

Summary
Repeat expansions in the C9orf72 gene are the most common genetic cause of (ALS) and frontotemporal dementia (FTD). Like other genetic forms of neurodegeneration, pinpointing the precise mechanism(s) by which this mutation leads to neuronal death remains elusive, and this lack of knowledge hampers the development of therapy for C9orf72-related disease. We used an agnostic approach based on genomic data (n = 41,273 ALS and healthy samples, and n = 1,516 C9orf72 carriers) to overcome these bottlenecks. Our drug-repurposing screen, based on gene- and expression-pattern matching and information about the genetic variants influencing onset age among C9orf72 carriers, identified acamprosate, a γ-aminobutyric acid analog, as a potentially repurposable treatment for patients carrying C9orf72 repeat expansions. We validated its neuroprotective effect in cell models and showed comparable efficacy to riluzole, the current standard of care. Our work highlights the potential value of genomics in repurposing drugs in situations where the underlying pathomechanisms are inherently complex.
Video abstract
Keywords: C9orf72, amyotrophic lateral sclerosis, frontotemporal dementia, age at onset, drug repurposing, acamprosate, translation
Graphical abstract
Highlights
-
•
Repeat expansions in C9orf72 are the most common genetic cause of ALS and FTD
-
•
The genetic risk of general ALS modifies the age at onset in C9orf72 cases
-
•
We performed a drug screen based on the genetic variants influencing age at onset
-
•
We identified acamprosate, a GABA analog, as a potential treatment for C9orf72
Saez-Atienzar et al. identify genetic variants influencing the age at onset among patients carrying C9orf72 repeat expansions. A drug screen based on these variants revealed acamprosate, a GABA analog, as a potentially repurposable treatment for C9orf72-related disease. The work underscores the potential of leveraging large-scale genomic data for drug repurposing.
Introduction
A repeat expansion within the C9orf72 gene is a common cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), two neurological disorders that result in the deaths of ∼17,000 Americans and Europeans annually.1,2,3,4 This genetic disease accounts for 1 in 10 ALS and FTD cases of European descent, and many carriers in the general population have a near-complete chance of manifesting symptoms during their lifetime.5,6 The exact processes by which this repeat expansion leads to neuronal death are not fully understood, although several mechanisms, such as dipeptide production, RNA toxicity, disruption of nucleocytoplasmic transport, and haploinsufficiency, have been suggested.7 This lack of molecular knowledge is a common theme across neurodegenerative disorders, where there is often a complex interplay among multiple pathways and cellular processes.8,9,10,11
The molecular complexity underlying neurodegenerative diseases also hampers drug discovery; ameliorating a single aspect of a cellular network may not be beneficial as it does not address the other pathological processes co-occurring within the cell.12 Traditional linear drug-discovery efforts, where millions of compounds are tested against a single target, are likely to fail in the face of such multidimensional conditions, and this disconnect alone may account for the high failure rate observed among clinical trials in neurodegenerative diseases.13 Genomic and transcriptomic data offer a potential solution, as these data types inherently capture the multifaceted nature of neurological diseases. We can exploit this information to match drugs that restore entire networks and systems, even when the target pathways or the mechanism of action of the drug are not fully understood.14,15 Compounds supported by genetic evidence are also more likely to succeed in clinical trials and to gain drug approval.16,17
In this context, we have used a massive genomic dataset to identify the genetic variants influencing age at onset among patients carrying the C9orf72 repeat expansion. Building on this genomic information, we have leveraged gene- and expression-pattern matching and pathway modeling to nominate potentially repurposable drugs for C9orf72-related ALS/FTD. We focused on onset age because of the wide range observed in this common genetic form of neurodegeneration, spanning from the fourth to the tenth decade of life, representing a natural experiment within the ALS population.5,18 Genetic factors are known to play a role in this variable age-at-onset presentation,19 and research in other neurological diseases shows that the phenotypic manifestations of high-risk pathogenic variants are influenced by minor effect variants elsewhere in the genome.20,21
Our pipeline nominated acamprosate, an oral medication used to manage alcohol use disorder, as a potential repurposable drug for slowing progression among symptomatic individuals and delaying disease onset among C9orf72 carriers. In vitro data demonstrated a neuroprotective effect of acamprosate in motor neurons derived from these patients. Crucially, our discovery approach evolved from the notion that genomics can nominate specific medications in an agnostic, data-driven manner, even when precise knowledge about disease mechanisms or drug pharmacodynamics is lacking.
Results
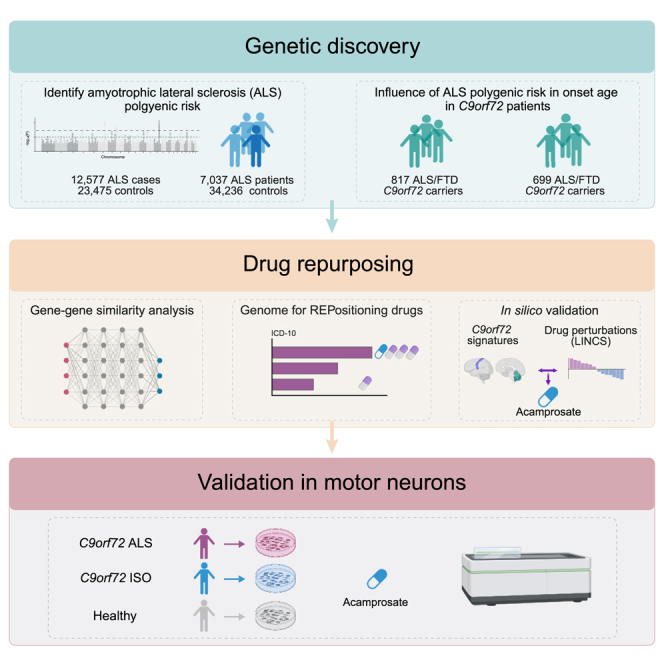
Figure 1 shows a graphical representation of our genetic workflow and drug-repurposing pipeline.
Figure 1.

Schematic illustration of the analytical workflow
The genetic risk score for sporadic ALS was generated using large cohorts as the reference and training sets. This general ALS genetic risk score was then calculated for a sizable cohort of C9orf72 carriers. Follow-up analyses included pathway analysis and the identification of individual loci with a major contribution to the age at onset. Using the information obtained from these genetic analyses, we performed drug repurposing based on gene-gene-pattern matching and expression-pattern matching to identify drugs that may delay symptom onset among C9orf72 carriers. In vitro drug validation confirmed the neuroprotective effect of the drug nominated by this approach.
Sporadic ALS risk variants modify the onset age among C9orf72-related ALS/FTD
We built a polygenic risk profile for general ALS using 7,037 cases not carrying the C9orf72 repeat expansion and 34,236 healthy controls. The best-fit model included 161 SNPs (odds ratio for the model = 1.125, 95% confidence interval [CI] = 1.093–1.156, p = 1.5 × 10−16). Figure 2 and Table S1 describe the genetic variants and mapped genes that make up the polygenic risk score for general ALS.
Figure 2.

Genetic variants influencing symptom onset age among ALS/FTD patients carrying the C9orf72 repeat expansion
The ideograms show the 161 ALS genetic risk loci making up the general ALS polygenic risk score. Labels with red text denote the 16 SNPs making up decile 10. The nearest genes to the variants are displayed. The colors of the circles correspond to the gene type: dark blue, RNA gene; light blue, protein-coding gene; red, pseudogene; black, intergenic. The numbers at the top indicate the chromosome. Interg, intergenic.
See also Table S1.
Next, using the 161 SNPs, we investigated the association of the general ALS genetic risk score with age at onset in a cohort of 1,516 ALS/FTD C9orf72 expansion carriers. We found the risk of general ALS (i.e., independent of the C9orf72 repeat expansion) was significantly associated with onset age among the C9orf72 expansion carriers (meta-analysis p = 1.5 × 10−3, β = −0.781, 95% CI = −1.263 to −0.299; Figures 3A–3C; Table S2). We observed similar results when analyzing ALS C9orf72 and FTD C9orf72 carrier patients alone (Figure S1). In contrast, the general ALS genetic risk score did not influence the age at onset among patients without C9orf72 repeat expansions (p = 0.437, β = 0.115, 95% CI = −0.175 to 0.404; Figures 3C and S2; Table S2).
Figure 3.

The ALS genetic risk score significantly influences onset age in C9orf72 carriers
(A and B) The regression lines show the association of the ALS genetic risk scores and age at onset in (A) the test dataset (n = 817 ALS/FTD C9orf72 carriers, p = 0.024, β = −0.765, 95% CI = −1.429 to −0.101) and (B) the replication dataset (n = 699, p = 0.026, β = −0.799, 95% CI = −1.499 to −0.099). The shadow areas represent the 90% confidence interval of the regression model.
(C) The forest plot shows the results of the meta-analysis of the test and replication datasets (p = 1.5 × 10−3, β = −0.781, 95% CI = −1.263 to −0.299).
See also Figures S1 and S2 and Table S2.
A subset of 16 SNPs drives the early-onset age among C9orf72-related ALS/FTD
Having established that the general ALS risk influenced the age at onset among C9orf72 patients, we next determined which of the 161 SNPs in the model was driving the effect.21 To do this, we performed a leave-one-out analysis to evaluate the contribution of each SNP to the onset age. This enabled us to rank the 161 SNPs and group the variants into 10 deciles (Table S3). We recalculated the genetic risk scores using the SNPs from each decile and used a linear regression model to test these refined genetic risk scores for association with age at onset.22,23,24,25,26,27 Figure S3 shows a schematic representation of this approach.
Decile 10 contained the 16 SNPs with the most significant association with earlier onset age (Figures 4A and 4B). A 1-SD increase in the genetic risk score of the decile 10 SNPs corresponded to a decrease of 2.17 years (95% CI = 1.52–2.81) in the age at onset, and there was a 4-SD difference between C9orf72 carriers at the extremes of the genetic risk score distribution. This difference implies that individuals at the highest end of the genetic risk score distribution developed the disease, on average, 8 years (8.68 years, 95% CI = 6.08–11.24; Figure 4B) earlier than their counterparts at the lowest end. In contrast, these loci did not alter the onset age among ALS patients not carrying the C9orf72 repeat expansion (Figure S4).
Figure 4.

Contribution of individual SNPs to age at symptom onset among C9orf72 patients
(A) The forest plot shows the effect size of each decile obtained by ranking the 161 individual SNPs based on their effect on age at onset in C9orf72 patients.
(B) The regression lines show the association between the ALS genetic risk scores and age at onset in 817 ALS/FTD C9orf72 carriers based on the 16 SNPs of decile 10 (n = 817 ALS/FTD C9orf72 carriers, p = 8.97 × 10−11, β = −2.17, 95% CI = −2.81 to −1.52). The shadow area represents the 90% confidence interval of the regression model.
(C) Sankey diagram showing the functional enrichment of decile 10 genes, based on Gene Ontology terms. Only gene lists that contain between 5 and 500 genes were selected for the analysis. The significant threshold was an false discovery rate (FDR)-corrected p < 0.05.
See also Figures S3–S5 and Tables S3, S4, and S7.
Cytoskeletal and axonal transport pathways influence C9orf72 age at onset
Enrichment analysis of the SNPs in decile 10 revealed that the cytoskeletal and axonal transport pathways influenced the age at onset among patients carrying the C9orf72 repeat expansion (Figures 4C and S5; Table S4). These data suggested that these biological processes are essential in determining the onset of C9orf72-related disease.
The two-step design of our drug-repurposing pipeline
We used our genetic data to perform a drug-repositioning analysis to discover medications that may delay the age at disease onset among C9orf72 patients. This approach harnesses our insights into the genetic factors influencing the variable age at onset in C9orf72-related disease and existing databases containing information on drug effects.28 A critical advantage of our pipeline is that it identifies drugs with broad, heterogeneous effects for use in complex human diseases like neurodegenerative conditions. Crucially, this approach is not based on a single change within the cell but instead relies on gene-pattern and expression-pattern matching to select medications that correct the disrupted networks.
The pipeline comprises two complementary parts (see Figure 1 for a graphical representation of these steps). In the first step, we performed gene-drug-pattern matching. To do this, we used the g:SNPense function of g:Profiler229 to identify the genes related to the SNPs in decile 10. These genes were then used as search terms (defined as seed genes in Table S5) in the Geneshot web server (accessed October 2021) to identify a broader list of functionally related genes.30 This database identifies genes associated with the search term based on their co-occurrence in publications and gene-gene similarity from human RNA sequencing (RNA-seq) data (ARCHS4).30 The resulting list of genes plus the seed genes (Table S5) was then used as input for the Genome for REPositioning drugs (GREP) analysis (version 1.0.0), which is a software package designed to identify drugs that target the gene set based on their enrichment in clinical indication categories.31 The gene-drug software used Fisher’s exact test to perform pharmacological enrichment and output the names of drugs associated with the gene set.
In the second step of our pipeline, we used gene expression-pattern matching to refine and narrow the candidate drug list. In essence, this approach identifies compounds that reverse the transcription patterns observed in brain tissue obtained from C9orf72 patients (n = 44 cases and 76 healthy controls). This method is widely used in drug repositioning to assess how well drugs can counteract disease-related gene expression patterns by comparing their effects to a disease gene signature.32 To do this, the drug perturbations were queried using the Library of Integrated Network-Based Cellular Signatures database,33 containing the differential expression analysis of 12,328 genes from 8,140 compound treatments of 30 cell lines. A bidirectional weighted Kolmogorov-Smirnov enrichment statistic test of gene expression ranks in the disease and the expression values of the drug signatures was used to assign a connectivity map (CMap) score to each drug, reflecting the degree to which the drug “flips” the gene expression signature of the disease.
Step 1: Drug prioritization using gene-drug-pattern matching
The genomic-based GREP analysis nominated 52 medications approved for human use that could be repurposed to delay onset among C9orf72 carriers (Figure 5A; Table S6). To investigate the targets of the nominated medications, we explored the significant biomedical terms associated with these drugs in the Drugmonizome database (accessed October 12, 2022).34 These therapies were enriched for CNS targets and are typically prescribed for anxiety disorders and epilepsy; exploring their mechanisms of action revealed the γ-aminobutyric acid (GABA) receptor as potentially relevant (Figure 5B).
Figure 5.

Repurposing drugs to delay onset among C9orf72 carriers
The figure shows the results obtained from our drug-repositioning pipeline.
(A) Enriched terms derived from the GREP software package, which is based on the Anatomical Therapeutic Chemical classification. Blue indicates the significant “other nervous system drugs” category. Some of the drugs within this category are shown (see Table S6 for a complete list).
(B) Lollipop plots depict drug enrichment analysis for different categories, such as disease indication, gene target, and mechanism of action. Information was obtained from the Drugmonizome database, and the x axis depicts the enrichment corrected p value, which uses Bonferroni correction for the disease indication and the gene target plots and FDR for the mechanism of action.
(C) Drug perturbation data were obtained from the LINCS database. The graphs show the CMap scores for the selected drugs across cell types. CMap scores are determined using a bidirectional weighted Kolmogorov-Smirnov enrichment statistic test, which compares gene expression changes in the disease and drug signatures to quantify the extent to which the drug effectively reversed (flipped) the gene expression signature associated with the disease. Lower scores indicate a more substantial potential for therapeutic effectiveness. An average CMap (diamond shape) was calculated using the normalized connectivity score to evaluate the overall effect of each drug across the tested cell lines. Drugs with a reversal potential were selected if (1) they depicted a negative average CMap and (2) they showed a negative or neutral (measured as 0) CMap score for each cell line (circle shape). A375, ASC, FIBRNPC, HCC515, HT29, NEU, A549, NPC, HA1E, PC3, MCF7, PHH, SKB, and VCAP refer to the cell line types available in the LINCS database (see STAR Methods for details).
(D) The Venn diagram shows the drugs that fulfilled these criteria in the motor cortex and cerebellum. Of these, acamprosate was selected for additional in vitro validation.
See also Figures S5 and S6.
Step 2: Drug prioritization using expression-pattern matching
The drug-disease expression-pattern matching Library of Integrated Network-Based Cellular Signatures (LINCS) analysis found that 3 of our 52 selected drugs demonstrated reversal of the C9orf72 transcriptomic disease signature across multiple cell lines: acamprosate, chlordiazepoxide, and alprazolam (Figures 5C and 5D). Acamprosate was chosen from this list for the following reasons. First, acamprosate demonstrated neuroprotective properties in a SOD1G93A rat spinal cord model of ALS.35 Second, and perhaps most important, in contrast to chlordiazepoxide and alprazolam, acamprosate is not associated with sedation or respiratory depression, which are potentially hazardous side effects in the ALS population.36,37
Acamprosate is neuroprotective in a patient-derived C9orf72 cell model
We assessed dose-response curves and potential levels of toxicity of acamprosate in motor neurons derived from induced pluripotent stem cells (iPSCs) obtained from two ALS patients carrying the C9orf72 repeat expansion, two healthy individuals, and one isogenic control line (see Figure 6A for an outline of the workflow and Figure S6 for a representative immunohistochemistry staining of cell cultures). The drug did not exert a toxic effect on C9orf72 or healthy motor neurons, even at the higher doses (30 μM for 72 h, viability measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay; Figures S7 and S8). Using cleaved caspase-3, an established biochemical proxy for cell survival in C9orf7238,39,40 and other genetic forms of ALS,41 acamprosate strongly reduced cell death in C9orf72-derived motor neurons; the percentage of cleaved caspase-3+ cells was 1.8-fold lower at 10 μM, and 2.3-fold lower at 30 μM (half-maximal effective concentration = 0.271 μM; Figures 6B, 6C, and S9).
Figure 6.

Acamprosate is neuroprotective in iPSC-derived motor neurons from C9orf72 patients
(A) Schematic representation of the experiments to validate the effect of acamprosate.
(B) The bar graph depicts the percentage of cells showing cleaved caspase-3 (caspase-3+ cells) after acamprosate treatment. Minor dots represent biological replicates, averaging from three technical replicates each. In contrast, the bordered dots represent the mean effect in iPSC-derived motor neurons from two healthy donors, two C9orf72 ALS patients, and one isogenic line. Data are mean ± SD. Comparisons within the control and the ALS-C9orf72 groups were performed using a two-way ANOVA, with a Tukey’s post hoc test (n = 2). Only p < 0.05 and comparisons with the vehicle group are displayed in the graph.
(C) Representative images of the motor neurons showing cleaved caspase-3 staining (green), MAP2 staining (red), and DAPI (blue). Scale bar, 50 μm. All molecular phenotypes were confirmed in a minimum of 3 technical replicates, and at least 25 fields were randomly selected and scanned per well of a 96-well plate in triplicate.
(D) Acamprosate effective doses (10 and 30 μM) were confirmed in additional iPSC-derived motor neurons, totaling cells from four healthy donors, five C9orf72 ALS patients, and two isogenic lines. Each point represents the mean effect per cell line. Data are mean ± SD. Comparisons within the control, the ALS-C9orf72, and the ISO-C9orf72 groups were performed using a two-way ANOVA, with Tukey’s post hoc test (control, n = 4; ALS-C9orf72, n = 5; ISO-C9orf72, n = 2). p < 0.05 are annotated.
(E) The graph shows the percentage of cleaved caspase-3+ motor neurons derived from C9orf72 patients treated with riluzole (10 μM) and riluzole plus acamprosate (10 and 30 μM). Dots represent the mean effect of each line. Data are mean ± SD. Comparisons were performed using a one-way ANOVA with Tukey’s post hoc test (n = 5). p < 0.05 are annotated as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.001, ∗∗∗∗∗p < 0.0001.
See also Figures S6–S9 and Table S10.
Based on this initial screening data, we selected the highest efficacious doses (10 and 30 μM) for additional testing. We confirmed the efficacy of acamprosate at protecting motor neurons in additional cell lines (five ALS patients carrying the C9orf72 repeat expansion, four healthy individuals, and two isogenic control lines; Figure 6D). Again, the drug had minimal effects on healthy motor neurons and isogenic controls (Figures 6B and 6C).
Effect of acamprosate is comparable to riluzole, the current standard of care
Next, we compared the efficacy of acamprosate in preventing cell death to riluzole, the most widely prescribed drug for ALS, which extends life expectancy by an average of 3–6 months.42 As expected, riluzole treatment reduced cell death in C9orf72 motor neurons (1.4-fold decrease at 10 μM; Figure 6E). Notably, acamprosate exerted an average of 30% higher protection, either alone (Figure 6D) or combined with riluzole, than that observed with riluzole (Figure 6E).
Discussion
We used our genetic and transcriptomic information to nominate medications that could be repurposed to delay symptom onset among C9orf72 carriers, and our data support further investigation of acamprosate as a potential treatment for this common form of neurodegeneration. Acamprosate is a US Food and Drug Administration (FDA)-approved medication with a favorable safety profile that is prescribed to maintain alcohol abstinence. The drug modulates glutamate receptors,43 a mechanism overlapping with riluzole, a commonly prescribed medication for ALS patients that slows clinical progression, at least in part, through its anti-glutamate properties.42,44 Interestingly, acamprosate, alone or in combination with riluzole, exceeded the neuroprotective properties of riluzole in our cell-based model.
Our work supports previous research suggesting that a combination of baclofen and acamprosate may be a plausible therapeutic strategy for ALS; the combination prevented TDP-43 stress granule formation in a human osteosarcoma U2OS cell line overexpressing human TDP-43 and displayed neuroprotective effects in primary motor neurons derived from SOD1G93A rat embryos.35 However, our work nominating acamprosate was an independent effort based on large-scale human genomic data. It is also the first time that acamprosate has been nominated as a personalized treatment for C9orf72-related ALS-FTD. These studies offer evidence that acamprosate may benefit ALS patients, warranting further consideration for the development of acamprosate as a treatment within this population. Our cell-based assays also suggested that a combination of riluzole and acamprosate treatment is more effective than acamprosate alone. Such combination therapy has emerged as a promising approach in neurological diseases, perhaps reflecting the need to ameliorate multiple pathways to produce a clinical benefit.45
Drug repositioning has become an attractive option for drug discovery, as using de-risked compounds lowers development costs and shortens time lines.46 Systematic approaches based on genetic and transcriptomic data integration are also promising.47,48 An emerging theme in the pharmaceutical industry is that drugs targeting proteins and pathways with genetic evidence are more likely to succeed.16,17 The effect of acamprosate on C9orf72-related disease may be due to its known glutaminergic and GABAergic properties, but this is currently unproven. Instead, our work reinforces the value of genomic data to rapidly nominate drugs for repurposing in situations where the underlying molecular mechanisms of disease are complex and where the mode of action of a drug is unclear. In support of this data-driven, mechanism-free approach, we highlight that acamprosate was first approved for use in alcohol-dependence syndrome 35 years ago, and yet precisely how the drug works in this condition remains speculative. This situation is hardly unique among CNS drugs. Despite decades of patient use, the exact molecular mechanism by which riluzole confers its survival advantage in patients with ALS is unknown. Delaying clinical trials until the mechanism of action of acamprosate is determined may be unnecessary, especially considering the current uncertainty about how the C9orf72 repeat expansion causes disease and the lack of reliable animal models.
Our findings also partially explain the variable age at onset observed in C9orf72 patients and confirm that genetic modifiers are crucial in determining the onset age in this patient population.6,49 Individuals with C9orf72-related disease with a high genetic risk of sporadic ALS developed symptoms approximately 3 years earlier than patients with a low genetic risk. Interestingly, the genetic risk attributable to general ALS did not affect onset age among patients not carrying the C9orf72 repeat expansion, as confirmed recently in a multicenter study.50 Our findings support the hypothesis that ALS is a multistep process51,52,53,54 and suggest that a personalized approach will be necessary to treat the various genetic forms of ALS and neurodegenerative disorders.
We identified “neuronal cytoskeleton” and “axonal transport” as the central pathways influencing the age at onset among C9orf72 carriers. Interestingly, arginine-containing dipeptides generated from C9orf72 repeat expansions are known to impede microtubule-based motility and axonal transport machinery in cells.55 This pathogenic mechanism involves physical interactions of the dipeptides with the kinesin-containing motor complexes. Reinforcing this research, we found the rs113247976 variant within this gene to be among the most influential on onset age, with C9orf72 patients carrying the KIF5A variant experiencing symptoms nearly 3.5 years earlier than patients who did not carry this variant (Table S7). These observations indicate that axonal transport is centrally involved in C9orf72-related neurodegeneration, and this cellular process may be a credible therapeutic target. Notwithstanding, our data also suggest that multiple cellular processes are disrupted by C9orf72 mutation, underscoring the importance of our more comprehensive genomic approach to drug repurposing.
Previous research has investigated disease modifiers of C9orf72. However, these efforts were focused on individual loci and ignored the collective contribution of the ALS polygenic inheritance.19,56,57,58 This knowledge gap was primarily due to the limited availability of large C9orf72 datasets required for this type of analysis. In contrast, our approach was based on genome-wide research involving 1,516 ALS/FTD C9orf72 carriers and 7,037 ALS non-carriers, making it one of the largest cohorts reported to date. We also focused on genomic variants outside the C9orf72 locus, which are likely more amenable to therapeutic intervention with small molecules than the repeat expansion itself.
Our research underscores the critical contribution of common risk factors as modifiers of monogenic forms of ALS.35 Unfortunately, data were unavailable to allow a similar analysis of other highly penetrant genetic mutations that cause ALS, such as SOD1 and TBK1. However, the onset age among SOD1 mutation carriers, such as the Ala5Val variant, is much more circumscribed than the variable age penetrance observed among C9orf72 patients.59 Nevertheless, our study provides a foundation for future research exploring how the common risk factors of ALS affect other monogenic forms of the disease and leveraging this information to repurpose medications.
In conclusion, we found that the risk of sporadic ALS is a critical determinant of the age at symptom onset among patients carrying the C9orf72 repeat expansion. We identified various pathways and genes that play pivotal roles in the underlying cellular processes determining onset timing. We integrated our genomic data with transcriptomics to nominate drugs that could be repurposed to delay disease onset, an approach that has previously prioritized medications for chronic conditions such as obesity60 and osteoporosis.61 Acamprosate emerged as the primary contender from these analyses, and we subsequently confirmed that this oral, inexpensive medication reduces C9orf72 motor neuron death in cell-based assays. Future research will explore how acamprosate may be exerting this effect. Nevertheless, our work pinpoints acamprosate for future exploration as a treatment to slow the manifestation of symptoms in this common genetic form of neurodegeneration. Our innovative and multidisciplinary approach could also fill the gap in therapeutic discovery in other complex neurodegenerative conditions, and we have made our computational pipeline publicly available to facilitate such work.
Limitations of the study
Our study has limitations. There was only a 3-year difference in onset age between the patients with the highest and the lowest genetic risk of sporadic ALS, representing a modest portion of the 40-year spread reported for C9orf72 onset age. Despite this, the magnitude of this effect was comparable to the results reported for the TMEM106B locus in C9orf72 patients.19 Interestingly, the outcome we observed was independent of this locus. Our capacity to predict age at onset in this group of patients, as well as our ability to assess the contribution of ALS genetic risk on disease progression, will improve as larger datasets with comprehensive phenotypic data become available.
Another limitation of our study is that the underlying mechanism of the neuroprotective effect of acamprosate in C9orf72-derived motor neurons remains unknown. Although exploring such mechanisms was beyond the scope of this paper, future research should carefully address this gap, especially considering the implications it may have for the broader ALS patient population. Previously published data suggested that acamprosate may also have a neuroprotective effect on non-C9orf72 ALS patients. A combination of acamprosate and baclofen (PXT864) has been proposed as a treatment for sporadic ALS. However, no clinical trial has been registered, and the company has discontinued its development, citing financial reasons.62 We anticipate our work will lead to a renewed interest in the otherwise overlooked acamprosate as a treatment for this uniformly fatal neurodegenerative disease. Such repurposing of FDA-approved drugs for unmet medical needs has strategic advantages that shorten the development time line—preclinical safety testing can be shortened as the safety profile and pharmacokinetic profile of existing drugs are already established. The standard daily dose (1,998 mg) of acamprosate is known to cross the blood-brain barrier and alter brain physiology; patients with alcohol dependence who were treated with acamprosate showed highly significant suppression of glutamate levels in the anterior cingulate gyrus over 4 weeks.63 These observations provide a quantitative biomarker of target engagement. It also supports the hypothesis that the protective effect observed in our cell-based assays at the relatively low dose of 1 μM will translate into human patients at the well-tolerated standard dose.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bryan Traynor (traynorb@mail.nih.gov).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The summary statistics of the reference dataset genome-wide association study (GWAS) are publicly available at http://databrowser.projectmine.com/. The individual-level data for the training and test datasets are available on dbGaP (accession nos. phs000101.v5.p1 and phs001963.v1.p1). The data for the replication dataset were from a different study (principal investigator: Christopher Shaw, King’s College London) and are available upon reasonable request. RNA-seq data from the motor cortex were obtained from the New York Genome Center, and RNA-seq data from the cerebellum were obtained from GEO (GEO: GSE67196). The figures were created using BioRender and Inkscape. The Sankey diagram was created using Visual Paradigm. The programming code used in this paper is available at https://github.com/sarasaezALS/C9orf72_AAO and https://zenodo.org/records/13259646 (https://doi.org/10.5281/zenodo.13259646) to facilitate the application of this methodology to other disorders. The individual-level polygenic risk scores generated for patients with the C9orf72 mutation are available at https://zenodo.org/uploads/13769448. The connectivity scores for the drugs evaluated as treatments for C9orf72-related disease are available at https://zenodo.org/records/13769483.
Consortia
The members of the International ALS Genomics Consortium are Robert H. Baloh, Robert Bowser, Christopher B. Brady, Alexis Brice, James Broach, William Camu, Ruth Chia, Adriano Chiò, John Cooper-Knock, Daniele Cusi, Jinhui Ding, Carsten Drepper, Vivian E. Drory, Travis L. Dunckley, Eva Feldman, Mary Kay Floeter, Pietro Fratta, Glenn Gerhard, J. Raphael Gibbs, Summer B. Gibson, Jonathan D. Glass, Stephen A. Goutman, John Hardy, Matthew B. Harms, Terry D. Heiman-Patterson, Lilja Jansson, Janine Kirby, Hannu Laaksovirta, John E. Landers, Francesco Landi, Isabelle Le Ber, Serge Lumbroso, Claire Guissart, Daniel JL. MacGowan, Nicholas J. Maragakis, Gabriele Mora, Kevin Mouzat, Liisa Myllykangas, Richard W. Orrell, Lyle W. Ostrow, Stuart Pickering-Brown, Erik P. Pioro, Stefan M. Pulst, John M. Ravits, Alan E. Renton, Wim Robberecht, Ekaterina Rogaeva, Jeffrey D. Rothstein, Erika Salvi, Sonja W. Scholz, Michael Sendtner, Pamela J. Shaw, Katie C. Sidle, Zachary Simmons, David J. Stone, Pentti J. Tienari, Bryan J. Traynor, John Q. Trojanowski, Juan C. Troncoso, Miko Valori, Philip Van Damme, Vivianna M. Van Deerlin, Ludo Van Den Bosch, and Lorne Zinman. The members of the ITALSGEN Consortium are Stefania M. Angelocola, Francesco P. Ausiello, Marco Barberis, Ilaria Bartolomei, Stefania Battistini, Enrica Bersano, Giulia Bisogni, Giuseppe Borghero, Maura Brunetti, Corrado Cabona, Andrea Calvo, Fabrizio Canale, Antonio Canosa, Teresa A. Cantisani, Margherita Capasso, Claudia Caponnetto, Patrizio Cardinali, Paola Carrera, Federico Casale, Adriano Chiò, Tiziana Colletti, Francesca L. Conforti, Amelia Conte, Elisa Conti, Massimo Corbo, Stefania Cuccu, Eleonora Dalla Bella, Eustachio D'Errico, Giovanni DeMarco, Raffaele Dubbioso, Carlo Ferrarese, Pilar M. Ferraro, Massimo Filippi, Nicola Fini, Gianluca Floris, Giuseppe Fuda, Salvatore Gallone, Giulia Gianferrari, Fabio Giannini, Maurizio Grassano, Lucia Greco, Barbara Iazzolino, Alessandro Introna, Vincenzo La Bella, Serena Lattante, Giuseppe Lauria, Rocco Liguori, Giancarlo Logroscino, Francesco O. Logullo, Christian Lunetta, Paola Mandich, Jessica Mandrioli, Umberto Manera, Fiore Manganelli, Giuseppe Marangi, Kalliopi Marinou, Maria Giovanna Marrosu, Ilaria Martinelli, Sonia Messina, Cristina Moglia, Maria Rosaria Monsurrò, Gabriele Mora, Lorena Mosca, Maria R. Murru, Paola Origone, Carla Passaniti, Cristina Petrelli, Antonio Petrucci, Angelo Pirisi, Susanna Pozzi, Maura Pugliatti, Angelo Quattrini, Claudia Ricci, Giulia Riolo, Nilo Riva, Massimo Russo, Mario Sabatelli, Paolina Salamone, Marco Salivetto, Fabrizio Salvi, Marialuisa Santarelli, Luca Sbaiz, Riccardo Sideri, Isabella Simone, Cecilia Simonini, Rossella Spataro, Raffaella Tanel, Gioacchino Tedeschi, Anna Ticca, Antonella Torriello, Stefania Tranquilli, Lucio Tremolizzo, Francesca Trojsi, Rosario Vasta, Veria Vacchiano, Giuseppe Vita, Paolo Volanti, Marcella Zollino, and Elisabetta Zucchi. The members of the SLAGEN Consortium are Vincenzo Silani, Isabella Fogh, Nicola Ticozzi, Antonia Ratti, Cinzia Tiloca, Silvia Peverelli, Cinzia Gellera, Giuseppe Lauria Pinter, Franco Taroni, Viviana Pensato, Barbara Castellotti, Giacomo P. Comi, Stefania Corti, Roberto Del Bo, Cristina Cereda, Mauro Ceroni, Stella Gagliardi, Lucia Corrado, Letizia Mazzini, Gianni Sorarù, Flavia Raggi, Gabriele Siciliano, Costanza Simoncini, Annalisa Lo Gerfo, Massimiliano Filosto, Maurizio Inghilleri, and Alessandra Ferlini. The members of Project MinE ALS Sequencing Consortium are Philip Van Damme, Philippe Corcia, Philippe Couratier, Patrick Vourc’h, Orla Hardiman, Russell McLaughlin, Marc Gotkine, Vivian Drory, Nicola Ticozzi, Vincenzo Silani, Jan H. van den Veldink, Leonard H. Berg, Mamede de Carvalho, Jesus S. Mora Pardina, Monica Povedano, Peter Andersen, Markus Weber, Ayşe Nazlı Başak, Ammar Al-Chalabi, Chris Shaw, Pamela J. Shaw, Karen E. Morrison, John E. Landers, and Jonathan D. Glass. The members of the American Genome Center are Adelani Adeleye, Camille Alba, Dagmar Bacikova, Clifton L. Dalgard, Daniel N. Hupalo, Elisa McGrath Martinez, Anthony R. Soltis, Gauthaman Sukumar, Coralie Viollet, and Matthew D. Wilkerson.
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institutes of Health, the National Institute on Aging (ZIA-AG000933, ZIA-AG000934), the National Institute of Neurological Disorders and Stroke (NINDS), the National Center for Advancing Translational Sciences, and by Merck Sharp & Dohme Corp. (a subsidiary of Merck & Co., Kenilworth, NJ). B.J.T. received additional support from the Centers for Disease Control and Prevention, the Muscular Dystrophy Association, Microsoft Research, the Packard Center ALS Research at Johns Hopkins, and the ALS Association. L.F. is supported by the UK Medical Research Council (MR/V027735/1 and MR/V000470/1) and the Motor Neurone Disease Association (MND019801). A.N.B., C. Tunca, and E.B. gratefully acknowledge the use of the services and facilities of the Koç University Research Center for Translational Medicine (KUTTAM), funded by the Presidency of Turkey, Head of Strategy and Budget. R.S. is supported by the Barrow Neurological Foundation. This work was also generated within the European Reference Network for Neuromuscular Diseases (ERN EURO-NMD, https://ern-euro-nmd.eu). This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health. The authors would like to thank the Project MinE GWAS Consortium. This study also used genotype and clinical data from the Wellcome Trust Case Control Consortium and the HyperGenes Consortium. We thank the patients and research subjects who contributed samples for this study, Dr. Ruth Pfeiffer, Dr. Justin Kwan, the Laboratory of Neurogenetics staff, and the NINDS Neurodegeneration Clinic staff for their collegial support. The authors also acknowledge the Target ALS Human Post-mortem Tissue Core, the New York Genome Center for Genomics of Neurodegenerative Disease, the ALS Association, and the TOW Foundation. Additional acknowledgments may be found in the supplemental information.
Author contributions
S.S.-A. and B.J.T. conceived and designed the study, acquired and analyzed the data, and wrote the paper. C.d.S.S., R. Chia, S.N.B., I.L., R.H., J.L., C.B., J.D., J.R.G., A.J., R.D., V.P., S.P., L.C., J.J.F.A.v.V., W.v.R., C. Tunca, E. Bayraktar, M.X., A.I., A.S., C. Tiloca, N.T., F.V., L.M., K.K., A.A.K., S.O.-M., F.R., M.F., S.C.P., A.P., S. Gagliardi, M.I., A.F., R.V., A. Calvo, C.M., A. Canosa, U.M., M.G., J.M., G.M., C.L., R.T., F.T., P.C., S. Gallone, M.B., D.G., M.S., C.F., E.S., G.P.C., S.C., R.D.B., M.C., G.L.P., F. Taroni, E.D.B., E.B., C.J.C., S.H.L., R. Chung, H.P., K.E.M., J.C.-K., P.J.S., G.B., R.J.B.D., C.L.D., S.W.S., A.A.-C., L.H.v.d.B., R.M., O.H., C.C., G. Sorarù, S.D., S. Chandran, S. Pal, A.R., C.G., K.J., T.D.-O., N.P., T.W., A.N., G. Siciliano, V.S., A.N.B., J.H.V., W.C., J.D.G., J.E.L., A. Chiò, R.S., C.E.S., L.F., and I.F. acquired and analyzed the data and edited the manuscript.
Declaration of interests
B.J.T. holds patents on clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of C9orf72.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-goat (donkey) | Thermo Fisher | Catalog # A21432, RRID: AB_2535853 |
| Anti-guinea pig (goat) | Thermo Fisher | Catalog # A21450, RRID: AB_2535867 |
| Anti-mouse (donkey) | Thermo Fisher | Catalog # A10037, RRID: AB_2534013 |
| Anti-mouse (donkey) | Thermo Fisher | Catalog # A21202, RRID: AB_141607 |
| Anti-rabbit (donkey) | Thermo Fisher | Catalog # A21206, RRID: AB_2535792 |
| Anti-rabbit (donkey) | Thermo Fisher | Catalog # A10042, RRID: AB_2534017 |
| Beta III tubulin (mouse) | Biolegend | Catalog # 801201, RRID: AB_2728521 |
| Caspase-3 (rabbit) | Merck Millipore | Catalog # AB3623, RRID: AB_91556 |
| ChAT (goat) | Merck Millipore | Catalog # AB144P, RRID: AB_2079751 |
| Islet ½ (rabbit) | Abcam | Catalog # ab109517, RRID: AB_10866454 |
| MAP-2 (guinea pig) | Synaptic systems | Catalog # 188004, RRID: AB_2138181 |
| NeuN (mouse) | Merck Millipore | Catalog # MAB377, RRID: AB_2298772 |
| Chemicals, peptides, and recombinant proteins | ||
| Acamprosate calcium | Sigma-Aldrich | Catalog # A6981 |
| Accutase | StemCell Technologies | Catalog # 07922 |
| All-trans retinoic acid | Merck Millipore | Catalog # 554720 |
| B27 supplement | Thermo Fisher | Catalog # 17504044 |
| Brain Derived Neurotrophic Factor (BDNF) | Peprotech | Catalog # 450-02 |
| Camptothecin (CPT) | Cell Signaling Technology | Catalog # 13637 |
| CHIR 99021 | Tocris Biosciences | Catalog # 4423 |
| Ciliary Neurotrophic Factor (CNTF) | Peprotech | Catalog # 450-13 |
| Compound E | Merck Millipore | Catalog # 530509 |
| D(−)-2-Amino-5-phosphonopentanoic acid (D-AP5) | Sigma-Aldrich | Catalog # A8054 |
| 4,6-diamidino-2-phenylindole (DAPI) | Merck Millipore | Catalog # 508741 |
| Dimethyl Sulfoxide (DMSO) | Merck Millipore | Catalog # 317275 |
| Distilled H2O | Merck Millipore | Catalog # EM3234 |
| Dorsomorphin homolog 1 (DMH-1) | Tocris Biosciences | Catalog # 4126 |
| GlutaMAX | Thermo Fisher | Catalog # 35050061 |
| Hank’s balanced salt solution (HBSS) | Thermo Fisher | Catalog # 14175 |
| Insulin-like Growth Factor-I (IGF-1) | Peprotech | Catalog # 100-11 |
| KnockOut DMEM/F-12 | Thermo Fisher | Catalog # A1370801 |
| mTeSR Plus Basal Medium | StemCell Technologies | Catalog # 100-0276 |
| N2 supplement | Thermo Fisher | Catalog # 17502048 |
| Paraformaldehyde | Thermo Fisher | Catalog # 047392.9M |
| Penicillin/streptomycin | Merck Millipore | Catalog # 516106 |
| Phosphate buffered saline (PBS) | Merck Millipore | Catalog # 6504 |
| Purmorphamine (PMN) | Merck Millipore | Catalog # 540220 |
| ReLeSR Passaging Reagent | StemCell Technologies | Catalog # 100-0484 |
| Riluzole | Merck Millipore | Catalog # 557324 |
| SB431542 | Tocris Biosciences | Catalog # 1614 |
| Triton X-100 | Merck Millipore | Catalog # 648464 |
| Vitronectin XF | StemCell Technologies | Catalog # 07180 |
| Y27632 dihydrochloride | Tocris Biosciences | Catalog # 1254 |
| Critical commercial assays | ||
| MTT assay | Thermo Fisher | Catalog #V13154 |
| SNP beadchip genotyping array | Illumina | Catalog #: InfiniumOmni2-5-8v1-4_A1 |
| Deposited data | ||
| ALS GWAS summary statistics | Van Rheenen et al.50 | https://www.projectmine.com/research/download-data/ |
| The individual-level data for the training and test datasets | Dewan et al.64 | dbGaP (accession # phs001963.v1.p1), RRID: SCR_002709 |
| The individual-level data for the training and test datasets | Nicolas et al.65 | dbGaP (accession # phs000101.v5.p1), RRID: SCR_002709 |
| The individual-level control data for the training and test datasets | https://dbgap.ncbi.nlm.nih.gov | dbGaP (accession # phs000001, phs000007, phs000187, phs000196, phs000292, phs000304, phs000315, phs000368, phs000372, phs000394, phs000397, phs000404, phs000421, phs000428, phs000615, phs000675, phs000801, and phs000869), RRID:SCR_002709 |
| The individual-level data used as the replication dataset in this study. | King’s College London, unpublished study | Available from study principal investigator (Christopher Shaw, King’s College London) upon reasonable request. |
| The individual-level polygenic risk scores generated for the C9orf72 patients. | This study | https://zenodo.org/uploads/13769448 |
| LINCS dataset | Subramanian et al.33 |
https://lincsproject.org, RRID: SCR_006454 |
| The LINCS connectivity scores for the drugs evaluated as treatments for C9orf72-related disease. | This study | https://zenodo.org/records/13769483 |
| RNA-sequencing data from the motor cortex | New York Genome Center | https://www.nygenome.org |
| RNA-sequencing data from the cerebellum | Prudencio et al.66 | GEO (accession # GSE67196), RRID: SCR_005012 |
| Experimental models: Cell lines | ||
| Human: iPS cell line | Cedars-Sinai | CS14iCTR-nxx |
| Human: iPS cell line | Coriell Biorepository | GM23338, RRID: CVCL_F182 |
| Human: iPS cell line | University of Sheffield | MIFF1, RRID: CVCL_1E69 |
| Human: iPS cell line | Cedars-Sinai | CS02iCTR-NTn1 |
| Human: iPS cell line | University of Sheffield | ALS-183-C9 |
| Human: iPS cell line | University of Sheffield | ALS-78 |
| Human: iPS cell line | Cedars-Sinai | CS28iALS-C9nxx |
| Human: iPS cell line | Cedars-Sinai | CS29iALS-C9nxx |
| Human: iPS cell line | Cedars-Sinai | CS52iALS-C9nxx |
| Human: iPS cell line | Cedars-Sinai | CS29iALS-C9n1.ISOxx |
| Human: iPS cell line | Cedars-Sinai | CS52iALS-C9n6.ISOxx |
| Software and algorithms | ||
| Codes and scripts | This paper | https://github.com/sarasaezALS/C9orf72_AAO; https://zenodo.org/records/13259646 |
| Drugmonizome | Kropiwnicki et al.34 | https://maayanlab.cloud/drugmonizome/#/ |
| ExperimentHub | https://www.bioconductor.org/packages/release/bioc/html/ExperimentHub.html | https://mrcieu.github.io/TwoSampleMR |
| Geneshot | Lachmann et al.30 |
https://maayanlab.cloud/genesgen, RRID: SCR_017582 |
| g:Profiler2 | Kolberg et al.29 |
https://biit.cs.ut.ee/gprofiler/ggos, RRID: SCR_018190 |
| Harmony software | Perkin Elmer | Catalog # hh17000010, RRID: SCR_023543 |
| PLINK (version 1.9) | PLINK Working Group |
https://www.cog-genomics.org/plink/1.9/, RRID: SCR_001757 |
| Prism (version 10) | GraphPad Software |
https://www.graphpad.com, RRID: SCR_002798 |
| PRSice2 | Choi et al.67 | https://choishingwan.github.ii/PRSice/, RRID: SCR_017057 |
| Python | Python Team |
https://www.python.org, RRID: SCR_008394 |
| R (version 4.0.5) | R Core Team |
https://www.r-project.org, RRID: SCR_001905 |
| SignatureSearch (version 1.11.0) | Duan et al.68 |
https://bioconductor.org/packpack/release/bioc/vignettes/sigsignatureSea/inst/doc/signatursignatu.html, RRID: SCR_016177 |
Experimental model and study participant details
Human participants
Tables S8 and S9 list the source and clinical features of the cohorts used in this study. The ALS patients were diagnosed according to the El Escorial criteria69 for the training and test datasets, and the FTD patients were analyzed according to the Neary criteria.70 The C9orf72 repeat expansions were detected using a repeat-primed polymerase chain reaction (PCR) assay according to an established protocol.3 Written consent was obtained from all individuals enrolled in this study, and the institutional review board approved the study of the National Institute on Aging (protocol number 03-AG-N329). The C9orf72 patients of the replication cohort were from a different study conducted at the King’s College London and recruited by the SLAGEN Consortium, the University of Edinburgh, the Boğaziçi University, and Project MinE. Written consent was obtained from all individuals at their respective centers (see the Replication cohort section for details).
Datasets
Four independent datasets were used in the analysis, as is standard in genetic risk score analysis (see Figure 1 for the analysis workflow). The reference dataset consisted of summary statistics from a published GWAS based on 12,577 ALS cases and 23,475 control individuals (Table S9).65 The allele weights obtained from the reference dataset were used to construct the ALS genetic risk score model in the training dataset.
The training dataset was composed of 7,037 ALS individuals known not to carry C9orf72 repeat expansions and 34,235 controls (Table S9). The case samples were previously genotyped in the Laboratory of Neurogenetics, National Institutes of Health, using HumanOmniExpress SNP arrays (version 1.0, Illumina Inc., San Diego, CA).22,65 The US control samples had been previously genotyped on Illumina SNP arrays as part of other GWAS efforts. These data were downloaded from the dbGaP repository (accession numbers phs000001, phs000007, phs000187, phs000196, phs000292, phs000304, phs000315, phs000368, phs000372, phs000394, phs000397, phs000404, phs000421, phs000428, phs000615, phs000675, phs000801, and phs000869). Additional SNP array data from the HYPERGENES project and the Wellcome Trust Case Control Consortium were included as Italian and UK control subjects.71 All study participants were of European ancestry, and familial cases were included in the analysis. The individual-level data for the training dataset was used as input for the PRSice2 algorithm.
To assess the effect of the ALS genetic risk score on age at onset in C9orf72 carriers, we calculate the genetic risk score in the C9orf72 cohort. This test dataset consisted of 817 ALS/FTD cases that carry the C9orf72 gene (see Table S9 for detailed information). Of these, 666 (81.5% of the cohort) were genotyped on SNP arrays in the Laboratory of Neurogenetics,65 and 151 (18.4%) underwent whole-genome sequencing.64 Genotype data for the 161 SNPs making up the model were extracted from the SNP array and whole-genome sequence data.
We replicated our findings in an independent C9orf72 cohort obtained from a different study conducted at King’s College London. This replication dataset consisted of 699 ALS/FTD cases known to carry the C9orf72 repeat expansion and genotyped on SNP arrays using Illumina InfiniumOmni2-5-v1.72
Method details
SNP array-based genotyping data quality control procedures and imputation
Standard quality-control procedures were applied to the genotype data of the training dataset (n = 7,037 cases and 34,235 controls) and the test dataset (n = 666 cases) before input into the PRSice2 algorithm. Briefly, individuals with low call rates (<95%), heterozygosity outliers (F-statistic > −0.15 or < 0.15), and ancestry outliers (+/− 6 standard deviations from means of principal components 1 and 2 of the 1000 Genomes phase 3 Caucasian with European ancestry from Utah (CEU) and Toscani in Siena, Italy (TSI) populations) were excluded. Variants with a missingness rate of >5%, exhibiting deviation from Hardy–Weinberg Equilibrium in controls (p-value < 10−6), and palindromic SNPs were excluded. Cryptically related samples (defined as Pi_hat >0.125) were removed. The remaining sample genotypes were imputed using the Michigan Imputation Server pipeline using Minimac473 under default settings with Eagle (version 2.4) phasing based on Haplotype Reference Consortium (release 1.1 2016). Samples from the United States, Italy, the United Kingdom, Belgium, and France were imputed as a single group. Those variants were additionally filtered post-imputation to exclude variants with minor allele frequency <0.01, missing call rates >15%, and imputation quality R2 < 0.3.
The SNP array-based genotyping in the replication dataset section describes the quality control and imputation methodology used in the replication dataset. Samples in common between the test and replication datasets were identified using the checksum program id_geno_checksum.v2 and removed.
Whole-genome sequencing of the test dataset
In the test dataset, one hundred and fifty-one C9orf72 carriers had previously undergone 150 base pair, paired-end whole-genome sequencing on a HiSeq X Ten sequencer.64,74 Genotype data for the 161 SNPs making up the model were extracted from this whole-genome sequence data. They were merged with the SNP array genotype data for the remaining 666 C9orf72 carriers using PLINK (version 1.9).75,76
Genetic risk score generation and computation
The genome-wide genetic risk score was calculated using the training dataset based on the weighted allele dosages obtained from the reference dataset as implemented in PRSice2.67,77 This approach allows variants below the typical GWAS significance threshold of 5.0 x 10−8 to be included in the analysis, and the model selected a p-value threshold ≤ 0.0001 for SNP selection. For the training dataset, 1,000 permutations were used to generate empirical p-value estimates for each GWAS-derived p-value. Each permutation test in the training dataset provided a Nagelkerke’s pseudo-R2 value after adjusting for an estimated ALS prevalence of 5 per 100,000 of the population.78 To avoid accounting for the C9orf72 effect, 150 kb upstream and downstream of the C9orf72 top GWAS variant, rs3849943 (chr9: 9:27,543,382; GRCh37), were removed from the analysis. Sex, age at onset, and principal components one to twenty were included as covariates in the model.
The --score command implemented in PLINK (version 1.9)75 was used to test the general ALS genetic risk score’s contribution to the age of symptom onset among the C9orf72 expansion carriers. Risk allele dosages were counted, giving a dose of two if homozygous for the risk allele, one if heterozygous, and zero if homozygous for the reference allele. Linear regression was used to evaluate the association between the genetic risk scores and the age at onset, as implemented in R (version 4.0.3). Sex, disease diagnosis (ALS or FTD), and principal components one to twenty were included as covariates in the model. For replication, 147 out of 161 SNPs were used to build genetic risk scores. Sex and principal component 1 were used as covariates in the model. Genetic risk scores were transformed to Z scores based on cases (a Z score of one is equivalent to a single standard deviation of increase from the case population mean of the genetic risk score). There was a four-standard deviation difference from the genetic risk score mean between C9orf72 carriers at the top and the bottom of the genetic risk score distribution. We calculated the maximum age at onset that the genetic risk score can explain by multiplying the slope of the regression (beta) by this standard deviation difference. This approach determined the extent to which the genetic risk score can account for variations in the age at onset. Individuals within the extremes were defined as those within the 3% tails of the genetic risk score distribution. On average, the score at the extremes exceeded ±2 standard deviations from the data mean (Table S2).79
Leave-one-out analysis and decile generation
Leave-one-out analyses were performed by iteratively excluding one variant from the ALS genetic risk score (based on 161 predictors) and re-estimating the causal effect on age at onset (Table S3). The 161 variants were then ordered based on the regression coefficient (beta) from the leave-one-out analysis and regrouped in ten deciles. Thus, we generated ten ranked deciles composed of 16 variants each. Decile ten contains the 16 variants with a more significant contribution to early age at onset. Regression analyses to evaluate the contribution of each decile (16 variants) to age at onset were performed using PLINK (version 1.9) as described previously.75
Pathway analysis
Functional enrichment analysis was performed using the g:GOSt function of g:Profiler2.29 Briefly, SNPs from decile one and the C9orf72 gene name were input for g:Profiler2.29,80 Enrichment was performed against the biological process and molecular function from the Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases. Only gene lists that contain between 5 and 500 genes were selected for the analysis. The significant threshold was an FDR-corrected p-value less than 0.05. Pathways containing a single gene were removed from the study.
Drug prioritization via gene-drug pattern matching
The SNPs that compose decile ten (Table S3) were mapped to gene names using the g:SNPense function of g:Profiler2.29 These genes were used as search terms (defined as seed genes in Table S5) to identify functionally related genes based on previous knowledge and gene-gene co-expression data in the Geneshot webserver (accessed October 2021).30 This application identifies genes associated with our search term based on their co-occurrence in publications and gene-gene similarity from human RNA-seq data (ARCHS4) to predict associations between genes and search terms.30 The resulting list of genes plus the seed genes (Table S5) was then used as input for the GREP analysis (version 1.0.0), a pipeline identifying drugs that can be repurposed to target the gene set based on their enrichment in clinical indication categories.31
Finally, drugmonizome was used to identify significant biomedical terms within the specified drugs based on their indications (from the SIDER (Side Effect Resource) database, the mechanism of action (MOA, from the DrugRepurposingHub database), and the gene targets (from the Drugbank database) (data accessed 10/12/2022).34
Drug prioritization via gene expression-pattern matching
The nominated drugs were validated using the drug-repurposing algorithm called Connectivity map (CMap) through the SignatureSearch package using the LINCS search method (version 1.8.2).68 This algorithm uses two inputs: (i) a disease gene expression signature based on a list of the up- and down-regulated genes, and (ii) a drug perturbation dataset composed of differential expression profiles of each gene after drug treatment. A bi-directional weighted Kolmogorov–Smirnov enrichment statistic test of gene expression ranks in the disease and drug signatures was used to assign a weighted normalized connectivity score (referred to as a CMap score) to each drug, reflecting the degree to which the drug ‘flips’ the signature of the disease. The CMap scores reflect the similarity between the query drug’s signature and those in the database, suggesting that the drug has similar or dissimilar biological effects.
To build the C9orf72 disease signature in the motor cortex, differentially expressed genes in C9orf72 cases (n = 36) versus controls (n = 58) were selected at a Bonferroni-corrected p-value <0.05 (see supplemental information for details). This dataset was obtained from the New York Genome Center. To build the C9orf72 disease signature in the cerebellum, differentially expressed genes in C9orf72 cases (n = 8) versus controls (n = 8) were selected at FDR-corrected p-value <0.05. This dataset was downloaded as a raw count matrix from GEO (accession number GSE67196).66 Drug signatures from the LINCS database were accessed through ExperimentHub (version 2.2.0) in the form of moderated z-scores from differential expression (DE) analysis of 12,328 genes from 8,140 compound treatments of 30 cell lines corresponding to a total of 45,956 signatures. Drug signatures corresponding to the nominated drugs were extracted from the analyzed results and were available for the following cell lines: A375 (LINCS ID = LCL-1235, a human cell line exhibiting epithelial morphology isolated from the skin of a patient with malignant melanoma, provided by the American Type Culture Collection (ATCC)), ASC (LCL-2104, human adipose stem cells, Sciencell Research Laboratories), FIBRNPC (LSC-1021, iPSC), HCC515 (LCL-2084, human cell line isolated from lung adenocarcinoma, Broad Institute), HT29 (LCL-1180, a human cell line with epithelial morphology isolated from a patient with colorectal adenocarcinoma, ATCC), NEU (LDC-1033, differentiated cell), A549 (LCL-1601, a human cell line isolated from the lung of a patient with non-small cell lung carcinoma, ATCC), NPC (LDC-1021, normal stem fibroblast-derived iPSCs), HA1E (LCL-2090, a human cell line isolated from kidney, Broad Institute), PC3 (LCL-1299, a human cell line isolated from a patient with grade IV prostate adenocarcinoma, ATCC), MCF7 (LCL-2138, a human cell line isolated from a patient with breast adenocarcinoma, ATCC), PHH (primary human hepatocyte), SKBR3 (LCL-1475, human cell line isolated from a patient with breast adenocarcinoma, ATCC), and VCAP (LCL-1147, a human cell line that was isolated from a patient with prostate carcinoma, ATCC). Additional information about the cell lines is available at lincsportal.ccs.miami.edu.
Drug in vitro validation
Human induced pluripotent stem cells (iPSCs) were maintained in 6-well plates coated with vitronectin XF (10 μg/mL) in the complete mTeSR-Plus medium. Media was replaced every 48 h, and cells were passaged as clumps every four to six days using ReLeSR, according to the manufacturer’s instructions. IPSCs were used between passages 20 and 35, and all iPSCs were cultured in 5% O2 and 5% CO2 at 37°C. In this study, we used iPS cells derived from four unaffected controls (CS14iCTR-21nxx, MIFF1, CS02iCTR-NTn1, GM23338), five iPS lines derived from ALS patients harboring C9orf72 repeat expansions (ALS-183-C9, CS52iALS-C9nxx, ALS-78, CS28iALS-C9nxx, CS29iALS-C9nxx) and two isogenic control lines of CS52iALS-nxx and CS29iALS-C9nxx, respectively (CS52iALS-C9n6.ISOxx, CS29iALS-C9n1.ISOxx (C9orf72 HRE Corrected)) (Table S10). The cells were fed on alternate days with the neuronal medium until day 40. Cells were previously characterized at day 40 of differentiation and were found to express classical mature motor neuron markers (ChAT, SMI32, Islet 1/2, MAP2, NeuN) (Figure S6 and Table S11).
Drug treatments were applied for 72 h at day 40 of differentiation, and cells were assayed for viability (MTT) or fixed for subsequent immunocytochemistry assays. All the imaging was performed using the Opera Phenix High Content Screening System (PerkinElmer) at ×40 magnification. The supplementary material provides a detailed description of the methodology.
Quantification and statistical analysis
The statistics and graphs for the cell line experiments were generated using GraphPad Prism 10 (GraphPad Software, La Jolla, California USA, www.graphpad.com). Comparisons were performed using one-way ANOVA or two-way ANOVA with Tukey’s post hoc multiple comparisons test. Three technical replicates per treatment were averaged before plotting, and statistical analysis was performed using the percentage of cleaved caspase-3 mean across cell lines. Plots represent mean ± SD. p-values smaller than 0.05 are annotated unless otherwise specified in the figure legend.
Published: October 21, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100679.
A video abstract is available at https://doi.org/10.1016/j.xgen.2024.100679#mmc2.
Contributor Information
Sara Saez-Atienzar, Email: sara.saezatienzar@osumc.edu.
Bryan J. Traynor, Email: traynorb@mail.nih.gov.
The International ALS Genomics Consortium:
Robert H. Baloh, Robert Bowser, Christopher B. Brady, Alexis Brice, James Broach, William Camu, Ruth Chia, Adriano Chio, John Cooper-Knock, Daniele Cusi, Jinhui Ding, Carsten Drepper, Vivian E. Drory, Travis L. Dunckley, Eva Feldman, Mary Kay Floeter, Pietro Fratta, Glenn Gerhard, J. Raphael Gibbs, Summer B. Gibson, Jonathan D. Glass, Stephen A. Goutman, John Hardy, Matthew B. Harms, Terry D. Heiman-Patterson, Lilja Jansson, Janine Kirby, Hannu Laaksovirta, John E. Landers, Francesco Landi, Isabelle Le Ber, Serge Lumbroso, Claire Guissart, Daniel JL. MacGowan, Nicholas J. Maragakis, Gabriele Mora, Kevin Mouzat, Liisa Myllykangas, Richard W. Orrell, Lyle W. Ostrow, Stuart Pickering-Brown, Erik P. Pioro, Stefan M. Pulst, John M. Ravits, Alan E. Renton, Wim Robberecht, Ekaterina Rogaeva, Jeffrey D. Rothstein, Erika Salvi, Sonja W. Scholz, Michael Sendtner, Pamela J. Shaw, Katie C. Sidle, Zachary Simmons, David J. Stone, Pentti J. Tienari, Bryan J. Traynor, John Q. Trojanowski, Juan C. Troncoso, Miko Valori, Philip Van Damme, Vivianna M. Van Deerlin, Ludo Van Den Bosch, and Lorne Zinman
ITALSGEN Consortium:
Stefania M. Angelocola, Francesco P. Ausiello, Marco Barberis, Ilaria Bartolomei, Stefania Battistini, Enrica Bersano, Giulia Bisogni, Giuseppe Borghero, Maura Brunetti, Corrado Cabona, Andrea Calvo, Fabrizio Canale, Antonio Canosa, Teresa A. Cantisani, Margherita Capasso, Claudia Caponnetto, Patrizio Cardinali, Paola Carrera, Federico Casale, Adriano Chio, Tiziana Colletti, Francesca L. Conforti, Amelia Conte, Elisa Conti, Massimo Corbo, Stefania Cuccu, Eleonora Dalla Bella, Eustachio D'Errico, Giovanni DeMarco, Raffaele Dubbioso, Carlo Ferrarese, Pilar M. Ferraro, Massimo Filippi, Nicola Fini, Gianluca Floris, Giuseppe Fuda, Salvatore Gallone, Giulia Gianferrari, Fabio Giannini, Maurizio Grassano, Lucia Greco, Barbara Iazzolino, Alessandro Introna, Vincenzo La Bella, Serena Lattante, Giuseppe Lauria, Rocco Liguori, Giancarlo Logroscino, Francesco O. Logullo, Christian Lunetta, Paola Mandich, Jessica Mandrioli, Umberto Manera, Fiore Manganelli, Giuseppe Marangi, Kalliopi Marinou, Maria Giovanna Marrosu, Ilaria Martinelli, Sonia Messina, Cristina Moglia, Maria Rosaria Monsurrò, Gabriele Mora, Lorena Mosca, Maria R. Murru, Paola Origone, Carla Passaniti, Cristina Petrelli, Antonio Petrucci, Angelo Pirisi, Susanna Pozzi, Maura Pugliatti, Angelo Quattrini, Claudia Ricci, Giulia Riolo, Nilo Riva, Massimo Russo, Mario Sabatelli, Paolina Salamone, Marco Salivetto, Fabrizio Salvi, Marialuisa Santarelli, Luca Sbaiz, Riccardo Sideri, Isabella Simone, Cecilia Simonini, Rossella Spataro, Raffaella Tanel, Gioacchino Tedeschi, Anna Ticca, Antonella Torriello, Stefania Tranquilli, Lucio Tremolizzo, Francesca Trojsi, Rosario Vasta, Veria Vacchiano, Giuseppe Vita, Paolo Volanti, Marcella Zollino, and Elisabetta Zucchi
SLAGEN Consortium:
Vincenzo Silani, Isabella Fogh, Nicola Ticozzi, Antonia Ratti, Cinzia Tiloca, Silvia Peverelli, Cinzia Gellera, Giuseppe Lauria Pinter, Franco Taroni, Viviana Pensato, Barbara Castellotti, Giacomo P. Comi, Stefania Corti, Roberto Del Bo, Cristina Cereda, Mauro Ceroni, Stella Gagliardi, Lucia Corrado, Letizia Mazzini, Gianni Sorarù, Flavia Raggi, Gabriele Siciliano, Costanza Simoncini, Annalisa Lo Gerfo, Massimiliano Filosto, Maurizio Inghilleri, and Alessandra Ferlini
Project MinE ALS Sequencing Consortium:
Philip Van Damme, Philippe Corcia, Philippe Couratier, Patrick Vourc'h, Orla Hardiman, Russell McLaughlin, Marc Gotkine, Vivian Drory, Nicola Ticozzi, Vincenzo Silani, Jan H. van den Veldink, Leonard H. Berg, Mamede de Carvalho, Jesus S. Mora Pardina, Monica Povedano, Peter Andersen, Markus Weber, Ayşe Nazlı Başak, Ammar Al-Chalabi, Chris Shaw, Pamela J. Shaw, Karen E. Morrison, John E. Landers, and Jonathan D. Glass
The American Genome Center:
Adelani Adeleye, Camille Alba, Dagmar Bacikova, Clifton L. Dalgard, Daniel N. Hupalo, Elisa McGrath Martinez, Anthony R. Soltis, Gauthaman Sukumar, Coralie Viollet, and Matthew D. Wilkerson
Supplemental information
References
- 1.Larson T.C., Kaye W., Mehta P., Horton D.K. Amyotrophic Lateral Sclerosis Mortality in the United States, 2011-2014. Neuroepidemiology. 2018;51:96–103. doi: 10.1159/000488891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Logroscino G., Piccininni M. Amyotrophic Lateral Sclerosis Descriptive Epidemiology: The Origin of Geographic Difference. Neuroepidemiology. 2019;52:93–103. doi: 10.1159/000493386. [DOI] [PubMed] [Google Scholar]
- 3.Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J., et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majounie E., Renton A.E., Mok K., Dopper E.G.P., Waite A., Rollinson S., Chiò A., Restagno G., Nicolaou N., Simon-Sanchez J., et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy N.A., Arthur K.C., Tienari P.J., Houlden H., Chiò A., Traynor B.J. Age-related penetrance of the C9orf72 repeat expansion. Sci. Rep. 2017;7:2116. doi: 10.1038/s41598-017-02364-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balendra R., Isaacs A.M. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat. Rev. Neurol. 2018;14:544–558. doi: 10.1038/s41582-018-0047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sierksma A., Escott-Price V., De Strooper B. Translating genetic risk of Alzheimer's disease into mechanistic insight and drug targets. Science. 2020;370:61–66. doi: 10.1126/science.abb8575. [DOI] [PubMed] [Google Scholar]
- 9.Taymans J.M., Fell M., Greenamyre T., Hirst W.D., Mamais A., Padmanabhan S., Peter I., Rideout H., Thaler A. Perspective on the current state of the LRRK2 field. NPJ Parkinsons Dis. 2023;9:104. doi: 10.1038/s41531-023-00544-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cacabelos R. What have we learnt from past failures in Alzheimer's disease drug discovery? Expet Opin. Drug Discov. 2022;17:309–323. doi: 10.1080/17460441.2022.2033724. [DOI] [PubMed] [Google Scholar]
- 11.Wilson D.M., 3rd, Cookson M.R., Van Den Bosch L., Zetterberg H., Holtzman D.M., Dewachter I. Hallmarks of neurodegenerative diseases. Cell. 2023;186:693–714. doi: 10.1016/j.cell.2022.12.032. [DOI] [PubMed] [Google Scholar]
- 12.Mead R.J., Shan N., Reiser H.J., Marshall F., Shaw P.J. Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat. Rev. Drug Discov. 2023;22:185–212. doi: 10.1038/s41573-022-00612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gribkoff V.K., Kaczmarek L.K. The need for new approaches in CNS drug discovery: Why drugs have failed, and what can be done to improve outcomes. Neuropharmacology. 2017;120:11–19. doi: 10.1016/j.neuropharm.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamb J., Crawford E.D., Peck D., Modell J.W., Blat I.C., Wrobel M.J., Lerner J., Brunet J.P., Subramanian A., Ross K.N., et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 15.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochoa D., Karim M., Ghoussaini M., Hulcoop D.G., McDonagh E.M., Dunham I. Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs. Nat. Rev. Drug Discov. 2022;21:551. doi: 10.1038/d41573-022-00120-3. [DOI] [PubMed] [Google Scholar]
- 17.Hingorani A.D., Kuan V., Finan C., Kruger F.A., Gaulton A., Chopade S., Sofat R., MacAllister R.J., Overington J.P., Hemingway H., et al. Improving the odds of drug development success through human genomics: modelling study. Sci. Rep. 2019;9 doi: 10.1038/s41598-019-54849-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chio A., Borghero G., Restagno G., Mora G., Drepper C., Traynor B.J., Sendtner M., Brunetti M., Ossola I., Calvo A., et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain. 2012;135:784–793. doi: 10.1093/brain/awr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallagher M.D., Suh E., Grossman M., Elman L., McCluskey L., Van Swieten J.C., Al-Sarraj S., Neumann M., Gelpi E., Ghetti B., et al. TMEM106B is a genetic modifier of frontotemporal lobar degeneration with C9orf72 hexanucleotide repeat expansions. Acta Neuropathol. 2014;127:407–418. doi: 10.1007/s00401-013-1239-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodrich J.K., Singer-Berk M., Son R., Sveden A., Wood J., England E., Cole J.B., Weisburd B., Watts N., Caulkins L., et al. Determinants of penetrance and variable expressivity in monogenic metabolic conditions across 77,184 exomes. Nat. Commun. 2021;12:3505. doi: 10.1038/s41467-021-23556-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nalls M.A., Escott-Price V., Williams N.M., Lubbe S., Keller M.F., Morris H.R., Singleton A.B., International Parkinson's Disease Genomics Consortium IPDGC Genetic risk and age in Parkinson's disease: Continuum not stratum. Mov. Disord. 2015;30:850–854. doi: 10.1002/mds.26192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saez-Atienzar S., Bandres-Ciga S., Langston R.G., Kim J.J., Choi S.W., Reynolds R.H., International ALS Genomics Consortium, ITALSGEN. Dewan R., Abramzon Y., Ahmed S., et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abd9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bandres-Ciga S., Saez-Atienzar S., Kim J.J., Makarious M.B., Faghri F., Diez-Fairen M., Iwaki H., Leonard H., Botia J., Ryten M., et al. Large-scale pathway specific polygenic risk and transcriptomic community network analysis identifies novel functional pathways in Parkinson disease. Acta Neuropathol. 2020;140:341–358. doi: 10.1007/s00401-020-02181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim N.Y., Lee H., Kim S., Kim Y.J., Lee H., Lee J., Kwak S.H., Lee S. The clinical relevance of a polygenic risk score for type 2 diabetes mellitus in the Korean population. Sci. Rep. 2024;14:5749. doi: 10.1038/s41598-024-55313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plym A., Penney K.L., Kalia S., Kraft P., Conti D.V., Haiman C., Mucci L.A., Kibel A.S. Evaluation of a Multiethnic Polygenic Risk Score Model for Prostate Cancer. J. Natl. Cancer Inst. 2022;114:771–774. doi: 10.1093/jnci/djab058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sekimitsu S., Xiang D., Smith S.L., Curran K., Elze T., Friedman D.S., Foster P.J., Luo Y., Pasquale L.R., Peto T., et al. Deep Ocular Phenotyping Across Primary Open-Angle Glaucoma Genetic Burden. JAMA Ophthalmol. 2023;141:891–899. doi: 10.1001/jamaophthalmol.2023.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang F., Ghanzouri I., Leeper N.J., Tsao P.S., Ross E.G. Development of a polygenic risk score to improve detection of peripheral artery disease. Vasc. Med. 2022;27:219–227. doi: 10.1177/1358863X211067564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Mossevelde S., van der Zee J., Gijselinck I., Sleegers K., De Bleecker J., Sieben A., Vandenberghe R., Van Langenhove T., Baets J., Deryck O., et al. Clinical Evidence of Disease Anticipation in Families Segregating a C9orf72 Repeat Expansion. JAMA Neurol. 2017;74:445–452. doi: 10.1001/jamaneurol.2016.4847. [DOI] [PubMed] [Google Scholar]
- 29.Kolberg L., Raudvere U., Kuzmin I., Adler P., Vilo J., Peterson H. g:Profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update) Nucleic Acids Res. 2023;51:W207–W212. doi: 10.1093/nar/gkad347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lachmann A., Schilder B.M., Wojciechowicz M.L., Torre D., Kuleshov M.V., Keenan A.B., Ma'ayan A. Geneshot: search engine for ranking genes from arbitrary text queries. Nucleic Acids Res. 2019;47:W571–W577. doi: 10.1093/nar/gkz393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakaue S., Okada Y. GREP: genome for REPositioning drugs. Bioinformatics. 2019;35:3821–3823. doi: 10.1093/bioinformatics/btz166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lussier Y.A., Chen J.L. The emergence of genome-based drug repositioning. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., Gould J., Davis J.F., Tubelli A.A., Asiedu J.K., et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017;171:1437–1452.e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kropiwnicki E., Evangelista J.E., Stein D.J., Clarke D.J.B., Lachmann A., Kuleshov M.V., Jeon M., Jagodnik K.M., Ma'ayan A. Drugmonizome and Drugmonizome-ML: integration and abstraction of small molecule attributes for drug enrichment analysis and machine learning. Database. 2021;2021 doi: 10.1093/database/baab017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boussicault L., Laffaire J., Schmitt P., Rinaudo P., Callizot N., Nabirotchkin S., Hajj R., Cohen D. Combination of acamprosate and baclofen (PXT864) as a potential new therapy for amyotrophic lateral sclerosis. J. Neurosci. Res. 2020;98:2435–2450. doi: 10.1002/jnr.24714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plosker G.L. Acamprosate: A Review of Its Use in Alcohol Dependence. Drugs. 2015;75:1255–1268. doi: 10.1007/s40265-015-0423-9. [DOI] [PubMed] [Google Scholar]
- 37.Tariq A., Hill N.S., Price L.L., Ismail K. Incidence and Nature of Respiratory Events in Patients Undergoing Bronchoscopy Under Conscious Sedation. J. Bronchology Interv. Pulmonol. 2022;29:283–289. doi: 10.1097/LBR.0000000000000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dafinca R., Scaber J., Ababneh N., Lalic T., Weir G., Christian H., Vowles J., Douglas A.G.L., Fletcher-Jones A., Browne C., et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cell. 2016;34:2063–2078. doi: 10.1002/stem.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beckers J., Tharkeshwar A.K., Fumagalli L., Contardo M., Van Schoor E., Fazal R., Thal D.R., Chandran S., Mancuso R., Van Den Bosch L., Van Damme P. A toxic gain-of-function mechanism in C9orf72 ALS impairs the autophagy-lysosome pathway in neurons. Acta Neuropathol. Commun. 2023;11:151. doi: 10.1186/s40478-023-01648-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y.J., Jansen-West K., Xu Y.F., Gendron T.F., Bieniek K.F., Lin W.L., Sasaguri H., Caulfield T., Hubbard J., Daughrity L., et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128:505–524. doi: 10.1007/s00401-014-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hart M.P., Gitler A.D. ALS-associated ataxin 2 polyQ expansions enhance stress-induced caspase 3 activation and increase TDP-43 pathological modifications. J. Neurosci. 2012;32:9133–9142. doi: 10.1523/JNEUROSCI.0996-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lacomblez L., Bensimon G., Leigh P.N., Guillet P., Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347:1425–1431. doi: 10.1016/s0140-6736(96)91680-3. [DOI] [PubMed] [Google Scholar]
- 43.Kalk N.J., Lingford-Hughes A.R. The clinical pharmacology of acamprosate. Br. J. Clin. Pharmacol. 2014;77:315–323. doi: 10.1111/bcp.12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Debono M.W., Le Guern J., Canton T., Doble A., Pradier L. Inhibition by riluzole of electrophysiological responses mediated by rat kainate and NMDA receptors expressed in Xenopus oocytes. Eur. J. Pharmacol. 1993;235:283–289. doi: 10.1016/0014-2999(93)90147-a. [DOI] [PubMed] [Google Scholar]
- 45.Cummings J.L., Tong G., Ballard C. Treatment Combinations for Alzheimer's Disease: Current and Future Pharmacotherapy Options. J. Alzheimers Dis. 2019;67:779–794. doi: 10.3233/JAD-180766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C., et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- 47.Jarada T.N., Rokne J.G., Alhajj R. A review of computational drug repositioning: strategies, approaches, opportunities, challenges, and directions. J. Cheminf. 2020;12:46. doi: 10.1186/s13321-020-00450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reay W.R., Cairns M.J. Advancing the use of genome-wide association studies for drug repurposing. Nat. Rev. Genet. 2021;22:658–671. doi: 10.1038/s41576-021-00387-z. [DOI] [PubMed] [Google Scholar]
- 49.Beck J., Poulter M., Hensman D., Rohrer J.D., Mahoney C.J., Adamson G., Campbell T., Uphill J., Borg A., Fratta P., et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 2013;92:345–353. doi: 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Rheenen W., van der Spek R.A.A., Bakker M.K., van Vugt J.J.F.A., Hop P.J., Zwamborn R.A.J., de Klein N., Westra H.J., Bakker O.B., Deelen P., et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 2021;53:1636–1648. doi: 10.1038/s41588-021-00973-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al Khleifat A., Iacoangeli A., van Vugt J.J.F.A., Bowles H., Moisse M., Zwamborn R.A.J., van der Spek R.A.A., Shatunov A., Cooper-Knock J., Topp S., et al. Structural variation analysis of 6,500 whole genome sequences in amyotrophic lateral sclerosis. NPJ Genom. Med. 2022;7:8. doi: 10.1038/s41525-021-00267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Al-Chalabi A., Calvo A., Chio A., Colville S., Ellis C.M., Hardiman O., Heverin M., Howard R.S., Huisman M.H.B., Keren N., et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 2014;13:1108–1113. doi: 10.1016/S1474-4422(14)70219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chio A., Mazzini L., D'Alfonso S., Corrado L., Canosa A., Moglia C., Manera U., Bersano E., Brunetti M., Barberis M., et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology. 2018;91:e635–e642. doi: 10.1212/WNL.0000000000005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cady J., Allred P., Bali T., Pestronk A., Goate A., Miller T.M., Mitra R.D., Ravits J., Harms M.B., Baloh R.H. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 2015;77:100–113. doi: 10.1002/ana.24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fumagalli L., Young F.L., Boeynaems S., De Decker M., Mehta A.R., Swijsen A., Fazal R., Guo W., Moisse M., Beckers J., et al. C9orf72-derived arginine-containing dipeptide repeats associate with axonal transport machinery and impede microtubule-based motility. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abg3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Blitterswijk M., Mullen B., Wojtas A., Heckman M.G., Diehl N.N., Baker M.C., DeJesus-Hernandez M., Brown P.H., Murray M.E., Hsiung G.Y.R., et al. Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol. Neurodegener. 2014;9:38. doi: 10.1186/1750-1326-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang M., Ferrari R., Tartaglia M.C., Keith J., Surace E.I., Wolf U., Sato C., Grinberg M., Liang Y., Xi Z., et al. A C6orf10/LOC101929163 locus is associated with age of onset in C9orf72 carriers. Brain. 2018;141:2895–2907. doi: 10.1093/brain/awy238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barbier M., Camuzat A., Hachimi K.E., Guegan J., Rinaldi D., Lattante S., Houot M., Sánchez-Valle R., Sabatelli M., Antonell A., et al. SLITRK2, an X-linked modifier of the age at onset in C9orf72 frontotemporal lobar degeneration. Brain. 2021;144:2798–2811. doi: 10.1093/brain/awab171. [DOI] [PubMed] [Google Scholar]
- 59.Bali T., Self W., Liu J., Siddique T., Wang L.H., Bird T.D., Ratti E., Atassi N., Boylan K.B., Glass J.D., et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J. Neurol. Neurosurg. Psychiatry. 2017;88:99–105. doi: 10.1136/jnnp-2016-313521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu J., Lee J., Salazar Hernandez M.A., Mazitschek R., Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brum A.M., van de Peppel J., van der Leije C.S., Schreuders-Koedam M., Eijken M., van der Eerden B.C.J., van Leeuwen J.P.T.M. Connectivity Map-based discovery of parbendazole reveals targetable human osteogenic pathway. Proc. Natl. Acad. Sci. USA. 2015;112:12711–12716. doi: 10.1073/pnas.1501597112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pharnext . In: Pharnext refocuses its clinical trial programs on PXT3003, its most promising drug candidate, to optimize financial resources allocation. Elliott M.J., editor. 2023. [Google Scholar]
- 63.Umhau J.C., Momenan R., Schwandt M.L., Singley E., Lifshitz M., Doty L., Adams L.J., Vengeliene V., Spanagel R., Zhang Y., et al. Effect of acamprosate on magnetic resonance spectroscopy measures of central glutamate in detoxified alcohol-dependent individuals: a randomized controlled experimental medicine study. Arch. Gen. Psychiatr. 2010;67:1069–1077. doi: 10.1001/archgenpsychiatry.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dewan R., Chia R., Ding J., Hickman R.A., Stein T.D., Abramzon Y., Ahmed S., Sabir M.S., Portley M.K., Tucci A., et al. Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron. 2021;109:448–460.e4. doi: 10.1016/j.neuron.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nicolas A., Kenna K.P., Renton A.E., Ticozzi N., Faghri F., Chia R., Dominov J.A., Kenna B.J., Nalls M.A., Keagle P., et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron. 2018;97:1267–1288. doi: 10.1016/j.neuron.2018.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prudencio M., Belzil V.V., Batra R., Ross C.A., Gendron T.F., Pregent L.J., Murray M.E., Overstreet K.K., Piazza-Johnston A.E., Desaro P., et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015;18:1175–1182. doi: 10.1038/nn.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Choi S.W., O'Reilly P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience. 2019;8:giz082. doi: 10.1093/gigascience/giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duan Y., Evans D.S., Miller R.A., Schork N.J., Cummings S.R., Girke T. signatureSearch: environment for gene expression signature searching and functional interpretation. Nucleic Acids Res. 2020;48 doi: 10.1093/nar/gkaa878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brooks B.R. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial "Clinical limits of amyotrophic lateral sclerosis" workshop contributors. J. Neurol. Sci. 1994;124:96–107. doi: 10.1016/0022-510x(94)90191-0. [DOI] [PubMed] [Google Scholar]
- 70.Neary D., Snowden J.S., Gustafson L., Passant U., Stuss D., Black S., Freedman M., Kertesz A., Robert P.H., Albert M., et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 71.Salvi E., Kutalik Z., Glorioso N., Benaglio P., Frau F., Kuznetsova T., Arima H., Hoggart C., Tichet J., Nikitin Y.P., et al. Genomewide association study using a high-density single nucleotide polymorphism array and case-control design identifies a novel essential hypertension susceptibility locus in the promoter region of endothelial NO synthase. Hypertension. 2012;59:248–255. doi: 10.1161/HYPERTENSIONAHA.111.181990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Rheenen W., Shatunov A., Dekker A.M., McLaughlin R.L., Diekstra F.P., Pulit S.L., van der Spek R.A.A., Võsa U., de Jong S., Robinson M.R., et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016;48:1043–1048. doi: 10.1038/ng.3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Das S., Forer L., Schönherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chia R., Sabir M.S., Bandres-Ciga S., Saez-Atienzar S., Reynolds R.H., Gustavsson E., Walton R.L., Ahmed S., Viollet C., Ding J., et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 2021;53:294–303. doi: 10.1038/s41588-021-00785-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Euesden J., Lewis C.M., O'Reilly P.F. PRSice: Polygenic Risk Score software. Bioinformatics. 2015;31:1466–1468. doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mehta P., Kaye W., Raymond J., Punjani R., Larson T., Cohen J., Muravov O., Horton K. Prevalence of Amyotrophic Lateral Sclerosis - United States, 2015. MMWR Morb. Mortal. Wkly. Rep. 2018;67:1285–1289. doi: 10.15585/mmwr.mm6746a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leonenko G., Baker E., Stevenson-Hoare J., Sierksma A., Fiers M., Williams J., de Strooper B., Escott-Price V. Identifying individuals with high risk of Alzheimer's disease using polygenic risk scores. Nat. Commun. 2021;12:4506. doi: 10.1038/s41467-021-24082-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The summary statistics of the reference dataset genome-wide association study (GWAS) are publicly available at http://databrowser.projectmine.com/. The individual-level data for the training and test datasets are available on dbGaP (accession nos. phs000101.v5.p1 and phs001963.v1.p1). The data for the replication dataset were from a different study (principal investigator: Christopher Shaw, King’s College London) and are available upon reasonable request. RNA-seq data from the motor cortex were obtained from the New York Genome Center, and RNA-seq data from the cerebellum were obtained from GEO (GEO: GSE67196). The figures were created using BioRender and Inkscape. The Sankey diagram was created using Visual Paradigm. The programming code used in this paper is available at https://github.com/sarasaezALS/C9orf72_AAO and https://zenodo.org/records/13259646 (https://doi.org/10.5281/zenodo.13259646) to facilitate the application of this methodology to other disorders. The individual-level polygenic risk scores generated for patients with the C9orf72 mutation are available at https://zenodo.org/uploads/13769448. The connectivity scores for the drugs evaluated as treatments for C9orf72-related disease are available at https://zenodo.org/records/13769483.