

Abstract
Background and Objectives
Stiff person syndrome spectrum disorder (SPSD) is a rare autoimmune disorder characterized by progressive muscle stiffness and painful spasms with an estimated prevalence of 1–2 cases per million people. Population-based epidemiologic studies are lacking because of both poor patient capture and the lack of standardized diagnostic criteria. Objectives of this study were to describe the incidence and prevalence of SPSD within the University of Colorado Health (UCH) system and apply previously proposed published criteria for SPSD within this population.
Methods
We queried Health Data Compass, an electronic health data repository for a large academic health care system, from 2012 through 2022 for all patients older than 18 years with International Classification of Diseases, 10th Revision (ICD-10) codes pertaining to stiff person syndrome. Records were reviewed for diagnostic confirmation. We calculated yearly and period prevalence and incidence rates based on observable person-time exposure of our cohort. We applied previously published Mayo Clinic and Johns Hopkins criteria for SPSD and compared period prevalence based on each criterion and evaluated for agreement.
Results
Two hundred and seventy-three patients met the initial inclusion criteria using ICD-10 codes; 59 were confirmed to have SPSD. The mean age was 49.7 years (SD = 12.9), 59.3% were female, 59.3% were considered antibody positive. The total database population over the interval was 2,801,674 persons. The estimated prevalence of SPSD based on our UCH cohort was 2.11 (95% CI 1.57–2.64) per 100,000 persons. The average yearly incidence was 0.35 per 100,000 person-years (95% CI 0.27–0.46). Applying different clinical diagnostic criteria, the estimated prevalence ranged from 1.36 (95% CI 0.93–1.79) to 1.82 (95% CI 1.32–2.32) per 100,000 persons.
Discussion
We describe a prevalence of SPSD of 2.11 per 100,000 persons in our UCH cohort. Prevalence estimates differed depending on which clinical diagnostic criteria were applied and whether possible SPSD cases were included. Using the most stringent criteria for diagnosis, we report a prevalence of 1.36 per 100,000 persons. While our study uniquely captures many US demographic groups, limitations remain because this is a retrospective review of a single system. Additional studies are needed to determine whether these results are representative of a national or global population.
Introduction
Stiff person syndrome (SPS) is a rare immune-mediated neurologic disorder characterized by progressive muscle stiffness and painful spasms, often occurring primarily in the axial musculature. SPS was first described in 1956 in patients with primarily truncal and proximal muscles affected.1 The spectrum of disease has expanded, ranging from stiff limb syndrome to a severe form known as progressive encephalomyelitis with rigidity and myoclonus (PERM)2 and other neurologic manifestations collectively described as stiff person spectrum disorder (SPSD).3 SPSD is most associated with autoantibodies against glutamic acid decarboxylase, epitope 65 (GAD65),4-8 but other autoantibodies reported in similar clinical phenotypes include those directed against glycine receptor α1 subunit (GlyR-α1),9-12 γ-aminobutyric acid-B receptors,13 amphiphysin,14 gephyrin, and dipeptidyl peptidase-like protein 6 (DPPX).15 Seronegative patients likely make up approximately 14%–20% of SPSD cases.16,17 The hypothesized pathophysiology of SPSD suggests immune-mediated dysfunction of the γ-aminobutyric acid signaling, leading to motor hyperactivity. An immune-mediated mechanism is further supported by patient response to immunomodulatory therapy, including IV immunoglobulin (IVIg), plasma exchange (PLEX), and/or rituximab.18
Despite advances, much remains unknown about SPSD. Clinical diagnosis is based on a combination of clinical presentation, examination findings, and/or supportive serologic and electrophysiologic testing. The lack of standardized criteria, delay, and misdiagnosis are common. Authors from the Mayo Clinic found that misdiagnosis was threefold more common than the diagnosis of confirmed SPSD in 1 cohort.17 Both the Mayo Clinic17 and John Hopkins University19 have proposed diagnostic criteria, but it is unclear in practice whether these criteria are representative and sufficient for SPSD.
Owing to diagnostic challenges, evolving autoantibody associations, and limited diagnostic criteria, the epidemiology of SPSD is largely unknown. The disease's prevalence is often quoted as “one-in-a-million,” but accurate epidemiologic data have been hard to obtain. This quote originated from an estimated prevalence of 1 in 1,250,000 from a center in Heidelberg, Germany, serving 2 to 3 million people, with 20 cases over 10 years, noted by Meinck and Thompson in 2002.20,21 The British Neurological Surveillance Unit identified 119 cases among the UK population over 5 years (2000–2005), implying a prevalence of 1–2 cases per million.22 In 2018, a United States–based study found a point prevalence of 2.06 per million patients in a population of patients evaluated within the United States Veterans Affairs (VA) Health Administration.23 While this study was generally close to the estimate of 1–2 per million, the study had various limitations including population characteristics with poor generalizability—population was predominantly male and limited to GAD65 antibody positivity only—and methodological limitations of a lack of defined population inclusion (i.e., denominator). A 2024 study used a taxonomic approach to analyze Medicare claims data, reporting a diagnostic prevalence as high as 3 in 100,000.24 A neurologist recorded the diagnosis in a minority of cases, and a positive titer was noted only by the International Classification of Diseases, 10th Revision (ICD-10) code R76.0. Using taxonomic codes for antibody titers limits the ability to delineate high versus low titers. With current estimations limited by poor disease capture and previous study population characteristics with poor generalizability, the incidence and prevalence of SPSD remain unknown. In this study, we report the incidence and prevalence of SPSD using a unique data warehouse for a large US health system in Colorado. Furthermore, we apply and compare existing diagnostic criteria to evaluate their utility in capturing the diverse phenotypes of SPSD and compare agreement between criteria.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The Colorado Multiple Institutional Review Board approved this study as exempt for secondary data use (COMIRB, #22-1727). Our institutional ethical standards committee waived informed consent, given the retrospective design.
Study Design
This retrospective study includes all adult patients (18 years and older) who had any contact with the University of Colorado Health (UCH) system from January 1, 2012, to December 31, 2022. We queried Health Data Compass (HDC), the data repository of the UCH, for electronic health records (EHRs) to identify patients with ICD-10 codes for SPS or stiff man syndrome (G25.82). Two neurologists (P.D.C. and A.L.P.) reviewed records for diagnostic confirmation, with a third autoimmune neurologist (E.A.M.) providing tie-breaking adjudication. A subset of patients were evaluated and treated by the authors, further substantiating a likely diagnosis of SPSD. This included a clinical evaluation of 45 patients (76.3% of the entire cohort) by A.L.P. Diagnostic confirmation of SPSD was based on the inclusion criteria of cardinal features of muscle stiffness and episodic muscle spasms,25 antibody positivity (criteria given further), and exclusion if there was an alternative diagnosis. For antibody-negative cases, there was need for additional supportive features of SPSD including hyperstartle response, muscle spasms induced by various triggers, additional cerebellar or brainstem features, and exclusion of an alternative diagnosis after review of red flags.17 Antibody-negative patients or those with low-titer GAD65 with red flags were excluded to minimize the inclusion of patients with misdiagnosis. Red flags used in our exclusion criteria have been previously described and included muscle stiffness and spasms not being a major symptom, prominent muscle wasting and fasciculations, no documented improvement with benzodiazepines, no objective improvement with immunotherapy, chronic pain as the predominate feature, functional neurologic signs, and absence of increased tone noted on clinical examination.17 eTable 1 provides clinical symptoms and red flags in our cohort.
Patients with a verified diagnosis of SPSD were defined as the University of Colorado Health or “UCH” cohort. We applied the proposed Mayo Clinic criteria17 definitions for probable and definite SPSD and Johns Hopkins criteria19 for possible, probable, and definite SPSD and compared their agreement (Figure 1).
Figure 1. Study Design.

Two hundred and seventy-three patients were identified by a query of the HDC database. Two hundred and fourteen patients were removed from the cohort because of other GAD65-related syndromes, misattributed diagnostic codes, insufficient data in the chart, or confirmation of alternative diagnoses. The table summarizes the breakdown of these 214 patients. SPS diagnosis was substantiated in 59 patients by chart review. Fifty of these patients were personally evaluated by members of our Neuroimmunology group, and 45 of the patients were evaluated by faculty with specific training in Autoimmune Neurology. Proposed diagnostic criteria for SPS were then applied, with 38 patients meeting proposed Mayo Clinic criteria (8 definite, 30 probable) and 51 meeting Johns Hopkins criteria (29 definite, 12 probable, and 10 possible). GAD65 = glutamic acid decarboxylase 65-kilodalton isoform; HDC = Health Data Compass; SPS = stiff person syndrome; SPSD = stiff person spectrum disorder.
Antibody-positive SPSD cohort was defined as patients with either serum GAD65 IgG >20 nmol/L (radioimmunoprecipitation assay [RIA] at Mayo Clinic Laboratories), serum GAD65 IgG >10,000 IU/mL (ELISA), CSF GAD65 IgG positive, GlyR-α1 IgG positive (serum or CSF at Mayo Clinic Laboratories), amphiphysin IgG positive (serum or CSF), or DPPX IgG (serum or CSF) positive. For patients with more than 1 laboratory assay for GAD65, Mayo Clinic Laboratories' results determined high titer. Patients below the threshold of high-titer cutoff for GAD65 and CSF negativity were considered “antibody negative.”
Data Source
We used the HDC, which captures EHRs from UCH; billing data from participating provider groups; and additional structural data points from the Colorado All Payers Claim Database, the Colorado Department of Public Health and Environment (CDPHE), and others. Patients in the UCH system have been captured by this database since EHR integration with Epic, with all sites contributing full data sets from 2011 onward.
Epidemiology Analysis
The study period was January 1, 2012, to December 31, 2022. Period prevalence was calculated by dividing all patients identified as having SPSD at any time during the period by the total number of persons who had any contact with the health system during the observation period. Patients were considered present in the system after their first recorded encounter and until recorded death. Death data were obtained using the EHR and integrated into Compass from CDPHE. 95% CIs were calculated using either the Wald method for proportions when the number of SPSD cases was 10 or greater or the exact Clopper-Pearson method when the number of SPSD cases was less than 10. The same method was applied to each calendar year. Prevalence for the entire study period was separately calculated for the different diagnostic criteria. Proportions were expressed per 100,000 persons.
Incidence was calculated with Poisson count rate models, with log link, log exposure time offset, and robust standard errors. Counts were the number of newly diagnosed SPSD cases within the study period. Exposure time was determined by how long a person was in the system before SPSD diagnosis. Patients with SPSD diagnosed on their first encounter were not included because of zero exposure time. Diagnosis started at the time of the first use of the SPS ICD-10 code (G25.82). 95% CIs were calculated with the Wald method. Incidence was calculated for the entire study period and for each calendar year. Incidences were expressed per 100,000 person-years.
Additional Statistical Analysis
Summary statistics were obtained for SPSD patient demographics, diagnostic criteria, disease characteristics, comorbidities, antibody biomarkers, electrophysiology, and medications. Mayo Clinic and Johns Hopkins criteria were compared among the patients with SPSD with a 2-way frequency table, the McNemar test, and a κ statistic. The κ coefficient was used to measure agreement, with a κ of 1 indicating perfect agreement and a κ of 0 indicating agreement equivalent to chance. Demographic statistics were reported for the source patient population.
Additional data quality checks identified a small subset of patients (n = 573) with implausible ages (>120 years) likely because of unidentified individuals. We could not confirm whether these individuals were duplicate records, so we removed them from the total analysis. This did not affect the incidence or prevalence calculations.
Data Availability
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results
Summary of Patient Demographics
Based on our UCH cohort of 59 patients, the mean age of patients was 49.7 years (SD = 12.9) and 59.3% were female. 81.4% self-identified as White, 8.5% Black, 1.2% American Indian or Alaskan Native, and 6.8% other. 91.5% self-reported as non-Hispanic, Latino/a, or Spanish origin (Tables 1 and 2).
Table 1.
Demographics
| Demographics | UCH SPSD cohort (n = 59) | Total UCH population (n = 2,801,674) |
| Age,a y, mean (SD) | 49.7 (12.9) | 47.5 (18.7) |
| Sex assigned at birth, n (%) | ||
| Female | 35 (59.3) | 1,519,928 (54.3) |
| Male | 24 (40.7) | 1,281,064 (45.7) |
| Unknown | 0 (0) | 682 (0.02) |
| Race, n (%) | ||
| White | 48 (81.4) | 2,063,672 (75.6) |
| Black | 5 (8.5) | 162,273 (6.0) |
| American Indian or Alaskan Native | 1 (1.2) | 15,732 (0.6) |
| More than 1 race | — | 21,121 (0.8) |
| Other | 4 (6.8) | 365,518 (13.0) |
| Unknown | — | 173,358 (6.2) |
| Ethnicity, n (%) | ||
| Hispanic, Latino/a, or Spanish origin | 5 (8.5) | 384,125 (13.7) |
| Non-Hispanic, Latino/a, or Spanish origin | 54 (91.5) | 2,213,032 (79.0) |
| Unknown | — | 204,517 (7.3) |
| SPSD phenotype, n (%) | ||
| Classic SPS | 39 (66.1) | — |
| Partial or stiff limb | 1 (1.7) | — |
| SPS plus | 16 (27.1) | — |
| PERM | 3 (5.1) | — |
| Comorbidities, n (%) | ||
| Type 1 diabetes mellitus or latent autoimmune diabetes of adults or LADA | 9 (15.3) | — |
| Thyroid disease | 26 (44.1) | — |
| Vitamin B12 deficiency | 26 (44.1) | — |
| Vitiligo | 8 (13.6) | — |
| Epilepsy | 4 (6.8) | — |
| Neuropathy | 37 (62.7) | — |
| Rheumatoid arthritis | 3 (5.1) | — |
| Previous spinal surgery | 8 (13.6) | — |
| Anxiety | 39 (67.2) | — |
| Depression | 27 (46.6) | — |
| Chronic opioid useb | 15 (25.4) | — |
| Self-reported chronic alcohol usec | 3 (5.1) | — |
Abbreviations: LADA = latent autoimmune diabetes of adults; PERM = progressive encephalomyelitis with rigidity and myoclonus; SPS = stiff person syndrome; SPSD = stiff person spectrum disorder; UCH = University of Colorado Health.
Age was calculated at either SPS diagnosis or at the midpoint of their exposure for patients without an SPS diagnosis.
Defined as >3 months of continuous use.
Defined as >7 standard drinks in a consecutive 7-day period.
Table 2.
Clinical and Diagnostic Data
| Patients with positive results/patients tested (%) | |
| Serum and CSF analysis | |
| Serum GAD65 IgG >20 nmol/L RIA | 23/57 (40.4) |
| Serum GAD65 IgG >10,000 IU/mL ELISA | 1/28 (3.6) |
| Serum GAD65 IgG >250 IU/mL ELISAa | 15/28 (53.6) |
| Serum GAD65 IgG >20 nmol/L RIA or serum GAD 65 IgG >10,000 IU/L ELISA | 27/59 (45.8)b |
| CSF GAD65 IgG positive | 8/26 (30.8) |
| Glycine receptor IgG positive | 10/32 (31.3) |
| Amphiphysin IgG positive | 1/48 (2.1) |
| Antibody positivec | 35/59 (59.3)b |
| Elevated total nucleated cells >5 cells/µL | 5/39 (12.8) |
| Elevated CSF protein >45 mg/dL | 12/39 (30.8) |
| Presence of CSF-restricted oligoclonal bands | 8/37 (21.6) |
| Elevated IgG index >0.66 | 2/34 (5.9) |
| Electrophysiologic testing | |
| EMG completed | 43/59 (72.9) |
| Meets at least 1 of the following 4 criteriad | 19/36 (52.8) |
| 1: inability to relax paraspinal muscles on EMG | 8/22 (36.4) |
| 2: exaggerated acoustic or exteroceptive responses by surface EMG | 0/1 (0.0) |
| 3: co-contraction of agonist/antagonist muscles by EMG | 4/5 (80.0) |
| 4: continuous motor unit activity in affected muscles | 14/30 (46.7) |
| Symptomatic treatment used during SPS disease coursee | |
| Diazepam | 52/59 (88.1) |
| Clonazepam | 22/59 (37.3) |
| Lorazepam | 12/59 (20.3) |
| Alprazolam | 4/59 (6.8) |
| Baclofen | 53/59 (89.8) |
| Botulinum toxin injection | 10/59 (16.9) |
| Immune therapy used during SPS disease course | |
| IVIg | 55/59 (93.2) |
| Plasma exchange | 23/59 (39.0) |
| Rituximab | 36/59 (61.0) |
| Mycophenolate | 16/59 (27.1) |
| Corticosteroidsf | 11/59 (18.6) |
| Azathioprine | 3/59 (5.1) |
| Cyclophosphamide | 4/59 (6.8) |
| aHSCT | 4/59 (6.8) |
Abbreviations: aHSCT = autologous hematopoietic stem cell transplant; GAD65 = glutamic acid decarboxylase 65-kilodalton isoform; IVIg = IV immunoglobulin G; RIA = radioimmunoprecipitation assay; SPS = stiff person syndrome; SPSD = stiff person spectrum disorder.
Serum GAD65 IgG titers obtained through ELISA testing methods may have limited diagnostic utility if they are reported >250 IU/mL. Because the laboratory does report titers beyond 250 IU/mL, it is difficult to interpret whether the patient has a high titer (defined as 10,000 IU/mL using ELISA).
Proportions calculated based on the total population; however, not all patients underwent comprehensive testing that included each antibody outside GAD65 antibody testing alone.
Antibody positive indicates that the patient had one of either serum GAD65 IgG >20 nmol/L, serum GAD65 IgG >10,000 IU/mL, CSF GAD 65 IgG, glycine receptor IgG, or amphiphysin IgG.
Most EMGs obtained before our evaluation did not always include comprehensive testing that could be used with applying proposed diagnostic criteria.
Additional benzodiazepines used at lower frequencies included temazepam and chlordiazepoxide.
Chronic steroid use for SPSD was defined as ≥3 months consecutively.
Antibody Status
In this UCH cohort, 59.3% (35/59) were considered antibody positive. Of patients who had antibody testing on the RIA at Mayo Clinic Laboratories, 40.4% (23/57) were positive at >20 nmol/L. One patient was identified to have a high titer (>10,000 IU/mL) on ELISA with a titer of 25,000 IU/mL. Three additional patients had limited GAD65 testing (i.e., laboratory testing limited to a titer of >250 IU/mL on ELISA testing, without further testing to determine whether values were either >20 nmol/L [RIA] or >10,000 IU/mL [other ELISA methods]). These 3 patients did not meet criteria for high-titer GAD65 and, therefore, were considered “antibody negative.” Twenty-four patients were considered “antibody-negative.” Fifteen of these patients had low-titer GAD65 accounting for 62.5% of what we considered our “antibody-negative” population. Nine patients were true seronegative, when accounting for low-titer GAD65 antibodies (thus a true seronegative rate of 15.3%). Low-titer GAD65 was considered only at the time of diagnosis and not counted if this was positive after IVIg. Of patients who were tested for GlyR-α1 IgG, 31.3% (10/32) were positive in the serum. GlyR-α1 IgG testing was not commercially available until 2020. Some patients who were GlyR-α1 IgG positive were identified starting in 2017 through research testing available per special request (Mayo Clinic Laboratories). One of 48 (2.1%) was amphiphysin IgG positive (in both serum and CSF). Owing to reports of SPSD-like presentations in DPPX autoimmunity, we assessed for this antibody in our cohort, but no cases were identified.
CSF Analysis
Lumbar puncture was completed in 67.8% (40/49) of our cohort. Elevated CSF protein (>45 mg/dL) was the most common finding in 30.8% (12/39), followed by the presence of CSF-restricted oligoclonal bands in 21.6% (8/37), a pleocytosis (>5 white blood cells/µL) in 12.8% (5/39), and an elevated IgG index (>0.66) in 5.9% (2/34). Of those who were tested, 30.8% (8/26) were GAD65 antibody positive in the CSF. Of 9 tested, none had GlyR-α1 antibodies in the CSF.
Electrophysiology
EMG was completed in 72.9% (43/59) with 52.8% (19/36) meeting at least 1 of the following criteria on EMG: (1) inability to relax paraspinal muscle, (2) co-contraction of agonist/antagonist muscles, (3) continuous motor unit activity in affected muscles, and (4) exaggerated acoustic or exteroceptive responses by surface EMG.
Comorbid Disorders
Medical, neurologic, and psychiatric comorbidities are summarized further.
Coexisting Autoimmunity
Type 1 diabetes or latent autoimmune diabetes of adults (LADA) was found in 15.3% (9/59), thyroid disease in 44.1% (26/59), vitamin B12 deficiency in 44.1% (26/59), vitiligo in 13.6% (8/59), and rheumatoid arthritis in 5.1% (3/59).
Neurologic and Psychiatric Comorbidities
Epilepsy was reported in 6.8% (4/59), neuropathy in 62.7% (37/59), anxiety in 67.2% (39/59), and depression in 46.6% (27/59).
Substance Use
Self-reported chronic alcohol use defined as >7 standard drinks in 7 consecutive days was reported in 5.1% (3/59). Chronic use of opioids (>3 months) was reported in 25.4% (16/59).
History of Spinal Surgery
A history of spinal surgery was reported in 13.6% (8/59).
Summary of Treatments
Treatment data collected from chart review included immune therapy and symptomatic treatments administered during the disease course. Common symptomatic therapies and immunotherapies were investigated for each disease phenotype (Figure 2).
Figure 2. Immune Therapy and Symptomatic Treatments by Disease Phenotype.

Treatment data were collected from chart review. We highlighted some common immune therapies and symptomatic treatments administered during the disease course among the various SPSD phenotypes. The use of steroids was defined as chronic steroids for ≥3 consecutive months and included 18.6% (11/59). IVIg was administered to most patients regardless of disease phenotype (93.2% [55/59]), followed by rituximab (61.0% [36/59]) and then PLEX (40.0 [23/59]). Cyclophosphamide was administered to a total of 4 patients (3 patients with SPS plus and 1 patient with classic SPS). Four patients underwent autologous hematopoietic stem cell transplant (2 patients with classic SPS, 2 patients with SPS plus, and 1 patient with pure cerebellar ataxia). Symptomatic treatment included baclofen (89.8% [53/59]), diazepam (89.8% [53/59]), and less frequently other benzodiazepines (clonazepam, lorazepam, temazepam, alprazolam, and/or chlordiazepoxide). Botulinum toxin was also used in the treatment of a minority of patients. aHSCT = autologous hematopoietic stem cell transplant; IVIg = IV immunoglobulin G; PERM = progressive encephalomyelitis with rigidity and myoclonus; PLEX = plasma exchange; SPS = stiff person syndrome; SPSD = stiff person spectrum disorder.
Symptomatic Treatment
In our cohort, 88.1% (52/59) of patients were treated with diazepam, 37.3% (22/59) of patients were treated with clonazepam, and a minority of patients also received an alternative benzodiazepine (including lorazepam, temazepam, alprazolam, and/or chlordiazepoxide). Baclofen was used in 89.8% (53/59) and botulinum toxin injections in 16.9% (10/59).
Immune Therapy Treatment
In our cohort, 93.2% (55/59) received IVIg, 61.1% (36/59) received rituximab, and 39.0% (23/59) received PLEX. Less commonly used immune therapies included chronic steroids (defined as ≥3 consecutive months) in 18.6% (11/59), mycophenolate mofetil or mycophenolate acid in 27.1% (16/59), azathioprine in 5.1% (3/59), and cyclophosphamide in 6.8% (4/59). Four patients (6.8%) underwent autologous hematopoietic stem cell transplant.
Epidemiology
The estimated prevalence of SPSD within this UCH cohort was 2.11 (95% CI 1.57–2.64) per 100,000 persons (Figure 3). Applying the proposed diagnostic criteria, the estimated prevalence was 1.36 (95% CI 0.93–1.79) per 100,000 persons for all patients meeting probable or definite Mayo Clinic criteria; 1.82 (95% CI 1.32–2.32) per 100,000 persons for all patients meeting possible, probable, or definite Johns Hopkins criteria; and 1.46 (95% CI 1.02–1.91) per 100,000 persons for all patients meeting probable or definite Johns Hopkins criteria (Figure 4). The average yearly incidence rate between 2012 and 2022 was 0.35 per 100,000 person-years (95% CI 0.27–0.46). Data demonstrate minimal year-to-year variability in the calculation of incidence and prevalence (Figure 3).
Figure 3. Yearly Incidence and Prevalence of Stiff Person Spectrum Disorder.

Yearly incidence and prevalence were calculated based on observable person-year exposure to the University of Colorado Healthcare system. The upper bounds of the first 2 years on the yearly incidence rate graph are removed to allow for enhanced visibility of subsequent years. The upper bound of 2012 and 2013 are, respectively, (6.7965–3.7280).
Figure 4. Diagnosis Prevalence Based on Diagnostic Criteria.

The estimated prevalence of SPSD within this cohort is 2.11 (95% CI 1.79–2.92) per 100,000 persons for the UCH cohort. Accounting for differences in diagnostic criteria, the estimated prevalence was calculated for the Mayo Clinic criteria at 1.36 per 100,000 (95% CI 0.92–1.78), Johns Hopkins criteria (including probable/definite only) at 1.46 per 100,000 (95% CI 1.02–1.91), and Johns Hopkins criteria (possible/probable/definite) at 1.82 per 100,000 (95% CI 1.32–2.32). SPSD = stiff person spectrum disorder; UCH = University of Colorado Health.
Diagnosis and Application of Clinical Criteria
Among the 59 identified in the UCH cohort, 52 patients (88.1%) satisfied either the proposed Mayo Clinic criteria for definite or probable SPSD or the proposed Johns Hopkins criteria for definite, probable, or possible SPSD. Cohort phenotypes included 66.1% (39/59) with classic SPS, 1.7% (1/59) with partial (or stiff limb) syndrome, 27.1% (16/59) with SPS plus, and 5.1% (3/59) with PERM. The latency between patient-reported symptoms and diagnosis was a mean of 4.52 (95% CI 3.24–5.81) years and median of 3.50 years (interquartile range 1.00–5.75 years with 95% CI 2.00–4.61 years). The latency time ranged from 0.08 to 21.74 years. Application of the 2 proposed diagnostic criteria (possible/probable/definite Johns Hopkins vs probable/definite Mayo Clinic) in this real-world data set explored the agreement between these criteria, with a κ coefficient of 0.36 (95% CI 0.13–0.58). Accounting for differences in diagnostic classification, we report a κ statistic of 0.73 (95% CI 0.55–0.92) when comparing the Johns Hopkins criteria for probable/definite cases. When only considering classic phenotypes, we calculated a κ of 0.30 (95% CI 0.05–0.55) with inclusion of the possible cases and 0.74 (95% CI 0.53–0.95) with exclusion of the possible cases in the Johns Hopkins criteria (Figure 5).
Figure 5. Criteria Agreement.
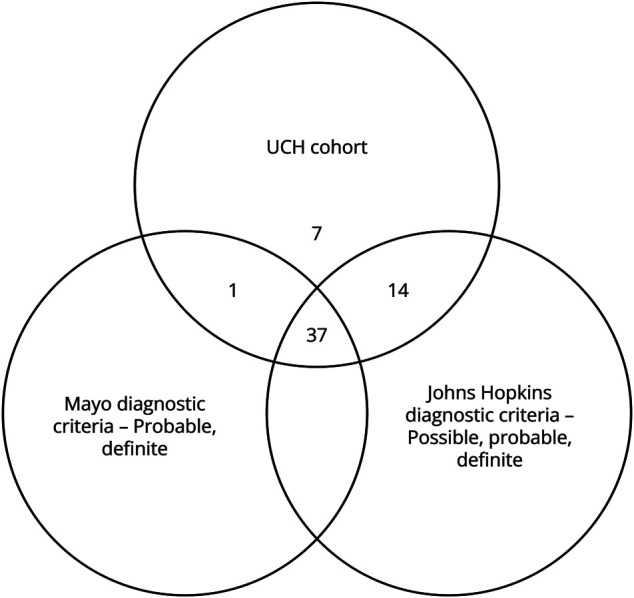
Proposed diagnostic criteria were applied to the 59 patients in the UCH cohort. Among them, 38 patients met the Mayo Clinic criteria (8 definite and 30 probable). Notably, many patients not meeting the definite criteria lacked EMG analysis. Furthermore, 51 patients fulfilled the Johns Hopkins criteria (29 definite, 12 probable, and 10 possible). Application of the 2 proposed diagnostic criteria (possible/probable/definite Johns Hopkins vs probable/definite Mayo) in this real-world data set explores the agreement between these criteria, with a κ coefficient of 0.36 (95% CI 0.13–0.58). With removal of the “possible” cases in the Johns Hopkins criteria, we saw a κ statistic of 0.73 (95% CI 0.55–0.92). When only considering classical phenotypes, we calculated a κ of 0.30 (95% CI 0.05–0.55) with inclusion of the possible cases and 0.74 (95% CI 0.53–0.95) with exclusion of the possible cases in the Johns Hopkins criteria. UCH = University of Colorado Health.
The 7 cases not satisfying the abovementioned criteria were adjudicated to be consistent with a diagnosis of possible SPSD before formal application of proposed SPSD based on our inclusion criteria and on clinical history, neurologic examination, diagnostic evaluation, and therapeutic response to treatment. These cases were further reviewed in depth, and all were found to have a range of neurologic symptoms resembling SPSD with variations in clinical presentation, diagnostic findings, and treatment responses. One case was consistent with a PERM phenotype, 4 with a classic SPS phenotype, and 2 with a SPS plus phenotype. Among these cases, 5 had a positive GAD65 antibody test, with values ranging from 0.06 to 745 nmol/L. These cases failed to meet either diagnostic criterion because of the presence of other possible competing comorbidities (i.e., not meeting the definition for exclusion of alternative diagnosis), inability to obtain comprehensive EMG testing, and/or low-positive antibody titers. Treatment responses varied, but all had some degree of improvement with immunotherapy. Notably, 1 patient case had clear clinical worsening with discontinuation of IVIg during a formal dependency trial while holding the dose for 12 weeks to obtain antibody testing. With resumption of IVIg, symptoms yet again improved. This has been well described in SPSD,26 but not part of the current proposed criteria. Details of each clinical case are available in eAppendix.
Discussion
Our study captured a large United States–based population with a diverse representation of sex, race, and ethnic subgroups in SPSD, providing novel information and enhancing existing epidemiology data in multiple ways. The estimated prevalence of SPSD within this cohort was 2.11 (95% CI 1.57–2.64) per 100,000 persons. Considering differences in diagnostic criteria, the estimated prevalence ranged from 1.36 (95% CI 0.93–1.79) to 1.82 (95% CI 1.32–2.32) per 100,000 persons when compared with our UCH SPSD cohort (Figure 4). The estimated disease prevalence in our large US population–based UCH cohort was 1 in 47,486. Even when considering the most stringent criteria for classic SPS, applying the Mayo Clinic criteria, the estimated disease prevalence was 1 in 73,744. The average yearly incidence rate between 2012 and 2022 was 0.35 per 100,000 person-years (95% CI 0.27–0.46) (Figure 3), or roughly 3.5 new cases per million people per year. Data from this United States–based cohort differ from the frequently quoted “one-in-a-million,” based on an estimated prevalence of 1 in 1,250,000 from a single center in Heidelberg, Germany,21 and a more recent prevalence estimate of 2.06 per million patients in a population of patients evaluated within the VA system.23
Our methodology for calculating incidence and prevalence in this chronic disease has several advantages. We used recommended definitions of prevalent and incident patients and observed population.27 Our population (denominator) consisted of any patient with any contact within our health system, from their first observed day, and who was assumed to contribute observation time until the end of the cohort, unless they expired (“complete period” population). Compass integrates data from the CDPHE including death records, helping to minimize missed deaths occurring outside the health system. Patients who died out of state or out of the country could potentially be missed, leading to an overestimation of person-time and underestimation of incidence. However, this methodology mitigates the risk of inflating incidence and prevalence of chronic diseases. Expert-driven chart reviews in all cases limited the risk of misidentifying prevalent cases because provider narratives provide critical information regarding the disease onset, clinical features, and timing of diagnostics.
Of 273 patients identified from HDC data search, only 59 were considered to clinically fit with a diagnosis of SPSD based on record review. The G25.82 code for SPSD was not a reliable marker with a calculated positive predictive value of 17.77% (95% CI 13.66%–21.88%).
Aligned with other reports, we found that SPSD is more common in women (59.3%). Comorbid autoimmunity was common in 64.4% (38/59) of patients diagnosed with type 1 diabetes mellitus, LADA, vitamin B12 deficiency, thyroid disease, vitiligo, and/or rheumatoid arthritis. A significant proportion of our cohort exhibited elevated CSF protein (12/39, 30.8%) and CSF restricted oligoclonal bands (8/37, 21.6%). These findings can be nonspecific in inflammatory CNS conditions, and their specificity in SPSD is yet to be determined.
Although proposed diagnostic criteria exist, there are no standardized international consensus criteria. This is a limitation in our ability to define and standardize the diagnosis of SPSD across institutions. We applied 2 proposed diagnostic criteria in this real-world data set and explored agreement between them. A κ coefficient of 0.36 (95% CI 0.13–0.58) between the Mayo Clinic and Johns Hopkins criteria noted poor agreement between these 2 criteria when applied to our UCH SPSD cohort, mostly due to the inclusion of possible SPSD cases. Removing possible SPSD cases from the Johns Hopkins criteria, we see improved agreement with a κ statistic of 0.73 (95% CI 0.53–0.95). There was no significant difference in the κ statistic when only considering probable/definite cases in the John Hopkins criteria versus the Mayo Clinic criteria. Separately, we had 7 patients who did not meet either criterion. Key features among these cases included a clinical response to immunotherapy (most IVIg), response to benzodiazepines, and a positive IVIg dependency trial (worsening with discontinuation and improvement with restarting). Many did not meet criteria based on lack of available clinical data (e.g., comprehensive EMG not completed) or possible competing comorbidities (not clearly meeting the definition for exclusion of alternative diagnosis).
In the development of formal consensus criteria for SPSD, how to capture the heterogeneous spectrum of the disease outside the classic SPS phenotype, such as SPS plus, PERM, and stiff limb (partial), will need to be considered. There will be a need to clearly define a high-titer GAD65 level. In our study, we used the widely accepted definition of serum GAD65 >20 nmol/L on RIA and >10,000 IU/mL on ELISA, which has been published previously. Serum GAD65 IgG titers obtained through ELISA testing methods may have limited diagnostic utility if reported >250 IU/mL because levels <10,000 IU/mL have not historically correlated with disease phenotype or treatment response.7,28,29 Anecdotally, we found many providers order GAD65 IgG ELISAs and interpret the reported >250 IU/mL as diagnostic (coding as ICD-10 G25.82) although they may be well below the suggested threshold of 10,000 IU/mL, leading to inappropriate diagnosis and treatment. Likewise, other studies have demonstrated low-titer GAD65 IgG testing commonly led to misdiagnosis.17 Our study identified EMG criteria as an area of discordance between the 2 proposed diagnostic criteria. Obtaining comprehensive EMG testing in community centers is uncommon, and it may necessitate evaluation at specialized centers. While abnormal acoustic startle and exteroceptive responses have a high specificity for SPSD, these specialized tests are only available at few institutions internationally and have low sensitivity with benzodiazepine use.30,31 Given this limitation, only 1 patient in 59 had this testing completed and it was negative, possibly due to concomitant benzodiazepine use. As such, this may not be a practical application in the standardized diagnostic criteria. Other specific abnormalities were reviewed on EMG including continuous motor unit activity in the paraspinal muscles and other affected muscle groups and co-contraction of agonist/antagonist muscles. While a limited number of patients had the full battery of specialized EMG procedures completed, the sensitivity of continuous motor unit activity in the paraspinal muscle within our cohort was 36.4% (95% CI 17.2%–59.3%), the sensitivity of continuous motor unit activity in the affected muscle outside paraspinals was 46.7% (95% CI 28.8%–64.5%), and the sensitivity of co-contraction of agonist/antagonist muscles was 80% (95% CI 28.4%–99.5%). Electrophysiologic findings can support the diagnosis, but how to integrate this into formal consensus criteria usable across various institutions will be critical. Electrophysiologic criteria, and more broadly SPSD consensus criteria, should be validated in a prospective, multicenter study.
Another challenge in the current proposed criteria includes the interpretation of exclusion of other causes. This can pose a challenge when competing diagnoses also involve some exclusionary criteria, such as fibromyalgia and functional neurologic disorders. It is not uncommon for patients to have multiple autoimmune or medical comorbidities, as seen in our cohort, which can pose a challenge in the diagnosis of SPSD in real-world cohorts. However, it is important to also recognize that there is a high rate of misdiagnosis of SPSD, particularly when red flags17 (eTable 1) are present, emphasizing the diagnostic challenge of this disease. Careful consideration of these red flags were evaluated in our UCH population, and patients who were considered antibody negative were excluded if red flags were present.
We used the ICD-10 code G25.82 for patient capture, followed by chart review required for diagnostic confirmation. Limitations include the retrospective single system design of our study. Despite substantial capture of many demographic groups, the results in our population may not be fully generalizable to the US population. One major limitation of this study was the lack of capturing an individual in the system if this diagnostic code was not used. Of 273 patients identified by G25.82, 217 were excluded because of not meeting criteria for SPSD based on adjudicated chart review, which may have included the lack of limited follow-up and diagnostic data. Both these limitations could potentially underestimate our total cases of SPSD. Separately, we did not include those younger than 18 years in our study. It has been suggested that pediatric-onset SPSD represents 5%–8% of total SPSD cases based on a study at the Mayo Clinic from 1984 to 2012.32,33 Additional studies are needed to characterize the disease in the pediatric population. Another limitation is the potential for bias to see rare diseases, such as SPSD, in academic health systems with training and experience in complex neuroimmunologic diseases. To help minimize this limitation, we used best practices for epidemiologic studies within health care data to minimize the risk of overestimating disease incidence and prevalence. Calculation of incidence was dependent on exposure time (i.e., determined by how long a person was in the system before the SPSD diagnosis), and therefore, SPSD cases diagnosed on their first encounter were not included because their exposure time was zero, minimizing the concern for referral bias. Finally, inclusion in our cohort was dependent on recognition of SPSD symptoms and/or serologic testing for associated autoantibodies. It is likely that our capture in later years of observation was incomplete because of the significant latency from clinical onset to the first serologic test or the SPSD-related diagnostic code.
Identification and inclusion of cases were dependent on expert review (A.L.P., P.D.C. with adjudication of tie-breaking cases [E.A.M.]). While inclusion criteria were used as guidelines for ruling in SPSD in the chart review, there are limitations based on data available in the EMR and the lack of in-person evaluations to confirm SPSD cases. However, this data set is unique in that 76.3% (45/59) of patients were clinically evaluated by a single autoimmune neurologist (A.L.P.) and 84.7% (50/59) were evaluated by other subspecialists in our neuroimmunology group. Patients evaluated by the same person or group with expertise in SPSD help minimize concern for misdiagnosis and inclusion of false cases, which could inflate our incidence and prevalence, but this remains a limitation of the study. In addition, this method of chart review can also underestimate incidence and prevalence because some cases reviewed did not have enough data to clearly rule in SPSD and were excluded from the analysis (34 cases with insufficient data to make a diagnosis; Figure 1).
We describe a prevalence of SPSD of 2.11 per 100,000 persons, or an estimate of 1 in 47,486, in a large Colorado population–based cohort. Our study uniquely captures significant numbers of many US demographic groups, making this more generalizable across the US population. Additional studies are needed to determine whether these results are also representative of a national or global population. Understanding the true disease burden of SPSD is fundamental to ensuring optimal patient care, allocating appropriate resources for this rare disease, and improving current standards. Formal consensus diagnostic criteria will lead to higher quality treatment guidelines based on clinical trials in well-defined cohorts.
Glossary
- CDPHE
Colorado Department of Public Health and Environment
- DPPX
dipeptidyl peptidase-like protein 6
- GAD65
glutamic acid decarboxylase 65-kilodalton isoform
- EHR
electronic health record
- GlyR-α1
glycine receptor α1 subunit
- HDC
Health Data Compass
- ICD-10
International Classification of Diseases, 10th Revision
- IVIg
IV immunoglobulin G
- LADA
latent autoimmune diabetes of adults
- PERM
progressive encephalomyelitis with rigidity and myoclonus
- PLEX
plasma exchange
- RIA
radioimmunoprecipitation assay
- SPS
stiff person syndrome
- SPSD
stiff person spectrum disorder
- UCH
University of Colorado Health
- VA
Veterans Affairs
Appendix. Authors
| Name | Location | Contribution |
| Paul Daniel Crane, MD | Department of Neurology, University of Colorado Anschutz Medical Campus, Aurora | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Stefan Sillau, PhD | Department of Neurology, and Department of Biostatistics & Informatics, University of Colorado Anschutz Medical Campus, Aurora | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Renee Dreher, BS | University of Colorado School of Medicine Anschutz Medical Campus, Aurora | Major role in the acquisition of data |
| Rikki Fix, BS | University of Colorado School of Medicine, Aurora; School of Osteopathic Medicine, Kanas City University, MO | Major role in the acquisition of data |
| Phillip Winters, BS | University of Colorado School of Medicine Anschutz Medical Campus, Aurora | Major role in the acquisition of data |
| Russell Van Coevering, MD | University of Colorado School of Medicine Anschutz Medical Campus; School of Medicine, Aurora, CO; Renown Health, Reno, NV | Major role in the acquisition of data |
| Eric Engebretson, MS | University of Colorado School of Medicine Anschutz Medical Campus, Aurora | Major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Brooke Valdez, BA | University of Colorado School of Medicine Anschutz Medical Campus, Aurora | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design |
| Elizabeth Matthews, MD | University of Colorado School of Medicine Anschutz Medical Campus, Aurora | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; analysis or interpretation of data |
| Kavita V. Nair, PhD | University of Colorado School of Medicine Anschutz Medical Campus; Skaggs School of Pharmacy and Pharmaceutical Sciences, University of Colorado Anschutz Medical Campus; Rocky Mountain MS Center, University of Colorado School of Medicine, Aurora | Drafting/revision of the manuscript for content, including medical writing for content; study concept or design |
| Aaron M. Carlson, MD | University of Colorado School of Medicine Anschutz Medical Campus, Aurora | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
| Amanda L. Piquet, MD | University of Colorado School of Medicine Anschutz Medical Campus, Aurora | Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data; study concept or design; analysis or interpretation of data |
Study Funding
This work was funded by the Rocky Mountain Multiple Sclerosis Center (RMMSC) and supported by Health Data Compass (HDC) Data Warehouse project (healthdatacompass.org).
Disclosure
P.D. Crane, S. Sillau, R. Dreher, R. Fix, P. Winters, R.V. Coevering, E. Engebretson, B. Valdez, and E.A. Matthews report no disclosures. K.V. Nair reports research grants from Genentech, PhRMA Foundation, Bristol Myers Squibb, Alexion, Squibb, Novartis, Horizon Biogen, and National Institute of Neurological Disorders and Stroke, and consulting fees from Bristol Myers Squibb, Novartis, Biogen, TG Therapeutics, Genentech and EMD Serono. In addition, she reports speakers bureau affiliation with Sanofi-Genzyme, Amgen and AJCM speaker series. A.M. Carlson reports unrelated grant funding from Horizon Therapeutics and recent service on the Health Services Subcommittee for the American Academy of Neurology. A.L. Piquet reports research grants from the University of Colorado, research funding from the Endowed Chair supported by the Céline Dion Foundation, Rocky Mountain MS Center, and the Foundation for Sarcoidosis; consulting fees from Genentech/Roche, UCB, EMD Serono, Kyverna and Alexion; honoraria from MedLink; and publication royalties from Springer as co-editor of a medical textbook. Go to Neurology.org/N for full disclosures.
References
- 1.Moersch FP, Woltman HW. Progressive fluctuating muscular rigidity and spasm (“stiff-man” syndrome); report of a case and some observations in 13 other cases. Proc Staff Meet Mayo Clin. 1956;31(15):421-427. [PubMed] [Google Scholar]
- 2.Barker RA, Revesz T, Thom M, Marsden CD, Brown P. Review of 23 patients affected by the stiff man syndrome: clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatry. 1998;65(5):633-640. doi: 10.1136/jnnp.65.5.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Hu C, Aljarallah S, et al. Expanding clinical profiles and prognostic markers in stiff person syndrome spectrum disorders. J Neurol. 2024;271(4):1861-1872. doi: 10.1007/s00415-023-12123-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ariño H, Höftberger R, Gresa-Arribas N, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol. 2015;72(8):874-881. doi: 10.1001/jamaneurol.2015.0749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalakas MC, Li M, Fujii M, Jacobowitz DM. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology. 2001;57(5):780-784. doi: 10.1212/wnl.57.5.780 [DOI] [PubMed] [Google Scholar]
- 6.Martinez-Hernandez E, Ariño H, McKeon A, et al. Clinical and immunologic investigations in patients with stiff-person spectrum disorder. JAMA Neurol. 2016;73(6):714-720. doi: 10.1001/jamaneurol.2016.0133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McKeon A, Tracy JA. GAD65 neurological autoimmunity. Muscle Nerve. 2017;56(1):15-27. doi: 10.1002/mus.25565 [DOI] [PubMed] [Google Scholar]
- 8.Li L, Hagopian WA, Brashear HR, Daniels T, Lernmark A. Identification of autoantibody epitopes of glutamic acid decarboxylase in stiff-man syndrome patients. J Immunol. 1994;152(2):930-934. doi: 10.4049/jimmunol.152.2.930 [DOI] [PubMed] [Google Scholar]
- 9.Hinson SR, Lopez-Chiriboga AS, Bower JH, et al. Glycine receptor modulating antibody predicting treatable stiff-person spectrum disorders. Neurol Neuroimmunol Neuroinflamm. 2018;5(2):e438. doi: 10.1212/NXI.0000000000000438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKeon A, Martinez-Hernandez E, Lancaster E, et al. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol. 2013;70(1):44-50. doi: 10.1001/jamaneurol.2013.574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piquet AL, Khan M, Warner JEA, et al. Novel clinical features of glycine receptor antibody syndrome: a series of 17 cases. Neurol Neuroimmunol Neuroinflamm. 2019;6(5):e592. doi: 10.1212/nxi.0000000000000592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swayne A, Tjoa L, Broadley S, et al. Antiglycine receptor antibody related disease: a case series and literature review. Eur J Neurol. 2018;25(10):1290-1298. doi: 10.1111/ene.13721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raju R, Rakocevic G, Chen Z, et al. Autoimmunity to GABAA-receptor-associated protein in stiff-person syndrome. Brain. 2006;129(pt 12):3270-3276. doi: 10.1093/brain/awl245 [DOI] [PubMed] [Google Scholar]
- 14.Murinson BB, Guarnaccia JB. Stiff-person syndrome with amphiphysin antibodies: distinctive features of a rare disease. Neurology. 2008;71(24):1955-1958. doi: 10.1212/01.wnl.0000327342.58936.e0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balint B, Jarius S, Nagel S, et al. Progressive encephalomyelitis with rigidity and myoclonus: a new variant with DPPX antibodies. Neurology. 2014;82(17):1521-1528. doi: 10.1212/wnl.0000000000000372 [DOI] [PubMed] [Google Scholar]
- 16.Dalakas MC. Stiff-person syndrome and GAD antibody-spectrum disorders: GABAergic neuronal excitability, immunopathogenesis and update on antibody therapies. Neurotherapeutics. 2022;19(3):832-847. doi: 10.1007/s13311-022-01188-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chia NH, McKeon A, Dalakas MC, et al. Stiff person spectrum disorder diagnosis, misdiagnosis, and suggested diagnostic criteria. Ann Clin Transl Neurol. 2023;10(7):1083-1094. doi: 10.1002/acn3.51791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dalakas MC. Therapies in stiff-person syndrome: advances and future prospects based on disease pathophysiology. Neurol Neuroimmunol Neuroinflamm. 2023;10(3):e200109. doi: 10.1212/nxi.0000000000200109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newsome SD, Johnson T. Stiff person syndrome spectrum disorders; more than meets the eye. J Neuroimmunol. 2022;369:577915. doi: 10.1016/j.jneuroim.2022.577915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Espay AJ, Chen R. Rigidity and spasms from autoimmune encephalomyelopathies: stiff-person syndrome. Muscle Nerve. 2006;34(6):677-690. doi: 10.1002/mus.20653 [DOI] [PubMed] [Google Scholar]
- 21.Meinck HM, Thompson PD. Stiff man syndrome and related conditions. Mov Disord. 2002;17(5):853-866. doi: 10.1002/mds.10279 [DOI] [PubMed] [Google Scholar]
- 22.Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. 2011;11(5):272-282. doi: 10.1136/practneurol-2011-000071 [DOI] [PubMed] [Google Scholar]
- 23.Galli JR, Austin SD, Greenlee JE, Clardy SL. Stiff person syndrome with anti-GAD65 antibodies within the national veterans affairs health administration. Muscle Nerve. 2018;58(6):801-804. doi: 10.1002/mus.26338 [DOI] [PubMed] [Google Scholar]
- 24.Hogans B, Siaton BC, Katzel LI, Sorkin JD. Stiff person syndrome: taxonomic analysis supports use of large data methods to appraise major comorbidities of a rare disorder. Biomed J Sci Tech Res. 2024;55(2). doi: 10.26717/BJSTR.2024.55.008673 [DOI] [Google Scholar]
- 25.Dalakas MC, Fujii M, Li M, McElroy B. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology. 2000;55(10):1531-1535. doi: 10.1212/wnl.55.10.1531 [DOI] [PubMed] [Google Scholar]
- 26.Yi J, Dalakas MC. Long-term effectiveness of IVIg maintenance therapy in 36 patients with GAD antibody-positive stiff-person syndrome. Neurol Neuroimmunol Neuroinflamm. 2022;9(5):e200011. doi: 10.1212/nxi.0000000000200011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rassen JA, Bartels DB, Schneeweiss S, Patrick AR, Murk W. Measuring prevalence and incidence of chronic conditions in claims and electronic health record databases. Clin Epidemiol. 2019;11:1-15. doi: 10.2147/clep.S181242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Budhram A, Sechi E, Flanagan EP, et al. Clinical spectrum of high-titre GAD65 antibodies. J Neurol Neurosurg Psychiatry. 2021;92(6):645-654. doi: 10.1136/jnnp-2020-325275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muñoz-Lopetegi A, de Bruijn M, Boukhrissi S, et al. Neurologic syndromes related to anti-GAD65: clinical and serologic response to treatment. Neurol Neuroimmunol Neuroinflamm. 2020;7(3):e696. doi: 10.1212/nxi.0000000000000696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meinck HM, Ricker K, Conrad B. The stiff-man syndrome: new pathophysiological aspects from abnormal exteroceptive reflexes and the response to clomipramine, clonidine, and tizanidine. J Neurol Neurosurg Psychiatry. 1984;47(3):280-287. doi: 10.1136/jnnp.47.3.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsumoto JY, Caviness JN, McEvoy KM. The acoustic startle reflex in stiff-man syndrome. Neurology. 1994;44(10):1952-1955. doi: 10.1212/wnl.44.10.1952 [DOI] [PubMed] [Google Scholar]
- 32.Clardy SL, Lennon VA, Dalmau J, et al. Childhood onset of stiff-man syndrome. JAMA Neurol. 2013;70(12):1531-1536. doi: 10.1001/jamaneurol.2013.4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeshokumar AK, Sun LR, Newsome SD. Defining the expanding clinical spectrum of pediatric-onset stiff person syndrome. Pediatr Neurol. 2021;114:11-15. doi: 10.1016/j.pediatrneurol.2020.09.007 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.