

Abstract
Aim
Although the antidepressant effect of ketamine on treatment‐resistant depression (TRD) has been frequently reported in North American and European countries, evidence is scarce among the Asian population. We aimed to evaluate the efficacy and safety of intravenous ketamine in Japanese patients with TRD.
Methods
In this double‐blind randomized placebo‐controlled trial, 34 Japanese patients with TRD were randomized to receive either intravenous ketamine (0.5 mg/kg) or placebo, administered over 40 min, twice a week, for 2 weeks. The primary outcome was the change in the Montgomery Åsberg Depression Rating Scale (MADRS) total score from baseline to post‐treatment. Secondary outcomes included changes in other depressive symptomatology scores and remission, response, and partial response rates. We also examined the association between baseline clinical demographic characteristics and changes in the MADRS total score.
Results
Intention‐to‐treat analysis indicated no significant difference in the decrease in MADRS total score between the groups (−8.1 ± 10.0 vs −2.5 ± 5.2, t[32] = 2.02, P = 0.052), whereas per‐protocol analysis showed a significant reduction in the ketamine group compared to the placebo group (−9.1 ± 10.2 vs −2.7 ± 5.3, t[29] = 2.22, P = 0.034). No significant group differences were observed in other outcomes. Adverse events were more frequent in the ketamine group than in the placebo group, and no serious adverse events were reported. A higher baseline MADRS total score and body mass index were associated with a greater reduction in the MADRS total score.
Conclusion
Intravenous ketamine outperformed placebo in Japanese patients with TRD who completed the study, suggesting that ketamine could alleviate depressive symptoms of TRD across diverse ethnic populations.
Keywords: ethnicity, ketamine, major depressive disorder, repeated doses, treatment‐resistant depression
Major depressive disorder (MDD) is highly prevalent and often associated with significant morbidity, mortality, and substantial societal costs. 1 , 2 , 3 Despite recent advances in the pharmacological and nonpharmacological treatment of MDD, approximately 30% of patients still fail to respond to drug therapy, 4 which is referred to as treatment‐resistant depression (TRD).
Ketamine, an N‐methyl‐d‐aspartate glutamate receptor antagonist initially approved as an anesthetic, has shown rapid and strong antidepressant effects in numerous clinical trials and meta‐analyses since the first clinical trial of ketamine for TRD in 2000. 5 , 6 , 7 Previous open‐label studies have suggested that some patients require repeated infusions beyond a single ketamine infusion to achieve a complete response. 8 , 9 , 10 A double‐blind randomized placebo‐controlled trial (DBRCT) by Singh et al., 11 showed that repeated intravenous administration of ketamine two or three times a week for 15 days outperformed placebo in 67 outpatients with TRD. However, recent data from RCTs evaluating repeated ketamine infusions have shown inconsistent antidepressant effects. 11 , 12 , 13 A DBRCT by Ionescu et al., 12 found that repeated ketamine infusions twice a week for 3 weeks as adjunctive treatment to ongoing antidepressants were not superior to adjunctive placebo in 26 outpatients with TRD. Another DBRCT demonstrated no significant difference in the antidepressant efficacy of ketamine between the group receiving six ketamine infusions and the group receiving five midazolam infusions as active placebo followed by a single ketamine infusion, administered over 12 days in 54 outpatients with TRD. 13
Most studies that evaluated ketamine's antidepressant effect have been reported in North America and Europe, with limited evidence among the Asian population. 14 , 15 An individual participant data meta‐analysis assessing RCTs of intravenous ketamine reported that 579 of 645 patients (89.8%) were non‐Asian. 16 Although open‐label studies from China and Taiwan suggest that repeated ketamine infusions potentially exhibit a higher response rate than a single infusion in patients with TRD, 15 , 17 no RCTs have been conducted in the Asian population. In particular, because ketamine is strictly regulated as a narcotic in Japan, no clinical trials on ketamine administration among Japanese patients with TRD have been reported, except for one nasal esketamine study that showed negative results. 18
Ethnic differences could affect the presentation of depressive symptoms and response to ketamine. According to a cross‐sectional study among a community sample in Hawaii, Japanese Americans reported lower positive affect on the Center for Epidemiologic Studies Depression Scale (CES‐D) than European Americans, although the CES‐D total score and the subdomain scores, including depressed affect, somatic symptoms, and interpersonal problems, did not significantly differ across populations. 19 Thus, Japanese Americans may present lower positive affect even when the severity of depressive symptoms is similar. Moreover, different ethnic populations demonstrated different expressions of polymorphic cytochrome P450 liver enzymes that metabolize ketamine, such as CYP3A4 and CYP2B6. 20 , 21 Although the total P450 content was higher in Caucasians than in Japanese people, the principal individual forms of P450, including CYP3A and CYP2C, showed no significant differences between the groups. 21 However, clinical data on whether the different polymorphic expressions of cytochrome enzymes influence response to ketamine are limited.
To fill the gap in evidence on RCTs of repeated ketamine infusions in Asian populations, we conducted a DBRCT that examined the antidepressant effect of ketamine infusions twice a week for 2 weeks compared to placebo, in Japanese patients with TRD. Furthermore, we evaluated the characteristics of patients with TRD who responded to ketamine infusion. Considering the favorable outcomes of repeated ketamine infusions in Asian patients from China and Taiwan observed in open‐label studies, as well as in Caucasian patients from North America as observed in the DBRCT by Singh et al., we hypothesized that intravenous ketamine would show superior efficacy to placebo in Japanese patients with TRD.
Methods
A list of abbreviations is provided in the Supporting Information (Table S1). This manuscript was reported in accordance with the CONSORT checklist (Table S2).
Study design and settings
This DBRCT, followed by an extension open‐label single‐arm trial, was conducted at the Keio University Hospital and Yokohama City University Hospital in Japan between August 2021 and October 2023. This trial was approved by the Certified Review Board of Keio in accordance with the Clinical Trials Act of Japan and was registered in the Japan Registry of Clinical Trials in May 2021 (jRCTs031210124). Written informed consent was obtained from all participants after a full explanation of the study was provided. This study was conducted in accordance with the ethical principles of the Declaration of Helsinki and was consistent with Good Clinical Practices and applicable regulatory requirements.
Participants
Inclusion criteria were as follows: (i) inpatients or outpatients 20–59 years of age who met the Diagnostic and Statistical Manual of Mental Disorders Fifth Edition (DSM‐5) 22 criteria for MDD using the structured clinical interview for DSM‐5, the Research Version (SCID‐5‐RV), 23 (ii) inadequate response defined as <50% subjective improvement to approved doses of at least two antidepressants in the current episode based on a visual analogue scale ranging from 0 (no improvement) to 100 (complete improvement), 12 , 24 , 25 , 26 (iii) a total score of ≥22 on the Montgomery Åsberg Depression Rating Scale (MADRS) 27 at screening, and (iv) sufficient decision‐making capacity as confirmed by the MacArthur Competence Tool for Clinical Research. 28 Patients were excluded if they were pregnant, were nursing or wished to get pregnant, had a history of epilepsy, met the criteria for substance abuse according to DSM‐5 within 6 months before the study, had a positive urine drug screen for illicit drugs, had contraindications for magnetic resonance imaging scan, had previously received either esketamine or ketamine, had a history of hypersensitivity to ketamine, had depression with psychotic features based on DSM‐5, had received electroconvulsive therapy within 3 months before enrollment, had a significant neurological or general medical condition, or showed abnormal laboratory test values of serum creatinine ≥1.5 mg/dL, AST ≥ 150 IU/L, or ALT ≥ 150 IU/L.
Sample size
According to the DBRCT by Singh et al., 11 evaluating the effects of repeated ketamine infusions (i.e. four times in 2 weeks) in US patients with TRD, the MADRS total score changes (mean ± standard deviation [SD]) in the ketamine and placebo groups were −18.4 ± 12.0 and −5.7 ± 10.2, respectively, with the estimated effect size of 1.14. Assuming the same effect size in this study involving Japanese patients with TRD, 14 patients per group were required for obtaining 80% power using a two‐sided t‐test at a significance level of 0.05. Considering the dropout rate of approximately 14.7% (5 of 34), the target sample size was 17 per group (34 patients in total).
Procedures
Enrolled participants were randomly allocated to one of the two treatment groups in a 1:1 ratio using a computer‐generated randomization scheme, balanced by two or four randomly permuted blocks with no stratification. The principal investigator (HU) and the investigator designated by the principal investigator (HT) conveyed the assignment by e‐mail only to the investigator responsible for the preparation of the study drug (T. Yatomi), an anesthesiologist (T. Yamada) responsible for supervision during study drug administration, and pharmacists in charge of dispensing. During the double‐blind period, neither clinical assessors nor participants were notified of the assignment. Anyone who knew about the assignment was not involved in data entry for the case report form until final data fixation. During the double‐blind period, baseline assessments were performed before the first administration of the study drug. In the ketamine group, intravenous ketamine (0.5 mg/kg) was administered over 40 min twice a week for 2 weeks, whereas in the placebo group, intravenous placebo (0.9% NaCl) was administered over 40 min twice a week for 2 weeks; both ketamine and placebo groups received study drug infusion four times in total. The number and frequency of doses were based on previous studies: an open‐label study indicated that repeated ketamine infusions had cumulative antidepressant effects and achieved approximately 70% response rate after four doses, 10 while a DBRCT demonstrated no significant difference in the antidepressant effects between the groups receiving ketamine twice a week and three times a week. 11 Clinical assessments were performed at two time points: baseline and post‐treatment. Repeated assessments were avoided to minimize the risks of type I error from multiple testing, participant burden, and practice effects. Post‐treatment assessments were performed within 14 days of the fourth administration of the study drug, and the participants were then notified of their treatment group. While participants assigned to the ketamine group completed the study, those allocated to the placebo group were offered the opportunity to receive open‐label intravenous ketamine treatment. During this open‐label treatment period, the participants received intravenous ketamine (0.5 mg/kg) for 40 min twice a week for 2 weeks (i.e. four times in total), and received clinical assessments before and after this course of treatment. Participants continued any antidepressant medications they had been receiving at the time of screening, at the same dosages, throughout the study. When clinically indicated, lorazepam and zolpidem were prescribed for anxiety and insomnia, respectively.
Assessment measures
In the double‐blind period, the following assessments were performed by assessors blinded to the allocation at baseline and post‐treatment: MADRS, the 17‐item Hamilton Depression Rating Scale (HDRS‐17), 29 and Quick Inventory of Depressive Symptoms Self‐Rated (QIDS‐SR). 30 Autism‐spectrum Quotient‐Japanese version (AQ‐J), 31 , 32 Japanese Adult Reading Test (JART), 33 and Posttraumatic Diagnostic Scale (PDS) 34 were assessed solely at baseline, to evaluate the degree of autism spectrum disorder traits, intelligence, or psychological trauma for characterization of the participants. These scales were assessed only at baseline because these background characteristics were not expected to change after treatment. During the open‐label period, the following assessments were performed before and after treatment: MADRS, HDRS‐17, and QIDS‐SR. Safety was assessed based on treatment‐emergent adverse events and vital signs, including pulse oximetry monitoring and physical examination. The following information was also collected: age, sex, ethnicity, height, weight, age at onset, duration of illness, duration of treatment, number of failed antidepressant trials, history of suicide attempts and ideation, treatment history, and concomitant medications.
Statistical analysis
All statistical analyses were conducted using spss version 29.0, for Windows (IBM Corporation, Armonk, NY, USA). The Shapiro–Wilk test was used to ensure the normality of the data distribution. Baseline demographic and clinical characteristics were compared between the ketamine and placebo groups using Pearson's chi‐squared test or Fisher's exact test for categorical variables and Student's t‐test or Mann–Whitney U‐test for continuous variables.
The primary outcome measure was the change in MADRS total score from baseline to post‐treatment assessment in the double‐blind phase. The secondary outcome measures were changes from baseline to post‐treatment assessment in the HDRS‐17 total score and QIDS‐SR total score, and rates of remission, response, and partial response, defined as a MADRS total score of ≤10 at the post‐treatment assessment, a ≥50%, and ≥25% reduction in the MADRS total score from baseline to post‐treatment assessment, respectively. We also assessed the three MADRS subdomains: dysphoria, retardation, and vegetative symptoms based on factor analysis in Japanese patients with MDD. 35 The dysphoric domain includes pessimistic thoughts, suicidal thoughts, and reported sadness; the retardation domain includes lassitude, inability to feel, apparent sadness, and concentration difficulties; and the vegetative domain includes reduced sleep, reduced appetite, and inner tension. Outcome values were compared between the two groups using Pearson's chi‐square test or Fisher's exact test for categorical variables and Student's t‐test or Mann–Whitney U‐test for continuous variables on intention‐to‐treat (ITT) or per‐protocol (PP) basis. Baseline values were used as endpoints for dropouts in ITT analysis, whereas only completers were included in PP analysis. By merging data from patients who underwent ketamine treatment in both double‐blind and open‐label phases, the outcome values were compared between the combined ketamine and placebo groups. A sensitivity analysis was performed to test the primary outcome by adjusting for the baseline MADRS total score.
Moreover, multiple regression analysis was performed to examine factors associated with changes from baseline to post‐treatment in the MADRS total score in patients who received intravenous ketamine infusions either in the double‐blind or open‐label phase. The following explanatory variables were included: age, sex, body mass index (BMI), and baseline MADRS total score. Logistic regression analyses were conducted to examine factors associated with remission, response, or partial response to ketamine using the same explanatory variables. Furthermore, the associations between the three baseline MADRS subdomain scores and the MADRS total score changes were tested using a linear regression model with age and sex as covariates. Each model with covariates was analyzed in PP analysis. A two‐tailed P‐value of <0.05 was considered statistically significant for all tests. The Bonferroni correction was applied to adjust for multiple comparisons when analyzing the three MADRS subdomains, with a P‐value <0.016 (i.e. 0.05/3).
Results
Participants
Of the 140 patients screened for eligibility, 34 were enrolled in the study, all Japanese outpatients. The participants were randomized into ketamine or placebo treatment groups (17 each) in the double‐blind phase (Fig. 1). A total of 31 (91.2%) underwent all four administrations of the study drug and both baseline and post‐treatment assessments in the double‐blind phase. Post‐treatment assessments were performed 3.2 ± 1.5 days (range: 2–7 days) after the fourth infusion. Three participants discontinued participation during the double‐blind phase because of their intention to withdraw (n = 2 in the ketamine group) or fatigue (n = 1 in the placebo group). All 16 participants who completed the placebo administration in the double‐blind phase entered the optional 2‐week open‐label phase. One participant discontinued study participation during the open‐label phase because of a treatment‐emergent adverse event (i.e. allergic skin rash).
Fig. 1.
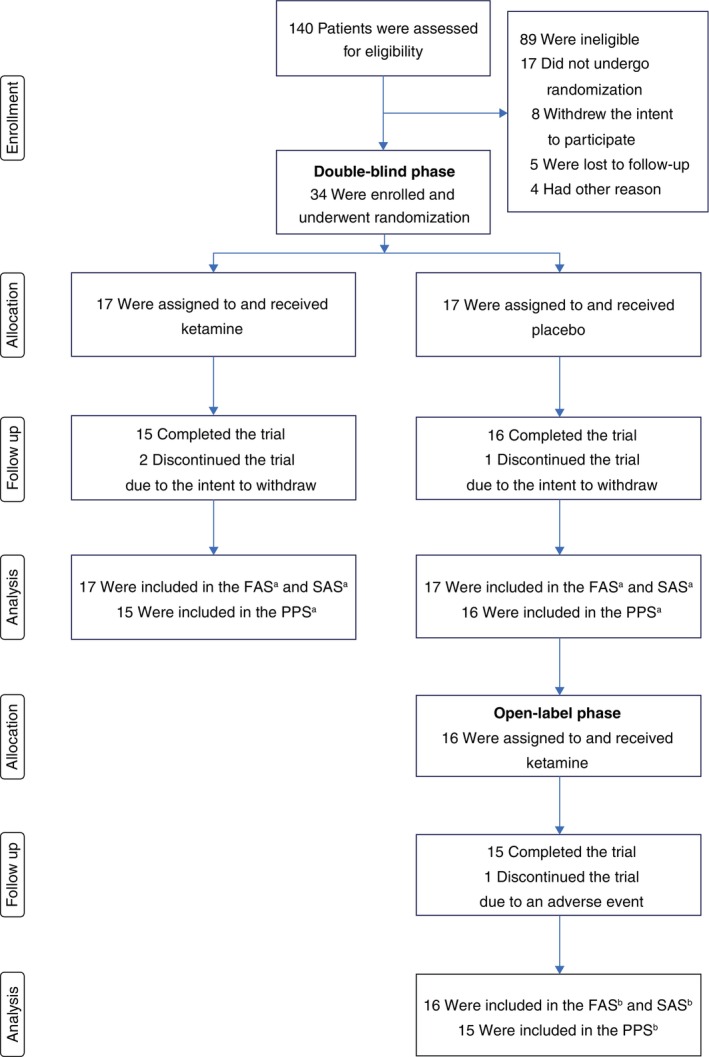
CONSORT flow diagram. aAnalysis sets in the double‐blind phase. bAnalysis sets in the open‐label phase. FAS, full analysis set; PPS, per protocol set; SAS, safety analysis set.
Baseline demographic and clinical characteristics did not differ significantly between the two groups (Table 1). In the entire sample, the mean ± SD age was 41.4 ± 9.4 years, and 11 participants (32.4%) were females. The mean ± SD MADRS total score at baseline was 28.1 ± 7.6. The most frequently used antidepressants were venlafaxine (n = 9), escitalopram (n = 7), clomipramine (n = 6), vortioxetine (n = 4), and mirtazapine (n = 4). The baseline AQ‐J, JART, and PDS scores were 25.4 ± 7.6, 111.5 ± 8.6, and 4.2 ± 8.5, respectively, indicating that autistic traits, intellectual decline, or psychological trauma were not prominent.
Table 1.
Demographic and clinical characteristics of the study sample
| Total (n = 34) | Ketamine (n = 17) | Placebo (n = 17) | ||
|---|---|---|---|---|
| Characteristics | Mean ± SD | Mean ± SD | Mean ± SD | P‐value |
| Age, years | 41.4 ± 9.4 | 39.9 ± 9.5 | 42.9 ± 9.1 | 0.38 |
| Body mass index | 25.0 ± 3.8 | 24.1 ± 3.3 | 25.8 ± 4.2 | 0.20 |
| Age of onset, years | 29.9 ± 10.4 | 29.1 ± 10.6 | 30.8 ± 10.1 | 0.66 |
| Duration of illness, years | 11.5 ± 8.1 | 10.8 ± 8.2 | 12.1 ± 7.9 | 0.65 |
| Duration of treatment, years | 11.1 ± 8.1 | 10.4 ± 8.3 | 11.7 ± 7.8 | 0.64 |
| Failed antidepressant trials | 3.8 ± 1.7 | 3.8 ± 1.7 | 3.9 ± 1.6 | 0.84 |
| Baseline MADRS total score | 28.1 ± 7.6 | 29.2 ± 7.9 | 26.9 ± 7.1 | 0.38 |
| Baseline AQ‐J score | 25.4 ± 7.6 | 23.1 ± 8.0 | 27.7 ± 6.4 | 0.09 |
| Baseline JART score | 111.5 ± 8.6 | 109.8 ± 7.5 | 113.2 ± 9.3 | 0.26 |
| Baseline PDS total score | 4.2 ± 8.5 | 6.7 ± 10.2 | 1.8 ± 5.4 | 0.10 |
| n (%) | n (%) | n (%) | ||
|---|---|---|---|---|
| Female | 11 (32.4) | 6 (35.2) | 5 (29.4) | 0.71 |
| Past history of self‐harm | 7 (20.6) | 4 (23.5) | 3 (17.7) | 1.00 § |
| Past history of suicidal attempts | 11 (32.4) | 5 (29.4) | 6 (35.3) | 0.71 |
| Past history of ECT | 7 (20.6) | 3 (17.7) | 4 (23.5) | 1.00 § |
| Concomitant use of benzodiazepines | 23 (67.7) | 11 (64.7) | 12 (70.6) | 0.71 |
Values are shown as mean ± SD or n (%).
Fisher's exact test.
AQ‐J, Autism‐spectrum Quotient‐Japanese version; ECT, electroconvulsive therapy; JART, Japanese Adult Reading Test; MADRS, Montgomery Åsberg Depression Rating Scale; PDS, Posttraumatic Diagnostic Scale.
Primary outcomes
Intention‐to‐treat analysis showed no significant difference in MADRS total score reduction between the ketamine (n = 17) and placebo (n = 17) groups (−8.1 ± 10.0 vs −2.5 ± 5.2, t[32] = −2.02, P = 0.052, d = 0.69) (Table 2 and Fig. 2a); however, PP analysis indicated a significantly greater reduction in the ketamine group (n = 15) than that in the placebo group (n = 16) (−9.1 ± 10.2 vs −2.7 ± 5.3, t[29] = −2.22, P = 0.034, d = 0.80) (Table 2 and Fig. 2b). Sensitivity analysis adjusting for the baseline MADRS total score showed similar results (Table S3).
Table 2.
Changes in outcome measures in the double‐blind phase
| Outcomes | N | Change | P‐value |
|---|---|---|---|
| MADRS | |||
| Ketamine | 17 (ITT) | −8.1 ± 10.0 (ITT) | 0.052 (ITT) |
| Placebo | 17 (ITT) | −2.5 ± 5.2 (ITT) | |
| Ketamine | 15 (PP) | −9.1 ± 10.2 (PP) | 0.034* (PP) |
| Placebo | 16 (PP) | −2.7 ± 5.3 (PP) | |
| HDRS‐17 | |||
| Ketamine | 17 (ITT) | −4.4 ± 6.3 (ITT) | 0.12 (ITT) |
| Placebo | 17 (ITT) | −1.4 ± 4.2 (ITT) | |
| Ketamine | 15 (PP) | −4.9 ± 6.5 (PP) | 0.09 (PP) |
| Placebo | 16 (PP) | −1.5 ± 4.3 (PP) | |
| QIDS‐SR | |||
| Ketamine | 17 (ITT) | −5.3 ± 4.9 (ITT) | 0.10 (ITT) |
| Placebo | 17 (ITT) | −2.4 ± 3.2 (ITT) | |
| Ketamine | 15 (PP) | −5.1 ± 4.0 (PP) | 0.06 (PP) |
| Placebo | 16 (PP) | −2.9 ± 3.0 (PP) | |
| MADRS dysphoric domain | |||
| Ketamine | 17 (ITT) | −2.8 ± 4.2 (ITT) | 0.16 (ITT) |
| Placebo | 17 (ITT) | −1.1 ± 2.1 (ITT) | |
| Ketamine | 15 (PP) | −3.1 ± 4.4 (PP) | 0.12 (PP) |
| Placebo | 16 (PP) | −1.2 ± 2.1 (PP) | |
| MADRS retardation domain | |||
| Ketamine | 17 (ITT) | −4.3 ± 4.5 (ITT) | 0.013 † (ITT) |
| Placebo | 17 (ITT) | −0.8 ± 3.2 (ITT) | |
| Ketamine | 15 (PP) | −4.9 ± 4.5 (PP) | 0.007 † (PP) |
| Placebo | 16 (PP) | −0.8 ± 3.3 (PP) | |
| MADRS vegetative domain | |||
| Ketamine | 17 (ITT) | −1.0 ± 2.5 (ITT) | 0.66 (ITT) |
| Placebo | 17 (ITT) | −0.6 ± 2.1 (ITT) | |
| Ketamine | 15 (PP) | −1.1 ± 2.6 (PP) | 0.61 (PP) |
| Placebo | 16 (PP) | −0.7 ± 2.2 (PP) | |
| Outcomes | N | n (%) | P‐value |
|---|---|---|---|
| Remission | |||
| Ketamine | 17 (ITT) | 3 (17.6) (ITT) | 0.60 § (ITT) |
| Placebo | 17 (ITT) | 1 (5.9) (ITT) | |
| Ketamine | 15 (PP) | 3 (20.0) (PP) | 0.33 § (PP) |
| Placebo | 16 (PP) | 1 (6.3) (PP) | |
| Response | |||
| Ketamine | 17 (ITT) | 3 (17.6) (ITT) | 0.23 § (ITT) |
| Placebo | 17 (ITT) | 0 (0) (ITT) | |
| Ketamine | 15 (PP) | 3 (20.0) (PP) | 0.10 § (PP) |
| Placebo | 16 (PP) | 0 (0) (PP) | |
| Partial response | |||
| Ketamine | 17 (ITT) | 7 (41.2) (ITT) | 0.47 (ITT) |
| Placebo | 17 (ITT) | 5 (29.4) (ITT) | |
| Ketamine | 15 (PP) | 7 (46.7) (PP) | 0.38 (PP) |
| Placebo | 16 (PP) | 5 (31.3) (PP) | |
Values are shown as mean ± SD or n (%).
P < 0.05;
P < 0.016 (multiple comparison correction).
Fisher's exact test.
HDRS‐17, 17‐item Hamilton Depression Rating Scale; ITT, intention‐to‐treat; MADRS, Montgomery Åsberg Depression Rating Scale; PP, per‐protocol; QIDS‐SR, Quick Inventory of Depressive Symptoms Self‐Rated; SD, standard deviation.
Fig. 2.
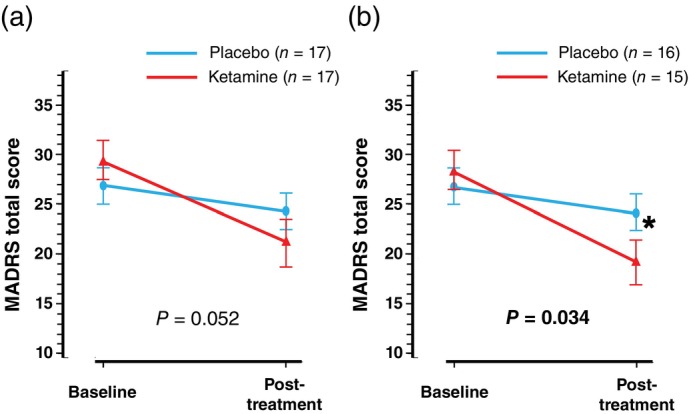
Comparison of the MADRS total score changes between the ketamine and placebo groups in ITT and PP analyses. The MADRS total score changes from baseline to post‐treatment assessment in the ketamine and placebo groups during the double‐blind phase are demonstrated. Results of ITT and PP analyses are shown in panel (a) and panel (b), respectively. Error bars indicate ±1 SE. *P < 0.05. ITT, intention‐to‐treat; MADRS, Montgomery Åsberg Depression Rating Scale; PP, per‐protocol; SE, standard error.
Secondary outcomes
There were no significant differences in total score changes from baseline to post‐treatment assessment in HDRS‐17 or QIDS‐SR between the two groups in either ITT or PP analyses (Table 2). There was a significantly greater reduction in the MADRS retardation domain score from baseline to post‐treatment in the ketamine group compared to the placebo group both in ITT and PP analyses (−4.3 ± 4.5 vs −0.8 ± 3.2, t[32] = −2.63, P = 0.013, d = 0.90 and −4.9 ± 4.5 vs −0.8 ± 3.3, t[29] = −2.88, P = 0.007, d = 1.03, respectively). In contrast, ITT or PP analyses showed no significant differences in the changes in MADRS dysphoric or vegetative domain scores between the two groups and found no significant group differences in the remission, response, or partial response rates at the post‐treatment assessment (Table 2).
Intention‐to‐treat analysis showed that when the data regarding ketamine treatment were combined, regardless of whether it was administered in a double‐blind or open‐label fashion, the combined ketamine group (n = 33) was superior to the placebo group (n = 17) in terms of MADRS total score reduction, MADRS retardation domain score reduction, and response rates. PP analysis indicated that the combined ketamine group (n = 30) was superior to the placebo group (n = 16) in MADRS and HDRS‐17 total score reductions, MADRS retardation domain score reductions, response rates, and partial response rates (Table 3).
Table 3.
Changes in outcome measures when the data of treatment with ketamine was combined regardless it was administered in the double‐blind or open‐label phase
| Outcomes | N | Change | P‐value |
|---|---|---|---|
| MADRS | |||
| Ketamine ¶ | 33 (ITT) | −9.2 ± 9.5 (ITT) | 0.010* (ITT) |
| Placebo | 17 (ITT) | −2.5 ± 5.2 (ITT) | |
| Ketamine ¶ | 30 (PP) | −10.1 ± 9.5 (PP) | 0.006** (PP) |
| Placebo | 16 (PP) | −2.7 ± 5.3 (PP) | |
| HDRS‐17 | |||
| Ketamine ¶ | 33 (ITT) | −4.5 ± 5.7 (ITT) | 0.057 (ITT) |
| Placebo | 17 (ITT) | −1.4 ± 4.2 (ITT) | |
| Ketamine ¶ | 30 (PP) | −4.9 ± 5.8 (PP) | 0.045* (PP) |
| Placebo | 16 (PP) | −1.5 ± 4.3 (PP) | |
| QIDS‐SR | |||
| Ketamine ¶ | 32 (ITT) | −4.4 ± 4.8 (ITT) | 0.10 (ITT) |
| Placebo | 17 (ITT) | −2.2 ± 3.2 (ITT) | |
| Ketamine ¶ | 29 (PP) | −4.9 ± 4.8 (PP) | 0.07 (PP) |
| Placebo | 16 (PP) | −2.4 ± 3.2 (PP) | |
| MADRS dysphoric domain | |||
| Ketamine ¶ | 33 (ITT) | −3.1 ± 3.7 (ITT) | 0.052 (ITT) |
| Placebo | 17 (ITT) | −1.1 ± 2.1 (ITT) | |
| Ketamine ¶ | 30 (PP) | −3.4 ± 3.8 (PP) | 0.039 (PP) |
| Placebo | 16 (PP) | −1.2 ± 2.1 (PP) | |
| MADRS retardation domain | |||
| Ketamine ¶ | 33 (ITT) | −4.8 ± 4.9 (ITT) | 0.004 † (ITT) |
| Placebo | 17 (ITT) | −0.8 ± 3.2 (ITT) | |
| Ketamine ¶ | 30 (PP) | −5.3 ± 4.9 (PP) | 0.002 †† (PP) |
| Placebo | 16 (PP) | −0.8 ± 3.3 (PP) | |
| MADRS vegetative domain | |||
| Ketamine ¶ | 33 (ITT) | −1.3 ± 2.8 (ITT) | 0.38 (ITT) |
| Placebo | 17 (ITT) | −0.6 ± 2.1 (ITT) | |
| Ketamine ¶ | 30 (PP) | −1.5 ± 2.9 (PP) | 0.35 (PP) |
| Placebo | 16 (PP) | −0.7 ± 2.2 (PP) | |
| Outcomes | N | n (%) | P‐value |
|---|---|---|---|
| Remission | |||
| Ketamine ¶ | 33 (ITT) | 7 (21.2) (ITT) | 0.24 § (ITT) |
| Placebo | 17 (ITT) | 1 (5.9) (ITT) | |
| Ketamine ¶ | 30 (PP) | 7 (23.3) (PP) | 0.23 § (PP) |
| Placebo | 16 (PP) | 1 (6.3) (PP) | |
| Response | |||
| Ketamine ¶ | 33 (ITT) | 8 (24.2) (ITT) | 0.039*, § (ITT) |
| Placebo | 17 (ITT) | 0 (0) (ITT) | |
| Ketamine ¶ | 30 (PP) | 8 (26.7) (PP) | 0.037*, § (PP) |
| Placebo | 16 (PP) | 0 (0) (PP) | |
| Partial response | |||
| Ketamine ¶ | 33 (ITT) | 19 (57.6) (ITT) | 0.059 (ITT) |
| Placebo | 17 (ITT) | 5 (29.4) (ITT) | |
| Ketamine ¶ | 30 (PP) | 19 (63.3) (PP) | 0.038* (PP) |
| Placebo | 16 (PP) | 5 (31.3) (PP) | |
Values are shown as mean ± SD or n (%).
P < 0.05.
P < 0.01.
P < 0.016.
P < 0.0033 (multiple comparison correction).
Ketamine group was combined either administered in the double‐blind or open‐label phase.
Fisher's exact test.
HDRS‐17, 17‐item Hamilton Depression Rating Scale; ITT, intention‐to‐treat; MADRS, Montgomery Åsberg Depression Rating Scale; PP, per‐protocol; QIDS‐SR, Quick Inventory of Depressive Symptoms Self‐Rated; SD, standard deviation.
Safety
During the double‐blind phase, treatment‐emergent adverse events were more frequently observed in the ketamine group than in the placebo group (ketamine, 100.0%; placebo, 17.6%) (Table 4). No serious adverse events were observed in either group. The most common treatment‐emergent adverse event in the ketamine group was dissociation (88.2%), followed by increased blood pressure (52.9%), dizziness (41.2%), somnolence (23.5%), and headache (23.5%). During the open‐label phase, the observed adverse events were similar in frequency (i.e. 93.8%) and pattern (i.e. dissociation, increased blood pressure, dizziness, and somnolence) to those observed during the double‐blind phase (Table 4). These adverse events typically occurred on the day of dosing and generally dissipated within 2 h from the start of dosing. One participant withdrew during the open‐label phase because of an allergic rash relevant to ketamine, which resolved completely within 2 days.
Table 4.
Treatment‐emergent adverse events in safety analysis set
| Double‐blind phase | Open‐label phase | ||
|---|---|---|---|
| Adverse events | Ketamine (n = 17) | Placebo (n = 17) | Ketamine (n = 16) |
| Patients with ≥1 any events | 17 (100) | 3 (17.6) | 15 (93.8) |
| Dissociation | 15 (88.2) | 0 (0) | 14 (87.5) |
| Increased blood pressure § | 9 (52.9) | 0 (0) | 9 (56.3) |
| Dizziness | 7 (41.2) | 1 (5.9) | 5 (31.3) |
| Somnolence | 4 (23.5) | 0 (0) | 4 (25.0) |
| Headache | 4 (23.5) | 0 (0) | 3 (18.8) |
| Nausea | 3 (17.6) | 0 (0) | 4 (25.0) |
| Anxiety | 3 (17.6) | 0 (0) | 2 (12.5) |
| Dysesthesia | 3 (17.6) | 0 (0) | 1 (6.3) |
| Euphoria | 3 (17.6) | 0 (0) | 1 (6.3) |
| Blurred vision | 2 (11.8) | 0 (0) | 0 (0) |
| Feeling of drunkenness | 1 (5.9) | 1 (5.9) | 1 (6.3) |
| Malaise | 1 (5.9) | 1 (5.9) | 0 (0) |
| Increased heart rate ¶ | 0 (0) | 0 (0) | 3 (18.8) |
| Agitation | 0 (0) | 0 (0) | 1 (6.3) |
| Skin rash | 0 (0) | 0 (0) | 1 (6.3) |
| Vomiting | 0 (0) | 0 (0) | 1 (6.3) |
| Subcutaneous hemorrhage | 0 (0) | 1 (5.9) | 0 (0) |
Values are shown as n (%).
Safety analysis set consisted of all the participants to whom study drugs were administered at least once.
Increased blood pressure was counted if systolic blood pressure increased by ≥30/15 mmHg, or if the peak systolic/diastolic blood pressure ≥160/110 mmHg during the study drug administration.
Increased heart rate was counted if heart rate increased by ≥20 beats per minute, or if the peak heart rate ≥110 beats per minute during the study drug administration.
Factors associated with ketamine's treatment effect
All the following models with covariates were analyzed in PP analysis. Baseline MADRS total score and BMI were negatively associated with the MADRS total score change (β = −0.51, P = 0.003; β = −0.38, P = 0.038) (Table 5). The multiple regression model was statistically significant (F[4, 25] = 5.37, P = 0.003), explaining 37.6% of the variance in MADRS total score changes. However, logistic regression analyses failed to reveal any factors associated with remission, response, or partial response (Table 5). Moreover, the baseline MADRS retardation domain score was negatively associated with the change in MADRS total score in the linear regression model with age and sex as covariates (β = −0.48, P = 0.012) (Table 6). This model was statistically significant (F[3, 26] = 2.73, P = 0.044), accounting for 16.2% of the explained variability in MADRS total score changes. However, the baseline MADRS dysphoric and vegetative domain scores were not associated with MADRS total score changes.
Table 5.
Factors associated with the change in the MADRS total score and the presence of remission, response, or partial response in patients who received ketamine
| Factors associated with the change in the MADRS total score (n = 30) | ||||
|---|---|---|---|---|
| Variables | B | β | 95% CI | P‐value |
| Age, years | 0.058 | 0.058 | −0.33 to 0.42 | 0.82 |
| Sex, female | 6.32 | 0.29 | −0.62 to 13.3 | 0.068 |
| Body mass index | −0.94 | −0.38 | −1.80 to −0.07 | 0.038* |
| Baseline MADRS total score | −0.66 | −0.51 | −1.09 to −0.24 | 0.003** |
| Factors associated with the presence of remission (n = 30) | ||||
|---|---|---|---|---|
| Variables | B | OR | 95% CI | P‐value |
| Age, years | 0.04 | 1.04 | 0.92–1.17 | 0.57 |
| Sex, female | −1.08 | 0.34 | 0.03–4.15 | 0.40 |
| Body mass index | 0.20 | 1.22 | 0.92–1.63 | 0.17 |
| Baseline MADRS total score | 0.03 | 1.03 | 0.90–1.19 | 0.64 |
| Factors associated with the presence of response (n = 30) | ||||
|---|---|---|---|---|
| Variables | B | OR | 95% CI | P‐value |
| Age, years | 0.03 | 1.03 | 0.91–1.15 | 0.67 |
| Sex, female | −1.58 | 0.21 | 0.02–2.58 | 0.22 |
| Body mass index | 0.21 | 1.23 | 0.91–1.68 | 0.18 |
| Baseline MADRS total score | 0.08 | 1.09 | 0.94–1.25 | 0.25 |
| Factors associated with the presence of partial response (n = 30) | ||||
|---|---|---|---|---|
| Variables | B | OR | 95% CI | P‐value |
| Age, years | 0.002 | 1.00 | 0.90–1.12 | 0.98 |
| Sex, female | −1.38 | 0.27 | 0.02–3.32 | 0.31 |
| Body mass index | 0.15 | 1.16 | 0.85–1.59 | 0.36 |
| Baseline MADRS total score | 0.09 | 1.09 | 0.93–1.28 | 0.28 |
P < 0.05.
P < 0.01.
CI, confidence interval; MADRS, Montgomery Åsberg Depression Rating Scale; OR, odds ratio.
Table 6.
The association between the baseline MADRS domain scores and the MADRS total score change in patients who received ketamine
| Variables | B | β | 95% CI | P‐value |
|---|---|---|---|---|
| Baseline MADRS dysphoric domain score | −1.51 | −0.46 | −2.81 to −0.22 | 0.024 |
| Baseline MADRS retardation domain score | −1.29 | −0.48 | −2.26 to −0.31 | 0.012† |
| Baseline MADRS vegetative domain score | −1.43 | −0.44 | −2.58 to −0.28 | 0.017 |
P < 0.016 (multiple comparison correction).
Age and sex were controlled as covariates in the linear regression model.
CI, confidence interval; MADRS, Montgomery Åsberg Depression Rating Scale.
Discussion
To the best of our knowledge, this is the first DBRCT followed by an extended single‐arm open‐label study, to evaluate the efficacy and safety of repeated intravenous ketamine infusions in Asian patients with TRD. In the double‐blind phase, significant superiority was observed in reductions in MADRS total score in the ketamine group compared to the placebo group in PP analysis, but not in ITT analysis. The sensitivity analysis adjusting for the baseline MADRS total score showed similar results. The effect size (Cohen's d) of ketamine treatment was 0.69 and 0.80 in ITT and PP analysis, respectively. Moreover, both ITT and PP analyses showed that ketamine outperformed placebo in changes in MADRS retardation domain score in the double‐blind phase. Treatment‐emergent adverse events were more frequent in the ketamine group than in the placebo group, whereas no serious adverse events were observed. Multiple regression analysis revealed that a higher MADRS total score and BMI at baseline were associated with a greater reduction in the MADRS total score after ketamine infusion.
This study showed a significant improvement in depressive symptoms in the ketamine group compared to the placebo group, in PP analysis but not in ITT analysis, suggesting that intravenous ketamine treatment outperformed placebo in alleviating depressive symptoms in patients who completed the study. This DBRCT demonstrated the benefit of repeated ketamine infusions in the Asian population, as observed in the previous case‐series study in China. 15 Contrarily, this DBRCT could not detect the superiority of ketamine over placebo in ITT analysis. Given the low dropout rate (3 of 34, 8.8%) compared to previous RCTs (9.3%–30.8%), 10 , 11 , 12 it is possibly because the effect size in this study was not large enough. It was estimated to be 0.69–0.80, which was lower than 1.14 reported in the study by Singh et al. 11 Post‐hoc analysis indicated that an effect size of at least 0.71 was needed for statistical significance in ITT analysis; however, that of 0.69 in our study missed this threshold. There are several possible reasons to explain this difference. First, the number of ketamine infusions in this study may have been too small for patients to fully benefit from the treatment. Some previous studies showed a cumulative and sustained antidepressant effect following six or more infusions. 8 , 9 , 10 Phillips et al., 10 conducted an open‐label study, and reported that the third of six infusions showed the highest response (41.4%), while the sixth infusion showed the second highest response (19.0%). A higher number of infusions may have increased the number of responders in this study. Second, low subjective expectations of Japanese participants regarding ketamine may have negatively impacted treatment response. Some studies have shown that patient expectancy is a predictor of response 36 , 37 and can lead to overestimating effect sizes in RCTs of psychedelics, including ketamine. 38 , 39 Lii et al., 39 conducted a triple‐blind RCT of intravenous ketamine during surgical anesthesia in 40 patients with MDD. They found that a single ketamine infusion did not show significant efficacy compared to a placebo in reducing the severity of depressive symptoms when differential subject‐expectancy bias was minimized with successful masking. Given the social background that ketamine is strictly regulated as a narcotic in Japan, it is possible that the participants' fear and defensive stance toward ketamine administration reduced their subjective expectations and negatively affected the therapeutic effects of this study. However, this study had a relatively high completion rate. High participation rates have been reported to be associated with frequent site visits 40 and a patient‐centered approach, where investigators demonstrate care and empathy, 41 which may have had psychotherapeutic effects on the patients in our placebo group. This may have reduced the ability to detect efficacy signals, although the subjective expectations were not assessed. Third, we used the last observation carried forward method to impute missing data in the ITT analysis. Although commonly used, this conservative method has a disadvantage regarding statistical power. In particular, clinical evaluations were conducted at only two time points (i.e. baseline and post‐treatment). Therefore, even in cases where participants dropped out after several doses, the imputation used the baseline data rather than the final point during the repeated infusions. This might have further underestimated the treatment effect.
The ketamine group demonstrated a significantly greater reduction in the MADRS‐retardation domain score than the placebo group, whereas no significant difference was observed in the score changes of MADRS dysphoric or vegetative domains between the two groups. This suggests that ketamine improved mood and interest‐activity symptoms. 42 Moreover, patients with more retardation symptoms at baseline had greater improvement in MADRS total scores following ketamine treatment (Table 6). In contrast, Uher et al., 43 found that patients with MDD with high interest‐activity symptoms at baseline were less likely to respond to citalopram or nortriptyline. Therefore, ketamine may be especially effective in alleviating the behavioral and emotional symptoms of TRD that are not responsive to conventional antidepressants.
The safety and tolerability of ketamine in this study were favorable and consistent with previous studies on patients with TRD. 6 , 8 , 44 There were no serious adverse events during this study. More than 90% of patients receiving ketamine treatment had at least one adverse event (e.g. dissociation, increased blood pressure, dizziness, and somnolence). These acute transient symptoms resolved within 2 h, which is consistent with earlier reports. 6 , 44 Only one participant withdrew from the study because of an adverse event (allergic rash), which completely resolved in 2 days.
Multiple regression analysis indicated that a higher baseline MADRS total score and BMI predicted a greater reduction in depressive symptoms. These findings align with previous studies. Some meta‐analyses assessing the efficacy of antidepressants for MDD found that the effect size of antidepressants compared to placebo increased with the baseline severity of depressive symptoms. 45 , 46 Moreover, previous clinical studies 47 , 48 and a meta‐analysis 16 of RCTs on ketamine's antidepressant effects reported that BMI predicted response to ketamine. Thus, severely depressed patients with a higher BMI at baseline might be suitable candidates for ketamine treatment. These features may be useful in decision‐making when ketamine is considered a treatment option.
This study achieved statistically significant results in the PP analysis of the MADRS for the primary outcome but did not show significant results for HDRS‐17 or QIDS‐SR as secondary outcomes. One possible reason is that the sample size of this study was calculated based on the primary outcome (i.e. the MADRS) and may not have been sufficient to provide reliable estimates for the secondary outcomes. Another possible reason included the different sensitivities to capture symptoms among the scales. MADRS mainly detects changes in core depressive symptoms, whereas HDRS‐17 relatively covers a broader range of symptoms, including anxiety, sleep disturbances, and somatic symptoms. QIDS‐SR is a self‐report scale assessing patient's subjective experiences. Thus, the sample size in this study may have lacked the statistical power necessary to detect significant changes in the secondary outcomes, potentially due to type II errors.
We combined ketamine treatment data from the double‐blind and open‐label periods. As shown in Table S4, the MADRS total score in the placebo group in ITT analysis decreased from 26.9 ± 7.3 to 24.4 ± 7.2 but returned to 26.9 ± 8.7 at the entry of the open‐label extension period. Similar score changes were observed in the MADRS total score in the PP analysis and the HDRS‐17 total score in both ITT and PP analyses. Given these score trajectories, we considered that placebo effects observed in the double‐blind phase were unlikely to carry over and had a minimum risk of affecting treatment data in the open‐label phase.
This study had several limitations. First, the small sample size may have resulted in type II errors in the ITT analysis, although a formal sample size calculation was performed. Post‐hoc analysis indicated that a sample size of 35 was required to achieve statistical significance. Second, no restrictions were placed on the use of other psychotropic medications (i.e. antipsychotics, anticonvulsants, and benzodiazepines), which could have influenced the treatment effect. Third, because this study was conducted at two sites in Japan, the generalizability of the results may be limited. Fourth, the inclusion criteria of inadequate response were based on subjective perception rather than longitudinal assessments using rating scales such as MADRS and HDRS‐17. Fifth, post‐treatment assessment was conducted only once in this study, preventing us from obtaining information on longitudinal changes in clinical severity during repeated infusions. Finally, an active placebo was not used in this study. The adverse events associated with ketamine may have unblinded actual drug administration to the participants. In contrast, while midazolam has been used as an active control in previous ketamine trials, 44 , 49 benzodiazepines may exert antidepressant effects by modulating GABAergic neural transmission. 50 , 51
Conclusion
The present study demonstrated that repeated administration of intravenous ketamine outperformed the placebo in alleviating depressive symptoms in Japanese patients with TRD who completed the study, particularly in severely depressed patients with a higher BMI at baseline. This study suggests that intravenous ketamine infusion may relieve symptoms in individuals with TRD across diverse ethnic populations, including Asian populations. Since ethnic differences could affect clinical response, further investigations with larger sample sizes, including diverse ethnic populations, are warranted.
Disclosure statement
Hiroyuki Uchida is the Editorial Board member of Psychiatry and Clinical Neurosciences and co‐author of this article. To minimize the bias, they were excluded from all editorial decision‐making. No conflict of interest to disclose for other authors. Y.O. received manuscript fees from Sumitomo Pharma within the past 3 years. H.T. received manuscript or speaker fees from Sumitomo Pharma, Janssen Pharmaceutical, Otsuka Pharmaceutical, Takeda, Wiley Japan, and Yoshitomi Yakuhin within the past 3 years. K.N‐T. received manuscript fees from Sumitomo Pharma and Wiley Japan within the past 3 years. T. Yatomi received grants from Japan Society for the Promotion of Science (21K07508, 23KJ1898), and The Keio University Doctorate Student Grant‐in‐Aid Program from Ushioda Memorial Fund (Graduate school recommendation) within the past 3 years. K.Y. received manuscript fees from Sumitomo Pharma and Wiley Japan within the past 3 years. S.T. received a fellowship from Nakatani Foundation and grants from Japan Society for the Promotion of Science (22KJ2687). N.N. received manuscript or speaker fees from Sumitomo Pharma and Wiley Japan within the past 3 years. K.K. received speaker fees from Janssen and Eisai Pharmaceutical within the past 3 years. S.H. received the JSPS Research Fellowship for Young Scientists (DC1), and The Keio University Doctorate Student Grant‐in‐Aid Program from Ushioda Memorial Fund. S.N. received grants from Japan Society for the Promotion of Science (18H02755, 22H03002), Japan Agency for Medical Research and development (AMED), Japan Research Foundation for Clinical Pharmacology, Naito Foundation, Takeda Science Foundation, and Uehara Memorial Foundation within the past 3 years. S.N. also received research support, manuscript fees or speaker's honoraria from Dainippon Sumitomo Pharma, Meiji Seika Pharma, Otsuka Pharmaceutical, Shionogi, and Yoshitomi Yakuhin within the past 3 years. T. Yamada received speaker fees from Maruishi Pharmaceutical within the past 3 years. S.I. received a grant from Japan Society for the Promotion of Science (23K14824) within the past 3 years. Y.F. received a grant from Japan Science and Technology Agency (JPMJFS2140) within the past 3 years. H.U. received grants from Daiichi Sankyo, Eisai, Mochida, Otsuka, and Sumitomo Pharma; speaker's fees from Eisai, Lundbeck, Meiji Seika Pharma, Otsuka, Boehringer Ingelheim Japan, MSD, and Sumitomo Pharma; and advisory board fees from Lundbeck, Sumitomo Pharma, Takeda Pharmaceutical Company, and Boehringer Ingelheim Japan for the past 3 years. Other authors have nothing to disclose.
Author contributions
Y.O., H.T.: conception and design of the study, acquisition and analysis of data, interpretation of results, drafting the manuscript, and review of the manuscript. K.N‐T., T.Y. (Taisuke Yatomi), K.Y., S.T., N.N., K.K., S.H., S.M.: acquisition and analysis of data, interpretation of results, and review of the manuscript. S.N.: conception and design of the study, interpretation of results, and review of the manuscript. T.Y. (Takashige Yamada), H.M., Y.I., M.J.: acquisition and analysis of data and review of the manuscript. K.Y.: conception and design of the study and review of the manuscript. T.E., S.I., Y.F., T.M.: acquisition and analysis of data and review of the manuscript. T.T., H.U.: conception and design of the study, acquisition and analysis of data, interpretation of results, and review of the manuscript.
Supporting information
Table S1. List of Abbreviations.
Table S2. CONSORT checklist.
Table S3. Changes in the MADRS total score in the double‐blind phase between the two groups using analysis of covariance, adjusting for the baseline MADRS total score.
Table S4. Depression rating scale scores measured at baseline and post‐treatment during the double‐blind and open‐label phase.
Acknowledgments
This study was supported by the Japan Society for the Promotion of Science KAKENHI under grant numbers 22H03001 (H.U.) and 22K15793 (H.T.), Keio Next‐Generation Research Project Program (H.U.), SENSHIN Medical Research Foundation (H.U.), Japan Research Foundation for Clinical Pharmacology (H.T.) and Takeda Science Foundation (T.M.).
[Correction added on 9 September 2024, after first online publication: The copyright line was changed.].
Contributor Information
Takuya Takahashi, Email: takahast@yokohama-cu.ac.jp.
Hiroyuki Uchida, Email: hiroyuki_uchida@keio.jp.
Data availability statement
The trial protocol or data that support the findings of this study are available from the corresponding author upon reasonable request. The institutional review board did not grant the deposit of raw data in a publicly accessible data archive or repository at the time of approval since the procedure was not included in the protocol or informed consent document.
References
- 1. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators . Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet 2017; 390: 1211–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. COVID‐19 Mental Disorders Collaborators . Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID‐19 pandemic. Lancet 2021; 398: 1700–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. The Lancet Global Health . Mental health matters. Lancet Glob. Health 2020; 8: e1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fava M. Diagnosis and definition of treatment‐resistant depression. Biol. Psychiatry 2003; 53: 649–659. [DOI] [PubMed] [Google Scholar]
- 5. Berman RM, Cappiello A, Anand A et al. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000; 47: 351–354. [DOI] [PubMed] [Google Scholar]
- 6. Zarate CA Jr, Singh JB, Carlson PJ et al. A randomized trial of an N‐methyl‐d‐aspartate antagonist in treatment‐resistant major depression. Arch. Gen. Psychiatry 2006; 63: 856–864. [DOI] [PubMed] [Google Scholar]
- 7. Newport DJ, Carpenter LL, McDonald WM et al. Ketamine and other NMDA antagonists: Early clinical trials and possible mechanisms in depression. Am. J. Psychiatry 2015; 172: 950–966. [DOI] [PubMed] [Google Scholar]
- 8. Murrough JW, Perez AM, Pillemer S et al. Rapid and longer‐term antidepressant effects of repeated ketamine infusions in treatment‐resistant major depression. Biol. Psychiatry 2013; 74: 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. aan het Rot M, Collins KA, Murrough JW et al. Safety and efficacy of repeated‐dose intravenous ketamine for treatment‐resistant depression. Biol. Psychiatry 2010; 67: 139–145. [DOI] [PubMed] [Google Scholar]
- 10. Phillips JL, Norris S, Talbot J et al. Single, repeated, and maintenance ketamine infusions for treatment‐resistant depression: A randomized controlled trial. Am. J. Psychiatry 2019; 176: 401–409. [DOI] [PubMed] [Google Scholar]
- 11. Singh JB, Fedgchin M, Daly EJ et al. A double‐blind, randomized, placebo‐controlled, dose‐frequency study of intravenous ketamine in patients with treatment‐resistant depression. Am. J. Psychiatry 2016; 173: 816–826. [DOI] [PubMed] [Google Scholar]
- 12. Ionescu DF, Bentley KH, Eikermann M et al. Repeat‐dose ketamine augmentation for treatment‐resistant depression with chronic suicidal ideation: A randomized, double blind, placebo controlled trial. J. Affect. Disord. 2019; 243: 516–524. [DOI] [PubMed] [Google Scholar]
- 13. Shiroma PR, Thuras P, Wels J et al. A randomized, double‐blind, active placebo‐controlled study of efficacy, safety, and durability of repeated vs single subanesthetic ketamine for treatment‐resistant depression. Transl. Psychiatry 2020; 10: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Su TP, Chen MH, Li CT et al. Dose‐related effects of adjunctive ketamine in Taiwanese patients with treatment‐resistant depression. Neuropsychopharmacology 2017; 42: 2482–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng W, Zhou YL, Liu WJ et al. Rapid and longer‐term antidepressant effects of repeated‐dose intravenous ketamine for patients with unipolar and bipolar depression. J. Psychiatr. Res. 2018; 106: 61–68. [DOI] [PubMed] [Google Scholar]
- 16. Price RB, Kissel N, Baumeister A et al. International pooled patient‐level meta‐analysis of ketamine infusion for depression: In search of clinical moderators. Mol. Psychiatry 2022; 27: 5096–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen MH, Wu HJ, Li CT et al. Is one or two infusions better in the first week of low‐dose ketamine treatment for medication‐resistant depression? A post hoc pooled analysis of randomized placebo‐controlled and open‐label trials. J. Psychiatr. Res. 2021; 144: 448–454. [DOI] [PubMed] [Google Scholar]
- 18. Takahashi N, Yamada A, Shiraishi A, Shimizu H, Goto R, Tominaga Y. Efficacy and safety of fixed doses of intranasal esketamine as an add‐on therapy to oral antidepressants in Japanese patients with treatment‐resistant depression: A phase 2b randomized clinical study. BMC Psychiatry 2021; 21: 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanazawa A, White PM, Hampson SE. Ethnic variation in depressive symptoms in a community sample in Hawaii. Cultur. Divers. Ethnic Minor. Psychol. 2007; 13: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hijazi Y, Boulieu R. Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N‐demethylation of ketamine in human liver microsomes. Drug Metab. Dispos. 2002; 30: 853–858. [DOI] [PubMed] [Google Scholar]
- 21. Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P‐450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994; 270: 414–423. [PubMed] [Google Scholar]
- 22. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders: DSM‐5, 5th edn. American Psychiatric Association, Washington, DC, 2013. [Google Scholar]
- 23. First M, Williams J, Karg R, Spitzer R. Structured Clinical Interview for DSM‐5―Research Version (SCID‐5 for DSM‐5, Research Version; SCID‐5‐RV). American Psychiatric Association, Arlington, VA, 2015. [Google Scholar]
- 24. Fava M, Freeman MP, Flynn M et al. Double‐blind, placebo‐controlled, dose‐ranging trial of intravenous ketamine as adjunctive therapy in treatment‐resistant depression (TRD). Mol. Psychiatry 2020; 25: 1592–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chandler GM, Iosifescu DV, Pollack MH, Targum SD, Fava M. RESEARCH: Validation of the Massachusetts General Hospital Antidepressant Treatment History Questionnaire (ATRQ). CNS Neurosci. Ther. 2010; 16: 322–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Desseilles M, Witte J, Chang TE et al. Assessing the adequacy of past antidepressant trials: A clinician's guide to the antidepressant treatment response questionnaire. J. Clin. Psychiatry 2011; 72: 1152–1154. [DOI] [PubMed] [Google Scholar]
- 27. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br. J. Psychiatry 1979; 134: 382–389. [DOI] [PubMed] [Google Scholar]
- 28. Appelbaum PS, Grisso T, Frank E, O'Donnell S, Kupfer DJ. Competence of depressed patients for consent to research. Am. J. Psychiatry 1999; 156: 1380–1384. [DOI] [PubMed] [Google Scholar]
- 29. Hamilton M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry 1960; 23: 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rush AJ, Gullion CM, Basco MR, Jarrett RB, Trivedi MH. The inventory of depressive symptomatology (IDS): Psychometric properties. Psychol. Med. 1996; 26: 477–486. [DOI] [PubMed] [Google Scholar]
- 31. Baron‐Cohen S, Wheelwright S, Skinner R, Martin J, Clubley E. The autism‐spectrum quotient (AQ): Evidence from Asperger syndrome/high‐functioning autism, males and females, scientists and mathematicians. J. Autism Dev. Disord. 2001; 31: 5–17. [DOI] [PubMed] [Google Scholar]
- 32. Kurita H. Reliability and validity of the Japanese version of the autism spectrum quotient Japanese version (AQ‐J). Jpn. J. Clin. Psychiatry 2003; 32: 1235–1240. [Google Scholar]
- 33. Matsuoka K, Uno M, Kasai K, Koyama K, Kim Y. Estimation of premorbid IQ in individuals with Alzheimer's disease using Japanese ideographic script (Kanji) compound words: Japanese version of National Adult Reading Test. Psychiatry Clin. Neurosci. 2006; 60: 332–339. [DOI] [PubMed] [Google Scholar]
- 34. Foa EB, Cashman L, Jaycox L, Perry K. The validation of a self‐report measure of posttraumatic stress disorder: The Posttraumatic Diagnostic Scale. Psychol. Assess. 1997; 9: 445–451. [Google Scholar]
- 35. Suzuki A, Aoshima T, Fukasawa T et al. A three‐factor model of the MADRS in major depressive disorder. Depress. Anxiety 2005; 21: 95–97. [DOI] [PubMed] [Google Scholar]
- 36. Rutherford BR, Marcus SM, Wang P et al. A randomized, prospective pilot study of patient expectancy and antidepressant outcome. Psychol. Med. 2013; 43: 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rutherford BR, Wall MM, Brown PJ et al. Patient expectancy as a mediator of placebo effects in antidepressant clinical trials. Am. J. Psychiatry 2017; 174: 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muthukumaraswamy SD, Forsyth A, Lumley T. Blinding and expectancy confounds in psychedelic randomized controlled trials. Expert Rev. Clin. Pharmacol. 2021; 14: 1133–1152. [DOI] [PubMed] [Google Scholar]
- 39. Lii TR, Smith AE, Flohr JR et al. Randomized trial of ketamine masked by surgical anesthesia in patients with depression. Nat. Ment. Health 2023; 1: 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Posternak MA, Zimmerman M. Therapeutic effect of follow‐up assessments on antidepressant and placebo response rates in antidepressant efficacy trials: Meta‐analysis. Br. J. Psychiatry 2007; 190: 287–292. [DOI] [PubMed] [Google Scholar]
- 41. Blasini M, Peiris N, Wright T, Colloca L. The role of patient‐practitioner relationships in placebo and nocebo phenomena. Int. Rev. Neurobiol. 2018; 139: 211–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Uher R, Farmer A, Maier W et al. Measuring depression: Comparison and integration of three scales in the GENDEP study. Psychol. Med. 2008; 38: 289–300. [DOI] [PubMed] [Google Scholar]
- 43. Uher R, Perlis RH, Henigsberg N et al. Depression symptom dimensions as predictors of antidepressant treatment outcome: Replicable evidence for interest‐activity symptoms. Psychol. Med. 2012; 42: 967–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Murrough JW, Iosifescu DV, Chang LC et al. Antidepressant efficacy of ketamine in treatment‐resistant major depression: A two‐site randomized controlled trial. Am. J. Psychiatry 2013; 170: 1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kirsch I, Deacon BJ, Huedo‐Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: A meta‐analysis of data submitted to the Food and Drug Administration. PLoS Med. 2008; 5: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fournier JC, DeRubeis RJ, Hollon SD et al. Antidepressant drug effects and depression severity: A patient‐level meta‐analysis. JAMA 2010; 303: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Freeman MP, Hock RS, Papakostas GI et al. Body mass index as a moderator of treatment response to ketamine for major depressive disorder. J. Clin. Psychopharmacol. 2020; 40: 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Niciu MJ, Luckenbaugh DA, Ionescu DF et al. Clinical predictors of ketamine response in treatment‐resistant major depression. J. Clin. Psychiatry 2014; 75: e417–e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grunebaum MF, Galfalvy HC, Choo TH et al. Ketamine for rapid reduction of suicidal thoughts in major depression: A midazolam‐controlled randomized clinical trial. Am. J. Psychiatry 2018; 175: 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Petty F, Trivedi MH, Fulton M, Rush AJ. Benzodiazepines as antidepressants: Does GABA play a role in depression? Biol. Psychiatry 1995; 38: 578–591. [DOI] [PubMed] [Google Scholar]
- 51. Rudolph U, Knoflach F. Beyond classical benzodiazepines: Novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 2011; 10: 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of Abbreviations.
Table S2. CONSORT checklist.
Table S3. Changes in the MADRS total score in the double‐blind phase between the two groups using analysis of covariance, adjusting for the baseline MADRS total score.
Table S4. Depression rating scale scores measured at baseline and post‐treatment during the double‐blind and open‐label phase.
Data Availability Statement
The trial protocol or data that support the findings of this study are available from the corresponding author upon reasonable request. The institutional review board did not grant the deposit of raw data in a publicly accessible data archive or repository at the time of approval since the procedure was not included in the protocol or informed consent document.