

Abstract
Tomato yellow leaf curl virus (Family Geminiviridae, Genus Begomovirus) is a serious menace in the cultivation of tomato causing Tomato leaf curl disease (ToLCD). Recently, we presented the TYLCV isolates having additional genomic features (nucleotides insertions) characterized from the tomato fields of Kuwait adding up to the genetic diversity repertoire of these viruses. The widespread prevalence of disease in tropics across the continents, emergence of genetic variants and ever increasing complete genome sequences of virus isolates in public database warrant a global analysis to infer the genetic diversity, evolutionary pattern so as to devise suitable disease control stratagems. Molecular phylogeny suggested the monophyletic origin of the TYLCV Kuwait isolates and TYLCV reported from Asian countries revealing their genetic relatedness. Though genetic diversity of TYLCV reported from elsewhere (TYLCV others) is two folds higher (0.11765) than TYLCV Kuwait, a relatively high number of polymorphic sites in the latter imply their inherent genetic diversity. Neutrality tests of TYLCV as a whole suggest the operation of phenomenon of purifying selection indicating deleterious mutations are being weeded out from the population. Recombination analysis discloses that TYLCV Kuwait isolates are potential genetic recombinants or involved in the generation of potential recombinants as parents. The results of the neutrality tests were further strengthened by the outcome of codon substitution analysis indicating the operation of purifying selection in the codons of C1 ORF of TYLCV population as a whole and in the sub-group TYLCV Kuwait. The implications of these results are discussed.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12985-024-02540-6.
Introduction
Tomato yellow leaf curl virus belonging to family Geminiviridae and genus Begomovirus is a serious threat to the production of tomato across the world since infection due to this virus could cause a yield loss even to the extent of 100% [1]. The pervasiveness and rapid rate of spread of the TYLCV causing economically considerable damages to the cultivation of tomato warrants an immediate attention [2]. Availability of infected plant materials and presence of the insect vectors, whiteflies (Bemisia tabacci), in the tropical conditions have led to the rampant spread of the virus across middle eastern countries, China, Australia, East Asia, Western Mediterranean regions to Caribbean and America. It has assumed a pathogen of global significance [3]. The disease has been reported to occur not only in tropical conditions but also in subtropical, temperate, and even semi-arid regions, worldwide [4]. In addition, the ability of vector Bemisia tabacci, which is a cryptic species characterized as a complex of closely related interbreeding strains or 39 species, to spread rapidly and its wide host range in the tropics have contributed to the extensive spread of the virus from Middle East to other parts of the world [5].
TYLCV genome is characterized by a closed circular single stranded DNA of 2.7–2.8 kb in length, with monopartite or bipartite genome compositions [6]. The monopartite viruses carry single DNA component coding for proteins involved in regulatory (C1 through C4) and structural functions (V2 and V2) such as replication, vector transmission and viral suppressors of RNA silencing [1]. In bipartite viruses, another DNA component called DNA B encodes for proteins involved in inter and intracellular movement functions within host cellular system [7]. However, in monopartite viruses lacking DNA B, the movement functions of the viruses are regulated by coat protein and pre-coat protein [8].
Intense breeding efforts to evolve cultivars or host plants resistant to TYLCV infection are a pragmatic approach to thwart the virus infection. Nevertheless, introduction of host-mediated virus resistance in crops have led to the emergence of extremely adaptive viral quasispecies and/or viral gene pools encompassing a multiple adaptive traits [9]. Hence, it is imperative to analyze the genetic variability and population architecture of economically important plant viruses to comprehend the features of viral evolution and to unravel the genetic basis of virus-host plant interactions. Above all these approaches would yield way forward in devising effective disease management strategies that are not only durable but also ecologically viable [10]. Recently we reported genetically distinct isolates of TYLCV from Kuwait and observed that a few of the Kuwait isolates exhibited an extra 19 nucleotides insertion in its genome [11]. As a follow up we present herein the genetic diversity and molecular evolutionary genomic analysis of TYLCV isolates reported elsewhere from the world and compare it with that of Kuwait isolates. Such information can aid in developing effective control against emerging variant in the future.
Materials and methods
Data source of complete genome sequences of TYLCV
Complete genome sequences of 234 isolates of Tomato yellow leaf curl virus (TYLCV) reported across the globe were obtained from the sequence repository, GenBank. TYLCV whole genome sequences analyzed were reported from 39 countries (Kuwait 34; Japan 9; Israel 3; South Korea 20; Dominican Republic 2; Portugal 1; Iran 21; Cuba 2; Spain 3; Reunion Island 1; China 53; Sudan 1; Puerto Rico 1; Egypt 1; Mexico 9; Oman 17; Lebanon2; Jordan 4; USA14; Morocco 1; The Netherlands 1; Cameroon 1; Hawaii 1; Guatemala 1; Iraq 1; Turkey1, New Caledonia 1; Sweden 1; Estonia 1; Mauritius 2; Azerbaijan1; French Polynesia 1; Trinidad and Tobago 2; Costa Rica 2; Saudi Arabia 6; Pakistan 5; Australia 1; Italy 4; India 2) representing diverse continents, Asia, Africa, South and North America, Europe, Australia. Only complete genome sequences of TYLCV isolated reported were used for analysis.
DNA polymorphism, evaluation of neutral evolution
DnaSP software was used in determining the nucleotide diversity and DNA polymorphism in the complete genome sequences of TYLCV [12]. To test the theory of neutral evolution, the test statistics such as Tajimas’s D [13], Fu and Li’s D, and Fu and Li’s F [14, 15] were computed in DnaSP software [12, 16, 17].
Genetic differentiation and gene flow estimates
DnaSP was used to compute nucleotide test statistics such as Ks, Kst (Kst value close to zero indicate no differentiation), Snn (Snn value close to one indicates differentiation) [18] and haplotype statistics Hs, Hst [19, 20]. These tests estimate genetic differentiation within the populations of IYSV genotypes. Fst statistics was used to estimate the extent of the gene flow (panmixia or free gene flow has values close to zero whereas infrequent gene flow attains values close to one) [16, 17, 20].
Population selection studies and neutrality test
Mean rates of non-synonymous (dN) and synonymous substitutions (dS) in the coding regions of C1 ORF (encoding replication associated protein which is important for reprograming the host cell cycle and mediating the viral genome rolling circle replication) were calculated using codon-based maximum likelihood methods, i.e., SLAC (single like ancestor counting), and FEL (fixed effects likelihood). DATAMONKEY server [21] was used to calculate dN/dS ratio. FEL analysis was performed on the sequence alignment [22]. Statistical significance is evaluated based on the asymptotic χ2 distribution and it includes site to site synonymous rate variation. Similarly, an adaptive branch-site REL (aBSREL) test for episodic diversification, which is an improved version of the commonly-used “branch-site” models, used to test if positive selection has occurred on a proportion of branches, was performed [23]. Also, a gene-wide model for selection analysis was performed using BUSTED (Branch-site Unrestricted Statistical Test for Episodic Diversification) module.
Molecular phylogeny construction
Multiple sequence alignment (MSA) was performed using MUSCLE algorithm [24]. Best-fit model of nucleotide substitution was determined using MODEL TEST in MEGA 11. Aligned sequence relatedness was evaluated using the Maximum Likelihood phylogenetic analysis that can accurately infer evolutionary relationships among viral sequences (default parameters with 1,000 bootstrap replicates) method based on Tamura parameter 3 model (T92) with Gamma distributed (G) available in MEGA 11 [25].
Recombination detection analysis
Unaligned sequences were loaded in SDT v1.2 program, pairwise scan was performed with the MUSCLE, and the sequence data was saved with minimum identity of 70% and maximum of 100% to ensure sequences were properly aligned. The aligned TYLCV sequences were then used as an input query and analyzed for recombination events using Recombination Detection Program (RDP) v 4.0 [26], BOOTSCAN [27], 3SEQ, GENECONV [28], MAXCHI [29], CHIMAERA [30] and SISCAN [31] available in RDP 4 Beta 4.88. Default settings for the different recombination detection methods and a Bonferroni corrected P-value cut-off of 0.05 was used for analysis [16, 17].
Results
Molecular phylogeny of TYLCV isolates
Phylogenetic tree of the complete genome sequences of TYLCV constructed based on the aligned nucleotide sequences (Fig. 1) using Maximum Likelihood method reveals that all the 34 TYLCV isolates reported from Kuwait formed a distinct clade. Interestingly, most of the isolates reported from Iran, Oman, Saudi Arabia, Pakistan, and some isolates from Japan, Portugal along with Kuwait isolates exhibited monophyletic origin suggesting their genetic relatedness. A clade comprising isolates from Mauritius, Reunion Island, New Caledonia, The Netherlands, Spain was also formed. Another Asiatic clade comprised isolates reported from South Korea, Japan, Israel, Jordan and Egypt. Another clade of the tree is made up on TYLCV isolates reported from USA, Cuba, Trinidad and Tobago, Gautemala, and Mexico. Most of the Chinese isolates were basal to these above mentioned clades suggesting their genetic distinctness.
Fig. 1.
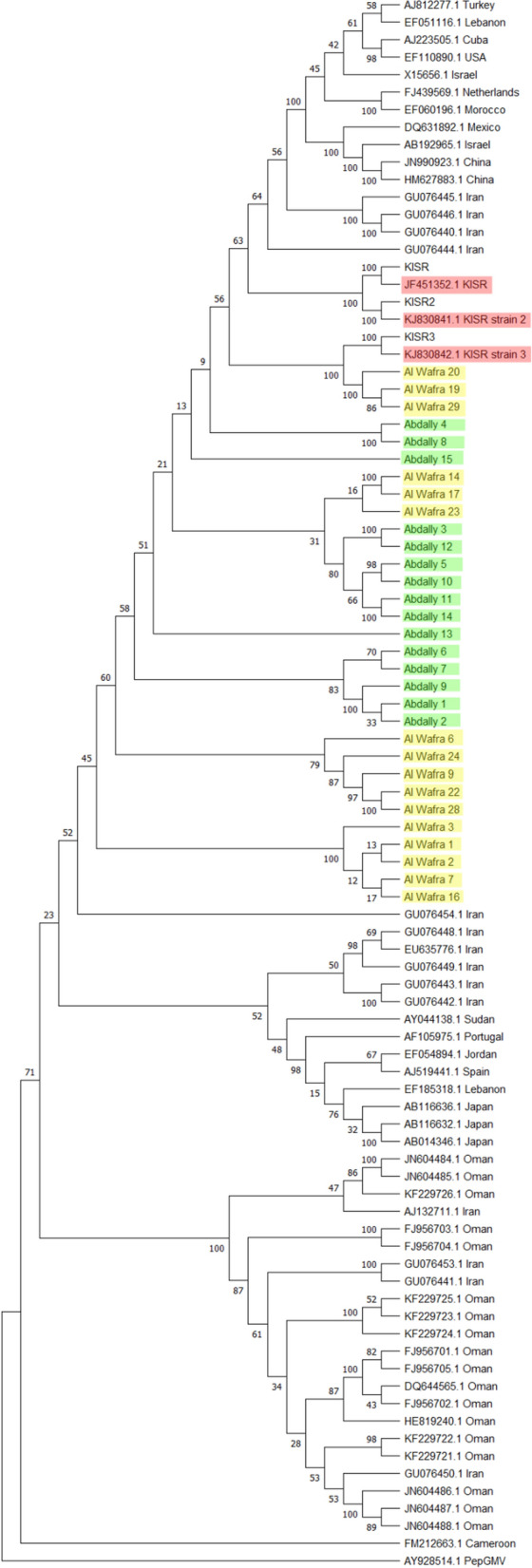
Molecular phylogeny based on the complete nucleotide sequences of Tomato yellow leaf curl virus (TYLCV) isolates reported from Kuwait and other parts of the world. GenBank accession number and place of origin are the indicators of each TYLCV isolate. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test are shown next to the branches
Genetic diversity of TYLCV isolates
The complete genome sequences of TYLCV isolates were used for the determination of genetic diversity, and DNA polymorphism (Table 1). The nucleotide diversity of (π) of TYLCV isolates reported from elsewhere (0.11765) is two folds high than that observed in TYLCV isolates of Kuwait (0.04784) suggesting the genetic relatedness of the Kuwait TYLCV isolates reported. Similarly, haplotype diversity (H)—a measure of probability that two sampled alleles are genetically different, of Kuwait isolates is relatively low (0.9660) than other isolates (0.9968). However, TYLCV isolates of Kuwait show relatively high number (493) of polymorphic sites (S) than isolates reported from other parts of world (432) implying the inherent diversity of Kuwait isolates (Table 1).
Table 1.
Genetic diversity test of Tomato yellow leaf curl virus genotypes reported from Kuwait and elsewhere
| Genotype | N | S | π | Hd |
|---|---|---|---|---|
| Kuwait | 34 | 493 | 0.04784 | 0.9660 |
| Others | 198 | 432 | 0.11765 | 0.9968 |
| All | 232 | 438 | 0.13196 | 0.9963 |
N, Number of isolates; S, Number of polymorphic (segregating) sites; Hd, haplotype diversity; π, nucleotide diversity
Neutrality test
The test of neutral evolution analyzed based on the total number of mutations and segregating sites, revealed negative values of test statistic Tajimas’s D for all the TYLCV isolates (Table 2). It suggests the phenomenon of operation of purifying selection and population expansion in TYLCV. Similarly, negative values of other test statistics such as Fu and Li’s D and Fu and Li’s F also revealed the parallel characteristic feature of TYLCV isolates. Hence, neutrality test analysis of TYLCV isolates reveal that any deleterious mutations in their genomes are removed from the population so as to pave way for population expansion.
Table 2.
Neutrality test of Tomato yellow leaf curl virus genotypes based on total number of mutations
| Genotypes | N | Tajimas’s D | Fu &Li’s D | Fu & Li’s F |
|---|---|---|---|---|
| Kuwait | 34 | − 0.23200 | − 0.21375 | − 0.26161 |
| Others | 198 | − 1.10415 | − 1.41829 | − 1.49866 |
| All | 232 | − 0.92093 | − 1.61201 | − 1.48999 |
Calculated using total number of mutations. *Statistically significant at P < 0.01
Genetic differentiation and gene flow
Haplotype-based statistics (Hs and Hst) and nucleotide-based statistics (Ks, Kst, Snn) were estimated to evaluate inherent genetic differentiation between TYLCV isolates of Kuwait and those reported elsewhere (Table 3). The observance of nucleotide statistic value (Snn) close to one (0.99138) and Kst value (0.05112) denotes greater genetic differentiation between the TYLCV isolates of Kuwait and genomes reported from other parts of the world. Similarly, Fst—a measure of extent of gene flow, reveal that (0.25128) there exists free gene flow or panmixis (Table 3).
Table 3.
Gene flow and genetic differentiation of Tomato yellow leaf curl virus genotypes (TYLCV KUWAIT VS TYLCV OTHERS)
| Genotypes | Hs | Hst | χ2 | P value | Kt | Ks* | Kst* | Snn | Z* | Fst |
|---|---|---|---|---|---|---|---|---|---|---|
| KUWAIT VS OTHERS | 0.99131 | 0.00502 | 232.000 | 0.0063 | 82.47309 | 3.60106 | 0.05112 | 0.99138 | 9.07501 | 0.25128 |
Hs, Hst—measure genetic differentiation based on haplotype statistics
Ks, Kst, Snn, Z—measure genetic differentiation based on nucleotide statistics
Fst—measures extent of gene flows
*Statistically significant at P < 0.05
Genetic recombination among the TYLCV isolates
In order to examine any potential recombination event(s) among the various isolates of TYLCV, Recombination Detection Programme 4 (RDP 4) was used (Table 4). After discarding the potentially weak and inadvertent recombinant signals, eight recombination events (event # 85, 132,65, 58, 88, 63, 76 and 128) were identified. These recombination events were detected by atleast three recombination detection modules in the RDP 4. TYLCV isolates of Kuwait (Al Wafra 19, Al Wafra 20, AlWafra 29, KR108214 Kuwait KISR) were found to be recombinant isolates detected as recombination event #63. TYLCV isolate AB613208-South Korea and KY996463 Trinidad & Tobago served as major and minor parent of this recombination event, respectively (Table 4). Also, KR108214 Kuwait (KISR) isolate was found to be a major parent involved in the generation of TYLCV isolates (EF054894 Jordan and KJ913683 Dominican Republic). Similarly, Kuwait TYLCV isolate (Al Wafra 17) was found to be a minor parent of isolates (EU085423 Iran and KF435136 Saudi Arabia) (Table 4).
Table 4.
Recombination events in Tomato yellow leaf curl virus genomes as detected by Recombination detection program (RDP)
| Recombinant isolate | Parental isolates | Recombination detection program | Recombination event # | p-values | |
|---|---|---|---|---|---|
| Major | Minor | ||||
| AB192965 Japan and AJ812277 Turkey | KT355023 Saudi Arabia | KY965867 USA | GENECONV, MaxChi, Chimaera, SiScan | 85 | 8.192 × 10–11–2.803 × 10–3 |
| AB613209 South Korea | AB636412 South Korea | HQ260984 South Korea | Chimaera, SiScan, 3Seq | 132 | 6.229 × 10–3–4.080 × 10–2 |
| AJ132711 Iran | KU836750 Mexico | HE819240 Oman | GENECONV, Boot Scan, MaxChi, Chimaera, SiScan, 3Seq | 65 | 3.716 × 10–26–2.141 × 10–2 |
| EF054894 Jordan and KJ913683 Dominican Republic | KR108214 Kuwait (KISR) | KX347172 Mauritius | GENECONV, Boot Scan, MaxChi, Chimaera, SiScan, 3Seq | 58 | 3.332 × 10–24–1.407 × 10–12 |
| EU085423 Iran and KF435136 Saudi Arabia | EU635776 Iran | Al Wafra 17 | MaxChi, Chimaera, SiScan, 3Seq | 88 | 6.180 × 10–5–1.162 × 10–2 |
| GU076440 Iran, KY412552 Azerbaijan, Al Wafra 19, Al Wafra 20, AlWafra 29, GU076446 Iran, KC106647 Iran, KR108214 Kuwait KISR, GU076445 Iran | AB613208 South Korea | KY996463 Trinidad & Tobago | GENECONV, Boot Scan, MaxChi, Chimaera, SiScan, 3Seq | 63 | 8.236 × 10–12–1.356 × 10–7 |
| GU076441 Iran, GU076450 Iran | EU085423 Iran | EU635776 Iran | Boot Scan, MaxChi, Chimaera, SiScan, 3Seq | 76 | 2.900 × 10–16–2.643 × 10–5 |
| HE819240 Oman | KY996463 Trinidad & Tobago | KM435324 China | MaxChi, Chimaera, 3Seq | 128 | 6.323 × 10–12–2.896 × 10–7 |
Natural selection analyses
Codons of gene encoding replication associated protein (C1) that are in positive or negative selection pressure would provide knowledge regarding its molecular evolution pattern. A comprehensive analysis was performed using individual site based models such as FEL (fixed effects likelihood), SLAC (single like ancestor counting) and individual branch site models aBRSEL (an adaptive branch-site REL) and gene-wide model BUSTED (Branch-site Unrestricted Statistical Test for Episodic Diversification). FEL analysis revealed that 161 sites of C1 ORF are under purifying selection in TYLCV isolates reported from elsewhere whereas 88 codon sites are undergoing purifying selection in TYLCV isolates reported from Kuwait. Similarly, the number of codon sites undergoing diversifying positive selection is at 16 for TYLCV others compared to 3 for TYLCV from Kuwait (Table 5).
Table 5.
Codon substitution studies in the replication associated protein gene (C1) of TYLCV isolates [dN, the number of non-synonymous substitutions per non-synonymous site; dS, the number of synonymous substitutions per synonymous site]
| Genotype | Sites under diversifying/positive selection at p ≤ 0.1 | Sites under purifying/negative selection at p ≤ 0.1 | ω = dN/dS |
|---|---|---|---|
| Fixed Effects Likelihood (FEL) analysis | |||
| TYLCV others | 16 | 161 | – |
| TYLCV Kuwait | 3 | 88 | – |
| Single-Likelihood Ancestor Counting (SLAC) | |||
| TYLCV others | 0 | 131 | 0.331 |
| TYLCV Kuwait | 0 | 25 | 0.239 |
| adaptive Branch Site (aBSREL) | |||
| TYLCV others | 13 | – | – |
| TYLCV Kuwait | 4 | – | – |
| Fast Unconstrained Bayesian AppRoximation (FUBAR) | |||
| TYLCV others | 8 | 165 | – |
| TYLCV Kuwait | 5 | 60 | – |
| Branch-site Unrestricted Statistical Test for Episodic Diversification (BUSTED) | |||
| TYLCV Others | 129 | – |
0.2466 (constrained) 0.5145 (unconstrained) |
| TYLCV Kuwait | 14 | – |
0.1426 (constrained) 1.246 (unconstrained) |
SLAC analysis utilizes a combination of maximum-likelihood (ML) and counting approaches to infer non-synonymous (dN) and synonymous (dS) substitution rates on a per-site basis for a given coding alignment and corresponding phylogeny. A global MG94xREV model fit was performed to optimize branch length and nucleotide substitution parameters before proceeding to hypothesis testing along with Nucleotide GTR model in SLAC analysis. SLAC analysis revealed that 131 sites of C1 ORF are under purifying selection in TYLCV isolates reported from elsewhere whereas 25 codon sites are undergoing purifying selection in TYLCV isolates reported from Kuwait (Supplementary Figs. 1 and 2).
A total of 273 branches were formally tested for diversifying selection among the TYLCV isolates reported from elsewhere and aBSREL found evidence of episodic diversifying selection on 13 out of 273 branches in the phylogeny. Significance was assessed using the Likelihood Ratio Test at a threshold of p ≤ 0.05, after correcting for multiple testing. Full adaptive model aBSREL which infers an optimized number of ω rate categories per branch estimated 636 parameters with a log likelihood of model fit (− 20,786.21). Similarly, a total of 29 branches were formally tested for diversifying selection when TYLCV isolates from Kuwait were studied. Evidence of episodic diversifying selection was found on 4 out of 29 branches in the phylogeny. Full adaptive model aBSREL estimated 84 parameters with a log likelihood of model fit (− 4811.18). Also, based on the likelihood ratio test, there is evidence of episodic diversifying selection in the TYLCV isolates of Kuwait (p = 1.710e−13). Among the coding sequences of TYLCV Kuwait isolates, 14 sites were found to have undergone positive selection with evidence ratio threshold > 10, whereas 129 sites were found to have positive selection when TYLCV others sequences were analyzed. Application of Bayesian approach to identify nonsynoymous (dN) and synonymous (dS) substitution rates via Fast Unconstrained Bayesian AppRoximation (FUBAR) revealed that 165 and 60 codon sites are under purifying selection in TYLCV others and TYLCV Kuwait isolates, respectively. FUBAR found 8 and 5 codon sites were undergoing positive selection in TYLCV others and TYLCV Kuwait isolates, respectively suggesting negative selection is predominant.
Discussion
Genetic diversity analysis of viral species has attained greater interests since the molecular evolutionary features of the viruses directly affect the realm of host-virus interactions [32, 33]. Genetic diversity analysis of TYLCV in plants and insect vectors has provided molecular insights regarding the spectrum of viral genomic variations [34–36]. In general, mutations, genome segment reassortment, genetic drift, genetic recombination, migration are the important sources of evolutionary modifications in defining the genetic architecture of viral populations [37–40]. Determination of geographical distribution of viral population and the inherent genetic variation, via phylo-geographic evolutionary analysis is also rewarding and has become an important analytical module in the field of viral genomics [39, 41, 42] (Ifthikhar et al. 2014). Recently we published molecular sequence features of TYLCV isolates characterized from the tomato fields of Kuwait (BMC research notes ref). The TYLCV genomes of Kuwait isolates reported earlier showed considerable genetic diversity. As a follow up, molecular evolutionary genomic analysis of TYLCV isolates reported across the globe is compared with that of isolates reported from Kuwait. Unlike the partial or complete gene sequence-based phylogenetic analysis reported earlier, complete viral genomes are quite informative for evolutionary analysis [39, 43] (Ifthikhar et al. 2014).
Genetic structure of begomovirus population results from the interplay of the evolutionarily important adaptations both in various plants and strains of vectors which immensely determine the geographical distribution and dispersion of the viral species. Identification of factors driving genetic variability within the population is crucial for estimating the population expansion and devising suitable control measure or disease management tactics. Earlier studies on genetic diversity and population structure of TYLCV in Spain, during an 8 year period, showed low genetic diversity and high genetic stability [44] in Italy, co-existence of TYLCV and TYLCSV [45], and TYLCV population structure in Mediterranean basin was shaped due to recombination [46]. A global analysis of tomato yellow leaf curl virus in Iran and the Arabian Peninsula using complete genome sequences of TYLCV strains has revealed that TYLCV-IL isolates from southern Iran possessed greater genetic variability than the northeastern isolates and identified recombination hot spots [10]. It also suggested that molecular evolution could be attributed due to the geographical isolation of the virus isolates and Iran was proposed as a TYCLV centre of diversity [10]. The geographic proximity of the Kuwait TYLCV isolates (BMC research notes) with that of Iran isolates and genetic distinctness of Kuwait isolates from others reported warrant an in depth molecular evolutionary genomic analysis. The results of this study reveal the monophyletic origin of Kuwait isolates with that from Middle East further corroborates the assertion that the region is centre of TYLCV genetic diversity (Fig. 1) [3, 10]. Despite the centre of diversity of TYLCV, the geographic isolation of the TYLCV strains (Fig. 1) confirm that these isolates do not pose a serious threat to global epidemiology of TYLCV infection [3]. This fact is also further confirmed from the genetic or nucleotide diversity of TYLCV isolates reported from Kuwait which is relatively low than that of nucleotide diversity of TYLCV others. Findings of neutrality tests and gene flow estimates suggest that any deleterious mutations in the genomes of TYLCV Kuwait isolates is removed so that purifying selection functions paving way for population explosion. It is further supported due to the high degree of gene flow between Kuwait and other isolates of TYLCV. The number of sites under positive selection and sites under purifying selection among the TYLCV isolates reveal that most of the genetic elements of C1 ORFs are neither under positive nor negative selection suggesting the neutral evolution of these codons.
Recombination analysis has been at the forefront of detection of genetic changes in viral genomes. Unlike recombination events detected in a relatively short genetic elements of a particular gene [16, 39], eight potential recombination events detected among the TYLCV isolates mostly involving Kuwait origin either as a recombinant or as the parents in the generation of recombinant strains suggest that the Middle Eastern region continues to contribute to the diversity of the TYLCV strains globally. Nevertheless, the fitness advantage of these recombinants vis-à-vis inter-species TYLCV recombinants reported earlier would be an interesting area of research [47]. A recent analysis of C1 protein of TYLCV sequences derived from five major geographically distinct Asian countries (India, China, Iran, Oman and S. Korea) reveal a significant differences among the gene sequences across geographical regions. Also, the pressures of selection and mutation shape the codon usage bias of the C1 gene of TYLCV [48]. Similar analysis of codon usage bias in TYLCV C1 genes reported worldwide could provide novel insights. TYLCV C1 ORF codon substitution analysis utilizing multiple algorithms also imply that most of the codons are under purifying or negative selection so that deleterious changes in the gene sequences are not tolerated.
Conclusion
The inherent genetic diversity of TYLCV Kuwait isolates and its contribution to the generation of potential genetic recombinant isolates are presented. Analysis of population structure of TYLCV across the globe reveals that it is under purifying selection and the phenomenon of population expansion is occurring. This report thus forms a groundwork for further investigation of molecular evolutionary genomics of one of the most devastating plant Geminiviruses.
Supplementary Information
Author contributions
Abrar Akbar: wrote the original draft of manuscript and performed bioinformatic analysis Hanadi Al hashash: contributed to experiment and data generation Ebtisam Al ali: manuscript proof reading, discussion and experimental design.
Funding
This project was funded by Kuwait foundation for the advancement of science (Grant no: PR18-12SL-09).
Availability of data and materials
The viral genome sequence datasets generated and described in this research are deposited in the NCBI GenBank database (ncbi.nlm.nih.gov) and are available under accession numbers OM691678 to OM691692 and OL890666 to OL890680). Data is available in the manuscript.
Declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Prasad A, Sharma N, Hari-Gowthem G, Muthamilarasan M, Prasad M. Tomato yellow leaf curl virus: impact, challenges, and management. Trends Plant Sci. 2020;25(9):897–911. [DOI] [PubMed] [Google Scholar]
- 2.Mabvakure B, Martin DP, Kraberger S, Cloete L, van Brunschot S, Geering AD, Thomas JE, Bananej K, Lett JM, Lefeuvre P, Varsani A. Ongoing geographical spread of Tomato yellow leaf curl virus. Virology. 2016;498:257–64. [DOI] [PubMed] [Google Scholar]
- 3.Lefeuvre P, Martin DP, Harkins G, Lemey P, Gray AJ, Meredith S, Lakay F, Monjane A, Lett JM, Varsani A, Heydarnejad J. The spread of tomato yellow leaf curl virus from the Middle East to the world. PLoS Pathog. 2010;6(10): e1001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moriones E, Navas-Castillo J. Tomato yellow leaf curl virus, an emerging virus complex causing epidemics worldwide. Virus Res. 2000;71:123–34. [DOI] [PubMed] [Google Scholar]
- 5.Czosnek H, Hariton-Shalev A, Sobol I, Gorovits R, Ghanim M. The incredible journey of begomoviruses in their whitefly vector. Viruses. 2017;9(10):273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee H, Song W, Kwak HR, Kim JD, Park J, Auh CK, Kim DH, Lee KY, Lee S, Choi HS. Phylogenetic analysis and inflow route of Tomato yellow leaf curl virus (TYLCV) and Bemisia tabaci in Korea. Mol Cells. 2010;30(5):467–76. [DOI] [PubMed] [Google Scholar]
- 7.Briddon RW, Patil BL, Bagewadi B, Nawaz-ul-Rehman MS, Fauquet CM. Distinct evolutionary histories of the DNA-A and DNA-B components of bipartite begomoviruses. BMC Evol Biol. 2010;8(10):97. 10.1186/1471-2148-10-97.PMID:20377896;PMCID:PMC2858149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poornima Priyadarshini CG, Ambika MV, Tippeswamy R, Savithri HS. Functional characterization of coat protein and V2 involved in cell to cell movement of Cotton leaf curl Kokhran virus-Dabawali. PLoS ONE. 2011;6(11): e26929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.García-Arenal F, Fraile A, Malpica JM. Variability and genetic structure of plant virus populations. Ann Rev Phytopathol. 2001;39:157–86. [DOI] [PubMed] [Google Scholar]
- 10.Hosseinzadeh MR, Shams-Bakhsh M, Osaloo SK, et al. Phylogenetic relationships, recombination analysis, and genetic variability among diverse variants of tomato yellow leaf curl virus in Iran and the Arabian Peninsula: further support for a TYLCV center of diversity. Arch Virol. 2014;159:485–97. [DOI] [PubMed] [Google Scholar]
- 11.Al-Ali E, Al-Hashash H, Akbar A, et al. Genetic recombination among tomato yellow leaf curl virus isolates in commercial tomato crops in Kuwait drives emergence of virus diversity: a comparative genomic analysis. BMC Res Notes 2023;16:71. 10.1186/s13104-023-06319-w [DOI] [PMC free article] [PubMed]
- 12.Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol Biol Evol. 2017;34:3299–302. 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- 13.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu Y-X, Li W-H. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu Y-X. Statistical tests of neutrality against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iftikhar R, Ramesh SV, Bag S, Ashfaq M, Pappu HR. Global analysis of population structure, spatial and temporal dynamics of genetic diversity, and evolutionary lineages of Iris yellow spot virus (Tospovirus: Bunyaviridae). Gene. 2014;547(1):111–8. 10.1016/j.gene.2014.06.036 [DOI] [PubMed] [Google Scholar]
- 17.Tabassum S, Adnan S, Khan FR. Gingival retraction methods: a systematic review. J Prosthodont. 2017;26(8):637–43. 10.1111/jopr.12522. [DOI] [PubMed] [Google Scholar]
- 18.Hudson RR. A new statistic for detecting genetic differentiation. Genetics. 2000;155(4):2011–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hudson RR, Boos DD, Kaplan NL. A statistical test for detecting geographic subdivision. Mol Biol Evol. 1992a;9(1):138–51. [DOI] [PubMed] [Google Scholar]
- 20.Hudson RR, Slatkin M, Maddison WP. Estimation of levels of gene flow from DNA sequence data. Genetics. 1992b;132:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weaver S, Shank SD, Spielman SJ, Li M, Muse SV, Kosakovsky Pond SL. Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol Biol Evolut. 2018;35(3):773–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kosakovsky Pond SL, Frost SD. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol. 2005;22(5):1208–22. [DOI] [PubMed] [Google Scholar]
- 23.Smith MD, Wertheim JO, Weaver S, Murrell B, Scheffler K, Kosakovsky Pond SL. Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol Biol Evol. 2015;32(5):1342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. 10.1093/nar/gkh340.PMID:15034147;PMCID:PMC390337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamura K, Stecher G, Kumar S. MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol. 2021;38:3022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin D, Rybicki E. RDP: detection of recombination amongst aligned sequences. Bioinformatics. 2000;16:562–3. [DOI] [PubMed] [Google Scholar]
- 27.Salminen MO, Carr JK, Burke DS, McCutchan FE. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res Hum Retroviruses. 1995;11(11):1423–5. [DOI] [PubMed] [Google Scholar]
- 28.Sawyer SA. GENECONV: A computer package for the statistical detection of gene conversion. 1999.
- 29.Maynard SJ. Analyzing the mosaic structure of genes. J Mol Evol. 1992;34:126–9. [DOI] [PubMed] [Google Scholar]
- 30.Posada D, Crandall KA. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci U S A. 2001;98(24):13757–62. 10.1073/pnas.241370698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibbs MJ, Armstrong JS, Gibbs AJ. Sister‐scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000;16:573–582. http://www.anu.edu.au/BoZo/software/. [DOI] [PubMed]
- 32.Gibbs A, Ohshima K. Potyviruses and the digital revolution. Annu Rev Phytopathol. 2010;48:205–23. [DOI] [PubMed] [Google Scholar]
- 33.Sacristán S, García-Arenal F. The evolution of virulence and pathogenicity in plant pathogen populations. Mol Plant Pathol. 2008;9(3):369–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al-Abedy AN, Kadhim JH, Abdalmoohsin RG, Al-Taey DK. Genetic diversity of tomato yellow leaf curl virus isolates and the effect of virus on the hormones content of tomato (Solanum lycopersicum) plants. Res Crops. 2021;22(2):347–55. [Google Scholar]
- 35.Hassan SQ, Abedy AN, Kadhim JH, Sahi GM. Genetic diversity of tomato yellow leaf curl virus (TYLCV) and its control. Indian J Ecol. 2020;47:164–70. [Google Scholar]
- 36.Yang X, Wang B, Luan J, et al. Molecular variation of tomato yellow leaf curl virus in the insect vector Bemisia tabaci. Sci Rep. 2017;7:16427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Butkovic A, Gonzalez R, Elena SF. Revisiting Orthotospovirus phylogeny using full-genome data and testing the contribution of selection, recombination and segment reassortment in the origins of members of new species. Arch Virol. 2021;166:491–9. [DOI] [PubMed] [Google Scholar]
- 38.Moya A, Holmes EC, Gonzalez-Candelas F. The population genetics and evolutionary epidemiology of RNA viruses. Nat Rev Microbiol. 2004;2:279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tabassum A, Ramesh SV, Zhai Y, Iftikhar R, Pappu HR. Viruses without borders: global analysis of the population structure, haplotype distribution, and evolutionary pattern of Iris yellow spot (Family Tospoviridae, Genus Orthotospovirus). Front Microbio. 2021. 10.3389/fmicb.2021.633710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iftikhar R, Ramesh SV, Bag S, Ashfaq M, Pappu HR. Global analysis of population structure, spatial and temporal dynamics of genetic diversity, and evolutionary lineages of Iris yellow spot virus (Tospovirus: Bunyaviridae). Gene. 2014;547(1):111-8. 10.1016/j.gene.2014.06.036. [DOI] [PubMed]
- 41.Chen S, Xing Y, Su T, Zhou Z, Dilcher DL, Soltis DE. Phylogeographic analysis reveals significant spatial genetic structure of Incarvillea sinesisas a product of mountain building. BMC Plant Biol. 2012;12:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hewitt GM. Genetic consequences of climatic oscillations in the quaternary. Philos Trans R Soc Lond B Biol Sci. 2004;359:183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benítez-Galeano MJ, Castells M, Colina R. The evolutionary history and spatiotemporal dynamics of the NC lineage of citrus Tristeza Virus. Viruses. 2017;9:272. 10.3390/v9100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sánchez-Campos S, Díaz JA, Monci F, Bejarano ER, Reina J, Navas-Castillo J, Aranda MA, Moriones E. High genetic stability of the begomovirus Tomato yellow leaf curl Sardinia virus in southern Spain over an 8-year period. Phytopathology. 2002;92(8):842–9. [DOI] [PubMed] [Google Scholar]
- 45.Davino S, Napoli C, Davino M, Accotto GP. Spread of Tomato yellow leaf curl virus in Sicily: partial displacement of another geminivirus originally present. Eur J Plant Pathol. 2006;114(3):293–9. [Google Scholar]
- 46.García-Andrés S, Accotto GP, Navas-Castillo J, Moriones E. Founder effect, plant host, and recombination shape the emergent population of begomoviruses that cause the tomato yellow leaf curl disease in the Mediterranean basin. Virology. 2007;359(2):302–12. [DOI] [PubMed] [Google Scholar]
- 47.Urbino C, Regragui ZF, Granier M, Peterschmitt M. Fitness advantage of inter-species TYLCV recombinants induced by beneficial intra-genomic interactions rather than by specific mutations. Virology. 2020;542:20–7. [DOI] [PubMed] [Google Scholar]
- 48.Mn M, Suresh KPH, Patil SS, Indrabalan UB, Beelagi MS, Pradeep S, Paramanandham K, Jacob SS, Srinivasa C, Kollur SP, Achar RR. Research Article A new informatics framework for evaluating the codon usage metrics, evolutionary models and phylogeographic reconstruction of Tomato yellow leaf curl virus (TYLCV) in different regions of Asian countries. Int J Health Allied Sci. 2022;11(1):15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The viral genome sequence datasets generated and described in this research are deposited in the NCBI GenBank database (ncbi.nlm.nih.gov) and are available under accession numbers OM691678 to OM691692 and OL890666 to OL890680). Data is available in the manuscript.