

Summary
Water-soluble Fe4L44− cages can be synthesized in a multicomponent self-assembly process exploiting functionalized trigonal ligands, FeII salts, and water-soluble sulfonated formylpyridine components. The cages are soluble in purely aqueous solution and display an overall 4− charge, but are capable of binding suitably sized non-coordinating anions in the host cavity despite their anionic nature. Anions such as PF6− or AsF6− occupy the internal cavity, whereas anions that are too small (BF4−) or too large (NTf2−) are not encapsulated. The external anionic charge and sterically blocked ligand cores limit the exchange rate of bound anions, as no exchange is seen over a period of weeks with the anion-filled cages, and internalization of added PF6− by an empty cage takes multiple weeks, despite the strong affinity of the cavity for PF6− ions. In the future, this recognition mechanism could be used to control release of anions for environmental applications.
Subject areas: Chemistry, Supramolecular chemistry
Graphical abstract
Highlights
-
•
Water-soluble self-assembled cages can be formed with external anionic groups
-
•
The anionic water-soluble cages bind suitably sized anions in their cavity
-
•
In/out exchange is restricted, with release only possible by destroying the cage
Chemistry; Supramolecular chemistry
Introduction
Molecular recognition in water is vital for the application of synthetic receptors in biological environments and for environmental remediation.1,2 Different types of substrates require variability in receptor design: molecular recognition of neutral species in water is far more effective than in organic solvents, as one can exploit hydrophobic driving forces to favor binding.3,4 Recognition of soft, lipophilic cations is also very well-explored, as CH-π and cation-π forces favor recognition.5 However, anion recognition in water is much more challenging, as hydrophobic interactions are generally minimal, and anions (unlike metal cations) are not receptive to interactions with properly oriented lone pairs. Most importantly, dehydration of anions is energetically unfavorable, which must be compensated by strong host:anion interactions, so affinities in water are often lower than in organic solvents.6,7 Examples of selective anion recognition in water can be seen with rigid cavity-containing receptors,8,9 self-assembling macrocycles,10,11,12,13 and hosts that exploit defined cavities with properly positioned hydrogen bond donors.14,15,16,17,18,19,20 Alternate strategies such as coordination to rare earth centers are also effective.21
An alternative method to create defined binding cavities is to use self-assembly. Self-assembled metal-ligand cage complexes are highly versatile, and have myriad applications in molecular recognition, catalysis and cargo transport, among others.22,23,24,25 While many complexes are restricted to organic solvents, there are a number of examples of self-assembled cages that are soluble in, and stable to water.26 Assembly in water confers greater target scope for molecular recognition, as the hosts can take advantage of hydrophobic effects to bind neutral species. Aqueous hosts have often been used to bind neutral guests and soft cations such as ferrocenium or tetraalkylammonium salts: the affinity is driven by either cation-π interactions between the guest and the aromatic host walls, by favorable charge matching between cationic guests and anionic hosts, or both.5,27,28,29
Strategies to confer water-solubility on hosts fall into three general categories: take advantage of high charge in the assembly, either cationic or anionic, to favor dissolution (as seen with Raymond’s Ga4L612− cages,30,31,32 Fujita’s MxLyn+ Pd-pyridyl assemblies,33,34,35,36 Ward’s cationic Co-pyridylpyrazole cages,37,38 as well as others39,40,41); incorporate charged or polyethylene glycol (PEG) groups to the periphery of normally organic-soluble cages42,43,44,45; or exploit counterion effects to drive solubility of moderately charged cages.46,47,48,49 These latter two strategies have been used by Nitschke to assemble water-soluble metal-iminopyridine cages of a variety of sizes and shapes, as well as performing a detailed analysis of the effects of cage structure and metal ion on the stability of the cages in water.50 Despite the presence of hydrolysable iminopyridine motifs coordinated to cationic metals, these cages can show resistance to hydrolysis for months, depending on coordinating metal.
Cationic self-assembled cages are well-known to bind anions in organic solution,51 including challenging targets such as sulfate,52 as well as halides53 and non-coordinating anions.54,55 There are also some examples that extend this recognition to purely aqueous solution, but they are far rarer,55,56,57,58,59 often requiring internally positioned H-bonding groups as well as overall cationic charge. The common anionic cages do not show affinity for anions, as might be expected.30 Here, we show that an anionic receptor can strongly bind non-coordinating anions in aqueous solution, and this external negative charge acts as a barrier to guest exchange. Water-soluble Fe4L4 complexes can be assembled by multicomponent assembly of neutral tris-amine ligands, Fe2+ salts, and sulfonate-containing formylpyridines, and these overall anionic cages can bind non-coordinating anions strongly, in purely aqueous solution, with no observable guest exchange seen over weeks at ambient temperature.
Results
Water-soluble cage synthesis and characterization
The first priority for aqueous anion recognition is to create hosts that are soluble in water. We have previously shown that the two ligands L1 and L2 (Figure 1) can be easily converted to M4L4 tetrahedra 3 and 4 upon multicomponent self-assembly with Zn salts and 2-formylpyridine (E2), and the complexes bind anions on the cage interior in CD3CN.60 Other work by the Kramer and Nitschke groups showed similar behavior for the unfunctionalized variants.61,62 Despite the 8+ charge of the Zn4L4 complexes, they are insoluble in water. To convert the organic-soluble complex to a water-soluble system, one could change the core ligand to incorporate solubilizing groups,42 modify the formylpyridine “endcap,”63 or exploit alternative counterions such as SO42−.46 In this system, two of these strategies were unsuccessful: formation of the carboxylate variant of ester L2 proved challenging, and while self-assembly of L1 with FeSO4 in CH3CN/H2O was possible, the complex proved quite sensitive, and the reaction was poorly repeatable. We therefore turned our attention to derivatizing the formylpyridine endcap. Aldehyde E1 has been previously used to form water-soluble ML3 fragments by Nitschke,63 and was easily synthesized by combining 3-hydroxy-6-formylpyridine with propylenesultone.
Figure 1.

Self-assembled cage synthesis
Self-assembly process for the formation of water-soluble cages 1 and 2.
The organic components E1 and L1 were reacted with different Fe2+ salts in 1:1 CH3CN:H2O and heated for 50°C for 16 h. When Fe(NTf2)2 was used, evidence of cage formation was seen, but the 1H NMR spectrum showed multiple different products, although no unreacted components E1 or L1 were observed. When the process was repeated with ester ligand E2, there was no evidence of cage formation at all from the 1H NMR spectrum. However, when the syntheses were performed with Fe(NTf2)2 in the presence of AsF6− (10 mol-eq of NaAsF6 with respect to Fe2+) in the reaction mixture, cage assembly was successful. When the components were reacted in a 3:1 aldehyde:ligand:metal ratio, sharp peaks for Fe4L4 cage 1 were seen in the product NMR, but a substantial amount of unreacted E1 was present. The water-soluble aldehyde E1 proved challenging to separate from the water-soluble cage 1, so it was used as limiting reagent. When a component ratio E1:Fe2+:L1 = 1.5:1:1 was used, clean 1 was formed in high conversion. As can be seen in Figures 2B and 2C, the M4L4 complex 1 formed cleanly in the optimized conditions, and only one anionic species can be seen in the 19F spectrum, that of bound AsF6−—no peaks for NTf2- are present (see Figures S6–S13 for full characterization). This observation mirrored that seen with the partial formation of empty complex 1 with Fe(NTf2)2 alone—in that case, no signals for NTf2- were observed in the 19F NMR spectrum at all. The reaction requires a mixture of 1:1 CH3CN:H2O to minimize decomposition of Fe2+ to iron oxide during the reaction: the assembly can be performed in pure water, but the mass recovery was much lower and no product was observed upon reaction in CH3CN alone, as complex 1 is insoluble in CH3CN.
Figure 2.

Structure and characterization of anion-bound cage
(A) Minimized structure of 1⋅AsF6 (SPARTAN 20).
(B) Observed and calculated isotope pattern for [Fe4L4⋅AsF6]5- ions in the ESI-MS spectrum of 1⋅AsF6.
(C) 1H NMR spectrum of 1⋅AsF6 (D2O, 400 MHz, 298K; NOTE—peak Hf overlaps the D2O peak, see Figure S10 for COSY spectrum).
(D) 19F NMR spectrum of 1⋅AsF6 (D2O, 376 MHz, 298K).
As there are four Fe2+ cations in the cage architecture, the absence of the NTf2− counteranions was slightly unexpected, but the reason was quickly established by ESI-MS analysis. Both the impure sample of 1 and the pure 1⋅AsF6 required negative mode to observe discrete peaks, and only negative ions were observed. Cage 1 is overall anionic in water—the observed charge state is 4−, indicating that all 12 sulfonate groups are anionic. The added NTf2- anions are evidently washed away during isolation. In the presence of AsF6−, only a single peak for AsF6− is seen in the 19F NMR, and only the mono-AsF6 complex 1⋅AsF65− can be seen in the ESI-MS, along with some empty 14−. No evidence for any NTf2− or poly-AsF6− complexes could be seen. Acquisition of M− peaks from the empty 1 complex required lower spray voltage to obtain a clean spectrum, and this complex was far more prone to fragmentation (see Figures S4 and S5), but the only peaks for intact cage were the 14− ion, with no NTf2− species present.
These data suggest that suitably sized anions are bound inside the cavity of cages 1 and 2 in aqueous solution—the anionic host binds anions, which is certainly surprising. There are few hosts known with anionic pendant groups that are capable of binding anions in water, and they tend to be macrocycles that exploit directed H-bonds in the cavity, or show low binding affinities.8,64,65 We were unsuccessful in obtaining crystals that were suitable for scXRD, presumably due to the flexible arms at the periphery, but the minimized structure of 1⋅AsF6 is shown in Figure 2A, illustrating the tight fit of the AsF6− anion in the cavity of 14−. While the binding of anions such as AsF6− in organic-soluble cages such as 3 or 4 in CH3CN is known, those cages are cationic, and that positive charge is an important driving force for target binding: similarly sized neutral guests have a significantly lower Ka than anions.61,62 Encapsulating anions in aqueous solution requires overcoming the anion hydration energy, which is substantial (−71 kJ mol−1 for PF6−, −205 kJ mol−1 for ClO4−, and −400 kJ mol−1 for SO42−).66,67,68 In addition, the overall 4− charge of cage 1 provides a charge mismatch: while the localized environment of the cavity is cationic due to the Fe centers, the overall complex charge is anionic. Other examples of water-soluble cages with anionic peripheries and cationic metal centers do not bind anions in water, to our knowledge.30,31,32,55,56,57,58,59
Anion-binding scope
The scope of the assembly process was then tested, varying the ligand (L1 and L2) and added counterion, using the optimized component ratio with E1 as limiting reagent. Ester ligand L2 was slightly less amenable to assembly than L1—the empty cage 2 did not form with Fe(NTf2)2 alone, but the PF6-bound complex 2⋅PF6 was cleanly formed in the presence of NaPF6. Formation of cage 1 was successful in the presence of NaPF6, NaAsF6 and NaSbF6 in the reaction mixture and the 1H NMR spectra of the 1⋅PnF6 variants displayed identical numbers of proton peaks at very similar shifts (see Figures 2C, S14, and S23; PnF6 is used here as a collective label for PF6, AsF6, or SbF6). However, there were some noticeable differences in the broadness of the peaks, as well as in the 19F spectra.
The clearest spectral evidence for internalized anions was with the PF6 and AsF6 complexes (see Figures 2, 3, S16, and S9). While the proton NMR signals varied only slightly, clear evidence for bound PF6 could be seen in the 19F spectra. Two sets of 19F doublets were seen, with the bound peaks 1.5 ppm upfield of the free PF6− (Figure 3A, referenced to added hexafluoroisopropanol [HFIP]). When NaPF6 was added to the sample, the free PF6− signals were enhanced, with no change to the bound peaks. The signal for bound AsF6− were more challenging to determine due to the broader signals for AsF6− and the smaller changes in shift upon binding, but the As-coupled quartet for bound guest showed an upfield shift of 0.2 ppm. The 19F spectra of SbF6 were unhelpful, due to the broadness and complex coupling pattern of the SbF6− anion, but the 1H NMR spectra of 1⋅SbF6 showed clear differences with the PF6/AsF6 spectra, indicating that the SbF6− anion is internally encapsulated—this is consistent with prior work, which indicated that SbF6− was the most strongly bound substrate for cages 3 and 4.60 In addition, the ESI-MS spectrum was very clean, showing only peaks for 14− and 1⋅SbF65− (see Figure S25): all these data suggest that SbF6− is internalized in the same manner as PF6− or AsF6−.
Figure 3.

Size-selective anion encapsulation
(A) 19F NMR spectra of templated cage 1⋅PF6, along with spectra for cage + added NaPF6.
(B) 19F NMR spectra of unoccupied cage 1 with residual BF4− along with spectra for cage + added NaBF4 showing no encapsulation of anion (D2O, 298K, 376 MHz).
The assembly process was also tested with Fe(BF4)2 and Fe(PF6)2 (see Figures S63–S66). The 1H and 19F NMRs of the 14− and 1⋅PF6 complexes formed this way showed peaks at identical shifts to the cages formed by reaction with Fe(NTf2)2 and NaBF4/PF6, although some additional line broadening was seen in the NMR spectra. This sheds light on the nature of the cations in the system—the majority of the cations upon isolation are Na+ salts, as the added Fe2+ is mainly used in the assembly, although use of excess Fe2+ leads to residual Fe2+ in the system, and these paramagnetic ions broaden the NMR. Use of Fe(NTf2)2/NaPnF6 minimizes this issue, favoring the sodium salt of the cages. There was no observed difference when KPnF6 was substituted for NaPnF6.
Notably, PF6−, AsF6−, and SbF6− are all highly similar in structure and properties, so would be expected to behave similarly. The scope of the anion binding was tested with other related anions, SO42−, ClO4−, and BF4−. These anions differ in size (slightly) from the successfully bound PnF6− ions, but more importantly have much higher dehydration energies. Reaction of L1 with FeSO4 was unsuccessful, and no evidence for M4L4 assembly was seen in the NMR, only broad peaks for uncoordinated ligand. Successful formation of cage 1 was possible with both Fe(ClO4)2 and Fe(NTf2)2/NaBF4. As ClO4− has no 19F signature, obtaining unambiguous evidence for internalization was difficult without a scXRD structure, but the ESI-MS spectrum showed the same peak distribution as 1⋅AsF6, with only 14− and 1⋅ClO45− peaks present, indicating that ClO4− is likely bound in the cage. In contrast, while cage 1 could be formed in the presence of NaBF4, there was no evidence of the smaller BF4− being bound on the cage interior, as can be seen in Figure 3B. The 1H spectrum is consistent with Fe4L4 cage formation, but no evidence for internalized BF4− was seen in the 19F spectrum. A small amount of residual free BF4− is present, but no discrete peak for bound BF4− can be seen. When excess NaBF4 was added, only one species is seen in solution, free BF4−. In addition, the ESI-MS analysis indicated a strong peak for the unoccupied [M4L4]4- ion, with only miniscule peaks for [M4L4⋅BF4]4- present (Figure S33). This leads to a conclusion that BF4− is too small to bind effectively on the cage interior, whereas hydrated SO42− is too large to effect templation: the “cutoff” for dehydration energy that can be overcome in cage templation likely lies around that of ClO4− (−205 kJ mol−1).66,67,68
Anion exchange
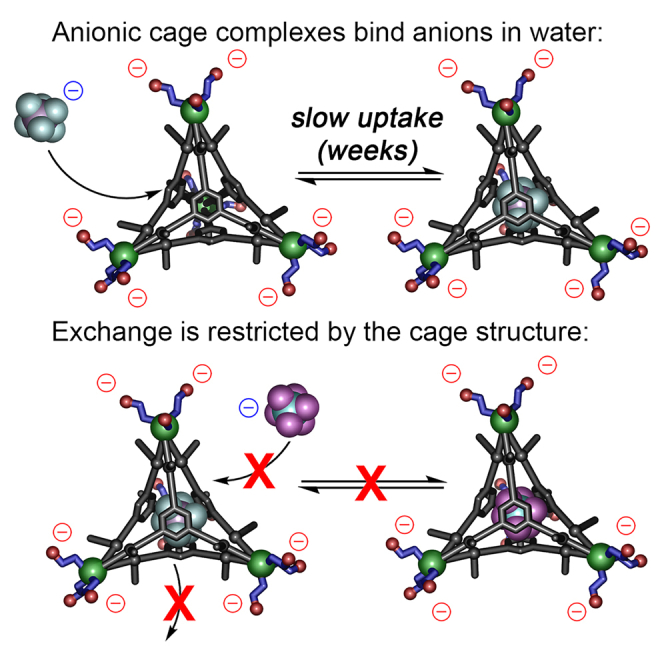
While the anionic self-assembled cages 1 and 2 can be synthesized with suitably sized anions bound on the interior, it was unclear what the effects of the peripheral anions on the cage exterior would have on the kinetics of guest exchange in solution. The organic-soluble cages 3 and 4 showed highly variable exchange properties, depending on the presence or absence of an anion on the cage interior.60 Exchange times ranged from multiple weeks at 50°C (when one bound anion was displaced by another) to seconds at 23°C when adding anion to empty cage. The pendant functional groups on the ligands provide a blockage to guest egress and ingress, slowing exchange.
We therefore tested whether guest exchange was possible with various combinations of cages 1⋅X and 2⋅X in water (see Figures 4 and S50–S61). As the possible exchange rates were highly variable, we performed two sets of experiments to access multiple different exchange regimes, both short (msec–sec) and long (hours–weeks). Cage 1⋅PF6 (1 mM, D2O), synthesized from L1, E1, Fe(NTf2)2 and NaPF6, which contains small amounts of free PF6− as well as cage-bound PF6, was subjected to a 19F–19F EXSY experiment (Figure 4A, mixing time = 300 ms). Zero evidence of anion exchange was observed during this short-timespan experiment, indicating that if any exchange occurs, it is far slower than the EXSY time scale. Therefore, solutions of cages 1⋅PF6, 1⋅AsF6 and 2⋅PF6 (1 mM, D2O) were treated with 10 mM anion (NaSbF6, NaAsF6, NaPF6, and NaBF4) and the systems monitored over time by both 1H and 19F NMR at ambient temperature. In all cases, no exchange was observed at all over a period of 2 weeks—no changes were seen in either the 1H or 19F spectra. The 1⋅PF6 complex was also heated at 70°C for 16 h in the presence of 10 equivalents of NaSbF6, which did not cause any exchange. Some cage decomposition was observed at these elevated temperatures, but the intact cage retained the bound PF6− ion (see Figure S57).
Figure 4.

Restricted anion exchange in the cage
(A) 19F–19F EXSY spectrum of 1⋅PF6+ PF6−, indicating no exchange on the NMR timescale (2 mM, D2O, 298K, 376 MHz, 300 ms mixing time).
(B) 19F NMR spectra of 10 mM NaAsF6 added to a solution of 1mM 1⋅PF6+ PF6− over time, indicating no exchange over a period of weeks (D2O, 298K, 376 MHz, see Figure S51 for full spectra).
The lack of exchange between PF6 and AsF6 is not likely to be due to one anion binding more strongly than the other, as no evidence of exchange was seen in either direction, i.e., adding PF6 to 1⋅AsF6, or AsF6 to 1⋅PF6. Evidently, the egress of anion is highly restricted in this system, even more so than was observed in CD3CN. Other guests were also tested, such as cyclohexane, which has been previously shown to bind in related M4L4 assemblies by Nitschke,51 but when excess cyclohexane was added to the empty 14− complex in D2O, no evidence of hydrocarbon encapsulation was seen. The ESI-MS spectra do offer some evidence of differential rates of anion release: the ratio of [1]4− to [1⋅PnF6]5− varies with anion size, with [1⋅PnF6]5− peaks for the larger PnF6− ions being more prevalent (see Figures S11, S19, and S25). This may suggest that the smaller anions (e.g., PF6−) are more easily expelled upon Coulombic explosion in the ESI, which is somewhat consistent with the observation that small anions (e.g., BF4−) are not retained in aqueous solution. Even so, no expulsion of larger PnF6- anions was seen in solution by NMR.
The next question was whether added anions could enter the cavity of previously synthesized cages at all, or whether the affinity was solely a templation effect in the self-assembly. The empty 14− cage (1 mM) was treated with 10 mM NaPF6 and the 19F spectrum monitored over time. As can be seen in Figure 5, added PF6− could indeed bind in the empty 14− cage, but very slowly—incomplete encapsulation was observed after 2 weeks at 23°C. This extremely slow exchange rate prevents determination of an accurate binding affinity, as equilibrium is not reached in a suitable amount of time. More forcing conditions (elevated temperature) lead to some cage decomposition, also preventing accurate analysis. However, it is clear that cage 14− strongly restricts anion egress, as no loss of bound anion is seen in any of the samples tested.
Figure 5.

Slow anion exchange into empty cage 1
19F NMR spectra of 10 mM NaPF6 added to a solution of 1mM 14− over time, indicating slow formation of 1⋅PF6 over a period of weeks (D2O, 298K, 376 MHz).
Finally, we attempted to release the anions by disassembling the cage complex (Figure 6): excess tren (tris-(2-aminoethyl)amine, 10 mM) was added to a 1⋅PF6 solution (1 mM, D2O). The tren nucleophile is a well-precedented method of disassembling M-iminopyridine cages via transimination, allowing cargo release,69 and the process usually occurs very rapidly. In this case, however, while some transimination occurred over a period of hours, ∼50% 1⋅PF6 remained intact after 2 weeks reaction, indicating unusual stability of the anion-bound M4L4 cage in aqueous solution. Indeed, no solvolysis of the 1⋅X complexes was seen over a period of months at ambient temperature in D2O—this is in contrast with other M4L6 and M4L4 M-iminopyridine complexes in water, which show decomposition over a period of minutes to days in aqueous solution. The anion recognition properties of 1 are dependent on two facets: size- and shape complementarity, and anion dehydration energy. Suitably sized PF6−, AsF6−, SbF6−, and ClO4− are strongly encapsulated in the cage. If the anion is too big, such as NTf2−, or too small, such as BF4− the empty cage can be formed with no encapsulated anion. Also, the strongly solvated sulfate ion SO42−⋅6H2O is far too large to bind inside the cavity, despite it being of the correct size to fit on the interior after desolvation. The more weakly solvated ions can displace their waters in the assembly process, allowing recognition.
Figure 6.

Cage disassembly and anion release
19F NMR spectra of 10 mM tris-(2-aminoethyl)amine (tren) added to a solution of 1mM [1⋅PF6]5- over time, indicating incomplete transimination of the cage and PF6− release after a period of weeks (D2O, 298K, 376 MHz).
Discussion
This leads to the question of why the exchange is so slow with the fully formed assembly. Two possibilities present themselves (Figure 7): (1) the aqueous solution could solvate the anions more strongly than in CD3CN, thus requiring a larger desolvation penalty to pass through the portals of the host; (2) the external anionic environment could repel the entering anions, or both factors are important. There is evidence for both factors: the templating anions are resistant to displacement by any other guest, be they anions of better size matching or neutral hydrophobic species. Egress of a bound anion does not require desolvation, so this suggests a repelling effect by the anionic exterior. On the other hand, binding of PF6− is possible with the empty cage 14−, albeit slowly, suggesting that the repulsive effect is not absolute, and anions can enter an empty cage, dependent on binding affinity. The very slow rate of this process compared to exchange in CD3CN60 indicates that anion desolvation is an additional barrier to exchange in aqueous solution. It is possible that anion exchange requires decomplexation of the ligands to the Fe2+ centers, but this exchange mechanism is very uncommon for Fe-iminopyridine complexes50 and the high stability of this complex in water makes it unlikely.
Figure 7.

Anion binding mechanism
Illustration of the molecular recognition process: (A) anions can template the formation of anionic cage 1, but (B) the external anionic slows guest entry and severely restricts guest egress.
Overall, the pendant functional groups on cages 1 and 2 both provide blockages to guest exchange: the ligand-centered groups act as doorstops to the revolving phenyl groups, slowing exchange when compared to the unfunctionalized variants, and the anionic groups at the periphery act as more of a “bouncer,” preventing entry except in limited circumstances. While the fully intact cages limit exchange, the templating effect occurs before assembly, so anions do not need to get past the bouncer to enter the cavity.
Limitations of the study
The limitations observed in this system lie mainly in the fragility of the cages before complete self-assembly. Reaction must occur in a CH3CN:water mix for solubility, and the free Fe2+ ions are prone to competitive reaction with water, depositing as iron oxide in the reaction mixture. While the cages are highly stable once formed, the accessible yield is relatively low due to this side reaction. In addition, the extremely slow exchange process makes determining binding affinities very challenging, as the system does not reach equilibrium over a period of weeks. Finally, using fluorous anions in water is challenging, as small amounts of hydrolysis byproducts are often present and complicate NMR analysis.
Conclusion
In conclusion, we have shown that self-assembled water-soluble Fe4L4 cages can be synthesized by a multicomponent assembly process exploiting Fe2+ salts, anionic formylpyridine endcaps and trigonal functionalized tris-aniline ligands. Despite the overall 4− charge of the self-assembled cages, the lack of directed H-bonds in the interior, and the challenges of desolvating anions in aqueous solution, these anionic cages strongly bind suitably sized anions in water. Strongly solvated anions are not bound, but mildly solvated ClO4− are, as well as poorly solvated PnF6− ions. The pendant anionic groups do not prevent anion binding, but they do add an additional layer of resistance to guest exchange, as no exchange can be seen between occupied cages and added anions over a period of weeks, and only very slow ingress of anions is seen with unoccupied cages: the anions act as a bouncer at the door, not letting other anions past. In addition, changing the ligand functional groups (from methyl groups in 1 to esters in 2) significantly reduces the effectiveness of anion binding, suggesting future possibilities with these hosts for triggered, selective anion release in water.
Resource availability
Lead contact
Requests for further information and resources should be directed to and will be fulfilled by the lead contact, Prof. Richard J. Hooley (richard.hooley@ucr.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Acknowledgments
The authors would like to thank the National Science Foundation (CHE-2303142 to R.J.H.) and the National Institutes of Health (1R01AG066626 to R.R.J.) for funding.
Author contributions
N.B.Z. and B.d.C. performed the synthesis and NMR characterization, C.C. performed the mass spectrometric analysis, R.R.J. and R.J.H. coordinated the project and designed the experiments. R.J.H. and N.B.Z. wrote the paper, and all authors contributed to the final draft.
Declaration of interests
The authors declare no competing interests.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Cyanuric Chloride | Sigma Aldrich | Cat#C95501 |
| Iron(II) chloride | Fisher Scientific | Cat#AC389350250 |
| Iron(II) trifluoromethanesulfonate | Strem Catalog | Cat#26-2830 |
| Iron(III) perchlorate hydrate | Millipore Sigma | Cat#309281 |
| Sodium hexafluorophosphate | Millipore Sigma | Cat#208051 |
| Sodium hexafluoroarsenate(V) | Millipore Sigma | Cat#223719 |
| Sodium hexafluoroantimonate(V) | Millipore Sigma | Cat#237981 |
| 2-Methyl-4-nitrophenol | Millipore Sigma | Cat#422908 |
| Iron(II) tetrafluoroborate hexahydrate | Combi-Blocks | Cat#QC-0482 |
| Methyl 5-nitrosalicylate | Combi-Blocks | Cat#OR-0249 |
| Potassium tert-butoxide | Fisher Scientific | Cat#AC168880250 |
| Palladium on Carbon | Spectrum | Cat#LF-100 |
| Hydrogen Gas | Airgas | Cat#HYR300 |
| Silver hexafluorophosphate | Fisher Scientific | Cat#AC211120250 |
Note – all NMR spectra for all newly synthesized compounds described in STAR★METHODS can be found in the Supplementary Information.
Experimental model and study participant details
No human participants or cell lines were used in this study.
Method details
General information
3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate was synthesized according to literature procedure.63 Ligands 4,4′,4''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-methylaniline) and trimethyl 6,6',6''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-aminobenzoate) were synthesized as previously reported.60 Fe(PF6)2 was synthesized through metathesis with AgPF6 and FeCl2. Acetonitrile and tetrahydrofuran were dried through a commercial solvent purification system (Pure Process Technologies, Inc.). All commercial reagents were used as received. Cyanuric chloride, Fe(NTf2)2, FeClO4, NaPF6, NaAsF6, NaSbF6 and 2-methyl-4-nitrophenol were purchased from Sigma Aldrich. Fe(BF4)2 and methyl 2-hydroxy-5-nitrobenzoate were purchased from Combi-Blocks. Potassium tert-butoxide, AgPF6 and FeCl2 were purchased from Fisher Scientific. 10% palladium on activated carbon, and hydrogen gas were purchased from Spectrum and Airgas, respectively. 1H, 19F, 13C and 2D NMR spectra were recorded on Bruker Avance NEO 400 MHz and 600 MHz NMR spectrometers. The spectrometers were automatically tuned and matched to the correct operating frequencies. Proton (1H) and carbon (13C) chemical shifts are reported in parts per million (δ) with respect to tetramethylsilane (TMS, δ = 0). Fluorine (19F) chemical shifts are reported in parts per million (δ) and referenced internally to hexafluoroisopropanol (δ = 76.65). Deuterated NMR solvents were obtained from Cambridge Isotope Laboratories, Inc., Andover, MA, and used without purification. Spectra were digitally processed (phase and baseline corrections, integration, peak analysis) using Bruker Topspin 1.3 and MestreNova.
Synthesis of cage 1⋅AsF6
4,4′,4''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-methylaniline) (30 mg, 0.070 mmol), Fe(NTf2)2 (42 mg, 0.070 mmol), 3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate (29 mg, 0.10 mmol), and NaAsF6 (143 mg, 0.68 mmol) were added to a 25 mL flask, equipped with a magnetic stir bar. Next, CH3CN (2 mL) and H2O (2 mL) were added. The flask, equipped with a reflux condenser, was brought to 50°C in a sand bath and allowed to stir overnight. The flask was then taken out of the sand bath and allowed to cool to room temperature. The cooled solution was removed from the reaction flask, leaving behind residual iron oxide. Acetone (100 mL) was then added to the mixture, and a fine pink powder precipitated out of solution. The mixture was sonicated and then centrifuged. The pink-purple solid collected was left to dry under vacuum overnight (14 mg, 16.4%).
Synthesis of cage 1⋅PF6
4,4′,4''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-methylaniline) (30 mg, 0.070 mmol), Fe(NTf2)2 (42 mg, 0.070 mmol), 3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate (29 mg, 0.10 mmol), and NaPF6 (113 mg, 0.68 mmol) were added to a 25 mL flask, equipped with a magnetic stir bar. Next, CH3CN (2 mL) and H2O (2 mL) were added. The flask, equipped with a reflux condenser, was brought to 50°C in a sand bath and allowed to stir overnight. The flask was then taken out of the sand bath and allowed to cool to room temperature. The cooled solution was removed from the reaction flask, leaving behind residual iron oxide. Acetone (100 mL) was then added to the mixture, and a fine pink powder precipitated out of solution. The mixture was sonicated and then centrifuged. The pink-purple solid collected was left to dry under vacuum overnight (43 mg, 50.6%).
Synthesis of cage 1⋅SbF6
4,4′,4''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-methylaniline) (20 mg, 0.050 mmol), Fe(NTf2)2 (28 mg, 0.050 mmol), 3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate (19 mg, 0.070 mmol), and NaSbF6 (116 mg, 0.45 mmol) were added to a 25 mL flask, equipped with a magnetic stir bar. Next, CH3CN (2 mL) and H2O (2 mL) were added. The flask, equipped with a reflux condenser, was brought to 50°C in a sand bath and allowed to stir overnight. The flask was then taken out of the sand bath and allowed to cool to room temperature. The cooled solution was removed from the reaction flask, leaving behind residual iron oxide. Acetone (100 mL) was then added to the mixture, and a fine pink powder precipitated out of solution. The mixture was sonicated and then centrifuged. The pink-purple solid collected was left to dry under vacuum overnight (19 mg, 33.1%).
Synthesis of cage 1⋅ClO4
4,4′,4''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-methylaniline) (40 mg, 0.090 mmol), FeClO4 (23 mg, 0.090 mmol) and 3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate(38 mg, 0.13 mmol) were added to a 25 mL flask equipped with a magnetic stir bar. Next, CH3CN (2 mL) and H2O (2 mL) were added. The flask, equipped with a reflux condenser, was brought to 50°C in a sand bath and allowed to stir overnight. The flask was then taken out of the sand bath and allowed to cool to room temperature. The cooled solution was removed from the reaction flask, leaving behind residual iron oxide. Acetone (100 mL) was then added to the mixture, and a fine pink powder precipitated out of solution. The mixture was sonicated and then centrifuged. The pink-purple solid collected was left to dry under vacuum overnight (38 mg, 34.3%).
Synthesis of 1, made with Fe(NTf2)2 and NaBF4
4,4′,4''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-methylaniline) (30 mg, 0.070 mmol), Fe(NTf2)2 (42 mg, 0.070 mmol), 3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate (29 mg, 0.10 mmol), and NaBF4 (74 mg, 0.68mmol) were added to a 25 mL flask, equipped with a magnetic stir bar. Next, CH3CN (2 mL) and H2O (2 mL) were added. The flask, equipped with a reflux condenser, was brought to 50°C in a sand bath and allowed to stir overnight. The flask was then taken out of the sand bath and allowed to cool to room temperature. The cooled solution was removed from the reaction flask, leaving behind residual iron oxide. Acetone (100 mL) was then added to the mixture, and a fine pink powder precipitated out of solution. The mixture was sonicated and then centrifuged. The pink-purple solid collected was left to dry under vacuum overnight (52 mg, 52.3%).
Synthesis of cage 1
4,4′,4''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-methylaniline) (30 mg, 0.070 mmol), Fe(NTf2)2 (42 mg, 0.070 mmol), and 3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate (29 mg, 0.10 mmol) were added to a 25 mL flask, equipped with a magnetic stir bar. Next, CH3CN (2 mL) and H2O (2 mL) were added. The flask, equipped with a reflux condenser, was brought to 50°C in a sand bath and allowed to stir overnight. The flask was then taken out of the sand bath and allowed to cool to room temperature. The cooled solution was removed from the reaction flask, leaving behind residual iron oxide. Acetone (100 mL) was then added to the mixture, and a fine pink powder precipitated out of solution. The mixture was sonicated and then centrifuged. The pink-purple solid collected was left to dry under vacuum overnight (12 mg, 14.8%). The sample was impure – see Figure S45 for the 1H spectrum.
Synthesis of cage 2⋅AsF6
Trimethyl 6,6′,6''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-aminobenzoate) (30 mg, 0.050 mmol), Fe(NTf2)2 (32 mg, 0.050 mmol), 3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate (22 mg, 0.080 mmol), and NaAsF6 (110 mg, 0.52 mmol) were added to a 25 mL flask, equipped with a magnetic stir bar. Next, CH3CN (2 mL) and H2O (2 mL) were added. The flask, equipped with a reflux condenser, was brought to 50°C in a sand bath and allowed to stir overnight. The flask was then taken out of the sand bath and allowed to cool to room temperature. The cooled solution was removed from the reaction flask, leaving behind residual iron oxide. Acetone (100 mL) was then added to the mixture, and a fine pink powder precipitated out of solution. The mixture was sonicated and then centrifuged. The pink-purple solid collected was left to dry under vacuum overnight (29 mg, 40.2%).
Synthesis of cage 2⋅PF6
Trimethyl 6,6′,6''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-aminobenzoate) (30 mg, 0.050 mmol), Fe(NTf2)2 (32 mg, 0.050 mmol), 3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate (22 mg, 0.080 mmol), and NaPF6 (87 mg, 0.52 mmol) were added to a 25 mL flask, equipped with a magnetic stir bar. Next, CH3CN (2 mL) and H2O (2 mL) were added. The flask, equipped with a reflux condenser, was brought to 50°C in a sand bath and allowed to stir overnight. The flask was then taken out of the sand bath and allowed to cool to room temperature. The cooled solution was removed from the reaction flask, leaving behind residual iron oxide. Acetone (100 mL) was then added to the mixture, and a fine pink powder precipitated out of solution. The mixture was sonicated and then centrifuged. The pink-purple solid collected was left to dry under vacuum overnight (41 mg, 56.7%).
Synthesis of 2, made with Fe(NTf2)2 and NaBF4
Trimethyl 6,6′,6''-((1,3,5-triazine-2,4,6-triyl)tris(oxy))tris(3-aminobenzoate) (30 mg, 0.050 mmol), Fe(NTf2)2 (32 mg, 0.050 mmol), 3-((6-formylpyridin-3-yl)oxy)propane-1-sulfonate (22 mg, 0.080 mmol), and NaBF4 (57 mg, 0.52 mmol) were added to a 25 mL flask, equipped with a magnetic stir bar. Next, CH3CN (2 mL) and H2O (2 mL) were added. The flask, equipped with a reflux condenser, was brought to 50°C in a sand bath and allowed to stir overnight. The flask was then taken out of the sand bath and allowed to cool to room temperature. The cooled solution was removed from the reaction flask, leaving behind residual iron oxide. Acetone (100 mL) was then added to the mixture, and a fine pink powder precipitated out of solution. The mixture was sonicated and then centrifuged. The pink-purple solid collected was left to dry under vacuum overnight (11 mg, 14.9%).
Mass spectrometric methods
The mass spectrometric sample of cages was prepared in 50:50 CH3CN:H2O and infused into a Thermo Orbitrap Velos Pro mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) with a HESI source. Thermo Xcalibur was used to analyze MS data and prepare the predicted isotope patterns.
ESI-MS spectrum of cage 1
Flow rate, sheath gas flow rate, aux gas flow rate, spray voltage, capillary temperature, and the S-lens RF level were set to be 3 μl/min, 5 arb, 10 arb, 2.8 kV, 215 C, and 40% respectively. Full mass spectra were acquired with a resolution of r = 30,000.
ESI-MS spectrum of cage 1⋅AsF6
Flow rate, sheath gas flow rate, aux gas flow rate, spray voltage, capillary temperature, and the S-lens RF level were set to be 5 μl/min, 10 arb, 12 arb, 2.8 kV, 200 C, and 40% respectively. Full mass spectra were acquired with a resolution of r = 60,000.
ESI-MS spectrum of cage 1⋅PF6
Flow rate, sheath gas flow rate, aux gas flow rate, spray voltage, capillary temperature, and the S-lens RF level were set to be 3 μl/min, 5 arb, 10 arb, 3.5 kV, 200 C, and 40% respectively. Full mass spectra were acquired with a resolution of r = 30,000.
ESI-MS spectrum of cage 1⋅SbF6
Flow rate, sheath gas flow rate, aux gas flow rate, spray voltage, capillary temperature, and the S-lens RF level were set to be 5 μl/min, 5 arb, 10 arb, 4 kV, 200 C, and 20% respectively. Full mass spectra were acquired with a resolution of r = 15,000.
ESI-MS spectrum of cage 1⋅ClO4
Flow rate, sheath gas flow rate, aux gas flow rate, spray voltage, capillary temperature, and the S-lens RF level were set to be 5 μl/min, 5 arb, 10 arb, 3.5 kV, 200 C, and 20% respectively. Full mass spectra were acquired with a resolution of r = 15,000.
ESI-MS spectrum of cage 1,made with Fe(NTf2)2 and NaBF4
Flow rate, sheath gas flow rate, aux gas flow rate, spray voltage, capillary temperature, and the S-lens RF level were set to be 3 μl/min, 5 arb, 10 arb, 3.2 kV, 200 C, and 40% respectively. Full mass spectra were acquired with a resolution of r = 30,000.
ESI-MS spectrum of cage 2⋅PF6
Flow rate, sheath gas flow rate, aux gas flow rate, spray voltage, capillary temperature, and the S-lens RF level were set to be 3 μl/min, 5 arb, 10 arb, 3.5 kV, 200 C, and 50% respectively. Full mass spectra were acquired with a resolution of r = 30,000.
Quantification and statistical analysis
No statistical methods or analyses were used in this study.
Additional resources
No additional resources were used in this study.
Published: November 8, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.111348.
Supplemental information
References
- 1.Escobar L., Ballester P. Molecular Recognition in Water Using Macrocyclic Synthetic Receptors. Chem. Rev. 2021;121:2445–2514. doi: 10.1021/acs.chemrev.0c00522. [DOI] [PubMed] [Google Scholar]
- 2.Kubik S. When Molecules Meet in Water-Recent Contributions of Supramolecular Chemistry to the Understanding of Molecular Recognition Processes in Water. ChemistryOpen. 2022;11 doi: 10.1002/open.202200028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hof F., Craig S.L., Nuckolls C., Rebek J., Jr. Molecular encapsulation. Angew. Chem. Int. Ed. 2002;41:1488–1508. doi: 10.1021/ja034535e. [DOI] [PubMed] [Google Scholar]
- 4.Jordan J.H., Gibb B.C. Molecular containers assembled through the hydrophobic effect. Chem. Soc. Rev. 2015;44:547–585. doi: 10.1039/C4CS00191E. [DOI] [PubMed] [Google Scholar]
- 5.Beatty M.A., Hof F. Host–guest binding in water, salty water, and biofluids: general lessons for synthetic, bio-targeted molecular recognition. Chem. Soc. Rev. 2021;50:4812–4832. doi: 10.1039/D0CS00495B. [DOI] [PubMed] [Google Scholar]
- 6.Langton M.J., Serpell C.J., Beer P.D. Anion Recognition in Water: Recent Advances from a Supramolecular and Macromolecular Perspective. Angew. Chem. Int. Ed. 2016;55:1974–1987. doi: 10.1002/anie.201506589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans N.H., Beer P.D. Advances in Anion Supramolecular Chemistry: From Recognition to Chemical Applications. Angew. Chem. Int. Ed. 2014;53:11716–11754. doi: 10.1002/anie.201309937. [DOI] [PubMed] [Google Scholar]
- 8.Jordan J.H., Gibb C.L.D., Wishard A., Pham T., Gibb B.C. Ion–Hydrocarbon and/or Ion–Ion Interactions: Direct and Reverse Hofmeister Effects in a Synthetic Host. J. Am. Chem. Soc. 2018;140:4092–4099. doi: 10.1021/jacs.8b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sokkalingam P., Shraberg J., Rick S.W., Gibb B.C. Binding Hydrated Anions with Hydrophobic Pockets. J. Am. Chem. Soc. 2016;138:48–51. doi: 10.1021/jacs.5b10937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parks F.C., Sheetz E.G., Stutsman S.R., Lutolli A., Debnath S., Raghavachari K., Flood A.H. Revealing the Hidden Costs of Organization in Host-Guest Chemistry using Chloride-Binding Foldamers and their Solvent Dependence. J. Am. Chem. Soc. 2022;144:1274–1287. doi: 10.1021/jacs.1c10758. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y., Parks F.C., Sheetz E.G., Chen C.-H., Flood A.H. Polarity-Tolerant Chloride Binding in Foldamer Capsules by Programmed Solvent-Exclusion. J. Am. Chem. Soc. 2021;143:3191–3204. doi: 10.1021/jacs.0c12562. [DOI] [PubMed] [Google Scholar]
- 12.Parks F.C., Liu Y., Debnath S., Stutsman S.R., Raghavachari K., Flood A.H. Allosteric Control of Photofoldamers for Selecting between Anion Regulation and Double-to-Single Helix Switching. J. Am. Chem. Soc. 2018;140:17711–17723. doi: 10.1021/jacs.8b10538. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y., Zhao W., Chen C.-H., Flood A.H. Chloride capture using a C–H hydrogen-bonding cage. Science. 2019;365:159–161. doi: 10.1126/science.aaw5145. [DOI] [PubMed] [Google Scholar]
- 14.Butler S.J., Jolliffe K.A. Anion Receptors for the Discrimination of ATP and ADP in Biological Media. ChemPlusChem. 2021;86:59–70. doi: 10.1002/cplu.202000567. [DOI] [PubMed] [Google Scholar]
- 15.Berry S.N., Qin L., Lewis W., Jolliffe K.A. Conformationally adaptable macrocyclic receptors for ditopic anions: analysis of chelate cooperativity in aqueous containing media. Chem. Sci. 2020;11:7015–7022. doi: 10.1039/D0SC02533J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borissov A., Marques I., Lim J.Y.C., Félix V., Smith M.D., Beer P.D. Anion Recognition in Water by Charge-Neutral Halogen and Chalcogen Bonding Foldamer Receptors. J. Am. Chem. Soc. 2019;141:4119–4129. doi: 10.1021/jacs.9b00148. [DOI] [PubMed] [Google Scholar]
- 17.Docker A., Tse Y.C., Tay H.M., Taylor A.J., Zhang Z., Beer P.D. Anti-Hofmeister Anion Selectivity via a Mechanical Bond Effect in Neutral Halogen-Bonding [2]Rotaxanes. Angew. Chem. Int. Ed. 2022;61 doi: 10.1002/anie.202214523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Docker A., Marques I., Kuhn H., Zhang Z., Félix V., Beer P.D. Selective Potassium Chloride Recognition, Sensing, Extraction, and Transport Using a Chalcogen-Bonding Heteroditopic Receptor. J. Am. Chem. Soc. 2022;144:14778–14789. doi: 10.1021/jacs.2c05333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mosquera J., Zarra S., Nitschke J.R. Aqueous Anion Receptors through Reduction of Subcomponent Self-Assembled Structures. Angew. Chem. Int. Ed. 2014;53:1556–1559. doi: 10.1002/anie.201308117. [DOI] [PubMed] [Google Scholar]
- 20.Jing L., Deplazes E., Clegg J.K., Wu X. A charge-neutral organic cage selectively binds strongly hydrated sulfate anions in water. Nat. Chem. 2024;16:335–342. doi: 10.1038/s41557-024-01457-5. [DOI] [PubMed] [Google Scholar]
- 21.Ramakrishnam Raju M.V., Harris S.M., Pierre V.C. Design and applications of metal-based molecular receptors and probes for inorganic phosphate. Chem. Soc. Rev. 2020;49:1090–1108. doi: 10.1039/C9CS00543A. [DOI] [PubMed] [Google Scholar]
- 22.McTernan C.T., Davies J.A., Nitschke J.R. Beyond Platonic: How to Build Metal–Organic Polyhedra Capable of Binding Low-Symmetry, Information-Rich Molecular Cargoes. Chem. Rev. 2022;122:10393–10437. doi: 10.1021/acs.chemrev.1c00763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chakrabarty R., Mukherjee P.S., Stang P.J. Supramolecular Coordination: Self-Assembly of Finite Two- and Three-Dimensional Ensembles. Chem. Rev. 2011;111:6810–6918. doi: 10.1021/cr200077m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rizzuto F.J., von Krbek L.K.S., Nitschke J.R. Strategies for binding multiple guests in metal–organic cages. Nat. Rev. Chem. 2019;3:204–222. doi: 10.1038/s41570-019-0085-3. [DOI] [Google Scholar]
- 25.Ward M.D., Raithby P.R. Functional behaviour from controlled self-assembly: challenges and prospects. Chem. Soc. Rev. 2013;42:1619–1636. doi: 10.1039/c2cs35123d. [DOI] [PubMed] [Google Scholar]
- 26.Percástegui E.G., Ronson T.K., Nitschke J.R. Design and Applications of Water-Soluble Coordination Cages. Chem. Rev. 2020;120:13480–13544. doi: 10.1021/acs.chemrev.0c00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dougherty D.A. The Cation−π Interaction. Acc. Chem. Res. 2013;46:885–893. doi: 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pluth M.D., Bergman R.G., Raymond K.N. Proton-mediated chemistry and catalysis in a self-assembled supramolecular host. Acc. Chem. Res. 2009;42:1650–1659. doi: 10.1021/ar900118t. [DOI] [PubMed] [Google Scholar]
- 29.Smithrud D.B., Sanford E.M., Chao I., Ferguson S.B., Carcanague D.R., Evanseck J.D., Houk K.N., Diederich F. Solvent effects in molecular recognition. Pure Appl. Chem. 1990;62:2227–2236. doi: 10.1351/pac199062122227. [DOI] [Google Scholar]
- 30.Caulder D.L., Raymond K.N. Supermolecules by Design. Acc. Chem. Res. 1999;32:975–982. doi: 10.1021/ar970224v. [DOI] [Google Scholar]
- 31.Hong C.M., Bergman R.G., Raymond K.N., Toste F.D. Self-Assembled Tetrahedral Hosts as Supramolecular Catalysts. Acc. Chem. Res. 2018;51:2447–2455. doi: 10.1021/acs.accounts.8b00328. [DOI] [PubMed] [Google Scholar]
- 32.Bierschenk S.M., Pan J.Y., Settineri N.S., Warzok U., Bergman R.G., Raymond K.N., Toste F.D. Impact of Host Flexibility on Selectivity in a Supramolecular Host-Catalyzed Enantioselective aza-Darzens Reaction. J. Am. Chem. Soc. 2022;144:11425–11433. doi: 10.1021/jacs.2c04182. [DOI] [PubMed] [Google Scholar]
- 33.Fujita M., Tominaga M., Hori A., Therrien B. Coordination assemblies from a Pd(II)-cornered square complex. Acc. Chem. Res. 2005;38:369–378. doi: 10.1021/ar040153h. [DOI] [PubMed] [Google Scholar]
- 34.Ueda Y., Ito H., Fujita D., Fujita M. Permeable Self-Assembled Molecular Containers for Catalyst Isolation Enabling Two-Step Cascade Reactions. J. Am. Chem. Soc. 2017;139:6090–6093. doi: 10.1021/jacs.7b02745. [DOI] [PubMed] [Google Scholar]
- 35.Fujita D., Suzuki K., Sato S., Yagi-Utsumi M., Yamaguchi Y., Mizuno N., Kumasaka T., Takata M., Noda M., Uchiyama S., et al. Protein encapsulation within synthetic molecular hosts. Nat. Commun. 2012;3:1093–2099. doi: 10.1038/ncomms2093. [DOI] [PubMed] [Google Scholar]
- 36.Zaffaroni R., Orth N., Ivanović-Burmazović I., Reek J.N.H. Hydrogenase Mimics in M12L24 Nanospheres to Control Overpotential and Activity in Proton-Reduction Catalysis. Angew. Chem. Int. Ed. 2020;59:18485–18489. doi: 10.1002/anie.202008298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tidmarsh I.S., Faust T.B., Adams H., Harding L.P., Russo L., Clegg W., Ward M.D. Octanuclear Cubic Coordination Cages. J. Am. Chem. Soc. 2008;130:15167–15175. doi: 10.1021/ja805605y. [DOI] [PubMed] [Google Scholar]
- 38.Cullen W., Metherell A.J., Wragg A.B., Taylor C.G.P., Williams N.H., Ward M.D. Catalysis in a Cationic Coordination Cage Using a Cavity-Bound Guest and Surface-Bound Anions: Inhibition, Activation, and Autocatalysis. J. Am. Chem. Soc. 2018;140:2821–2828. doi: 10.1021/jacs.7b11334. [DOI] [PubMed] [Google Scholar]
- 39.He Y.P., Yuan L.B., Chen G.H., Lin Q.P., Wang F., Zhang L., Zhang J. Water-Soluble and Ultrastable Ti4L6 Tetrahedron with Coordination Assembly Function. J. Am. Chem. Soc. 2017;139:16845–16851. doi: 10.1021/jacs.7b09463. [DOI] [PubMed] [Google Scholar]
- 40.Roy B., Zangrando E., Mukherjee P.S. Self-assembly of a redox active water soluble Pd6L8 ‘molecular dice. Chem. Commun. 2016;52:4489–4492. doi: 10.1039/C6CC00042H. [DOI] [PubMed] [Google Scholar]
- 41.Liu G., Di Yuan Y., Wang J., Cheng Y., Peh S.B., Wang Y., Qian Y., Dong J., Yuan D., Zhao D. Process-Tracing Study on the Post-assembly Modification of Highly Stable Zirconium Metal-Organic Cages. J. Am. Chem. Soc. 2018;140:6231–6234. doi: 10.1021/jacs.8b03517. [DOI] [PubMed] [Google Scholar]
- 42.Mal P., Schultz D., Beyeh K., Rissanen K., Nitschke J.R. An unlockable-relockable iron cage by subcomponent self-assembly. Angew. Chem. Int. Ed. 2008;47:8297–8301. doi: 10.1002/anie.200803066. [DOI] [PubMed] [Google Scholar]
- 43.Bolliger J.L., Belenguer A.M., Nitschke J.R. Enantiopure water-soluble [Fe4L6] cages: host-guest chemistry and catalytic activity. Angew. Chem. Int. Ed. 2013;52:7958–7962. doi: 10.1002/anie.201302136. [DOI] [PubMed] [Google Scholar]
- 44.Kishi N., Li Z., Yoza K., Akita M., Yoshizawa M. An M2L4 molecular capsule with an anthracene shell: encapsulation of large guests up to 1 nm. J. Am. Chem. Soc. 2011;133:11438–11441. doi: 10.1021/ja2037029. [DOI] [PubMed] [Google Scholar]
- 45.Symmers P.R., Burke M.J., August D.P., Thomson P.I.T., Nichol G.S., Warren M.R., Campbell C.J., Lusby P.J. Non-equilibrium cobalt(iii) “click” capsules. Chem. Sci. 2015;6:756–760. doi: 10.1039/C4SC03036B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolliger J.L., Ronson T.K., Ogawa M., Nitschke J.R. Solvent Effects upon Guest Binding and Dynamics of a FeII4L4 Cage. J. Am. Chem. Soc. 2014;136:14545–14553. doi: 10.1021/ja5077102. [DOI] [PubMed] [Google Scholar]
- 47.Percástegui E.G., Mosquera J., Nitschke J.R. Anion Exchange Renders Hydrophobic Capsules and Cargoes Water-Soluble. Angew. Chem. Int. Ed. 2017;56:9136–9140. doi: 10.1002/anie.201705093. [DOI] [PubMed] [Google Scholar]
- 48.Grommet A.B., Hoffman J.B., Percástegui E.G., Mosquera J., Howe D.J., Bolliger J.L., Nitschke J.R. Anion Exchange Drives Reversible Phase Transfer of Coordination Cages and Their Cargoes. J. Am. Chem. Soc. 2018;140:14770–14776. doi: 10.1021/jacs.8b07900. [DOI] [PubMed] [Google Scholar]
- 49.Zhang D., Ronson T.K., Lavendomme R., Nitschke J.R. Selective Separation of Polyaromatic Hydrocarbons by Phase Transfer of Coordination Cages. J. Am. Chem. Soc. 2019;141:18949–18953. doi: 10.1021/jacs.9b10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Percástegui E.G., Mosquera J., Ronson T.K., Plajer A.J., Kieffer M., Nitschke J.R. Waterproof architectures through subcomponent self-assembly. Chem. Sci. 2019;10:2006–2018. doi: 10.1039/C8SC05085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang D., Ronson T.K., Nitschke J.R. Functional Capsules via Subcomponent Self-Assembly. Acc. Chem. Res. 2018;51:2423–2436. doi: 10.1021/acs.accounts.8b00303. [DOI] [PubMed] [Google Scholar]
- 52.Custelcean R., Bosano J., Bonnesen P.V., Kertesz V., Hay B.P. Computer-Aided Design of a Sulfate-Encapsulating Receptor. Angew. Chem. Int. Ed. 2009;48:4025–4029. doi: 10.1002/anie.200900108. [DOI] [PubMed] [Google Scholar]
- 53.Riddell I.A., Ronson T.K., Nitschke J.R. Mutual stabilisation between MII4L6 tetrahedra and MIIX42− metallate guests. Chem. Sci. 2015;6:3533–3537. doi: 10.1039/C5SC01083G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai Y.-L., Xie M., Zhou X.-C., Wang X.-Z., Zhu X.-W., Luo D., Zhou X.-P., Li D. Precise Post-Synthetic Modification of Heterometal-Organic Capsules for Selectively Encapsulating Tetrahedral Anions. Angew. Chem. Int. Ed. 2024;63 doi: 10.1002/anie.202402829. [DOI] [PubMed] [Google Scholar]
- 55.Zhang D., Ronson T.K., Mosquera J., Martinez A., Guy L., Nitschke J.R. Anion Binding in Water Drives Structural Adaptation in an Azaphosphatrane-Functionalized FeII4L4. J. Am. Chem. Soc. 2017;139:6574–6577. doi: 10.1021/jacs.7b02950. [DOI] [PubMed] [Google Scholar]
- 56.Custelcean R., Bonnesen P.V., Duncan N.C., Zhang X., Watson L.A., Van Berkel G., Parson W.B., Hay B.P. Urea-Functionalized M4L6 Cage Receptors: Anion-Templated Self-Assembly and Selective Guest Exchange in Aqueous Solutions. J. Am. Chem. Soc. 2012;134:8525–8534. doi: 10.1021/ja300677w. [DOI] [PubMed] [Google Scholar]
- 57.Sawada T., Fujita M. A Single Watson−Crick G·C Base Pair in Water: Aqueous Hydrogen Bonds in Hydrophobic Cavities. J. Am. Chem. Soc. 2010;132:7194–7201. doi: 10.1021/ja101718c. [DOI] [PubMed] [Google Scholar]
- 58.Plajer A.J., Percástegui E.G., Santella M., Rizzuto F.J., Gan Q., Laursen B.W., Nitschke J.R. Fluorometric Recognition of Nucleotides within a Water-Soluble Tetrahedral Capsule. Angew. Chem. Int. Ed. 2019;58:4200–4204. doi: 10.1002/anie.201814149. [DOI] [PubMed] [Google Scholar]
- 59.Sudan S., Chen D.W., Berton C., Fadaei-Tirani F., Severin K. Synthetic Receptors with Micromolar Affinity for Chloride in Water. Angew. Chem. Int. Ed. 2023;62 doi: 10.1002/anie.202218072. [DOI] [PubMed] [Google Scholar]
- 60.da Camara B., Ziv N.B., Carta V., Mota Orozco G.A., Wu H.-T., Julian R.R., Hooley R.J. Gated, Selective Anion Exchange in Functionalized Self-Assembled Cage Complexes. Chem-Eur. J. 2023;29 doi: 10.1002/chem.202203588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferguson A., Staniland R.W., Fitchett C.M., Squire M.A., Williamson B.E., Kruger P.E. Variation of guest selectivity within [Fe4L4]8+ tetrahedral cages through subtle modification of the face-capping ligand. Dalton Trans. 2014;43:14550–14553. doi: 10.1039/C4DT02337D. [DOI] [PubMed] [Google Scholar]
- 62.Xu L., Zhang D., Ronson T.K., Nitschke J.R. Improved Acid Resistance of a Metal–Organic Cage Enables Cargo Release and Exchange between Hosts. Angew. Chem. Int. Ed. 2020;59:7435–7438. doi: 10.1002/anie.202001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryan H.P., Haynes C.J.E., Smith A., Grommet A.B., Nitschke J.R. Guest Encapsulation within Surface-Adsorbed Self-Assembled Cages. Adv. Mater. 2021;33 doi: 10.1002/adma.202004192. [DOI] [PubMed] [Google Scholar]
- 64.Lisbjerg M., Jessen B.M., Rasmussen B., Nielsen B.E., Madsen A.Ø., Pittelkow M. Discovery of a cyclic 6 + 6 hexamer of d-biotin and formaldehyde. Chem. Sci. 2014;5:2647–2650. doi: 10.1039/C4SC00990H. [DOI] [Google Scholar]
- 65.Yawer M.A., Havel V., Sindelar V. A Bambusuril Macrocycle that Binds Anions in Water with High Affinity and Selectivity. Angew. Chem. Int. Ed. 2015;54:276–279. doi: 10.1002/anie.201409895. [DOI] [PubMed] [Google Scholar]
- 66.Liu Y., Sengupta A., Raghavachari K., Flood A.H. Anion Binding in Solution: Beyond the Electrostatic Regime. Chem. 2017;3:411–427. doi: 10.1016/j.chempr.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 67.Smith D.W. Ionic Hydration Enthalpies. J. Chem. Educ. 1977;54:540. doi: 10.1021/ed054p540. [DOI] [Google Scholar]
- 68.Collins K.D. Ion hydration: Implications for cellular function, polyelectrolytes, and protein crystallization. Biophys. Chem. 2006;119:271–281. doi: 10.1016/j.bpc.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 69.Castilla A.M., Ronson T.K., Nitschke J.R. Sequence-Dependent Guest Release Triggered by Orthogonal Chemical Signals. J. Am. Chem. Soc. 2016;138:2342–2351. doi: 10.1021/jacs.5b13016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.