

Abstract
SPINT1, a membrane-anchored serine protease inhibitor, regulates cascades of pericellular proteolysis while its tissue-specific functions remain incompletely characterized. In this study, we generate Spint1-lacZ knock-in mice and observe Spint1 expression in embryonic pancreatic epithelium. Pancreas-specific Spint1 disruption significantly diminishes islet size and mass, causing glucose intolerance and downregulation of MAFA and insulin. Mechanistically, the serine protease HEPSIN interacts with SPINT1 in β cells, and Hepsin silencing counteracts the downregulation of Mafa and Ins1 caused by Spint1 depletion. Furthermore, we demonstrate a potential interaction between HEPSIN and GLP1R in β cells. Spint1 silencing or Hepsin overexpression reduces GLP1R-related cyclic AMP levels and Mafa expression. Spint1-disrupted mice also exhibit a significant reduction in Exendin-4-induced insulin secretion. Moreover, SPINT1 expression increases in islets of prediabetic humans compared to non-prediabetic groups. The results unveil a role for SPINT1 in β cells, modulating glucose homeostasis and insulin production via HEPSIN/MAFA signaling.
Subject terms: Proteases, Type 2 diabetes, Extracellular signalling molecules
Pancreatic Spint1 disruption causes glucose intolerance, reduced islet size, and decreased insulin synthesis and secretion. SPINT1 regulates HEPSIN’s modification of GLP1R/MAFA activation, vital for glucose control by β cells, linking it to diabetes.
Introduction
Serine Protease Inhibitor, Kunitz Type 1 (SPINT1) acts as a type I transmembrane serine protease inhibitor, inhibiting proteolytic activity through its Kunitz domain 11. SPINT1 has been reported to play a crucial role in forming and maintaining epithelial integrity in specific organs1. However, more research is needed to explore the function of SPINT1 in non-epithelium cells. Our previous studies have demonstrated the upregulation of human SPINT1 in the ductular reactions of the liver with cholangiopathy, impeding the differentiation of hepatic progenitors and enhancing liver fibrosis2. Despite these insights, the roles in physiological homeostasis of SPINT1 and their downstream effectors in other organs/tissues during development remain largely unexplored.
Membrane-anchored serine proteases (MASPs) have been identified as a target protein for SPINT13. MASPs are crucial in several homeostasis, development, and cancer progression3. Well-studied enzymes within the MASPs include HEPSIN, matriptase (MTX), and transmembrane serine protease 2 (TMPRSS2). These proteases are known to proteolytically activate their substrates in the pericellular milieu, encompassing G protein-coupled receptors (GPCRs), adhesion molecules, ion channels, proteases, and so on3.
GPCRs exhibit widespread distribution across diverse tissues and play a vital role in physiological activities by sensing the extracellular environment4,5. Glucagon-like peptide 1 receptor (GLP1R, one of the GPCRs) has been elucidated to exert an essential part in insulin secretion and production. Physiologically, GLP1R in pancreatic β cells, after being activated by glucagon-like peptide 1 (GLP1), stimulates insulin production4,5. Several GLP1R agonists have been developed and clinically applied in diabetes treatment6,7. It is intriguing to determine whether there is a pericellular or membrane-associated proteolytic modification of GLP1R, similar to the protease-activated receptor (PAR) family8. This subfamily relies on the proteolytic function of various serine proteases, such as MASPs, to activate their function. Although proteolytic processing is a significant mechanism for regulating GLP1 activity9, it remains unclear whether there is a proteolytic modification of GLP1R, akin to PARs, that regulates the signal transduction or insulin production in pancreatic β cells.
The transcription of the insulin-encoding genes is predominantly regulated by several primary transcription factors10,11, including pancreatic and duodenal homeobox 1 (PDX1)10, neuronal differentiation 1 (NEUROD1)11, and MAF bZIP transcription factor A (MAFA)10, which are responsible for insulin production through the transcription of insulin-encoding genes10. Hierarchically, these transcription factors respond to upstream signaling pathways that mediate environmental cues12. For example, GLP1 or GLP1-like drugs have been shown to stimulate the expression of Mafa in β cells13,14.
In this study, we generated Spint1-lacZ transgenic mice and observed heightened Spint1 expression in the fetal pancreas as early as embryonic day 12.5 (E12.5). Deficiency of Spint1 diminished pancreatic β cell proliferation and insulin expression, leading to impaired glucose tolerance. Furthermore, we identified HEPSIN as the primary SPINT1-targeted protease, which can downregulate the expression of MAFA and insulin in pancreatic β cells through the proteolytic processing of GLP1R. These findings indicate that SPINT1 is crucial in maintaining glucose homeostasis by positively regulating insulin production in pancreatic β cells via the HEPSIN/GLP1R/MAFA signaling pathway.
Results
Spint1 was highly expressed in the embryonic pancreatic epithelia
To investigate the physiological function of SPINT1, we initiated our study by examining the spatiotemporal expression of Spint1 in the mouse embryonic stage. We created Spint1lacZ/+ transgenic mice by inserting a Spint1-lacZ-neo reporter cassette into the first intron of the Spint1 genomic locus (Supplementary Fig. 1a). Southern blotting and PCR genotyping analysis validated the correctly targeted clones (Supplementary Fig. 1b). To trace the spatiotemporal expression of Spint1 in the embryonic stage, the E10.5, E12.5, and E14.5 embryos of Spint1lacZ/+ mice were collected and stained with X-gal. The results showed that Spint1-lacZ was expressed on the surface of the nose, mouth, limbs, abdomen, and genital area in the E10.5, E12.5, and E14.5 embryos (Supplementary Fig. 1c). Notably, serial histological sections of the embryo revealed that the primordial pancreas exhibited the earliest expression of Spint1-lacZ, beginning at E12.5 in Spint1lacZ/+ embryos (Fig. 1a, left panel and Supplementary Fig. 1d). The intestine, stomach, lung, and liver showed minimal lacZ signals at this stage (Supplementary Figs. 1d, 2a, b). Remarkably, by E14.5, the entire primordial pancreatic duct in Spint1lacZ/+ embryos exhibited the highest level of Spint1, including the acinar-like structures in the ductal tip region, which would subsequently become pancreatic endocrine cells (Fig. 1a, right panel and Supplementary Fig. 2c). Thus, the results implied that SPINT1 may play a role in the endocrine pancreas.
Fig. 1. Expression patterns of Spint1-lacZ in Spint1lacZ/+ mouse embryonic pancreas and the islet phenotypes resulting from pancreas-specific Spint1-deficiency in 8-week-old mice.

a Representative LacZ-stained sections showed distinctive Spint1-lacZ expression patterns in the pancreas of E12.5 and E14.5 Spint1lacZ/+ embryos. Notably, prominent LacZ signals were observed in the E14.5 pancreas, encompassing the primordial pancreatic duct, its branches, and the acini-like structures budding from the ductal tip region. The bottom panel illustrated images of Spint1+/+ counterparts. Scale bar, 100 μm for 200× panels and 20 μm for 400× panels. b Validation of tissue-specific disruption of Spint1 in the mouse pancreatic islets using Q-RT-PCR. RNAs were extracted from the 8-week-old Spint1fl/fl and Spint1−/− mouse pancreatic islets (left panel) and hypothalamus (HTH., right panel) and subjected to reverse transcription and Q-RT-PCR with normalization to Gapdh. Data were obtained from three independent experiments (three mice per group). c Immunohistochemical analysis of SPINT1 in the pancreatic islets of 8-week-old Spint1fl/fl and Spint1−/− mice. Scale bar, 20 μm. d Quantification of islet area percentage in 8-week-old Spint1fl/fl and Spint1−/− mice. Each pancreas was serially sectioned (300 sections per pancreas, 5 μm per section), and one out of every 60 serial sections (300 μm intervals) was taken for H&E staining to reveal islet areas. ImageJ determined the percentage of islet area in a whole pancreas area based on the merged full-view microscopic images of 6 sections per mouse (four mice per group). e Analysis of islet mass in 8-week-old Spint1fl/fl and Spint1−/− mice. The islet mass was calculated by multiplying the islet area percentage in (d) by pancreas weight in Supplementary Fig. 3g (n = 4 per group). f Measurement of islet numbers in the pancreas of 8-week-old Spint1fl/fl and Spint1−/− mice. The islets with a diameter below 100 μm were defined as small islets, those with a diameter between 100 and 200 μm as medium islets, and those with a diameter above 200 μm as large islets. The islet numbers with different diameters were measured using microscopic images from H&E-stained sections using ImageJ (one out of every 300 μm interval from serial sections of each pancreas, 6 sections per mouse, 5 mice per group). g Quantification of islets sizes in 8-week-old Spint1fl/fl and Spint1−/− mice. Pancreatic islets were isolated after collagenase perfusion (details in the Methods section) and stained with dithizone solution (7.5 mg/mL). The islet images (left panel, 4 low-power fields per pancreas) were taken under a dissecting microscope and subjected to size measurement (average islet area in pixels per islet) using ImageJ (right panel, n = 8 per group). Statistical significance was assessed by a two-tailed Student’s t-test for all quantifications. For the bar plot, bars are represented as mean ± SEM. In the box plots, the boxes span from the 25th to the 75th percentiles, with a line indicating the median. Whiskers extend to values within 1.5 times the interquartile range, defined as the difference between the 25th and 75th percentiles. Scale bar, 50 μm. *P < 0.05; **P < 0.01; ****P < 0.0001. Below the asterisks are the precise statistical results. Source data are provided as a Source Data file.
Pancreas-specific Spint1 deficiency caused the reduction of serum insulin levels, islet mass, and the proliferation of β cells
To assess SPINT1’s impact on the endocrine pancreas, we generate pancreas-specific Spint1-deficient mice (Spint1−/−) from Spint1lacZ/+ mice (Supplementary Fig. 3a). Genomic PCR determined control (Spint1fl/fl) and Spint1−/− mouse genotypes (Supplementary Fig. 3b). Pancreas and livers from 1-week-old mice were subjected to quantitative real-time polymerase chain reaction (Q-RT-PCR) analysis to validate the pancreas-specific deficiency of Spint1. The results showed that Spint1 mRNA was explicitly depleted in the Spint1−/− pancreas compared with the Spint1fl/fl mice, while its expression in the liver showed no significant difference between the two groups (Supplementary Fig. 3c). Moreover, Spint1 deficiency did not affect the neonatal mouse body weights (Supplementary Fig. 3d). Remarkably, the measurement of the serum insulin level in 1-week-old Spint1fl/fl and Spint1−/− mice showed that Spint1 deficiency significantly decreased the mouse serum insulin levels (Supplementary Fig. 3e). To characterize further the phenotypes of Spint1 deficiency in the endocrine pancreas, we analyzed the morphology and function of pancreatic islets in 8-week-old adult mice. Q-RT-PCR verified that Spint1 mRNA levels were significantly depleted in the pancreatic islets but not in the hypothalamus (HTH.) (Fig. 1b) of the Spint1−/− mice compared to Spint1fl/fl mice. The hypothalamus was included because it has been found to express Pdx1 and can sense and regulate the glucose level15. Immunohistochemistry (IHC) also showed that the SPINT1 protein level was dramatically decreased in Spint1−/− pancreatic islets compared to Spint1fl/fl islets (Fig. 1c).
Our study further conducted a morphometric analysis to quantify the area, mass, size, and number of 8-week-old mouse islets. The results unveiled a significant decrease in the islet area and mass in the Spint1−/− mice (Fig. 1d, e) compared to Spint1fl/fl mice, while their body weights and pancreas weights showed no significant difference from those of the Spint1fl/fl mice (Supplementary Fig. 3f, g). Due to variations in islet size, we quantified and categorized pancreatic islets based on their diameters (>200 μm, 100–200 μm, and <100 μm) within whole pancreas sections at 300 μm intervals. The results indicated that Spint1 deficiency significantly reduced the number of islets with a diameter above 200 μm. In comparison, it had no noticeable effect on the number of islets with a diameter below 100 μm or between 100 and 200 μm (Fig. 1f). To avoid the omission of small islets in our calculation, we also utilized a series of whole pancreas sections at 100 μm intervals to evaluate islet size, mass, and number. Similarly, islets in Spint1−/− mice exhibited smaller area, mass, and number (in the category with diameters above 200 μm) than those in Spint1fl/fl mice (Supplementary Fig. 3h–j). Concordantly, both the number and size of purified, dithizone-stained Spint1−/− pancreatic islets were significantly reduced compared to those of Spint1fl/fl mice (Fig. 1g). However, we found that Spint1 deficiency had only a minor effect on the size and number of mouse islets at an early age (1-week-old, Supplementary Fig. 3k).
Immunostaining microscopy revealed that SPINT1 was expressed in both α and β cells (Supplementary Fig. 3l). However, the β cell area and mass of Spint1−/− mice were significantly decreased compared to that of the Spint1fl/fl mice [Fig. 2a, b (calculated by 300-μm intervals), Supplementary Fig. 3m, n (calculated by 100-μm intervals), and Supplementary Fig. 4]. In contrast, there was no significant difference in the percentage of the pancreatic glucagon-positive area (α cells) between Spint1fl/fl and Spint1−/− mice (Supplementary Fig. 5a). To explore further whether Spint1 deficiency suppressed β cell proliferation, which might lead to a reduction in pancreatic islet and β cell area, mass, and number, we analyzed the Ki67 expression levels in the β cells of the 8-week-old mouse pancreas. The results showed that Spint1 deficiency significantly diminished the Ki67-positive percentage in insulin-positive areas with diameters greater than 200 μm in the mouse pancreas (Fig. 2c, d and Supplementary Fig. 5b), while it had no significant effect on diameters below 200 μm (Supplementary Fig. 5c). In addition, Spint1 deficiency had no significant effect on the apoptosis of β cells (Supplementary Fig. 5d, middle panels, assayed by caspase 3 positivity). As a control, the Ki67-positive or caspase 3-positive area percentages showed no significant difference in the lymph nodes between Spint1fl/fl and Spint1−/− mice (Supplementary Fig. 5d, upper and lower panels). Furthermore, we used a bromodeoxyuridine (BrdU) incorporation assay to confirm the reduced β cell proliferation in the larger islets of Spint1−/− mice. Similar to the Ki67 result, Spint1 deficiency diminished the percentage of β cells positive for BrdU in the islets with diameters greater than 200 μm (Supplementary Fig. 5e). Additionally, given the reported depletion of Spint1 leading to defects in tight junction formation in the mouse intestine16, and considering the role of tight junction proteins in β cell proliferation17, we assessed the changes of tight junction proteins in Spint1-depleted pancreas. Our findings revealed diminished zonula occludens-1 (ZO-1) and OCCLUDIN levels in Spint1−/− mice β cells compared to Spint1fl/fl mice (Supplementary Fig. 5f, g). This observation suggested that the Spint1 deficiency might compromise the expression of tight junction proteins, thereby contributing to the decrease in β cell proliferation. Thus, these results collectively indicated that the reduction in islet area, mass, and number in Spint1−/− mice was at least partly attributed to the decreased β cell proliferation.
Fig. 2. Dysfunction of β cells and glucose tolerance in pancreas-specific Spint1-deficiency mice.

a Percentages of insulin-positive area in 8-week-old Spint1fl/fl and Spint1−/− mice. Mouse pancreas sections were immunohistochemically stained using an anti-insulin antibody. The merged full-view microscopic images (Supplementary Fig. 4) were analyzed to obtain the percentage of the insulin-positive area over the total pancreas area using ImageJ (one out of every 300 μm interval from serial sections per pancreas, 6 sections per mouse, 5 mice per group). b Analysis of β cell mass between 8-week-old Spint1fl/fl and Spint1−/− mice. The β cell mass was calculated by multiplying the insulin-positive area percentage in (a) by the pancreas weight in Supplementary Fig. 3g. (5 mice per group). c Representative immunofluorescence images of Ki67+ β cells in large islets (>200 μm in diameters) of 8-week-old Spint1fl/fl and Spint1−/− mice. Pancreatic sections were subjected to immunofluorescence microscopy to detect Ki67 (green) and insulin (red, β cells). Nuclei were counterstained with DAPI (blue). White arrows mark the Ki67+ β cells in the Ki67 and merged images. Higher magnification images are shown in the inset at the lower left corner of each panel. Scale bar, 20 μm. d Percentages of Ki67+ β cells in large islets (>200 μm in diameters) of Spint1fl/fl (n = 6) and Spint1−/− (n = 7) mice (6 sections per pancreas). The percentages of Ki67+ β cells in middle and small islets were shown in Supplementary Fig. 5c. e Analysis of glucose tolerance in 8-week-old Spint1fl/fl and Spint1−/− mice. Mice were fasted for 8 h and then intraperitoneally injected with 20% glucose solution (2 g glucose/kg body weight). The tail blood was then taken at 15, 30, 60, and 120 min after injection and examined for glucose level using a glucometer (n = 11 per group). f Measurement of glucose-induced serum insulin in 8-week-old Spint1fl/fl and Spint1−/− mice. Serum was collected from the submandibular region before (0 min), 15, and 30 min after the oral gavage of glucose (2 g/kg body weight), and insulin levels were measured using an ELISA kit (n = 4 per group). g Kinetics of insulin secretion in perfused islets from 8-week-old Spint1fl/fl and Spint1−/− mice. Pancreatic islets were isolated from mice using collagenase perfusion, and 80 pancreatic islets per mouse were handpicked. The islets were first perfused with a low concentration of glucose (2.8 mM) for 120 min and then challenged with a high concentration of glucose (16.7 mM) for 40 min (details in the Methods section). The secreted insulin levels in the conditioned media were measured every 2 min using an ELISA kit (n = 12 for Spint1fl/fl mice and n = 9 for Spint1−/− mice). h Analysis of the secreted levels of insulin in Spint1-silenced MIN6 cells. Cells were transfected with siRNAs (siSpint1) to knock down Spint1. Control cells were transfected with scrambled siRNAs (scramble). Cells were then incubated in a medium containing 2.8 mM glucose for 2 h, followed by culture in a medium containing 16.7 mM glucose for 40 min. The conditioned media were collected, diluted 200 times, and examined for insulin levels using an ELISA kit. These results were statistically analyzed from three independent experiments (n = 3). i Early onset of diabetes in streptozotocin-induced 8-week-old Spint1−/− mice. Mice were intraperitoneally injected with streptozotocin (40 mg/kg body weight) for 5 consecutive days and provided with 10% sucrose drinking water. Ante cibum blood glucose levels (AC glucose) were measured before streptozotocin treatment (day 0) and on days 6, 8, 15, and 18 after the treatment (n = 5 per group). These results were statistically analyzed from three independent experiments. j Aggravated glucose intolerance in streptozotocin-treated 8-week-old Spint1−/− mice compared to Spint1fl/fl mice. The streptozotocin-treated mice in (i) were fasted and intraperitoneally injected with 20% glucose solution (2 g/kg body weight). Blood glucose levels were measured at 15, 30, 60, and 120 min after glucose injection using a glucometer (n = 5 per group). k Analysis of β cell mass between streptozotocin-treated 8-week-old Spint1fl/fl and Spint1−/− mice. Mouse pancreas sections were immunohistochemically stained using an anti-insulin antibody, and β cell mass was calculated using the protocol in (b). Statistical significance was assessed by a two-tailed Student’s t-test for (a, b, d, k), and a two-way ANOVA followed by Sidak’s multiple comparison analysis for (e–j). All data were represented as mean ± SEM. For bar plots, bars are represented as mean ± SEM. In the box plots, the boxes span from the 25th to the 75th percentiles, with a line indicating the median. Whiskers extend to values within 1.5 times the interquartile range, defined as the difference between the 25th and 75th percentiles. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Below the asterisks are the precise statistical results. Source data are provided as a Source Data file.
Impaired glucose tolerance and early-onset deterioration of hyperglycemia in mice with pancreatic Spint1 deficiency
Since the β cell mass declined in the Spint1−/− mouse pancreas, we further examined whether Spint1 deficiency might affect glucose tolerance and insulin production using the intraperitoneal glucose tolerance test (IGTT) and glucose-stimulated insulin secretion (GSIS) test. The results showed that the blood glucose levels of the Spint1−/− mice were significantly higher than those of the Spint1fl/fl mice at 30 min after glucose administration (Fig. 2e). Moreover, the serum insulin levels at 15 min after glucose stimulation were also lower in the Spint1−/− mice (Fig. 2f). The ex vivo glucose-stimulated insulin secretion assay also demonstrated that isolated Spint1−/− pancreatic islets exhibited significantly flattened peaks in both phases of insulin secretion, which generally occurred at the time points of 124 and 136 min after the high-glucose administration, respectively (Fig. 2g). We also found that Spint1 silencing significantly reduced insulin secretion in the mouse insulinoma cell line MIN6 (Fig. 2h). These results provide compelling evidence for the role of SPINT1 in maintaining normal glucose tolerance and proper insulin secretion.
We also determined whether Spint1 deficiency increased the risk of developing diabetes. After a 5-day treatment with streptozotocin (STZ), Spint1−/− mice, but not Spint1fl/fl mice, developed diabetes (fasting glucose level >250 mg/dL along with polyuria and polydipsia) on day 6, while the diabetic syndrome in Spint1fl/fl mice initially occurred after day 8 (Fig. 2i). Moreover, the hyperglycemia at 30 and 60 min during IGTT was significantly aggravated in the streptozotocin-treated Spint1−/− mice (Fig. 2j). Furthermore, the β cell mass was also marginally decreased in streptozotocin-treated Spint1−/− mice compared to their counterpart Spint1fl/fl mice (Fig. 2k). To clarify SPINT1’s role in the STZ-induced immunological response, we examined the infiltration of immune cells in the diabetic Spint1fl/fl and Spint1−/− mouse islets after STZ treatment using IF microscopy. There was no significant difference in infiltrated immune cell density within β cell regions (Supplementary Fig. 5h). Collectively, the results indicated that Spint1 deficiency in the mouse pancreas impaired glucose tolerance, induced early-onset diabetes, and exacerbated the severity of hyperglycemia in diabetic mice.
In vivo proteomic insights into the dysregulated signal pathways induced by Spint1 deficiency in mouse pancreatic islets
To delve deeper into the signal pathways or signatures influenced by Spint1 in pancreatic islets, we employed the proteomic technique of stable isotope labeling by amino acids in cell culture (SILAC) to measure the disparities in the protein levels between the Spint1fl/fl and Spint1−/− mouse pancreatic islets (Fig. 3a). The in vivo SILAC analysis revealed that 809 proteins were differentially expressed between Spint1−/− and Spint1fl/fl islets. The gene ointology (GO) analysis showed that several pathways, such as those for insulin processing and the succinate metabolic process were downregulated in the Spint1−/− pancreatic islets, while other processes, including those for the valine catabolic process and l-serine biosynthetic process, were upregulated in them (Fig. 3b). Furthermore, results from the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed that insulin-related pathways such as those for insulin secretion, G-coupled receptors (GPCR), and cAMP-related proteins were downregulated in the Spint1−/− islet cells, while oxidative stress pathways and endoplasmic reticulum stress pathways were upregulated in these cells (Fig. 3c). Ingenuity pathway analysis (IPA) further showed that insulin secretion signaling was significantly decreased in Spint1−/− pancreatic islet cells (Fig. 3d and Supplementary Fig. 5i). Notably, among those 809 altered proteins in the IPA, 309 proteins were related to endocrine system disorders (Fig. 3e). These findings suggest that Spint1 deficiency might affect the function of pancreatic β cells by reducing the insulin-processing/secretion pathways and downregulating GPCR pathways.
Fig. 3. Quantitative proteomic analysis of Spint1fl/fl and Spint1−/− islets using an alternative SILAC method.

a SILAC workflow for the proteomic analysis of Spint1fl/fl and Spint1−/− islets (4 mice per group). b Gene ontology (GO) analysis of the biological processes for differentially regulated proteins in the SILAC analysis of Spint1fl/fl and Spint1−/− islets. The biological processes significantly affected the downregulated and upregulated protein groups in Spint1−/− pancreatic islets compared to Spint1fl/fl islets listed in the upper and lower panels, respectively. The fractions in the histograms represent the proportion of identified proteins (numerator) in our dataset that correspond to those pathways, with the denominator indicating the total number of proteins in each pathway. c Protein abundance differences between Spint1fl/fl and Spint1−/− islets. The graph was generated using the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of the biological pathways for the differentially regulated proteins between Spint1fl/fl and Spint1−/− islets. The differentially regulated proteins were divided into the downregulated (upper panel) and upregulated (lower panel) groups in Spint1−/− islets. The biological pathways with significant alteration in this study (e.g., insulin-related pathways) are highlighted with color dots in the graphs. d Analysis of the most highly regulated signal pathways using ingenuity pathway analysis (IPA) to examine the proteins differentially expressed in Spint1−/− islets relative to Spint1fl/fl islets. The top six differentially regulated signal pathways were identified using the threshold of −log (P value) >3. Downregulated pathways are represented by blue bars, while upregulated pathways are indicated by orange bars in Spint1−/− islets compared to Spint1fl/fl islets. The fractions in the histograms show the proportion of the identified proteins (numerator) in our database that correspond to those pathways, with the denominator indicating the total number of proteins in each pathway. e Top five diseases and disorders identified through IPA of the differentially regulated proteins in Spint1−/− islets compared to Spint1fl/fl islets. The fractions in the table represent the number of the identified proteins associated with various diseases and disorders (numerator) relative to the total number of proteins for the corresponding disease and disorder in the IPA dataset (denominator). Statistical analysis was performed using a two-sided Fisher’s exact test, and the false discovery rate was controlled using the Benjamini–Hochberg procedure to correct p values (P) for (b, d, e). Source data are provided as a Source Data file.
HEPSIN as a SPINT1-targeted protease to downregulate MAFA and insulin in Spint1-deficient pancreatic islet cells
Based on the SILAC data, we further explored whether Spint1 plays a role in regulating insulin production. Q-RT-PCR showed that Spint1 deficiency and silencing significantly decreased the gene expression of Ins1 and Ins2 in 8-week-old mouse pancreatic islets and NIT-1 cells, respectively (left panels of Fig. 4a, b). Similar results were observed in 1-week-old Spint1−/− and Spint1fl/fl mouse pancreatic islets (Supplementary Fig. 6a). Given that MAFA, PDX1, and NEUROD1 primarily regulate insulin gene transcription18, we then analyzed the effects of Spint1 depletion on the protein expression of these transcription factors. Spint1 deficiency led to a dramatic decrease in MAFA protein levels (Fig. 4a, right panel), accompanied by a differential augmentation of PDX1 and NEUROD1 proteins in the mouse islets, potentially acting as a compensatory mechanism19. Similar results were also observed in the Spint1-silencing NIT-1 and MIN6 cells (Fig. 4b, right panels; c). In quantitative IF microscopy, it showed that the protein levels of MAFA were decreased in the pancreatic islets of the Spint1−/− mice (Fig. 4d). Furthermore, using a public RNA sequencing data from human islet donors, classified along with their glycaemic continuum from normoglycaemia to type 2 diabetes20, revealed a moderate correlation between SPINT1 expression and the expression levels of MAFA and INS in human pancreatic islets (Supplementary Fig. 6b). The findings together illustrated that the depletion of Spint1 in pancreatic β cells resulted in reduced MAFA protein level, leading to downregulation of insulin. To further confirm SPINT1’s role in MAFA expression in β cells, we overexpressed SPINT1 protein in NIT-1 cells. The results showed that Spint1 overexpression could increase MAFA protein levels in NIT-1 cells (Fig. 4e).
Fig. 4. Downregulation of MAFA in Spint1-depleted mouse β cells accompanied by HEPSIN upregulation.

a Expression levels of Ins1/2 mRNA, three key transcription factors for insulin expression, and a glucose sensor in Spint1fl/fl and Spint1−/− mouse islets. The expression levels of insulin (Ins1 and Ins2) in mouse islets were examined using Q-RT-PCR with normalization to Gapdh (n = 3 per group, left panels). The protein levels of transcription factors (MAFA, PDX1, and NEUROD1) for insulin expression and a glucose sensor (glucokinase, HK4) were analyzed in collected mouse islets using immunoblot assays (n = 6 mice per group). α-tubulin was used as a loading control. b Effect of Spint1 silencing on the expression levels of Ins1, Ins2, MAFA, PDX1, NEUROD1, HK4, and α-tubulin in NIT-1 cells (n = 3 per group). c Effect of Spint1 silencing on the expression levels of MAFA, PDX1, NEUROD1, HK4, and α-tubulin in MIN6 cells (n = 3 per group). d Immunofluorescence staining of MAFA and insulin in 8-week-old Spint1fl/fl and Spint1−/− mouse islets. Pancreatic sections were subjected to immunofluorescence staining of MAFA (green) and insulin (red). The images were then taken using a fluorescence microscope. Scale bar, 20 μm. The right histogram represented the relative intensity of MAFA in β cells (three sections per pancreas, four mice for Spint1fl/fl, five mice for Spint1−/− mice). e Effect of Spint1 overexpression on MAFA expression in NIT-1 cells. Cells were transiently transfected with Spint1 plasmids with a MYC tag and control vectors (PLKO). Cell lysates were subjected to immunoblot analysis using anti-MYC and anti-MAFA antibodies. α-tubulin was used as a loading control. MAFA protein levels were then quantified and statistically calculated from three independent experiments with normalization to α-tubulin and relative to PLKO control (right panel, n = 3). f Effect of a broad serine protease inhibitor (aprotinin) on the expression of MAFA in Spint1-silencing NIT-1 cells. siSpint1- or scrambled siRNA-transfected NIT-1 cells were treated with or without 40 μg/mL aprotinin for 24 h. Cell lysates were subjected to immunoblot analysis using anti-MAFA and anti-PDX1 antibodies. α-tubulin was used as a loading control. MAFA and PDX1 protein levels were statistically calculated from three independent experiments and shown at the bottom of each image. g Effects of recombinant mouse SPINT1 proteins (rt-mSPINT1) on the MAFA expression in Spint1-depleted NIT-1 cells. Cells were transfected with siSpint1 and scramble siRNAs, cultured for 24 h, and then treated with 0, 0.4, 0.8, and 1.6 μg/mL rt-mSPINT1 for another 24 h. Cell lysates were subjected to immunoblot analysis using anti-MAFA and anti-His-tag antibodies. Vinculin was used as a loading control. MAFA protein levels were statistically calculated from three independent experiments with normalization to vinculin (right panel). The recombinant protein and anti-His-tag antibody products are listed in Supplementary Table 3. h Analysis of Hepsin expression in 8-week-old Spint1fl/fl and Spint1−/− mouse islets using Q-RT-PCR. Results were statistically calculated from three independent experiments. i Quantification of HEPSIN IHC intensity in Spint1fl/fl and Spint1−/− mouse islets. HEPSIN protein levels in Spint1fl/fl and Spint1−/− mouse islets were examined using IHC and shown in Supplementary Fig. 6e. HEPSIN intensities in Spint1fl/fl and Spint1−/− mouse islets were statistically calculated using ImageJ from three sections per mouse pancreas (n = 4 for Spint1fl/fl mice, n = 5 for Spint1−/− mice). j Immunoblot analysis of HEPSIN and matriptase (MTX) in mouse islets, NIT-1, and MIN6 cells. Lysates were collected from Spint1fl/fl and Spint1−/− mouse islets (left panel), NIT-1 and MIN6 cells (middle and right panels) with or without Spint1 silencing (siSpint1 versus scramble) and subjected to immunoblot analysis using anti-HEPSIN and anti-MTX antibodies. α-tubulin or β-actin was used as a loading control. The histogram (right panel) represents the quantification result of HEPSIN levels in the left panels. Data were statistically calculated with normalization to β-actin or α-tubulin from three independent experiments (n = 3 per group of mice or cell cultures). Statistical significance was assessed by a two-tailed Student’s t-test for all panels. All data were represented as mean ± SEM. *P < 0.05; **P < 0.01; ****P < 0.0001. Below the asterisks are the precise statistical results. Source data are provided as a Source Data file.
As SPINT1 functions as a membrane-anchored serine protease inhibitor, we further explored whether a serine protease was involved in SPINT1-mediated MAFA expression. We analyzed the effects of aprotinin (a broad serine protease inhibitor) on the MAFA expression in Spint1-silencing NIT-1 cells. The results showed that aprotinin could restore the MAFA protein levels in Spint1-depleted NIT-1 cells (Fig. 4f and Supplementary Fig. 6c). Concordantly, purified recombinant mouse SPINT1 proteins (rt-mSPINT1) increased MAFA expression in a dose-response manner (Fig. 4g). These findings suggest that there might be SPINT1-targeted MASPs regulating MAFA expression in pancreatic β cells. To identify which MASP was essential for SPINT1-mediated MAFA expression, we used Q-RT-PCR to analyze the expression of 18 members of the MASPs in 8-week-old mouse pancreatic islets. This analysis was based on previous observations indicating that some serine proteases, when relieved from the inhibition by their cognate inhibitors or activated by their upstream serine proteases21, may further positively regulate their mRNA levels. The results showed that six of these protease-encoding genes had higher expression levels (Ct values <31), including Tmprss15, Hepsin, St14 (matriptase), Tmprss7, Tmprss2, and Tmprss4: among them, the Hepsin mRNA level was upregulated approximately six folds due to Spint1 deficiency (Fig. 4h and Supplementary Fig. 6d). Quantitative IHC analysis further revealed an elevation in HEPSIN expression within Spint1−/− pancreatic islets (Fig. 4i and Supplementary Fig. 6e). Consistent with this finding, western blotting analysis showed that Spint1 deficiency increased the HEPSIN protein levels in mouse pancreatic islets, NIT-1, and MIN6 cells (Fig. 4j). To examine further whether HEPSIN played an essential role in SPINT1-modulated MAFA expression in β cells, we analyzed the effects of Hepsin silencing on the expression of MAFA and insulin after Spint1 depletion. Q-RT-PCR and western blot analysis showed that Hepsin silencing restored the expression of Mafa/MAFA and Ins1/insulin in Spint1-silencing NIT-1 cells (Fig. 5a, b). Similarly, Hepsin silencing alone significantly increased MAFA expression in NIT-1 cells (Supplementary Fig. 6f). In human primary islet cells, SPINT1-silencing decreased the mRNA levels of MAFA, while HEPSIN deduction rescued the MAFA and INS expression in the SPINT1-silencing cells (Fig. 5c). Besides, Hepsin knockdown further rescued the MAFA expression and insulin secretion, which were downregulated by Spint1 silencing in the MIN6 cells (Fig. 5d, e). The collective findings support the involvement of HEPSIN in SPINT1-mediated insulin secretion in pancreatic β cells. Consequently, the results imply that the depletion of Spint1 leads to an increase in HEPSIN levels, and Hepsin silencing counteracts the downregulation of Mafa and Ins1 mRNAs and their protein levels induced by Spint1 silencing in mouse pancreatic β cells. This scenario might also apply to human counterparts.
Fig. 5. Hepsin involvement in Spint1-mediated expression of Mafa, Ins1, and their proteins, as well as insulin secretion.

a Q-RT-PCR analysis of Spint1, Hepsin, Mafa, and Ins1 expression in NIT-1 cells with depletion of Spint1, Hepsin, or both. Cells were transfected with siSpint1, siHepsin, both, or scramble siRNAs (control) and then subjected to RNA extraction and Q-RT-PCR, with normalization to Gapdh. Results were obtained from statistical calculations of three independent experiments (n = 3). b Immunoblot analysis of HEPSIN, MAFA, and insulin in NIT-1 cells with Spint1, Hepsin, or both silencing. The protein levels of MAFA (middle panel) and insulin (right panel) were statistically calculated with normalization to α-tubulin (n = 5 for MAFA, n = 3 for insulin). c Q-RT-PCR analysis of SPINT1, HEPSIN, MAFA, and INS1 expression in human primary islet cells with silencing of SPINT1, HEPSIN, or both. Cells were transfected with siSPINT1, siHEPSIN, both, or scramble siRNAs (control) and then subjected to RNA extraction and Q-RT-PCR, with normalization to GAPDH. Results were statistically calculated from three independent experiments (n = 3). d Effect of Spint1 or Hepsin silencing on MAFA and insulin expression in MIN6 cells. The histogram represents the quantitation data of western blots in the left panel. All data were statistically calculated from four independent experiments (n = 4), with normalization to α-tubulin. e Effect of Spint1 or Hepsin silencing on the glucose-stimulated insulin secretion in MIN6 cells. MIN6 cells were transfected with siSpint1 or siHepsin and then treated with 2.8 mM glucose or 16.7 mM glucose. The experimental details are described in the Methods section. The secreted insulin levels were statistically calculated from three independent experiments (n = 3) with normalization to the control (scramble, 2.8 mM glucose). Statistical significance was assessed using a two-tailed Student’s t-test for (d), a one-way ANOVA with Tukey’s post hoc test for (a–c), and a two-way ANOVA followed by Sidak’s multiple comparison analysis for (e). All data were represented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Below the asterisks are the precise statistical results. Source data are provided as a Source Data file.
Identification of GLP1R as a potential HEPSIN effector in SPINT1-regulated Mafa and Ins1 expression
To identify which membrane protein(s) were the downstream effector(s) of HEPSIN for SPINT1 to regulate the levels of MAFA and insulin in pancreatic β cells, we searched the interactome database of the STRING website22 for identifying potentially interacting partners of HEPSIN. As shown in Supplementary Fig. 7a, among the interacting proteins of HEPSIN, GLP1R attracted our attention, as it functions as a transmembrane GPCR to modulate insulin synthesis and secretion via CREB-MAFA signaling13,23. To investigate whether SPINT1, HEPSIN, and GLP1R could form a complex in cells, we performed co-immunoprecipitation (Co-IP) assays, and the results showed that SPINT1, HEPSIN, and GLP1R could create a complex in HEK293 cells (Supplementary Fig. 7b). Furthermore, we conducted proximity ligation assay (PLA) in human primary islet cells and human pancreatic tissues to validate the Co-IP results. We observed that SPINT1, HEPSIN, and GLP1R could interact with each other in a complex in human islet cells (Fig. 6a, b and Supplementary Fig. 7c, d).
Fig. 6. SPINT1 inhibited HEPSIN-induced GLP1R cleavage and preserved GLP1R activity.

a Analysis of SPINT1, HEPSIN, and GLP1R interactions in human primary islet cells using proximity ligation assay (PLA). Human islet cells were incubated with three pairs of primary antibodies (anti-GLP1R and anti-SPINT1, anti-HEPSIN and anti-SPINT1, anti-GLP1R and anti-HEPSIN antibodies), with each antibody as a control. The interaction signals after PLA (red puncta) and insulin signals (green) were visualized using immunofluorescence microscopy. Nuclei were counterstained with DAPI (blue). Scale bar, 5 μm. b Quantitation of PLA signals in human islet cells. The numbers of red puncta in the insulin-positive area in (a) were counted and statistically calculated from three independent experiments, with three to eight cells counted for each independent experiment. c Western blot analysis of GLP1R in Glp1r-overexpressing NIT-1 cells with or without silencing of Spint1 or Hepsin. Glp1r-overexpressing NIT-1 cells were transfected with siSpint1, siHepsin, or both and then incubated with 35 μg/mL MG132 for 3 h. Cell lysates were then immunoblotted using anti-FLAG (GLP1R) and anti-HEPSIN antibodies. The ratios of cleaved to full-length (c/f) GLP1R band intensities from four independent experiments (n = 4), normalized to GAPDH and presented as the amount relative to MG132 treatment alone, were statistically analyzed (bottom panel). d Western blot analysis of GLP1R in Glp1r-overexpressing NIT-1 cells with overexpression of Spint1, Hepsin, or both. Glp1r-overexpressing NIT-1 cells were transfected with the indicated plasmids (Hepsin, Spint1, or both) and then incubated with 35 μg/mL MG132 for 3 h. Cell lysates were then subjected to immunoblot analysis using anti-FLAG (GLP1R), anti-SPINT1, and anti-MYC-tag (HEPSIN) antibodies. The ratios of cleaved to full-length (c/f) GLP1R band intensities from three independent experiments (n = 3), normalized to GAPDH and presented as the amount relative to MG132 treatment alone, were statistically analyzed (bottom panel). e Immunoblot analysis of SPINT1, HEPSIN, and GLP1R in Glp1r-overexpressing HEK293T cells with or without the plasmids encoding cDNA for Hepsin, protease-null Hepsin mutant (mt.) or Spint1. Cells were transiently transfected with the indicated plasmids (Glp1r, Hepsin, protease-null Hepsin mutant [S353A], and Spint1). Cell lysates were then collected and subjected to immunoblotting using anti-FLAG (GLP1R), anti-SPINT1, and anti-MYC-tag (HEPSIN) antibodies. GAPDH and α-tubulin were used as loading controls. f The histogram shows the quantitative GLP1R cleavage results from (e). The ratios of cleaved to full-length (c/f) GLP1R band intensities were statistically calculated from three independent experiments (n = 3) with normalization to GAPDH and presented as the relative values to that of Glp1r overexpression alone. g–i Analysis of Spint1’s role in cellular cAMP levels in the islets from Spint1fl/fl and Spint1−/− mice and in NIT-1 cells. All samples underwent treatment with 2.5 mM PAR2 antagonist to block background cAMP, followed by stimulation of 25 nM GLP1R agonist (Exendin-4, Ex4). Lysates were collected and subjected to cAMP level measurement using an ELISA kit. g NIT-1 cells were transiently transfected with siSpint1. Control cells were transfected with scramble siRNA. These results were statistically analyzed from three independent experiments (n = 3). h Mouse islets were collected from 8-week-old Spint1fl/fl and Spint1−/− mice (n = 5 per mouse group). i NIT-1 cells were transiently transfected with Spint1 plasmid and PLKO empty vector. These results were statistically analyzed from three independent experiments (n = 3). j Analysis of the roles of SPINT1 and HEPSIN in the GLP1R activity in HEK293T cells by measuring the production of cyclic AMP (cAMP). HEK293T cells were transiently transfected with plasmids containing the cDNA for Glp1r, Spint1, and Hepsin. These results were statistically analyzed from four independent experiments (n = 4). k Analysis of in vivo GLP1R function on stimulating insulin production in Spint1fl/fl and Spint1−/− mice after Ex4 treatment. Each mouse was intraperitoneally injected with 24 nmol/kg Ex4 for 30 min before an oral gavage of glucose (2 g/kg body weight). Blood samples were then collected and subjected to insulin measurement using ELISA. Control mice were injected with PBS (n = 4 per group). l Analysis of serum insulin levels normalized to β cell mass in Spint1fl/fl and Spint1−/− mice after Ex4 treatment. The Ex4-induced upregulation of insulin levels in each mouse in panel (k) was calculated as the insulin AUC (area under the curve) divided by their respective β cell mass. The connecting lines show the change in insulin levels for each mouse before and after Ex4 treatment (n = 4 per group). Statistical significance was determined using a one-way ANOVA with Tukey’s post hoc test for (c, d, f, j), a two-way ANOVA followed by Sidak’s multiple comparison analysis for (k), a two-tailed Student’s t-test for (b, g, h, i), and a two-tailed paired Student’s t-test for (l). For bar plots, bars are represented as mean ± SEM. In the box plots, the boxes span from the 25th to the 75th percentiles, with a line indicating the median. Whiskers extend to values within 1.5 times the interquartile range, defined as the difference between the 25th and 75th percentiles. *P < 0.05; ***P < 0.001; ****P < 0.0001. Below the asterisks are the precise statistical results. Source data are provided as a Source Data file.
Since HEPSIN was a transmembrane serine protease, we analyzed whether HEPSIN could modify GLP1R via a proteolytic cleavage in β cells. The results demonstrated that Spint1 knockdown could enhance the proteolytic cleavage of exogenous GLP1R in NIT-1 cells, and Hepsin silencing could reduce the GLP1R cleavage induced by Spint1 knockdown (Fig. 6c). Concordantly, Hepsin overexpression increased the cleaved GLP1R level, and Spint1 overexpression suppressed HEPSIN-induced GLP1R cleavage in NIT-1 and HEK293 cells (Fig. 6d–f). Since GLP1R activation is known to promote the production of secondary messenger cyclic AMP (cAMP)24, we then examined the role of SPINT1 in the cAMP synthesis in NIT-1 cells. The result showed that Spint1 silencing significantly tempered the induction effect of GLP1R agonist Exendin-4 (Ex4) on cAMP production (Fig. 6g). Similar results were also observed in Spint1fl/fl and Spint1−/− mouse pancreatic islets, where Spint1 deficiency led to a significant reduction in cAMP levels (Fig. 6h). Conversely, Spint1 overexpression enhanced cAMP levels in NIT-1 cells (Fig. 6i). We further examined the roles of SPINT1 and HEPSIN in GLP1R-induced cAMP production by analyzing cAMP levels in Spint1-, Hepsin- and Glp1r-overexpressing HEK293 cells. The results showed that Hepsin overexpression reduced GLP1R-induced cAMP production (Fig. 6j).
To determine whether Spint1 depletion affects in vivo GLP1R function, we analyzed serum insulin levels in Spint1fl/fl and Spint1−/− mice following treatment of Ex4 and glucose oral gavage, as serum insulin levels are commonly used as an indicator for Ex4-induced GLP1R activity in vivo25. The result showed that Spint1 deficiency suppressed the upregulation of serum insulin levels by Ex4 (Fig. 6k, l and Supplementary Fig. 8a, b), suggesting that GLP1R signaling was likely impaired in Spint1−/− mice. Given that GLP1R activation can elevate MAFA expression through the cAMP-CREB pathway13, we assessed CREB phosphorylation levels in Spint1-depleted NIT-1 cells under the Ex4 treatment. The result revealed that Spint1 knockdown decreased the Ex4-induced CREB phosphorylation and MAFA protein levels (Fig. 7a), as well as the Mafa and Ins1 mRNA levels (Fig. 7b). Conversely, Hepsin knockdown restored the stimulating effects of Ex4 on CREB phosphorylation, MAFA, and insulin levels (Fig. 7a, b). Similarly, the GLP1R antagonist (Ex9–36) suppressed the Hepsin silencing-induced MAFA expression in NIT-1 cells (Supplementary Fig. 6f). Thus, the results together indicate that GLP1R is involved in the action of SPINT1/HEPSIN axis on MAFA-mediated insulin production.
Fig. 7. Effect of Spint1 or Hepsin depletion on GLP1R signaling-induced phospho-CREB, Mafa, and Ins1 in β cells.

a Roles of Spint1 and Hepsin in Ex4-induced CREB phosphorylation and MAFA expression in NIT-1 cells. Cells were transfected with siSpint1 and siHepsin. Control cells were transfected with scramble siRNAs. After transfection, cells were starved for 24 h and then treated with or without 25 nM Ex4 or 25 nM Exendin 9–36 (Ex9–36, a GLP1R antagonist) for 24 h. Cell lysates were then collected and subjected to western blot analysis using anti-phospho-CREB, anti-CREB, and anti-MAFA antibodies. GAPDH served as a loading control. The ratios of phospho-CREB to total CREB levels (middle panel) and MAFA protein levels (right panel), normalized to GAPDH, were statistically analyzed across four independent experiments (n = 4). b Analysis of Spint1, Hepsin, Mafa, and Ins1 expression in NIT-1 cells silenced for Spint1, Hepsin, or both, with or without Ex4 treatment. Cells were transfected with siSpint1, siHepsin, or both. Controls were transfected with scramble siRNAs. Transfectants were serum-starved for 24 h and then treated with or without 25 nM Ex4 for another 24 h. RNA was then extracted and subjected to Q-RT-PCR analysis. Results were statistically analyzed by normalizing to Gapdh relative to the control from three independent experiments (n = 3). Statistical significance was determined using a two-tailed Student’s t-test for all panels. All data were represented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Below the asterisks are the precise statistical results. Source data are provided as a Source Data file.
To exclude the possibility that SPINT1 regulated MAFA through other signaling pathways of GLP1R in β cells, we analyzed the changes of other potential downstream pathways of GLP1R in Spint1-silenced NIT-1 cells26. Spint1 knockdown had a mild effect on ERK signaling and did not affect PI3K, AKT, and GSK3α signaling (Supplementary Fig. 8c). To further ascertain whether MAFA was regulated by GLP1R signaling in β cells, we examined the effect of Glp1r silencing on Ex4-induced MAFA expression. Western blotting revealed that Glp1r knockdown reduced Ex4 induction on MAFA in NIT-1 cells (Supplementary Fig. 8d), confirming the involvement of GLP1R in MAFA expression within β cells. These results were corroborated by the IPA analysis of mouse in vivo SILAC data, revealing that Spint1 deficiency significantly decreased GPCR, CREB, and PKA signaling (Supplementary Fig. 5i). Since Spint1−/− mice exhibited a decline in Ki67-positive β cells (Fig. 2d), we investigated the activity-relevant phosphorylation changes of insulin receptor (INR), epidermal growth factor receptor (EGFR), and insulin growth factor 1 receptor (IGF1R) in Spint1-depleted NIT-1 cells due to their involvement in β cell proliferation27–29. Western blotting demonstrated that Spint1 silencing reduced the tyrosine phosphorylation levels of INR, IGF1R, and EGFR in NIT-1 cells (Supplementary Fig. 8e-f). The results indicate that Spint1 modulates the GLP1R-CREB-MAFA signaling axis and the signaling pathways of INR, EGFR, and IGF1R, potentially regulating β cell proliferation.
Potential involvement of SPINT1 in human diabetes
Additionally, to elucidate the clinical relevance of SPINT1 in diabetes, we assessed the SPINT1 mRNA levels in the set of human transcriptome data available in a public database. We categorized human donors’ islets into three groups according to the corresponding HbA1c levels (<5.7%, 5.7–6.4%, and >6.4%), as serum HbA1c levels serve as a crucial clinical index for the risk of diabetes30. The results showed that SPINT1 was significantly expressed at higher levels in donors with HbA1c levels of 5.7–6.4% compared to the other two groups (Supplementary Fig. 9a). Consistently, IHC showed a trend of higher SPINT1 levels in pancreas islets of prediabetic individuals (HbA1c between 5.7 and 6.4%) compared to non-diabetic ones (Supplementary Fig. 9b). Considering the reported hyperinsulinemia syndrome in prediabetic patients due to insulin resistance31, the increased expression of SPINT1 in β cells of these patients might contribute to elevated insulin synthesis aimed at maintaining glucose homeostasis.
Furthermore, our study delved into the potential regulation of GLP1R signaling by SPINT1 and HEPSIN via dipeptidyl peptidase 4 (DPP4), a well-known serine protease that degrades GLP132. We measured the DPP4 activity in Spint1- or Hepsin-silencing NIT-1 cells and found that SPINT1 and HEPSIN had no significant effects on DPP4 activity in cells (Supplementary Fig. 9c). Collectively, the results suggest that the pericellular proteolysis axis of SPINT1 and HEPSIN plays a significant role in regulating the signaling pathway of GLP1R-CREB-MAFA for maintaining insulin production (Fig. 8).
Fig. 8. Schematic illustration of the mechanism for SPINT1-regulated insulin synthesis.
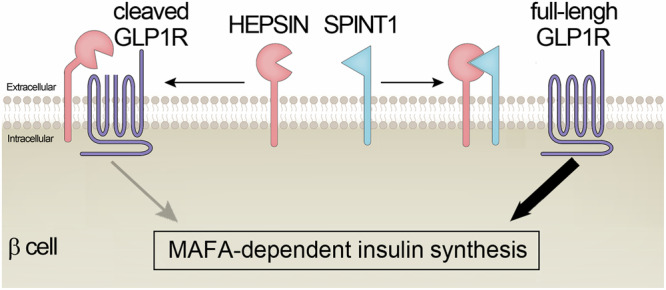
The serine protease inhibitor SPINT1 upholds MAFA-dependent insulin production by targeting the serine protease HEPSIN. Without SPINT1, overactivity of HEPSIN leads to proteolytic processing of its membrane substrate, such as GLP1R, suppressing the MAFA-insulin pathway in β cells. SPINT1 can inhibit HEPSIN’s activity in this regard, thereby augmenting MAFA-dependent insulin expression.
Discussion
Limited information is available regarding the roles of SPINT1 and its cognate protease(s) in pancreatic β cells or diabetes. One report showed that Hepsin-deficient mice are resistant to the condition, predisposing them to hyperglycaemia or diabetes, which is attributed to the action of HEPSIN on enhancing liver glucose metabolism33. Our results further indicated that HEPSIN played a role in inhibiting the insulin production of pancreatic β cells and that SPINT1 acted against HEPSIN to promote insulin synthesis. These two studies together suggest that HEPSIN may play a systemic role in increasing serum glucose levels via both the enhancement of glucose metabolism in the liver and the downregulation of insulin production in pancreatic β cells, with SPINT1 acting as a crucial cognate inhibitor of HEPSIN in the latter process.
Our current examination of pancreas-specific Spint1-deficient mice has provided valuable insights. It suggested that the deficiency of the Spint1 gene may increase the risk of diabetes. However, no cases of human diabetes patients with homozygous mutations in SPINT1 have been reported. This could be due to the lethal nature of germline homozygous deficiency of this gene during embryonic development, a phenotype reported in mice34 and likely applicable to humans as well. However, future investigation is needed to ascertain whether less severe mutations or single-nucleotide polymorphisms in the human SPINT1 gene might predispose individuals to impaired glucose tolerance or diabetes.
In humans, elevated serum protease inhibitor levels have been associated with diabetes and several metabolic syndromes35,36, suggesting that increased expression of serine protease inhibitor may serve as a compensatory response to dysregulated glucose homeostasis35,36. This phenomenon is also observed in SPINT1 expression in β cells at different stages of diabetes (Supplementary Fig. 9a, b). In this study, we hypothesize that under normal conditions, SPINT1 inhibits HEPSIN, thereby maintaining GLP1R activity and increasing the synthesis of MAFA and insulin in β cells. Conversely, the lack of SPINT1 may render HEPSIN over-activated, leading to the decrease of GLP1R function and increasing the risk of diabetes. Interestingly, Spint1 deficiency enhanced HEPSIN expression, thereby causing GLP1R cleavage. This cleavage extinguishes the Ex4-induced GLP1R activity, reducing the Mafa/Ins synthesis pathway. HEPSIN has been identified as a critical regulator in the liver and brown adipose tissue37. At the same time, SPINT1, as its cognate inhibitor, may directly alter HEPSIN-regulated energy metabolism in β cells. Therefore, SPINT1 might be a promising candidate for developing a biosimilar drug to treat diabetes by inhibiting HEPSIN.
GLP1R belongs to the GPCR family and plays a critical role in β cell functions through interacting with its ligand GLP138. Evidence shows that GLP1R knockout diminishes the effect of the GLP1R agonist, Ex4, on insulin secretion, and Pdx1-driven GLP1R expression can restore Ex4-induced insulin secretion in Glp1r−/− mice25, indicating that pancreatic GLP1R is vital for Ex4-induced insulin secretion. Similarly, our findings suggest that Spint1−/− mice lost their response to Ex4-induced insulin secretion and reduced MAFA/insulin synthesis, at least partially, due to HEPSIN overactivity, which may enhance the cleavage of GLP1R and impair GLP1R signaling. Cleavage of GPCR has been reported in other GPCR family members39,40, including PARs41 and adhesion GPCR42, and also occurs during the shedding process of multiple GPCR members43,44. In our case, the cleavage of GLP1R might negatively impact β cell function and reduce insulin production. However, the detailed mechanism by which HEPSIN cleaves GLP1R remains unclear and warrants further investigation.
Multiple lines of evidence indicate the significance of proteases and their corresponding protease inhibitors in maintaining pancreatic β cell function. For example, trypsin facilitates pancreatic islet expansion in db/db mice by modulating components of the islet capsule matrix45. Similarly, α1-antitrypsin has been reported to protect β cell function by mitigating inflammatory responses46,47. Furthermore, SERPINB1 and SERPINB8 can enhance β cell proliferation and shield against β cell exhaustion in high-fat diet conditions36,48,49. We include SPINT1 and HEPSIN in this list by demonstrating that SPINT1 inhibits GLP1R cleavage by suppressing HEPSIN’s proteolytic function in β cells. Since GLP1R signaling enhances pancreatic β cell function50, SPINT1 reveals a critical role in β cells by ensuring proper GLP1R activity. This mechanism underscores the importance of maintaining a delicate balance between serine proteases and their inhibitors to prevent β cell malfunction and hyperglycaemia.
Clinically, GLP1R agonists have been widely applied in treating type 2 diabetes6,7. Although these drugs have shown clinical benefits, they pose the risk of hyperactivating GLP1R signaling, leading to exhaustion or even damage of β cells over prolonged use51. Thus, it is vital to develop a novel approach that can adequately enhance GLP1R signaling for diabetes with less damage to β cells. Here, our findings suggest the potential for developing a therapeutic approach by modulating pericellular serine protease activities to enhance GLP1R action, thereby achieving therapeutic efficacy in diabetes with reduced side effects.
Regarding the mechanism underlying the reduced proliferation of β cells in large islet Spint1-deficient mice, we propose several possibilities to explain this phenomenon. First, β cells in larger islets are less functionally competent than those in smaller islets and produce less insulin52–54. Such a difference in local insulin levels might be further enlarged when the insulin synthesis in β cells was decreased by Spint1 deficiency. Since insulin can stimulate β cell proliferation27, the larger islets in Spint1−/− mice might significantly reduce Ki67 signals compared to Spint1fl/fl mice, possibly due to decreased insulin availability for β cells. Second, islet endothelial cells are known to promote β cell proliferation by releasing hepatocyte growth factors and other growth factors55, depending on the paracrine support from insulin and vascular endothelial growth factor (VEGF) secreted from β cells55. It has been shown that smaller islets express more VEGF-A56 than larger islets, potentially granting their endothelial cells increased resistance to the detrimental effects of decreased insulin production caused by the deficiency of Spint1. Consequently, Spint1−/− β cells in larger islets might experience a more significant reduction in growth factor secretion by islet endothelial cells compared to those in smaller islets, contributing to reduced proliferation. Third, GLP1R can enhance the proliferation of β cells through its effects on EGFR57. The decreased levels of GLP1R seem to have a more remarkable effect on large islets because Glp1r−/− mice have fewer large pancreatic islets than Glp1r+/+ mice58. This aligns with our results indicating a decrease in the number of large islets in Spint1−/− mice and reduced GLP1R signaling in Spint1-deficient or -depleted β cells. We speculate that larger islets, which contain β cells that are less functionally competent52–54, might be more susceptible to alterations in GLP1R signaling due to a lack of efficient compensation. Consequently, the relatively lower GLP1R signaling in larger islets of Spint1−/− mice would likely cause a significant reduction in β cell proliferation compared to Spint1fl/fl mice.
In line with our findings, which revealed a critical role of SPINT1 in β cells, we observed a high expression of HEPSIN in α cells and similar expression levels of SPINT1 between α and β cells in immunohistochemistry analysis (Supplementary Fig. 9d–f). Given that the balance of a protease and its cognate inhibitor varies across different tissues/cells59, the higher ratio of HEPSIN to SPINT1 in α cells than β cells is reasonable, as it might create a cell environment unsuitable for the expression of GLP1R and insulin in α cells. However, whether there is another role of SPINT1/HEPSIN in α cells remains to be determined.
In sum, our results highlight the roles of Spint1 in pancreatic β cells and glucose homeostasis. Mechanistically, SPINT1 maintains MAFA-mediated insulin production by inhibiting HEPSIN, preventing its proteolytic cleavage on GLP1R in β cells. These findings offer insights into the role of pericellular proteolysis in insulin production and suggest that SPINT1 and HEPSIN may serve as promising therapeutic targets for developing novel drugs against diabetes.
Methods
Animal study approval
All animal experiments complied with the Guidebook for the Care and Use of Laboratory Animals (published by The Chinese-Taipei Society for Laboratory Animal Sciences) and approved by the Institutional Animal Care and Use Committee at National Taiwan University, College of Medicine.
Generation of Spint1lacZ/+ transgenic mice
The knockout-first targeting vector (ID: PRPGS00114_B_E12) for the mouse Spint1 gene was obtained from the European Mouse Mutant Cell Repository (Munich, Germany). The targeting vector contained a lacZ-PGK-neo reporter that was composed of a splice acceptor site from mouse Engrailed 2 (En2SA), an encephalomyocarditis virus internal ribosomal entry site (IRES), the lacZ cDNA, and the SV40 polyadenylation element (pA), followed by a neomycin selection cassette comprising the neomycin-resistant gene (neo) cDNA driven by the PGK promoter. To generate Spint1lacZ/+ transgenic mice (formal name: C57BL/6-Spint1tm1a(EUCOMM)Wtsi), the targeting vector was linearized and transfected into C57BL/6 mouse embryonic stem (ES) cells to insert the lacZ-PGK-neo reporter into the first intron of the mouse Spint1 genomic locus. The selection process was as follows: neomycin was used to select the correctly targeted ES clones, which PCR and Southern blotting then confirmed before being injected into blastocysts. The obtained chimeric mice were mated to C57B/6 mice to generate germline transmission and heterozygous mice (Spint1lacZ/+), which, after F3, were used to trace the expression pattern of Spint1. For Southern blotting, mouse DNA was cleaved by the NdeI restriction enzyme, separated using agarose gel electrophoresis, and transferred to a nitrocellulose (NC) membrane. The probe for Southern blotting was localized between the NdeI site and Spint1 exon 1. The detailed procedures used for the Southern blotting have been described elsewhere60. For mouse genotyping, mouse tail snips were taken and digested with Protease K digestion at 55 °C overnight, followed by DNA extraction and PCR. The probe and primer are listed in Supplementary Table 1.
Generation of Spint1fl/fl and Spint1−/− mice
To generate pancreas-specific Spint1-deficient mice and their control mice, the Spint1lacZ/+ transgenic mice described above were mated with flippase (FLP)-expressing transgenic mice (Flp/+, strain: Tg(CAG-FLPe)37Ito; RIKEN BioResource Research Center) to remove the flippase recognition target (FRT) site-flanked reporter cassette (lacZ-PGK-neo). The derived heterozygous loxP-floxed mice (Spint1fl/+) were mated with each other to obtain the homozygous floxed mice (Spint1fl/fl, formal name: C57BL/6-Spint1tm1c(EUCOMM)Wtsi). The Spint1fl/fl mice were then crossed with Pdx1-Cre transgenic mice (Pdx1-Cre/+, strain: B6.FVB-Tg(Pdx1-cre)6Tuv/J; The Jackson Laboratory, ME) to generate Pdx1-Cre/+::Spint1fl/+ mice, which were then crossed with Pdx1-Cre/-::Spint1fl/fl mice (namely, Spint1fl/fl) to generate Pdx1-Cre/+::Spint1fl/fl mice (namely, Spint1−/−, formal name: C57BL/6-Spint1tm1d(EUCOMM)Wtsi,Tg(Pdx1-cre)6Tuv/J), and two other types of littermates (Pdx1-Cre/+::Spint1fl/+ and Pdx1-Cre/-::Spint1fl/+). The experiments in this study utilized only male mice, a decision based on the fact that their hormonal dynamics are more stable than female mice. This choice was made to ensure the reliability and consistency of our results. For genotyping, mouse tail snips were taken and subjected to genomic DNA extraction as described above; PCR was performed using the primer pair (WT-F and WT-R) to distinguish the wild-type Spint1 and recombinant Spint1-loxP alleles based on size difference, and the Cre cDNA-specific primer pair to detect the Cre allele. All primer sequences are listed in Supplementary Table 1.
Cell culture
NIT-1 and HEK293T cells were originally obtained from the American Type Culture Collection. NIT-1 cells were cultured in RPMI 1640 media (Thermo Fisher), and HEK293T cells were maintained in DMEM media (Thermo Fisher), supplemented with 10% fetal bovine serum (FBS, Hyclone) and 1% l-glutamine (Sigma-Aldrich). MIN6 cells were cultured in low-glucose DMEM (Sigma-Aldrich) with 15% FBS and 1% l-glutamine. The primary islet cells (ABC-TC4286, ACCEGEN) were cultured in DMEM media (Sigma-Aldrich) with 10% FBS and 1% l-glutamine. All cells were cultured in 5% CO2 at 37 °C in a humidified incubator, and the media were refreshed every 2 days.
Intraperitoneal glucose tolerance test (IGTT) and glucose-stimulated insulin secretion test (GSIS)
After an 8-hour fasting period, IGTT was performed by intraperitoneal injection of 2 mg/g d-glucose (Sigma-Aldrich) into the mice. Tail vein blood samples were taken after the glucose injection and subjected to measuring blood glucose levels using a blood glucometer (Abbott). For the GSIS test, blood samples were collected from the submandibular region after oral gavage of 2 mg/g d-glucose. To assess the effects of GLP1R agonist exendin-4 (Ex4, Sigma-Aldrich) on GSIS, Ex4 was administered via intraperitoneally injected at a dose of 24 nmol/kg mice body weight for 30 min before glucose challenge, with volume-matched PBS serving as control. These blood samples were then meticulously kept at room temperature for 10 min for clotting and centrifuged at 2000 × g for 10 min. The samples were then stored at a −80 °C refrigerator until further analysis. The supernatants were finally extracted to measure insulin levels using an ELISA kit (Mercodia, Sweden).
Pancreatic islet isolation and islet perfusion
For pancreatic islet isolation, eight-week-old male mice were gently injected with 4 ml pancreas lysis buffer (1.5 mg/ml collagenase V [Thermo Fisher] in Hank’s Balanced Salt Solution [HBSS, Corning]) into the mouse bile tube using a 30G needle. After the injection, the swelled pancreata were isolated and incubated with 2 ml of pancreas lysis buffer in a 50-ml tube with a 37 °C water bath for 15 min. Meanwhile, the samples were shaken every five minutes. Then, samples were vortexed for 10 s to release the islets from the pancreas completely, and the proteolytic reaction of collagenase V was inhibited using 20 ml of 10% FBS RPMI media. Samples were sieved with an opening diameter of 1 mm to remove undissolved tissue after centrifugation at 300 × g at 4 °C for 5 min. The pellets were dissolved in HBSS and repeated the sieving and centrifugation. Next, the pellets were suspended in 1077 Histopaque solution (Sigma-Aldrich), which was gently added drop by drop into the RPMI media to produce a density gradient. After centrifugation at 1200 × g for 25 min, islets that stayed in the region between the RPMI and 1077 Histopaque solution were collected, stained by 7.5 mg/ml dithizone (Sigma-Aldrich) for 2 min, and handpicked into phosphate buffered saline (PBS) with a density of 80 islets per 200 μl of PBS.
The islet perfusion assay used the Pump 11 Elite/Pico Elite Plus (70-4506, Harvard Apparatus). In general, 80 islets were first inoculated in the perfusion media (110 mM NaCl, 4.6 mM KCl, 1 mM MgCl2. 6H2O, 5 mM NaHCO3, 20 mM HEPES, pH7.4) containing non-stimulatory 2.8 mM glucose for 120 min. The insulin secretion of the islets was then stimulated by the treatment of 16.7 mM glucose perfusion media. The initial perfusion samples from the 2.8 mM glucose-treated conditioned media were collected for 150 μl/min flow rate in five tubes (each per 2 min, as controls) before the treatment with 16.7 mM glucose perfusion media. After the stimulation of 16.7 mM glucose, each flow-through of the conditioned media was collected every two minutes for 40 min61. Each sample was diluted 100 times using perfusion media and subjected to the measurement of the secreted insulin levels using the insulin ELISA kit (Mercodia).
X-gal staining in embryos and nuclear fast red counterstaining
The Spint1lacZ/+ embryos (E10.5, E12.5, and E14.5) were dissected from pregnant mice. The isolated embryos were rinsed in PBS and then fixed in 2% paraformaldehyde at 4 °C for 2 h. The fixed embryos were washed in PBS and incubated in 5-bromo-4-chloro-3-indolyl-beta-d-galacto-pyranoside (X-gal, Thermo Fisher) staining solution [10 mg/ml X-gal with 3.6 mM K4Fe(CN)6 and 3.1 mM K3Fe(CN)6] at room temperature overnight. Non-X-gal staining embryos were used as negative controls. After X-gal staining, embryos were washed with PBS, dehydrated, embedded in paraffin, and sliced at a thickness of 5 μm. The wax in the sliced embryo samples was washed using Histoclear solution (National Diagnostics). The samples were then hydrated in ddH2O and counterstained with 0.1% nuclear fast red (Sigma-Aldrich). The slides were dehydrated, covered with the mounting medium (Sigma-Aldrich), and imaged using a microscope.
Quantitative real-time polymerase chain reaction (Q-RT-PCR)
Total RNA from 1-week-old mouse pancreas, mouse β cell lines, and human primary islet cells were extracted using Trizol (Thermo Fisher) and chloroform (Sigma-Aldrich). We utilized an RNA purification kit (Geneaid) for the 8-week-old mouse pancreatic islets. Five micrograms of the total RNA per sample were performed using a cDNA synthesis kit (Thermo Fisher), following the manufacturer’s protocol. The gene expression analysis was conducted by measuring the expression levels of each gene using Q-RT-PCR with SYBR Green (Thermo Fisher) and the Step One real-time PCR system (Applied Biosystems). The pair of primers for each gene are listed in Supplementary Table 2.
Immunohistochemistry and immunofluorescence microscopy
Mouse pancreases were isolated, fixed with 4% formaldehyde, and embedded in paraffin. Sections of the mouse and human pancreas were cut at a thickness of 5 μm. The human pancreas sample was derived from the Department of Pathology, National Taiwan University Hospital (details in Supplementary Methods section). Sections were de-waxed in xylene (Thermo Fisher) and hydrated in ddH2O. Antigen retrieval was performed in the retrieval buffer (20 mM Tris, pH 8.0) using a pressure cooker at full pressure for 10 min. Specifically, for the anti-SPINT1 antibody, a protease K solution (50 mM Tris Base, 0.5% Triton X-100, 20 μg/ml, PH 8.0) was applied, following the Protease K antigen retrieval protocol at 37 °C for 15 min16.
For the IHC assay, after antigen retrieval, samples were rinsed with 0.1% triton X-100 for 20 min, and the endogenous peroxidase was blocked by 3% H2O2 for 10 min, followed by incubation with the blocking buffer and primary antibody at 4 °C overnight. The following antibodies were used: anti-mouse SPINT1 (AF1141, R&D), anti-human SPINT1 (AF1048, R&D), anti-MAFA (NB400-137, Novus), anti-HEPSIN (ab73133, Abcam), anti-Ki67 (GTX16667, GeneTex), and anti-active caspase 3 (#9661, Cell Signaling). Then, the samples were washed with PBS three times and incubated with HRP-conjugated secondary antibodies [anti-rabbit and anti-mouse IgG (K5007, Dako); anti-goat IgG (MP-7405, Vector Laboratories)] for 30 min. Next, the samples were washed with PBS three times, and the images were visualized by incubation with 3,3’-diaminobenzidine (DAB; K5007, Dako) for the required time. The samples were then counterstained with hematoxylin for 1 min, dehydrated, and covered with a mounting reagent (Sigma-Aldrich).
For IF, after antigen retrieval, samples were rinsed with 0.1% Triton X-100 for 20 min and incubated with a blocking buffer for 1.5 h, followed by incubation with the primary antibody in the blocking buffer. The following antibodies were used: anti-MAFA antibody (NB400-137, Novus), anti-Ki67 (GTX16667, GeneTex), anti-Glucagon (67286-1-lg, ProteinTech), anti-ZO-1 (33-9100, Thermo Fisher), anti-OCCLUDIN (71-1500, Thermo Fisher), anti-CD3 (GTX16669, GeneTex), and anti-insulin antibody (I2018, Merck Millipore). Antibodies were applied to the pancreas sections at 4 °C overnight. After washing with PBS three times, samples were incubated with fluorescent secondary antibodies (A11008, A11011, and A11004, Thermo Fisher) for 30 min. After washing with PBS three times, samples were incubated with 0.25% Sudan Black B (ab146284, Abcam) in 70% EtOH for 90 min to reduce the autofluorescence. Then, samples were washed with PBS thrice and covered with fluorescence mounting reagent (0100-01, Southern Biotech).
Due to the positive result for anti-SPINT1 and anti-HEPSIN in the IHC assay but not in the IF assay, we implemented the tissue stripping protocol62 to combine IF and IHC assay in the same section. The samples were washed with ddH2O to remove the mounting reagent from IF slides, immersed in a stripping buffer at 55 °C for 30 min, and washed for 1 h in ddH2O. The samples then underwent the IHC procedure62.
Protein–protein interaction assay in tissue slides using proximity ligation assay (PLA)
Human pancreatic tissues on slides were incubated with a pair of primary antibodies [anti-GLP1R antibody (NBP1-97308, Nonus) vs. anti-human SPINT1, and anti-GLP1R antibody vs. anti-HEPSIN]. Twenty-four hours after incubation, samples were washed with PBS three times and then subjected to the examination of protein–protein interaction using the PLA kit (DUO92002, DUO92004, and DUO92008, Thermal Fisher) following the manufacturer’s protocol. Other samples incubated with individual primary antibodies were used as controls. Before covering with fluorescence mounting reagent, samples were incubated with an anti-insulin antibody overnight, washed three times with PBS, and then incubated with a secondary antibody for 30 min.
For human primary islet cells, cells were seeded at a density of 1 × 104 cells per well in eight-well chamber slides (171080, Thermal Fisher), cultured for 2 days, and fixed in 4% paraformaldehyde. Cells were then subjected to PLA analysis using a pair of primary antibodies (anti-GLP1R antibody vs. anti-human SPINT1, anti-GLP1R antibody vs. anti-HEPSIN, and anti-HEPSIN antibody vs. anti-human SPINT1) and the subsequent procedures as described in the previous section.
Islet morphometry
Islet morphometry was performed according to the published methods63. Each mouse pancreas was sectioned at 5 μm in thickness, and one of every 20 serial sections (100 intervals, at least 12 sections per pancreas) or every 60 serial sections (300 intervals, at least 6 sections per pancreas) was subjected to hematoxylin and eosin (H&E) staining. Microscopic images (40×) were taken and tiled using Adobe Photoshop to obtain an entire cross-section of the pancreas. The islet area percentage was determined by dividing the islet area (in pixel values) by the total pancreas area using ImageJ software. The islet mass was obtained by multiplying the islet area percentage by the pancreas weight measured via a microbalance. To obtain the β cell area percentage, pancreatic sections were first immunohistochemically stained for insulin, and the full-view pancreas images were obtained as described above. The β cell area percentage was then determined by dividing the insulin-positive area (in pixel values) by the total pancreas area using ImageJ. The β cell mass was calculated by multiplying the percentage of the β cell area by the pancreas weight.
Stable isotope labeling by amino acids in cell culture (SILAC)
The NIT-1 cells underwent stable isotope labeling through cultivation by seven doubling times in 13C6-lysine/13C6-arginine-containing RPMI 1640 media (Thermo Fisher), which served as a heavy isotope-labeled group because it is challenging to label islet cells in living organisms by isotopes64. To measure the level of stable isotope incorporation after seven doubling times, a mass spectrometer was applied to verify the level of heavy isotope incorporation. The results showed that the level of heavy isotope incorporation in the cells was 97–98%. The Spint1fl/fl and Spint1−/− mouse islets were classified as light isotope-labeled groups (n = 4 in each group of mice). Stable heavy isotope-labeled NIT-1 cells and mouse pancreatic islets were individually lysed in RIPA buffer (0.5% Triton X-100, 0.5% NP-40, and 0.1% SDS in 50 mM Tris buffer, pH 7.8) and centrifuged at 17,500 × g at 4 °C for 15 min. The protein concentrations of each sample were measured using Bradford assay (Bio-Rad). The islet cell lysates (Spint1fl/fl or Spint1−/−) were gently mixed with heavy isotope-labeled NIT-1 cell lysates at a protein ratio of 1:1. The lysate mixture was subjected to SDS-PAGE and the running gel was then divided into 15 fractions. The proteins in each gel fraction were incubated in 5 mM of dithiothreitol (Sigma-Aldrich) at 60 °C for 20 min and 20 mM of iodoacetamide (Sigma-Aldrich) at room temperature for 10 min. For protein in-gel trypsinization, the proteins in each gel fraction were incubated in Sequencing Grade Modified Trypsin (10 μg/ml trypsin, Promega) at 37 °C overnight. The samples were acidified using 1% trifluoroacetic acid (TFA; Thermo Fisher), and the peptides were extracted using 3, 50, and 100% acetonitrile (ACN; Thermo Fisher). The extracts were dried by a SpeedVac vacuum (Thermo Fisher). The peptides were resuspended in 0.1% TFA and purified using the ZipTip C18 column (Merck Millipore). The peptides in the column were washed three times with 10 μl of 0.1% TFA and then eluted using 30 μl of 50% ACN with 0.1% TFA65.
Recombinant DNA and siRNA transfection
For recombinant DNA transfection, HEK293T cells were seeded at a density of 5 × 105 cells per well of six-well plates in DMEM, and NIT-1 cells were seeded at a density of 1 × 106 cells per well of six-well plates in RPMI. The next day, the cells were transfected with control pcDNA3.1 (PLKO) or the pcDNA3.1 containing 3xFlag-Glp1r-myc, Spint1-his-myc, or Hepsin-his-myc cDNA using Lipofectamine 3000 (STEM00015, Thermo Fisher). In constructing the FLAG-tagged GLP1R, a GLP1R cDNA (RC211333, OriGene Technologies) was inserted into a mammalian expression vector (p3xFLAG-Myc-CMV-24), which contained three FLAG tags at the N-terminal of GLP1R, along with a c-Myc tag at the C-terminal. DNA sequencing analysis confirmed the GLP1R sequence integrity and ensured nucleotides were in the correct open reading frame. For siRNA transfection, human primary islet cells, MIN6, and NIT-1 cells were seeded at a density of 1 × 106 cells per well of six-well plates and transfected with scramble siRNA and siSpint1 or siHepsin (D-001210, M-042266 and M-046311, Dharmacon) using Lipofectamine 3000. Transfected cells were grown in their media with 10% FBS for 48 h.
Intracellular cAMP detection
Cells or 100 mouse islets were pre-treated for intracellular cAMP detection with 2.5 mM PAR2 antagonist (nt-YRLLSF-ct, Mission Biotech) for 4 h with serum-free media to block the pre-treatment endogenous cAMP background. Cells were treated with 25 nM of the GLP1R agonist (Ex4) for 3 min. Cells were washed with PBS twice and lysed using 100 μl lysis buffer (1% Triton X-100 with 0.1 M HCl). Cell lysates were centrifuged at 17,500 × g at 4 °C for 15 min. The supernatants were collected and subjected to the measurement of the protein concentrations using a Bradford assay (Bio-Rad). Cell lysates with equal protein amounts per sample were taken to detect cAMP levels using the cAMP ELISA kit (Enzo), following the manufacturer’s protocol. The cAMP levels were statistically calculated from three independent experiments.
Insulin secretion measurement
The siRNA-transfected MIN6 cells were refreshed with low-glucose (2.8 mM glucose) DMEM and cultured for two hours. One set of cells was then treated with high-glucose DMEM (16.7 mM glucose). The other set of cells was refreshed with low-glucose DMEM as a control. Forty minutes after medium refreshment, the conditioned media of the transfectants were collected and centrifuged at 600 × g for 5 min. The supernatants were then collected, diluted 200 times, and subjected to the measurement of the insulin levels using an insulin ELISA kit (Mercodia).
Statistical analysis
A one-way ANOVA with Tukey’s post hoc test, a two-way ANOVA followed by Sidak’s multiple comparison analysis, and a two-sided Student’s t-test and Welch’s t-test was performed using GraphPad Prism 8 (GraphPad Software, CA). Data were statistically calculated from at least three independent experiments and presented as mean ± SEM; p values less than 0.05 were considered significant in all studies. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
We are grateful for the technical services of the Transgenic Mouse Model Core Facility of the National Core Facility for Biopharmaceuticals, National Science and Technology Council, Taiwan, and the Gene Knockout Mouse Core Laboratory of the National Taiwan University Center of Genomic Medicine. We appreciate the services of the First Core Laboratory of the National Taiwan University College of Medicine. We thank Mr. Cheng-Ju Wang for mouse maintenance; Dr. Shian-Wei Chen, Dr. Siow-Wey Hee, Dr. Hsiao-Lin Lee, and Chen-Wei Lin for technical help in isolating and culturing islet cells; Dr. Lee-Ming Chuang (Department of Internal Medicine, National Taiwan University Hospital) and Dr. Susumu Seino (Division of Molecular and Metabolic Medicine, Kobe University Graduate School of Medicine) for providing MIN6 cells, and Dr. Yi-Wen Chen (Graduate Institute of Biomedical Sciences, China Medical University, Taiwan) for providing primary human islet cells. We appreciate the proteomics services that were provided by the Academia Sinica Common Mass Spectrometry Facilities for Proteomics and Protein Modification Analysis located at the Institute of Biological Chemistry, Academia Sinica, supported by Academia Sinica Core Facility and Innovative Instrument Project (AS-CFII-111-209). We want to thank the Department of Medical Research staff for providing clinical data from the NTUH-integrative Medical Database. We thank Tzu-Yu Lo for their assistance with the illustrations. M.-S. Lee was supported by National Health Research Institutes (NHRI) grants NHRI-EX109-10725BI and NHRI-EX113-11318BI, the Ministry of Science and Technology (MOST) grants MOST (105-2911-I-002-521, 106-2320-B-002-046-MY3, 108-2320-B-002-024-MY3, 110-2320-B-002-067-MY3, and NSTC 113-2320-B-002-020), National Taiwan University (NTU) grants NTU (105R89612, 107L890504, 110L893503), and NTUH Grant UN110-029, H.-P. Huang was supported by MOST (103-2321-B-002-095, 104-2314-B-002-010-MY3, 107-2320-B-002-058-MY3, 110-2320-B002-060, 111-2320-B-002-050-MY3), S.-W. Lin was supported by MOST (107-2319-B-002-001, 108-2319-B-002-002, 109-2740-B-002-004, 110-2740-B-002-004, 111-2740-B-002-004). I.-S. Yu was supported by MOST (103-2314-B-002-006-MY2, 104-2320-B-002-059-MY3).
Author contributions
H.-H.L. executed this study and wrote the manuscript. I.-S.Y. and S.-W.L. generated the Spint1-deficient mice and animals for this study. M.-S.C. assisted with embryo isolation and histochemistry. T.-J.C. provided MIN6 cells, devices, and experimental skills consultation. Y.-C.C., H.-Y.L., C.-J.K., and P.-H.C. provided concept consultation. T.-C.H. assisted in designing the SILAC protocol and analysis. S.-Y.H. provides human pancreas tissue. K.-W.K. and T.-Y.C. assisted in maintaining the human primary islet cells. H.-P.H. and M.-S.L. conceptualized and supervised the study and edited the manuscript. All authors approved the final manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
The mass spectrometry proteomics data have been deposited and are available in the ProteomeXchange Consortium via the PRIDE66 partner repository with the dataset identifier PXD039190. Public datasets used in this study include dataset GSE164416 from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164416) under the accession code GSE164416, and the data from the STRING without an accession code (https://string-db.org/cgi/network?taskId=bhlFnsy12Vby&sessionId=buNtCnsKYrNR). Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hsiang-Po Huang, Email: hphuang691290@g.ntu.edu.tw.
Ming-Shyue Lee, Email: mslee2006@ntu.edu.tw.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-54927-2.
References
- 1.Kirchhofer, D. et al. Tissue expression, protease specificity, and Kunitz domain functions of hepatocyte growth factor activator inhibitor-1B (HAI-1B), a new splice variant of HAI-1. J. Biol. Chem.278, 36341–36349 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Huang, H.-P. et al. Persistent elevation of hepatocyte growth factor activator inhibitors in cholangiopathies affects liver fibrosis and differentiation. Hepatology55, 161–72 (2012). [DOI] [PubMed]
- 3.Antalis, T. M., Bugge, T. H. & Wu, Q. Membrane-anchored serine proteases in health and disease. Prog. Mol. Biol. Transl. Sci.99, 1–50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chambers, A. P. et al. The role of pancreatic preproglucagon in glucose homeostasis in mice. Cell Metab.25, 927–934.e923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song, Y. et al. Gut-proglucagon-derived peptides are essential for regulating glucose homeostasis in mice. Cell Metab.30, 976–986.e973 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan, Q. et al. Recent advances in incretin-based pharmacotherapies for the treatment of obesity and diabetes. Front. Endocrinol.13, 838410 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klen, J. & Dolzan, V. Glucagon-like peptide-1 receptor agonists in the management of type 2 diabetes mellitus and obesity: the impact of pharmacological properties and genetic factors. Int. J. Mol. Sci.23, 3451 (2022). [DOI] [PMC free article] [PubMed]
- 8.Han, X. & Nieman, M. T. The domino effect triggered by the tethered ligand of the protease activated receptors. Thromb. Res.196, 87–98 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh, A.-K., Yadav, D., Sharma, N. & Jin, J.-O. Dipeptidyl peptidase (DPP)-IV inhibitors with antioxidant potential isolated from natural sources: a novel approach for the management of Diabetes. Pharmaceuticals14, 586 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Docherty, H. M. et al. Relative contribution of PDX-1, MafA and E47/beta2 to the regulation of the human insulin promoter. Biochem. J.389, 813–820 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naya, F. J. et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev.11, 2323–2334 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaneto, H. & Matsuoka, T.-a Role of pancreatic transcription factors in maintenance of mature β-cell function. Int. J. Mol. Sci.16, 6281–6297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanchet, E. et al. Feedback inhibition of CREB signaling promotes beta cell dysfunction in insulin resistance. Cell Rep. Artic.10, 1149–1157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marinho, T. S., Martins, F. F., Cardoso, L. E. M., Aguila, M. B. & Mandarim-de-Lacerda, C. A. Pancreatic islet cells disarray, apoptosis, and proliferation in obese mice. The role of semaglutide treatment. Biochimie193, 126–136 (2022). [DOI] [PubMed] [Google Scholar]
- 15.Wicksteed, B. et al. Conditional gene targeting in mouse pancreatic ß-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes59, 3090–3098 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawaguchi, M. et al. Membrane-bound serine protease inhibitor HAI-1 is required for maintenance of intestinal epithelial integrity. Am. J. Pathol.179, 1815–1826 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikolova, G. et al. The vascular basement membrane: a niche for insulin gene expression and Beta cell proliferation. Dev. Cell10, 397–405 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Zhu, Y., Liu, Q., Zhou, Z. & Ikeda, Y. PDX1, neurogenin-3, and MAFA: critical transcription regulators for beta cell development and regeneration. Stem Cell Res. Ther.8, 240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwaoka, R. & Kataoka, K. Glucose regulates MafA transcription factor abundance and insulin gene expression by inhibiting AMP-activated protein kinase in pancreatic β-cells. J. Biol. Chem.293, 3524–3534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Wigger, L. et al. Multi-omics profiling of living human pancreatic islet donors reveals heterogeneous beta cell trajectories towards type 2 diabetes. Nat. Metab.3, 1017–1031 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Kataoka, H., Kawaguchi, M., Fukushima, T. & Shimomura, T. Hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2): Emerging key players in epithelial integrity and cancer. Pathol. Int68, 145–158 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Szklarczyk, D. et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res.47, D607–D613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang, X. et al. The identification of novel proteins that interact with the GLP-1 receptor and restrain its activity. Mol. Endocrinol.27, 1550–1563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baggio, L. L. & Drucker, D. J. Biology of incretins: GLP-1 and GIP. Gastroenterology132, 2131–2157 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Lamont, B. J. et al. Pancreatic GLP-1 receptor activation is sufficient for incretin control of glucose metabolism in mice. J. Clin. Invest.122, 388–402 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marzook, A., Tomas, A. & Jones, B. The interplay of glucagon-like peptide-1 receptor trafficking and signalling in pancreatic beta cells. Front. Endocrinol.12, 678055 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beith, J. L., Alejandro, E. U. & Johnson, J. D. Insulin stimulates primary beta-cell proliferation via Raf-1 kinase. Endocrinology149, 2251–2260 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rachdaoui, N. Insulin: the friend and the foe in the development of type 2 diabetes mellitus. Int. J. Mol. Sci. 21, 1770 (2020). [DOI] [PMC free article] [PubMed]
- 29.Song, Z. et al. Epidermal growth factor receptor signaling regulates β cell proliferation in adult mice. J. Biol. Chem.291, 22630–22637 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherwani, S. I., Khan, H. A., Ekhzaimy, A., Masood, A. & Sakharkar, M. K. Significance of HbA1c test in diagnosis and prognosis of diabetic patients. Biomark. Insights11, 95–104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tabák, A. G., Herder, C., Rathmann, W., Brunner, E. J. & Kivimäki, M. Prediabetes: a high-risk state for diabetes development. Lancet379, 2279–2290 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barnett, A. DPP-4 inhibitors and their potential role in the management of type 2 diabetes. Int. J. Clin. Pr.60, 1454–1470 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Li, S. et al. Hepsin enhances liver metabolism and inhibits adipocyte browing in mice. Proc. Natl Acad. Sci. USA22, 12359-12367 (2020). [DOI] [PMC free article] [PubMed]
- 34.Tanaka, H. et al. Hepatocyte growth factor activator inhibitor type 1 (HAI-1) is required for branching morphogenesis in the chorioallantoic placenta. Mol. Cell. Biol.25, 5687–5698 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kryvalap, Y. & Czyzyk J. The role of proteases and serpin protease inhibitors in β-cell biology and diabetes. Biomolecules12, 67 (2022). [DOI] [PMC free article] [PubMed]
- 36.El Ouaamari, A. et al. SerpinB1 promotes pancreatic β cell proliferation. Cell Metab.23, 194–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li, S. et al. Hepsin enhances liver metabolism and inhibits adipocyte browning in mice. Proc. Natl Acad. Sci. USA117, 12359–12367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tei, E. et al. Synergistic endocrine induction by GLP-1 and TGF-beta in the developing pancreas. Pancreas31, 138–141 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Goth, C. K. et al. Site-specific O-glycosylation by polypeptide N-acetylgalactosaminyltransferase 2 (GalNAc-transferase T2) co-regulates beta(1)-adrenergic receptor N-terminal cleavage. J. Biol. Chem.292, 4714–4726 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klenk, C., Schulz, S., Calebiro, D. & Lohse, M. J. Agonist-regulated cleavage of the extracellular domain of parathyroid hormone receptor type 1. J. Biol. Chem.285, 8665–8674 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dale, C. & Vergnolle, N. Protease signaling to G protein-coupled receptors: implications for inflammation and pain. J. Recept Signal Transduct. Res.28, 29–37 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Nieberler, M., Kittel, R. J., Petrenko, A. G., Lin, H. H. & Langenhan, T. Control of adhesion GPCR function through proteolytic processing. Handb. Exp. Pharm.234, 83–109 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Tafesse, F. G. et al. GPR107, a G-protein-coupled receptor essential for intoxication by Pseudomonas aeruginosa exotoxin A, localizes to the Golgi and is cleaved by furin. J. Biol. Chem.289, 24005–24018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mattila, S. O. et al. GPR37 is processed in the N-terminal ectodomain by ADAM10 and furin. FASEB J.35, e21654 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Egozi, A., Bahar Halpern, K., Farack, L., Rotem, H. & Itzkovitz, S. Zonation of pancreatic acinar cells in diabetic mice. Cell Rep.32, 108043 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park, S. S. et al. Therapeutic potential of alpha-1 antitrypsin in type 1 and type 2 diabetes mellitus. Medicina57, 397 (2021). [DOI] [PMC free article] [PubMed]
- 47.Kalis, M., Kumar, R., Janciauskiene, S., Salehi, A. & Cilio, C. M. alpha 1-antitrypsin enhances insulin secretion and prevents cytokine-mediated apoptosis in pancreatic beta-cells. Islets2, 185–189 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Kryvalap, Y. & Czyzyk, J. The role of proteases and serpin protease inhibitors in beta-cell biology and diabetes. Biomolecules12, 67 (2022). [DOI] [PMC free article] [PubMed]
- 49.Kassem, D. H., Adel, A., Sayed, G. H. & Kamal, M. M. A novel SERPINB1 single-nucleotide polymorphism associated with glycemic control and beta-cell function in Egyptian type 2 diabetic patients. Front. Endocrinol.11, 450 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yusta, B. et al. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab.4, 391–406 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Abdulreda, M. H., Rodriguez-Diaz, R., Caicedo, A. & Berggren, P. O. Liraglutide compromises pancreatic β cell function in a humanized mouse model. Cell Metab.23, 541–546 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Farhat, B. et al. Small human islets comprised of more beta-cells with higher insulin content than large islets. Islets5, 87–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lehmann, R. et al. Superiority of small islets in human islet transplantation. Diabetes56, 594–603 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Huang, H. H., Novikova, L., Williams, S. J., Smirnova, I. V. & Stehno-Bittel, L. Low insulin content of large islet population is present in situ and in isolated islets. Islets3, 6–13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hogan, M. F. & Hull, R. L. The islet endothelial cell: a novel contributor to beta cell secretory dysfunction in diabetes. Diabetologia60, 952–959 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li, W. et al. Small islets transplantation superiority to large ones: implications from islet microcirculation and revascularization. J. Diabetes Res.2014, 192093 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buteau, J., Foisy, S., Joly, E. & Prentki, M. Glucagon-like peptide 1 induces pancreatic β-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes52, 124–132 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Ling, Z. et al. Glucagon-like peptide 1 receptor signaling influences topography of islet cells in mice. Virchows Arch.438, 382–387 (2001). [DOI] [PubMed] [Google Scholar]
- 59.Xu, H. et al. Mechanisms for the control of matriptase activity in the absence of sufficient HAI-1. Am. J. Physiol. Cell Physiol.302, C453–C462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang, H. P. et al. Gene targeting and expression analysis of mouse Tem1/endosialin using a lacZ reporter. Gene Expr. Patterns11, 316–326 (2011). [DOI] [PubMed] [Google Scholar]
- 61.González-Mariscal, I. et al. Human CB1 receptor isoforms, present in hepatocytes and β-cells, are involved in regulating metabolism. Sci. Rep.6, 33302 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ehrenberg, A. J. et al. A manual multiplex immunofluorescence method for investigating neurodegenerative diseases. J. Neurosci. Methods339, 108708 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marinho, Td. S., Aguila, M. B. & Mandarim-de-Lacerda, C. A. Pancreatic islet stereology: estimation of beta cells mass. Int. J. Morphol.37, 1331–1334 (2019). [Google Scholar]
- 64.Ong, S. E. The expanding field of SILAC. Anal. Bioanal. Chem.404, 967–976 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Zhong, J. et al. TSLP signaling network revealed by SILAC-based phosphoproteomics. Mol. Cell. Proteom.11, M112.017764 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res.50, D543–D552 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The mass spectrometry proteomics data have been deposited and are available in the ProteomeXchange Consortium via the PRIDE66 partner repository with the dataset identifier PXD039190. Public datasets used in this study include dataset GSE164416 from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164416) under the accession code GSE164416, and the data from the STRING without an accession code (https://string-db.org/cgi/network?taskId=bhlFnsy12Vby&sessionId=buNtCnsKYrNR). Source data are provided with this paper.