

Abstract
Large language models trained on sequence information alone can learn high level principles of protein design. However, beyond sequence, the three-dimensional structures of proteins determine their specific function, activity, and evolvability. Here we show that a general protein language model augmented with protein structure backbone coordinates can guide evolution for diverse proteins without needing to model individual functional tasks. We also demonstrate that ESM-IF1, which was only trained on single chain structures, can be extended to engineer protein complexes. Using this approach, we screened ~30 variants of two therapeutic clinical antibodies used to treat SARS-CoV-2 infection and achieved up to 25-fold improvement in neutralization and 37-fold improvement in affinity against antibody-escaped viral variants-of-concern BQ.1.1 and XBB.1.5, respectively. These findings highlight the advantage of integrating structural information to identify efficient protein evolution trajectories without requiring any task-specific training data.
Introduction
Evolution generates diverse proteins at the level of biological sequences by exploring a vast search space of potential mutations and acquiring those that improve fitness. However, it is the three-dimensional structure encoded by these sequences that ultimately determines the function and activity of a protein. Consequently, as proteins accumulate mutations, they undergo corresponding structural changes, which in turn facilitate functional adaptations (1).
In the laboratory, this tendency for greater sequence change to cause structural divergence poses a major challenge to engineering better proteins via a stepwise evolutionary process. Mutations added in sequential rounds of artificial evolution are increasingly likely to destabilize the structure and therefore diminish the protein’s evolvability (2). Identifying functionally beneficial mutations is also challenged by the fact that almost all mutations to a prototypical protein are deleterious, or at best neutral, and only a rare subset are beneficial on its fitness landscape (3–8). In total, these phenomena can often reduce the evolutionarily accessible paths and make evolution more susceptible to local fitness optima (9, 10), further complicating attempts to increase fitness.
To address both the structural constraints of protein design and the high dimensionality of the mutational search space, we utilized a general protein language model augmented with structural information and trained across millions of non-redundant single sequence-structure pairs on the sequence recovery task, ESM-IF1 (11). Most simply, the model considers the inverse task of that performed by many of the recent powerful structure-prediction tools, including AlphaFold and ESMFold (12, 13): prediction of a sequence that will adopt the fold of a desired target structure (Figure 1a). This is accomplished by predicting the identity of an amino acid given both the preceding amino acid sequence (referred to as autoregressive modeling) and the entire structure’s backbone coordinates (Methods). Thus, sequences assigned high likelihood scores by the structure-informed language model are expected to fold into the backbone of the input structure with high confidence (Figure 1b).
Figure 1: Guiding evolution of diverse proteins with a structure-guided language model.
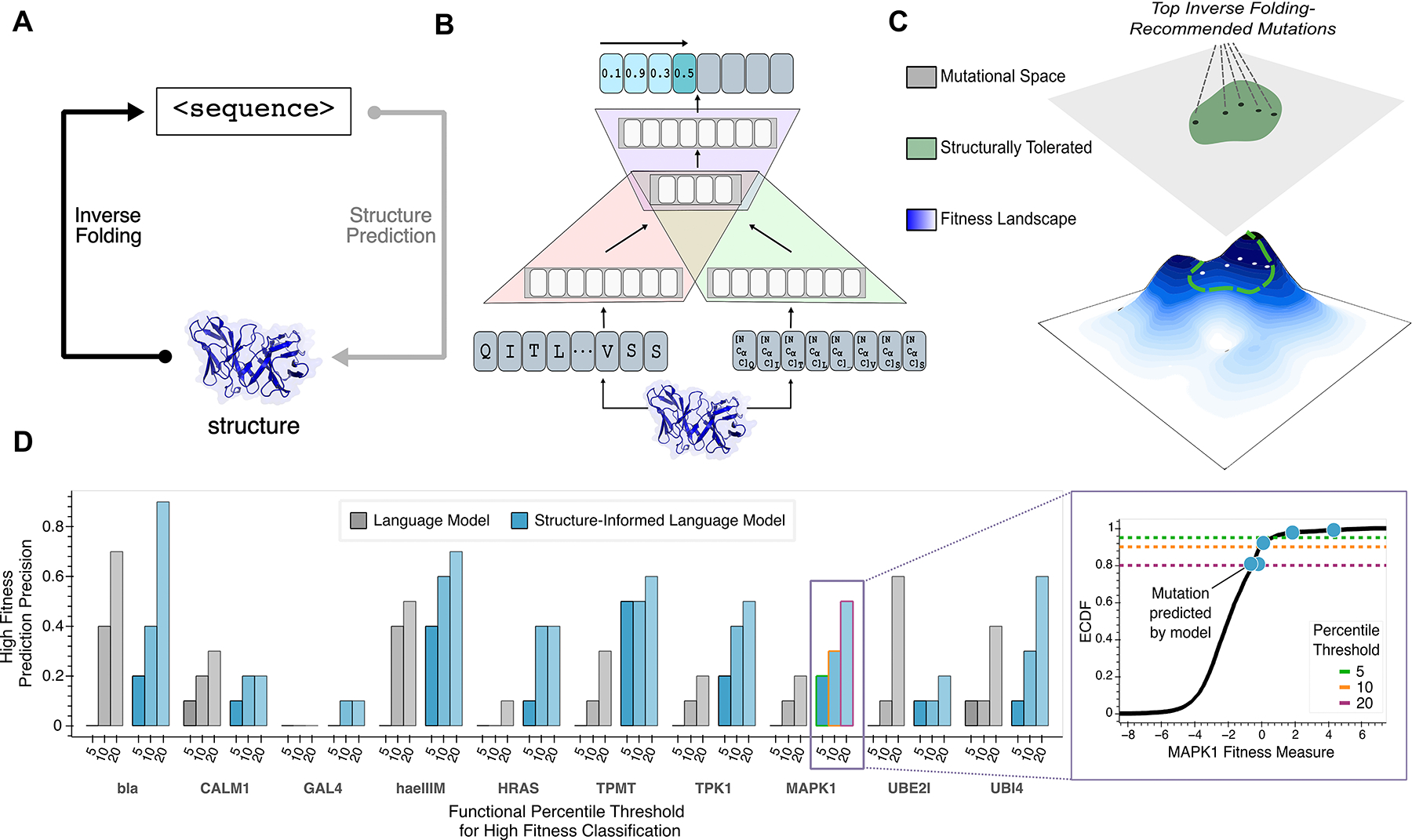
(A) The sequence design problem refers to the prediction of a protein amino acid sequence that will adopt the fold of a given three-dimensional backbone structure; this is conceptually analogous to the inverse problem solved by structure prediction tools like AlphaFold (12). (B) A hybrid autoregressive model (11) integrates amino acid values and backbone structural information to evaluate the joint likelihood over all positions in a sequence. Amino acids from the protein sequence are tokenized (red), combined with geometric features extracted from a structural encoder (green), and modeled with an encoder-decoder transformer (purple). (C) Our structure-guided framework for protein design indirectly explores the underlying fitness landscape, without modeling a specific definition of fitness or requiring any task-specific training data, by constraining the search space to regions where the backbone fold preserved. (D) High fitness sensitivity analysis reveals that multimodal input improves language model performance compared to sequence-only input across 10 proteins from diverse protein families (left). ‘High Fitness Prediction Precision’ is the fraction of the top ten single amino acid substitution predictions that are experimentally determined to confer high protein fitness, defined as having an activity level above the specified percentile threshold among all experimentally screened variants. A representative plot (right) demonstrates this metric for assessing enrichment of high-fitness MAPK1 mutations. Given the vastness of the search space, finding any function-enhancing variant is valuable for most practical settings, and thus only successfully predicted mutations highlighted (blue) on the empirical cumulative density function (ECDF) of the experimental data (black). The three different thresholds, as defined by percentiles, are also shown as dashed lines. Structure-informed language model predictions are more enriched, on average, for high fitness variants across various tested thresholds for high fitness classification. bla, Beta-lactamase TEM; CALM1, Calmodulin-1; haeIIIM, Type II methyltransferase M.HaeIII; HRAS, GTPase HRas; MAPK1, Mitogen-activated protein kinase; TMPT, Thiopurine S-methyltransferase; TPK1, Thiamin pyrophosphokinase 1; UBI4, Polyubiquitin; UBE2I, SUMO-conjugating enzyme UBC9
The problem of designing a sequence for a desired target structure, such as in inverse folding, is considered only in terms of protein folding (14–16), and thus does not guarantee a functional protein (17). A key barrier to finding an optimal solution for this sequence design problem is that often many sequences can fold into a given backbone conformation (18). Our framework for protein design does not model an explicit protein function or definition of protein fitness. Rather, using a structure-guided paradigm, we leverage this sequence-structure degeneracy to indirectly explore the underlying fitness landscape by focusing exploration to regions where the backbone fold of the protein is preserved. We hypothesize that constraining evolution to regimes of high sequence likelihood can serve as an effective prior for high-fitness variants, and thereby improve the efficiency of evolution (Figure 1c).
We reasoned that this approach may be particularly valuable for the evolution of human antibodies, which are used clinically to treat a broad range of diseases (19). Antibodies offer protection by selectively binding to a target antigen involved in pathogenesis and modifying or disrupting its function (20). An important optimization step in the development of most therapeutic human antibodies involves an intensive process to identify amino acid substitutions that further enhance potency and efficacy. Here, we demonstrate this task can be accomplished efficiently with machine learning using an inverse folding model. A central concept of this study is to use the complete structure of the antibody-antigen complex to guide evolution. By conditioning the structure-informed language model on the entire antibody-antigen complex, we sought to enable the discovery of mutations that preserve or enhance the stability of the entire complex, and thus that improve antibody function.
Results
Enriching sequence exploration for high function protein variants across diverse tasks with a structure-informed language model
We evaluated whether adding structural information to a language model can be used to guide protein evolution by predicting mutations that improve a protein’s activity for a specified property without training on or explicitly modeling the task itself. Accordingly, for 10 proteins from diverse families among four organisms, and with functions ranging from enzyme catalysis (TPMT) to oncogenesis (HRAS) to transcriptional regulation (GAL4), we scored variants profiled in large deep mutational scanning experiments (21–30) against the target backbone of the wild-type protein (31–40) to compute sequence log likelihoods (Methods, Table S1). Importantly, these predictions are made in a completely unsupervised setting, with the model never having been trained on any experimental data. To demonstrate the utility for a practical user who wishes to find the most beneficial mutations, we assessed prediction precision by comparing the top scoring variants to their experimentally determined functional activity in the relative context of the entire sequence-fitness landscape.
Within just the set of top ten predictions, we identified numerous high fitness protein variants, out of the thousands of tested for each protein, with experimentally determined activities ranking in the top percentiles of the entire deep mutational scanning screen (Figure 1d). Our analysis demonstrates that conditioning on structural information serves to improve predictive capabilities of protein language models as we successfully recover mutations in the top fifth percentile for 9 out of the 10 proteins compared to just 2 proteins using a state-of-the-art general protein language model trained only on sequence information and specifically for variant prediction (ESM-1v) (41) (Figure 1d). This improvement in prediction also holds with increasingly relaxed thresholds for classification as high-fitness variants.
Based on this experiment, we conclude that structure-based sequence design offers a promising alternative to brute force experimental searches for functionally beneficial mutations. Notably, some of the top mutations predicted are also the same ones discovered from exhaustive experimental exploration. For example, for restriction enzyme haeIIIM, variant Q18E is recommended as one of the top five single amino acid predictions and experimentally ranks as the second-best substitution (and > 5 standard deviations above the mean) out of the nearly 2000 substitutions screened across the endonuclease (30). Another key advantage of our task-independent framework, in addition to being broadly applicable across diverse proteins, is the ability to improve a single protein for multiple desired properties without needing to develop specialized high-throughput assays to screen each independently. From just the top 10 predictions for MAPK1, we identify substitutions Q105M and Y64D, which are experimentally shown to confer resistance to two different oncogenic-targeting MAPK1 kinase inhibitors (24).
Structural information enables state-of-the-art zero-shot antibody mutational effect prediction for language models
To analyze the effectiveness of augmenting a general protein language model with structural information, specifically for antibody variant prediction, we compared the likelihoods of sequences for three antibodies across entire mutational landscapes to corresponding experimental fitness values from a total of five existing mutagenesis datasets. The mutational landscapes of the first two antibodies were evaluated by measuring the scFv equilibrium dissociation constants (KD) of all possible evolutionary intermediates between the inferred germline and somatic sequence of naturally affinity-matured influenza broadly neutralizing antibodies (bnAbs) CR9114 and CR6261, which bind the conserved stem epitope of influenza surface protein hemagglutinin (HA) (42). For both bnAbs, only mutations in the heavy chain, which is responsible for antigen binding, were characterized. The profiled mutational landscape of CR9114 includes all possible combinations of 16 substitutions, whereas that of CR6261 includes all possible combinations of 11 substitutions, totaling 216 = 65,536 and 211 = 2,048 variant antibody sequences respectively. Each of these libraries were screened for binding against two distinct influenza HA subtypes (H1 and H3 for CR9114 and H1 and H9 for CR6261). The fifth dataset assesses the effects of all possible single amino acid substitutions with a deep mutational scan profiling 4,275 mutations in the variable regions for both heavy chain (VH) and light chain (VL) of antibody G6.31 to binding with its ligand, vascular endothelial growth factor A (VEGF-A) (43).
For each dataset, we computed the Spearman correlation between the log likelihood estimated by the structure-informed language model and the experimentally determined binding measure for a given antigen, across all sequences in the mutational library. We scored the likelihood of each candidate sequence in the library using the backbone coordinates of a structure with the mature antibody bound to its target antigen (44–46).
Across all five experimental binding datasets, we found that the structure-informed language model performs better than three other sequence-based methods: i) a general protein language model trained across diverse protein sequences, ESM-1v (41), ii) a specialized antibody language model trained exclusively on sequences sampled from the Observed Antibody Space (OAS) database, AbLang (47), and iii) a site-independent model of mutational frequency curated with extensive antibody sequence alignments, abYsis (48). In nearly all experimental scenarios, supplementing sequence information with the backbone coordinates of the antibody alone, without providing antigen information as input, is sufficient to outperform other sequence-only methods. A notable feature of the autoregressive architecture is that it computes the joint likelihood over all positions in a sequence, making it well-suited to score combinatorial sequence changes. We find that this method can capture complex epistatic interactions, or potential interdependence among individual amino acids, as it performs well on the CR9114 and CR6261 libraries composed of sequences with multiple mutations (Figure 2a,b).
Figure 2: Prediction of antibody-antigen complexes resolves mutational landscapes by implicitly learning features of binding and protein epistasis.
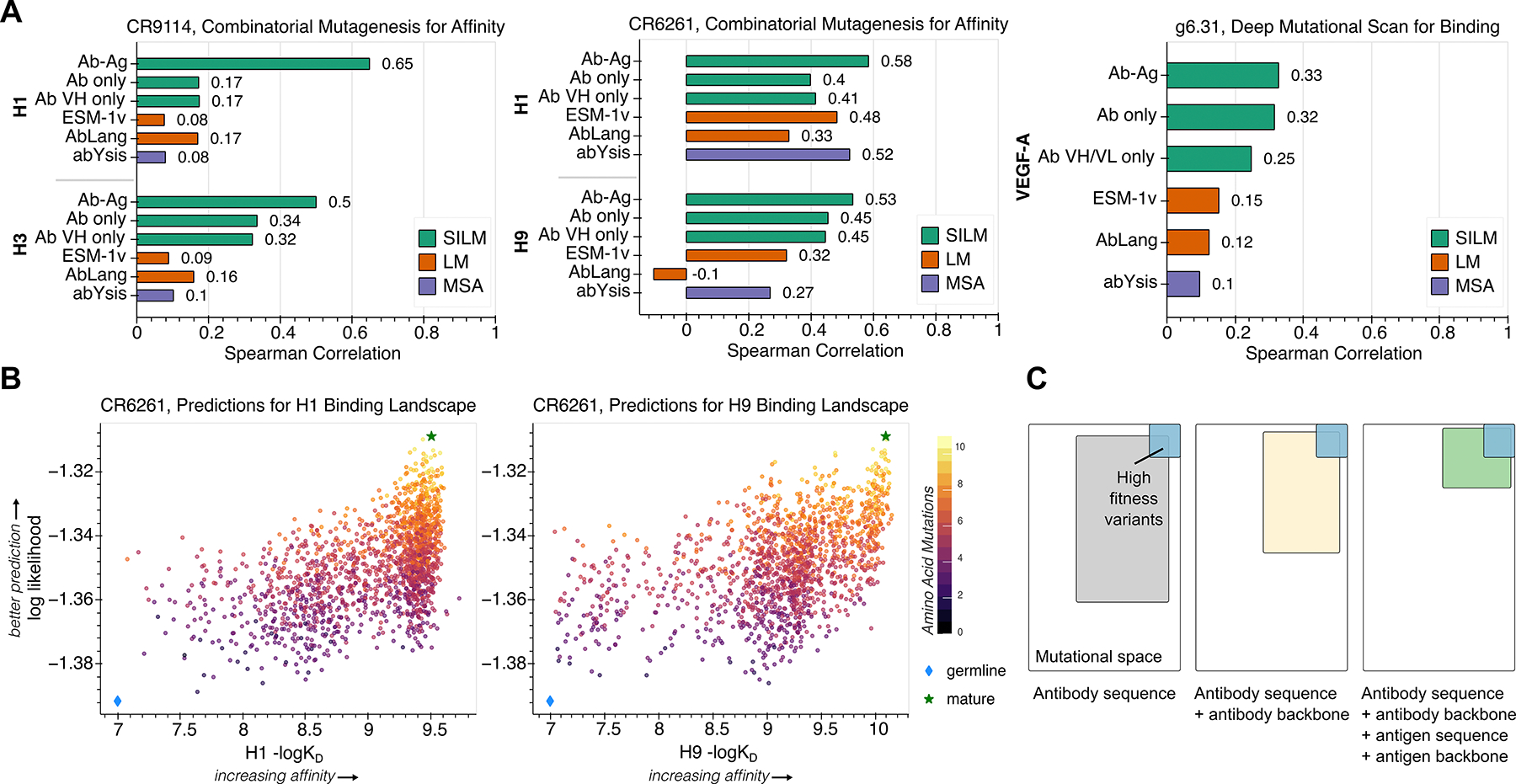
(A) Spearman correlation using the structure-informed language model as well as sequence-based modeling approaches ESM-1v (41), AbLang (47), and abYsis (48) reported for three antibodies screened with corresponding antigens. Bars are colored by the type of model used: SILM, Structure-informed Language Model (green); LM, Language Model (orange); and MSA, Multiple Sequence Alignment (purple). The structure-informed language model was evaluated in three different settings: i) providing the entire antibody variable region and antigen complex (Ab-Ag) ii) providing only the antibody variable region (Ab only), and iii) providing only the single antibody variable region of the chain responsible for binding or being mutated (Ab VH only or Ab VH/VL only). Antibody sequences scored by the structure-informed language model with antigen information were computed using input complexes of CR9114 with H5 HA (PDB 4FQI (44)), CR6261 with H1 HA (PDB 3GBN (45)), and g6.31 with VEGF-A (PDB 2FJG (46)). B) Scatter plots showing predictions against experimentally determined dissociation constants of CR6261 against HA-H1(left) and HA-H9 (right). The germline and mature sequences are highlighted on all plots as indicated in the legend. For visualization, all scatter plots omit points on the lower limit of quantitation. (C) Conceptual illustration of protein language model performance with improved priors. Providing sequence and structural information of both the antibody and antigen enables the structure-informed language model to most efficiently enrich for high fitness antibody variants (top right, blue square) by identifying and guiding focused sequence exploration (green square) away from regimes of mutations destabilizing to the complex.
We achieved the greatest improvement in performance on all five experimental screens by incorporating the structure of both the antibody and antigen (Figure 2a), indicating that the structure-informed model can implicitly learn features of binding (Figure 2c). This result is notable given that the model is only trained on single-chain protein structures, whereas the antibody-antigen complexes we use as inputs are composed of either three (G6.31) or four (CR9114, CR6261) protein chains. The most substantial contribution of antigen information is observed in the case of CR9114-H1, for which the correlation increases from 0.17 with only antibody information to 0.65 with sequence and backbone coordinates of the entire complex. In contrast, this same performance improvement is not observed when the sequence-only general protein language model is provided with additional context of the paired antibody chain or antigen sequence (Figure S4). We find that extending our model beyond the monomeric structures seen during training to protein complexes also performs better for antibody prediction than ProteinMPNN (49), an alternate structure-based deep learning method, which was trained on a dataset that includes multichain protein structures (Figure S5).
Remarkably, these results show that we could even predict the effects of mutations to a cross-reactive antibody on binding to a strain of influenza different than the one used as input to the model. (Figure 2a,b). Despite using a target antibody structure in complex with HA from H5N1 influenza to score CR9114 variants, we obtain correlations of 0.65 and 0.50 with experimental binding data for H1 and H3, respectively. This is particularly striking since the antibody epitope, which spans both HA subunits, only has 67% sequence identity between the H5 strain of the structure used to make predictions (A/Vietnam/1203/2004) and the H1 strain experimentally tested against (A/New Caledonia/20/99) (Figure S6, Table S2). This same cross-reactive predictive capability is observed with CR6261 (Figure 2a), for which the experimentally tested H9 (A/Hong Kong/1073/1999) differs at over a third of the residues in the epitope from the1918 H1N1 influenza strain used in the structure (A/Brevig Mission/1/1918). Although the structure-informed language model cannot learn explicit chemical rules of binding (e.g., hydrogen bonding or disulfide bridge formation) since it does not have access to amino acid side chain atomic coordinates, these results suggest that structural principles like interface packing or potential steric interference are not only implicitly accessible from residue identities, but are also informative for binding prediction.
Our model’s top recommended mutations are made independent of a specific definition of fitness; they simply represent a set of variants with a high likelihood of folding into the input backbone structure. Therefore, our model’s recommendations may also help identify mutations that improve other useful biochemical properties beyond affinity. Impressively, for example, the top recommended mutation to the VL of G6.31 is F83A, which was identified in the original screening study to be particularly interesting as it confers a three-fold increase in VEGF-A binding affinity and a 5°C improvement in melting temperature, despite being 25Å from the antigen and in the antibody framework region. It was determined that the VL F83A substitution induces more compact packing and the site serves as a conformational switch that affects biological activity at the antibody-antigen interface by modulating both interdomain and elbow angle dynamics (43). However, while our model successfully enriches for high fitness variants across many settings, an associated consequence of this structure-based framework is the limited ability to identify mutations that impart beneficial effects by modifying the backbone of the mature antibody.
Engineering therapeutic antibodies for increased potency and resilience
Finally, we aimed to assess if the structure-augmented language model’s predictive capabilities could not only resolve trends on large sets of experimental data, but also enable efficient and successful directed evolution campaigns while testing only a small number (on the order of tens) of variants. To do so, we considered the task of improving the potency and resilience (effectiveness against a virus as it mutates over time) of two mature, clinical monoclonal antibody therapies.
LY-CoV1404 (Bebtelovimab) was isolated from a COVID-19 convalescent donor and binds to the receptor binding domain (RBD) of the SARS-CoV-2 Spike protein (50). It was approved by the U.S. F.D.A. on February 11, 2022 given its activity against both the original Wuhan and Omicron SARS-CoV-2 variants and was the last remaining approved monoclonal antibody therapy withstanding viral evolution (51) until its discontinuation on November 30, 2022 due to antibody evasion by VOC BQ.1.1. (52)
SA58 (BD55–5840) was isolated from a vaccinated individual and is one of two RBD-targeting neutralizing antibodies (NAb) in a rationally developed antibody cocktail. SA58 alone retained efficacy against all Omicron subvariants, including in vivo protection against BA.5 (53, 54) and was shown to be effective as a post-exposure prophylaxis in a clinical study (55).
For both antibody engineering campaigns, we used the structure-informed language model to compute likelihoods of all ~4,300 possible single-residue substitutions in the VH or VL regions of the antibody. In the first round of evolution, we selected only the top ten predictions at unique residues in each chain for experimental validation. An important practical benefit of our method is the ability to optimize against measures of fitness most relevant to the protein’s downstream function, such as viral neutralization or receptor agonism, rather than being limited to indirect surrogate measures like affinity that are more amenable to high-throughput screening (4, 56). We leverage this advantage to directly evolve these antibodies for their ability to more potently neutralize SARS-CoV-2 pseudotyped lentivirus.
Variants recommended by the structure-informed language model were assessed by comparing the half-maximal inhibitory concentration (IC50) relative to the wild-type antibody. Remarkably, although we chose to only test 20 single-site substitutions for each of the two clinical monoclonal antibody therapies, approximately one-third of them improved neutralizing potency. Notably, several of these antibody variants improve neutralization IC50 by over 3-fold with just a single amino acid change (Figure 3a, Supplementary Data 1). We also observe greater variance in changes to neutralization for SA58 than Ly-1404, which may be reflective of intrinsic differences in the number of residues critical for and participating in neutralization and binding, even beyond the antigen interface.
Figure 3. Evolution of antibodies with a structure-informed language model improves neutralization potency and resilience.
(A) Each point represents the fold-change in IC50 of pseudovirus neutralization for antibody variants with single amino acid mutations. Antibodies are tested against the viral strain represented in the input structure (Ly1404- Wuhan, SA58-BA.1 Omicron). A dashed line is shown at fold-change of 1 corresponding to no change. Improved antibody potency is defined as 1.1-fold or higher improvement in IC50 compared to wild-type. (B) Conceptual representation of viral evolution. Selection for immune evasion drives antibody escape, which fundamentally represents a dynamic change in the underlying fitness landscape for the antibody. This antigenic drift displaces a potent antibody from a peak on the previous fitness landscape (left) to a new starting point at lower activity (right). (C) Strip plots visualizing antibody evolution across two rounds. Each point shows the corresponding fold-change in IC50 of pseudovirus neutralization for a designed variant and is colored according to the number of mutations it has (1–8). Consistent with preserving backbone fold, all 55 designed variants across both antibody evolutionary campaigns could be expressed. All round 1 variants are only composed of only single amino acid changes while beneficial mutations are combined in round 2. All round 2 variants have improved neutralization activity compared to their respective wild-type antibody (dotted line). (D) Pseudovirus neutralization curves are shown for the most potent evolved antibody variant, consisting of mutations annotated to the left. The top LY-CoV1404 variant, bearing seven amino acid substitutions in VH, achieves a 25-fold improvement in neutralization against BQ.1.1 (top). The top SA58 variant, bearing single amino acid mutations in both VH and VL, achieves a 14-fold improvement in neutralization against BQ.1.1 (bottom). (E) Residues at which mutations improve neutralization against either the structure-encoded strain, BQ.1.1, or both viral strains are highlighted with spheres for antibodies LY-CoV1404 (PDB 7MMO (50)) and SA58 (PDB 7Y0W (54)). Notably, beneficial mutations are identified both within the binding interface as well distal to the antigen. Neutralization enhancing mutations are labeled in Figure S10.
Prompted by recent evidence showing that conservation of the overall RBD structure is robust to SARS-CoV-2 evolution (57), we next sought to determine whether we could also evolve the previously mature antibodies against SARS-CoV-2 BQ.1.1, the variant responsible for diminished therapeutic efficacy. Although the antibodies were previously effective, a change in antigen conceptually represents a fundamental shift in the underlying fitness landscape (Figure 3b. Accordingly, the baseline neutralization activity against BQ.1.1 for LY-1404 and SA58 dropped to IC50 values in the nanomolar range. However, from the same set of 20 single amino acid substitutions to LY-CoV1404, we found that nearly half improve neutralization of variant BQ.1.1. In addition to a high success rate, we also found multiple of these mutations provided a large magnitude of improvement. Several single amino acid substitutions to LY-CoV1404 individually result in over a 4-fold improvement while the most beneficial mutation to SA58 results in a nearly 5-fold improvement (Figure 3c).
Taken together, approximately two-third and one-half of tested single amino acid substitutions to LY-CoV1404 and SA58, respectively, were beneficial for neutralization of either the original strain or BQ.1.1. Interestingly, for both antibodies, the most potent single amino acid mutations were distinct to the two different strains tested (Figure S7). These results reinforce that, despite all being predicted to have the same backbone fold, the top set of designed variants feature functional diversity and can be used for distinct notions of protein fitness.
A common challenge in directed evolution is contending with the combinatorial explosion of possible sequences that emerges from trying to combine a set of individually beneficial mutations. In the second round of evolution, we simply use the structure-informed model again to acquire up to five top-scoring combinations of mutations to each antibody chain (Methods). Notably, across both evolutionary trajectories, all 11 LY-CoV1404 and 5 SA58 antibody designs with multiple mutations have IC50 values better than wild-type, with many designs showing synergistic effects upon combination. For example, just a single amino acid mutation in each of the two chains of SA58 leads to over an 14-fold improvement (Figure 3c,d). Similarly, the most potent evolved design of LY-CoV1404 is a combination of seven of the eight beneficial single amino acid substitution to the VH and improves neutralization 25-fold (Figure 3d). Critically, these improvements to neutralizing potency against BQ.1.1 do not sacrifice potency against the original strains. We found that the top SA58 design against BQ.1.1 after the second round of evolution also improves BA.1 neutralization over 3-fold (Supplementary Data 1).
To rigorously evaluate the benefit of adding structural information, we also performed identical evolutionary campaigns for both LY-CoV1404 and SA58 using an ensemble of general sequence-only protein language models to recommend variants (58) (Methods), an approach which has previously been experimentally validated in applications for antibody engineering and serves as a competitive unsupervised baseline. Consistent with the computational results, we found that the structure-informed language model leads to final antibody designs with substantially greater overall magnitudes in improvement (25-fold vs 2-fold for LY-CoV1404, 14-fold vs 4-fold for SA58) (Figure S8). We particularly observe that, in comparison to our structurally-informed evolution campaigns, combinations of language model-recommended beneficial mutations have limited additive effects. These results further underscore the value of selecting mutations from the outset that are known to be structurally compatible, and thereby enable a more efficient ascent up the fitness landscape.
Additional characterization of evolved antibodies
To further characterize the basis for enhanced neutralization of SARS-CoV-2 VOC BQ.1.1, we tested the binding affinity of all variant antibodies to RBD as bivalent IgG using biolayer interferometry (BLI) to obtain the apparent dissociation constant (KD,app). For LY-CoV1404, a total of 23 designs across both rounds of evolution exhibited improved viral neutralization, and each of these improved antibodies were confirmed to have increased apparent binding affinities, up to ~27-fold. Interestingly, however, we found improved apparent affinity to not be a sufficient condition for improved neutralization potency as four additional model-recommended mutations, which were neutral or deleterious to neutralization, actually improved binding. Across all variants there is a Spearman correlation of 0.45 between fold-change in IC50 and fold-change in KD,app (Figure 4a,b).
Figure 4: Antibodies evolved for high potency also exhibit improved affinity.
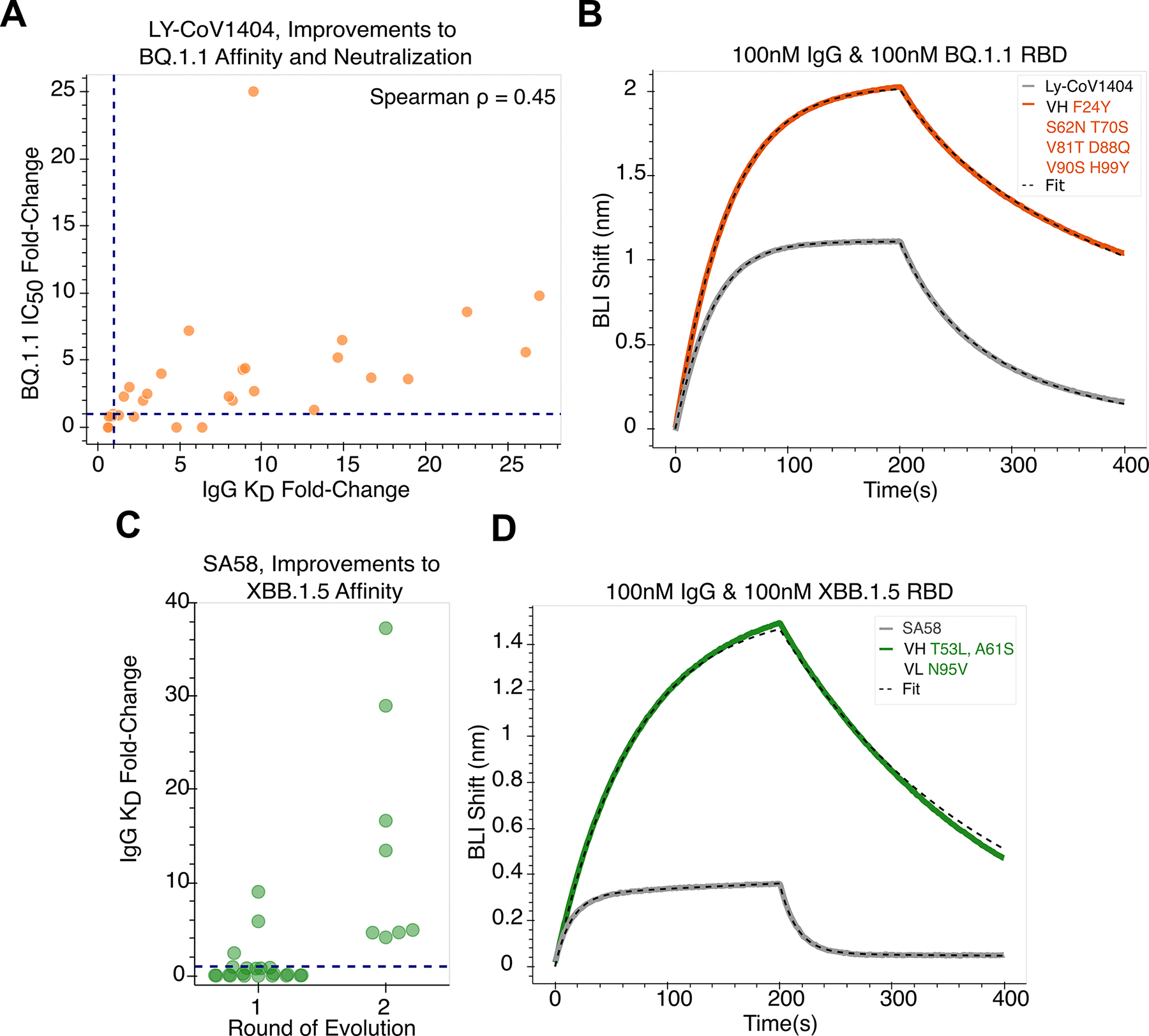
(A) LY-CoV1404 antibody variants show a Spearman correlation of 0.45 between apparent affinity fold-change and potency fold-change. Improved affinity is observed to be necessary but not sufficient for improved neutralization activity. Four variants exhibit improved affinity but do not enhance neutralization. All variants with improved neutralization also display improved affinity. The top LY-CoV1404 design with a 25-fold improvement in neutralization has a 9.5-fold improvement in affinity to BQ.1.1 RBD, as measured using BLI. (C) SA58 antibodies evolved for improved potency against BQ.1.1 also exhibit improved affinity against VOC XBB.1.5, up to 37-fold. (B, D) Representative traces of BLI binding assays for LY-CoV1404 and SA58 variants with improved affinity.
We similarly screened the SA58 variants for binding to the RBD of BQ.1.1. However, since the KD,app of the wildtype antibody as IgG was already sub-picomolar, further improvements to binding were below the limit of quantitation and indistinguishable using this measure. Given this strong binding affinity of wildtype SA58 to BQ.1.1 RBD, we also screened this same set of variants against emerging VOC XBB.1.5 and observe improvements in KD,app up to 37-fold (Figure 4c,d).
By testing several top affinity-matured designs in a polyspecificity assay, we also confirmed that improvements in binding are not mediated by generalized enhancements of non-specific interactions (Figure S9a). In this assay, we observed no substantial changes in off-target binding of the evolved antibodies to membrane soluble proteins, particularly within a therapeutically viable range (as defined by controls of clinically approved antibodies with recorded high and low polyspecificity). Furthermore, we found no correlation between fold-change in polyspecificity and affinity fold-change (Figure S9b).
Analysis of evolutionary exploration
Confronted by the large number of possible mutations, traditional experiment-based methods for antibody affinity maturation often restrict the mutational search space to only a few regions of the antibody. Specifically, binding optimization efforts are typically focused within the complementarity determining regions (CDR), which are hotspots for natural somatic hypermutation. However, using our unbiased approach to consider all regions of the variable domain allows for many discoveries that may be less intuitive to a rational designer. For example, the most beneficial substitutions to LY-CoV1404, VH F24Y and VH V90S, are located within framework regions and positioned distally from the binding interface (Figure S10, Table S3). Interestingly, they both improve neutralization of BQ.1.1 by over 4-fold and are not deleterious to Wuhan neutralization. In other cases, the structure-informed language model also successfully predicts beneficial substitutions using residues rarely observed among human antibody sequences. Substitution VL N95V in SA58, which improves neutralization approximately 5-fold against BQ.1.1, is mediated by the incorporation of a valine observed in only 0.7% of human antibody sequences at that position and enhances antibody-antigen contact. While the model is capable of successfully making novel predictions, in some instances it also does suggest reverting residues to ones frequently selected for in natural somatic hypermutation. Mutation VL F51Y in LY-CoV1404 changes a phenylalanine observed in just 5% of sequences to a tyrosine observed in 86% of sequences. However, this variant results in no change to Wuhan neutralization. Overall, these results highlight the value in augmenting a language model with structural information to evolve antibodies and proteins complexes.
Discussion
The discovery of mutations that improve protein function is inherently challenging due to the large sequence search space and complex rules that govern the relationship between sequence and function, such as stability or environmental selection pressures. We show that an inverse folding protein language model informed with the sequence and backbone structural coordinates of a protein can considerably improve directed evolution efforts by serving as an improved prior compared to sequence-only deep learning methods. Importantly, we highlight that a structure-guided approach can interrogate protein fitness landscapes indirectly, without needing to explicitly model individual functional tasks or properties, making it broadly applicable to proteins across diverse settings ranging from enzyme catalysis to antibiotic and chemotherapy resistance (Figure 1d). We also demonstrate the structure-informed language model generalizes to multimeric proteins, despite being trained only on single-chain proteins, through its ability to implicitly learn features of binding. This result is particularly remarkable considering the model has no access to amino acid side chain atoms, coordinates, or bond information.
Equipped with these capabilities, we evolve clinical therapeutic antibodies and identify several mutations which act synergistically to improve antibody potency and resilience against emerging variants of concern. In the context of pandemics and emergency-use situations, where monoclonal antibody therapies are limited in supply and vulnerable to resistance from viral evolution, the ability to rapidly make improvements in potency with a general method could have major clinical and economic implications.
Machine learning has transformed protein engineering across several design objectives. Methods that design sequences for de novo proteins with specified folds have enabled entirely new capabilities to address previously intractable problems in many settings (49, 59–62). Here, we consider the directed evolution problem of improving a desired function of an existing protein. In comparison to fourteen other promising machine learning-guided protein evolution methods used to experimentally guide directed evolution campaigns on various proteins (8, 56, 58, 63–73), our success rates of generating designs with functional activity better than the wildtype protein compare favorably, while not requiring any assay-labeled fitness data to use for training or task-specific model supervision (Figure S11, Supplementary Data 5). These results support the notion that the protein’s structure, itself, may be used in lieu of learned surrogate functions of fitness. By eliminating the reliance on any initial data collection or prior knowledge of the protein, our structure-informed method has the potential to accelerate entire evolutionary campaigns.
Computational methods like the one we propose have the opportunity to democratize protein engineering efforts. Not only is our approach more efficient than conventional resource-intensive techniques that experimentally test the effects of all single-residue changes on biochemical functions like binding affinity, but consequently it enables directed evolution based on properties that are not easily measured at scale or that are incompatible with high-throughput screening. Overcoming these limitations, we anticipate our structure-based paradigm will be useful for evolving proteins across many domains.
Supplementary Material
Acknowledgments
We would like to thank Duo Xu, Soohyun Kim, and the members of the Kim lab for helpful discussions on this project. We are also grateful for assistance from D. Xu with protein graphics.
Funding:
V.R.S acknowledges the support of the Stanford University Medical Scientist Training Program grants (T32-GM007365 and T32-GM145402). V.R.S. and T.U.J.B. are both supported by the Sarafan ChEM-H Chemistry/Biology Interface Training Program. T.U.J.B. is also supported by the Knight-Hennessy Graduate Scholarship Fund and a CIHR Doctoral Foreign Study Award (FRN:170770). B.L.H is supported by the Stanford Science Fellows Program. This work was supported by the Virginia & D.K. Ludwig Fund for Cancer Research and the Chan Zuckerberg Biohub.
Footnotes
Competing interests
V.R.S., B.L.H., and P.S.K. are named as inventors on a patent application applied for by Stanford University and the Chan Zuckerberg Biohub entitled “Antibody Compositions and Optimization Methods”. B.L.H acknowledges outside interest in Prox Biosciences as a scientific co-founder.
Data, code, and materials availability:
Open-source code is provided that allows a user to easily evaluate the structure-informed language model on protein complexes of interest and predict beneficial amino acid substitutions at https://github.com/varun-shanker/structural-evolution. This is implemented as a simple call to a python script with a pdb/cif file and the target chain to be mutated as the main arguments. Code and scripts used in this study have been deposited to Zenodo at https://doi.org/10.5281/zenodo.11260509. Raw data and files used in this study have deposited to Zenodo at https://doi.org/10.5281/zenodo.11260318.
References
- 1.Chothia C, Lesk AM, The relation between the divergence of sequence and structure in proteins. EMBO J. 5, 823–826 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bloom JD, Labthavikul ST, Otey CR, Arnold FH, Protein stability promotes evolvability. Proc. Natl. Acad. Sci. 103, 5869–5874 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Axe DD, Foster NW, Fersht AR, A Search for Single Substitutions That Eliminate Enzymatic Function in a Bacterial Ribonuclease. Biochemistry 37, 7157–7166 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Romero PA, Arnold FH, Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 10, 866–876 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shafikhani S, Siegel RA, Ferrari E, Schellenberger V, Generation of large libraries of random mutants in Bacillus subtilis by PCR-based plasmid multimerization. BioTechniques 23, 304–310 (1997). [DOI] [PubMed] [Google Scholar]
- 6.Guo HH, Choe J, Loeb LA, Protein tolerance to random amino acid change. Proc. Natl. Acad. Sci. 101, 9205–9210 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rennell D, Bouvier SE, Hardy LW, Poteete AR, Systematic mutation of bacteriophage T4 lysozyme. J. Mol. Biol. 222, 67–88 (1991). [DOI] [PubMed] [Google Scholar]
- 8.Ogden PJ, Kelsic ED, Sinai S, Church GM, Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 366, 1139–1143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poelwijk FJ, Kiviet DJ, Weinreich DM, Tans SJ, Empirical fitness landscapes reveal accessible evolutionary paths. Nature 445, 383–386 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Wittmann BJ, Yue Y, Arnold FH, Informed training set design enables efficient machine learning-assisted directed protein evolution. Cell Syst. 12, 1026–1045.e7 (2021). [DOI] [PubMed] [Google Scholar]
- 11.Hsu C, Verkuil R, Liu J, Lin Z, Hie B, Sercu T, Lerer A, Rives A, “Learning inverse folding from millions of predicted structures” in Proceedings of the 39th International Conference on Machine Learning (PMLR, 2022; https://proceedings.mlr.press/v162/hsu22a.html), pp. 8946–8970. [Google Scholar]
- 12.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, Smetanin N, Verkuil R, Kabeli O, Shmueli Y, dos Santos Costa A, Fazel-Zarandi M, Sercu T, Candido S, Rives A, Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023). [DOI] [PubMed] [Google Scholar]
- 14.Yue K, Dill KA, Inverse protein folding problem: designing polymer sequences. Proc. Natl. Acad. Sci. 89, 4163–4167 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS, High-Resolution Protein Design with Backbone Freedom. Science 282, 1462–1467 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Dahiyat BI, Mayo SL, De Novo Protein Design: Fully Automated Sequence Selection. Science 278, 82–87 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Lim WA, Sauer RT, Alternative packing arrangements in the hydrophobic core of λrepresser. Nature 339, 31–36 (1989). [DOI] [PubMed] [Google Scholar]
- 18.Lau KF, Dill KA, Theory for protein mutability and biogenesis. Proc. Natl. Acad. Sci. 87, 638–642 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter PJ, Lazar GA, Next generation antibody drugs: pursuit of the “high-hanging fruit.” Nat. Rev. Drug Discov. 17, 197–223 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Schroeder HW, Cavacini L, Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 125, S41–S52 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livesey BJ, Marsh JA, Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations. Mol. Syst. Biol. 16, e9380 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weile J, Sun S, Cote AG, Knapp J, Verby M, Mellor JC, Wu Y, Pons C, Wong C, van Lieshout N, Yang F, Tasan M, Tan G, Yang S, Fowler DM, Nussbaum R, Bloom JD, Vidal M, Hill DE, Aloy P, Roth FP, A framework for exhaustively mapping functional missense variants. Mol. Syst. Biol. 13, 957 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bandaru P, Shah NH, Bhattacharyya M, Barton JP, Kondo Y, Cofsky JC, Gee CL, Chakraborty AK, Kortemme T, Ranganathan R, Kuriyan J, Deconstruction of the Ras switching cycle through saturation mutagenesis. eLife 6, e27810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brenan L, Andreev A, Cohen O, Pantel S, Kamburov A, Cacchiarelli D, Persky NS, Zhu C, Bagul M, Goetz EM, Burgin AB, Garraway LA, Getz G, Mikkelsen TS, Piccioni F, Root DE, Johannessen CM, Phenotypic Characterization of a Comprehensive Set of MAPK1/ERK2 Missense Mutants. Cell Rep. 17, 1171–1183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, Kircher M, Khechaduri A, Dines JN, Hause RJ, Bhatia S, Evans WE, Relling MV, Yang W, Shendure J, Fowler DM, Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet. 50, 874–882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mishra P, Flynn JM, Starr TN, Bolon DNA, Systematic Mutant Analyses Elucidate General and Client-Specific Aspects of Hsp90 Function. Cell Rep. 15, 588–598 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roscoe BP, Bolon DNA, Systematic exploration of ubiquitin sequence, E1 activation efficiency, and experimental fitness in yeast. J. Mol. Biol. 426, 2854–2870 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitzman JO, Starita LM, Lo RS, Fields S, Shendure J, Massively parallel single-amino-acid mutagenesis. Nat. Methods 12, 203–206, 4 p following 206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stiffler MA, Hekstra DR, Ranganathan R, Evolvability as a Function of Purifying Selection in TEM-1 β-Lactamase. Cell 160, 882–892 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Rockah-Shmuel L, Tóth-Petróczy Á, Tawfik DS, Systematic Mapping of Protein Mutational Space by Prolonged Drift Reveals the Deleterious Effects of Seemingly Neutral Mutations. PLOS Comput. Biol. 11, e1004421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hewib WM, Lountos GT, Zlotkowski K, Dahlhauser SD, Saunders LB, Needle D, Tropea JE, Zhan C, Wei G, Ma B, Nussinov R, Waugh DS, Schneekloth JS Jr., Insights Into the Allosteric Inhibition of the SUMO E2 Enzyme Ubc9. Angew. Chem. Int. Ed. 55, 5703–5707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho LT-Y, Alexandrou AJ, Torella R, Knafels J, Hobbs J, Taylor T, Loucif A, Konopacka A, Bell S, Stevens EB, Pandit J, Horst R, Withka JM, Pryde DC, Liu S, Young GT, An Intracellular Allosteric Modulator Binding Pocket in SK2 Ion Channels Is Shared by Multiple Chemotypes. Structure 26, 533–544.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Klink BU, Goody RS, Scheidig AJ, A newly designed microspectrofluorometer for kinetic studies on protein crystals in combination with x-ray diffraction. Biophys. J. 91, 981–992 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward RA, Colclough N, Challinor M, Debreczeni JE, Eckersley K, Fairley G, Feron L, Flemington V, Graham MA, Greenwood R, Hopcroft P, Howard TD, James M, Jones CD, Jones CR, Renshaw J, Roberts K, Snow L, Tonge M, Yeung K, Structure-Guided Design of Highly Selective and Potent Covalent Inhibitors of ERK1/2. J. Med. Chem. 58, 4790–4801 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Wu H, Horton JR, Battaile K, Allali-Hassani A, Martin F, Zeng H, Loppnau P, Vedadi M, Bochkarev A, Plotnikov AN, Cheng X, Structural basis of allele variation of human thiopurine-S-methyltransferase. Proteins Struct. Funct. Bioinforma. 67, 198–208 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer P, Prodromou C, Liao C, Hu B, Mark Roe S, Vaughan CK, Vlasic I, Panaretou B, Piper PW, Pearl LH, Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 23, 511–519 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grishin AM, Condos TEC, Barber KR, Campbell-Valois F-X, Parsot C, Shaw GS, Cygler M, Structural Basis for the Inhibition of Host Protein Ubiquitination by Shigella Effector Kinase OspG. Structure 22, 878–888 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Hong M, Fitzgerald MX, Harper S, Luo C, Speicher DW, Marmorstein R, Structural Basis for Dimerization in DNA Recognition by Gal4. Structure 16, 1019–1026 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minasov G, Wang X, Shoichet BK, An Ultrahigh Resolution Structure of TEM-1 β-Lactamase Suggests a Role for Glu166 as the General Base in Acylation. J. Am. Chem. Soc. 124, 5333–5340 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Didovyk A, Verdine GL, Structural Origins of DNA Target Selection and Nucleobase Extrusion by a DNA Cytosine Methyltransferase *. J. Biol. Chem. 287, 40099–40105 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meier J, Rao R, Verkuil R, Liu J, Sercu T, Rives A, Language models enable zero-shot prediction of the effects of mutations on protein function. bioRxiv [Preprint] (2021). 10.1101/2021.07.09.450648. [DOI] [Google Scholar]
- 42.Phillips AM, Lawrence KR, Moulana A, Dupic T, Chang J, Johnson MS, Cvijovic I, Mora T, Walczak AM, Desai MM, Binding affinity landscapes constrain the evolution of broadly neutralizing anti-influenza antibodies. eLife 10, e71393 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koenig P, Lee CV, Walters BT, Janakiraman V, Stinson J, Patapoff TW, Fuh G, Mutational landscape of antibody variable domains reveals a switch modulating the interdomain conformational dynamics and antigen binding. Proc. Natl. Acad. Sci. 114, E486–E495 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, van der Vlugt R, Lamrani M, Korse HJWM, Geelen E, Sahin Ö, Sieuwerts M, Brakenhoff JPJ, Vogels R, Li OTW, Poon LLM, Peiris M, Koudstaal W, Ward AB, Wilson IA, Goudsmit J, Friesen RHE, Highly Conserved Protective Epitopes on Influenza B Viruses. Science 337, 1343–1348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ekiert DC, Bhabha G, Elsliger M-A, Friesen RHE, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA, Antibody Recognition of a Highly Conserved Influenza Virus Epitope. Science 324, 246–251 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuh G, Wu P, Liang W-C, Ultsch M, Lee CV, Moffat B, Wiesmann C, Structure-Function Studies of Two Synthetic Anti-vascular Endothelial Growth Factor Fabs and Comparison with the Avastin™ Fab *. J. Biol. Chem. 281, 6625–6631 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Olsen TH, Moal IH, Deane CM, AbLang: an antibody language model for completing antibody sequences. Bioinforma. Adv. 2, vbac046 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swindells MB, Porter CT, Couch M, Hurst J, Abhinandan KR, Nielsen JH, Macindoe G, Hetherington J, Martin ACR, abYsis: Integrated Antibody Sequence and Structure—Management, Analysis, and Prediction. J. Mol. Biol. 429, 356–364 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Dauparas J, Anishchenko I, Bennett N, Bai H, Ragotte RJ, Milles LF, Wicky BIM, Courbet A, de Haas RJ, Bethel N, Leung PJY, Huddy TF, Pellock S, Tischer D, Chan F, Koepnick B, Nguyen H, Kang A, Sankaran B, Bera AK, King NP, Baker D, Robust deep learning based protein sequence design using ProteinMPNN. Science 378, 49–56 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Westendorf K, Žentelis S, Wang L, Foster D, Vaillancourt P, Wiggin M, Lovett E, van der Lee R, Hendle J, Pustilnik A, Sauder JM, Kraft L, Hwang Y, Siegel RW, Chen J, Heinz BA, Higgs RE, Kallewaard NL, Jepson K, Goya R, Smith MA, Collins DW, Pellacani D, Xiang P, de Puyraimond V, Ricicova M, Devorkin L, Pritchard C, O’Neill A, Dalal K, Panwar P, Dhupar H, Garces FA, Cohen CA, Dye JM, Huie KE, Badger CV, Kobasa D, Audet J, Freitas JJ, Hassanali S, Hughes I, Munoz L, Palma HC, Ramamurthy B, Cross RW, Geisbert TW, Menachery V, Lokugamage K, Borisevich V, Lanz I, Anderson L, Sipahimalani P, Corbett KS, Yang ES, Zhang Y, Shi W, Zhou T, Choe M, Misasi J, Kwong PD, Sullivan NJ, Graham BS, Fernandez TL, Hansen CL, Falconer E, Mascola JR, Jones BE, Barnhart BC, LY-CoV1404 (bebtelovimab) potently neutralizes SARS-CoV-2 variants. Cell Rep. 39, 110812 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takashita E, Yamayoshi S, Simon V, van Bakel H, Sordillo EM, Pekosz A, Fukushi S, Suzuki T, Maeda K, Halfmann P, Sakai-Tagawa Y, Ito M, Watanabe S, Imai M, Hasegawa H, Kawaoka Y, Efficacy of Antibodies and Antiviral Drugs against Omicron BA.2.12.1, BA.4, and BA.5 Subvariants. N. Engl. J. Med. 387, 468–470 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Center for Drug Evaluation and Research, FDA Announces Bebtelovimab is Not Currently Authorized in Any US Region. FDA; (2022). [Google Scholar]
- 53.Cao Y, Yisimayi A, Jian F, Song W, Xiao T, Wang L, Du S, Wang J, Li Q, Chen X, Yu Y, Wang P, Zhang Z, Liu P, An R, Hao X, Wang Y, Wang J, Feng R, Sun H, Zhao L, Zhang W, Zhao D, Zheng J, Yu L, Li C, Zhang N, Wang R, Niu X, Yang S, Song X, Chai Y, Hu Y, Shi Y, Zheng L, Li Z, Gu Q, Shao F, Huang W, Jin R, Shen Z, Wang Y, Wang X, Xiao J, Xie XS, BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 608, 593–602 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cao Y, Jian F, Zhang Z, Yisimayi A, Hao X, Bao L, Yuan F, Yu Y, Du S, Wang J, Xiao T, Song W, Zhang Y, Liu P, An R, Wang P, Wang Y, Yang S, Niu X, Zhang Y, Gu Q, Shao F, Hu Y, Yin W, Zheng A, Wang Y, Qin C, Jin R, Xiao J, Xie XS, Rational identification of potent and broad sarbecovirus-neutralizing antibody cocktails from SARS convalescents. Cell Rep. 41 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song R, Zeng G, Yu J, Meng X, Chen X, Li J, Xie X, Lian X, Zhang Z, Cao Y, Yin W, Jin R, Post-exposure prophylaxis with SA58 (anti-SARS-COV-2 monoclonal antibody) nasal spray for the prevention of symptomatic COVID-19 in healthy adult workers: a randomized, single-blind, placebo-controlled clinical study*. Emerg. Microbes Infect. 12, 2212806 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makowski EK, Kinnunen PC, Huang J, Wu L, Smith MD, Wang T, Desai AA, Streu CN, Zhang Y, Zupancic JM, Schardt JS, Linderman JJ, Tessier PM, Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space. Nat. Commun. 13, 3788 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Starr TN, Greaney AJ, Hannon WW, Loes AN, Hauser K, Dillen JR, Ferri E, Farrell AG, Dadonaite B, McCallum M, Matreyek KA, Corti D, Veesler D, Snell G, Bloom JD, Shifting mutational constraints in the SARS-CoV-2 receptor-binding domain during viral evolution. Science 377, 420–424 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hie BL, Shanker VR, Xu D, Bruun TUJ, Weidenbacher PA, Tang S, Wu W, Pak JE, Kim PS, Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol, 1–9 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ingraham J, Baranov M, Costello Z, Frappier V, Ismail A, Tie S, Wang W, Xue V, Obermeyer F, Beam A, Grigoryan G, “Illuminating protein space with a programmable generative model” (preprint, Bioinformatics, 2022); 10.1101/2022.12.01.518682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, Ahern W, Borst AJ, Ragotte RJ, Milles LF, Wicky BIM, Hanikel N, Pellock SJ, Courbet A, Sheffler W, Wang J, Venkatesh P, Sappington I, Torres SV, Lauko A, De Bortoli V, Mathieu E, Ovchinnikov S, Barzilay R, Jaakkola TS, DiMaio F, Baek M, Baker D, De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sumida KH, Núñez-Franco R, Kalvet I, Pellock SJ, Wicky BIM, Milles LF, Dauparas J, Wang J, Kipnis Y, Jameson N, Kang A, De La Cruz J, Sankaran B, Bera AK, Jiménez-Osés G, Baker D, Improving Protein Expression, Stability, and Function with ProteinMPNN. J. Am. Chem. Soc. 146, 2054–2061 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goudy OJ, Nallathambi A, Kinjo T, Randolph NZ, Kuhlman B, In silico evolution of autoinhibitory domains for a PD-L1 antagonist using deep learning models. Proc. Natl. Acad. Sci. 120, e2307371120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Biswas S, Khimulya G, Alley EC, Esvelt KM, Church GM, Low-N protein engineering with data-efficient deep learning. Nat. Methods 18, 389–396 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Bedbrook CN, Yang KK, Robinson JE, Mackey ED, Gradinaru V, Arnold FH, Machine learning-guided channelrhodopsin engineering enables minimally invasive optogenetics. Nat. Methods 16, 1176–1184 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu Z, Kan SBJ, Lewis RD, Wittmann BJ, Arnold FH, Machine learning-assisted directed protein evolution with combinatorial libraries. Proc. Natl. Acad. Sci. 116, 8852–8858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cadet F, Fontaine N, Li G, Sanchis J, Ng Fuk Chong M, Pandjaitan R, Vetrivel I, Offmann B, Reetz MT, A machine learning approach for reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes. Sci. Rep. 8, 16757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saito Y, Oikawa M, Nakazawa H, Niide T, Kameda T, Tsuda K, Umetsu M, Machine-Learning-Guided Mutagenesis for Directed Evolution of Fluorescent Proteins. ACS Synth. Biol. 7, 2014–2022 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Biswas S, Kuznetsov G, Ogden PJ, Conway NJ, Adams RP, Church GM, Toward machine-guided design of proteins. bioRxiv [Preprint] (2018). 10.1101/337154. [DOI] [Google Scholar]
- 69.Romero PA, Krause A, Arnold FH, Navigating the protein fitness landscape with Gaussian processes. Proc. Natl. Acad. Sci. 110, E193–E201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bedbrook CN, Yang KK, Rice AJ, Gradinaru V, Arnold FH, Machine learning to design integral membrane channelrhodopsins for efficient eukaryotic expression and plasma membrane localization. PLOS Comput. Biol. 13, e1005786 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao J, Warmuth MK, Govindarajan S, Ness JE, Wang RP, Gustafsson C, Minshull J, Engineering proteinase K using machine learning and synthetic genes. BMC Biotechnol. 7, 16 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Repecka D, Jauniskis V, Karpus L, Rembeza E, Rokaitis I, Zrimec J, Poviloniene S, Laurynenas A, Viknander S, Abuajwa W, Savolainen O, Meskys R, Engqvist MKM, Zelezniak A, Expanding functional protein sequence spaces using generative adversarial networks. Nat. Mach. Intell. 3, 324–333 (2021). [Google Scholar]
- 73.Shin J-E, Riesselman AJ, Kollasch AW, McMahon C, Simon E, Sander C, Manglik A, Kruse AC, Marks DS, Protein design and variant prediction using autoregressive generative models. Nat. Commun. 12, 2403 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu T-Y, Torrey J, Thomas J, Bobrowicz P, Vásquez M, Wittrup KD, Krauland E, Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel. 26, 663–670 (2013). [DOI] [PubMed] [Google Scholar]
- 75.Makowski EK, Wu L, Desai AA, Tessier PM, Highly sensitive detection of antibody nonspecific interactions using flow cytometry. mAbs 13, 1951426 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crawford KHD, Eguia R, Dingens AS, Loes AN, Malone KD, Wolf CR, Chu HY, Tortorici MA, Veesler D, Murphy M, Pe e D, King NP, Balazs AB, Bloom JD, Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 12, 513 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rogers TF, Zhao F, Huang D, Beutler N, Burns A, He W, Limbo O, Smith C, Song G, Woehl J, Yang L, Abbott RK, Callaghan S, Garcia E, Hurtado J, Parren M, Peng L, Ramirez S, Ricketts J, Ricciardi MJ, Rawlings SA, Wu NC, Yuan M, Smith DM, Nemazee D, Teijaro JR, Voss JE, Wilson IA, Andrabi R, Briney B, Landais E, Sok D, Jardine JG, Burton DR, Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369, 956–963 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Andrews SF, Raab JE, Gorman J, Gillespie RA, Cheung CSF, Rawi R, Cominsky LY, Boyington JC, Creanga A, Shen C-H, Harris DR, Olia AS, Nazzari AF, Zhou T, Houser KV, Chen GL, Mascola JR, Graham BS, Kanekiyo M, Ledgerwood JE, Kwong PD, McDermott AB, A single residue in influenza virus H2 hemagglutinin enhances the breadth of the B cell response elicited by H2 vaccination. Nat. Med. 28, 373–382 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Open-source code is provided that allows a user to easily evaluate the structure-informed language model on protein complexes of interest and predict beneficial amino acid substitutions at https://github.com/varun-shanker/structural-evolution. This is implemented as a simple call to a python script with a pdb/cif file and the target chain to be mutated as the main arguments. Code and scripts used in this study have been deposited to Zenodo at https://doi.org/10.5281/zenodo.11260509. Raw data and files used in this study have deposited to Zenodo at https://doi.org/10.5281/zenodo.11260318.