

Abstract
N6-methyladenosine (m6A) is an abundant mRNA modification in the brain which plays important roles in neurodevelopment and brain function. However, due to technical limitations, global profiling of m6A sites within the individual cell types that make up the brain has not been possible. Here, we develop a mouse model that enables transcriptome-wide m6A detection in any tissue of interest and at single-cell resolution. We use these mice to map m6A across different brain regions and within single cells of the mouse cortex and discover a high degree of shared methylation across brain regions and cell types. However, we also identify a small number of differentially methylated mRNAs in neurons that encode important regulators of neuronal signaling, and we discover that microglia have lower levels of m6A compared to other cell types. Finally, we perform single-cell m6A mapping in aged mice and identify many transcripts with age-dependent changes in m6A.
Introduction
m6A is a widespread RNA modification which plays critical roles in regulating RNA processing and gene expression. Although m6A is found in all tissues, it is particularly abundant in the brain, where it has been shown to contribute to diverse processes such as neurodevelopment, learning and memory, and reward signaling1–11. In addition, m6A dysregulation has been implicated in a variety of brain diseases, such as glioblastoma, depression, and aging-associated neurodegenerative disease8,12–14. Transcriptome-wide m6A profiling studies have been instrumental for understanding how m6A functions in the brain and have shown that m6A can be dynamically regulated in the brain under a variety of conditions, including fear conditioning, stress, synaptic activity, and neuronal injury8,15–19. However, since most m6A mapping methods require large amounts of input RNA, such studies have been limited to profiling of bulk brain tissue. As a result, we have a limited understanding of how m6A is regulated and distributed within the distinct cell types that make up the brain. This gap in knowledge is a major barrier to our understanding of how m6A contributes to brain function in both healthy and disease states.
Recently, our lab developed DART-seq, a method for m6A profiling that utilizes a fusion protein consisting of the m6A-binding YTH domain fused to the cytidine deaminase APOBEC120. When APOBEC1-YTH is expressed in cells, it directs C-to-U mutations at cytidine residues that invariably follow m6A sites. C-to-U mutations are then identified from RNA-seq data to determine the location of m6A residues transcriptome-wide. Unlike other m6A profiling methods, DART-seq does not require high amounts of input RNA. Indeed, we recently demonstrated the ability of single-cell DART-seq (scDART-seq) to profile m6A in the transcriptomes of individual cells21, which revealed previously unknown features of m6A biology.
Here, we leverage the power of the scDART-seq technology to investigate the distribution of m6A within single cells of the mouse brain. We developed DART transgenic mice expressing inducible APOBEC1-YTH in all tissues and used them to profile m6A across distinct brain regions and within single cells of the cortex. We find that RNA methylation is generally consistent across distinct brain regions and cortical cell types. However, we also observe differential methylation of a subset of functionally important RNAs, suggesting the presence of cell type-specific m6A signatures within distinct transcripts. In addition, we uncover a surprising paucity of m6A in microglial mRNA. Furthermore, we find that neurons can be distinguished by their RNA methylation patterns independent of gene expression, implying that these differences in methylation contribute to a distinct cellular heterogeneity.
Finally, we perform single-cell m6A mapping in the cortex of aged mice and identify thousands of sites that undergo differential methylation with age in specific cell types. Several of the differentially methylated mRNAs we uncovered encode proteins implicated in neuronal activity or age-related neurodegenerative diseases22–26, processes which have previously been linked to m6A14,15,19. Altogether, our studies provide a new mouse model which enables m6A mapping in vivo and uncover previously unknown features of m6A distribution in single cells of the mouse brain which have been missed by bulk m6A mapping approaches. We have compiled our data into a searchable online database (http://dartsource.duke.edu/) which is freely available to the research community.
Results
Identification of m6A sites across brain regions
To perform scDART-seq in the mouse cortex, we generated transgenic mice with ubiquitous, tamoxifen-inducible expression of APOBEC1-YTH (DART mice), as well as a mouse line that expresses an m6A binding-deficient version (APOBEC1-YTHmut)20 to control for background editing (Fig. 1a). We confirmed tamoxifen-dependent transgene expression in major tissues (brain, liver, lung, kidney, and heart) and in three different brain regions (cortex, hippocampus, and cerebellum) (Fig. 1b,c and Extended Data Fig. 1). We observed robust C-to-U editing adjacent to m6A sites in all tissues following tamoxifen treatment (Extended Data Fig. 2). Editing is absent without tamoxifen and is greatly reduced in APOBEC1-YTHmut mice (Extended Data Fig. 3), indicating that C-to-U editing in vivo depends on m6A recognition, as in cultured cells20,21. Since the rate of C-to-U editing (%C2U) adjacent to m6A correlates with m6A abundance20,21, we also examined %C2U values across tissues. We identified m6A sites with similar %C2U levels across tissues as well as those with variable editing, suggesting the potential for both consistent and tissue-specific levels of m6A at shared sites across tissues (Extended Data Fig. 2b,c).
Fig. 1 – Identification of m6A sites in vivo using DART mice.
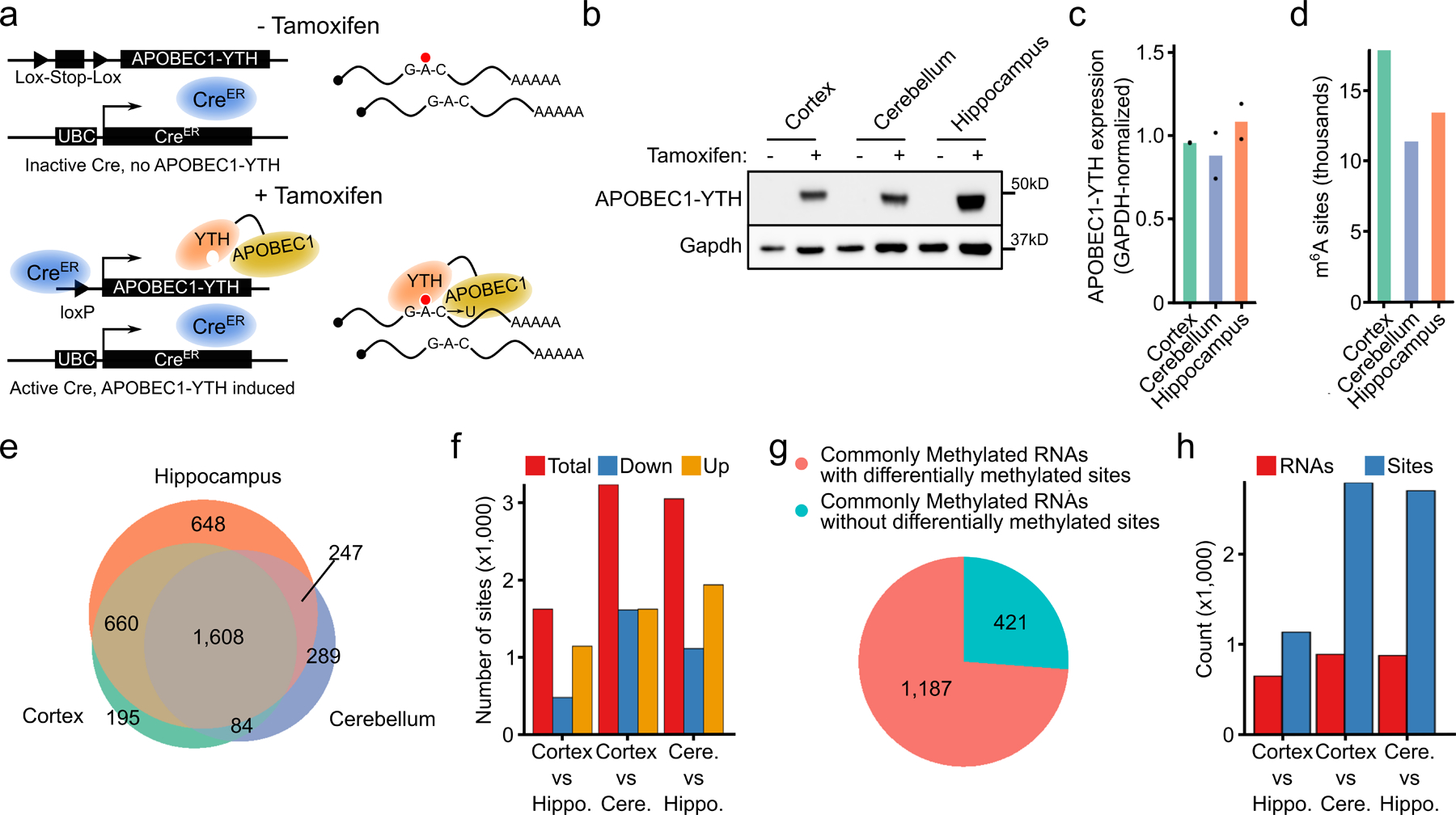
a, Schematic of transgene expression in DART mice. APOBEC1-YTH is induced in all tissues upon tamoxifen treatment. b, Western blot showing induction of the APOBEC1-YTH and APOBEC1-YTHmut proteins after tamoxifen treatment in 3 different brain regions. n = 2 biological replicates. c, Quantification of APOBEC1-YTH protein from (b) by densitometry. GAPDH was run concurrently on a separate identical blot. d, Number of m6A sites identified in each brain region. e, Overlap of methylated RNAs identified in the cortex, hippocampus, and cerebellum. Only RNAs expressed in all three brain regions are shown. f, Total number of differentially methylated sites between each pair of brain regions. Red indicates total sites, blue indicates sites with a relative decrease in %C2U in the second listed region, and orange indicates sites with a relative increase in %C2U in the second listed region. Cere. = Cerebellum, Hippo. = Hippocampus. g, Number of RNAs that are methylated in all 3 brain regions that contain differentially methylated sites (pink), or no differentially methylated sites (blue). h, Total number of differentially methylated sites (blue) and RNAs (red) between each pair of brain regions within RNAs that are methylated in all 3 brain regions. Cere. = Cerebellum, Hippo. = Hippocampus.
We next sought to identify RNAs that are methylated in distinct brain regions. We used DART mice to perform in vivo DART-seq in the cortex, hippocampus, and cerebellum. We observed highly similar transcriptome-wide gene expression profiles between DART mice and wild type mice, as well as between APOBEC1-YTH and APOBEC1-YTHmut mice, demonstrating that expression of the m6A-binding APOBEC1-YTH protein does not substantially alter gene expression patterns (Extended Data Fig. 4a–d). Furthermore, quantitative mass spectrometry showed that APOBEC1-YTH induction does not generally lead to major changes in protein abundance across tissues (Supplementary Table 1). We then identified m6A sites using Bullseye21, which accounts for read depth and background editing in APOBEC1-YTHmut samples. We identified 17,878 m6A sites in the cortex, 11,391 sites in the cerebellum, and 13,461 sites in the hippocampus (Fig. 1d, Extended Data Fig. 4e, and Supplementary Table 2). These sites exhibit a strong enrichment near the stop codon and in long internal exons, consistent with the distribution of m6A (Extended Data Fig. 4f,g). Furthermore, the methylated RNAs we identified have a high degree of overlap with those identified in previous studies in the same brain regions (Extended Data Fig. 4h). Additionally, we tested randomly selected sites using an RT-qPCR-based method for m6A detection27 and validated the presence of m6A at each site tested (Extended Data Fig. 4i). Altogether, these data indicate that DART mice can be used to identify m6A sites transcriptome-wide in vivo.
Previous studies have suggested that some RNAs undergo brain region-specific methylation8,28–30, while others have found few changes28,31,32. However, these studies used antibody-based m6A profiling, which is inherently prone to stochastic peak calling that can make it difficult to identify true differential methylation events33,34. Therefore, we utilized our in vivo DART-seq datasets to find differential methylation among commonly expressed RNAs in the cortex, cerebellum, and hippocampus. We found that the majority of commonly expressed and m6A-modified RNAs are methylated in all three brain regions (Figure 1e and Extended Data Fig. 4j). Of the transcripts that exhibit brain region-specific methylation, the greatest number was observed in the hippocampus. This is unlikely to be due to higher expression of APOBEC1-YTH, as both protein and RNA levels are comparable across all three brain regions (Fig. 1b,c and Extended Data Fig. 4k). Thus, although some distinct methylation at the whole transcript level exists across brain regions, most m6A-containing transcripts are methylated across all three brain regions. We also used a t-distributed stochastic neighbor embedding (tSNE) and hierarchical clustering analysis to examine how the methylated RNAs that we identified in the brain compares to those previously reported in other tissues28. We found that the brain m6A methylome is slightly unique compared to other tissues, and we observed more shared methylated transcripts across brain regions compared to other tissue types (Extended Data Fig. 4l,m). Interestingly, the expression of m6A writer, eraser, and m6A regulatory components is similar between the brain and other tissues, suggesting that other factors may account for these differences (Extended Data Fig. 4n). However, it is possible that the expression or activity of these proteins may account for the observed differences.
We next analyzed differential methylation at the level of individual m6A sites (see Methods). This revealed between 1,000–3,000 differentially methylated sites across each pair of brain regions, a subset of which we validated using RT-qPCR-based m6A quantification (Fig. 1f, Supplementary Table 3 and Extended Data Fig. 4o). This differential methylation of individual sites is not restricted to RNAs that are uniquely methylated in each brain region, as 73% of the RNAs that are methylated in all three brain regions contain one or more uniquely methylated sites (Fig. 1g). Interestingly, consistent with a previous study29, we identified more differentially methylated sites in the cerebellum compared to other regions (Fig. 1f,h). Differentially methylated RNAs across brain regions are enriched for genes involved in nervous system development, synaptic transmission, and neuronal signaling, consistent with known roles of m6A in neuronal development, synaptic transmission, and learning and memory4,5,9,15,16,18,35 (Supplemental Table 3).
Landscape of m6A across cell types in the mouse cortex
To date, all m6A profiling in the brain has been done using bulk tissue and does not provide cell type-specific methylation information. Thus, our knowledge of how m6A profiles might differ across cell types within the brain is very limited. To address this, we performed scDART-seq to map m6A in 7,702 single cells in the mouse cortex (Fig. 2a and Extended Data Fig. 5a–e). Using Bullseye, we identified 405,714 m6A sites (28,412 distinct methylated residues) across all cells (Supplementary Table 4). These sites are enriched near the stop codon and in long internal exons (Fig. 2b and Extended Data Fig. 5f), and there is high overlap of methylated RNAs identified using scDART-seq with those identified in the mouse cortex using bulk DART-seq and MeRIP-seq (Extended Data Fig. 5g). Additionally, similar to cultured cells21, we found that most methylated sites are present in a low proportion of cells in the population (Extended Data Fig. 5h–k).
Fig. 2 – Single-cell m6A profiling in the mouse cortex.
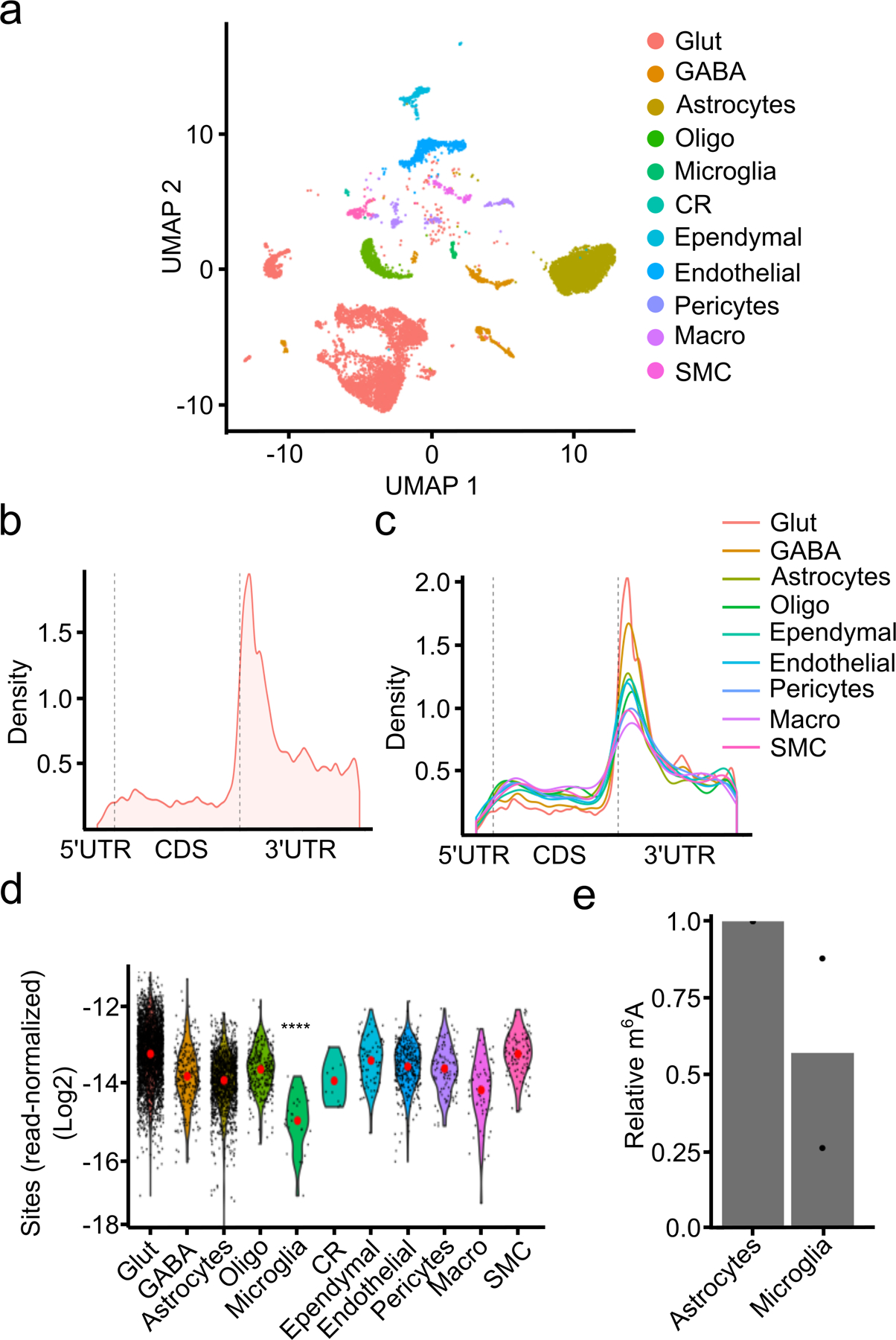
a, UMAP visualization of cell types identified in the adult mouse cortex. b, Metagene analysis of m6A sites identified by scDART-seq across all cell types in the mouse cortex. c, Metagene analysis of m6A sites identified within each cell type classification. Cell types represented by fewer than 100 cells are omitted. d, Number of m6A sites identified in each cell type normalized by the total number of reads in the cell. Red dot indicates the mean. Significance was determined using a two-sided Wilcoxon rank-sum test comparing microglia to all other cells. **** = p < 0.0001. p = 2.73e−14. e, UPLC-MS data showing decreased m6A/A ratio in mRNA purified from microglia compared to astrocytes. n = 2 biological replicates. Glut = Glutamatergic Neuron; GABA = GABAergic Neuron; Oligo = Oligodendrocyte; Micro = Microglia; CR = Cajal-Retzius Cell; Macro = Peripheral Macrophage; SMC = Smooth Muscle Cell.
Since distal processes in neurons may be lost during tissue dissociation36, the full population of methylated transcripts identified using scDART-seq may not be represented. To investigate this, we performed DART-seq on nuclear RNAs from sorted neurons. 94% of the methylated nuclear transcripts were also found in single neurons (Extended Data Fig. 5l). Furthermore, 26% of methylated transcripts previously found in neuronal processes16 were also methylated in single neurons (Supplementary Table 4), suggesting that our preparation may be retaining at least some RNAs from neuronal processes.
We next compared global m6A methylation patterns across all identified cell types in the cortex. We found that m6A exhibits the same general topology within mRNAs across all cell types (Fig. 2c), but surprisingly we found that microglia have substantially fewer m6A sites compared to other cell types (Figure 2d, Extended Data Fig. 6a–b). The average %C2U value among called sites in microglia is similar to other cell types, suggesting that although fewer sites are methylated in microglia, there is not a general shift to decreased methylation of these sites (Extended Data Fig. 6c). The decrease in m6A sites is not a sampling effect, as random sampling of equivalent numbers of glutamatergic neurons and astrocytes with similar read coverage also revealed fewer m6A sites in microglia (Extended Data Fig. 6d–g). Furthermore, APOBEC1-YTH mRNA and protein levels are similar across cell types, indicating that this is not an artifact of APOBEC1-YTH expression (Extended Data Fig. 6h–l).
To confirm this paucity of methylation in microglia, we used GLORI37 to quantify the number of m6A sites in microglia and astrocytes purified from DART mice. This analysis validated the relative hypomethylation observed in microglia (Supplementary Table 5). Additionally, we used UPLC-MS to quantify m6A in polyA+ RNA purified from microglia and astrocytes isolated from wild type mice. This revealed substantially less m6A in microglia, further confirming that this effect is not restricted to DART mice (Fig. 2e). Interestingly, analysis of our scRNA-seq data did not reveal major differences in the expression of m6A writers, readers, erasers, or exon junction complex (EJC) components in microglia (Extended Data Fig. 6m and Supplementary Table 6). However, differences in protein expression or activity of any of these m6A regulatory proteins could account for the relative hypomethylation observed in microglia. Overall, our data suggest that most cell types in the adult cortex have similar numbers and distribution of m6A sites, but that microglia have substantially reduced m6A methylation.
Comparison of methylation profiles across cell types
We next investigated whether specific RNAs are differentially methylated across cell types in the cortex by comparing average single-cell %C2U values (see Methods). In total, we found 502 differentially methylated RNAs (DM-RNAs) across cell types (Supplementary Table 7). Interestingly, we observed a modest positive correlation between changes in RNA abundance and %C2U (Fig. 3a, Extended Data Fig. 6n–r and Extended Data Fig. 7a,b), suggesting that RNAs with cell type-specific hypermethylation are more likely to have increased expression in that cell type.
Fig. 3 – Differential methylation of RNA subsets across cell types in the mouse cortex.
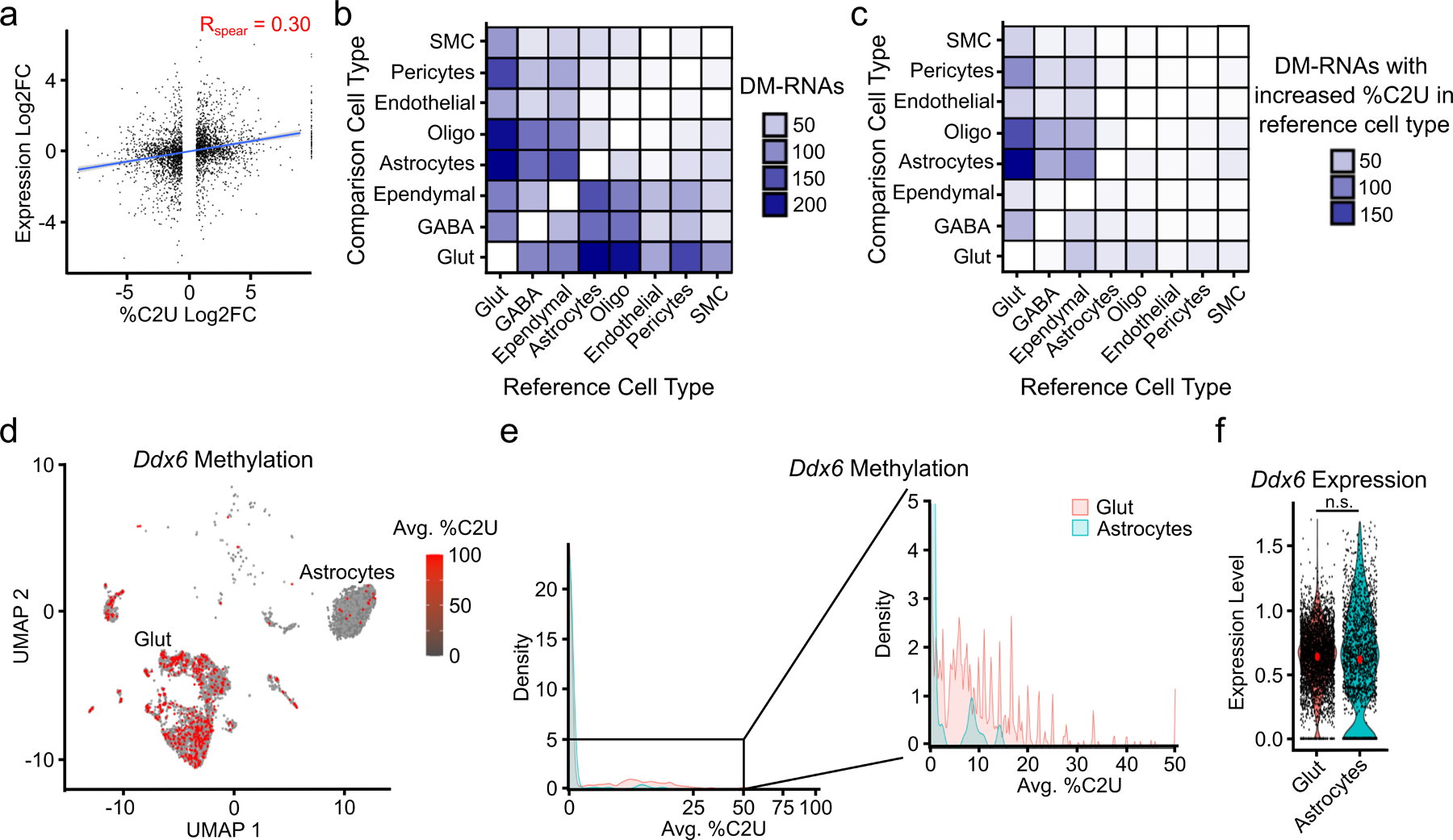
a, Correlation between changes in methylation and gene expression for all 502 DM-RNAs identified across cell types. Blue line indicates linear regression, gray band represents 95% confidence interval. Spearman correlation coefficient is shown. p < 2.2 x 10−16. n = 2,872. b, Number of DM-RNAs identified between each pair of cell types in the cortex. Heatmap is colored by the number of DM-RNAs identified in the reference cell type relative to the comparison cell type. c, Heatmap showing the number of DM-RNAs with increased average %C2U in the reference cell type relative to the comparison cell type, colored by the number of DM-RNAs. d, UMAP visualization colored by the mean %C2U across all identified m6A sites in the Ddx6 mRNA, showing higher %C2U in glutamatergic neurons compared to astrocytes. Only cells with expression and coverage of Ddx6 are shown (n = 4,082). e, Density plot showing the distribution of average Ddx6 %C2U values within single cells in glutamatergic neurons and astrocytes. Region in black box is highlighted on the right. f, Violin plot showing the normalized expression of Ddx6 in glutamatergic neurons and astrocytes. n.s. = not statistically significant. Glut = Glutamatergic Neuron; GABA = GABAergic Neuron; Oligo = Oligodendrocyte; Micro = Microglia; CR = Cajal-Retzius Cell; Macro = Peripheral Macrophage; SMC = Smooth Muscle Cell.
In general, we found relatively few DM-RNAs when comparing pairs of different cell types, with 70% of comparisons having fewer than 20 DM-RNAs (Fig. 3b). However, we found over 200 DM-RNAs between glutamatergic neurons and astrocytes (Fig. 3b). Interestingly, nearly all of these DM-RNAs have higher %C2U values in neurons (Fig. 3c), suggesting that neurons have elevated m6A levels relative to astrocytes and is consistent with a previous study29. Additionally, in contrast to all DM-RNAs, %C2U changes of DM-RNAs identified between glutamatergic neurons and astrocytes are not correlated with gene expression changes (Extended Data Fig. 7c–e). These DM-RNAs are enriched for genes important for neurodevelopment and neuronal signaling, one example being the DEAD-box protein Ddx6 (Fig. 3d–f and Extended Data Fig. 7f–l). Altogether, these data show that while most RNAs are consistently methylated across cell types, some transcripts exhibit cell type-specific methylation. Additionally, compared to other cell types, glutamatergic neurons have a high number of hypermethylated transcripts.
We next wondered whether subtypes of glutamatergic neurons differ by their methylation profiles. Neurons that reside in different cortical layers form subpopulations with distinct input and output connections and can be distinguished by their gene expression patterns38,39. Using this strategy, we identified global patterns of methylation across neuronal subtypes, which revealed a similar distribution and number of m6A sites across subtypes (Fig. 4a–c and Extended Data Fig. 8a). We next investigated whether individual transcripts exhibit neuronal subtype-specific methylation. We found relatively few DM-RNAs, with some neuronal subtypes showing no evidence for differentially methylated RNAs (Fig. 4d and Supplementary Table 8). However, we did identify several functionally important mRNAs that are differentially methylated across neurons in different cortical layers. This includes several immediate early genes, such as Egr3, Fos, and Nr4a2. (Fig. 4e–g and Extended Data Fig. 8b–e). Importantly, we observed similar expression of the APOBEC1-YTH transgene at the RNA and protein level within different layers of the cortex (Extended Data Fig. 8f and Extended Data Fig. 1b), indicating that these differences are not due to differential expression of the transgene. Additionally, the majority of DM-RNAs have little to no difference in gene expression between neuronal subtypes, indicating that changes in %C2U are not due to differential detection ability based on RNA abundance (Extended Data Fig. 8g). Indeed, we observed a slight negative correlation between change in %C2U and RNA abundance (Extended Data Fig. 8g–i). Altogether, these data suggest that m6A methylation of most mRNAs is highly consistent across glutamatergic neuron subtypes, but that some differential methylation exists within functionally important regulators of neuronal signaling.
Fig. 4 – Patterns of m6A methylation across neuronal subtypes.
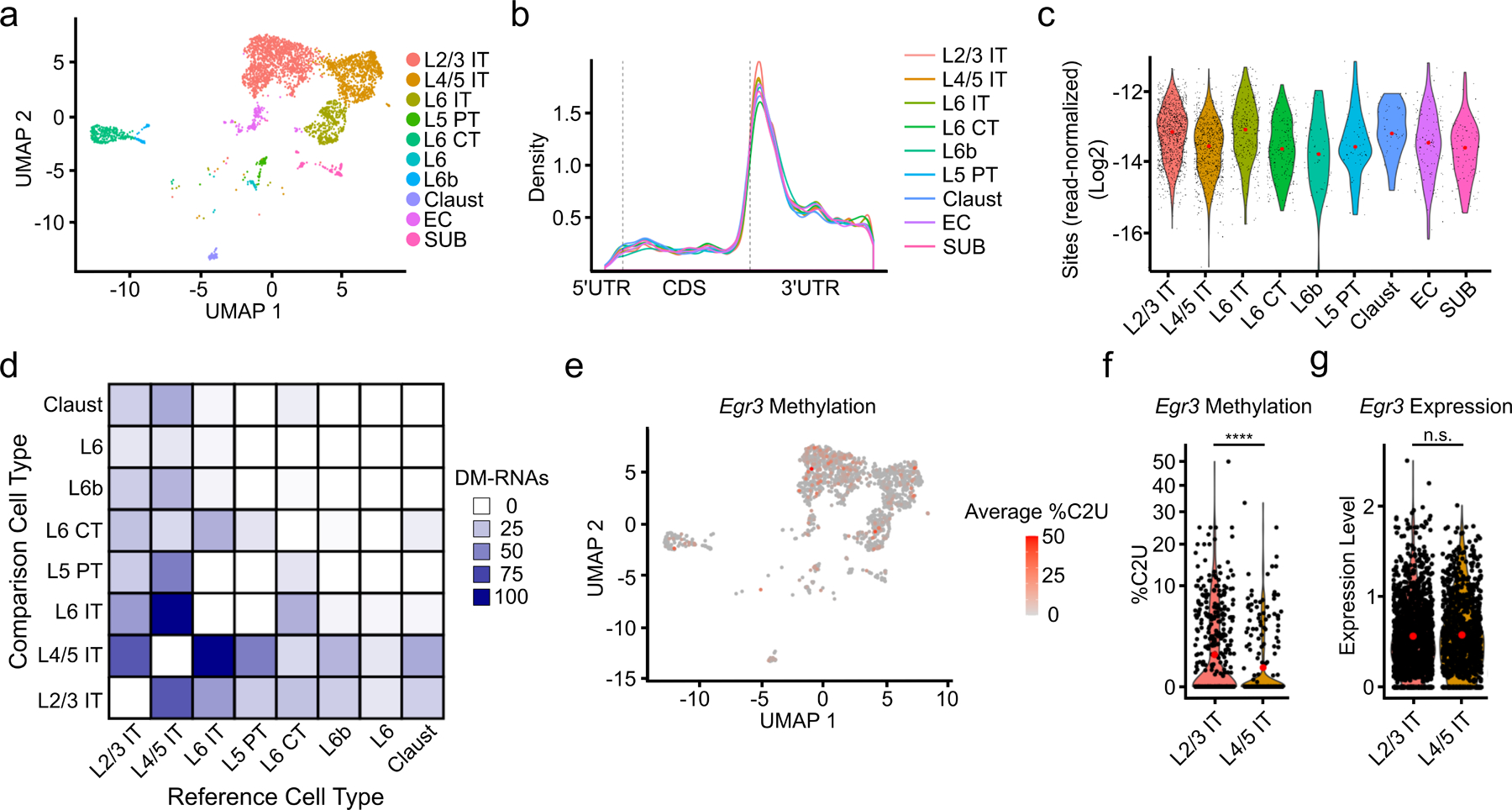
a, UMAP visualization of neuronal subtypes within the cortex. b, Metagene analysis of all m6A sites found in each neuronal subtype. c, Number of m6A sites identified in each cell normalized by the total number of reads in the cell and grouped by neuronal subtype. Red dot indicates mean value. d, Matrix showing the number of DM-RNAs identified between each pair of neuronal subtypes, ranging from 0 to 102. Boxes are colored by the number of DM-RNAs in the reference cell type relative to the comparison cell type e, UMAP visualization of neuronal subtypes colored by average %C2U of all sites identified in the Egr3 mRNA. Only cells with Egr3 expression and coverage are shown (n = 1,517). f, Violin plot showing the average single-cell %C2U values for all sites in the Egr3 mRNA in L2/3 IT and L4/5 IT neurons. Red dots indicate mean values. **** = p < 0.0001. Significance was determined using a two-sided Wilcoxon rank sum test with an FDR p-value adjustment. 5.79e−05. g, Violin plot showing the normalized gene expression values of Egr3 in L2/3 IT and L4/5 IT neurons. Red dots indicate mean values. Significance was determined using FindMarkers in Seurat (two-sided Wilcoxon rank-sum test was used, with Bonferroni p-value adjustment). p = 0.512. L2/3 = Layer 2/3; L4/5 = Layer 4/5; L6 = Layer 6; IT = Intratelencephalic; PT = Pyrimidal tract; CT = Corticothalamic tract; Claust = Claustrum; EC = Entorhinal cortex; SUB = Subiculum.
m6A contributes to molecular heterogeneity of neurons
Differential gene expression patterns are traditionally used to find biological heterogeneity within cellular populations in scRNA-seq data. However, it is unknown whether differences in mRNA methylation contribute to cellular heterogeneity as well. To investigate this, we clustered glutamatergic neurons based on their m6A methylation profiles by adapting an algorithm originally designed to group cells using chromatin accessibility (scATAC-seq) data40. This revealed 18 distinct m6A clusters (Fig. 5a). These clusters do not merely reflect RNA abundance, since they are distinct from gene expression-based clustering and since neuronal subtypes identified by gene expression are not enriched within specific m6A clusters (Fig. 5b,c and Extended Data Fig. 9a,b). Additionally, there are fewer than 20 differentially expressed RNAs in any given m6A cluster (Extended Data Fig. 9c), suggesting that m6A-dependent heterogeneity is independent of underlying gene expression patterns. None of the m6A writers, readers, erasers, or EJC components are differentially expressed within specific m6A clusters, and m6A sites have a similar distribution within mRNAs in each cluster (Extended Data Fig. 9d and Supplementary Table 9). To ensure that m6A clusters reflect true biological heterogeneity and not random noise, we randomly redistributed the single-cell RNA methylation values across all glutamatergic neurons and repeated the clustering analysis. However, we did not identify distinct clusters or differentially methylated sites, confirming that the m6A clusters reflect true cellular heterogeneity (Extended Data Fig. 9e).
Fig. 5 – Distinguishing cells based on their m6A profiles.
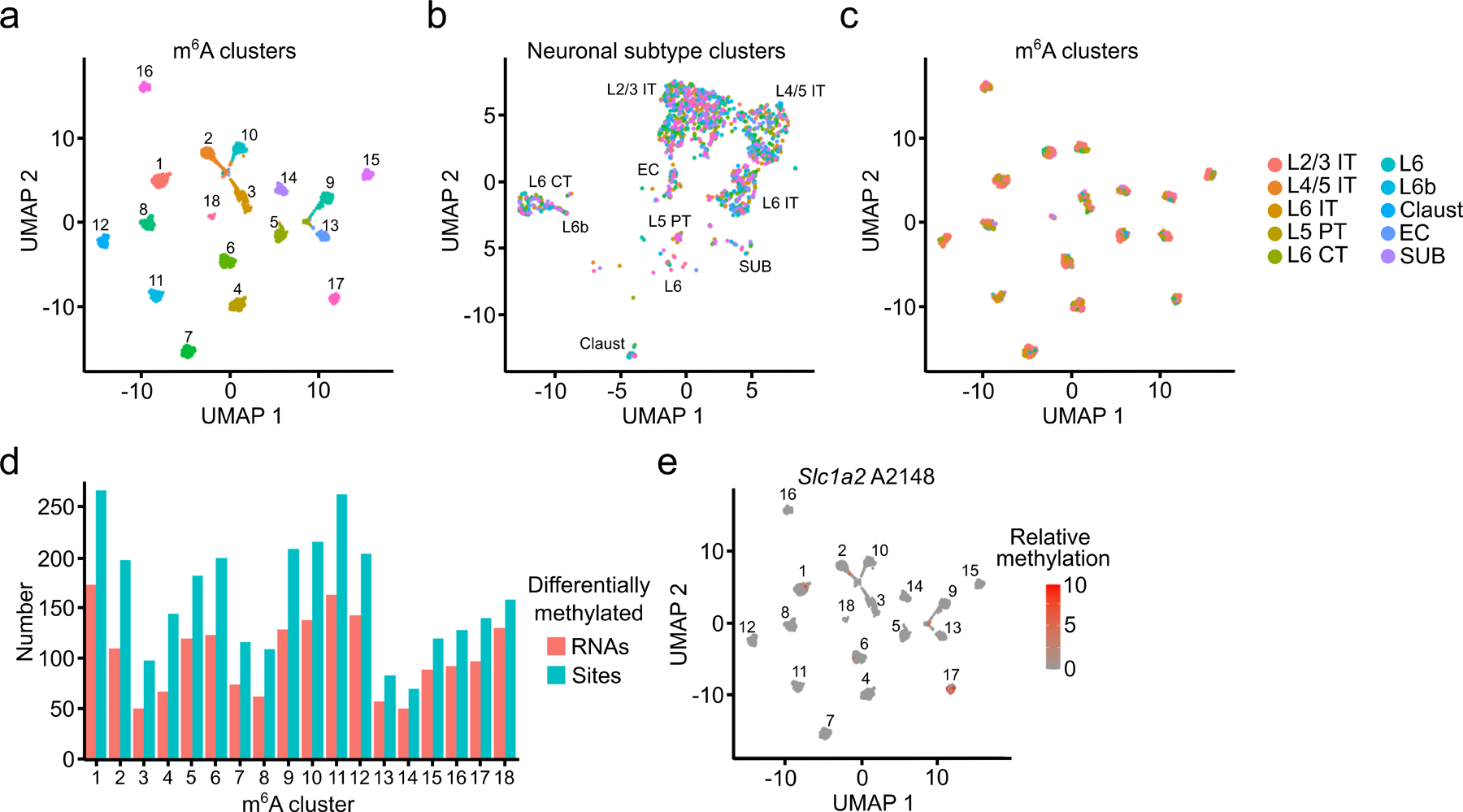
a, UMAP visualization of glutamatergic neurons clustered by m6A methylation (n = 2,246). b, UMAP visualization of glutamatergic neurons clustered by gene expression but colored by m6A cluster (n = 2,246). c, UMAP visualization of glutamatergic neurons clustered by m6A but colored by neuronal subtype (n = 2,246). d, Number of differentially methylated sites and RNAs identified within each m6A cluster. e, UMAP visualization of relative %C2U values of A2148 in the Slc1a2 mRNA within individual cells of m6A clusters (n = 2,246). A2148 is the most significantly differentially methylated site in Slc1a2.
We next identified the differentially methylated sites driving the m6A-based clustering (see Methods). Each m6A cluster has between 70–250 differentially methylated sites, representing 50–200 parent RNAs (Fig. 5d and Supplementary Table 9). These RNAs are enriched for regulators of synaptic transmission and neuronal signaling, processes in which m6A has previously been implicated8,9,15,41 (Extended Data Fig. 9f and Supplementary Table 9). Although most parent RNAs contain just one or two differentially methylated sites, there are 24 RNAs with at least 10 sites differentially methylated across multiple m6A clusters (Extended Data Fig. 9g). Interestingly, this includes important regulators of glutamatergic signaling like Grin2b, Gria2, and Slc1a2 (Fig. 5e and Extended Data Fig. 9h–k). These RNAs are not differentially expressed (Extended Data Fig. 9l–n), suggesting that differential methylation is not a major driver of their stability. However, since m6A can affect mRNA localization and translation in neurons9,42,43, it may impact these processes for these transcripts.
Age-dependent differential methylation in the mouse cortex
Altered levels of m6A writers and erasers have been associated with neurodegenerative disease30,44. However, it is unclear how m6A changes in the brain during normal aging and whether there are cell type-specific differences in methylation with age. To investigate this, we performed scDART-seq on the cortex from aged (14–15 month old) mice and integrated the sequencing data with the young (8–9 week old) mouse cortex dataset to examine age-specific changes in methylation across cell types (Fig. 6a, Extended Data Fig. 10a,b and Supplementary Table 4). While some cell types (microglia, ependymal cells, pericytes, and smooth muscle cells) have little to no differential methylation during aging (<20 sites), others (astrocytes, oligodendrocytes, and GABAergic neurons) have hundreds of DM-RNAs (Fig. 6b). Glutamatergic neurons have substantially more DM-RNAs than other cell types (837), ~75% of which show increased methylation in the aged brain (Fig. 6b, Extended Data Fig. 10c–e and Supplementary Tables 4,10), which is consistent with previous studies45. This hypermethylation in aged neurons is not due to differences in levels of APOBEC1-YTH or of m6A writers, readers, erasers, or EJC components (Extended Data Fig. 10f,g and Supplementary Table 10).
Fig. 6 – Analysis of differential methylation in the cortex during aging.
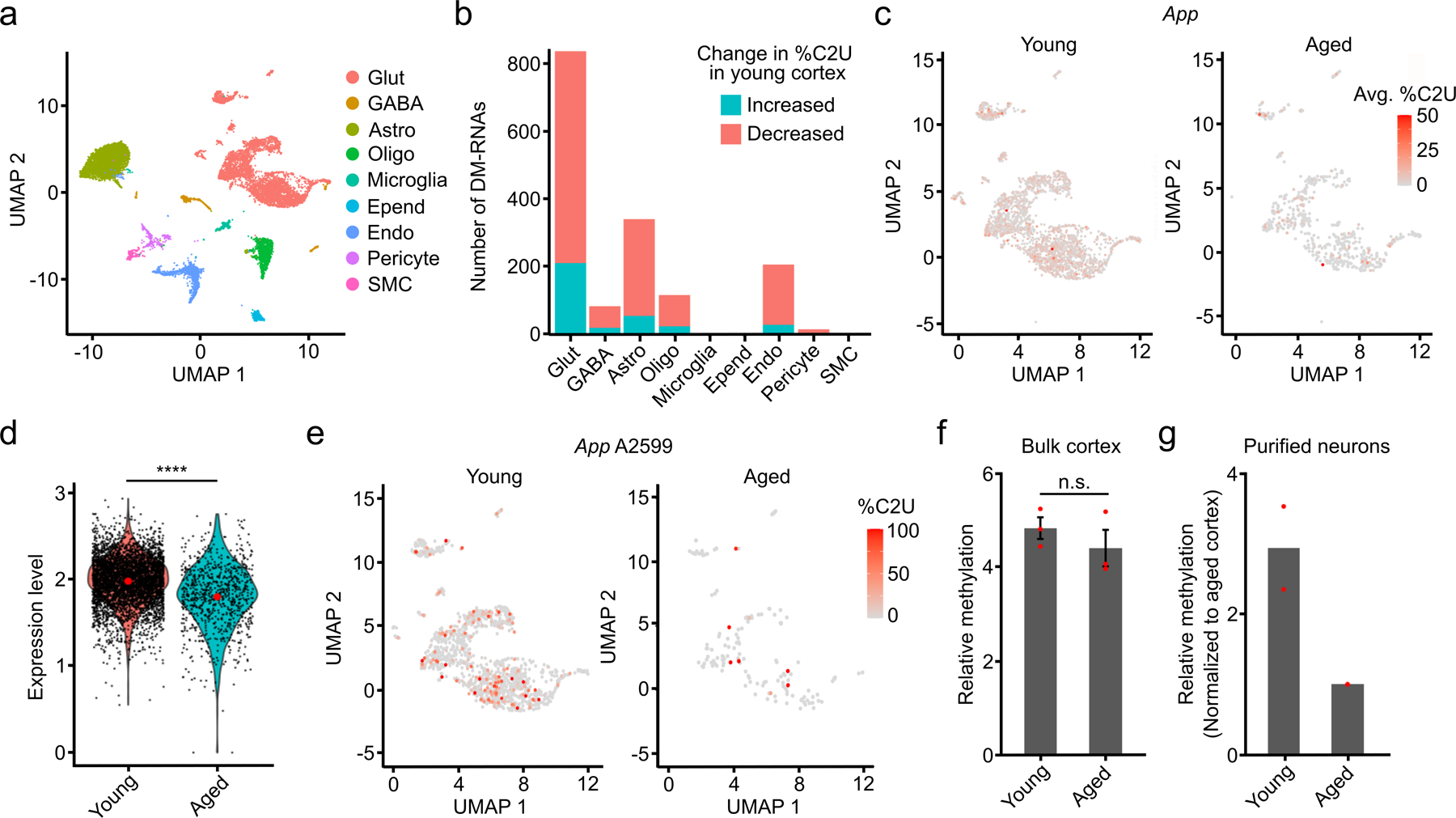
a, UMAP visualization of cortical cell types using integrated datasets from young and aged mice. b, Number of up- and downregulated DM-RNAs during aging in the indicated cell types. c, UMAP visualization of glutamatergic neurons from young and aged mice, colored by the average %C2U of all m6A sites identified in the App mRNA. Only cells with App expression and coverage are shown (young: n = 3,770; aged: n = 1,296). d, Normalized expression of App in young and aged neurons. Expression log2-fold change = −0.22 (young: n = 4,554; aged: n = 846). Significance was determined using FindMarkers in Seurat (two-sided Wilcoxon rank-sum test was used, with Bonferroni p-value adjustment). e, UMAP visualization of glutamatergic neurons from young and aged mice, colored by the %C2U value for site A2599 in the App mRNA. Only cells with App expression and coverage are shown (young: n = 1,876; aged: n = 335). f, Quantification of relative methylation at App A2599 in bulk cortex using RT-qPCR-based m6A quantification. n = 3 biological replicates. Significance was determined using a two-sided Wilcoxon rank-sum test. Error bars represent standard error. p = 1. g, RT-qPCR-based quantification of relative methylation at App A2599 in cortical neurons purified from young and aged mice. n = 2 biological replicates.
Interestingly, several of the top DM-RNAs we identified are associated with autism and intellectual disability (Gria2 and Atp2b2)22–24 or with age-related neurodegenerative diseases like Alzheimer’s disease (App and Lrp1)25,26. We focused further on the App mRNA due to its robust age-dependent differential methylation and its links to Alzheimer’s disease25 (Extended Data Fig. 10h). App contains 15 differentially methylated sites, 14 of which have decreased methylation in the aged brain despite similar RNA expression (Fig. 6c–e and Extended Data Fig. 10i,j). Additionally, App is methylated in substantially fewer individual neurons in aged mice compared to young mice (Fig. 6c). App is not differentially methylated in non-neuronal cell types, indicating that this effect is cell-type specific (Extended Data Fig. 10j). Consistent with this, we performed in vitro DART-seq on bulk cortical tissue and did not observe a significant difference in App methylation between young and aged mice (Extended Data Fig. 10k). To further confirm the neuron-specific hypermethylation of App in young neurons, we applied RT-qPCR-based m6A quantification to RNA isolated from bulk cortical tissue and purified neurons. As expected, we observed no change in m6A content in bulk cortex during aging, but we saw decreased methylation in App in purified neurons from aged mice (Fig. 6f–g). Altogether, this confirms the decrease in App methylation with age specifically in neurons and demonstrates the power of scDART-seq to reveal cell type-specific differential methylation that is missed by analyzing m6A in bulk tissue.
Discussion
m6A is a critical regulator of gene expression in the brain, but transcriptome-wide m6A profiling studies to date have been limited to examining m6A in bulk tissues. As a result, our understanding of how m6A is distributed and regulated in the diverse cell types that make up the brain is limited. Here, we leverage the scDART-seq technology by developing a transgenic mouse line that enables in vivo labeling of m6A sites and global profiling of m6A at single-cell resolution in the mouse brain.
Previous studies have shown that m6A is largely static across brain regions, with subsets of RNAs exhibiting brain region-specific methylation. We used DART mice to profile m6A in the cortex, cerebellum, and hippocampus and found that indeed there is a high degree of shared methylation across the three brain regions, as well as evidence for region-specific methylation. Interestingly, the cerebellum has the most differential methylation, which is consistent with previous studies29,45. These findings and their agreement with previous work further demonstrate the accuracy of m6A mapping using DART mice and their utility as a novel tool for studies of m6A in vivo.
We used DART mice to map m6A at single-cell resolution in the mouse cortex and uncovered several previously unknown features of m6A distribution. First, we found that mRNAs in the brain contain more m6A sites than previously estimated and that most of these sites are in only a subset of cells. This is similar to our previous observations in HEK293T cells21, suggesting that this is likely to be a general feature of m6A across cell types. Additionally, we made the surprising discovery that microglia have significantly less m6A than other cell types in the cortex. This observation was initially based on fewer m6A sites identified with scDART-seq but then validated using GLORI and mass spectrometry measurements of m6A in mRNA from purified microglia. This microglial-specific hypomethylation does not appear to be due to altered expression of major m6A regulatory proteins, so future studies will be needed to further explore the reason for this reduced methylation. Additionally, since microglia undergo transcriptional and post-transcriptional changes following their activation46–48, it will be interesting to perform scDART-seq in the context of inflammation or other conditions to determine whether microglial activation stimulates m6A methylation to dynamically regulate gene expression. Indeed, previous studies using cultured cells have discovered distinct m6A signatures in activated microglia49 and altered METTL3 expression in response to inflammation 50. Further work will be needed to investigate the degree to which m6A is dynamically regulated in microglia and its biological consequences.
Our comparison of methylation profiles across cortical cell types revealed remarkable similarity in the distribution of m6A within transcripts, the number of m6A sites per cell, and the transcripts containing methylation. This is consistent with recent work showing that m6A deposition is mostly “hard-coded” by sequence and gene architecture32,51–53. However, we did identify subsets of differentially methylated transcripts across cell types, including functionally important regulators of neuronal activity. In addition, we found m6A-dependent cellular heterogeneity among glutamatergic neurons, largely driven by differential methylation of RNAs involved in synaptic function. Importantly, this heterogeneity is independent of underlying gene expression patterns and is not correlated with RNA abundance, suggesting that the main role of these methylation events may not be to control RNA stability. The role of m6A in regulating translation in the brain has been well-established9,43, so it is possible that m6A-mediated regulation of protein production or RNA localization helps drive functional outputs of the cellular heterogeneity we observed.
Our single-cell m6A profiling in young and aged mice revealed hundreds of transcripts that undergo age-dependent methylation. This is most pronounced in neurons and includes RNAs that have both hyper- and hypomethylation with age. Several differentially methylated mRNAs encode proteins implicated in age-associated neurodegenerative disease, such as the App mRNA, which has substantially reduced m6A methylation in the aged brain. We did not observe major changes in App abundance during aging, suggesting that m6A may not impact its stability. However, it is possible that App methylation impacts protein production or RNA localization. It is also possible that m6A has distinct effects on App in a pathological context, so it will be interesting to profile m6A methylation in the aged brain from AD or other disease models and investigate whether differential methylation of App and other disease-associated transcripts impacts their expression. Indeed, previous studies have implicated m6A in AD and have identified both hyper- and hypomethylated transcripts in the aging brain14,30,45,54–58. These data, combined with our findings of cell type-specific methylation during healthy aging, further underscore the importance of investigating how m6A is altered in single cells in the context of aging-related neurodegenerative disease.
The DART mice developed here represent the first genetically encoded model for global m6A profiling. Since APOBEC1-YTH-mediated editing effectively acts as an m6A recorder, these mice will likely be useful for a wide variety of studies investigating the effects of drug treatments, environmental or behavioral perturbations, or other natural or exogenous influences on m6A methylation. Additionally, although we used DART mice to examine m6A in the brain, we anticipate that these mice will be useful for investigating m6A in other tissues as well. Further, these mice can be crossed with Cre lines of interest to study m6A in cell or tissue-specific contexts or in the presence of specific genetic perturbations. Thus, we anticipate that DART mice will be a highly useful resource for the RNA and epitranscriptomics communities and will enable numerous studies of m6A methylation in a wide range of contexts.
Limitations of the Study
The mice developed here rely on expression of APOBEC1-YTH to map m6A sites. Therefore, expression levels of the transgene and accessibility of m6A sites to the APOBEC1-YTH protein can potentially impact m6A identification. We demonstrate here and in previous studies21,59 that subtle differences in APOBEC1-YTH protein expression across cells has little correlation with the number of m6A sites identified. We have also shown that conducting DART-seq in vitro identifies similar numbers of m6A sites as when APOBEC1-YTH is expressed in cells59, suggesting that ribosomes and other RBPs do not substantially impact APOBEC1-YTH accessibility in vivo. . However, we cannot completely rule out the possibility that some sites remain inaccessible due to these or other factors, such as RNA structure. In addition, the use of DART mice involves induction of the APOBEC1-YTH transgene, so potential biological effects of prolonged APOBEC1-YTH expression should be considered. We do not observe any effects of transgene induction on DART mice health, behavior, or brain morphology, but induction may need to be optimized based on study design.
The process of single cell isolation used for our single-cell m6A profiling can damage cells, potentially leading to incomplete capture of distal cellular compartments60,61. Thus, the m6A sites we identified may not reflect the full complement of m6A residues present in neurons and other cell types with extended projections. DART-seq is compatible with any single cell isolation and scRNA-seq library preparation method, so alternative methods for single cell isolation will be interesting to explore in future studies.
Methods
Mice
All housing and procedures involving mice were approved by the Duke Institutional Animal Care and Use Committee (IACUC). Mice were housed in a laboratory facility with a 12h light/dark cycle with temperatures between 70–74°F and 30–70% relative humidity. Wild type C57BL/6 and UBC-CreER mice were purchased from The Jackson Laboratory and genotyped using recommended protocols. DART transgenic mice were generated in collaboration with the Duke Transgenic and Knockout Mouse Shared Resource. The APOBEC1-YTH-HA sequence from pCMV-APOBEC1-YTH (Addgene 131636) was cloned into Ai9 (Addgene 22799) to allow insertion of a lox-stop-lox-APOBEC1-YTH-HA into the Rosa26 locus. Embryonic stem cells were transfected using a 4 mm cuvette with 0.8 mLs ES medium using a Biorad Gene Pulser X Cell and CE module. Exponential program was used with 250 Volts and 500 UF capacitance. 35μg of linearized construct was transfected into about 20 million G4cells (passage 13) and plated onto 4 x 10 CM plates with feeders. Plates were changed every day and selected with 175μg/mL G418 for 7 days. Clones were picked and analyzed for the desired mutation by PCR and then expanded and frozen. After expansion, further confirmation of expanded clones by molecular analysis was made before injection into morulae of ICR mice to make chimeras. Presence of the transgene was confirmed using PCR. Mice were backcrossed a minimum of 5 generations with C57BL/6 before use in experiments. UBC-CreER mice were then crossed with DART transgenic mice and both alleles were maintained as hemizygous.
Tissue collection for Sanger sequencing, immunoblotting, and bulk DART-seq
6–8 weeks old DART transgenic mice (8–10 weeks old at collection) were injected with corn oil or 20mg/kg of tamoxifen (Sigma-Millipore) intraperitoneally every 24 hours for 5 days. 7 days after the final injection, mice were euthanized with CO2 before tissues (liver, lung, kidney, heart, whole cortex, hippocampus, and cerebellum) were harvested and stored in liquid nitrogen. Samples were homogenized in TRIzol (Invitrogen). Protein and total RNA was purified.
Determination of %C2U editing with Sanger sequencing
RNA was isolated from the bulk cortex of individual mice, with a mix of male and female mice represented within each young and aged sample group. For Extended Data Fig. 2a–c, n = 3 biological replicates (3 females) were used. For Extended Data Fig. 3a, 1 male DART transgenic mouse was used. For Extended Data Fig. 3b–f, 3 male APOBEC1-YTHmut DART transgenic mice used. All mice used in Extended Data Figs. 2–3 were 8–10 weeks old. For Extended Data Fig. 10k, young replicates consist of n = 5 biological replicates (2 males, 3 females, aged 8–10 weeks), while aged replicates consist of n = 4 biological replicates (1 male, 3 females, aged 14–16 months). cDNA libraries were generated using 300ng of total RNA with iScript Reverse Transcription Supermix (Bio-rad, 1708840) in 20μL reactions according to the manufacturer’s protocol. The cDNA was then diluted 1:5 by adding 80μL of water. Regions surrounding potential m6A sites were amplified by PCR using CloneAmp 2x HiFi MasterMix (Takara) using the following protocol per reaction: 7.5μL CloneAmp Mastermix, 0.75μL each of 10μM forward and reverse primers, 5μL water, and 1μL diluted cDNA. The PCR reaction was run as follows: 1) 98°C for 3 minutes, 2) 98°C for 10 sec., 3) 55–62°C for 10 sec., 4) 72°C for 5 sec., 5) Repeat 2–5 for 30 cycles total, 6) 72°C for 5 minutes, 7) Hold at 4°C. After PCR, samples were run on a 1% agarose gel and bands were extracted and sent for Sanger sequencing using the forward or reverse primers used in PCR amplification. %C2U values were quantified using EditR62. Chromatogram images were generated using Snapgene. All oligo sequences are provided in an excel table as Supplementary Data.
Immunoblotting
For Fig. 1b and Extended Data Fig. 1a, 3 female APOBEC1-YTH DART transgenic mice, 8–10 weeks old were used. For Extended Data Fig. 1e, 6 DART transgenic mice, 8–10 weeks old, were used (3 male, 3 female). For bulk mouse tissue samples, total protein was isolated using TRIzol according to the manufacturer’s protocol and solubilized in 1% SDS. Protein concentration was quantified using the Qubit Protein Assay (Thermo Fisher) on the Qubit 4 Fluorometer using 1:5-fold dilutions of the samples in water. For sorted cells, pellets were homogenized in lysis buffer (25 mM Tris-HCl, pH7.4; NaCl 150 mM; Triton X-100 1% (v/v); sodium dodecyl sulfate 0.1% (v/v)) and incubated on ice for 10 minutes prior to centrifugation at 15,000xg for 10 minutes at 4°C to remove insoluble material. Samples were then prepared with 1μg/μL total protein in 1x NuPAGE LDS Sample Buffer (Invitrogen) and 0.1 M DTT (VWR). SDS-PAGE was performed using 4–12% SDS-PAGE gels (Invitrogen) and transferred for 60 minutes at 100V in Towbin transfer buffer (25 mM Tris Base, 192 mM Glycine, 20% methanol (v/v)) to a nitrocellulose membrane (GE Amersham). Then, the membrane was blocked in PBST (1x PBS (Invitrogen) with 0.1% Tween 20 (Sigma-Aldrich)) containing 5% milk (w/v) (Quality Biological) for 1 hour with shaking at 22°C. The membranes were then incubated with primary antibody (anti-GAPDH, 1:1,000: Proteintech, 10494–1-AP; anti-HA, 1:1,000: Cell Signaling Technologies, 3724) overnight at 4°C with shaking. Primary antibodies were diluted in PBST with 5% milk (w/v) with 0.2% sodium azide (Sigma-Aldrich). Then, the blots were washed 3 times for 5 minutes each in PBST with shaking at 22°C. Blots were then incubated with secondary antibody (goat anti-rabbit IgG conjugated with HRP, 1:5,000: Abcam, ab6721; goat anti-mouse IgG conjugated with HRP, 1:2,500: Invitrogen, 62–6520) for 1 hour in PBST with shaking at 22°C. Blots were then washed 3 times for 5 minutes each in PBST with shaking at 22°C. Amersham ECL Prime Reagent was used to visualize bands, which were imaged using a Chemidoc MP (BioRad).
Quantitative mass spectrometry for protein expression analysis
Tissue samples from 8–10 week old corn oil- and tamoxifen-treated DART mice were homogenized in TRIzol. Tissue was collected from whole cortex, heart, liver, lung, and kidney. There are n = 3 biological replicates (n = 6 DART transgenic mice in total, 3 females, 3 males) for each tissue and treatment condition (30 samples total), each of which consists of tissue derived from an individual mouse. Total protein was isolated using TRIzol according to the manufacturer’s protocol and solubilized in 1% SDS, 10mM Tris. 10 ug of each sample was reduced and alkylated followed by trypsin digestion using an S-trap. After lyophilization and reconstitution of the sample in 20 uL, 1–2 uL of each was analyzed by LC-MS/MS using a 30 min gradient on a Thermo Vanquish Neo coupled to a Thermo Orbitrap Astral using a DIA method. Data was analyzed in Spectronaut.
Bulk DART-seq library preparation
4 APOBEC1-YTH (4 females) and 2 APOBEC1-YTHmut (2 males) DART transgenic mice (6–8 weeks old, 8–10 weeks old at the time of collection) were treated with tamoxifen to induce transgene expression as described above. The cortex, hippocampus, and cerebellum from each mouse was isolated as an independent biological replicate. 1μg of total RNA from each replicate was used as input for polyA purification (NEB) followed by library preparation with the NEBNext Ultra II Directional RNA Library Prep Kit (NEB) and indexed using NEBNext Multiplex Oligos for Illumina (NEB). Samples were then sequenced on a NovaSeq6000 (Illumina).
Sample preparation for tissue sectioning and immunofluorescence
6–8 week old DART transgenic mice (n = 2, 1 male and 1 female) were injected with 20mg/kg of tamoxifen (Sigma-Millipore) or corn oil intraperitoneally every 24 hours for 5 days. 7 days after the final injection, mice were deeply anesthetized with isoflurane (Covetrus), perfused with 1x PBS and fixed with 4% paraformaldehyde (Thermo-Fisher) in 1x PBS (PFA). Brains were collected and postfixed in PFA for 24 hours and transferred to 30% sucrose for cryoprotection for 48–72 hours. Brains were cryosectioned using a sliding microtome (Leica, CM3050S-3-1-1) into 30 μm sections and thaw mounted onto positively charged glass slides. The tissue was permeabilized for 1 hour in 0.2% Triton X-100 (Sigma-Millipore) in 1x TBS (0.2% TBST). The tissue was incubated in a humidity chamber at RT in ~50 μL of blocking solution (10% Normal Goat Serum (Abcam) in 0.2% TBST (NGST)) for 1 hour. Primary antibodies (see below) were prepared in 5% NGST with 0.02% sodium azide (Sigma). The tissue sections were incubated in ~50 μL of primary antibody solution in a humidity chamber (Closed container with wet kimwipes placed inside) overnight at 22°C. The slides were washed 3 times in 0.2% TBST for 5 minutes each. Secondary antibodies were prepared in 5% NGST with 1mg/mL DAPI (1:1000; Sigma-Millipore). The tissue sections were incubated in ~50 μL of secondary antibody for 2 hours at 22°C in a humidity chamber. The slides were washed 3 times in 0.2% TBST for 5 minutes each, then allowed to dry (~3 minutes). The tissue sections were mounted in ~100 μL of aqueous mounting media (Vectashield Vibrance Antifade Mounting Medium; Vector Laboratories) with a glass coverslip.
Image acquisition for immunofluorescence
The following antibodies and concentrations were used: rabbit anti-HA (Cell Signaling, C29F4, 1:250), chicken anti-Iba1 (Synaptic Systems, 234 009, 1:500), guinea pig anti-GFAP (Synaptic Systems, 173 004, 1:1500), AlexaFluor488-conjugated goat anti-chicken (Thermo-Fisher, A-11039, 1:500), AlexaFluor568-conjugated goat anti-guinea pig (Invitrogen, A-11075, 1:500), AlexaFluor647-conjugated goat anti-rabbit (Thermo-Fisher, A-21245, 1:500). Samples were examined using a Leica DMi8 inverted STED and confocal with a motorized scanning stage and Galvo scanner using a 93x NA 1.3 glycerol with motorized correction collar WD 0.3mm objective. Images were collected using 405nm diode and 470–670nm 70% pulsed white light laser lines for excitation and blue (A = x340–380, dichroic 400, LP425), green (I3 = xBP470–490, dichroic 510, mLP515), and red (N2.1 = x515–560, dichroic 580, mLP590) conventional fluorescence filters with high sensitivity hybrid spectral detectors for DAPI, Alexa488, Alexa568, and Alexa647 fluorophores with pinholes set to 1 airy unit for each channel, line averaging of 1, 2248x2248 image format, and 1x optical zoom. Z-stacks were acquired with an interval of 0.4 μm and the gain and offset optimized for the brightest central planes of the stack. The lasers and detectors to acquire each fluorophore are as follows: HA: 645 nm at 4% with 60 gain (HyD 5), GFAP: 570 nm at 2% with 40 gain (HyD 4), Iba1: 495 nm at 4% with 65 gain (HyD 2), and DAPI: 405 nm at 3% with 700 gain (PMT 1). The Lecia LasX software (Version 3.5.5.19976) was used for image acquisition and initial processing. 3D stacks were deconvoluted in the Hyugens Professional software (Version 20.10.1p3 64b). A logarithmic vertical mapping function and an automatic estimation of background was utilized in the lowest mode with a 0.7μm area radius. All images were processed with 0% relative background, less than 0.055 absolute background, and a CMLE deconvolution algorithm. The maximum number of iterations used spanned between 40 and 60 and the signal to noise ratio was set between 10 and 20; these settings were optimized for each image to maintain a 0.05 quality threshold. An optimized iteration mode and automatic brick layout was utilized for all images.
Tiled samples were examined using a 10x NA 0.4 HC PL APO dry WD 2.2 mm objective. Images were collected using 405nm diode and 470–670nm 70% pulsed white light laser lines for excitation and blue (A = x340–380, dichroic 400, LP425), green (I3 = xBP470–490, dichroic 510, mLP515), and red (N2.1 = x515–560, dichroic 580, mLP590) conventional fluorescence filters with high sensitivity hybrid spectral detectors for DAPI and Alexa647 fluorophores with pinholes set to 2 airy unit for each channel, line averaging of 1, 512x512 image format, and 0.91x optical zoom. Z-stacks were acquired with an interval of 4 μm and the gain and offset optimized for the brightest central planes of the stack. The lasers and detectors to acquire each fluorophore are as follows: HA: 645 nm at 4% with 30 gain (HyD 5) and DAPI: 405 nm at 1% with 750 gain (PMT 1). The Lecia LasX software (3.5.5.19976) was used for image acquisition and initial processing. In the tile scan feature, coordinate positions were marked around the boundaries of each tissue sample to define where scanning should take place. The software automatically calculates the number of tiles needed to cover the defined area. Tile images were auto-stitched and linearly blended using a mosaic merge. 3D stacks were input into FIJI (1.54d) to create maximum intensity projection images. Brightness and contrast settings were adjusted accordingly and applied to all images for each channel.
Image quantification and analysis
Deconvoluted 3D stacks were used as input into Imaris x64 (Version 9.6.0) for quantification of fluorescence intensity. Channels were used to generate isosurfaces for astrocytes, microglia, and HA-tagged cells using a surface detail of 0.111 μm and absolute intensity to discriminate individual cells. Cells were manually filtered by number of voxels above 10 in each image. Microglia and astrocyte 3D isosurfaces were defined and HA signal within the isosurfaces was quantified. Statistics for each cell were exported and visualized in RStudio running R (4.2.2 and 4.4.0).
Isolation of microglia and astrocytes for immunoblotting and mass spectrometry
For mass spectrometry, a pool of evenly mixed male and female cortexes (8–10 weeks old, purchased from The Jackson Laboratory) were used per replicate (n = 2). For immunoblotting, 2 male and 2 female DART transgenic mice (8–10 weeks old) were used per replicate (n = 1), treated with tamoxifen as described above to induce APOBEC1-YTH. To isolate microglia and astrocytes, mice were deeply anesthetized with isoflurane (Covetrus) and perfused with cold, oxygenated artificial cerebral spinal fluid (126mM NaCl, 3mM KCl, 1.25mM NaH2PO4, 20mM NaHCO3, 20mM D-glucose, 2mM MgSO4, 2mM CaCl2, oxygenated with carbogen) (oACSF). Perfused brains were dissected, sliced into 0.5mm coronal sections while submerged in oACSF, and cortical regions were isolated from each section. The cortical tissue from each mouse was resuspended in 500μL fresh oACSF and 200μL of neuronal isolation enzyme (Invitrogen) (Enzyme was solubilized in 2.5mL of oACSF prior to use) and incubated at 37°C for 35 minutes. Each cortex was washed 3 times with 1mL oACSF + 0.2% BSA. After the washes, each cortex was resuspended in 2mL oACSF + 0.2% BSA and gently triturated with 10mL serological pipettes 15 times. Large tissue chunks were allowed to briefly settle (~1 minute) and the supernatant was transferred to a 15mL centrifuge tube and centrifuged at 200xg for 7 minutes at 4°C. The supernatant was removed and the cells were suspended in 7mL oACSF + 0.2% BSA and 3mL SIP (90% Percoll (Sigma), 10% 10x ACSF) to make a 30% percoll solution and centrifuged at 250xg for 25 minutes at 4°C. The supernatant and debris were discarded and the cells were resuspended in 300μL of oACSF, passed through a 70μm filter (Corning), and counted on a hemacytometer with 1:1 Trypan Blue (Gibco) to estimate viability. Only animals with > 80% viability were used for sequencing. Cells were resuspended in 100μL oACSF + 0.2% BSA + 0.25μg TruStainFcX Plus (Biolegend) per 1 million cells and incubated on ice for 5 minutes. Cells were then incubated with anti-Cd11b (microglia) conjugated to Alexa Fluor 488 (1:50, Biolegend, 101217) and anti-Acsa2 (astrocytes) conjugated to APC (1:10, Miltenyi, 130-099-138) on ice for 45 minutes in the dark. Cells were then washed with 1mL oACSF (spin at 200xg for 10 minutes) and resuspended to a concentration between 1–10 million cells per mL and sorted using a FACS Aria A02. Cd11b+/Acsa2− cells were sorted as microglia and Acsa2+/Cd11b− cells were sorted as astrocytes. Immediately after sorting, cells were pelleted (1,000xg for 1 minute). For mass spectrometry, 1mL of TRIzol was added and RNA was extracted according to manufacturer’s instructions. For immunoblots, the supernatant was removed and pelleted cells were stored at −80°C until further processing.
Mass spectrometry for m6A quantification
Total RNA was extracted from sorted microglia and astrocytes from a total of 60 C57B6/J mice (8–12 weeks old, 30 males and 30 females) using TRIzol (Invitrogen) according to the manufacturer’s instructions followed by treatment with DNaseI (NEB) for 15 minutes at 37°C. PolyA+ RNA was isolated by performing two rounds of purification ith the Dynabeads mRNA Purification Kit (Invitrogen). Resulting polyA+ RNA was digested with 2U of Nuclease P1 (Sigma) in 50μL of nuclease buffer (2.5mM ZnCl2, 25mM NaCl 2h at 37°C. Samples were then treated with 5U Antarctic phosphatase (NEB) for 2h at 37°C and processed on the Xevo TQ-S system.
RT-qPCR-based m6A identification from bulk brain regions
This method is based off the previously published protocol27 with minor modifications. Samples consist of one animal per replicate. For Fig. 6f, each replicate consists of material from a single young C57BL/6 animal (n = 3, all female, aged 8–10 weeks) or a single aged C57BL/6 animal (n = 3, 1 male and 2 females, aged 14–16 months). Total RNA was extracted from samples using TRIzol (Invitrogen) according to the manufacturer’s protocol, then treated with DNAse I (NEB) for 15 minutes at 37°C and isolated again using RNeasy MinElute Cleanup Kit (Qiagen). 150ng of input RNA was used. Two reverse transcription reactions were assembled using BstI polymerase (NEB) or SuperScript III (Invitrogen). One reaction used a primer adjacent to the putative m6A site (+) for reverse transcription, while the other used a non-adjacent primer 40–70nt upstream. The presence of m6A impedes priming off of the adjacent primer, not the nonadjacent primer when using BstI. SuperscriptIII is not affected by the presence of m6A regardless of primer. Final BstI reactions consisted of 1x ThermoPol Buffer (NEB), 50μM dNTPs, 500nM primer, 150ng total RNA, and 10U BstI Polymerase. Samples were incubated in a thermocycler for 3 minutes at 25°C, 50 minutes at 50°C, and 3 minutes at 85°C. SuperScriptIII reactions were carried out according to the manufacturer’s instructions. 1μL of cDNA was used as template for quantitative PCR. Then, the number of cycles shifted using the (+) primer vs. the (-) primer in the BstI reactions was calculated, normalized to primer efficiency differences observed in SuperScriptIII reactions using the following formula:
The normalized shift in the number of cycles represents the m6A-dependent inhibition of BstI when performing reverse transcription with the adjacent (+) primer. To obtain relative methylation values, the number of cycles shifted was converted to a fold-change using the following formula:
Then, the relative methylation between regions was calculated by dividing the m6A value in the cortex by that observed in the cerebellum or hippocampus from the same animal. All oligo sequences are provided in an excel table as Supplementary Data.
Isolation of purified neurons from young and aged mice
Young adult C57/BL6J mice (n = 3, 8–10 weeks old with mixed sexes) and aged C57/BL6J mice (n = 2, 13–16 months old, 1 male and 1 female) were used for each biological replicate, in which neurons from 3 young and 2 aged mice were isolated in parallel (n = 2 biological replicates). Single cell suspensions were made and counted as described above for the isolation of purified astrocytes and microglia. After counting and confirming > 80% viability, cells were fixed in ethanol by increasing the volume of oACSF to 2mL and adding 2mL of 100% ice cold ethanol dropwise while gently vortexing. Then samples were incubated on ice for 15 minutes. Cells were pelleted at 1,700xg for 5 minutes at 4°C, washed in 2mL 1x PBS + 0.1% BSA, pelleted again, and resuspended in 100μL in oACSF + 0.2% BSA + 0.25μg TruStain FcX Plus (Biolegend) per 1 million cells and incubated on ice for 5 minutes. Cells were then incubated with anti-NeuN antibody conjugated to AlexaFluor 488 (1:100; Abcam, ab190195) for 45 minutes on ice in the dark. 1mL of cold 1x PBS + 0.1% BSA was added and cells were centrifuged 2,000xg for 5 minutes at 4°C, supernatant removed and washed once more with 1x PBS with 0.1% BSA. Young and aged cells were filtered through a 70μm filter and sorted for NeuN+ cells on a FACSymphony S6 (BD). After sorting, cells were immediately pelleted at 2,000xg for 5 minutes at 4°C and resuspended in 90μL of water and 10μL 10x SDS buffer (10mM EDTA, 5% SDS, 200mM Tris pH 7.4) and frozen at −80°C. The next day, lysates were thawed for 5 minutes at 37°C and 1mg/mL proteinase K (Invitrogen) was added and incubated for 30 minutes at 42°C to digest proteins. Nucleic acids were isolated by adding 1/10 volume 3M sodium acetate (Sigma-Millipore) and 2 volumes phenol:chloroform:isoamyl alcohol (25:24:1, Invitrogen). The samples were vortexed for 1 minute, then centrifuged at 15,000xg for 3 minutes. The top aqueous layer was transferred to a new tube in which 200μL of chloroform was added, vortexed for 1 minute, centrifuged for 3 minutes at 15,000xg, and the aqueous layer again transferred to a new tube. The solution was brought to 180μL with water, and 20μL of DNaseI buffer and 2μL of DNaseI (NEB) were added and incubated at 37°C for 15 minutes. The remaining RNA was then cleaned using an RNeasy MinElute Cleanup Kit (Qiagen).
RT-qPCR-based m6A identification from purified neurons
Relative methylation was performed as described above for bulk brain regions with the following modifications. First, 12ng of total RNA was used as input. Second, the relative methylation was calculated (after determining the CycShiftNorm and m6A value) by dividing the m6A value in the young neurons by that observed in the aged neurons from the same replicate, in which samples were processed at the same time. Bulk young vs. aged consist of RNA isolated from 3 young (8–9 week old) and 2 aged (13–16 month old) whole cortex. Normalized cycle shifts (CycShiftNorm) are shown. All oligo sequences are provided in an excel table as Supplementary Data.
Preparation of scDART-seq libraries from cortex of young mice
For young mice, 6–8 week old mice were treated with tamoxifen as described above to induce APOBEC1-YTH or APOBEC1-YTHmut expression. 2 APOBEC1-YTH biological replicates (5 cortexes total, 2 male and 3 female cortexes) and 2 APOBEC1-YTHmut biological replicates (3 cortexes total, 2 male and 1 female cortex) were used. Single cell suspensions were made and counted as described above for the isolation of purified astrocytes and microglia. After counting and confirming > 80% viability, cell suspensions were fixed using the Whole Cell Fixation Kit (Parse Biosciences) according to the manufacturer’s instructions. Split-pool barcoding and sequencing library preparation was performed according to the manufacturer's instructions using the Evercode WT Kit (Parse Biosciences). Sequencing was performed at the Duke University Sequencing and Genomic Technologies Core Facility using an Illumina NovaSeq6000 (two S4 flow cells).
Preparation of scDART-seq libraries from the cortex of aged mice
For aged mice 14–15 month old mice were treated with tamoxifen as described above to induce APOBEC1-YTH expression. 2 APOBEC1-YTH biological replicates (2 cortexes total, 1 69 week old male and 1 63 week old female) were used. Mice were sedated, perfused, and cortexes were isolated and processed as described above to make fixed single cell solutions. Split pool barcoding and sequencing library preparation was performed using the Evercode WT Mini Kit (Parse Biosciences). Samples were sequenced using an S4 flow cell on a NovaSeq 6000 (Illumina).
Isolation of purified neuronal nuclei for sequencing of nuclear RNA
DART mice (n = 2 biological replicates consisting of two mice; replicate A is 2 males and replicate B is 2 females) were treated with tamoxifen as described above to induce APOBEC1-YTH expression. Single cell suspensions were made and counted as described above for the isolation of purified astrocytes and microglia. Cell suspensions were gently homogenized in nuclear lysis buffer (100μL per 1 million cells) (10mM Tris-HCl pH 7.4, 10mM NaCl, 3mL MgCl2, 0.01% Tween-20, 0.01% Nonidet P40 Substitute, 0.001% Digitonin). Lysates were incubated on ice for 5 minutes and nuclei were pelleted at 500xg for 5 minutes at 4°C. Supernatant was removed and nuclei were washed with 1mL oACSF and pelleted again at 500xg for 5 minutes at 4°C. Nuclei were then fixed, stained, and sorted as above. Briefly, nuclei were resuspended in oACSF and fixed by adding an equivalent volume of cold ethanol and incubated on ice for 15 minutes. Nuclei were pelleted at 1,700xg for 5 minutes at 4°C, washed in 2mL 1x PBS + 0.1% BSA, and resuspended in 100μL in oACSF + 0.2% BSA + 0.25μg TruStain FcX Plus (Biolegend) per 1 million nuclei and incubated on ice for 5 minutes, then incubated with anti-NeuN antibody conjugated to AlexaFluor 488 (1:100; Abcam, ab190195) for 45 minutes on ice in the dark. Nuclei were washed twice with 1mL of cold 1x PBS + 0.1% BSA. Nuclei were filtered using a 40μm filter and sorted for NeuN+ cells on a FACSymphony S6 (BD). After sorting, cells were pelleted at 2,000xg for 5 minutes at 4°C and resuspended in 90μL of water and 10μL 10x SDS buffer (10mM EDTA, 5% SDS, 200mM Tris pH 7.4) and frozen at −80°C. Lysates were thawed for 5 minutes at 37°C, and 1mg/mL proteinase K (Invitrogen) was added and incubated for 30 minutes at 42°C to digest proteins. Nucleic acids were isolated by adding 1/10 volume 3M sodium acetate (Sigma-Millipore) and 2 volumes phenol:chloroform:isoamyl alcohol (25:24:1, Invitrogen). The samples were vortexed for 1 minute, then centrifuged at 15,000xg for 3 minutes. The top aqueous layer was transferred to a new tube in which 200μL of chloroform was added, vortexed for 1 minute, centrifuged for 3 minutes at 15,000xg, and the aqueous layer again transferred to a new tube. The solution was brought to 180μL with water, and 20μL of DNaseI buffer and 2μL of DNaseI (NEB) were added and incubated at 37°C for 15 minutes. The remaining RNA was then cleaned using an RNeasy MinElute Cleanup Kit (Qiagen). Illumina sequencing libraries were then prepared using the SMARTer® Stranded Total RNA-Seq Kit v3 -Pico Input Mammalian (Takara) using 25ng of input total RNA. Samples were sequenced using an Illumina NovaseqX.
m6A profiling using GLORI
Transcriptome-wide m6A analysis in astrocytes and microglia was performed using GLORI37. Briefly, DART transgenic mice (n = 4 mice, 3 males and 1 female) were subjected to tamoxifen injection to induce APOBEC1-YTH expression as described above. Astrocytes and microglia were then purified by flow cytometry as described above. Total RNA was then extracted using TRIzol, and 5–10 ng of total RNA was used for glyoxal and nitrite treatment. Libraries were prepared using the SMARTer Stranded Total RNA-seq Kit v3 – Pico Input Mammalian (Takara) kit and sequenced using an Illumina NovaseqX. Identification of m6A sites was performed using GLORI-tools as previously described37.
Bulk DART-seq data processing and gene expression analysis
Adapter sequences were trimmed using Flexbar63 (3.0.3), and then sequences were aligned to the mm10 genome using STAR64 (2.7.5a). PCR duplicates were marked using Samtools65 (1.11) fixmate followed by Samtools markdup, using the -r option to remove marked duplicates. A counts matrix for all genes across all samples was generated using featureCounts from the Subread66 (1.6.3) package and used for differential gene expression analysis using DESeq267.
Hierarchical clustering of bulk RNA-seq data
Differential expression across samples was assessed by using to the featureCounts matrix and performing the DESeq command (DESeq2), using the “LRT” option (Likelihood ratio test). The normalized counts were then log2-transformed using rlog (DESeq2). The transformed matrix was used to derive correlation coefficients for each pair of samples using cor(). Correlation was plotted using phheatmap.
Bulk DART-seq m6A site identification
Adapter sequences were trimmed using Flexbar (3.0.3), and then sequences were aligned to the mm10 genome using STAR (2.7.5a). PCR duplicates were marked using STAR –runMode inputAlignmentsFromBAM. Bullseye21 (https://github.com/mflamand/Bullseye) was then used to identify m6A sites. The parseBAM.pl script was used to parse the BAM files and create a counts matrix of the number of reads for each nucleotide at all positions with coverage. The Find_edit_site.pl script was then used to find C-to-U mutations with at least 25 reads of coverage, an edit ratio of 1%-95% (Treads/Totalreads), an edit ratio at least 1.5-fold higher than mutant control samples, and at least 2 C-to-U editing events observed. Those sites were further filtered to include only those occurring in an RAC (R = G/A) motif and in at least 2 of 3 biological replicates.
Differential methylation of m6A across brain regions
Differentially methylated sites were identified as previously described42. Briefly, all sites identified in each brain region that contained a minimum coverage of at least 20 reads were considered across four biological replicates. The number of total reads and the number of C-to-U mutations were used to determine the editing frequency for each site, which was then fit to a quasi-binomial general linear model. Significance was determined using a Wald’s test, followed by Independent Hypothesis Weighing (IHW) p-value adjustment for multiple comparisons. Only sites with an adjusted p-value of < 0.05 were considered differentially methylated.
Metagene analysis
metaPlotR68 was used to generate m6A metagenes from bulk and single-cell m6A datasets. RseQC (5.0.1)69 was used to generate RNA-seq coverage metagenes.
Enrichment of sites by exon length
A table containing all annotated exons in the mm10 genome build was downloaded from the UCSC Table Browser. First and last exons from each transcript were removed. Then, the number of exons containing methylation sites was normalized to the total number of exons within each group to obtain the enrichment factor.
Gene ontology
The Database for Annotation, Visualization and Integrated Discovery (DAVID)70,71 was used to perform gene ontology. For single-cell gene ontology, enrichment of RNAs within gene lists was measured against a background list consisting of all identified methylated RNAs from the scDART-seq dataset. For bulk sample gene ontology, enrichment within gene lists was measured against background lists consisting of all identified methylated RNAs from the brain regions being compared.
Initial processing of scDART-seq split-pool data
Fastq files assigned to each sublibrary from each sequencing lane were concatenated to generate a single fastq file for each sublibrary. The Parse Biosciences split-pipe (v1.1.0) pipeline was used to identify and quality control cellular barcodes and then align to the genome using STAR (2.7.5a). Options --outFilterScoreMinOverLread 0.5 and -- outFilterMatchNminOverLread 0.5 were used.
scDART-seq m6A site identification
PCR duplicates from bam files generated from the initial processing were marked using Picard MarkDuplicates (2.27.2) (http://broadinstitute.github.io/picard/). The –BARCODE_TAG option was used to only mark PCR duplicates within the same barcode (Cell ID). Then, Bullseye parseBAM.pl was used to make a nucleotide representation matrix for APOBEC1-YTH-expressing samples. The –removeDuplicates option was used to ignore PCR duplicate reads marked in the Picard step. This step generates a matrix consisting of the number of A,T,C,G,N, and total reads corresponding to each mapped nucleotide for every cell barcode. Then, the nucleotide representation matrix was made for the APOBEC1-YTHmut-expressing cells. For APOBEC1-YTHmut cells, the Bullseye parseBAM.pl with the –removeDuplicates option was used to generate a nucleotide representation matrix for each mapped nucleotide in each cell. Then, the collapse_matrix.pl script (https://github.com/mflamand/Bullseye) was used to combine the total reads at each nucleotide across all cells, generating the average representation across all cells. C-to-U mutations were found using the BullseyeFind_edit_site.pl script using the following site-calling criteria for single-cell m6A site identification: 0.5–95 %C2U, minimum coverage 3 reads in the cell, minimum of 2 C2U mutations must be observed in the cell). All sites found were filtered for only those occurring in RAC (R=G/A) motifs to enrich for m6A-adjacent editing events. Finally, sites found in fewer than 3 cells were eliminated to reduce false positive sites resulting from sequencing errors.
Identification of cell types using scDART-seq data
The split-pipe pipeline matrix, features, and barcodes files were used to generate Seurat objects in Seurat72 (4.3.0). The following quality control thresholds were used to filter out low quality cells: 100,000–5,000,000 reads per cell, ≥3,000 genes detected, < 10% reads mapping to mitochondrial RNAs. Normalization and clustering were performed using Seurat in the following way: NormalizeData, FindVariableFeatures (n = 3000 features), ScaleData, RunPCA (npcs = 40), RunUMAP (dims = 1:40), FindNeighbors (dims = 1:40), FindClusters (resolution = 1). Doublet finder73 was used to predict and eliminate doublets. A 10% doublet rate was assumed and the following settings were used: PCs 1:30, pN = 0.22, and pK = 0.3. After removing predicted doublets, clusters with similar gene expression patterns were merged. To do this, FindAllMarkers (Seurat) was used to determine differential expression (log2 fold-change > 0.58, adj. p-value < 0.01) for each cluster compared to the nearest three clusters (Euclidean distance between centers). A “descore” was calculated. Descore = sum(-log10(adj. p-value)) for all differentially expressed RNAs. Clusters with the closest descore < 1,000 were merged, with preference for merging given to the Euclidean nearest neighbor. Clusters can only be involved in 1 merge event per round. The process was repeated until no clusters had a descore < 1,000 with their nearest neighbors. Then, we performed differential expression analysis to identify the expression of known marker genes (Rbfox3, neurons; Slc17a7, glutamatergic neurons (n = 3,842 APOBEC1-YTH-expressing); Gad1, GABAergic neurons (n = 368 APOBEC1-YTH-expressing); Aqp4+/Aldoc, astrocytes (n = 1,952 APOBEC1-YTH-expressing); Mbp, oligodendrocytes (n = 497 APOBEC1-YTH-expressing); Hexb/Lyve1−/Mrc1−, microglia (n = 54 APOBEC1-YTH-expressing); Trp73+/Rln+, Cajal-Retzius cells (n = 21 APOBEC1-YTH-expressing); Dnah12/Foxj1+, ependymal cells (n = 125 APOBEC1-YTH-expressing); Cd34+/Flt1+, endothelial cells (n = 441 APOBEC1-YTH-expressing); Lyve1+/Mrc1+/Hexb−, peripheral macrophages (n = 89 APOBEC1-YTH-expressing); Pdgfrb+/Cspg4+, pericytes (n = 159 APOBEC1-YTH-expressing); Acta2+/Des+, smooth muscle cells (n = 154 APOBEC1-YTH-expressing))38,74,75. Clusters with significant expression of marker genes from multiple cell types were eliminated.
To identify neuronal subtypes, cells labeled as glutamatergic neurons were isolated as a new Seurat object. The cells underwent the same normalization and clustering process as described above. Then, descores were calculated and clusters merged as described above. Then, differential expression analysis was used to identify the expression of known marker genes (Slc30a3/Rasgrf2+, L2/3 IT (n = 1,182 APOBEC1-YTH-expressing); Slc30a3+/Rorb+, L4/5 IT (n = 784 APOBEC1-YTH-expressing); Slc30a3+/Osr1+, L6 IT (n = 371 APOBEC1-YTH-expressing); Bcl6+, L5 PT (n = 60 APOBEC1-YTH-expressing); Foxp2+, L6 CT (n = 230 APOBEC1-YTH-expressing); Cplx3+, L6b (n = 57 APOBEC1-YTH-expressing); Foxp2+/Bcl6+, L6 (n = 32 APOBEC1-YTH-expressing); Synpr+, Claustrum (n = 45 APOBEC1-YTH-expressing))38,74,75. Clusters with significant expression of marker genes from multiple subtypes were eliminated.
Differential methylation analysis of cell types and neuronal subtypes (%C2U analysis)
The single-cell mean %C2U across all m6A sites (Supplementary Table 4) within each RNA was found and used to generate a matrix consisting of the average %C2U values for each RNA within each cell. The distribution of %C2U values in each cell type was compared pairwise with all other cell types using a Wilcoxon rank-sum test. RNAs were considered significantly differentially methylated between pairs of cell types if they had an FDR-adjusted p-value < 0.05, the %C2U was greater than 0 in at least 10 cells between the cell types compared, and the log2 fold-change of the %C2U was greater than 0.58 (or less than −0.58), which corresponds to a 1.5-fold change.
Differential methylation between the young and aged cortex
As above, the single-cell mean %C2U across all m6A sites identified in the young cortex (Supplementary Table 4) within each RNA was found and used to generate a matrix consisting of the average %C2U values for each RNA within each cell. The distribution of %C2U values in each cell type from young cortex was compared with the same cell type from aged cortex using a Wilcoxon rank-sum test. RNAs were considered significantly differentially methylated between young and aged cell types if they had an FDR-adjusted p-value < 0.05, the %C2U was greater than 0 in at least 10 cells between the cell types compared, and the absolute value of the log2 fold-change of the %C2U was greater than 0.58, which corresponds to a 1.5-fold change.
For site-level analysis between young and aged neurons, a matrix consisting of the single-cell %C2U values for each site identified in young cortex (Supplementary Table 4) was generated. Then a Wilcoxon rank-sum test was used to compare the distribution of %C2U values between young and aged glutamatergic neurons. Sites were considered significantly differentially methylated between young and aged neurons if they had an FDR-adjusted p-value < 0.05, the %C2U was greater than 0 in at least 10 cells, and the absolute value of the log2 fold-change of %C2U was greater than 0.58.
Comparison of microglia to random subsets of neurons and astrocytes
A matrix consisting of the number of identified m6A sites per cell was used to compute the average number of m6A sites per cell. The matrix was then filtered to only include either all astrocytes or all glutamatergic neurons. 54 random cell IDs were selected using sample_n() and the mean number of m6A sites per cell was quantified. 54 was selected because there are 54 microglia in the final dataset. This calculation was performed 10,000 times. Then the astrocyte and glutamatergic neuron matrices were filtered to only include cell IDs from cells that had 200,000 or fewer reads. This was selected so the mean reads per cell would be as close to that observed in microglia as possible. The randomization and quantification of average m6A sites per cell was repeated 10,000 times again using this filtered matrix.
m6A-based clustering
A matrix of the raw %C2U values for all m6A sites (Supplementary Table 4) in each cell was used to create a chromatin assay file using Signac76 (options: min.cells = 10, min.features = 200). The chromatin assay file was converted to a Seurat object (“peaks” assay). Then, latent semantic indexing was used for dimension reduction (RunTFIDF(), FindTopFeatures(min.cutoff = ‘q0’), RunSVD()), followed by RunUMAP(dims = 1:30). FindNeighbors() and FindClusters() were then used to identify m6A clusters.
To control for quantitative artifacts arising from the use of sparse datasets, the %C2U values in the input matrix were randomized using sample(). This counts matrix was then processed using Signac as described above.
Differentially methylated sites were identified between m6A clusters using FindAllMarkers() on the peaks assay containing methylation information. Sites were considered differentially methylated if the absolute value of the average log2 fold-change was greater than 0.58 and the adjusted p-value was less than 0.05. To find differential expression, FindAllMarkers() was used on the RNA assay, containing scRNA-seq counts data. RNAs were considered differentially expressed if the absolute value of the average log2 fold-change was greater than 0.58 and the adjusted p-value was less than 0.05.
Statistical testing and visualization
No statistical methods were used to pre-determine sample sizes. Data collection and analysis were not performed blind to the conditions of the experiments. No formal randomization was performed, but animals/samples were assigned to conditions prior to the start of the experiment with no particular agenda, except for attempting to achieve as even of a sex distribution when possible. No data points were excluded from this study for any reason except technical failure of experiments. All Wilcoxon rank-sum tests and t-tests were performed as two-sided unless otherwise indicated. Data distribution was not formally tested, but t-tests were performed as heteroscedastic and non-parametric tests were used otherwise. Spearman correlation coefficients were calculated using the R function cor(). Data visualization was done in R with the ggplot2 (3.4.0) package. Venn diagrams are visualized using the eulerr (7.0.0) package in R.
Extended Data
Extended Data Fig. 1 – Tamoxifen-inducible APOBEC1-YTH expression in DART mice.
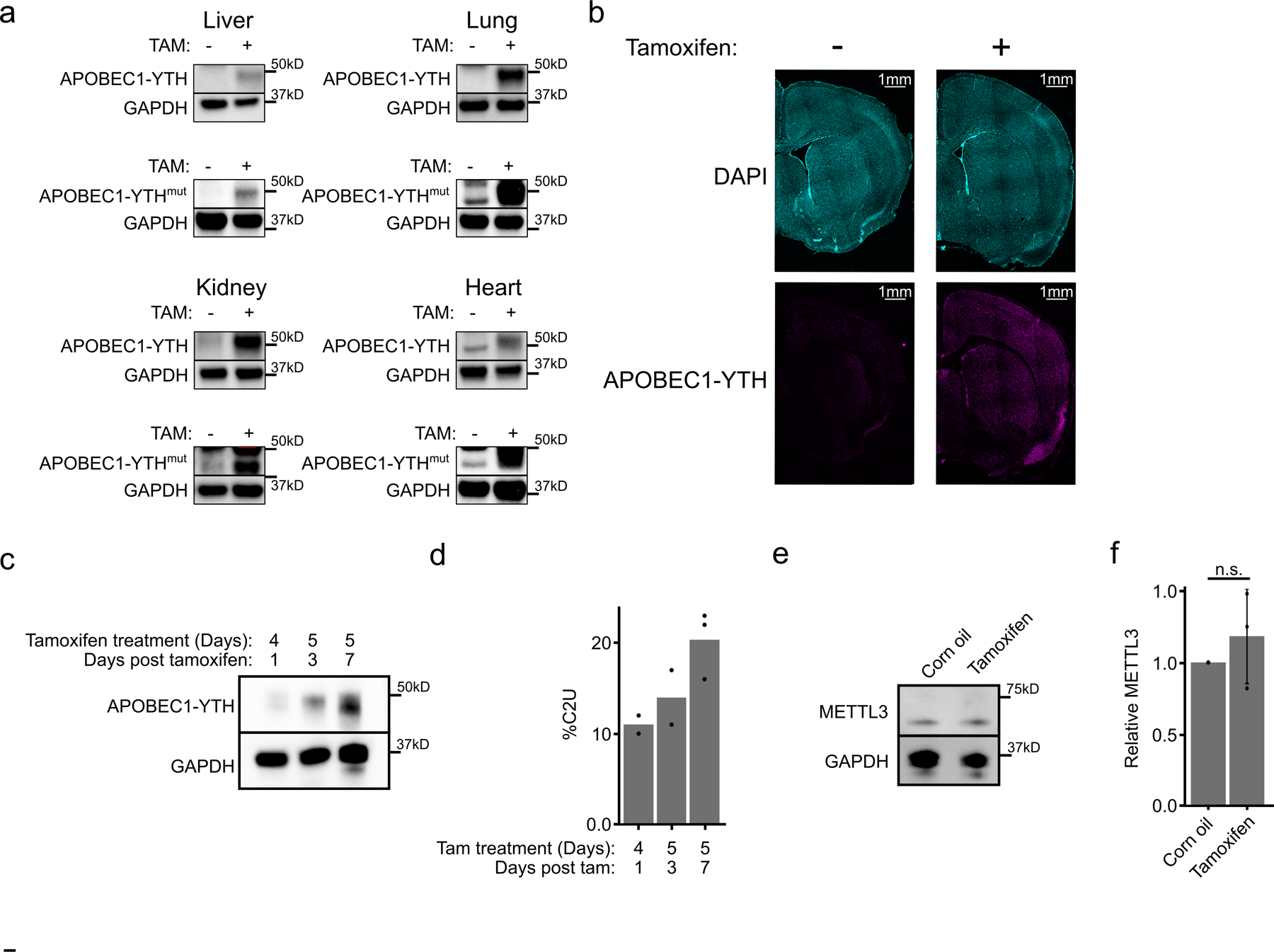
a, Western blots from liver, lung, kidney, and heart showing inducible transgene expression in APOBEC1-YTH mice (DART mice; top row) and APOBEC1-YTHmut mice (bottom row). Blots are representative of n = 2 biological replicates. GAPDH was run concurrently on a separate identical blot. b, Immunofluorescence of APOBEC1-YTH expression in a coronal DART mouse brain section. Expression is induced across all layers of the cortex. Images are representative of data from n = 2 vehicle-treated and n = 3 tamoxifen-treated mice, each biological replicates). c, Western blot comparing APOBEC1-YTH expression after different induction protocols. Blots are representative of n = 2 biological replicates. GAPDH was run concurrently on a separate identical blot. d, Quantification of %C2U adjacent to Zhx1 A2295 after each induction protocol (n = 2 biological replicates for 4 days tamoxifen +1 and 5 days tamoxifen + 3; n =3 biological replicates for 5 days tamoxifen + 7). e, Western blot detecting METTL3 from the cortex of mice treated with corn oil or tamoxifen. Image is representative of n = 3 biological replicates. GAPDH was run concurrently on a separate identical blot. f, Quantification of western blots from (f). n = 3 biological replicates. Corn oil mean = 1. Tamoxifen mean 1.18. p = 0.44. Error bars represent standard deviation.
Extended Data Fig. 2 – Characterization of tamoxifen-inducible C-to-U editing at m6A sites in vivo.
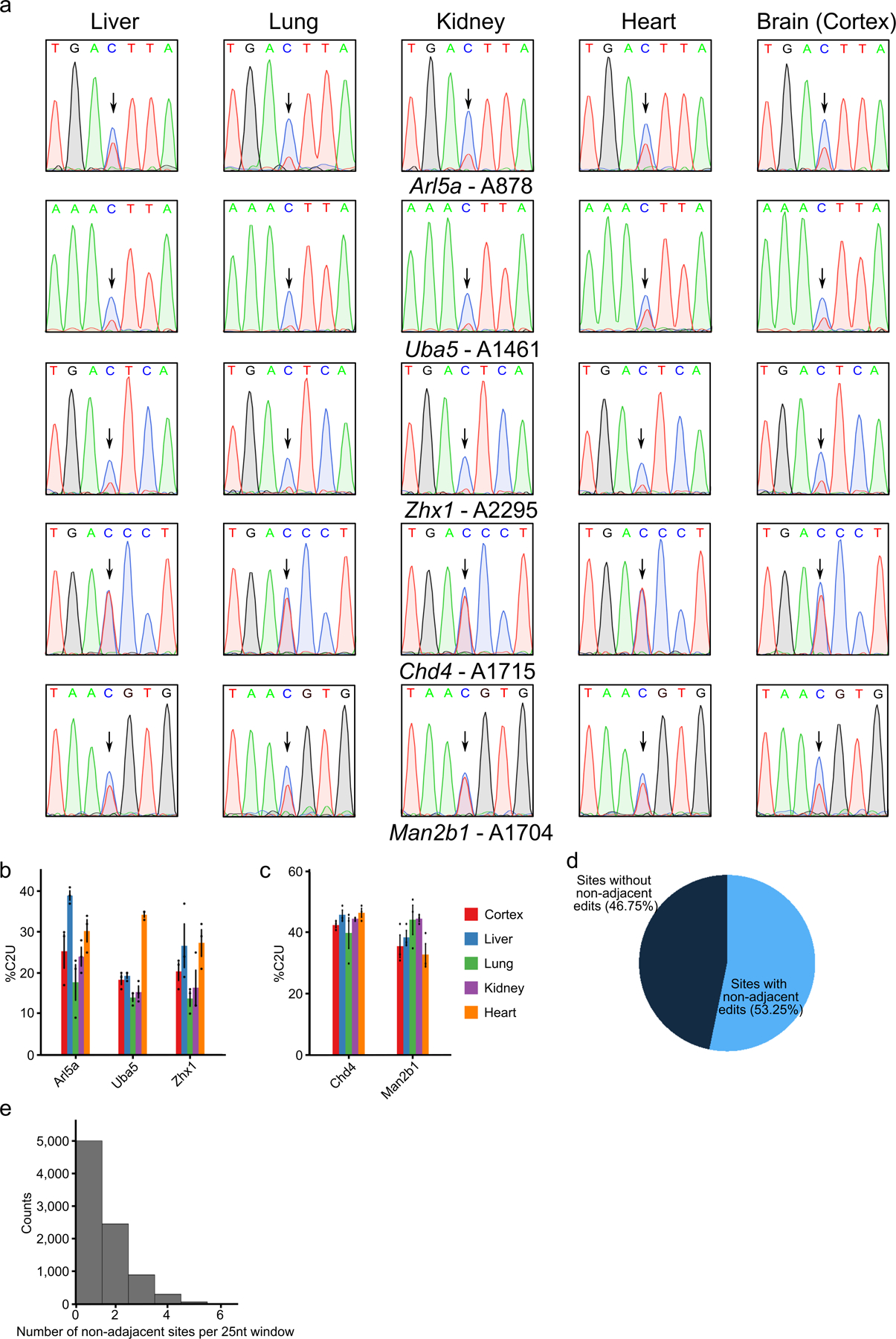
a, Sanger sequencing traces indicating C-to-U editing adjacent to m6A sites in five different mRNAs across tissue types following tamoxifen induction of APOBEC1-YTH. Arrows indicate cytidines adjacent to m6A sites. Data are representative of 3 biological replicates. b, Quantification of editing percentage (%C2U) in each tissue for sites in the Arl5a, Uba5, and Zhx1 mRNAs. Methylation at the indicated sites differs across tissues. Error bars represent standard error (n = 3 biological replicates). Mean values for Arl5a: Cortex = 25.33, Liver = 39.0, Lung = 17.67, Kidney = 24.0, Heart = 30.33. Mean values for Uba5: Cortex =18.33, Liver =19.33, Lung = 14.0, Kidney = 15.33, Heart = 34.33. Mean values for Zhx1: Cortex = 20.33, Liver = 26.67, Lung = 13.67, Kidney = 16.33, Heart = 27.33. c, Quantification of editing percentage (%C2U) in each tissue adjacent to m6A sites in the Chd4, and Man2b1 mRNAs. Methylation at the indicated sites is consistent across tissues. Error bars represent standard error (n = 3 biological replicates). Mean values for Chd4: Cortex = 42.67, Liver = 46.0, Lung = 40.0, Kidney = 44.67, Heart = 46.67. Mean values for Man2b1: Cortex = 35.67, Liver = 38.67, Lung = 44.33, Kidney = 44.67, Heart =33.0. d, Pie chart indicating the proportion of m6A sites in the cortex with non-adjacent editing events within a 25nt window. e, Histogram showing the number of non-adjacent editing events observed in each 25nt window for the 53% of m6A sites in (d) that contain non-adjacent edits.
Extended Data Fig. 3 – C-to-U editing in vivo requires APOBEC1-YTH induction and m6A binding.
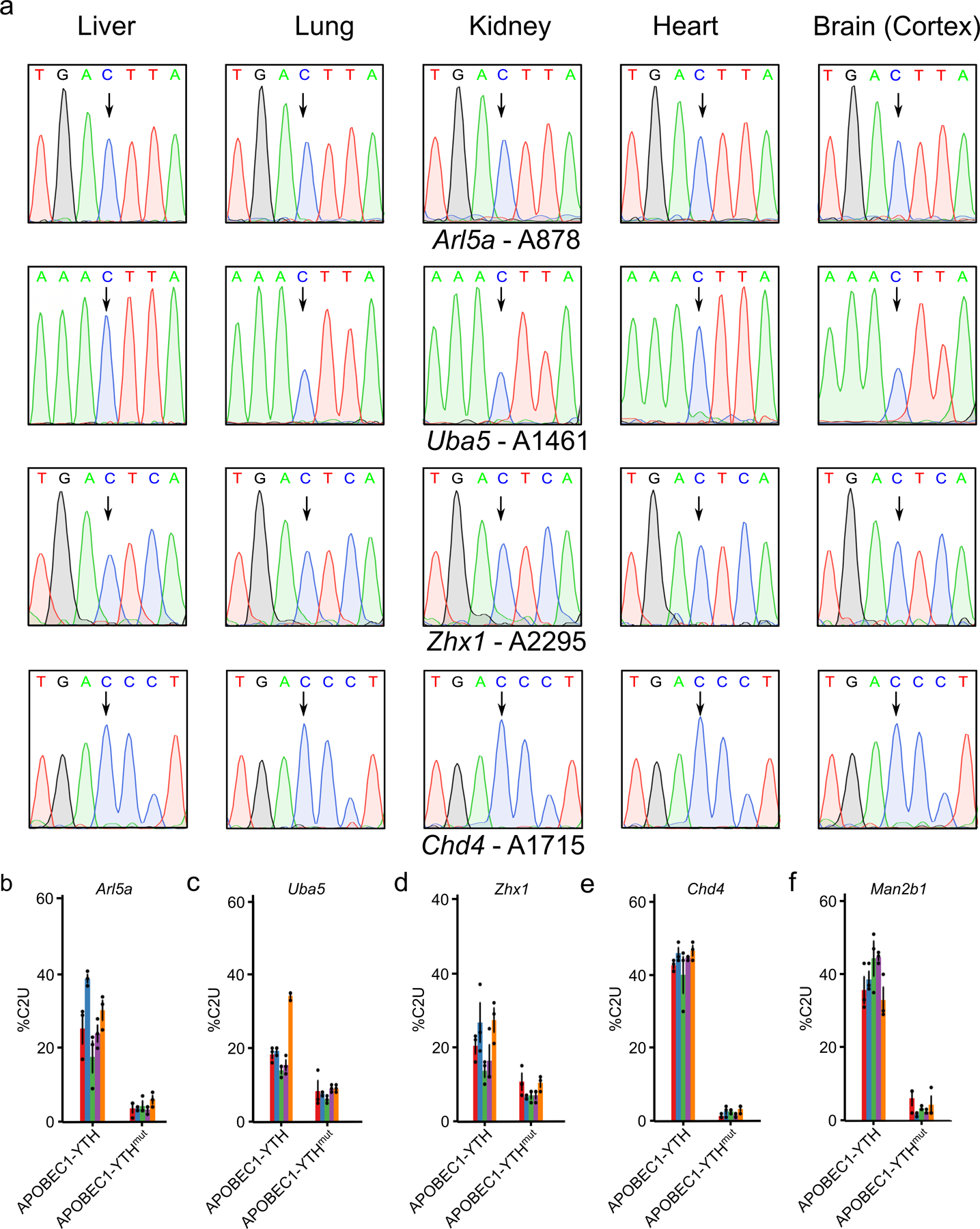
a, Sanger sequencing traces indicate a lack of C-to-U editing in the absence of APOBEC1-YTH induction. DART mice were injected with corn oil (vehicle control, n = 1 biological replicates) and RT-PCR/Sanger sequencing was performed to assess editing adjacent to m6A sites in four different mRNAs across tissue types. Arrows indicate cytidines adjacent to m6A sites. b-f, Quantification of %C2U values adjacent to m6A sites in cellular mRNAs from APOBEC1-YTHmut-expressing mice compared to APOBEC1-YTH-expressing mice. C-to-U editing is substantially reduced in APOBEC1-YTHmut-expressing mice, indicating that editing in DART mice depends on m6A recognition. Error bars represent standard error (n = 3 biological replicates). Mean values for APOBEC1-YTH samples are the same as Extended Data Fig. 2b.c. Mean values for APOBEC1-YTHmut samples are: Arl5a - Cortex = 3.67, Liver = 3.67, Lung = 4.33, Kidney = 3.33, Heart = 6.0; Uba5 - Cortex = 8.33, Liver = 7.67, Lung = 6.0, Kidney = 9.0, Heart = 9.0; Zhx1 - Cortex = 10.67, Liver = 6.67, Lung = 7.0, Kidney = 7.0, Heart = 10.33; Chd4 - Cortex = 1.33, Liver = 2.67, Lung = 2.67, Kidney = 1.67, Heart = 3.33; Man2b1 - Cortex = 6.0, Liver = 1.67, Lung = 3.33, Kidney = 2.33, Heart = 4.33.
Extended Data Fig. 4 – Characterization of gene expression and m6A methylation in the cortex, hippocampus, and cerebellum.
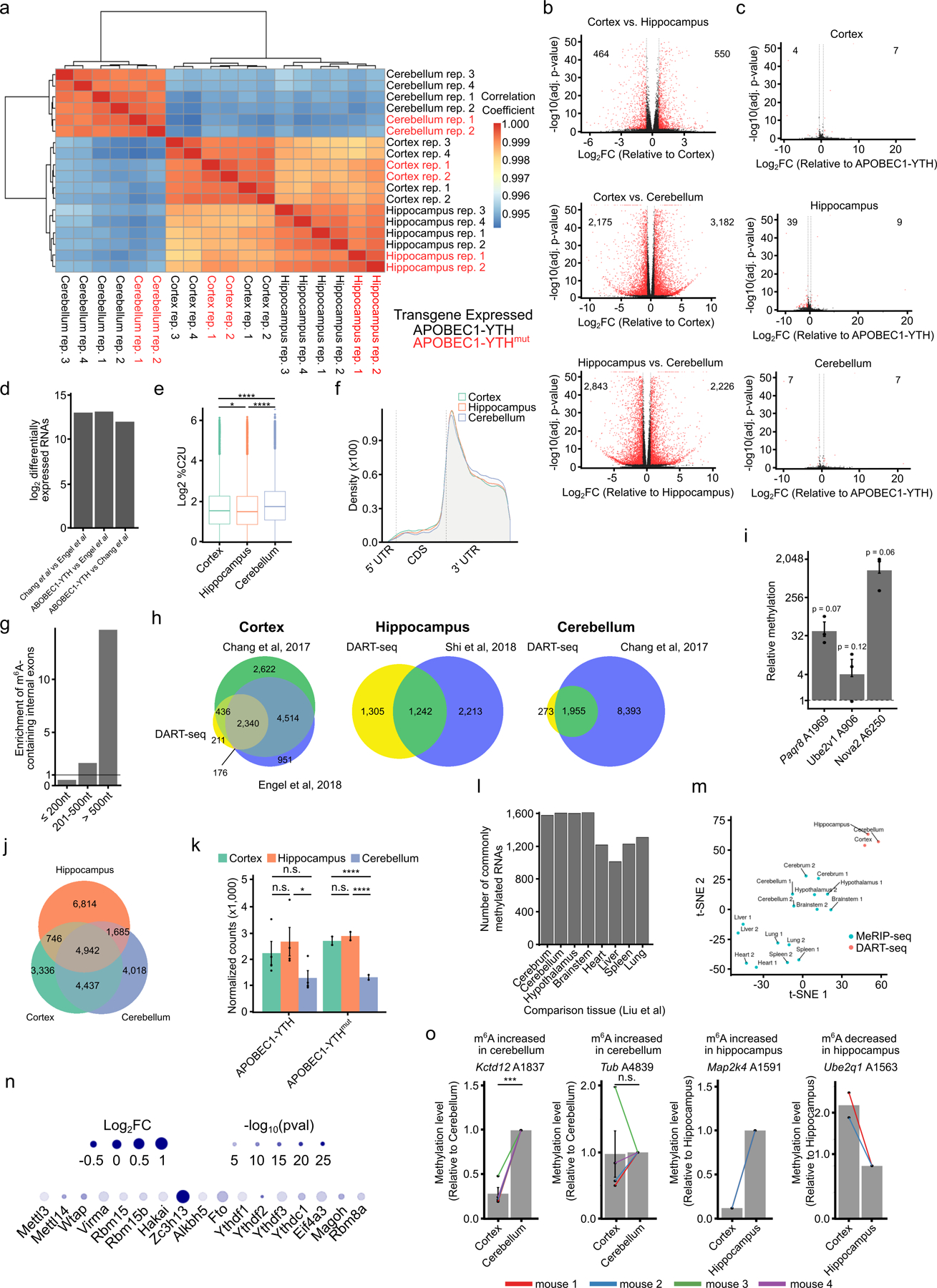
a, Hierarchical clustering of gene expression similarity scores across samples from each brain region expressing either APOBEC1-YTH or APOBEC1-YTHmut. Samples primarily cluster by brain region, not by which transgene is expressed. b, Volcano plots indicating the number of differentially expressed genes between APOBEC1-YTH-expressing brain regions. Significance was determined using DESeq2 (negative binomal regression followed by Wald test and FDR-based p-value adjustment). c, Volcano plots indicating the small number of gene expression differences between APOBEC1-YTH and APOBEC1-YTHmut-expressing cells in each brain region. Numbers indicate the number of up/downregulated genes. Significance was determined using DESeq2 (negative binomal regression followed by Wald test and FDR-based p-value adjustment). d, Number of differentially expressed RNAs between the cortex of DART mice and wild type mice29,62. There are similar numbers of differentially expressed RNAs across all samples, indicating that APOBEC1-YTH expression does not substantially alter gene expression patterns. e, Distribution of %C2U values at m6A sites identified in the cortex (n = 17,878 sites from 4 biological replicates), hippocampus (n = 13,461 sites from 4 biological replicates), and cerebellum (11,391 sites from 4 biological replicates). Significance was determined using a two-sided Wilcoxon rank-sum test. Center line in box represents median, while the box represents the 25th–75th percentile. Whiskers represent the highest and lowest values (1.5x interquartile range), with outliers shown as dots. Cortex vs. Hippocampus p-value = 0.004. Cortex vs. Cerebellum p-value = < 2.2e−16. Hippocampus vs. Cerebellum p-value = < 2.2e−16. * = p < 0.05. **** = p < 0.0001. f, Metagene analysis showing the distribution of m6A sites identified in DART mice in each brain region. Significance was determined using a two-sided t-test with no p-value adjustment. g, m6A sites identified in the cortex of DART mice are enriched in long internal exons. Barplot indicates the number of methylated internal exons relative to the total number of internal exons within each group. Solid line at 1 indicates no enrichment. h, Overlap of methylated RNAs identified by in vivo DART-seq and antibody-based m6A profiling in each brain region. i, Independent validation of methylation at randomly selected m6A sites using RT-qPCR-based relative m6A quantification. Dotted line at 1 indicates no detectable methylation. n = 3 biological replicates. Error bars represent standard error. Significance was determined using a one-sided t-test comparing to 1. Mean for Paqr8 = 49.41, mean for Ube2v1 = 6.31, mean for Nova2 = 1,375. j, Overlap of individual m6A sites identified across all three brain regions. k, Comparison of APOBEC1-YTH transgene RNA levels across brain regions. The number of normalized counts mapping to rat APOBEC1 is shown. Significance was determined using DESeq2 (negative binomal regression followed by Wald test and FDR-based p-value adjustment). Error bars represent standard error (n = 4 biological replicates). Mean for APOBEC1-YTH samples: Cortex = 2,218; Hippocampus = 2,644; Cerebellum = 1,278. Mean for APOBEC1-YTH samples: Cortex = 2,680; Hippocampus = 2,855; Cerebellum = 1,304. * = p < 0.05. **** = p < 0.0001. l, Bar chart showing the number of overlapping methylated RNAs between DART-seq in the cortex and MeRIP-seq datasets (Liu et al 2020) across mouse tissues. m, Clustering of m6A methylation patterns from mouse brain regions and other organs using the DART-seq and Liu et al 2020 datasets. n, Gene expression analysis of m6A writers, readers, erasers, and core EJC components derived from RNA-seq data from Liu et al 2020. Log2FC represents brain relative to other tissues. o, Validation of differential m6A levels using RT-qPCR-based m6A quantification. Two sites identified with in vivo DART-seq as being differentially methylated between the cortex and cerebellum (n = 4 biological replicates) and two sites differentially methylated between the cortex and hippocampus (n = 2 biological replicates) are validated. Data are plotted as the relative methylation level detected in cortex, normalized to either cerebellum or hippocampus from the same animal. Individual data points are shown with lines indicating brain regions from the same animal. Error bars represent standard error. Significance was determined using a one-sided t-test comparing values to 1. *** = p < 0.01.
Extended Data Fig. 5 – Characteristics of gene expression and RNA methylation in the adult mouse cortex.
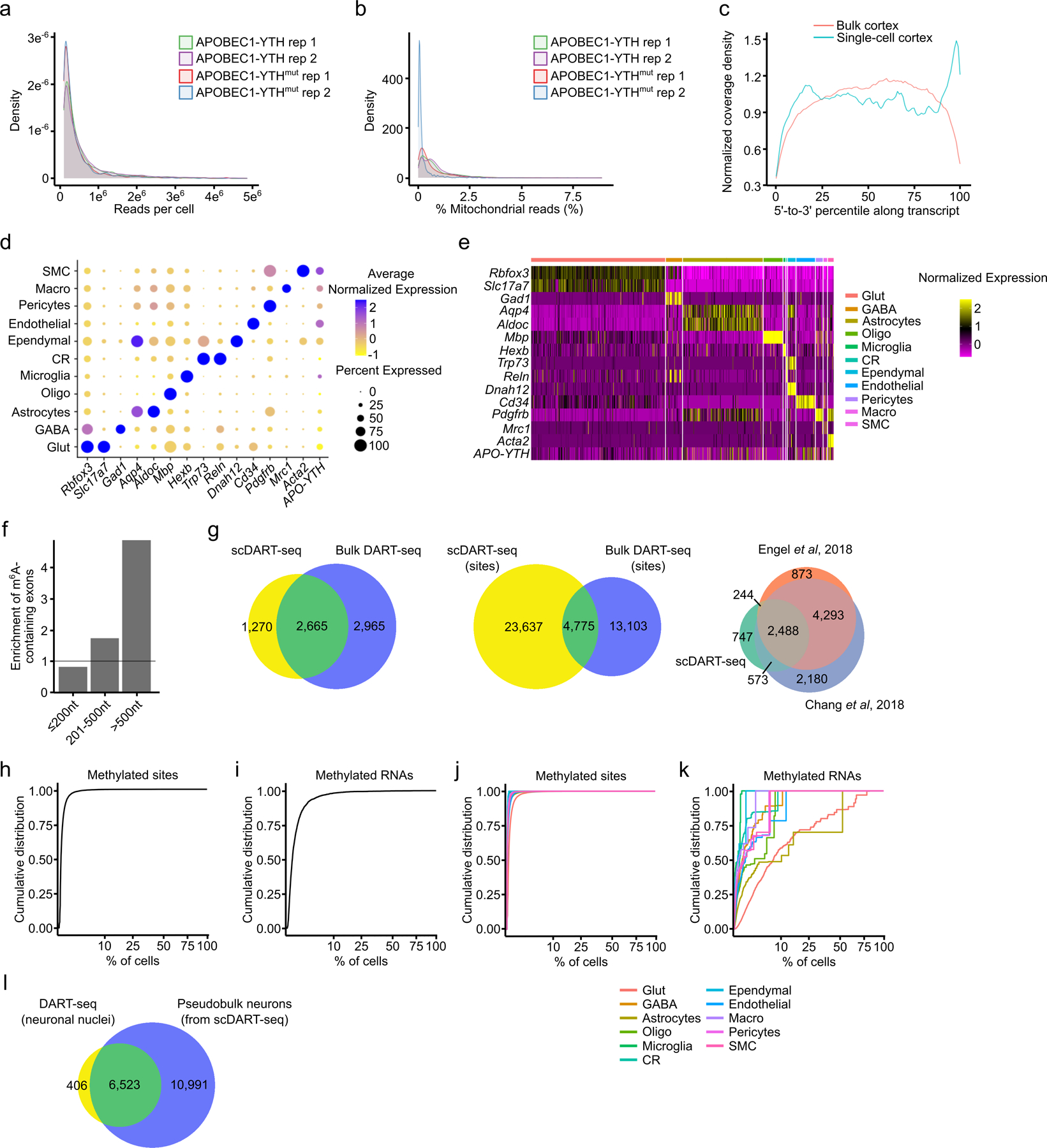
a, Density plot showing the number of total RNA-seq reads per cell from replicate mice with the indicated genotype. b, Density plot showing the percentage of reads mapping to mitochondrial mRNAs in replicate mice with the indicated genotype. The vast majority of cells in all replicates show very low mitochondrial representation (< 2%). c, Metagene analysis of RNA-seq reads from bulk and scDART-seq data using split-pool barcoding. Although there is an enrichment of reads at the 3’ end of transcripts using split-pool barcoding scRNA-seq, good coverage is obtained across transcript length. d, Marker gene expression across individual cell types in the cortex. Dot color indicates average normalized expression in the indicated cell type. Dot size indicates percentage of cells that express the marker gene. e, Heatmap showing marker gene expression within different cell types in the cortex. f, Barplot showing the enrichment of m6A sites in long internal exons from scDART-seq analysis of the mouse cortex. Enrichment indicates the number of methylated internal exons relative to the total number of internal exons within each group. g, Overlap of methylated RNAs identified by scDART-seq and bulk DART-seq in the mouse cortex (left). Overlap of methylated sites identified in the cortex using scDART-seq and bulk DART-seq (middle). Overlap of methylated RNAs identified by scDART-seq and MeRIP-seq in the mouse cortex (right). h, Cumulative distribution plot as in (d), but showing each cell type separately. i, Cumulative distribution plot of all m6A sites identified in the mouse cortex and the percentage of cells in which each site is methylated. Only cells expressing the parent RNA were considered (n = 27,400). j, Cumulative distribution plot of methylated RNAs identified in the mouse cortex and the percentage of cells in which the RNA is methylated. Cells that do not express the RNA were excluded from the analysis (n = 3,991). k, Cumulative distribution plot as in (c), but showing each cell type separately. l, Overlap of methylated RNAs identified by performing nuclear DART-seq on mouse cortical neurons and those identified in neurons from the scDART-seq dataset. Glut = Glutamatergic Neuron; GABA = GABAergic Neuron; Oligo = Oligodendrocyte; Micro = Microglia; CR = Cajal-Retzius Cell; Macro = Peripheral Macrophage; SMC = Smooth Muscle Cell.
Extended Data Fig. 6 – Analysis of m6A in microglia and other cortical cell types.
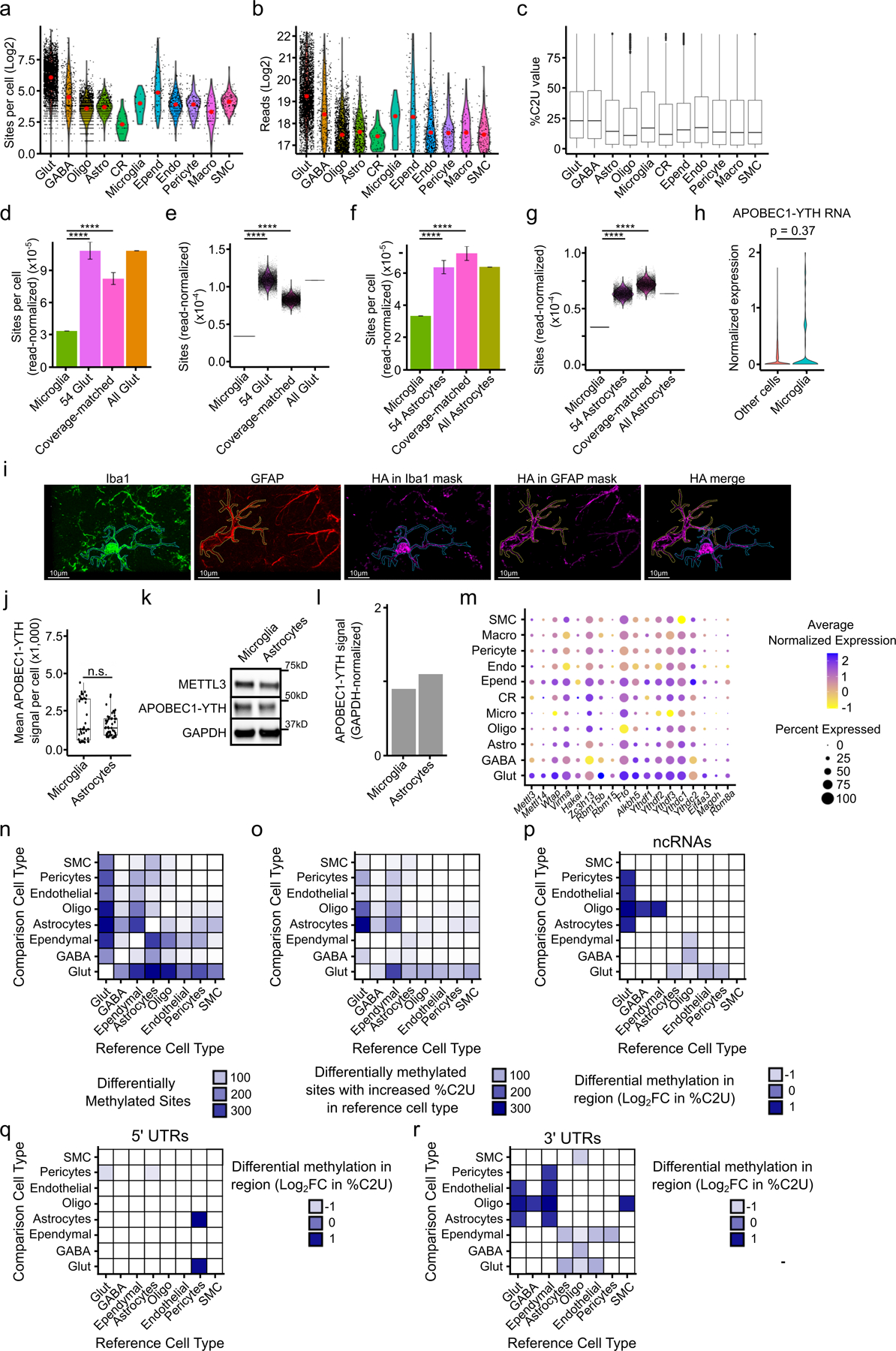
a, Total number of m6A sites per cell within the indicated cell types, not normalized to read counts. Red dot indicates the mean. b, Total number of reads detected per cell within each cell type. Red dot indicates the mean. c, All single-cell %C2U values for m6A sites identified within each cell type (Glut n = 280,790; GABA n = 11,214; Astro n = 19,370; Oligo n = 4,769; Micro n = 133; CR n = 211; Ependy n = 6,775; Endo n = 6,314; Peri n = 1,949; Macro n = 935; SMC n = 2,232). Boxes show 25–75th percentiles and the median, with whiskers for the highest and lowest values and outliers shown. d, Number of m6A sites per cell, normalized to read coverage, identified in microglia (n = 54 cells) and glutamatergic neurons (“All Glut”; n = 3,842 cells). 54 Glut represents 10,000 iterations of 54 randomly selected glutamatergic neurons within the scDART-seq dataset. Coverage-matched represents 10,000 iterations of 54 randomly selected glutamatergic neurons with similar reads per cell as microglia. Error bars represent standard deviation. Significance was determined using a two-sided Wilcoxon rank-sum test. p-value for all shown comparisons < 2.2e−16. **** = p < 0.0001. e, Violin plot of data in (d). Each dot represents the average number of read-normalized sites per cell obtained from 54 random glutamatergic neurons sampled. Significance was determined using a two-sided Wilcoxon rank-sum test. p-value for all shown comparisons < 2.2e−16. **** = p < 0.0001. f, Number of m6A sites per cell, normalized to read coverage, identified in microglia (n = 54 cells) and astrocytes (“All Astrocytes”; n = 1,952 cells). 54 Astrocytes represents 10,000 iterations of 54 random astrocytes within the scDART-seq dataset. Coverage-matched represents 10,000 iterations of 54 random astrocytes with similar reads per cell as microglia. Error bars represent standard deviation. Significance was determined using a two-sided Wilcoxon rank-sum test. p-value for all shown comparisons < 2.2e−16. **** = p < 0.0001. g, Violin plot of data in (f). Each dot represents the average read-normalized sites per cell obtained from each iteration of 54 random astrocytes sampled. h, Normalized expression of APOBEC1-YTH in microglia compared to all other cell types. Significance was determined using FindMarkers in Seurat (two-sided Wilcoxon rank-sum test was used, with Bonferroni p-value adjustment). i, Representative images of n = 2 biological replicates, comparing APOBEC1-YTH expression (magenta) in a microglial cell (marked by Iba1 staining, green) and an astrocyte (marked by GFAP staining, red). Merge shows the neighboring microglia and astrocyte, with external APOBEC1-YTH signal removed for clarity. j, Quantification of APOBEC1-YTH immunofluorescence intensity in microglia (n = 43 cells from 2 distinct animals) and astrocytes (n = 49 cells from 2 distinct animals) in DART mice. Significance was determined using a two-sided Wilcoxon rank-sum test. n.s. = not statistically significant. Boxes show 25–75th percentiles and the median, with whiskers for the highest and lowest values and outliers shown. k, Western blot showing APOBEC1-YTH and METTL3 expression within cortical microglia and astrocytes isolated by FACS. n = 1 biological replicate. GAPDH was run concurrently on a separate identical blot. l, Quantification of (k) showing the GAPDH-normalized expression of APOBEC1-YTH. m, Dot plot showing expression level and percentage of cells with expression of mRNAs for m6A methyltransferase components, m6A readers, m6A erasers, and EJC proteins. n, Heatmap showing the total number of differentially methylated sites identified between each pair of cell types in the cortex. Heatmap is colored by the number of differentially methylated sites identified in the reference cell type relative to the comparison cell type. o, Heatmap showing the number of differentially methylated sites with increased average %C2U in the reference cell type relative to the comparison cell type, colored by the number of differentially methylated sites. p, Heatmap showing the average log2 fold-change in %C2U for all sites found in non-coding RNAs (ncRNAs) within each region in the reference cell type relative to the comparison cell type. q, Heatmap showing the average log2 fold-change in %C2U for all sites found in 5’ UTRs within each region in the reference cell type relative to the comparison cell type. r, Heatmap showing the average log2 fold-change in %C2U for all sites found in 3’ UTRs in the reference cell type relative to the comparison cell type. There are no pairs of cell types with significant differential methylation within coding sequences.
Extended Data Fig. 7 – Differential methylation between neurons and astrocytes in the mouse cortex.
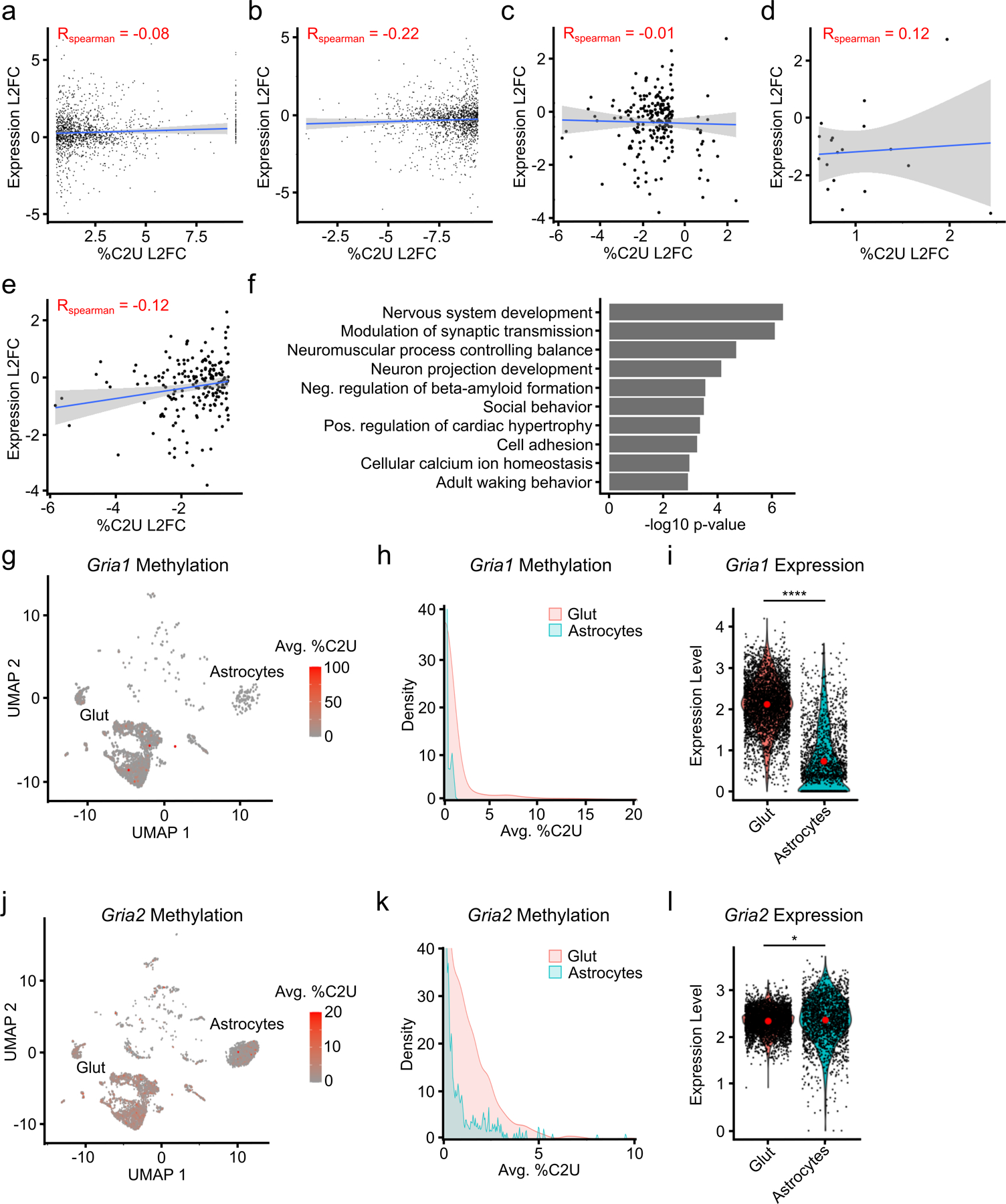
a, Correlation between changes in methylation and gene expression for all DM-RNAs with increased %C2U identified across cell types. Blue line indicates linear regression, gray band represents 95% confidence interval. Spearman correlation coefficient is shown. p = 0.0013. n = 1,436. b, As in (a), except only DM-RNAs with decreased %C2U. Blue line indicates linear regression, gray band represents 95% confidence interval. Spearman correlation coefficient is shown. p < 2.2 x 10−16. n = 1,436. c, Comparison of log2-fold change in gene expression and %C2U values for DM-RNAs identified between glutamatergic neurons and astrocytes. Blue line indicates linear regression, gray band represents 95% confidence interval. Spearman correlation coefficient is shown. p = 0.77. n = 214. d, As in (c), except only DM-RNAs with increased %C2U. Blue line indicates linear regression, gray band represents 95% confidence interval. Spearman correlation coefficient is shown. p = 0.6325. n = 18. e, As in (c), except only DM-RNAs with decreased %C2U. Blue line indicates linear regression, gray band represents 95% confidence interval. Spearman correlation coefficient is shown. p = 0.08654. n = 196. f, Top 10 most significant Gene Ontology terms enriched among DM-RNAs identified between glutamatergic neurons and astrocytes. g, UMAP visualization of glutamatergic neurons and astrocytes colored by the mean %C2U value across all m6A sites identified in the Gria1 mRNA (n = 2,506). h, Density plot showing the distribution of the average %C2U value for all sites in Gria1 in single cells in glutamatergic neurons and astrocytes. i, Violin plot showing the normalized expression of Gria1 in glutamatergic neurons and astrocytes. Red dot indicates the mean. Significance was determined using FindMarkers in Seurat (two-sided Wilcoxon rank-sum test was used, with Bonferroni p-value adjustment). n = 6,162. p < 2.2e−308. **** = p < 0.0001. j, UMAP visualization of glutamatergic neurons and astrocytes colored by the mean %C2U value across all m6A sites identified in the Gria2 mRNA (n = 4,818). k, Density plot showing the distribution of the average %C2U value for all sites in Gria2 in single cells in glutamatergic neurons and astrocytes. l, Violin plot showing the normalized expression of Gria2 in glutamatergic neurons and astrocytes. Red dot indicates the mean. Significance was determined using FindMarkers in Seurat (two-sided Wilcoxon rank-sum test was used, with Bonferroni p-value adjustment). n = 6,162. p = 0.01. * = p < 0.05. Glut = Glutamatergic neurons.
Extended Data Fig. 8 – Differential methylation in neuronal subtypes.
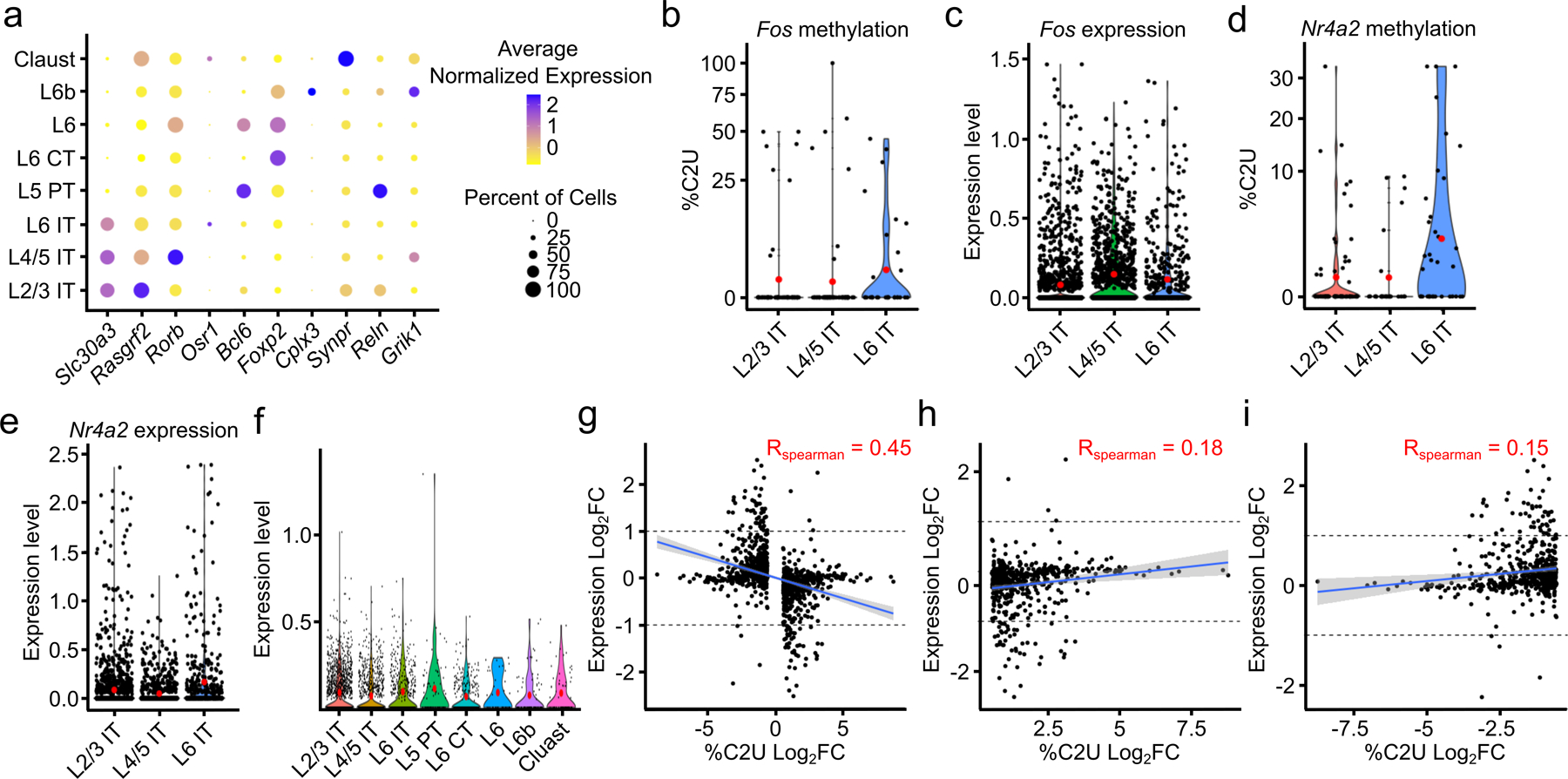
a, Expression of marker genes within each glutamatergic neuron subtype. Dot color indicates average expression level of the indicated mRNA. Dot size indicates percentage of cells in which the mRNA is expressed. b, Average %C2U value of all sites in the Fos mRNA in IT glutamatergic neurons from different cortical layers. Red dots indicate mean values. n = 320. c, Normalized gene expression of the Fos mRNA in IT glutamatergic neurons from different cortical layers. n = 2,930. d, Average %C2U value of all sites in the Nr4a2 mRNA in IT glutamatergic neurons from different cortical layers. Red dots indicate mean values. n = 360. e, Normalized gene expression of the Nr4a2 mRNA in IT glutamatergic neurons from different cortical layers. Red dots indicate mean values. n = 2,930. f, APOBEC1-YTH RNA expression within each neuronal subtype. Red dots indicate mean values. n = 3,823. g, Correlation of changes in %C2U and gene expression of DM-RNAs between neuronal subtypes. Spearman correlation coefficient is shown, blue indicates linear regression, gray band represents 95% confidence interval. p < 2.2 x 10−16. n = 1,034. h, As in (g), but only showing DM-RNAs with increased %C2U values in each neuronal subtype. p = 2.65 x 10−5. n = 512. Spearman correlation coefficient is shown, blue indicates linear regression, gray band represents 95% confidence interval. i, As in (g), but only showing DM-RNAs with decreased %C2U values in each neuronal subtype. p = 3.9 x 10−4. n = 522. Spearman correlation coefficient is shown, blue indicates linear regression, gray band represents 95% confidence interval. L2/3 = Layer 2/3; L4/5 = Layer 4/5; L6 = Layer 6; IT = Intratelencephalic; PT = Pyrimidal tract; CT = Corticothalamic tract; Claust = Claustrum.
Extended Data Fig. 9 – Clustering by m6A reveals heterogeneous methylation in glutamatergic neurons.
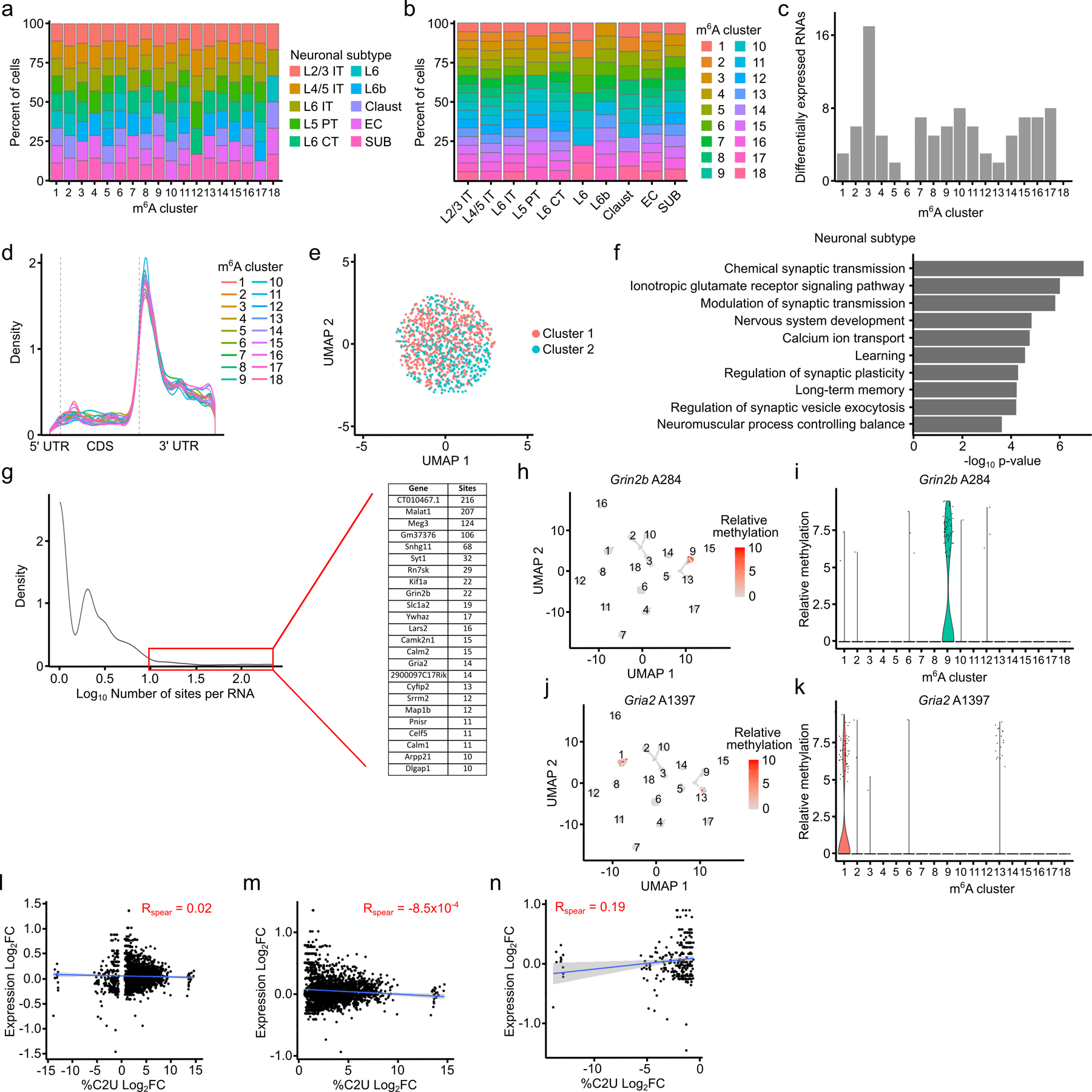
a, Stacked bar plot showing the percentage of cells within each m6A cluster that belong to the corresponding neuronal subtype. b, Stacked bar plot showing the percentage of cells within each neuronal subtype belonging to different m6A clusters. c, Number of differentially expressed RNAs within each m6A cluster. d, Metagene plot showing distribution of m6A sites in each m6A cluster. e, UMAP visualization of m6A clustering using shuffled methylation data. %C2U values for all m6A sites were randomly shuffled across all cells. f, Top 10 Gene Ontology terms among all RNAs containing differentially methylated sites across m6A clusters. g, Density plot showing the distribution of the number of differentially methylated sites identified within each parent RNA. Highlighted region and inset indicate RNAs with at least 10 differentially methylated sites. h, UMAP visualization of relative %C2U values for site A284, the most significantly differentially methylated site in Grin2b, across m6A clusters. n = 2,246. i, Violin plot showing relative %C2U values for each cell within each m6A cluster for site A284 in Grin2b. j, UMAP visualization of relative %C2U values for site A1397, the most significantly differentially methylated site in Gria2, across m6A clusters. n = 2,246. k, Violin plot showing relative %C2U values for each cell within each m6A cluster for site A1397 in Gria2. l, Correlation between the log2-fold change in parent gene expression and the log2-fold change in %C2U for all differentially methylated sites found across m6A clusters. Blue line shows linear regression, gray band represents 95% confidence interval. Spearman correlation coefficient is shown. p = 0.40. n = 2,794. m, As in (l), but only showing sites with increased %C2U within each methylation cluster. p = 0.96. n = 2,533. Blue line shows linear regression, gray band represents 95% confidence interval. n, As in (l), but only showing sites with decreased %C2U within each methylation cluster. p = 0.002. n = 261. Blue line shows linear regression, gray band represents 95% confidence interval.
Extended Data Fig. 10 – Differential methylation patterns in glutamatergic neurons during aging.
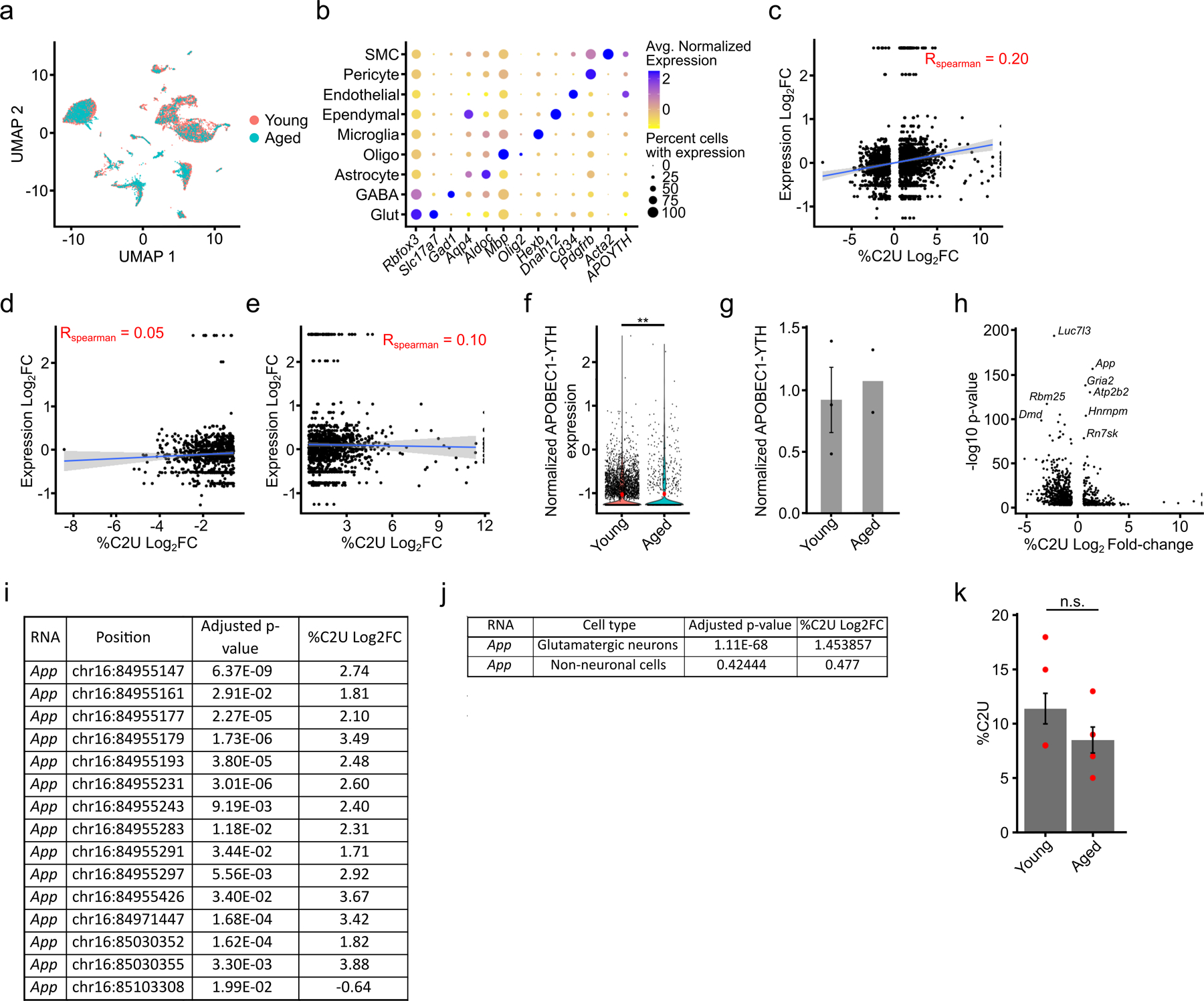
a, UMAP visualization showing that cortical glutamatergic neurons from young and aged mice cluster together based on marker gene expression. b, Dot plot showing marker gene expression in the indicated cell types for the integrated scDART-seq datasets from young and aged mice. c, Comparison of log2-fold change of gene expression and average %C2U values of DM-RNAs identified in glutamatergic neurons from young versus aged mice. Blue line indicates linear regression gray band represents 95% confidence interval. Spearman correlation coefficient is shown. p < 2.2 x 10−16. n = 2,055. d, As in (c), but only showing DM-RNAs with increased %C2U changes in neurons from aged mice. Spearman correlation coefficient is shown. Blue line indicates linear regression gray band represents 95% confidence interval. p = 0.149. n = 852. e, As in (c), but only showing DM-RNAs with increased %C2U changes in neurons from young mice. Spearman correlation coefficient is shown. Blue line indicates linear regression gray band represents 95% confidence interval. p = 0.000725. f, Normalized expression of APOBEC1-YTH RNA in glutamatergic neurons from young and aged mice. Significance was determined using FindMarkers in Seurat (two-sided Wilcoxon rank-sum test was used, with Bonferroni p-value adjustment). p = 0.003. ** = p <0.01. Red dots indicate mean values. Average log2 fold-change = 0.22. n = 5,401. g, Western blot densitometry quantification of APOBEC1-YTH protein level in young (n = 3 biological replicates) and aged (n = 2 biological replicates) DART mouse cortex, normalized to GAPDH. Error bars represent standard error. Mean for young cortex = 0.87. Mean for aged cortex = 1.29. h, Plot showing the −log10 of the adjusted p-value and %C2U log2-fold change from differential methylation analysis. The second most significant differentially methylated RNA is App. i, Adjusted p-values and %C2U log2 fold-changes for all differentially methylated sites in the App mRNA. j, Comparison of the adjusted p-value and %C2U log2-fold changes for differential methylation of App in glutamatergic neurons and non-neuronal cells between young and aged mice. k, In vitro DART-seq results showing the %C2U values adjacent to App A2599 in bulk cortex from young (n = 5) and aged (n = 4) mice. Significance was determined using a two-sided Wilcoxon rank-sum test with no p-value adjustment. p = 0.38. n.s. = not significant. Error bars represent standard error. Mean for young cortex = 11.4. Mean for aged cortex = 8.5.
Supplementary Material
Acknowledgments
We thank all members of the Meyer laboratory for helpful discussions. We are grateful to the Duke Center for Genomic and Computational Biology, the Duke Human Vaccine Institute Flow Cytometry Core, the Duke Light Microscopy Core Facility, and the Duke Proteomics and Metabolomics Shared Resource for providing the infrastructure and support for sequencing, sorting of single cells, confocal imaging, UPLC-MS/MS, and proteomics mass spectrometry. This work was supported by the Rita Allen Foundation Scholars Program to K.D.M., the Kinship Foundation Searle Scholars Program to K.D.M., and the National Institutes of Health (R01MH118366, DP1DA046584, and RM1HG011563 to K.D.M.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Code availability
All code for downloading, installing, and using Bullseye is available at https://github.com/mflamand/Bullseye. Other R code is available upon request. A searchable list of m6A sites is available at http://dartsource.duke.edu.
Availability of materials
All reasonable requests for materials generated during this study will be fulfilled upon contacting the corresponding author.
Competing Interests
K.D.M. has filed a patent application for the DART-seq technology through Duke University. All other authors declare no competing interests.
Data availability
All sequencing data generated by this study has been deposited to the Gene Expression Omnibus under Bioproject PRJNA996152. (Accession numbers: GSE237871 (bulk brain regions), GSE268159 (aged mouse scRNA-seq), GSE266876 (young cortex scRNA-seq), GSE267234 (sorted neuronal nuclei), GSE267627 (GLORI)). Proteomics data is deposited to the MassIVE Database (Accession: MSV000094375). Due to size limitations, images in the study are available upon request to the corresponding author.
References
- 1.Shi H, Wei J & He C Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 74, 640–650 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zaccara S, Ries RJ & Jaffrey SR Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol 20, 608–624 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Meyer KD et al. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 149, 1635–1646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y et al. N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci 21, 195–206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon K-J et al. Temporal Control of Mammalian Cortical Neurogenesis by m6A Methylation. Cell 171, 877–889.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu H et al. m6A mRNA Methylation Is Essential for Oligodendrocyte Maturation and CNS Myelination. Neuron 105, 293–309.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koranda JL et al. Mettl14 Is Essential for Epitranscriptomic Regulation of Striatal Function and Learning. Neuron 99, 283–292.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engel M et al. The Role of m6A/m-RNA Methylation in Stress Response Regulation. Neuron 99, 389–403.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi H et al. m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Widagdo J et al. Experience-Dependent Accumulation of N6-Methyladenosine in the Prefrontal Cortex Is Associated with Memory Processes in Mice. J. Neurosci. Off. J. Soc. Neurosci 36, 6771–6777 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hess ME et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat. Neurosci 16, 1042–1048 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Yang C et al. The role of m6A modification in physiology and disease. Cell Death Dis 11, 1–16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang L et al. Interaction of tau with HNRNPA2B1 and N6-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell 81, 4209–4227.e12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han M et al. Abnormality of m6A mRNA Methylation Is Involved in Alzheimer’s Disease. Front. Neurosci 14, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flamand MN & Meyer KD The epitranscriptome and synaptic plasticity. Curr. Opin. Neurobiol 59, 41–48 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flamand MN & Meyer KD m6A and YTHDF proteins contribute to the localization of select neuronal mRNAs. Nucleic Acids Res 50, 4464–4483 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weng Y-L et al. Epitranscriptomic m6A Regulation of Axon Regeneration in the Adult Mammalian Nervous System. Neuron 97, 313–325.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madugalle SU, Meyer K, Wang DO & Bredy TW RNA N6-Methyladenosine and the Regulation of RNA Localization and Function in the Brain. Trends Neurosci 43, 1011–1023 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livneh I, Moshitch-Moshkovitz S, Amariglio N, Rechavi G & Dominissini D The m6A epitranscriptome: transcriptome plasticity in brain development and function. Nat. Rev. Neurosci 21, 36–51 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Meyer KD DART-seq: an antibody-free method for global m6A detection. Nat. Methods 1275–1280 (2019) doi: 10.1038/s41592-019-0570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tegowski M, Flamand MN & Meyer KD scDART-seq reveals distinct m6A signatures and mRNA methylation heterogeneity in single cells. Mol. Cell 82, 868–878 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salpietro V et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun 10, 3094 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carayol J et al. Converging Evidence for an Association of ATP2B2 Allelic Variants with Autism in Male Subjects. Biol. Psychiatry 70, 880–887 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Yang W et al. The Evidence for Association of ATP2B2 Polymorphisms with Autism in Chinese Han Population. PLOS ONE 8, e61021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Brien RJ & Wong PC Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci 34, 185–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shinohara M, Tachibana M, Kanekiyo T & Bu G Role of LRP1 in the pathogenesis of Alzheimer’s disease: evidence from clinical and preclinical studies: Thematic Review Series: ApoE and Lipid Homeostasis in Alzheimer’s Disease. J. Lipid Res 58, 1267–1281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castellanos-Rubio A et al. A novel RT-QPCR-based assay for the relative quantification of residue specific m6A RNA methylation. Sci. Rep. 2019 91 9, 1–7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu J et al. Landscape and Regulation of m6A and m6Am Methylome across Human and Mouse Tissues. Mol. Cell 77, 426–440.e6 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Chang M et al. Region-specific RNA m6A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang H, Camats-Perna J, Medeiros R, Anggono V & Widagdo J Altered Expression of the m6A Methyltransferase METTL3 in Alzheimer’s Disease. eNeuro 7, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz S et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep 8, 284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uzonyi A et al. Exclusion of m6A from splice-site proximal regions by the exon junction complex dictates m6A topologies and mRNA stability. Mol. Cell 83, 237–251.e7 (2023). [DOI] [PubMed] [Google Scholar]
- 33.McIntyre ABR et al. Limits in the detection of m6A changes using MeRIP/m6A-seq. Sci. Rep 10, 6590 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grozhik AV & Jaffrey SR Distinguishing RNA modifications from noise in epitranscriptome maps. Nat. Chem. Biol 14, 215–225 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Choi SH et al. RBM45 is an m6A-binding protein that affects neuronal differentiation and the splicing of a subset of mRNAs. Cell Rep 40, 111293 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cuevas-Diaz Duran R, González-Orozco JC, Velasco I & Wu JQ Single-cell and single-nuclei RNA sequencing as powerful tools to decipher cellular heterogeneity and dysregulation in neurodegenerative diseases. Front. Cell Dev. Biol 10, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu C et al. Absolute quantification of single-base m6A methylation in the mammalian transcriptome using GLORI. Nat. Biotechnol 41, 355–366 (2023). [DOI] [PubMed] [Google Scholar]
- 38.Zeisel A et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Polioudakis D et al. A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 103, 785–801.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stuart T, Srivastava A, Madad S, Lareau CA & Satija R Single-cell chromatin state analysis with Signac. Nat. Methods 2021 1811 18, 1333–1341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Merkurjev D et al. Synaptic N6-methyladenosine (m6A) epitranscriptome reveals functional partitioning of localized transcripts. Nat. Neurosci 21, 1004–1014 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Flamand MN, Ke K, Tamming R & Meyer KD Single-molecule identification of the target RNAs of different RNA binding proteins simultaneously in cells. Genes Dev (2022) doi: 10.1101/gad.349983.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zou Z et al. FMRP phosphorylation modulates neuronal translation through YTHDF1. 2022.11.29.518448 Preprint at 10.1101/2022.11.29.518448 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McMillan M et al. RNA methylation influences TDP43 binding and disease pathogenesis in models of amyotrophic lateral sclerosis and frontotemporal dementia. Mol. Cell 83, 219–236.e7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shafik AM et al. N6-methyladenosine dynamics in neurodevelopment and aging, and its potential role in Alzheimer’s disease. Genome Biol 22, 17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Izzy S et al. Time-Dependent Changes in Microglia Transcriptional Networks Following Traumatic Brain Injury. Front. Cell. Neurosci 13, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolf SA, Boddeke HWGM & Kettenmann H Microglia in Physiology and Disease. Annu. Rev. Physiol 79, 619–643 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Yeh H & Ikezu T Transcriptional and epigenetic regulation of microglia in health and disease. Trends Mol. Med 25, 96–111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Q, Wen S, Ye W, Zhao S & Liu X The potential roles of m6A modification in regulating the inflammatory response in microglia. J. Neuroinflammation 18, 149 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wen L et al. The m6A methyltransferase METTL3 promotes LPS-induced microglia inflammation through TRAF6/NF-κB pathway. NeuroReport 33, 243 (2022). [DOI] [PubMed] [Google Scholar]
- 51.Yang X, Triboulet R, Liu Q, Sendinc E & Gregory RI Exon junction complex shapes the m6A epitranscriptome. Nat. Commun 13, 7904 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garcia-Campos MA et al. Deciphering the “m6A Code” via Antibody-Independent Quantitative Profiling. Cell 178, 731–747.e16 (2019). [DOI] [PubMed] [Google Scholar]
- 53.He PC et al. Exon architecture controls mRNA m6A suppression and gene expression. Science 379, 677–682 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keller L et al. The Obesity Related Gene, FTO, Interacts with APOE, and is Associated with Alzheimer’s Disease Risk: A Prospective Cohort Study. J. Alzheimers Dis 23, 461–469 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Castro-Hernández R et al. Conserved reduction of m6A RNA modifications during aging and neurodegeneration is linked to changes in synaptic transcripts. Proc. Natl. Acad. Sci 120, e2204933120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao F et al. METTL3-dependent RNA m6A dysregulation contributes to neurodegeneration in Alzheimer’s disease through aberrant cell cycle events. Mol. Neurodegener 16, 70 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H et al. FTO is involved in Alzheimer’s disease by targeting TSC1-mTOR-Tau signaling. Biochem. Biophys. Res. Commun 498, 234–239 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Huang H, Song R, Wong JJ-L, Anggono V & Widagdo J The N6-methyladenosine RNA landscape in the aged mouse hippocampus. Aging Cell 22, e13755 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu H, Yin X, Holley CL & Meyer KD Improved Methods for Deamination-Based m6A Detection. Front. Cell Dev. Biol 10, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kulkarni A, Anderson AG, Merullo DP & Konopka G Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol 58, 129–136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prakadan SM, Shalek AK & Weitz DA Scaling by shrinking: empowering single-cell ‘omics’ with microfluidic devices. Nat. Rev. Genet 18, 345–361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kluesner MG et al. EditR: A Method to Quantify Base Editing from Sanger Sequencing. CRISPR J 1, 239–250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roehr JT, Dieterich C & Reinert K Flexbar 3.0 – SIMD and multicore parallelization. Bioinformatics 33, 2941–2942 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao Y, Smyth GK & Shi W The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Olarerin-George AO & Jaffrey SR MetaPlotR: A Perl/R pipeline for plotting metagenes of nucleotide modifications and other transcriptomic sites. Bioinformatics 33, 1563–1564 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang L, Wang S & Li W RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012). [DOI] [PubMed] [Google Scholar]
- 70.Sherman BT et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res 50, W216–W221 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang DW, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGinnis CS, Murrow LM & Gartner ZJ DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tasic B et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yao Z et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184, 3222–3241.e26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stuart T, Srivastava A, Madad S, Lareau CA & Satija R Single-cell chromatin state analysis with Signac. Nat. Methods 2021 1811 18, 1333–1341 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated by this study has been deposited to the Gene Expression Omnibus under Bioproject PRJNA996152. (Accession numbers: GSE237871 (bulk brain regions), GSE268159 (aged mouse scRNA-seq), GSE266876 (young cortex scRNA-seq), GSE267234 (sorted neuronal nuclei), GSE267627 (GLORI)). Proteomics data is deposited to the MassIVE Database (Accession: MSV000094375). Due to size limitations, images in the study are available upon request to the corresponding author.