF488 (RB6-8C5) | BioLegend | Cat# 108417; RRID:AB_389309 |
| Anti-mouse NK1.1 AF488 (PK136) | BioLegend | Cat# 108718; RRID:AB_493183 |
| Anti-mouse CD11c PE-Cy7 (N418) | BioLegend | Cat# 117318; RRID:AB_493568 |
| Anti-mouse I-Ab eFluor450 (M5/114.15.12) | eBioscience (ThermoFisher) | Cat# 48-5321-82; RRID:AB_1272204 |
| Anti-mouse I-Ak BV421 (10–3.6) | BD Biosciences | Cat# 743471; RRID:AB_2741527 |
| Anti-mouse CD11b Percp-Cy5.5 (M1/70) | BD Biosciences | Cat# 550993; RRID:AB_394002 |
| Anti-mouse 33D1 APC (33D1) | BioLegend | Cat# 124914; RRID:AB_1227625 |
| Anti-mouse CD8a PE (53–6.7) | BioLegend | Cat# 100708; RRID:AB_312747 |
| Anti-mouse CD8a BV510 (53–6.7) | BioLegend | Cat# 100751; RRID:AB_2561389 |
| Anti-mouse CD8a AF488 (53–6.7) | BioLegend | Cat# 100723; RRID:AB_389304 |
| Anti-mouse CD8a Bv711 (53–6.7) | BioLegend | Cat# 100747; RRID:AB_11219594 |
| Anti-mouse Sirpa BUV805 (P84) | BD Biosciences | Cat# 741997; RRID:AB_2871296 |
| Anti-mouse XCR1 BV510 (ZET) | BioLegend | Cat# 148218; RRID:AB_2565231 |
| Anti-mouse CD103 APC (2E7) | BioLegend | Cat# 121414; RRID:AB_1227502 |
| Anti-mouse F4/80 BV711 (BM8) | BioLegend | Cat# 123147; RRID:AB_2564588 |
| Anti-mouse CD80 BV605 (16-10A1) | BioLegend | Cat# 104729; RRID:AB_11126141 |
| Anti-mouse CD86 BV650 (GL-1) | BioLegend | Cat# 105036; RRID:AB_2686973 |
| Anti-mouse CD40 PE-Dazzle594 (3/23) | BioLegend | Cat# 124630; RRID:AB_2572185 |
| Anti-mouse CD45.2 BUV395 (104) | BD Biosciences | Cat# 564616; RRID:AB_2738867 |
| Anti-mouse CD45.1 AF700 (A20) | BioLegend | Cat# 110723; RRID:AB_493732 |
| Anti-mouse IL-10R PE (1B1.3a) | BioLegend | Cat# 112705; RRID:AB_313518 |
| Anti-mouse CD73 PE-Cy7 (TY/11.8) | BioLegend | Cat# 127224; RRID:AB_2716103 |
| Anti-mouse FR4 PE-Dazzle594 (12A5) | BioLegend | Cat# 125015; RRID:AB_2721703 |
| TotalSeq-C0301 anti-mouse Hashtag 1 | BioLegend | Cat# 155861; RRID:AB_2800693 |
| TotalSeq-C0302 anti-mouse Hashtag 2 | BioLegend | Cat# 155863; RRID:AB_2800694 |
| TotalSeq-C0303 anti-mouse Hashtag 3 | BioLegend | Cat# 155865; RRID:AB_2800695 |
| Anti-mouse CTLA-4 (UC10-4F10-11) | Bio X Cell | Cat# BP0032 |
| Anti-glutathione S-transferase (PIP) | Bio X Cell | Cat# BE0260 |
| Bacterial and virus strains | ||
| XL-1 Blue E.coli | Daniel J. Campbell Lab | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Liberase TM | Roche | Cat# 05401127001 |
| DNase I | Sigma-Aldrich | Cat# D4513 |
| Collagenase Type V | Sigma-Aldrich | Cat# C9263 |
| 5(6)-Carboxyfluorescein diacetate N-succinimidyl ester | Sigma-Aldrich | Cat# 21888 |
| Enzyme-free cell dissociation buffer | Sigma-Aldrich | Cat# C5789 |
| Bafilomycin A1 | Sigma-Aldrich | Cat# SML1661 |
| ACK lysis buffer | ThermoFisher | Cat# A1049201 |
| LPS | Sigma-Aldrich | Cat# L4005 |
| Fixable Viability Dye eFluor780 | ThermoFisher | Cat# 65-0865-18 |
| Streptavidin-APC | BioLegend | Cat# 405207 |
| PD-1 Ig | Bio-Techne | Cat# 1021-PD |
| Critical commercial assays | ||
| CD11c MicroBeads, mouse | Miltenyi Biotec | Cat# 130-125-835 |
| CD4 MicroBeads, mouse | Miltenyi Biotec | Cat# 130-117-043 |
| CD4+ T cell Isolation Kit, mouse | Miltenyi Biotec | Cat# 130-104-454 |
| Dynabeads mouse T cell activator | ThermoFisher | Cat# 11456D |
| Cell Stimulation Cocktail (plus protein transport inhibitors) | ThermoFisher | Cat# 00-4975-03 |
| eBioscience Foxp3/Transcription Factor Staining Buffer Set | ThermoFisher | Cat# 00-5523-00 |
| SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing | Takara Bio | Cat# 634894 |
| NexteraXT DNA Library Preparation Kit | Illumina | Cat# FC-131-1024 |
| Library Construction Kit, 16 rxns | 10x Genomics | Cat# 1000190 |
| Chromium Single Cell Mouse TCR Amplification Kit, 16 rxns | 10x Genomics | Cat# 1000254 |
| Chromium Next GEM Single Cell 5′ Kit v2, 4 rxns | 10x Genomics | Cat# 1000265 |
| Chromium Next GEM Chip K Single Cell Kit, 16 rxns | 10x Genomics | Cat# 1000287 |
| 5′ Feature Barcode Kit, 16 rxns | 10x Genomics | Cat# 1000256 |
| Dual Index Kit TN Set A, 96 rxn | 10x Genomics | Cat# 1000250 |
| Dual Index Kit TT Set A 96 rxns | 10x Genomics | Cat# 1000215 |
| NextSeq 2000 P3 Reagents (100 Cycles) | Illumina | Cat# 20040559 |
| R-PE Lightning-Link Conjugation Kit | AbCam | Cat# AB102918 |
| Deposited data | ||
| Bulk and single-cell RNA-sequencing data | – | Gene Expression Omnibus (GEO) series GEO: GSE249482 |
| Code used for bulk and single-cell RNA-sequencing analyses | http://doi.org/10.5281/zenodo.13315646 | |
| Experimental models: Cell lines | ||
| Platinum-E | Shivani Srivastava Lab, Fred Hutchinson Cancer Center | N/A |
| NIH/3T3 | Daniel J. Campbell Lab | N/A |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jackson Laboratory | Cat# 000664 |
| B6.SJL-PtprcaPepcb/BoyJ | Jackson Laboratory | Cat# 002014 |
| B6.129S2-Il10rbtm1Agt/J | Jackson Laboratory | Cat# 005027 |
| C57BL/6-Foxp3tm1Flv/J | Jackson Laboratory | Cat# 008374 |
| NOD/ShiLtJ | Jackson Laboratory | Cat# 001976 |
| BALB/cJ | Jackson Laboratory | Cat# 000651 |
| CD80-mCherry.Rag2−/− | Lucy S.K. Walker and David M. Sansom Labs, University College London | N/A |
| Recombinant DNA | ||
| pCD86-EGFP | Addgene | Cat# 133858 |
| modified MSCV2.2 | Daniel J. Campbell Lab | N/A |
| Software and algorithms | ||
| FlowJo v10.8 and v10.9. Software | BD Life Sciences | N/A |
| R 4.0.3 | CRAN | N/A |
| limma_3.49.4 | https://bioconductor.org/packages/release/bioc/html/limma.html | N/A |
| ROAST | https://bioconductor.org/packages/release/bioc/html/limma.html | N/A |
| STAR v.2.4.2a | https://github.com/alexdobin/STAR | N/A |
| HTSeq 2.0.2 | https://pypi.org/project/HTSeq/ | N/A |
| Picard tools 1.134 | https://github.com/broadinstitute/picard | N/A |
| Seurat_v3 | https://satijalab.org/seurat/ | N/A |
| CellRanger | https://www.10xgenomics.com/support/software/cell-ranger/getting-started/cr-what-is-cell-ranger# | N/A |
| ComplexHeatmap | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html | N/A |
| EdgeR | https://bioconductor.org/packages/release/bioc/html/edgeR.html | N/A |
Experimental model and study participant details
Mice
C57BL/6 (B6), C57BL/6.CD45.1+ (JAXBoy, B6.CD45.1+), C57BL/6.CRFB4−/− (B6.Il10rb−/−) and NOD/ShiLtJ (NOD) mice were purchased from The Jackson Laboratory. C57BL/6.Foxp3-mRFP (B6.Foxp3-mRFP) mice were originally purchased from The Jackson Laboratory and bred in-house. The B6.Foxp3-mRFP mice have a monomeric red fluorescent protein (mRFP) knocked in downstream of Foxp3. NOD mice were confirmed to be have normal blood glucose at the time of analysis. The B6, B6.CD45.1, B6.Il10rb−/−, B6.Foxp3-mRFP, and NOD mice were maintained at the Benaroya Research Institute (BRI, Seattle, WA). Experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of BRI. Mice used in experiments at BRI were between 8 and 12 weeks of age and both male and female mice were used except for NOD mice that were exclusively female.
CD80-mCherry.Rag2−/− mice were generated and bred in-house at University College London (UCL, London, UK). CD80-mCherry was expressed under the control of the endogenous CD80 promoter, and mice were generated on a BALB/c background (C.J.W., D.M.S., and L.S.K.W., unpublished data). Wild-type BALB/c mice were originally purchased from The Jackson Laboratory and bred in-house at UCL. For experiments at UCL, mice were housed in individually ventilated cages with environmental enrichment (e.g., cardboard tunnels, paper houses, chewing blocks, and aspen wood wool nesting material) in a temperature- and humidity-controlled facility with a 14-h light/10-h dark cycle and ad libitum feeding. Experimental animals were located in the middle rows of the IVC rack to minimize the impact of differences in light exposure. All injections were carried out in the absence of anesthesia and analgesia, typically in the morning, and mice were returned to the home cage immediately following the procedure. The first injection for in vivo CTLA-4 transendocytosis experiments was typically between 3 p.m. and 6 p.m. The welfare of experimental animals was monitored regularly (typically immediately post-procedure, then at least every 2–3 days) and no procedure-related adverse events were noted. For the majority of experiments, animals were randomly assigned into treatment groups after being matched for age and no blinding was used. No data points were excluded. The number of replicates is provided in the figure legend. Animal work at UCL was performed in accordance with the relevant Home Office regulations following institutional ethical approval (University College London Animal Welfare Ethical Review Body). Mice used at UCL were both male and female and between 12 and 14 weeks of age.
Method details
Mouse treatments
The development of the murine Fc.IL-2 mutein (Fc.Mut24) and Fc.WT IL-2 was previously described.9 These molecules were produced and purified by Olympic Protein Technologies (Seattle, WA). Purified Fc.Mut24 and Fc.WT contained less than 15 endotoxin units (EU)/mL. For all experiments, 10 μg of Fc.WT or Fc.Mut24 was administered via intraperitoneal injection. LPS (Escherichia coli O55:B5) was administered to mice (2.5μg/mouse) by intraperitoneal injection. For blocking CTLA-4 in vivo, 750 μg of anti-CTLA-4 antibody clone UC10-4F10-11 (Bio X Cell) or hamster IgG isotype control antibody (clone PIP, Bio X Cell) was administered via intravenous injection on Day 2 after PBS or Fc.Mut24 treatment.
Single-cell RNA-seq of Foxp3+ TR cells
CD4+ T cells from the spleen of Foxp3-mRFP mice were enriched using a CD4 negative selection kit (Miltenyi Biotec) according to the manufacturer’s protocol. Enriched CD4+ T cells were stained with viability dye and labeled with anti-mouse CD16/32 (Biolegend). Samples were stained with TotalSeq-C anti-mouse hashtag antibodies (Biolegend), and 1 x 105 CD4+ Foxp3+ TR per sample were sorted (FACS Aria II, Becton Dickinson) into RPMI-10. Samples were pooled in groups of 3 for a total of four pooled samples (n = 12). Lineage antibodies to exclude populations during sorting included those targeting CD8 (cytotoxic T cells), CD19 (B cells), GR-1 (neutrophils), and NK1.1 (NK cells). A single cell suspension from each pooled sample was loaded in a single channel of the 10x Chromium Controller (10X Genomics). Sequencing libraries were generated using the NextGEM Single Cell 5′ Kit v2 kit. Gene expression, mouse TCR, and feature barcoding libraries were pooled and treated with Illumina Free Adapter Blocking Reagent (Illumina). Sequencing of pooled libraries was carried out on a NextSeq 2000 sequencer (Illumina), using two NextSeq P3 flowcells (Illumina) with the aim of capturing 1.5 x 104 cells per pooled sample and a target sequencing depth of 30,000/reads per cell.
Demultiplexing and alignment of single-cell RNA-seq data
Cell Ranger (version 6.1.1, 10x Genomics) mkfastq was used to demultiplex and produce raw fastq files for downstream analyses. Cell Ranger multi was used to align per-pool reads against the reference mouse transcriptome (mm10), in addition to barcoded hash tags, Cell Ranger vdj was called to assign TCR reads to cells. All three assays were aggregated across the four pools using Cell-Ranger aggr to produce raw count matrices for downstream analyses. Cells containing fewer than 500 genes were removed from this matrix, as well as any cell exceeding 4,500 features, with 12.5% and 5% of genes assigned to mitochondria and hemeglobin genes, respectively. Expression data of all assays were normalized (RNA: log-norm) and scaled to produce final matrices for downstream analyses.
Analysis of single-cell RNA-seq data
Principal component analysis (PCA) of RNA expression was performed in Seurat 2.91 The top 30 PCs from RNA were reduced into UMAP space for visualization, followed by Louvain clustering in Seurat for cluster assignment. Top genes for all clusters and cell identities reported in this study were calculated by Wilcoxon rank sum tests within Seurat to identify all genes with p < .05. The average expression of these genes across clusters was determined by averageExpression in Seurat, followed by scaling to convert to z-scores. Heatmaps of these z-scores were produced via the package ComplexHeatmap. Gene module scores for modules of interest were assigned via addModuleScore within Seurat, and cells were manually gated based on module scores into high- and low-scoring cells in order to produce violin plots. All plots were produced using ggplot2. All statistical analyses were performed in R version 4.2.1.
Tissue preparation for flow cytometry and cDC sorting
To isolate T cells from B6 mice, spleen samples were mashed through 70-μm strainers into RPMI 1640 plus 10% FBS (RPMI-10). To isolate CD11c+ cells from B6 mice, minced whole spleens were digested in basal RPMI 1640 supplemented with 25 μg/ml Liberase TM (Roche) and 25 μg/ml DNase I (Sigma-Aldrich) for 20 min under agitation at 37°C. Erythrocytes were lysed in ACK lysis buffer, and the remaining cells were washed with RPMI-10. CD11c+ cells were enriched using CD11c MicroBeads (Miltenyi Biotec) according to the manufacturer’s protocol. To isolate T cells and CD11c+ cells from non-obese diabetic (NOD) mice, minced whole spleens and pancreatic lymph nodes (pancLN) were digested in basal RPMI 1640 supplemented with 25 μg/ml Liberase TM (Roche) and 25 μg/ml DNase I for 20 min under agitation at 37°C. Erythrocytes were lysed in ACK lysis buffer for spleen samples. Minced pancreas samples from NOD mice were digested in RPMI-10 supplemented with 5 mg/mL Collagenase Type V (Sigma) and 10 μg/ml DNase I followed by incubation in enzyme-free cell dissociation buffer (Sigma) for 5–10 min at room temperature. CD11c+ cells were enriched from the spleen and pancreas, but not pancLN samples of NOD mice, using CD11c MicroBeads (Miltenyi Biotec) according to the manufacturer’s protocol.
Flow cytometry
For flow cytometric analysis, cells were stained with anti-mouse CD16/32 (Biolegend) for 10 min at 4°C to block nonspecific binding to Fc receptors, followed by cell surface staining in FACS buffer (PBS-2% BCS) for 20–30 min at 4°C. For staining CD11c+ samples, lineage antibodies included those targeting CD5 (T cells), CD19 (B cells), GR-1 (neutrophils), and NK1.1 (NK cells). To assess PD-1 binding, mouse PD-1-Fc fusion protein (Bio-Techne) was PE-labeled using the R-PE Lightning-Link kit (AbCAM) according to the manufacturer’s instructions and used to in the surface staining step to label CD11c+ enriched cells in standard FACS buffer. For intracellular antigens, cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience), followed by staining in permeabilization buffer for 20 min at room temperature. Before fixation, dead cells were labeled using Fixable Viability Dye eFluor780 (ThermoFisher). For measuring IL-10 expression, intracellular staining was performed for 2 h at 4°C after stimulation for 2 h with PMA/Ionomycin Cell Stimulation Cocktail containing protein transport inhibitors (ThermoFisher). Data were acquired on an Aurora (Cytek Biosciences) or Symphony (BD Biosciences) flow cytometer and analyzed with FlowJo (FlowJo LLC). Counting beads were added to samples to quantify cell numbers.
In vitro suppression assay
CD44loCD62Lhi cTR, CD44hiCD62Lhi ceTR, and CD44hiCD62Llo eTR were sorted from the spleens for B6.Foxp3-mRFP mice treated 3 days previously with 10ug Fc.Mut24. CD4+Foxp3-mRFP− Tconv were sorted from the spleens of untreated B6.Foxp3-mRFP mice, and subsequently labeled with 1μM carboxyfluorescein succinimidyl ester (CFSE, Sigma-Aldrich) at 37°C for 10m. Each of the sorted TR populations was then co-cultured in triplicate with 4x104 CFSE-labeled Tconv cells at the indicated ratios in a round-bottom 96 well plate and stimulated with 1x104 mouse T-activator anti-CD3/anti-CD28 Dynabeads (ThermoFisher) for 3 days. Cells were harvested and expression of CD44 and CFSE dilution by gated Foxp3-mRFP- Tconv cells was analyzed by flow cytometry. % suppression for cell proliferation and CD44 expression was calculated as:
| ((Response in absence of TR - Response with TR)/Response in absence of TR) ∗100. |
Bulk RNA-seq of cDCs
300 cells per population were sorted (FACS Aria II, Becton Dickinson) into lysis buffer, and cDNA was prepared using the SMART-Seq Ultra Low Input RNA Kit for Sequencing (Takara Bio). RNA-seq libraries were constructed using the NexteraXT DNA Library Preparation Kit (Illumina) with half the recommended volumes and reagents. Paired-end sequencing of pooled libraries was run on a NextSeq 2000 (Illumina) with 59-base reads and a target depth of 5 million reads per sample. After the run, base-calling and demultiplexing were performed automatically on BaseSpace (Illumina) to generate FASTQ files. The FASTQs were aligned to the University of California Santa Cruz (UCSC) Human Genome assembly version 19, using STAR v.2.4.2a, and gene counts were generated using htseq-count. QC and metrics analysis was performed using the Picard family of tools (v1.134). To detect differentially expressed genes between cell populations, the RNA-seq analysis functionality of the linear models for microarray data (Limma) R package was used, and the ROAST method within the Limma R package was used to perform gene set enrichment analysis.92,93 Expression counts were normalized using the TMM algorithm.94 A false discovery rate adjustment was applied to correct for multiple testing.
Ex vivo CTLA-4 cycling
Splenocytes from B6 mice were labeled with Allophycocyanin (APC)-conjugated anti-CTLA-4 antibody for 30 min at 4°C to detect surface CTLA-4, for 2 h at 37°C to detect cycling CTLA-4, or after fixation and permeabilization to detect total CTLA-4. The surface CTLA-4 and cycling CTLA-4 samples were washed with FACS buffer, and secondary staining was performed with biotinylated anti-APC antibody for 20 min at 4°C, followed by tertiary staining with Streptavidin-APC for 20 min at 4°C. The signal amplification approach was used to enhance the detection of CTLA-4 antigen, which is poorly expressed on the cell surface.
Ex vivo CTLA-4 transendocytosis
Human CD86 C-terminally tagged with enhanced GFP (pCD86-EGFP, Addgene) was sub-cloned into a modified version of the MSCV2.2 retroviral plasmid in which the IRES-GFP cassette was removed. This plasmid was transfected into Plat-E packaging cells to produce retrovirus that was used to transduce NIH/3T3 fibroblasts. The resultant NIH/3T3 transfectants were sorted (FACS Aria Fusion, Becton Dickinson) for uniform GFP expression. CD4+ T cells from the spleen of B6 mice were enriched using CD4 MicroBeads (Miltenyi Biotec) according to the manufacturer’s protocol. Enriched CD4+ T cells were co-cultured with CD86-GFP expressing NIH/3T3 cells at a 1:1 ratio for 2 h at 37°C. Where indicated, 100 μg/mL of anti-CTLA-4 antibody (clone UC10-4F10-11, Bio X Cell) or 25 nM Bafilomycin A1 (Sigma-Aldrich) was added to the culture. After incubation, NIH/3T3 cells remained adherent while CD4+ T cells were removed, stained, and analyzed by flow cytometry.
In vivo CTLA-4 transendocytosis
CD80-mCherry.Rag2−/− mice (12–14 weeks old, male and female) were injected intravenously on Day 0 with 8x106 CD4+ T cells purified from wild-type BALB/c lymph nodes. 500 μg of anti-CTLA-4 antibody (clone UC10-4F10-11, BioXCell) or hamster IgG isotype control antibody (clone PIP, Bio X Cell) was administered via intraperitoneal injection on days 0, 3, and 6. PBS or Fc.Mut24 was also administered via intraperitoneal injection on Day 3. Splenocytes were analyzed by flow cytometry on day 7.
Construction of mixed bone marrow chimeras
Recipient B6.CD45.1+ mice were irradiated with a split dose of 1200rad (2 × 600rad, separated by 4h) and given 5x106 total bone marrow cells harvested from B6.CD45.1+ (90%) and B6.Il10rb−/− (10%) donors. Mixed bone marrow chimeras were allowed to reconstitute all leukocyte subsets for 9 weeks before use.
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism version 9. When comparing two groups, p values were calculated by two-tailed Student’s t-tests, while multiple t-tests were used for comparing more than two groups. For multiple t-tests, the discovery was determined using the two-stage step-up method of Benjamini, Krieger, and Yekutieli, with Q = 1%. No formal sample size calculations were performed, and data distribution was assumed normal but not formally tested. The sample size, p values, number of replicates, and statistical tests used in each experiment are listed in the figure legends. In animal experiments, the experimental unit is an individual mouse.
Published: November 2, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114938.
Supplemental information
References
- 1.Nelson B.H. IL-2, regulatory T cells, and tolerance. J. Immunol. 2004;172:3983–3988. doi: 10.4049/jimmunol.172.7.3983. [DOI] [PubMed] [Google Scholar]
- 2.Kalia V., Sarkar S. Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2-A Balancing Act. Front. Immunol. 2018;9:2987. doi: 10.3389/fimmu.2018.02987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pepper M., Pagán A.J., Igyártó B.Z., Taylor J.J., Jenkins M.K. Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity. 2011;35:583–595. doi: 10.1016/j.immuni.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klatzmann D., Abbas A.K. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol. 2015;15:283–294. doi: 10.1038/nri3823. [DOI] [PubMed] [Google Scholar]
- 5.Kim H.T., Koreth J., Whangbo J., Nikiforow S., Reynolds C.G., Stowe P., Ho V.T., Cutler C., Antin J.H., Soiffer R.J., Ritz J. Organ-specific response after low-dose interleukin-2 therapy for steroid-refractory chronic graft-versus-host disease. Blood Adv. 2022;6:4392–4402. doi: 10.1182/bloodadvances.2022007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He J., Zhang R., Shao M., Zhao X., Miao M., Chen J., Liu J., Zhang X., Zhang X., Jin Y., et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2020;79:141–149. doi: 10.1136/annrheumdis-2019-215396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Humrich J.Y., Cacoub P., Rosenzwajg M., Pitoiset F., Pham H.P., Guidoux J., Leroux D., Vazquez T., Riemekasten G., Smolen J.S., et al. Low-dose interleukin-2 therapy in active systemic lupus erythematosus (LUPIL-2): a multicentre, double-blind, randomised and placebo-controlled phase II trial. Ann. Rheum. Dis. 2022;81:1685–1694. doi: 10.1136/ard-2022-222501. [DOI] [PubMed] [Google Scholar]
- 8.Peterson L.B., Bell C.J.M., Howlett S.K., Pekalski M.L., Brady K., Hinton H., Sauter D., Todd J.A., Umana P., Ast O., et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J. Autoimmun. 2018;95:1–14. doi: 10.1016/j.jaut.2018.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khoryati L., Pham M.N., Sherve M., Kumari S., Cook K., Pearson J., Bogdani M., Campbell D.J., Gavin M.A. An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci. Immunol. 2020;5 doi: 10.1126/sciimmunol.aba5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raeber M.E., Sahin D., Karakus U., Boyman O. A systematic review of interleukin-2-based immunotherapies in clinical trials for cancer and autoimmune diseases. EBioMedicine. 2023;90 doi: 10.1016/j.ebiom.2023.104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lykhopiy V., Malviya V., Humblet-Baron S., Schlenner S.M. IL-2 immunotherapy for targeting regulatory T cells in autoimmunity. Genes Immun. 2023;24:248–262. doi: 10.1038/s41435-023-00221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jamison B.L., Neef T., Goodspeed A., Bradley B., Baker R.L., Miller S.D., Haskins K. Nanoparticles Containing an Insulin-ChgA Hybrid Peptide Protect from Transfer of Autoimmune Diabetes by Shifting the Balance between Effector T Cells and Regulatory T Cells. J. Immunol. 2019;203:48–57. doi: 10.4049/jimmunol.1900127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz R.H. T cell anergy. Annu. Rev. Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 14.Vanasek T.L., Nandiwada S.L., Jenkins M.K., Mueller D.L. CD25+Foxp3+ regulatory T cells facilitate CD4+ T cell clonal anergy induction during the recovery from lymphopenia. J. Immunol. 2006;176:5880–5889. doi: 10.4049/jimmunol.176.10.5880. [DOI] [PubMed] [Google Scholar]
- 15.Martinez R.J., Zhang N., Thomas S.R., Nandiwada S.L., Jenkins M.K., Binstadt B.A., Mueller D.L. Arthritogenic self-reactive CD4+ T cells acquire an FR4hiCD73hi anergic state in the presence of Foxp3+ regulatory T cells. J. Immunol. 2012;188:170–181. doi: 10.4049/jimmunol.1101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haase C., Jørgensen T.N., Michelsen B.K. Both exogenous and endogenous interleukin-10 affects the maturation of bone-marrow-derived dendritic cells in vitro and strongly influences T-cell priming in vivo. Immunology. 2002;107:489–499. doi: 10.1046/j.1365-2567.2002.01529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avdic S., Cao J.Z., McSharry B.P., Clancy L.E., Brown R., Steain M., Gottlieb D.J., Abendroth A., Slobedman B. Human cytomegalovirus interleukin-10 polarizes monocytes toward a deactivated M2c phenotype to repress host immune responses. J. Virol. 2013;87:10273–10282. doi: 10.1128/JVI.00912-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wing K., Onishi Y., Prieto-Martin P., Yamaguchi T., Miyara M., Fehervari Z., Nomura T., Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 19.Qureshi O.S., Zheng Y., Nakamura K., Attridge K., Manzotti C., Schmidt E.M., Baker J., Jeffery L.E., Kaur S., Briggs Z., et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ovcinnikovs V., Ross E.M., Petersone L., Edner N.M., Heuts F., Ntavli E., Kogimtzis A., Kennedy A., Wang C.J., Bennett C.L., et al. CTLA-4-mediated transendocytosis of costimulatory molecules primarily targets migratory dendritic cells. Sci. Immunol. 2019;4 doi: 10.1126/sciimmunol.aaw0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chinen T., Kannan A.K., Levine A.G., Fan X., Klein U., Zheng Y., Gasteiger G., Feng Y., Fontenot J.D., Rudensky A.Y. An essential role for the IL-2 receptor in T. Nat. Immunol. 2016;17:1322–1333. doi: 10.1038/ni.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levine A.G., Arvey A., Jin W., Rudensky A.Y. Continuous requirement for the TCR in regulatory T cell function. Nat. Immunol. 2014;15:1070–1078. doi: 10.1038/ni.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smigiel K.S., Richards E., Srivastava S., Thomas K.R., Dudda J.C., Klonowski K.D., Campbell D.J. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J. Exp. Med. 2014;211:121–136. doi: 10.1084/jem.20131142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toomer K.H., Yuan X., Yang J., Dee M.J., Yu A., Malek T.R. Developmental Progression and Interrelationship of Central and Effector Regulatory T Cell Subsets. J. Immunol. 2016;196:3665–3676. doi: 10.4049/jimmunol.1500595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiedemann G.M., Grassmann S., Lau C.M., Rapp M., Villarino A.V., Friedrich C., Gasteiger G., O'Shea J.J., Sun J.C. Divergent Role for STAT5 in the Adaptive Responses of Natural Killer Cells. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakajima H., Liu X.W., Wynshaw-Boris A., Rosenthal L.A., Imada K., Finbloom D.S., Hennighausen L., Leonard W.J. An indirect effect of Stat5a in IL-2-induced proliferation: a critical role for Stat5a in IL-2-mediated IL-2 receptor alpha chain induction. Immunity. 1997;7:691–701. doi: 10.1016/s1074-7613(00)80389-1. [DOI] [PubMed] [Google Scholar]
- 27.ENCODE Project Consortium The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 28.Gattinoni L., Zhong X.S., Palmer D.C., Ji Y., Hinrichs C.S., Yu Z., Wrzesinski C., Boni A., Cassard L., Garvin L.M., et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yao C., Lou G., Sun H.W., Zhu Z., Sun Y., Chen Z., Chauss D., Moseman E.A., Cheng J., D'Antonio M.A., et al. BACH2 enforces the transcriptional and epigenetic programs of stem-like CD8. Nat. Immunol. 2021;22:370–380. doi: 10.1038/s41590-021-00868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun I.H., Oh M.H., Zhao L., Patel C.H., Arwood M.L., Xu W., Tam A.J., Blosser R.L., Wen J., Powell J.D. mTOR Complex 1 Signaling Regulates the Generation and Function of Central and Effector Foxp3. J. Immunol. 2018;201:481–492. doi: 10.4049/jimmunol.1701477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayatsu N., Miyao T., Tachibana M., Murakami R., Kimura A., Kato T., Kawakami E., Endo T.A., Setoguchi R., Watarai H., et al. Analyses of a Mutant Foxp3 Allele Reveal BATF as a Critical Transcription Factor in the Differentiation and Accumulation of Tissue Regulatory T Cells. Immunity. 2017;47:268–283.e9. doi: 10.1016/j.immuni.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 32.Neumann C., Blume J., Roy U., Teh P.P., Vasanthakumar A., Beller A., Liao Y., Heinrich F., Arenzana T.L., Hackney J.A., et al. c-Maf-dependent T. Nat. Immunol. 2019;20:471–481. doi: 10.1038/s41590-019-0316-2. [DOI] [PubMed] [Google Scholar]
- 33.Lam A.J., Uday P., Gillies J.K., Levings M.K. Helios is a marker, not a driver, of human Treg stability. Eur. J. Immunol. 2022;52:75–84. doi: 10.1002/eji.202149318. [DOI] [PubMed] [Google Scholar]
- 34.Burchill M.A., Yang J., Vogtenhuber C., Blazar B.R., Farrar M.A. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J. Immunol. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 35.Martinez G.J., Pereira R.M., Äijö T., Kim E.Y., Marangoni F., Pipkin M.E., Togher S., Heissmeyer V., Zhang Y.C., Crotty S., et al. The transcription factor NFAT promotes exhaustion of activated CD8⁺ T cells. Immunity. 2015;42:265–278. doi: 10.1016/j.immuni.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mognol G.P., Spreafico R., Wong V., Scott-Browne J.P., Togher S., Hoffmann A., Hogan P.G., Rao A., Trifari S. Exhaustion-associated regulatory regions in CD8. Proc. Natl. Acad. Sci. USA. 2017;114:E2776–E2785. doi: 10.1073/pnas.1620498114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seo H., Chen J., González-Avalos E., Samaniego-Castruita D., Das A., Wang Y.H., López-Moyado I.F., Georges R.O., Zhang W., Onodera A., et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8. Proc. Natl. Acad. Sci. USA. 2019;116:12410–12415. doi: 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisson S., Darrasse-Jèze G., Litvinova E., Septier F., Klatzmann D., Liblau R., Salomon B.L. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J. Exp. Med. 2003;198:737–746. doi: 10.1084/jem.20030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan C.L., Kuchroo J.R., Sage P.T., Liang D., Francisco L.M., Buck J., Thaker Y.R., Zhang Q., McArdel S.L., Juneja V.R., et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2021;218:e20182232. doi: 10.1084/jem.20182232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang G., Tajima M., Honjo T., Ohta A. STAT5 interferes with PD-1 transcriptional activation and affects CD8+ T-cell sensitivity to PD-1-dependent immunoregulation. Int. Immunol. 2021;33:563–572. doi: 10.1093/intimm/dxab059. [DOI] [PubMed] [Google Scholar]
- 41.Toomer K.H., Lui J.B., Altman N.H., Ban Y., Chen X., Malek T.R. Essential and non-overlapping IL-2Rα-dependent processes for thymic development and peripheral homeostasis of regulatory T cells. Nat. Commun. 2019;10:1037. doi: 10.1038/s41467-019-08960-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perry J.A., Shallberg L., Clark J.T., Gullicksrud J.A., DeLong J.H., Douglas B.B., Hart A.P., Lanzar Z., O'Dea K., Konradt C., et al. PD-L1-PD-1 interactions limit effector regulatory T cell populations at homeostasis and during infection. Nat. Immunol. 2022;23:743–756. doi: 10.1038/s41590-022-01170-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qureshi O.S., Kaur S., Hou T.Z., Jeffery L.E., Poulter N.S., Briggs Z., Kenefeck R., Willox A.K., Royle S.J., Rappoport J.Z., Sansom D.M. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J. Biol. Chem. 2012;287:9429–9440. doi: 10.1074/jbc.M111.304329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider H., Rudd C.E. Diverse mechanisms regulate the surface expression of immunotherapeutic target ctla-4. Front. Immunol. 2014;5:619. doi: 10.3389/fimmu.2014.00619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Serwas N.K., Hoeger B., Ardy R.C., Stulz S.V., Sui Z., Memaran N., Meeths M., Krolo A., Yüce Petronczki Ö., Pfajfer L., et al. Human DEF6 deficiency underlies an immunodeficiency syndrome with systemic autoimmunity and aberrant CTLA-4 homeostasis. Nat. Commun. 2019;10:3106. doi: 10.1038/s41467-019-10812-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janman D., Hinze C., Kennedy A., Halliday N., Waters E., Williams C., Rowshanravan B., Hou T.Z., Minogue S., Qureshi O.S., Sansom D.M. Regulation of CTLA-4 recycling by LRBA and Rab11. Immunology. 2021;164:106–119. doi: 10.1111/imm.13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hildner K., Edelson B.T., Purtha W.E., Diamond M., Matsushita H., Kohyama M., Calderon B., Schraml B.U., Unanue E.R., Diamond M.S., et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cancel J.C., Crozat K., Dalod M., Mattiuz R. Are Conventional Type 1 Dendritic Cells Critical for Protective Antitumor Immunity and How? Front. Immunol. 2019;10:9. doi: 10.3389/fimmu.2019.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy T.L., Murphy K.M. Dendritic cells in cancer immunology. Cell. Mol. Immunol. 2022;19:3–13. doi: 10.1038/s41423-021-00741-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mattei F., Schiavoni G., Belardelli F., Tough D.F. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J. Immunol. 2001;167:1179–1187. doi: 10.4049/jimmunol.167.3.1179. [DOI] [PubMed] [Google Scholar]
- 51.Liu J., Zhang X., Cheng Y., Cao X. Dendritic cell migration in inflammation and immunity. Cell. Mol. Immunol. 2021;18:2461–2471. doi: 10.1038/s41423-021-00726-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rawat K., Tewari A., Li X., Mara A.B., King W.T., Gibbings S.L., Nnam C.F., Kolling F.W., Lambrecht B.N., Jakubzick C.V. CCL5-producing migratory dendritic cells guide CCR5+ monocytes into the draining lymph nodes. J. Exp. Med. 2023;220 doi: 10.1084/jem.20222129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oderup C., Cederbom L., Makowska A., Cilio C.M., Ivars F. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology. 2006;118:240–249. doi: 10.1111/j.1365-2567.2006.02362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Onishi Y., Fehervari Z., Yamaguchi T., Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc. Natl. Acad. Sci. USA. 2008;105:10113–10118. doi: 10.1073/pnas.0711106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kastenmuller W., Gasteiger G., Subramanian N., Sparwasser T., Busch D.H., Belkaid Y., Drexler I., Germain R.N. Regulatory T cells selectively control CD8+ T cell effector pool size via IL-2 restriction. J. Immunol. 2011;187:3186–3197. doi: 10.4049/jimmunol.1101649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tekguc M., Wing J.B., Osaki M., Long J., Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2023739118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marangoni F., Zhakyp A., Corsini M., Geels S.N., Carrizosa E., Thelen M., Mani V., Prüßmann J.N., Warner R.D., Ozga A.J., et al. Expansion of tumor-associated Treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell. 2021;184:3998–4015.e19. doi: 10.1016/j.cell.2021.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sugiura D., Maruhashi T., Okazaki I.M., Shimizu K., Maeda T.K., Takemoto T., Okazaki T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science. 2019;364:558–566. doi: 10.1126/science.aav7062. [DOI] [PubMed] [Google Scholar]
- 59.Spencer S.D., Di Marco F., Hooley J., Pitts-Meek S., Bauer M., Ryan A.M., Sordat B., Gibbs V.C., Aguet M. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J. Exp. Med. 1998;187:571–578. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berg D.J., Kühn R., Rajewsky K., Müller W., Menon S., Davidson N., Grünig G., Rennick D. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J. Clin. Invest. 1995;96:2339–2347. doi: 10.1172/JCI118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Gassart A., Camosseto V., Thibodeau J., Ceppi M., Catalan N., Pierre P., Gatti E. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc. Natl. Acad. Sci. USA. 2008;105:3491–3496. doi: 10.1073/pnas.0708874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tze L.E., Horikawa K., Domaschenz H., Howard D.R., Roots C.M., Rigby R.J., Way D.A., Ohmura-Hoshino M., Ishido S., Andoniou C.E., et al. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J. Exp. Med. 2011;208:149–165. doi: 10.1084/jem.20092203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferris S.T., Carrero J.A., Mohan J.F., Calderon B., Murphy K.M., Unanue E.R. A minor subset of Batf3-dependent antigen-presenting cells in islets of Langerhans is essential for the development of autoimmune diabetes. Immunity. 2014;41:657–669. doi: 10.1016/j.immuni.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carrero J.A., McCarthy D.P., Ferris S.T., Wan X., Hu H., Zinselmeyer B.H., Vomund A.N., Unanue E.R. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc. Natl. Acad. Sci. USA. 2017;114:E10418–E10427. doi: 10.1073/pnas.1713543114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zakharov P.N., Hu H., Wan X., Unanue E.R. Single-cell RNA sequencing of murine islets shows high cellular complexity at all stages of autoimmune diabetes. J. Exp. Med. 2020;217:e20192362. doi: 10.1084/jem.20192362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kalekar L.A., Schmiel S.E., Nandiwada S.L., Lam W.Y., Barsness L.O., Zhang N., Stritesky G.L., Malhotra D., Pauken K.E., Linehan J.L., et al. CD4(+) T cell anergy prevents autoimmunity and generates regulatory T cell precursors. Nat. Immunol. 2016;17:304–314. doi: 10.1038/ni.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pletinckx K., Vaeth M., Schneider T., Beyersdorf N., Hünig T., Berberich-Siebelt F., Lutz M.B. Immature dendritic cells convert anergic nonregulatory T cells into Foxp3- IL-10+ regulatory T cells by engaging CD28 and CTLA-4. Eur. J. Immunol. 2015;45:480–491. doi: 10.1002/eji.201444991. [DOI] [PubMed] [Google Scholar]
- 68.Thomann A.S., Schneider T., Cyran L., Eckert I.N., Kerstan A., Lutz M.B. Conversion of Anergic T Cells Into Foxp3. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.704578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gravano D.M., Vignali D.A.A. The battle against immunopathology: infectious tolerance mediated by regulatory T cells. Cell. Mol. Life Sci. 2012;69:1997–2008. doi: 10.1007/s00018-011-0907-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Setoguchi R., Hori S., Takahashi T., Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 2005;201:723–735. doi: 10.1084/jem.20041982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.West E.E., Jin H.T., Rasheed A.U., Penaloza-Macmaster P., Ha S.J., Tan W.G., Youngblood B., Freeman G.J., Smith K.A., Ahmed R. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J. Clin. Invest. 2013;123:2604–2615. doi: 10.1172/JCI67008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ma S., So M., Ghelani A., Srivas R., Sahoo A., Hall R., Liu W., Wu H., Yu S., Lu S., et al. Attenuated IL-2 muteins leverage the TCR signal to enhance regulatory T cell homeostasis and response. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1257652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Duhen T., Duhen R., Lanzavecchia A., Sallusto F., Campbell D.J. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood. 2012;119:4430–4440. doi: 10.1182/blood-2011-11-392324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wing J.B., Tanaka A., Sakaguchi S. Human FOXP3. Immunity. 2019;50:302–316. doi: 10.1016/j.immuni.2019.01.020. [DOI] [PubMed] [Google Scholar]
- 75.Alegre M.L., Noel P.J., Eisfelder B.J., Chuang E., Clark M.R., Reiner S.L., Thompson C.B. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol. 1996;157:4762–4770. [PubMed] [Google Scholar]
- 76.Wang X.B., Zheng C.Y., Giscombe R., Lefvert A.K. Regulation of surface and intracellular expression of CTLA-4 on human peripheral T cells. Scand. J. Immunol. 2001;54:453–458. doi: 10.1046/j.1365-3083.2001.00985.x. [DOI] [PubMed] [Google Scholar]
- 77.Hayes E.T., Hagan C.E., Khoryati L., Gavin M.A., Campbell D.J. Regulatory T Cells Maintain Selective Access to IL-2 and Immune Homeostasis despite Substantially Reduced CD25 Function. J. Immunol. 2020;205:2667–2678. doi: 10.4049/jimmunol.1901520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hwang K.W., Sweatt W.B., Mashayekhi M., Palucki D.A., Sattar H., Chuang E., Alegre M.L. Transgenic expression of CTLA-4 controls lymphoproliferation in IL-2-deficient mice. J. Immunol. 2004;173:5415–5424. doi: 10.4049/jimmunol.173.9.5415. [DOI] [PubMed] [Google Scholar]
- 79.Raeber M.E., Rosalia R.A., Schmid D., Karakus U., Boyman O. Interleukin-2 signals converge in a lymphoid-dendritic cell pathway that promotes anticancer immunity. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aba5464. [DOI] [PubMed] [Google Scholar]
- 80.Eisenbarth S.C. Dendritic cell subsets in T cell programming: location dictates function. Nat. Rev. Immunol. 2019;19:89–103. doi: 10.1038/s41577-018-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schottelius A.J., Mayo M.W., Sartor R.B., Baldwin A.S. Interleukin-10 signaling blocks inhibitor of kappaB kinase activity and nuclear factor kappaB DNA binding. J. Biol. Chem. 1999;274:31868–31874. doi: 10.1074/jbc.274.45.31868. [DOI] [PubMed] [Google Scholar]
- 82.Zagorulya M., Yim L., Morgan D.M., Edwards A., Torres-Mejia E., Momin N., McCreery C.V., Zamora I.L., Horton B.L., Fox J.G., et al. Tissue-specific abundance of interferon-gamma drives regulatory T cells to restrain DC1-mediated priming of cytotoxic T cells against lung cancer. Immunity. 2023;56:386–405.e10. doi: 10.1016/j.immuni.2023.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moreno Ayala M.A., Campbell T.F., Zhang C., Dahan N., Bockman A., Prakash V., Feng L., Sher T., DuPage M. CXCR3 expression in regulatory T cells drives interactions with type I dendritic cells in tumors to restrict CD8. Immunity. 2023;56:1613–1630.e5. doi: 10.1016/j.immuni.2023.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anderson M.S., Bluestone J.A. The NOD mouse: a model of immune dysregulation. Annu. Rev. Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 85.Lai Y.P., Kuo L.C., Lin B.R., Lin H.J., Lin C.Y., Chen Y.T., Hsiao P.W., Chang H.T., Ko P.C.I., Chen H.C., et al. CD28 engagement inhibits CD73-mediated regulatory activity of CD8. Commun. Biol. 2021;4:595. doi: 10.1038/s42003-021-02119-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martínez-Llordella M., Esensten J.H., Bailey-Bucktrout S.L., Lipsky R.H., Marini A., Chen J., Mughal M., Mattson M.P., Taub D.D., Bluestone J.A. CD28-inducible transcription factor DEC1 is required for efficient autoreactive CD4+ T cell response. J. Exp. Med. 2013;210:1603–1619. doi: 10.1084/jem.20122387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nemeth Z.H., Lutz C.S., Csoka B., Deitch E.A., Leibovich S.J., Gause W.C., Tone M., Pacher P., Vizi E.S., Hasko G. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J. Immunol. 2005;175:8260–8270. doi: 10.4049/jimmunol.175.12.8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ben Addi A., Lefort A., Hua X., Libert F., Communi D., Ledent C., Macours P., Tilley S.L., Boeynaems J.M., Robaye B. Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the A(2B) receptor. Eur. J. Immunol. 2008;38:1610–1620. doi: 10.1002/eji.200737781. [DOI] [PubMed] [Google Scholar]
- 89.Cheng G., Yuan X., Tsai M.S., Podack E.R., Yu A., Malek T.R. IL-2 receptor signaling is essential for the development of Klrg1+ terminally differentiated T regulatory cells. J. Immunol. 2012;189:1780–1791. doi: 10.4049/jimmunol.1103768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yan J., Liu B., Shi Y., Qi H. Class II MHC-independent suppressive adhesion of dendritic cells by regulatory T cells in vivo. J. Exp. Med. 2017;214:319–326. doi: 10.1084/jem.20160629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu D., Lim E., Vaillant F., Asselin-Labat M.L., Visvader J.E., Smyth G.K. ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics. 2010;26:2176–2182. doi: 10.1093/bioinformatics/btq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Robinson M.D., Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Bulk and single-cell RNA-seq data have been deposited into the Gene Expression Omnibus under series GEO: GSE249482.
-
•
Code used for analyses can be found at http://doi.org/10.5281/zenodo.13315646.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.