

Abstract
Flavonoids, natural compounds ubiquitous in the human diet, are esteemed for their multifaceted pharmacological properties. The tyrosinase inhibitory capacity of five flavonoids from Alhagi graecorum was investigated through a comprehensive integration of in vitro and in silico methodologies. Molecular docking simulations demonstrated the proficient binding of the isolated compounds to the principal binding site of tyrosinase, akin to the standard tyrosinase inhibitor kojic acid. These compounds exhibited analogous binding affinities, among which compound 5 manifested notably heightened levels of polar bonds and hydrophobic interactions. Molecular dynamics (MD) simulations were utilized to explore the interaction dynamics between the isolated flavonoids and tyrosinase. The analysis of various MD parameters revealed consistent trajectories for the tested compounds, with compound 5 demonstrating notable energy equilibration. Strong hydrogen bonding interactions were observed between the flavonoids and the tyrosinase active site. The results of interaction energy calculations showed a balanced interaction mediated by hydrophobic interactions, with compound 5 exhibiting the lowest interaction energies. Additionally, the findings from MM/PBSA analysis demonstrated the lowest binding free energy for compound 5. Moreover, the in vitro tyrosinase inhibition assay revealed notable discrepancies among the studied flavonoids. Particularly, compound 5 demonstrated the most pronounced anti-tyrosinase activity, as evidenced by its lowest IC50 value. This experimental outcome is consistent with the results of computational predictions. Therefore, flavonoids of A. graecorum might be valuable for the development of tyrosinase inhibitors.
1. Introduction
Tyrosinase is a copper-containing enzyme that plays a key role in melanin biosynthesis. It is a crucial target in treating hyperpigmentation disorders and melanoma.1 The primary role of tyrosinase involves catalyzing the biosynthesis of melanin by facilitating the hydroxylation of L-tyrosine to L-DOPA, followed by the oxidation of L-DOPA to dopaquinone.2 Consequently, the polymerization of dopaquinone results in the production of melanin, which contributes to the pigmentation of skin, hair, and eyes.2 The dysregulation of tyrosinase function results in an abnormal upsurge in melanin synthesis, causing an accumulation of pigment within melanoma cells.1 As a result, these cells fail to transfer their melanin products to neighboring keratinocytes.1 Also, the distribution of tyrosinase activity could result in hyperpigmentation, which in turn causes serious conditions such as skin cancer, liver spots, and melasma.3 For these reasons, the production of novel tyrosinase inhibitors with improved efficacies has attracted significant attention in chemical and pharmaceutical industries.4 Given the growing demand for safer and more sustainable cosmetic and pharmaceutical ingredients, natural tyrosinase inhibitors have garnered significant attention and preference over their synthesized counterparts.1 This preference stems from their perceived efficacy, biocompatibility, and reduced environmental impact, making them promising candidates for various applications in skincare and medicine.1
The plant species Alhagi, commonly referred to as camelthorns or manna tree, belongs to the Fabaceae family. These herbs are extensively distributed across central, northern, and southern Africa, as well as in the Middle East, Europe, northwest China, and North America.5 The Latin name originates from the Arabic term “Alhag”, signifying the wise elder. A variety of Alhagi species exert a broad spectrum of biological activities for the folk medicine remedies of many diseases.5Alhagi graecorum Boiss. (A. graecorum), commonly referred to as al-akool, mannatree, or manna tree, is a perennial shrub belonging to the Fabaceae family, prevalent in arid and semiarid regions. Indigenous to Saudi Arabia, it is extensively distributed throughout the Nile region, the Mediterranean Basin, eastern and western deserts, the Red Sea coast, and the Sinai Peninsula.6 The scientific designation was authenticated using The Plant List and World Flora Online platforms. This versatile herb is distinguished by its diverse array of phytochemical constituents, encompassing flavonoids, resins, alkaloids, saponins, triterpenes, and phenolic compounds.5,7,8 The bioactive secondary metabolites present in this plant species play a significant role in its pharmacological applications, which include antioxidant, antimicrobial, anti-inflammatory, and hepatoprotective activities.5 Dried samples of this plant have been traditionally utilized in folk medicine as both a laxative and vermifuge, particularly for treating bilharziasis and rheumatic pains.5 Furthermore, A. graecorum is employed in addressing a spectrum of health issues, including liver disorders, urinary tract infections, and various gastrointestinal discomforts.9,10 Several studies have documented the extensive use of various parts of the plant in managing hemorrhoidal conditions.9,10 The abundant natural product diversity and potent biological activities of A. graecorum underscore its significance as a valuable medicinal plant with notable pharmacological potential.
In recent years, employing computational techniques such as molecular docking and molecular dynamics (MD) simulations with in vitro assays has emerged as a powerful strategy for elucidating the inhibitory activity of natural drugs.11−13 By combining computational predictions with experimental validation, researchers gain a comprehensive understanding of the molecular interactions between bioactive compounds and their target enzymes, such as tyrosinase.14,15 Molecular docking enables the exploration of potential binding modes and affinity between ligands and target proteins, providing valuable insights into their inhibitory mechanisms.16,17 Subsequently, MD simulations offer dynamic perspectives on the stability and flexibility of ligand-protein complexes over time, further refining our understanding of their inhibitory potency.18−20 Coupled with in vitro assays, which directly measure biological activity, this integrative approach not only enhances the reliability of computational predictions but also validates the inhibitory potential of natural drugs in a physiologically relevant context.21,22 Thus, this synergistic combination holds immense promise for accelerating the discovery and development of novel tyrosinase inhibitors from natural sources.
In this study, we adopted a multifaceted approach, combining computational and experimental methodologies, to assess the effectiveness of flavonoids derived from A. graecorum as inhibitors of tyrosinase activity. Employing a blend of in silico and in vitro techniques, our aim was to explore the potential of these phytochemicals in inhibiting tyrosinase, a key enzyme involved in melanin synthesis. Initially, we utilized docking virtual screening to predict the binding modes and estimate the binding affinities of the isolated flavonoids to the active site of tyrosinase. Subsequently, by employing MD simulations, we conducted an exhaustive analysis of the dynamic behavior exhibited by various complexes formed between the isolated flavonoids and tyrosinase. The inhibitory potential of the isolated compounds against the target enzyme was then validated through rigorous tyrosinase inhibitory activity in vitro assays. Through combining computational predictions with experimental data, this study offers an extensive understanding of the tyrosinase-inhibitory potential of flavonoids derived from A. graecorum. The results hold significant potential for advancing the creation of new melanogenesis inhibitors with versatile applications across multiple domains.
2. Experimental Section
2.1. General
The NMR spectral data of targeted phytochemicals were acquitted by using the renowned Bruker AM-500 spectrometer (500 and 100 MHz for both 1H NMR and 13C NMR, respectively). The Rudolph Autopol III polarimeter was utilized for assessing the optical rotation of isolated phytochemicals. The ultraviolet spectral data were estimated by utilizing a Shimadzu UV–Vis 160i spectrophotometer. HREIMS and EIMS spectra were obtained using a mass spectrometer of the type Finnigan MAT TSQ 700. FTIR spectral analysis was performed by using a Shimadzu FTIR-8400 instrument with KBr pellet preparation for sample analysis.
2.2. Plant Collection, Extraction, and Isolation of Flavonoids
The whole aerial parts of A. graecorum were harvested from Beni-Suef city in May 2022. Taxonomists from the Department of Botany and Microbiology at Beni-Suef University carried out the identification process of the plant under investigation as previously reported.11,12 The Voucher specimen was kept in our natural products lab under the number BU365180622. Subsequently, the collected A. graecorum material was dried, ground, and subjected to extraction using 70% ethanol. The resulting extract was concentrated in vacuo, yielding a viscous mass of the crude extract (374 g). A solvent–solvent partitioning technique was then employed on the obtained extract, using EtOAc, n-butanol, and n-hexane. Following two-dimensional paper chromatographic analysis, the EtOAc fraction (EAFAG) was chosen for further chromatographic exploration to isolate phytoconstituents. A 15 g portion of the EtOAc fraction was chromatographed over a silica gel column (100 cm × 3.5 cm, 0.5 kg) using the solvents chloroform, chloroform–methanol, and methanol in the order of increasing polarity. This fractionation process resulted in the collection of 25 subfractions which were combined into 14 fractions (A1–A14) according to their 2D paper chromatography profile. Subfraction A6 was eluted with 25% methanol in chloroform and was further purified over a polyamide 6S column (30 cm × 1 cm) eluted with methanol to give 5 main fractions after combination (Y1–Y5). Subfraction Y3 displayed two major spots upon TLC screening. This fraction was further purified over a Sephadex LH-20 column eluted with methanol to afford the purified compounds 2 (15 mg) and 5 (22 mg). Compound 4 (19 mg) was obtained after purification of subfraction Y4 on a Sephadex LH-20 column using methanol as a solvent. Fraction A7 separately underwent fractionation by means of a Sephadex LH-20 column eluted with the solvent system chloroform/ethanol followed by ethanol/methanol to afford the pure forms of compounds 1 (21 mg) and 3 (17 mg).
2.3. Molecular Docking Analysis
The binding affinities of isolated flavonoids for human tyrosinase were evaluated using molecular docking simulations performed with AutoDock Vina 1.5.6.23 The three-dimensional structure of the enzyme was obtained from the Protein Data Bank (PDB ID: 5M8Q). Autodock Tools (ADT) version 1.5.6 software was utilized for preparing the enzyme for docking through the addition of polar hydrogens, removal of solvent molecules, and setting the grid box to the coordinates of the native ligand. The geometrical structures of isolated flavonoids were optimized via optimization at the B3LYP level24−26 with the 6-311G (d, p) basis set27 using the Gaussian 16 software package.28 Subsequently, the docking protocol followed the previously reported methods.15,18,29
2.4. Molecular Dynamics Simulations
MD simulations of various flavonoid-tyrosinase complexes were performed by employing the PDB file of complexes with the lowest binding energy as determined from the molecular docking analysis. The 30 ns MD simulations involving both the enzyme in aqueous medium and various tyrosinase-isolated flavonoids complexes were executed using the GROMACS 2022.4 software30,31 The all-atom MD simulations of free enzyme and various phytochemical-enzyme systems were conducted using the CHARMM36m force field, which is known for its structure-balanced properties.32 The new CGenFF server was employed to generate the topology and geometric parameters of investigated flavonoids (https://cgenff.com/). In a setup involving periodic boundary conditions, both the tyrosinase enzyme and its complexes were contained within a dodecahedron-shaped enclosure, resulting in an adjusted volume of 515.79 nm3. The water model employed for the solvation process is the CHARMM-modified TIP3P,33 accompanied by the introduction of 12 sodium counterions to uphold electroneutrality. Mitigation of unfavorable thermodynamic interactions ensued through the application of the steepest descent energy minimization technique for a duration of 10 ps (ps).34 Subsequent to this step, a two-phase equilibration process was carried out, each spanning 100 ps at a temperature of 300 K, employing both the NVT (constant Number of particles, Volume, and Temperature) and NPT (constant Number of particles, Pressure, and Temperature) ensembles.35 Post-equilibration, the system underwent 30 ns MD simulations under standard conditions (300 K, 1 bar) using established protocols.36
2.5. Molecular Mechanics/Poisson–Boltzmann Surface Area (MM/PBSA) Analysis
To assess the binding free energies through MM/PBSA calculations between the flavonoids and tyrosinase, we employed the gmx_MMPBSA tool to analyze the MD trajectories.37 This computational approach provided insights into the energetic contributions driving the formation of enzyme–inhibitor complexes.
2.6. Tyrosinase Inhibitory Activity Assay
The inhibitory potential of investigated drugs against the target enzyme (tyrosinase) was assessed utilizing the adapted dopachrome method.38,39 Tested drugs and the reference drug and kojic acid were dissolved in DMSO and mixed with 31 U/mL of mushroom tyrosinase (1:1 v/v; Sigma, USA) and 5 mM L-DOPA (1:1 v/v; Sigma, USA) and 50 mM phosphate buffer (pH 6.8; 1:2 v/v). Following incubation for 10 min at 37 °C, the absorbance was measured at 475 nm on a Tecan plate reader (Tecan, USA). The percent inhibition of tyrosinase activity was calculated as follows:
AControl and ASample are the absorbance values in the absence and presence of the inhibitor. The IC50 value, a concentration giving 50% inhibition of tyrosinase activity, was determined by the interpolation of concentration–response curves. The experiment was conducted in triplicate.
3. Results and Discussion
3.1. Phytochemical Investigation
The ethanolic extract of A. graecorum was subjected to successive solvent partitioning, and the EtOAc fraction was successively chromatographed by using different solvent systems and stationary phases. This isolation process afforded five known flavonoid glycosides. The chemical structures of the isolated compounds were elucidated by utilizing a combination of spectroscopic analyses, comparison with authentic standards via TLC (Thin Layer Chromatography), and cross-referencing with data from previously reported compounds (see Supporting Information). Accordingly, the structures of isolated flavonoids (Figure 1) were identified as avicularin (1),40 isoquercitrin (2),41 isorhamnetin 3-O-α-L-arabinofuranoside (3),42 tamarixetin 3-O-α-L-rhamnoside (4),43 and thermopsoside (5).44
Figure 1.
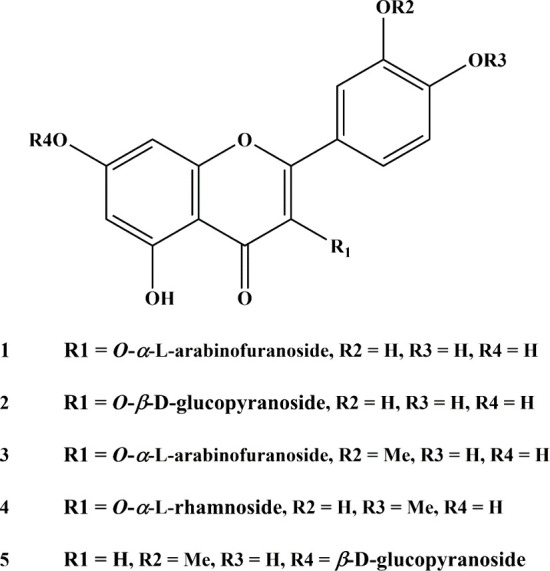
Structures of the isolated compounds.
3.2. Molecular Docking Analysis
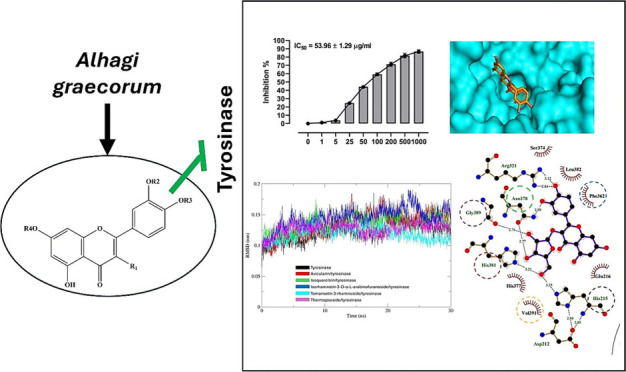
The binding affinities of A. graecorum phytochemicals (1–5) toward tyrosinase were studied by molecular docking assessments using AutoDock Vina. Table 1 shows the outcomes derived from our molecular docking computations, including binding affinities, polar bonds, and amino acid residues engaged in hydrophobic interactions. The docking pose with the highest energy favorability for each drug-enzyme complex was identified by considering both binding energies and the alignment of the drug within the binding site of the target enzyme. Figure 2 presents the results of the docking assessments, illustrating the spatial orientation of tested flavonoids within the binding cavity of the tyrosinase, alongside a surface representation of the ligand within the binding site. Furthermore, Figure 3 displays a ligplot estimation highlighting the involvement of various residues in both polar and hydrophobic interactions with different drugs.
Table 1. Results of the Molecular Docking Analysis of the Isolated Compounds against Tyrosinase.
| Compound | Binding affinity (kcal/mol) | Polar interacting residues | Hydrophobic interacting residues |
|---|---|---|---|
| 1 | –7.3 | His215, Arg321, Asn378, His381, and Gly389 | Glu216, Phe362, Ser374, His377, Leu382, and Val391 |
| 2 | –7.3 | Arg321, Asn378, and Ser394 | Phe362, His377, His381, Leu382, Gly389, and Val391 |
| 3 | –7.5 | Arg321, Val373, Asn378, and Ser394 | His215, Phe362, Ser374, His377, His381, Leu382, Gly389, and Val391 |
| 4 | –7.8 | Arg321, Gly389 and Val391 | His215, Phe362, His377, Asn378, His381, Leu382, and Val391 |
| 5 | –7.8 | Glu216, Arg321, Tyr348, Ser349, and Tyr369 | His215, Phe362, Pro371, His377, Asn378, His381, Leu382, and Val391 |
| Kojic acid | –5.8 | Asn378 and Ser394 | His192, His215, Phe362, His381, Gly388, Gly389, Gln390, and Val391 |
Figure 2.

Binding site interactions of compounds 1–5 and kojic acid with tyrosinase.
Figure 3.

A ligplot estimation of various residues engaged in both polar and hydrophobic interactions of compounds 1–5 and kojic acid with tyrosinase.
The binding affinities of investigated flavonoids against tyrosinase ranged closely from −7.3 to −7.8 kcal/mol. These low binding affinities observed for the isolated flavonoids suggest their potential activity against the target enzyme. Although the binding energies were closely clustered, compounds 4 and 5 displayed the lowest binding affinities (−7.8 kcal/mol), whereas compounds 1 and 2 showcased the highest binding affinities (−7.3 kcal/mol). Notably, all isolated flavonoids effectively docked into the identical binding site as the reference drug kojic acid, as depicted in Figure 2. Additionally, the amino acids engaged in both polar and hydrophobic interactions with kojic acid were similarly implicated in the binding mechanism of all tested ligands, as highlighted by circles in Figure 3. These residues, including Ph362, play a crucial role in the mechanism of binding of tyrosinase, facilitating thermodynamically favorable π–π interactions with all isolated phytochemicals.
The isolated drugs exhibited a notable binding profile characterized by a high extent of polar interactions, particularly evident in compounds 1 and 5, which displayed the highest number of polar bonds (five polar interacting residues). Conversely, the binding modes of all examined flavonoids revealed a dense array of hydrophobic interacting residues. Therefore, our docking calculations indicated a consistent binding mode for the tested ligands, implying a high degree of similarity in their activities against the investigated enzyme. However, the results of our virtual screening suggest that the activity of the isolated flavonoids against tyrosinase may be attributed to their low binding affinities and the significant existence of both polar and hydrophobic interactions.
3.3. Molecular Dynamics Simulations
Herein, we present the results of our MD simulations performed in this study to delve into the dynamic behaviors and various stabilities of the studied tyrosinase-isolated flavonoid complexes.45,46 MD simulation is considered a powerful tool to figure out the temporal evolution and structural dynamics of drug-enzyme interactions at the atomic level.47 Deep insights into stabilities, flexibilities and conformational variations of isolated flavonoids-tyrosinase complexes were gained by subjecting these complexes to simulated environmental conditions over time.47 By the results of these simulations, we seek to explore the molecular mechanism governing the catalyzed biotransformation of tested drugs within the binding pocket of tyrosinase.15 To assess the concordance of the tested flavonoids with the binding site of the target enzyme, we utilized 30 ns MD simulation runs. Specifically, complexes with a minimum binding affinity for each flavonoid were targeted for these simulations. Subsequently, an exhaustive examination of the MD trajectories spanning the 30 ns duration was undertaken. This analysis focused keenly on key parameters, including root-mean-square deviations (RMSDs), interaction energies, radius of gyration (Rg), root-mean-square fluctuations (RMSFs), solvent accessible surface area (SASA), and hydrogen bonding profiles. Both the free enzyme and various complexes formed with the isolated flavonoids underwent this thorough investigation.
3.3.1. Structural Stability and Dynamic Behavior
RMSD assessment provides valuable information on the structural stability and conformational changes of biological macromolecules during the course of MD simulations.20 Thus, we assessed the deviation of atomic positions of backbone RMSD values of various complexes with respect to unbound tyrosinase as a reference structure over the course of a 30 ns MD simulation. As illustrated in Figure 4A, within the first 7 ns of equilibration, the RMSD values of the protein backbone of both the unbound tyrosinase and the different flavonoid-enzyme systems exhibited an increasing pattern. Subsequently, these RMSD values stabilized and fluctuated within a range of about 0.10 to 0.19 nm throughout the time of simulation. Compound 4 displayed the lowest RMSD profile with an average of ≈0.12 nm, indicating a relatively stable conformation with minimal deviation from the reference enzyme, suggesting structural integrity and consistency along the simulation. Conversely, compound 3 displayed the highest RMSD values (≈0.15 nm) among tested flavonoids, indicating higher deviation from the reference enzyme and larger conformational variations. However, the RMSD fluctuations of compound 3 are still within the normal range of recognized stable systems. The average RMSD values of compounds 1, 2, and 5 are 0.134 0.135, and 0.127 nm, respectively, suggesting that these compounds exhibit relatively stable binding configurations with the target enzyme over the course of the MD simulation. The consistency in RMSD values across these compounds indicates that they maintain their structural integrity and do not undergo significant conformational changes during the simulation period. This stability in binding may imply a strong and consistent interaction between these flavonoids and tyrosinase, which could be indicative of their potential efficacy as enzyme inhibitors.
Figure 4.

MD simulation (30 ns) analysis of tyrosinase and isolated compounds-tyrosinase complexes: (A) Backbone RMSD of the unbound tyrosinase and isolated compounds-tyrosinase complexes; and (B) RMSD of isolated compounds.
To understand the independent behavior of the flavonoid molecules, we calculated root-RMSD values throughout the 30 ns simulation (Figure 4B). By aligning each simulation frame with the initial drug structure, we quantified the structural deviations of the flavonoids over time.48,49 This analysis provides valuable insights into the flexibility and stability of the drug molecules, which are critical considerations in drug development. As represented in Figure 4B, the RMSDs of various drugs displayed a high variation in their drug-to-drug RMSD profile. Similarly, compound 3 displayed the highest ligand RMSD profile among the tested drugs. This outcome suggests that the ligand exhibits substantial conformational changes, potentially adopting different orientations or conformations within the binding pocket of tyrosinase. These high fluctuations may affect the stability and interactions of the drug with the target enzyme, influencing its binding affinity and biological efficacy. The remaining drugs showed comparable RMSD behavior during the simulation time. While there was notable variability in the RMSD values among various flavonoids, the RMSD profiles of investigated drugs appeared to stabilize and exhibit normal fluctuations within the expected range.
Next, we assessed the RMSF profile for the free enzyme and different complexes with the objective of assessing the local protein mobility (Figure 5A). This graph was produced to depict the RMSF values plotted versus number of amino acid residues during the 30 ns simulation span.50 As expected, the RMSF values of tyrosinase-flavonoid complexes displayed the same behavior as the unbound tyrosinase, indicating the nonsignificant alteration of the mobility or flexibility of the enzyme residues within the binding site. Consequently, we can conclude that isolated phytochemicals induce no significant conformational change in the structure of the binding site of tyrosinase. The RMSF profile illustrated fluctuations occurring in the catalytic site of the target enzyme, spanning about 0.03 to 0.42 nm across all complexes. Interestingly, the RMSF profile of the free enzyme appeared lower compared with that of tyrosinase complexes with investigated flavonoids, implying that the interaction of the investigated drugs induces stabilization or decreased mobility within specific regions of the enzyme. This stabilization suggests that tested flavonoids interact with particular amino acids in the binding pocket, thus minimizing fluctuations or enhancing the flexibility in these binding sites. Additionally, limited fluctuations were detected at the binding cavity in the complexes formed by tested drugs with the target enzyme, in comparison to the free tyrosinase. The analysis indicates that the residues forming the primary binding site exhibit limited movement upon drug binding, suggesting a stable binding pocket architecture throughout the simulation period.
Figure 5.
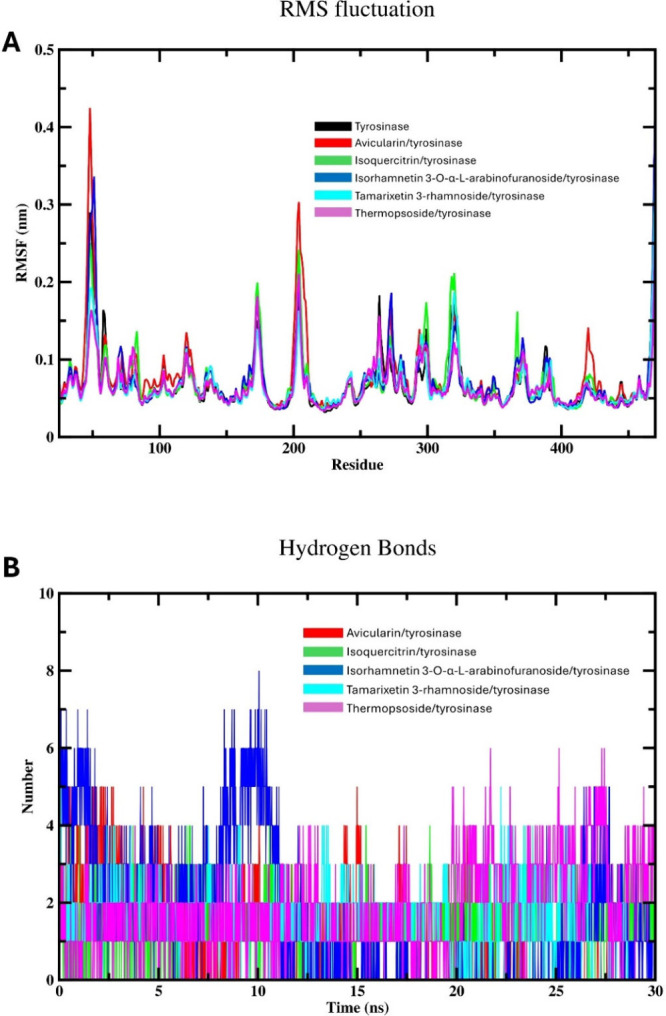
MD simulation (30 ns) analysis of tyrosinase and isolated phytochemicals complexes with tyrosinase: (A) Backbone RMSF per residue number for the free enzyme and the various compound-enzyme complexes; (B) Hydrogen bonding profile of isolated compounds-tyrosinase complexes.
The investigation of the binding attributes of examined flavonoids within the active site of tyrosinase involves evaluating the hydrogen bonding patterns of investigated systems over the 30 ns MD simulation period, as illustrated in Figure 5B. A detailed assessment of the hydrogen bonding profile indicated the robust presence of hydrogen bonding, consistent with the findings of molecular docking. Across the various complexes studied, a maximum of eight polar bonds with varying intensities were identified. This substantial level of polar interaction highlights the suitability of the investigated flavonoids with the active site of tyrosinase. Contrary to the results of molecular docking, compound 3 displayed the highest number of hydrogen bonds (eight bonds), yet the existence of these bonds is not intense along the 30 ns simulation time. Thus, compound 3 might reveal no consistent hydrogen bond formation mode with tyrosinase. This could indicate that the interaction between this compound and the enzyme is not governed by polar interactions, suggesting the existence of other types of interactions, such as hydrophobic or electrostatic interactions. On the other hand, the hydrogen bonding profiles of compounds 1 and 5 displayed a regular and intense behavior during the 30 ns simulation span, which is in agreement with the outputs of docking assessments. This outcome suggests strong and stable interactions between the tested flavonoids and the tyrosinase active site throughout the simulation duration. This consistent pattern of hydrogen bonding underscores the robustness of the drug-enzyme complex and implies a sustained inhibition potential against tyrosinase.
The dynamics and stabilities of various studies complexes could by assessed by examining the Rg values.51 These values act as an indicator for the enzyme compactness, reflecting a change in folding, unfolding, and conformational transitions over the simulation time.51 The calculated Rg values of free tyrosinase and various flavonoid complexes are represented in Figure 6A. The simulation results indicate that the systems exhibited stability after approximately 7 ns, indicating that both enzyme and the investigated complexes attained equilibrium around this time. Notably, the Rg values of free enzyme were found to be lower compared to those of the complexed rivals. This observation suggests that different drugs may induce enzyme compactness or constrain its flexibility. It is plausible that the binding of isolated flavonoids leads to a reduction in the overall size or flexibility of the enzyme. This phenomenon might be ascribed to the ligands prompting extra stabilization in particular regions, thereby eliciting conformational variations that facilitate a notable condensed state of the target enzyme. The results obtained are in agreement with prior MD studies on this enzyme, which have similarly suggested that the binding of some flavonoids minimizes the compactness of the enzyme while improving its structural compactness.52
Figure 6.
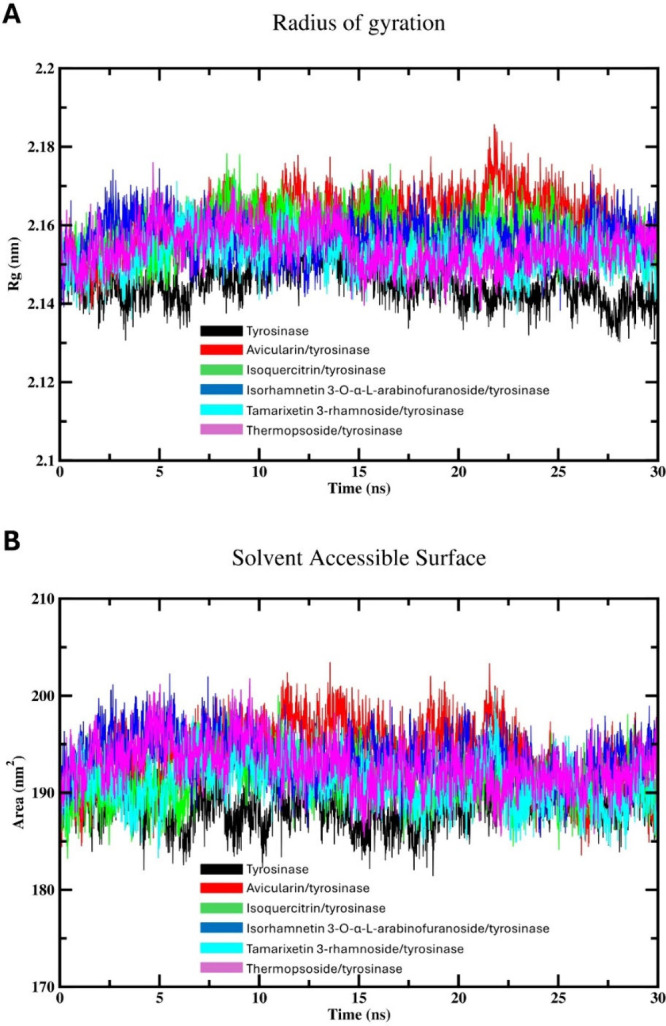
MD simulation (30 ns) analysis of tyrosinase and isolated phytochemical complexes with tyrosinase: (A) Protein radius of gyration for both free tyrosinase and various drugs-tyrosinase complexes; and (B) Protein SASA of the enzyme and various complexes.
Calculating SASA provides crucial information about the extent of surface exposure of biomolecules, aiding in understanding their interactions with solvent molecules, ligands, and other biomolecules.15 Additionally, SASA calculations can shed light on conformational changes, protein folding dynamics, and binding events during molecular simulations.15 The variations in SASA observed across all investigated systems throughout the simulation analysis are illustrated in Figure 6B. Compound 1 showed the highest SASA values, suggesting the binding of this flavonoid to tyrosinase may increase the exposure of tyrosinase to the surrounding water. Also, this could indicate that compound 1 binding results in conformational changes in the binding cavity of tyrosinase, leading to a more extended or flexible enzyme structure. The SASA profile of the unbound enzyme exhibits a lower value in contrast to tyrosinase complexed with flavonoids, suggesting that the interaction of these phytochemicals might decrease the exposure of the enzyme to the solvent environment. This implies that various drugs elicit conformational alterations in the enzyme’s active site, resulting in a more condensed or constrained geometry of the enzyme and effectively influencing protein dynamics or function. Importantly, the divergent patterns in SASA observed between tyrosinase in its unbound state and different flavonoid complexes align with the differences observed in the Rg values, confirming the accuracy and reliability of the MD calculations.
3.3.2. Interaction Energy Calculations
Computing the Coulombic-short-range (Coul-SR) interaction energy offers valuable insights into the electrostatic interactions between charged particles within a specified cutoff distance.53 Analyzing the Coul-SR values allows determination of the strength and stability of electrostatic interactions between different components of the complex.54 Variation in Coul-SR values along the simulation time can provide information about dynamic processes, such as ligand binding, protein conformational changes, or solvent reorganization, which are crucial for understanding the behavior and function of biomolecular systems.55 On the other hand, Lennard-Jones-short-range (LJ-SR) interaction energy estimates van der Waals interactions within a defined distance, important for the stability of biological macromolecules.56 Also, LJ-SR values assess the attractive and repulsive forces among nonbonded particles, assisting in system integrity.56Figure 7 illustrates the Coul-SR and LJ-SR interaction energies, delineating the interactions between tyrosinase and the tested flavonoids throughout the simulation time. Meanwhile, Table 2 outlines the average Coul-SR and LJ-SR interaction energies for the complexes formed between isolated phytochemicals and the target enzyme.
Figure 7.
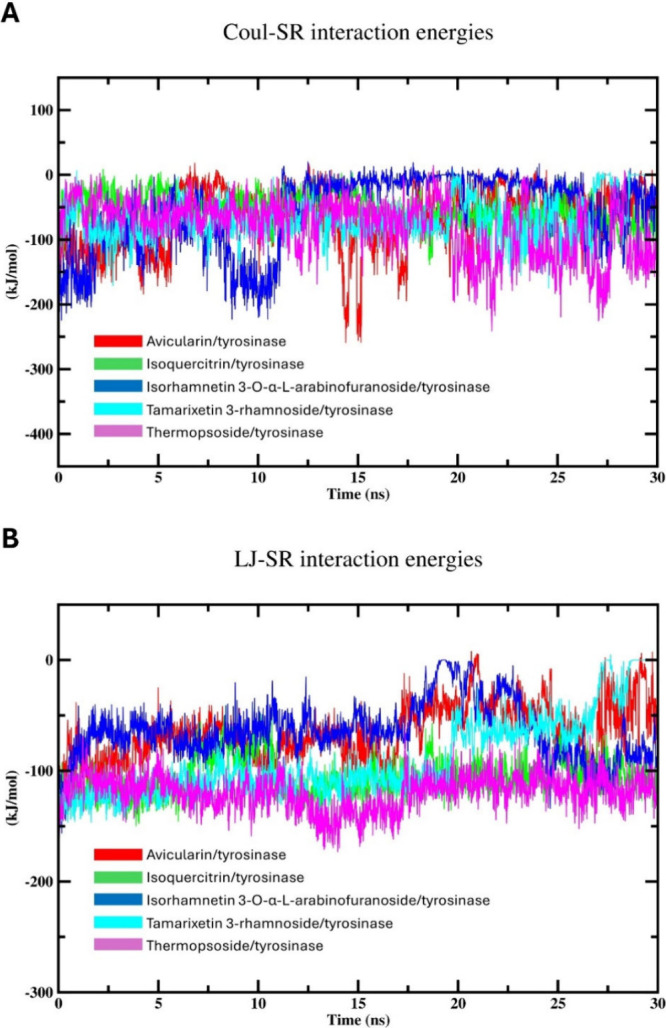
MD simulation (30 ns) analysis of tyrosinase and isolated phytochemical complexes with tyrosinase: (A) Coulomb-SR interactions energies of the enzyme amino acid residues with isolated compounds; and (B) Lennard-Jones-SR interaction energies of the enzyme amino acid residues with isolated compounds.
Table 2. Average Coul-SR and LJ-SR Interaction Energies of Isolated Flavonoids-Tyrosinase Complexes.
| Compound | Average Coul-SR interaction energy (kJ/mol) | Average LJ-SR interaction energy (kJ/mol) |
|---|---|---|
| 1 | –61.49 ± 20 | –64.55 ± 16 |
| 2 | –56.99 ± 5.3 | –106.011 ± 4.8 |
| 3 | –53.59 ± 21 | –54.42 ± 9.8 |
| 4 | –67.42 ± 5.8 | –89.90 ± 16 |
| 5 | –79.19 ± 12 | –119.35 ± 4.4 |
Figure 7A depicts the Coul-SR interaction energy values, revealing consistent fluctuations within the expected ranges. Notably, the interaction between compound 5 and tyrosinase exhibited the lowest energy values, averaging −79.19 ± 12 kJ/mol. This interaction reached equilibrium around 20 ns, maintaining fluctuations between approximately 7 and −240 kJ/mol thereafter. Conversely, compound 3 displayed the highest Coul-SR values after stabilizing around 11 ns. Comparable trends in interaction energy were noted for the other compounds, with minor discrepancies. Broadly, fluctuations in Coul-SR interaction energies for all compounds ranged from 20 to −250 kJ/mol. Higher negative values of the interaction energy denote increased stability in the formed complex.
Conversely, the LJ-SR energies (Figure 7B) were deemed reliable indicators for predicting binding interactions. Likewise, compound 5 exhibited the least LJ-SR interaction energy patterns, affirming its efficacy as a tyrosinase inhibitor. The average LJ-SR value for the compound 5 complex is −119.35 ± 4.4 kJ/mol and and it reached equilibrium after approximately 17 ns. Also, compound 3 showed the highest average LJ-SR value. These outcomes are in agreement with the outcome of docking assessment. Interestingly, compound 2 displayed an LJ-SR interaction energy profile comparable with that of compound 5. The increasingly negative LJ-SR values of these complexes suggested heightened van der Waals interactions among the atoms constituting the complex, suggesting an enhanced attraction between nonbonded particles. This attraction force increases the stability of the formed complex, affording a tightly bound complex. Additionally, the high fluctuations imply ongoing conformational adaptation and reorientation within the formed complex, indicating dynamic binding and the potential detachment of the drug from the active site throughout the simulation duration.
Therefore, the weaker Coul-SR interactions compared with their LJ-SR counterparts indicate a preference for van der Waals forces over electrostatic interactions among the atoms constituting the complexes. This indicates that certain complexes are mainly undergoing energy minimization through hydrophobic binding mechanisms, including dispersion forces. Furthermore, the usual fluctuations and increasingly negative interaction energy values suggest relatively stable interactions between the tested flavonoids and the target enzyme. As a result, the complexes exhibit a blend of kinetic and energetic favorability, without any one interaction type dominating. Additionally, minor adjustments in the conformation and fluctuations around the energetically favored state are expected for various complexes, suggesting a binding mode that is both stable and dynamically balanced.
3.3.3. MM/PBSA Analysis
The MMPBSA analysis of the five flavonoids interacting with tyrosinase reveals distinct binding affinities characterized by their van der Waals, electrostatic, and solvation energies (Table 3). Among the flavonoids, compound 5 exhibited the most favorable binding with a total binding energy (ΔGtotal) of −19.05 ± 2.81 kcal/mol, driven by strong gas phase interactions (ΔGgas = −54.21 ± 2.60 kcal/mol), comprising the highest van der Waals (−32.09 ± 1.87 kcal/mol) and electrostatic (−22.12 ± 1.80 kcal/mol) contributions. Compound 4 also demonstrated significant binding, with a ΔGtotal of −16.49 ± 1.76 kcal/mol, despite a higher solvation energy (36.42 ± 1.12 kcal/mol) that slightly offset its gas phase energy. In contrast, compound 1 showed the least favorable interaction, with a ΔGtotal of −6.23 ± 2.55 kcal/mol, indicating weaker overall binding affinity. These variations in binding energy highlight the differential interaction patterns of the flavonoids with tyrosinase, suggesting that compounds 5 and 4 may have more potent inhibitory effects, potentially due to their stronger van der Waals and electrostatic interactions, making them promising candidates for tyrosinase inhibition.
Table 3. MM/PBSA Calculation Results (kcal/mol).
| Complex | ΔEvdw | ΔEele | ΔGsolv | ΔGgas | ΔGtotal |
|---|---|---|---|---|---|
| 1 | – 15.27 ± 0.99 | –18.19 ± 1.99 | 27.23 ± 1.33 | –33.46 ± 2.21 | –6.23 ± 2.55 |
| 2 | –28.81 ± 1.23 | –20.75 ± 1.40 | 34.94 ± 1.69 | –49.56 ± 1.85 | –14.62 ± 2.53 |
| 3 | –24.26 ± 1.08 | –19.06 ± 1.33 | 35.25 ± 1.04 | –43.32 ± 1.71 | –8.07 ± 2.00 |
| 4 | –31.45 ± 0.89 | –21.46 ± 1.02 | 33.42 ± 1.12 | –52.91 ± 1.35 | –16.49 ± 1.76 |
| 5 | –32.09 ± 1.87 | –22.12 ± 1.80 | 35.16 ± 1.06 | –54.21 ± 2.60 | –19.05 ± 2.81 |
3.4. In Vitro Tyrosinase Inhibition
The in vitro tyrosinase inhibitory potential of flavonoids from A. graecorum was evaluated (Figure 8), revealing varying degrees of potency among the tested compounds. Compound 1 exhibited an IC50 value of 53.96 ± 1.29 μg/mL, indicating moderate inhibitory activity against tyrosinase. In comparison, compound 2 displayed a slightly higher potency with an IC50 of 43.87 ± 3.14 μg/mL, while compound 3 showed a similar level of activity with an IC50 of 47.09 ± 3.85 μg/mL. Notably, compound 4 demonstrated significantly enhanced inhibitory potential, yielding an IC50 value of 22.46 ± 2.21 μg/mL, suggesting its promising role as a potent tyrosinase inhibitor. Compound 5 emerged as the most potent inhibitor among the tested flavonoids, exhibiting an IC50 value of 14.65 ± 1.22 μg/mL, which was notably lower than that of the reference drug kojic acid (IC50 = 10.27 ± 0.93 μg/mL). These findings underscore the potential of flavonoids from A. graecorum as effective tyrosinase inhibitors, with compound 5 showing particular promise for further development as a therapeutic agent for hyperpigmentation disorders or other related conditions.
Figure 8.

Tyrosinase inhibitory activity and IC50 values of compounds 1–5 (A, E) and kojic acid (F). Data are mean ± SD, N = 3.
The empirical outcomes are consistent with the findings derived from the docking and MD simulations. Notably, compound 5 exhibited the most favorable binding configuration and the lowest GROMACS interaction energy values, reinforcing its efficacy as a tyrosinase inhibitor. The alignment observed between the outcomes of the experimental inhibitory activity assay and the results of in silico techniques offers robust validation for our computational approaches. This validation underscores the reliability and efficacy of our computational strategies in predicting the inhibitory potential of the drugs tested against the target enzyme. Additionally, it highlights the complementary nature of computational and experimental approaches in elucidating the inhibitory mechanisms of natural compounds. In summary, our collective findings strongly suggest that compound 5 holds considerable promise as a potent agent for curtailing tyrosinase activity, thus presenting prospective therapeutic avenues for conditions linked to irregular melanin production. Further studies elucidating the mechanism of action and in vivo efficacy of these compounds are warranted to fully assess their therapeutic potential and clinical applicability.
4. Conclusion
The chromatographic fractionation of the EtOAc extract from A. graecorum led to the isolation of five flavonoids, previously unreported in this plant. These flavonoids were evaluated for their tyrosinase inhibitory potential using both computational and experimental approaches. Molecular docking studies revealed that all compounds effectively bound to the active site of tyrosinase, with binding affinities comparable to those of kojic acid, a known standard inhibitor. Among them, compound 5 demonstrated superior interactions through a higher degree of polar bonds and hydrophobic interactions, marking it as a particularly potent inhibitor. MD simulations further supported these findings, showing stable trajectories and significant energy equilibration for compound 5. RMSF, Rg, and SASA analyses indicated that flavonoid binding enhanced the stability of the enzyme’s active site by reducing its flexibility and solvent accessibility. Interaction energy calculations corroborated the hydrophobic nature of the binding, with compound 5 displaying the lowest interaction energies. Consistently, in vitro assays confirmed that compound 5 had the most potent tyrosinase inhibitory activity, with the lowest IC50 value. The close alignment between the computational predictions and experimental results validates the reliability of the computational methodologies employed. Overall, these findings suggest that flavonoids from A. graecorum, particularly compound 5, hold promise as potential tyrosinase inhibitors. This provides a strong foundation for future research into developing these flavonoids as novel melanogenesis inhibitors with therapeutic applications in hyperpigmentation disorders and cosmetic formulations.
Acknowledgments
Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2024R381), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors also thank the Centro de Computación Científica of the UAM (CCC-UAM) for the generous allocation of computer time and continued technical support, and they acknowledge the support from project Y2020/EMT-6290 (PRIES-CM) of the Comunidad de Madrid and project PID2023-150717NB-I00 from Ministerio de Ciencia, Innovacion y Universidades in Spain.
Data Availability Statement
The manuscript and Supporting Information contain all data supporting the reported results.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c07624.
Cartesian coordinates of DFT optimized flavonoids at the B3LYP level of theory and 1H NMR and 13C NMR spectra of compounds 1–5 (PDF)
Author Contributions
Conceptualization, A.M.M., A-M.L., and E.M.K.; methodology, R.S.A., A.M.M., A-M.L., A.A.E., S.M.A., I.E., M.F.A., and E.M.K.; software, A-M.L. and E.M.K; validation, A.M.M., A-M.L., and E.M.K.; formal analysis, A.M.M., and E.M.K.; investigation, R.S.A., A.M.M., A-M.L., S.M.A., M.F.A., A.A.E., and E.M.K.; resources, R.S.A., S.M.A., M.F.A., and A-M.L.; data curation, R.S.A., E.M.K., and A.M.M.; writing—original draft preparation, E.M.K., and A.M.M.; writing—review and editing, A.M.M., and A-M.L.; visualization, E.M.K., and A.M.M.; supervision, A.M.M., and A-M.L.; project administration, R.S.A. and A.M.M.; funding acquisition, R.S.A. All authors have read and agreed to the published version of the manuscript.
Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2024R381), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
The authors declare no competing financial interest.
Supplementary Material
References
- Roulier B.; Pérès B.; Haudecoeur R. Advances in the Design of Genuine Human Tyrosinase Inhibitors for Targeting Melanogenesis and Related Pigmentations. J. Med. Chem. 2020, 63 (22), 13428–13443. 10.1021/acs.jmedchem.0c00994. [DOI] [PubMed] [Google Scholar]
- Kurpejović E.; Wendisch V. F.; Sariyar Akbulut B. Tyrosinase-based production of l-DOPA by Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2021, 105 (24), 9103–9111. 10.1007/s00253-021-11681-5. [DOI] [PubMed] [Google Scholar]
- Thawabteh A. M.; Jibreen A.; Karaman D.; Thawabteh A.; Karaman R. Skin Pigmentation Types, Causes and Treatment—A Review. Molecules 2023, 28 (12), 4839. 10.3390/molecules28124839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathee P.; Kumar S.; Kumar D.; Kumari B.; Yadav S. S. Skin hyperpigmentation and its treatment with herbs: an alternative method. Future Journal of Pharmaceutical Sciences 2021, 7 (1), 132. 10.1186/s43094-021-00284-6. [DOI] [Google Scholar]
- Muhammad G.; Hussain M. A.; Anwar F.; Ashraf M.; Gilani A.-H. Alhagi: A Plant Genus Rich in Bioactives for Pharmaceuticals. Phytotherapy Research 2015, 29 (1), 1–13. 10.1002/ptr.5222. [DOI] [PubMed] [Google Scholar]
- Awmack C. S.; Lock J. M. The Genus Alhagi (Leguminosae: Papilionoideae) in the Middle East. Kew Bulletin 2002, 57 (2), 435–443. (acccessed 2024/03/07/). JSTOR 10.2307/4111121. [DOI] [Google Scholar]
- Ali-Shtayeh M. S.; Jamous R. M.; Al-Shafie J. H.; Elgharabah W. A.; Kherfan F. A.; Qarariah K. H.; Khdair I. S.; Soos I. M.; Musleh A. A.; Isa B. A.; et al. Traditional knowledge of wild edible plants used in Palestine (Northern West Bank): A comparative study. Journal of Ethnobiology and Ethnomedicine 2008, 4 (1), 13. 10.1186/1746-4269-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan S. A.; Kamel E. M.; Ewais M. A.; Khowailed A. A.; Hassanein E. H.; Mahmoud A. M. Flavonoids of Haloxylon salicornicum (Rimth) prevent cisplatin-induced acute kidney injury by modulating oxidative stress, inflammation, Nrf2, and SIRT1. Environmental Science and Pollution Research 2023, 30 (17), 49197–49214. 10.1007/s11356-023-25694-2. [DOI] [PubMed] [Google Scholar]
- Awaad Amani A. S.; Maitland D. J.; Soliman G. A. Antiulcerogenic Activity of Alhagi maurorum. Pharmaceutical Biology 2006, 44 (4), 292–296. 10.1080/13880200600714160. [DOI] [Google Scholar]
- Harvey A. L. Medicines from nature: are natural products still relevant to drug discovery? *. Trends Pharmacol. Sci. 1999, 20 (5), 196–198. (acccessed 2024/03/07) 10.1016/S0165-6147(99)01346-2. [DOI] [PubMed] [Google Scholar]
- Alruhaimi R. S.; Mostafa-Hedeab G.; Abduh M. S.; Bin-Ammar A.; Hassanein E. H. M.; Kamel E. M.; Mahmoud A. M.. A flavonoid-rich fraction of Euphorbia peplus attenuates hyperglycemia, insulin resistance, and oxidative stress in a type 2 diabetes rat model. Frontiers in Pharmacology 2023, 14, 10.3389/fphar.2023.1204641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel E. M.; Ahmed N. A.; El-Bassuony A. A.; Hussein O. E.; Alrashdi B.; Ahmed S. A.; Lamsabhi A. M.; Arab H. H.; Mahmoud A. M. Xanthine oxidase inhibitory activity of Euphorbia peplus L. phenolics. Combinatorial Chemistry & High Throughput Screening 2022, 25 (8), 1336–1344. 10.2174/1386207324666210609104456. [DOI] [PubMed] [Google Scholar]
- Alruhaimi R. S.; Mahmoud A. M.; Elbagory I.; Ahmeda A. F.; El-Bassuony A. A.; Lamsabhi A. M.; Kamel E. M. Unveiling the tyrosinase inhibitory potential of phenolics from Centaurium spicatum: Bridging in silico and in vitro perspectives. Bioorganic Chemistry 2024, 147, 107397. 10.1016/j.bioorg.2024.107397. [DOI] [PubMed] [Google Scholar]
- Chang T.-S. An Updated Review of Tyrosinase Inhibitors. International Journal of Molecular Sciences 2009, 10 (6), 2440–2475. 10.3390/ijms10062440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel E. M.; Bin-Ammar A.; El-Bassuony A. A.; Alanazi M. M.; Altharawi A.; Ahmeda A. F.; Alanazi A. S.; Lamsabhi A. M.; Mahmoud A. M. Molecular modeling and DFT studies on the antioxidant activity of Centaurea scoparia flavonoids and molecular dynamics simulation of their interaction with β-lactoglobulin. RSC Adv. 2023, 13 (18), 12361–12374. 10.1039/D3RA01661G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alruhaimi R. S.; Hassanein E. H. M.; Abd El-Aziz M. K.; Siddiq Abduh M.; Bin-Ammar A.; Kamel E. M.; Mahmoud A. M. The melatonin receptor agonist agomelatine protects against acute pancreatitis induced by cadmium by attenuating inflammation and oxidative stress and modulating Nrf2/HO-1 pathway. International Immunopharmacology 2023, 124, 110833. 10.1016/j.intimp.2023.110833. [DOI] [PubMed] [Google Scholar]
- Kamel E. M.; Lamsabhi A. M. Water biocatalytic effect attenuates cytochrome P450-mediated carcinogenicity of diethylnitrosamine: A computational insight. Organic & Biomolecular Chemistry 2021, 19 (41), 9031–9042. 10.1039/D1OB01439K. [DOI] [PubMed] [Google Scholar]
- Kamel E. M.; Tawfeek A. M.; El-Bassuony A. A.; Lamsabhi A. M. Mechanistic aspects of reactive metabolite formation in clomethiazole catalyzed biotransformation by cytochrome P450 enzymes. Organic & Biomolecular Chemistry 2023, 21, 7158. 10.1039/D3OB01014G. [DOI] [PubMed] [Google Scholar]
- Kamel E. M.; Tawfeek A. M.; El-Bassuony A. A.; Lamsabhi A. M. Mechanistic insights into chloramphenicol-mediated inactivation of cytochrome P450 enzymes and their active site mutants. New J. Chem. 2023, 47, 16429. 10.1039/D3NJ02991C. [DOI] [Google Scholar]
- Kamel E. M.; Lamsabhi A. M. The quasi-irreversible inactivation of cytochrome P450 enzymes by paroxetine: a computational approach. Organic & Biomolecular Chemistry 2020, 18 (17), 3334–3345. 10.1039/D0OB00529K. [DOI] [PubMed] [Google Scholar]
- Beltran E.; Serafini M. R.; Alves I. A.; Aragón Novoa D. M. Novel Synthesized Tyrosinase Inhibitors: A Systematic Patent Review (2012-Present). Curr. Med. Chem. 2024, 31 (3), 308–335. 10.2174/0929867330666230203111437. [DOI] [PubMed] [Google Scholar]
- Kamel E. M.; Alwaili M. A.; Rudayni H. A.; Allam A. A.; Lamsabhi A. M. Deciphering the Molecular Mechanisms of Reactive Metabolite Formation in the Mechanism-Based Inactivation of Cytochrome p450 1B1 by 8-Methoxypsoralen and Assessing the Driving Effect of phe268. Molecules 2024, 29 (7), 1433. 10.3390/molecules29071433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37 (2), 785. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38 (6), 3098. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Hehre W. J.; Radom L.; Schleyer P. v. R.; Pople J. A.. Ab initio molecular orbital theory; Wiley New York, 1986. [Google Scholar]
- Gaussian 16 Rev. C.01; Wallingford, CT, 2016. [Google Scholar]
- Al-Amarat W.; Abukhalil M. H.; Althunibat O. Y.; Alfwuaires M. A.; Alnamshan M. M.; Alqosaibi A. I.; Ahmeda A. F.; Kamel E. M.; Arab H. H.; Mahmoud A. M. Galangin attenuates liver injury, oxidative stress and inflammation, and upregulates Nrf2/HO-1 signaling in streptozotocin-induced diabetic rats. Processes 2021, 9 (9), 1562. 10.3390/pr9091562. [DOI] [Google Scholar]
- Bauer P.; Hess B.; Lindahl E. GROMACS 2022.4 Manual. November 2022, 16, 2022. [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmüller H.; MacKerell A. D. Jr CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14 (1), 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D. Jr; Bashford D.; Bellott M.; Dunbrack R. L. Jr; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102 (18), 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4 (3), 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52 (12), 7182–7190. 10.1063/1.328693. [DOI] [Google Scholar]
- Lemkul J. A. From Proteins to Perturbed Hamiltonians: A Suite of Tutorials for the GROMACS-2018 Molecular Simulation Package [Article v1.0]. Living Journal of Computational Molecular Science 2019, 1 (1), 5068.(acccessed 2024/02/18) 10.33011/livecoms.1.1.5068. [DOI] [Google Scholar]
- Valdés-Tresanco M. S.; Valdés-Tresanco M. E.; Valiente P. A.; Moreno E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17 (10), 6281–6291. 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- Masuda T.; Yamashita D.; Takeda Y.; Yonemori S. Screening for Tyrosinase Inhibitors among Extracts of Seashore Plants and Identification of Potent Inhibitors from Garcinia subelliptica. Biosci., Biotechnol., Biochem. 2005, 69 (1), 197–201. (acccessed 3/17/2024) 10.1271/bbb.69.197. [DOI] [PubMed] [Google Scholar]
- Alam N.; Yoon K. N.; Lee K. R.; Shin P. G.; Cheong J. C.; Yoo Y. B.; Shim J. M.; Lee M. W.; Lee U. Y.; Lee T. S. Antioxidant Activities and Tyrosinase Inhibitory Effects of Different Extracts from Pleurotus ostreatus Fruiting Bodies. Mycobiology 2010, 38 (4), 295–301. 10.4489/MYCO.2010.38.4.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugheighele S. E.; Imafidon K. E.; Choudhary M. I.; Shakil A.; Okoro E. E. Isolation of Quercetin and Avicularin from Dennettia tripetala (G. Baker) Seeds, and Evaluation of the Oxidative Stress Management Capacity and Cytotoxic Activities of Its Acetone Extract and Fractions. Chemistry Africa 2022, 5 (5), 1275–1285. 10.1007/s42250-022-00413-5. [DOI] [Google Scholar]
- Li H.; Liu Y.; Yi Y.; miao Q.; Liu S.; Zhao F.; Cong W.; Wang C.; Xia C. Purification of quercetin-3-O-sophoroside and isoquercitrin from Poacynum hendersonii leaves using macroporous resins followed by Sephadex LH-20 column chromatography. Journal of Chromatography B 2017, 1048, 56–63. 10.1016/j.jchromb.2017.01.041. [DOI] [PubMed] [Google Scholar]
- Olszewska M. High-performance liquid chromatographic identification of flavonoid monoglycosides from Prunus serotina ehrh. Acta Polym. Pharm. 2005, 62 (6), 435–441. [PubMed] [Google Scholar]
- Son Y. K.; Lee M. H.; Han Y. N. A new antipsychotic effective neolignan fromfirmiana simplex. Archives of Pharmacal Research 2005, 28 (1), 34–38. 10.1007/BF02975132. [DOI] [PubMed] [Google Scholar]
- Khushbaktova Z. A.; Faizieva S. K.; Syrov V. N.; Yuldashev M. P.; Batirov É. K.; Mamatkhanov A. U. Isolation, Chemical Analysis, and Study of the Hypolipidemic Activity of the Total Flavonoid Extract from Thermopsis altherniaflora. Pharmaceutical Chemistry Journal 2001, 35 (3), 155–158. 10.1023/A:1010410029719. [DOI] [Google Scholar]
- Kamel E. M.; Alqhtani H. A.; Bin-Jumah M.; Rudayni H. A.; El-Bassuony A. A.; Mokhtar Lamsabhi A. Deciphering molecular mechanisms underlying the inhibition of β-glucuronidase by xanthones from Centaurium spicatum. Bioorganic Chemistry 2024, 150, 107609. 10.1016/j.bioorg.2024.107609. [DOI] [PubMed] [Google Scholar]
- Kamel E. M.; Aba Alkhayl F. F.; Alqhtani H. A.; Bin-Jumah M.; Rudayni H. A.; Lamsabhi A. M. Bridging in silico and in vitro perspectives to unravel molecular mechanisms underlying the inhibition of β-glucuronidase by coumarins from Hibiscus trionum. Biophys. Chem. 2024, 313, 107304. 10.1016/j.bpc.2024.107304. [DOI] [PubMed] [Google Scholar]
- Honarparvar B.; Govender T.; Maguire G. E. M.; Soliman M. E. S.; Kruger H. G. Integrated Approach to Structure-Based Enzymatic Drug Design: Molecular Modeling, Spectroscopy, and Experimental Bioactivity. Chem. Rev. 2014, 114 (1), 493–537. 10.1021/cr300314q. [DOI] [PubMed] [Google Scholar]
- Alwaili M. A.; Aba Alkhayl F. F.; Rudayni H. A.; Allam A. A.; Altoom N. G.; Lamsabhi A. M.; Kamel E. M. Unraveling molecular mechanisms of β-glucuronidase inhibition by flavonoids from Centaurea scoparia: integrated in silico and in vitro insights. New J. Chem. 2024, 48, 14236–14252. 10.1039/D4NJ02393E. [DOI] [Google Scholar]
- Kamel E. M.; Maodaa S.; Al-Shaebi E. M.; Lamsabhi A. M. Mechanistic insights into the metabolic pathways of vanillin: unraveling cytochrome P450 interaction mechanisms and implications for food safety. Organic & Biomolecular Chemistry 2024, 22, 6561–6574. 10.1039/D4OB00973H. [DOI] [PubMed] [Google Scholar]
- Kamel E. M.; Alkhayl F. F. A.; Alqhtani H. A.; Bin-Jumah M.; Rudayni H. A.; Lamsabhi A. M. Dissecting molecular mechanisms underlying the inhibition of β-glucuronidase by alkaloids from Hibiscus trionum: Integrating in vitro and in silico perspectives. Computers in Biology and Medicine 2024, 180, 108969. 10.1016/j.compbiomed.2024.108969. [DOI] [PubMed] [Google Scholar]
- Lobanov M. Y.; Bogatyreva N. S.; Galzitskaya O. V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42 (4), 623–628. 10.1134/S0026893308040195. [DOI] [PubMed] [Google Scholar]
- Li X.; Guo J.; Lian J.; Gao F.; Khan A. J.; Wang T.; Zhang F. Molecular Simulation Study on the Interaction between Tyrosinase and Flavonoids from Sea Buckthorn. ACS Omega 2021, 6 (33), 21579–21585. 10.1021/acsomega.1c02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprzeska-Zingrebe E. A.; Meyer S.; Roloff A.; Kunte H.-J.; Smiatek J. Influence of compatible solute ectoine on distinct DNA structures: thermodynamic insights into molecular binding mechanisms and destabilization effects. Phys. Chem. Chem. Phys. 2018, 20 (40), 25861–25874. 10.1039/C8CP03543A. [DOI] [PubMed] [Google Scholar]
- Padariya M.; Kalathiya U.; Baginski M. Molecular basis and potential activity of HIV-1 reverse transcriptase toward trimethylamine-based compounds. Biotechnology and Applied Biochemistry 2017, 64 (6), 810–826. 10.1002/bab.1543. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Qu H.; Wu G. Molecular insights into cucurbit[8]uril-mediated complexes: Enhanced interaction cooperation towards pseudostatic dynamics. J. Mol. Liq. 2023, 391, 123266. 10.1016/j.molliq.2023.123266. [DOI] [Google Scholar]
- Oliveira M. P.; Hünenberger P. H. Influence of the Lennard-Jones Combination Rules on the Simulated Properties of Organic Liquids at Optimal Force-Field Parametrization. J. Chem. Theory Comput. 2023, 19 (7), 2048–2063. 10.1021/acs.jctc.2c01170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The manuscript and Supporting Information contain all data supporting the reported results.