

Abstract
Aircraft observations have revealed ubiquitous new particle formation in the tropical upper troposphere over the Amazon1,2 and the Atlantic and Pacific oceans3,4. Although the vapours involved remain unknown, recent satellite observations have revealed surprisingly high night-time isoprene mixing ratios of up to 1 part per billion by volume (ppbv) in the tropical upper troposphere5. Here, in experiments performed with the CERN CLOUD (Cosmics Leaving Outdoor Droplets) chamber, we report new particle formation initiated by the reaction of hydroxyl radicals with isoprene at upper-tropospheric temperatures of −30 °C and −50 °C. We find that isoprene-oxygenated organic molecules (IP-OOM) nucleate at concentrations found in the upper troposphere, without requiring any more vapours. Moreover, the nucleation rates are enhanced 100-fold by extremely low concentrations of sulfuric acid or iodine oxoacids above 105 cm−3, reaching rates around 30 cm−3 s−1 at acid concentrations of 106 cm−3. Our measurements show that nucleation involves sequential addition of IP-OOM, together with zero or one acid molecule in the embryonic molecular clusters. IP-OOM also drive rapid particle growth at 3–60 nm h−1. We find that rapid nucleation and growth rates persist in the presence of NOx at upper-tropospheric concentrations from lightning. Our laboratory measurements show that isoprene emitted by rainforests may drive rapid new particle formation in extensive regions of the tropical upper troposphere1,2, resulting in tens of thousands of particles per cubic centimetre.
Subject terms: Atmospheric science, Climate change
Experiments performed in the CERN CLOUD chamber show that, under upper-tropospheric conditions, new atmospheric particle formation may be initiated by the reaction of hydroxyl radicals with isoprene emitted by rainforests.
Main
Aerosol particles are important for climate because they scatter and absorb incoming solar radiation and seed cloud droplets by acting as cloud condensation nuclei (CCN). More CCN make clouds more reflective and may increase their extent and lifetime. Around half of CCN globally, and almost all in the upper troposphere6, arise from new particle formation, which involves the spontaneous condensation of low-volatility vapours in the atmosphere to form liquid or solid particles (particle nucleation). The initial stable molecular clusters form at diameters slightly above 1 nm. To become CCN, the new particles should not be scavenged by pre-existing aerosol but grow by further vapour condensation to a size of around 50 nm and larger (particle growth). Although new particle formation has been extensively studied at ground-based sites7, little is known about the precursor vapours responsible for new particles in the remote upper troposphere and in marine regions. In particular, high concentrations of freshly formed particles are observed in the upper free troposphere over the Amazon1,2 and the tropical Atlantic and Pacific oceans3,4. Chemical-transport models indicate that new particle formation persists across the tropical upper troposphere over a latitude band covering about 40% of Earth’s surface3 and provides a global supply of CCN for low-altitude clouds in the sub-tropics and tropics3,8. However, the source of these particles has remained a puzzle for the past 20 years.
Early studies proposed that convective clouds could transport vapours from the boundary layer and form new particles in cold cloud outflows at high altitudes4,9. In the absence of an established mechanism, it was suggested that the oxidation products of isoprene (C5H8) could contribute to the high mass concentrations of freshly formed particles observed in the upper troposphere over the Amazon10. Recent modelling studies have speculated that pure biogenic new particle formation from monoterpenes is the source of these particles11,12, but insufficient monoterpene concentrations have been found to account for the high particle-number concentrations13.
On the other hand, recent satellite observations have revealed high concentrations of isoprene in the upper troposphere over tropical South America, Central Africa and Southeast Asia, reaching up to around 1 ppbv during night-time5 (1 ppbv is equivalent to around 0.6 × 1010 molecules cm−3 at 10 km altitude and 2.5 × 1010 molecules cm−3 at 0 km). The isoprene concentrations fall during daytime owing to high hydroxyl radical (OH) concentrations, which result in an isoprene lifetime of around 1–2 h. Furthermore, observations at 5,240 m altitude in the Bolivian Andes have found isoprene oxidation products in both gas and particle phases in air masses originating from the Amazon free troposphere14. Isoprene is emitted by vegetation, especially deciduous and broad-leaved evergreen trees, and is the most abundant hydrocarbon emitted into the atmosphere, after methane15. Median isoprene mixing ratios in the Amazon rainforest vary between 0.5 and 2.0 ppbv at night and 2.0–6.0 ppbv during the day16. Modelling studies show that isoprene is efficiently transported from the tropical boundary layer to the upper troposphere in deep convective clouds17.
Isoprene influences the oxidation capacity of the atmosphere18,19 and contributes to the formation of secondary organic aerosol20–24 particle mass, which—in turn—affects the climate25,26. The contribution of isoprene to secondary organic aerosol particle mass is at present believed to be dominated by the reactive uptake of isoprene dihydroxy epoxide (IEPOX)21,27 and other IP-OOM28. Lower contributions are thought to arise from the condensation of low-volatility compounds formed during oxidation of the first-generation product, isoprene hydroxy hydroperoxide (ISOPOOH)29–31. IEPOX and ISOPOOH are isomers of C5H10O3 and cannot be separated by normal chemical-ionization mass spectrometry. The ability of isoprene to form new particles is considered negligible24 and, moreover, isoprene inhibits new particle formation from monoterpenes under boundary-layer conditions32–34. However, it has so far remained unknown whether IP-OOM35 can form new particles in the upper troposphere, where it is extremely cold and scavenging losses are small, and—if so—what are the associated nucleation and growth rates.
CLOUD experiment
Here we report experiments performed in the CERN CLOUD chamber36 to study new particle formation from the reaction of OH with isoprene at upper-tropospheric concentrations and temperatures of −30 °C and −50 °C. The experiments were performed during the CLOUD15 and CLOUD16 campaigns, September–November 2022 and 2023, respectively. Before injection into the chamber, the isoprene vapour was passed through a cryo-trap at −53 °C to eliminate low-volatility contaminants (as confirmed by mass-spectrometer measurements). Further details of the CLOUD facility and its analysing instruments are provided in Methods.
The range of experimental parameters is summarized in Extended Data Table 1, together with the ambient upper-tropospheric conditions over the Amazon measured by Curtius et al.13 during research flight (RF) 19. To maximize the IP-OOM detection efficiency, we combined the measurements of three mass spectrometers that use ammonium, nitrate and bromide chemical-ionization (see Methods for further details), whereas the research flight uses nitrate ionization alone. In general, there is good overlap of the CLOUD experiments with the ambient conditions for this single flight (see Methods for further discussion). We note that, although the CLOUD chamber operates at atmospheric pressure, this does not affect the simulation of particle-nucleation dynamics at upper-tropospheric conditions. We performed experiments with nitrogen oxide (NO) concentrations varied between zero and around 7 × 109 cm−3, characteristic of the outflow from electrified deep convective clouds. Trace amounts of sulfuric acid (H2SO4), methanesulfonic acid (CH3SO3H) and iodine oxoacids37 (HIOx, x = 2, 3) also exist in the upper troposphere from the oxidation of vapours such as sulfur dioxide (SO2), dimethyl sulfide (DMS; CH3SCH3) and iodine (I2), respectively. For some experiments, therefore, we introduced upper-tropospheric concentrations of sulfuric acid or iodine oxoacids to explore their interactions with IP-OOM.
Extended Data Table 1.
Comparison of CLOUD experimental conditions with the ambient conditions during CAFE-Brazil research flight RF 19, periods T4 and T9 (Curtius et al.13)

The times of the CB flights are in local time. *Only CLOUD experiments with added NOx. †The definitions of IP0N and IP1-2N in the CB study refer to the measurement of the NO3− chemical ionization alone, whereas the CLOUD results refer to measurements using the combination of NO3−, Br− and NH4+ chemical-ionization methods. ‡Only CLOUD experiments with H2SO4. §The CLOUD limit-of-detection (LOD) for H2SO4 is 2 × 104 cm−3. ||The CB LOD for H2SO4 is 2 × 106 cm−3 at altitudes above 8 km. ¶Only experiments with HIO3. #The CLOUD LOD for NO is 2 × 108 cm−3. ☆Using equation 7 in ref. 35. **Using equations 1–4 in ref. 35.
IP-OOM
Hydroxyl radicals preferentially attack one of the terminal carbon atoms of isoprene, forming isoprene peroxy radicals (ISOPOO, C5H8(OH)(OO)). In the absence of NO, ISOPOO reacts with hydroperoxy radicals (HO2) to form ISOPOOH (C5H8(OH)(OOH))35,38. In the presence of NO, ISOPOO also reacts to form isoprene hydroxy nitrate (IHN, C5H8(OH)(ONO2)) and further products35. Further reaction of IHN with OH forms isoprene dihydroxy dinitrate (C5H8(OH)2(ONO2)2) and other second-generation products containing one or two nitrogen atoms. Further reactions of ISOPOOH with OH include the formation of its isomer, IEPOX21, as well as second-generation products containing zero or one nitrogen atom.
Oxidation by hydroxyl radicals of ISOPOOH29,30, IEPOX21,39,40 and IHN are the main channels that feed the second-generation IP-OOM, IP0-2N. Because the volatility of IP-OOM largely depends on their oxygen number41, we define IP0-2N as CiHjOkNl with the requirements that i, j ≥ 4, l = 0–2 and with a minimum oxygen number that takes the nitrogen content into account. As a nitrate group (-O-NO2) is considered to decrease the volatility of an organic compound by about the same factor as a hydroxyl group (-OH)41, we require (k − 2l) ≥ 4. We define IP0N, IP1N and IP2N as IP-OOM containing 0, 1 or 2 nitrogen atoms, respectively. In our study, therefore, IP0-2N (= IP0N + IP1N + IP2N) excludes ISOPOOH and IEPOX but may include some first-generation IP-OOM from ISOPOO35. The IP0-2N include IP-OOM monomers with up to five carbon atoms (C5) and dimers with up to ten carbons (C10), formed by reactions between two isoprene peroxy radicals (RO2), which produce covalently bound molecules.
As with other condensable vapours, both ISOPOOH and IEPOX deposit irreversibly on the CLOUD chamber walls at −50 °C (ref. 42) and also at −30 °C. The latter is confirmed by our measurements of a wall-loss lifetime for C5H10O3 at −30 °C, which indicates irreversible loss on impact (Extended Data Fig. 1a). Although we cannot distinguish ISOPOOH from IEPOX, chemical model calculations35,43 indicate that more than 80% of our C5H10O3 signal is ISOPOOH at 5.5 × 106 cm−3 OH (Methods and Extended Data Fig. 1b). Assuming this ISOPOOH fraction, we measure the total molar yield of IP0N from the reaction of hydroxyl radicals with C5H10O3 in the absence of nitrogen oxides (NOx) to be 46% at −30 °C and 55% at −50 °C, with a systematic uncertainty of a factor of two (Extended Data Fig. 2). In the presence of NOx, the IP0N yield falls to around 38% owing to RO2 termination by NO or by further reactions that produce IP1N and IP2N (Extended Data Fig. 2). At upper-tropospheric NO concentrations of up to 7 × 109 cm−3, we measure the nitrate IP-OOM fraction to be IP1-2N/IP0-2N = 23–88%, in which IP1-2N = IP1N + IP2N.
Extended Data Fig. 1. Wall-loss rates of C5H10O3, C5H12O6 and HIO3 at −30 °C and the IEPOX fraction of C5H10O3 at −30 °C and −50 °C.

a, Wall-loss lifetimes (1/(loss rate)) of C5H10O3, C5H12O6 and HIO3 at 100% mixing fan speed (increased loss rate). At stage 1, the mixing fan speed is set to 12% (standard operation). At stage 2, the mixing fan speed is set to 100% to increase the wall-loss rate. All three vapours have wall-loss rates consistent with that measured for H2SO4 (not shown), indicating irreversible loss on wall impact. b, Simulation of the fraction of IEPOX in C5H10O3 using the reduced Wennberg et al. mechanism35. The dashed grey line is a fit to the data of the form . Our simulation predicts that roughly 30% of the measured C5H10O3 signal is IEPOX under the conditions of panel a and that both the ISOPOOH and IEPOX (summed as C5H10O3) are lost to the chamber wall on impact, at −30 °C and at −50 °C. The experimental conditions for panel a are: isoprene = 0.13 ppbv (3.7 × 109 cm−3), O3 = 30 ppbv (8.6 × 1011 cm−3), I2 = 3.1 × 107 cm−3, SO2 = 3 × 108 cm−3, OH = 8.9 × 106 cm−3, HO2 = 1.8 × 108 cm−3, RH = 57% and temperature = −30 °C. The experimental conditions for panel b are: isoprene = 0.04–1.50 ppbv (0.1–4.2 × 1010 cm−3), O3 = 1–590 ppbv (3.7 × 1010 to 1.8 × 1013 cm−3), I2 = 0–7.5 × 107 cm−3, SO2 = 0–4.6 × 109 cm−3, OH = 0.11–6.90 × 107 cm−3, HO2 = 0.6–17.0 × 108 cm−3, HO2/OH ratio = 11–118, NO = 0–0.22 ppbv, NO2 = 0–0.77 ppbv, RH = 29–70% and temperature = −30 °C and −50 °C.
Extended Data Fig. 2. IP0N yield from OH reaction with C5H10O3.

Measured IP0N concentration versus reacted C5H10O3 (see Methods for details). The dashed lines show the predictions for several IP0N molar yields; a yield of 100% implies that each OH reaction with C5H10O3 produces one IP0N molecule. The data indicate that the yield is between 20% and 50%, with an overall systematic uncertainty of a factor of two. When NOx is added to the system, the yield of IP0N is reduced, owing to NO terminating isoprene peroxy radicals. The ratios of IEPOX and ISOPOOH in C5H10O3 are determined from the measured OH concentrations and the fit to the simulation shown in Extended Data Fig. 1b. For simplification, we apply a general reaction-rate coefficient of 10−10 and 10−11 cm3 s−1 for the ISOPOOH + OH and IEPOX + OH reactions, respectively35. The experimental conditions are: isoprene = 0.04–1.50 ppbv (0.1–4.2 × 1010 cm−3), O3 = 1–590 ppbv (3.7 × 1010 to 1.8 × 1013 cm−3), I2 = 0–7.5 × 107 cm−3, SO2 = 0–4.6 × 109 cm−3, OH = 0.11–6.90 × 107 cm−3, HO2 = 0.6–18.0 × 108 cm−3, HO2/OH ratio = 11–117, NO = 0–0.22 ppbv, NO2 = 0–0.77 ppbv, RH = 29–70% and temperature = −30 °C and −50 °C.
Example experiments
A typical run sequence in the absence of NOx is shown in Fig. 1. The experiment started at stage 1 by setting the internal mixing fans from 100% (high wall-loss rate) to 12% (standard operation) and switching on the ultraviolet (UV) light, which photolysed ozone to produce 2 × 106 cm−3 OH and 6 × 107 cm−3 HO2 radicals. Stages 1, 2 and 3 were, respectively, under different ionization conditions: neutral (all ions swept from the chamber by high-voltage electrodes); natural ionization from galactic cosmic rays (high voltage switched off); and pion beam (upper-tropospheric ion concentrations). The nucleation rate, J1.7, is the measured flux of particles passing a 1.7-nm threshold size. Because J1.7 varied little between these stages (Fig. 1b), it indicates relatively little sensitivity to ion concentrations. Before stage 5, the nucleation rate was 0.48–0.73 cm−3 s−1, whereas IP0N were around 1.6 × 107 cm−3 and sulfuric acid and iodine oxoacids were both below 105 cm−3. These nucleation rates far exceed those expected for HIOx and H2SO4, conservatively assuming 1 × 108 cm−3 contaminant ammonia (NH3) (refs. 37,44,45). As no other condensable vapours were present, this experiment shows that IP-OOM form new particles under upper-tropospheric conditions at −50 °C.
Fig. 1. Example new particle formation experiment from IP-OOM at −50 °C, without NOx.
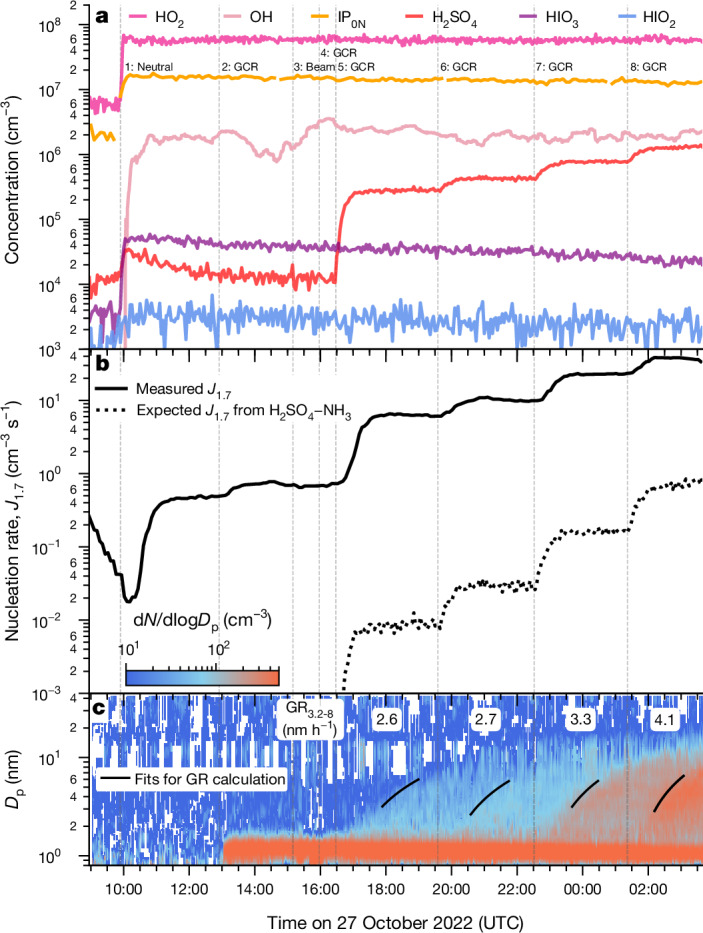
a–c, Evolution of vapour concentrations (a), particle-nucleation rates at 1.7 nm, J1.7 (b), and naturally charged negative particle number size distribution (dN/dlogDp) and growth rates measured between 3.2 and 8.0 nm, GR3.2-8 (nm h−1), for total (naturally charged + neutral) particles (c). The black lines in c depict the linear fits of 50% appearance time of particles between 3.2 and 8.0 nm. The vertical dashed lines and labels indicate the start of a new stage, at which the experimental conditions were adjusted. Trace sulfuric and iodic acid contaminants are present at the start of the run at concentrations of 1–5 × 104 cm−3. Sulfur dioxide is injected and progressively increased during stages 5–8, which produces steps in H2SO4. The dotted black curve in b shows the expected H2SO4–NH3 nucleation rate, conservatively assuming that NH3 is present at the 4 pptv limit of detection. Stages 1 and 3 are, respectively, under neutral (ion-free) and beam (ion-enhanced) conditions, whereas all of the other stages are under galactic cosmic ray (GCR; natural ion concentrations) conditions. The experimental conditions are: isoprene = 0.20–0.41 ppbv (6.2–13.0 × 109 cm−3), O3 = 84–96 ppbv (2.6–3.0 × 1012 cm−3), I2 = 3.7–23.0 × 105 cm−3, SO2 = 0–4 × 109 cm−3, OH = 1–3 × 106 cm−3, HO2 = 4.7–6.0 × 107 cm−3, HO2/OH ratio = 18–37, RH = 62%, NO less than limit of detection (7 pptv) and temperature = −49 °C.
During stages 5–8, SO2 was introduced into the chamber in steps, while keeping all other experimental conditions fixed (Fig. 1a). This increased the H2SO4 concentration from 1.5 × 104 cm−3 to between 2.8 × 105 and 1.3 × 106 cm−3. Despite these extremely low acid concentrations, which are equivalent to between 0.05 and 0.2 parts per trillion by volume (pptv) at 10 km altitude, the nucleation rate increased from 6.2 to 37 cm−3 s−1 between stages 5 and 8, respectively (Fig. 1b). Once again, these nucleation rates far exceed those expected for H2SO4–NH3. The particle growth rates between 3.2 and 8.0 nm increased from 2.6 to 4.1 nm h−1 between stages 5 and 8 (Fig. 1c). Because the expected particle growth rate for 1.3 × 106 cm−3 acid is around 0.15 nm h−1 (ref. 46), these high growth rates must largely result from condensation of IP0N.
In other experiments, we introduced small concentrations of HIOx in the presence of negligible H2SO4. Extended Data Fig. 3 shows an example at −50 °C. Here IP0N were held constant at 1.6 × 107 cm−3 s−1 and contaminant H2SO4 was less than 5.0 × 104 cm−3. The HIOx concentration was increased in steps from 1.2 × 105 to 8.6 × 105 cm−3 and the nucleation rate increased from 3.3 to 18 cm−3 s−1. These nucleation rates closely match those measured for the IP0N–H2SO4 system, suggesting that either inorganic acid plays a similar role in nucleation by stabilizing the embryonic molecular clusters.
Extended Data Fig. 3. Example new particle formation experiment at −50 °C with added HIOx.

Evolution of vapour concentrations (a), particle-nucleation rates at 1.7 nm, J1.7 (b), and naturally charged particle number size distribution (dN/dlogDp) and growth rate measured between 3.2 and 8.0 nm, GR3.2-8 (nm h−1) (c). The black line in panel c depicts the linear fit of 50% appearance time of particles between 3.2 and 8.0 nm. The expected nucleation rates for HIOx at the kinetic limit are less than 2 × 10−4 cm−3 s−1. The results show that HIOx enhances IP0N nucleation in a similar way as H2SO4. Discontinuous lines show missing data. The experimental conditions are: isoprene = 0.07–0.16 ppbv (2.1–4.9 × 109 cm−3), O3 = 95 ppbv (3 × 1012 cm−3), I2 = 0.4–6.7 × 107 cm−3, OH = 3.2 × 106 cm−3, HO2 = 7.4 × 107 cm−3, RH = 63% and temperature = −49 °C.
In Extended Data Fig. 4, we show two further examples at −50 °C, without (left panels) and with (right panels) NOx. Both experiments started with acid concentrations below the limit of detection (2 × 104 cm−3). Extended Data Fig. 4c shows a threefold enhancement in J1.7 with the transition from neutral to galactic cosmic ray conditions, reaching 0.7 cm−3 s−1 at 2.3 × 107 cm−3 non-nitrate IP-OOM (IP0N) but showing very little further increase at higher (beam) ionization rates. This indicates that ions can enhance the stability of IP-OOM molecular clusters at especially low concentrations of acids or IP-OOM. When NOx is present and under galactic cosmic ray conditions (Extended Data Fig. 4d), the initial nucleation rate is 1.7 cm−3 s−1 at 3.5 × 107 cm−3 IP0N plus 1.3 × 108 cm−3 IP1-2N. This comparison suggests that nitrate IP-OOM are less effective for nucleation than non-nitrate IP-OOM. In Extended Data Fig. 4b, sulfuric acid was increased in steps from 2 × 104 to 3 × 106 cm−3 and J1.7 increased from 1.7 to 43 cm−3 s−1, demonstrating that the synergy between IP-OOM and sulfuric acid also occurs in the presence of NOx.
Extended Data Fig. 4. Aerosol formation from pure IP0N products and mixture of IP0N, IP1-2N and H2SO4.

Evolution of vapour concentrations (a,b), particle-nucleation rates at 1.7 nm, J1.7 (c,d), and naturally charged particle number size distribution (dN/dlogDp) and growth rates measured between 3.2 and 8.0 nm, GR3.2-8 (nm h−1) (e,f). The black lines in panel f depict the linear fits of 50% appearance time of particles between 3.2 and 8.0 nm. The results show that pure IP0N aerosol formation is feasible and atmospheric ions can enhance the nucleation rate by a few times. The addition of low concentrations of H2SO4 further enhances IP0-2N nucleation by up to two orders of magnitude. The vertical dashed lines and labels indicate the start of a new stage in which the experimental conditions were adjusted. The dotted black curve in panel d shows the expected H2SO4–NH3 nucleation rate, conservatively assuming that NH3 is present at the 4 pptv limit of detection44,93. Stages 1 and 3 are, respectively, under neutral (ion-free) and beam (ion-enhanced) conditions, whereas all of the other stages are under galactic cosmic ray (GCR; natural ion concentrations) conditions. The SO2 concentration is increased in steps during stages 5–12. The experimental conditions for the right panels are: isoprene = 0.08–0.63 ppbv (2.4–20.0 × 109 cm−3), O3 = 189 ppbv (6 × 1012 cm−3), OH = 2.4 × 106 cm−3, HO2 = 1.8 × 108 cm−3, RH = 32% and temperature = −49 °C. The experimental conditions for the left panels are: isoprene = 0.05–0.50 ppbv (1.6–16.0 × 109 cm−3), O3 = 1.2 ppbv (3.8 × 1010 cm−3), SO2 = 0–1.9 × 108 cm−3, OH = 6 × 107 cm−3, HO2 = 1 × 109 cm−3, NO = 0.12–0.81 ppbv, NO2 = 0.57–0.84 ppbv, RH = 52% and temperature = −48 °C.
New particle formation rates
We show in Extended Data Fig. 5 our measurements of J1.7 versus IP0N, IP1-2N and IP0-2N at −50 °C, in the absence of acids (near or below the limit of detection). Extended Data Fig. 5a shows that the nucleation rates increase from around 0.006 cm−3 s−1 to 48 cm−3 s−1 by increasing IP0N from 6 × 106 to 9 × 107 cm−3. By contrast, there is a relatively weak dependence of the nucleation rate on IP1-2N (Extended Data Fig. 5b,c, which cover a larger IP1-2N range from 2.4 × 106 to 2.5 × 108 cm−3). Nevertheless, careful inspection of the data in Extended Data Fig. 5b at the lowest IP0N concentrations does show some dependence of J1.7 on nitrate IP-OOM. The combined measurements suggest that IP0N are more effective for nucleation than IP1-2N, or—equivalently—that nitrate IP-OOM have higher volatilities than non-nitrate IP-OOM. However, at colder temperatures in the upper troposphere, the volatilities of all IP-OOM will decrease and nitrate IP-OOM will contribute more strongly to nucleation.
Extended Data Fig. 5. Particle-nucleation rates from IP-OOM at −50 °C, in the absence of acids.

Nucleation rates at 1.7 nm, J1.7, versus IP-OOM without nitrogen, IP0N (a), with nitrogen, IP1N + IP2N (b) and the sum, IP0-2N (c). Panel a indicates a mild enhancement of the nucleation rates with increased IP1N + IP2N. Panels b and c confirm that the nucleation rates are only weakly dependent on IP1N + IP2N. These data show that non-nitrate IP-OOM are primarily responsible for particle nucleation, with a smaller contribution from nitrate-containing isoprene products. The experimental conditions are: isoprene = 0.04–0.50 ppbv (0.14–1.7 × 1010 cm−3), O3 = 1.2–590 ppbv (4 × 1010 to 1.8 × 1013 cm−3), OH = 0.14–6.40 × 107 cm−3, HO2 = 1.6–17.0 × 108 cm−3, HO2/OH ratio = 11–118, NO = 0–0.18 ppbv, NO2 = 0–0.74 ppbv, RH = 29–61% and temperature = −50 °C. All experiments are carried out under galactic cosmic ray ionization (ambient-boundary-layer conditions). The error bars represent the standard deviation of the measurement at steady state.
In Fig. 2, we present our measurements of J1.7 versus vapour concentrations at −30 °C and −50 °C. Here we consider only the IP0N component of IP0-2N, following the discussion above. In our experiments, the range of acid concentrations (mostly below 3 × 106 cm−3) is representative of the upper free troposphere47,48 and the isoprene concentrations (0.14–4.2 × 1010 cm−3) correspond to those measured in the upper troposphere over tropical rainforests5, as do the oxidant concentrations18 (OH = 0.11–6.9 × 107 cm−3, HO2 = 0.6–17 × 108 cm−3 and HO2/OH ratio = 11–118). These vapour concentrations are sufficient to drive rapid nucleation rates between 0.006 and 48 cm−3 s−1, which greatly exceed HIOx and H2SO4–NH3 nucleation under these conditions37,44,45. Such fast nucleation rates can readily account for the high particle-number concentrations of up to 20,000 cm−3 observed between 8 and 14 km over the Amazon2.
Fig. 2. Particle-nucleation rates from IP-OOM at −30 °C and −50 °C, with variable NOx.

a–d, Nucleation rates at 1.7 nm, J1.7, versus IP0N without NOx present (a), IP0N with NOx (b), HIOx + H2SO4 with and without NOx (c) and the product (HIOx + H2SO4) × IP0N with and without NOx (d). Measurements without NOx are indicated by diamonds (without acids) and circles (with acids). Measurements with NOx are indicated by triangles (without acids) and squares (with acids). Hollow symbols indicate −30 °C and solid symbols indicate −50 °C. In c, nucleation rates measured at contaminant acid concentrations are assigned the acid limit of detection (around 2 × 104 cm−3). The solid lines in c show the nucleation rates expected for H2SO4 with 4 pptv NH3 at −30 °C (red) and −50 °C (blue), both at 60% RH (ref. 44). The dashed and solid lines in d represent fits to the equation 10a×log10(x)+b, in which, for the dashed line, a = 1.241 and b = −15.065, and for the solid line, a = 1.505 and b = −19.948. Panels a–c show that both IP0N and total acid (HIOx + H2SO4) contribute to the nucleation rate. Panel d indicates that it is the product of IP0N and total acid (that is, the dimer formation rate) that best describes the nucleation rate, J1.7, as the data points cluster into two groups primarily characterized by temperature alone. IP1-2N also contribute to particle nucleation but they are less effective than IP0N (Extended Data Fig. 5). The experimental conditions are: isoprene = 0.04–1.50 ppbv (0.1–4.2 × 1010 cm−3), O3 = 1–590 ppbv (3.7 × 1010 to 1.8 × 1013 cm−3), I2 = 0–7.5 × 107 cm−3, SO2 = 0–4.6 × 109 cm−3, OH = 0.11–6.90 × 107 cm−3, HO2 = 0.6–17.0 × 108 cm−3, HO2/OH ratio = 11–118, NO = 0–0.22 ppbv, NO2 = 0–0.77 ppbv, RH = 29–70% and temperature = −30 °C and −50 °C. The error bars represent the standard deviation of the measurement at steady state. All measurements are made under galactic cosmic ray conditions (natural ionization amounts).
The nucleation rates depend on the concentrations of both IP0N (Fig. 2a,b) and total acid (HIOx + H2SO4; Fig. 2c). Figure 2d suggests that the nucleation rates are linearly dependent on the product of IP0N and total acid concentration, indicating that the critical step is dimer formation of an inorganic acid with a single IP0N. We note that the J1.7 measurements in Fig. 2d at −50 °C with acids and NOx (filled square symbols) are systematically around a factor of 2–3 lower than the measurements without NOx (filled circles). This is an artefact resulting from a systematic uncertainty in those measurements (filled square symbols), as it is absent when J2.5 is measured for the same events but with a different particle counter (Extended Data Fig. 6). We measure a 20-fold increase of J1.7 between −30 °C and −50 °C (Fig. 2d) owing to decreasing IP-OOM volatilities.
Extended Data Fig. 6. Particle-nucleation rates at 2.5 nm versus vapour concentrations.

Nucleation rates at 2.5 nm, J2.5, versus IP0N (a,b), HIOx + H2SO4 (c) and the product (HIOx + H2SO4) × IP0N (d). Panel a and b show experiments without and with added NOx. Panel c shows all experiments and panel d shows experiment with acids, both with and without NOx. IP0N excludes nitrogen-containing isoprene-oxygenated molecules. The solid lines in panel c show the nucleation rates at 1.7 nm expected for H2SO4 with 4 pptv NH3 at 60% RH (ref. 44). The dashed and solid lines in panel d represent fits to the equation 10a×log10(x)+b, in which, for the dashed line, a = 1.167 and b = −14.743, and for the solid line, a = 1.669 and b = −22.707. Panels a and b show that both total acid (HIOx + H2SO4) and IP0N contribute to the nucleation rate. The experimental conditions are: isoprene = 0.04–1.50 ppbv (0.1–4.2 × 1010 cm−3), O3 = 1–590 ppbv (3.7 × 1010 to 1.8 × 1013 cm−3), I2 = 0–7.5 × 107 cm−3, SO2 = 0–4.6 × 109 cm−3, OH = 0.11–6.90 × 107 cm−3, HO2 = 0.6–18.0 × 108 cm−3, HO2/OH ratio = 11–118, NO = 0–0.22 ppbv, NO2 = 0–0.77 ppbv, RH = 29–70% and temperature = −30 °C and −50 °C. All experiments are carried out under galactic cosmic ray ionization (ambient conditions). The error bars represent the standard deviation of the measurement at steady state.
Molecular content of nucleating clusters
We have confirmed the nucleation mechanism inferred from Fig. 2d by direct molecular measurements with an Atmospheric Pressure Interface Time-of-Flight (APi-TOF) mass spectrometer during nucleation events without acids at −50 °C (Fig. 3a,c). Negatively charged (ion-induced) nucleation involves sequential accretion of IP-OOM monomers (C5 band) or dimers (C10 band) to an initial CiHjOx− or NO3− ion. When NOx is present, a sharp reduction can be seen in the concentration of the C15 clusters compared with C10 (Fig. 3c). By contrast, the no-NOx data (Fig. 3a) show a smooth sequential growth of the clusters out to the detection limit. This indicates that the C15 clusters with NOx are relatively unstable and have a high evaporation rate back to C10 clusters, hence the high concentration of the latter. We infer that the IP1-2N have higher volatility than IP0N and are less effective for nucleation. Nevertheless, C10 and C15 molecular clusters are seen in Fig. 3c with more than 3N (including NO3− core ion), which must include contributions from IP1-2N as well as IP0N. This confirms that IP1-2N do indeed participate in ion-induced nucleation, but they are less effective than IP0N, which leads to a rate-limiting step from C10 to C15. This is consistent with the previous conclusions drawn from measurements of nucleation rate.
Fig. 3. Molecular composition of charged and neutral clusters during IP-OOM nucleation without acids at −50 °C.

a–d, Mass defect (difference from integer mass) versus m/z during IP-OOM nucleation events without acid and without added NOx (a,b) or with NOx (c,d). The data points are coloured by the number of carbon (a,b) or nitrogen (c,d) atoms. The symbol area in a and c is proportional to the normalized signal intensity by total signal, whereas that in b and d is proportional to IP-OOM concentrations. The charger ions (Br−, NO3− and NH4+) are removed from the molecular formula in b and d. The data show that IP-OOM—which are around C5 for the monomer or C10 for the dimer—nucleate at −50 °C. The experimental conditions in a–d are: isoprene = 0.19, 0.07, 0.28 and 0.07 ppbv, O3 = 196.0, 129.0, 1.9 and 1.8 ppbv, OH = 0.3, 0.3, 1.6 and 4.6 × 107 cm−3, HO2 = 1.8, 2.2, 3.4 and 6.8 × 108 cm−3, NO = 0, 0, 0.07 and 0.18 ppbv, NO2 = 0, 0, 0.10 and 0.54 ppbv, RH = 31, 57, 66 and 29% and temperature = −49, −49, −48 and −48 °C, respectively. The concentrations of IP0N were unmeasured (instrument was not available), 3.4 × 107, unmeasured and 2.5 × 107 cm−3 and the concentrations of IP1-2N were unmeasured, 0, unmeasured and 1.5 × 108 cm−3, respectively. The bromide chemical-ionization mass spectrometer was converted to measure charged clusters for the experiments shown in a and c, under ground-level and upper-tropospheric ion concentrations, respectively.
The composition of neutral clusters and molecules during IP-OOM nucleation without and with NOx is shown in Fig. 3b,d, respectively. Here no signal is detected above the C10 band owing to the relatively low charging efficiency of the chemical-ionization mass spectrometers, compared with unit efficiency for ions and ion-induced (charged) clusters in the APi-TOF mass spectrometer. Nevertheless, the neutral data reveal a clear shift towards (higher-mass) IP1N and IP2N compounds after the addition of NOx. Furthermore, comparison with the corresponding charged clusters (Fig. 3a,c) shows that nucleation favours the more highly oxygenated compounds with lower volatility.
In Extended Data Fig. 7, we show the molecular composition of charged clusters in the presence of trace amounts of sulfuric acid (Extended Data Fig. 7a,c,e) and iodic acid (Extended Data Fig. 7b,d) during IP-OOM nucleation at −50 °C. The data are without NOx (Extended Data Fig. 7a–e) and with NOx (Extended Data Fig. 7f). These measurements confirm that the same nucleation mechanism occurs after addition of acids as seen previously without acids, that is, sequential addition of IP-OOM monomers (C5) or dimers (C10) to a core acid ion. Here the negatively charged core ions comprise the pure monomer, dimer or trimer of sulfuric acid (Extended Data Fig. 7a,c) with HSO4−, or iodic acid with IO3− (Extended Data Fig. 7b,d), whereas the positively charged core ions (Extended Data Fig. 7e,f) comprise an IP-OOM+. The core acid anion serves only to stabilize the initial molecular cluster with a single IP-OOM; there is no evidence for accretion of further acid molecules as the clusters grow, as expected at these very low acid concentrations (3–7 × 105 cm−3).
Extended Data Fig. 7. Molecular composition of charged clusters with sulfuric acid, iodic acid and no acid during IP-OOM nucleation at −50 °C.

Mass defect (difference from integer mass) versus m/z (mass-to-charge ratio) during nucleation events for ions and negatively charged (a–d) and positively charged (e,f) molecular clusters measured with two APi-TOF instruments (see Methods), coloured by the number of carbon (a,b,e), sulfur (c), iodine (d) and nitrogen (f) atoms. The data are without NOx (a–e) and with NOx (f). In panel d, the (purple) clusters with no iodine contain a NO3− ion. The symbol area is proportional to the ion signal rate (cps, counts s−1). It should be noted that the APi-TOF used in this experiment is less sensitive than the MION2-APi-TOF and thus the absolute ion signals and cluster population should not be compared between the instruments. Together with Fig. 3, our results show that ion-induced nucleation occurs both with pure IP-OOM and with further H2SO4 and HIOx. The experimental conditions for a,c,e are: isoprene = 0.19 ppbv (5.9 × 109 cm−3), O3 = 93 ppbv (2.9 × 1012 cm−3), I2 = unmeasured, SO2 = unmeasured, OH = 3.0 × 106 cm−3, HO2 = unmeasured, RH = 63% and temperature = −49 °C. The concentrations of IP0N = unmeasured, H2SO4 = 3.0 × 105 cm−3 and HIOx = 2.8 × 104 cm−3. The experimental conditions for b,d are: isoprene = 0.09 ppbv (2.8 × 109 cm−3), O3 = 97 ppbv (3 × 1012 cm−3), I2 = 5.1 × 107 cm−3, SO2 = 0, OH = 3.4 × 106 cm−3, HO2 = 7.6 × 107 cm−3, RH = 63% and temperature = −49 °C. The concentrations of low-volatility vapours are IP0N = 1.3 × 107 cm−3, H2SO4 = 8.3 × 103 cm−3 and HIOx = 6.7 × 105 cm−3. The experimental conditions for f are: isoprene = 0.12 ppbv (3.7 × 109 cm−3), O3 = 1.2 ppbv (3.7 × 1010 cm−3), SO2 = 1.2 × 108 cm−3, OH = 6.2 × 107 cm−3, HO2 = 1 × 109 cm−3, RH = 42%, NO = 0.25 ppbv, NO2 = 0.78 ppbv and temperature = −48 °C. The concentrations of low-volatility vapours are IP0N = 1.2 × 107 cm−3, H2SO4 = 2.4 × 105 cm−3 and HIOx = 0 cm−3.
Particle growth rates
Our measurements indicate that IP0N (and, less effectively, IP1-2N) are rapidly nucleating together with H2SO4 and HIOx at −30 °C and −50 °C (Fig. 2). We therefore expect that IP-OOM will also drive rapid particle growth at larger sizes as the Kelvin (curvature) barrier falls and progressively higher-volatility compounds are able to condense onto the particles—as previously seen for new particle formation from α-pinene oxidation products49. We show in Fig. 4 our measurements of particle growth rates between 3.2 and 8.0 nm versus IP0N (Fig. 4a) and IP0-2N (Fig. 4b) for experiments with and without NOx at −30 °C and −50 °C. Figure 4a shows that IP1-2N are strongly contributing to early growth of particles; the measured growth rates cannot be explained by IP0N alone. This is confirmed in Fig. 4b, in which all data (with or without NOx and at either temperature) are consistent with having the same dependency of growth rate on IP0-2N.
Fig. 4. Particle growth rates and particle composition.

a,b, Particle growth rates from 3.2 to 8.0 nm, GR3.2-8, versus IP0N (a) and IP0-2N (b). The solid lines show the predicted GR3.2-8 at the kinetic limit assuming monomer condensation and without considering any dipole enhancement. The prediction assumes that IP-OOM have a general formula of C5H12O6 and a density of 1.34 g cm−3 (ref. 50). The dashed lines are linear fits to the experimental data of the form GR3.2-8 = 2.04 × 10−7 × IP0N (a) and GR3.2-8 = 1.23 × 10−7 × IP0-2N (b). c,d, Mass-defect plots showing the molecular composition of particles measured with the FIGAERO at −30 °C (Br-FIGAERO) and −50 °C (I-FIGAERO), with particle geometric mean sizes ranging from 6 to 20 nm. The colour legends indicate the number of atoms of carbon (c) and nitrogen (d). The symbol area is proportional to the normalized signal by the sum of measured signals. The charger ions are removed from the molecular formula. The annotations show the molecular formula of the particle-phase compounds. Our measurements show that both IP0N and IP1-2N drive rapid early particle growth and that C5H12O5-6 are the main condensing vapours in IP0N. The experimental conditions in a and b are: isoprene = 0.05–1.50 ppbv (0.15–4.40 × 1010 cm−3), O3 = 1–592 ppbv (3.6 × 1010 to 1.8 × 1013 cm−3), I2 = 0–1.1 × 108 cm−3, SO2 = 0–4.9 × 109 cm−3, OH = 0.09–7.40 × 107 cm−3, HO2 = 0.6–19.0 × 108 cm−3, NO = 0–0.26 ppbv, NO2 = 0–0.80 ppbv, RH = 29–72% and temperature = −30 °C and −50 °C. The experimental conditions for in c are: isoprene = 1.1 ppbv, O3 = 184 ppbv, I2 = 2.9 × 107 cm−3, SO2 = 2.7 × 107 cm−3, OH = 2.3 × 106 cm−3, HO2 = 0.8 × 108 cm−3, NO = 0 ppbv, NO2 = 0 ppbv, RH = 72%, temperature = −30 °C and DMS = 0 ppbv and those for in d are: isoprene = 0.2 ppbv, O3 = 1 ppbv, I2 = 0, SO2 = 1.2 × 108 cm−3, OH = 5.2 × 107 cm−3, HO2 = 1.2 × 109 cm−3, NO = 0.11 ppbv, NO2 = 0.65 ppbv, RH = 38%, temperature = −48 °C and DMS = 0.16 ppbv. The vertical error bars represent the statistical uncertainty in the appearance-time growth-rate measurements derived from the 95% confidence interval on the growth-rate fit. The horizontal error bars represent the standard deviation of measured IP0N or IP0-2N during the growth period.
We find that isoprene at upper-tropospheric concentrations will drive particle growth rates between 3 and 56 nm h−1. These rapid growth rates imply that new IP-OOM particles can reach several tens of nanometres in size within a few hours, which will help to prevent them from evaporation when descending to lower altitudes and warmer temperatures. Within measurement uncertainties, particle growth rates at all temperatures below −30 °C are linearly dependent on the IP0-2N concentration, and they reach the kinetic limit. As expected from their low concentrations, the growth rates show no correlation with H2SO4 and HIOx (not shown).
We have verified these observations by direct particle-phase measurements made with the Filter Inlet for Gases and AEROsols (FIGAERO; Fig. 4c,d). The main IP0-2N compounds in the particles are two second-generation oxidation products (C5H12O5 and C5H12O6) formed from reactions of ISOPOOH with OH, which together constitute 30% of the total signal. The same two compounds have been previously identified in the particle phase by isoprene experiments at room temperature30. The remaining particle-phase compounds are largely C10 IP-OOM dimers, in agreement with their measured low volatilities. The non-nitrate C10 dimers are suppressed by the presence of NOx, as seen by comparing Fig. 4c with Fig. 4d. We note that the nitrate IP-OOM in Fig. 4d are probably under-represented because we measured that they have lower evaporation temperatures and so may escape thermal-desorption measurement in the FIGAERO.
Upper-tropospheric particle formation
In summary, we find that IP-OOM rapidly form new particles at upper-tropospheric concentrations and temperatures below −30 °C. Moreover, the nucleation rates are up to 100 times faster in the presence of extremely low concentrations of sulfuric acid or iodine oxoacids, reaching rates around 30 cm−3 s−1 at −50 °C and acid concentrations of 106 cm−3. In the presence of NOx, a large fraction of IP-OOM—around 23–88%—are found to contain either one or two nitrogen atoms. We find that nitrate IP-OOM contribute relatively weakly to particle nucleation compared with non-nitrate IP-OOM at the same concentration. However, cooler temperatures will favour nucleation from nitrate isoprene products13. Both non-nitrate and nitrate IP-OOM are equally effective at driving rapid particle growth at several tens of nm h−1, at all temperatures below −30 °C. We present in Extended Data Fig. 8 four nucleation rate measurements that schematically encapsulate the effect of acids and NOx on IP-OOM nucleation.
Extended Data Fig. 8. Four experiments that demonstrate the influence of acids and NOx on IP-OOM nucleation at −50 °C.

Four experiments showing nucleation rates at 1.7 nm, J1.7, measured at similar IP0N concentrations, 1.2–3.3 × 107 cm−3, but with added acids, NOx and acids + NOx, respectively. The marker area is proportional to the IP0N concentration. The experimental conditions are shown next to the data points. Circles correspond to absence of acids and squares include acids. The data show that extremely low H2SO4 concentrations, 1.3–2.8 × 106 cm−3, strongly enhance the nucleation rates regardless of the absence or presence of NOx. On the other hand, additions of high nitrate IP-OOM concentrations around 108 cm−3 only produce relatively small increases in nucleation rates.
Our measured nucleation and growth rates provide the mechanistic missing link connecting the presence of abundant isoprene in the tropical upper troposphere5 with the high particle-number concentrations found at high altitudes over the Amazon2. Our findings reveal a new mechanism (Fig. 5) that switches on rapid particle nucleation in extensive regions of the upper troposphere. Isoprene emitted by tropical rainforests is efficiently transported by deep convective clouds and released at cloud outflows in the upper free troposphere10,17. During night-time, high isoprene concentrations build up in the upper troposphere5 as a result of relatively slow oxidation by ozone and nitrate radicals35. During daytime, the isoprene is rapidly oxidized by hydroxyl radicals and mixed with NOx from lightning to produce IP-OOM. These mix with trace ambient acids to drive rapid particle nucleation and growth at cold temperatures below around −30 °C. Peak IP-OOM concentrations—and therefore the fastest new particle formation rates—will occur shortly after sunrise when the isoprene accumulated during the night is oxidized during the first 1–2 h of daylight5. However, later in the day, the increase of OH and HO2 will accelerate reactions that form acids and favour production of non-nitrate IP-OOM from daytime-convected isoprene, which may lead to further particle nucleation. The newly formed particles grow over periods of several days by further condensation of low-volatility vapours, including acids. Model studies show that particles nucleated in the upper free troposphere over the Amazon are gradually transported downwards on horizontal scales much larger than 1,000 km (ref. 8).
Fig. 5. Schematic of new particle formation from isoprene in the upper troposphere.

Isoprene from forests is efficiently transported at night by deep convective clouds into the upper troposphere. During daylight, the isoprene accumulated overnight, together with daytime-convected isoprene, reacts with hydroxyl radicals and NOx from lightning to produce IP-OOM. The IP-OOM combine with trace ambient acids to produce high particle-number concentrations at cold temperatures below −30 °C. The newly formed particles grow rapidly over several hours to days while following the descending air masses. This mechanism may provide an extensive source of CCN for shallow continental and marine clouds, which strongly influence Earth’s radiative balance.
Isoprene is the most abundant non-methane hydrocarbon emitted into the atmosphere, but its ability to nucleate particles in the boundary layer is considered negligible. Our findings show, however, that isoprene emitted by forests can drive rapid particle nucleation and growth in the upper troposphere. After further growth and descent to lower altitudes, these particles may represent a globally important source of CCN for shallow continental and marine clouds, and so influence Earth’s radiative balance. Isoprene from forests may therefore provide a major source of biogenic particles in both the present-day and pre-industrial atmospheres that are at present unaccounted in atmospheric chemistry and climate models.
Methods
The CLOUD experiments
The CERN CLOUD chamber36 was used to conduct the experiments presented in this study. CLOUD is an electropolished, stainless-steel, 26.1-m3 chamber designed to study new particle formation under the full range of tropospheric and lower-stratospheric conditions. The thermal housing around the chamber is able to control the temperature from 208 to 373 K with high precision (±0.1 K)51. CLOUD was operated at a pressure of approximately 965 ± 5 mbar in this study. To avoid cross-contamination between different experimental programmes and to achieve extremely low NH3 concentrations, the chamber is cleaned by rinsing the chamber walls with ultrapure water and heating to 373 K for more than 24 h. To maintain cleanliness and ensure minimal contamination, ultrapure synthetic air—derived from mixing cryogenic liquids (21% oxygen and 79% nitrogen)—is continuously injected into the chamber. The chamber is characterized by a low loss rate, with condensation sink values comparable with those observed in pristine environments.
Various light sources are positioned in the CLOUD chamber to selectively drive photochemistry. OH production is initiated by illuminating O3 with a UV fibre-optic system, a combination of four 200-W Hamamatsu Hg-Xe lamps with wavelengths spanning 250 and 450 nm, a krypton fluoride (KrF) excimer UV laser at 248 nm and a 52-W low-pressure mercury lamp centred at 254 nm. As well as O3 photolysis, OH radicals are also produced by photochemical production from nitrous acid (HONO) and hydrogen peroxide (H2O2). Both the HONO and H2O2 generators were designed specifically for CLOUD experiments. Following the same principle as an earlier study52, a gas–liquid mixture of HONO is synthesized from continuous mixing of H2SO4 (Sigma Aldrich, 99%) with sodium nitrite (NaNO2, Sigma Aldrich, 99%) in a stainless-steel reactor53. HONO is transferred from liquid phase to gas phase by flowing nitrogen gas (1–2 l min−1) through the reactor. HONO is then introduced into the CLOUD chamber and photolysed by a UV light source centred at 385 nm to produce OH radicals and NO. The HONO reactor is continuously cooled to 5 °C and a cryo-trap is placed between the reactor and the chamber to remove excess water vapour and avoid ice blockage of the chamber input pipe. Gaseous H2O2 is produced from bubbling N2 gas through a H2O2 solution. The H2O2 solution is stored in a glass beaker contained in a stainless-steel container at a constant temperature of 5 °C. A different combination of UV sources is used to photolyse H2O2 to produce different amounts of OH radicals.
A green light sabre centred at 528 nm is used to photolyse molecular iodine (I2). All light systems are continuously monitored by a spectrometer and an array of photodiodes at the bottom of the chamber. Dedicated actinometry experiments allow quantitative determination of actinic fluxes of the light system at different intensities.
Particle formation under different ionization regimes is simulated by combining a strong electric field (±30 kV) and the pion beam produced by the CERN Proton Synchrotron. The electric field eliminates natural ions in less than 1 s, thus creating ion-free conditions (neutral experiments). The pion beam produced by the CERN Proton Synchrotron enhances ion production on top of the galactic cosmic rays. Two magnetically coupled stainless-steel fans mounted at the top and bottom of the chamber enable uniform spatial mixing of particles and vapours within a few minutes. Iodine is injected into the chamber from a temperature-controlled evaporator containing crystalline iodine (I2, Sigma-Aldrich, 99.999% purity) at the bottom of the chamber. The SO2 (Carbagas, 100 parts per million by volume (ppmv) in N2) and isoprene (PanGas, 1,000 ppmv in N2) are injected into the chamber from pressurized gas cylinders and the O3 is introduced to the chamber by passing O2 through an ozone generator.
The data presented in this study were collected in two consecutive CLOUD campaigns (CLOUD15 and CLOUD16). The CLOUD15 and CLOUD16 campaigns were carried out from September to November in 2022 and 2023, respectively. Because the experiments reported in this study were carried out at extremely low temperatures (−30 °C and −50 °C), heat-insulation systems (CLOUD15) and active cooling systems (CLOUD16) were used to reduce measurement systematic error. The heat-insulation systems were primarily made with thermal insulation foam to isolate the instrument inlet system from ambient air. The active cooling systems involved circulating the air inside the chamber thermal housing, at the same temperature as the chamber, around the inlet systems of different instruments. The active cooling systems were also wrapped with thermal insulation foam to allow for more effective inlet cooling. These cooling systems were applied to all mass spectrometers and particle counters, except a butanol condensation particle chamber (CPC; TSI 3776), a nano-scanning mobility particle sizer (nano-SMPS, TSI 3938) and a long-SMPS (TSI 3082), which used a heat-insulation system in both campaigns to act as a standard to avoid systematic errors resulting from changing from the heat-insulation system to the active cooling system.
Measurement of chemical composition
Ozone (O3)
O3 was monitored using a gas monitor (Thermo Environmental Instruments, TEI 49C).
Hydroxyl radicals (OH)
The OH radical was measured by HORUS54 (HydrOxyl Radical measurement Unit based on fluorescence Spectroscopy).
Hydroperoxyl radical (HO2)
The HO2 radical was primarily measured using the bromide chemical-ionization mass spectrometer coupled with a multi-scheme chemical-ionization inlet-2 (Br-MION2-CIMS)55 and HORUS in both CLOUD15 and CLOUD16 campaigns. HORUS measures HO2 by chemically converting it to OH by NO. However, the RO2 radical (organic peroxy radicals) produced from isoprene oxidation may also contribute to the HO2 signal measured by HORUS, as the reaction between RO2 + NO can also produce OH radicals. By contrast, the HO2 measurement by Br-MION2-CIMS is less ambiguous, as it is defined by the peak HO2Br− (ref. 55). However, the measurement of HO2 by Br-MION2-CIMS is severely affected by air–water content55, making offline calibration difficult. Therefore, the HO2 measurement by Br-MION2-CIMS was calibrated by HORUS under RO2 radical-free conditions. The online calibration was carried out for every absolute humidity condition reported in this manuscript. During a small section in which the primary ions of Br-MION2-CIMS were saturated by either HONO or H2O2, either the low-pressure bromide chemical-ionization mass spectrometer or HORUS was used to complement the HO2 measurement after intercomparing the data with Br-MION2-CIMS and HORUS during experiments without HONO and H2O2. The precision of OH and HO2 data acquired by the HORUS instrument is quantified at 13% and 7%, respectively, with uncertainties calculated at 1σ over a 10-min averaging period. Furthermore, the systematic error of the measurement is calculated to be 12% for OH and 30% for HO2.
Nitrogen oxide (NO) and dioxide (NO2)
NO was measured by detecting the chemiluminescence of NO and O3 using a chemiluminescence detector (ECO PHYSICS, CLD 780TR). This instrument was calibrated by a second NO monitor (ECO PHYSICS, CLD 780TR), which—in turn—was calibrated using the CMK5 Touch dilution system (Umwelttechnik MCZ GmbH) with a NO bottle (Praxair, 1.00 ppmv in N2) and synthetic air (Nippon Gases, hydrocarbon-free). The first detector, which provides data for this study, was found to contain background values that have been subtracted in this study. NO2 was measured by a cavity-attenuated phase-shift nitrogen dioxide monitor (CAPS NO2, Aerodyne Research Inc.). Hourly, the instrument undergoes a 5-min background measurement of pure N2 gas. During the 5-min background measurements, data have been interpolated to give a continuous time series. The NO2 monitor was calibrated using a custom-made cavity-enhanced differential optical absorption spectroscopy instrument56. After the subtraction of an average instrument background concentration, the final NO2 concentration was obtained.
Nitrous acid (HONO) and hydrogen peroxide (H2O2)
Both HONO and H2O2 were detected using bromide chemical-ionization mass spectrometry55, as they exhibit reasonable affinity with the bromide anion. Direct calibrations of these two species were not carried out on-site and the current estimation assumes that they share the same detection sensitivity as H2SO4 (a low-limit estimation). Because these species serve as the precursors of OH and NO radicals, which were reliably traced, the concentrations of HONO and H2O2 are not crucial to the reported results and are therefore omitted from this study.
Two bromide chemical-ionization systems were used to detect HONO and H2O2. The first system, Br-MION2-CIMS, offers sensitive detection of both species at concentrations below about 1010 cm−3, with a detection limit of around 6 × 106 cm−3 (H2O2) and 1.6 × 105 cm−3 (HONO). However, in some experiments, the estimated HONO and H2O2 concentrations exceeded 1010 cm−3. The second system, Br-AIM-CIMS, uses bromide chemical-ionization at low pressure in combination with an active water feedback loop to control the Br-hydration in the ion molecule reactor and avoids saturation. Br-AIM-CIMS was used to measure concentrations from above the detection limit of 4.8 × 107 cm−3 (HONO) and 3.3 × 107 cm−3 (H2O2), based on a calibration factor of 3 × 1012 for HONO and H2O2.
Sulfur dioxide (SO2)
To measure the concentration of SO2, a gas monitor (Thermo Fisher Scientific Model 42i-TLE) was used. However, as the SO2 concentrations in our experiments were usually below 5 × 109 cm−3 (150 pptv), we also used the Br-MION2-CIMS to measure SO2 (ref. 55) in both CLOUD15 and CLOUD16 campaigns. The measurement of SO2 by Br-MION2-CIMS is substantially affected by air–water content, so we conducted online SO2 calibration using the SO2 monitor at both −30 °C and −50 °C. The derived calibration factors are 1.7 × 1013 at −30 °C and 1.5 × 1011 at −50 °C for CLOUD15 and 3.1 × 1011 at −50 °C for CLOUD16. During the experiments, when the primary ions of Br-MION2-CIMS were saturated by either HONO or H2O2, the Br-AIM-CIMS was used to complement the SO2 measurement. With an active water sensitivity control, Br-AIM-CIMS measures SO2 concentrations from above the detection limit of 3 × 107 cm−3 with a constant calibration factor of 20 × 1012 at −30 °C and −50 °C.
Sulfuric acid (H2SO4)
To ensure the quality of the reported data, we monitored H2SO4 concentrations using two chemical-ionization mass spectrometers: the nitrate chemical-ionization mass spectrometer (NO3-CIMS) and the MION2-CIMS operating in bromide chemical-ionization mode (Br-MION2-CIMS55). Furthermore, isotopically labelled H15NO3 was used during the CLOUD16 campaign to distinguish the nitrogen atom originating from the analyte with the reagent ion. The H2SO4 calibration was carried out by two independent calibration systems. The first set-up used the original calibration box designed by Kürten et al.57 along with their in-house calibration scripts. The second set-up is similar to the original version but with different physical dimensions. Also, the recently developed open-source MARFORCE model is used to simulate H2SO4 production in both calibration set-ups55.
In total, we conducted seven calibration experiments at different stages of the CLOUD15 campaign, and each CIMS instrument was calibrated using both calibration set-ups. Two calibrations were performed for the Br-MION2-CIMS, resulting in equivalent H2SO4 calibration factors of 157% and 149%. For the NO3-CIMS, five calibrations were carried out, resulting in equivalent calibration factors of 88%, 100%, 95%, 154% and 164%. Given that the NO3-CIMS provided most of the H2SO4 concentration in this study, we use the calibration carried out immediately after the experiments for this study. This results in a calibration factor of 6.2 × 109 cm−3 for the NO3-CIMS and an equivalent calibration factor of 9.0 × 109 cm−3 for the Br-MION2-CIMS. We use the minimum and maximum of the seven calibrations, ranging from 88% to 164%, as the systematic error of the H2SO4 detection for CLOUD15. It is important to note that we had to change the optimal inlet flow rates of the Br-MION2-CIMS at −30 °C and −50 °C. The varying temperatures and flow rates result in different inlet loss rates, all of which have been accounted for in this dataset.
As well as the normal H2SO4 calibration, we conducted a set of iodine oxoacid nucleation experiments at −10 °C, similar to those presented in ref. 37. The nucleation rates in these experiments are comparable with all of our earlier experiments, further enhancing our confidence in the reported acid concentrations.
In the CLOUD16 campaign, a total of seven calibration experiments were carried out. Two calibration experiments were conducted for the Br-MION2-CIMS, before and after the presented experiments. The results yield equivalent H2SO4 calibration factors of 120% and 118%. For the labelled NO3-CIMS, six calibrations were performed in total, three before the isoprene experiments, resulting in equivalent calibration factors of 100%, 99% and 88%. It is important to note that, during the last few days of the isoprene experiments, the NO3-CIMS suffered from a pump failure that may have caused a shift (by up to 20%) in the calibration factor owing to a slight change in the sample flow. This potentially affects only two experiments in this study. To correct for this, we have assumed a linear correlation between the sample flow and calibration factor. The failing pumps were then replaced and the data from the rest of the experiments were calibrated after the presented experiments, with two calibrations that yielded equivalent calibration factors of 190% and 185%. This yields a calibration factor of 1 × 1010 cm−3 for the labelled NO3-CIMS and an equivalent calibration factor of 1.9 × 1010 cm−3 for the Br-MION2-CIMS. By considering all of the calibration experiments, the systematic error of H2SO4 detection for CLOUD16 is estimated to range from 88% to 120%. Furthermore, using these two instruments, after applying their respective calibration factors, we compared the measured methanesulfonic acid concentrations from the CLOUD chamber at −50 °C. This comparison demonstrated a good agreement, confirming the accuracy of the calibrations.
Iodine species
We measured iodic acid (HIO3) and iodous acid (HIO2) using both the NO3-CIMS and Br-MION2-CIMS and we use the same calibration factor as H2SO4 in the data analysis, similar to our earlier studies37,45,55,58,59. We used Br-MION2-CIMS to measure I2, which is detected at the collision limit, shown by our recent studies55,60.
Isoprene
Isoprene was measured by a proton transfer reaction mass spectrometer using the hydronium chemical-ionization method61 (H3O-PTR-MS). This particular instrument used in this study is an adapted version, which is explained in greater detail previously62.
ISOPOOH and IEPOX detection and separation
Measuring and distinguishing between ISOPOOH and IEPOX can be experimentally challenging owing to their identical molecular formula (C5H10O3). As a result, mass-spectrometric methods often detect them together at the same exact mass in the same peak35. To address this issue, techniques such as tandem mass spectrometry have been used to separate ISOPOOH and IEPOX from each other29.
In this study, these two isomeric compounds were measured both by the Br-MION2-CIMS and the proton transfer reaction mass spectrometer 3 (ref. 63) operating in ammonium chemical-ionization mode (NH4-PTR3-CIMS64). NH4-PTR3-CIMS measured ISOPOOH and IEPOX primarily as clusters with ammonium cation, as the proton affinity (see the ‘Quantum-chemical calculations’ section) of NH3 (204.25 kcal mol−1) is higher than that of 1,2-ISOPOOH (198.31 kcal mol−1), 4,3-ISOPOOH (195.51 kcal mol−1) and cis-β-IEPOX (204.11 kcal mol−1). In this study, we also aim to investigate the capability of the Br-MION2-CIMS in detecting ISOPOOH and IEPOX. We calculate the formation free enthalpies of 1,2-ISOPOOH (−27.5 kcal mol−1), 4,3-ISOPOOH (−26.9 kcal mol−1) and cis-β-IEPOX (−28.0 kcal mol−1) with the bromide anion, respectively. We find that the formation free enthalpies are almost equal to the value of hypoiodous acid (HOI) clustered with the bromide anion (26.9 kcal mol−1), as presented in ref. 55. Because the instrument used in ref. 55 and in this study is the same and the instrument tuning is identical, the fragmentation of these bromide anion cluster ions should be comparable. He et al.55 calibrated both the H2SO4 and the HOI, and the calibration factor of HOI was approximately two times larger than that of H2SO4. Therefore, the calibration factor used for C5H10O3 is two times the calibration factor for H2SO4 in this study.
As neither the NH4-PTR3-CIMS nor the Br-MION2-CIMS are able to distinguish between ISOPOOH and IEPOX, the reported C5H10O3 in this study is the sum of ISOPOOH and IEPOX. Earlier studies have shown that ISOPOOH is effectively lost to metal surfaces by converting it to methyl vinyl ketone (MVK) and methacrolein (MACR)42,65,66, whereas IEPOX is not affected by metal surfaces67. However, as the experiments in this study focus on extremely low temperatures (−30 °C and −50 °C), the chamber wall itself may also serve as a cryo-trap68 for both ISOPOOH and IEPOX. Therefore, it prevents us from using wall-loss-rate perturbation experiments to separate these two species at these temperatures.
To understand the distribution of ISOPOOH and IEPOX in C5H10O3, we carry out a kinetic simulation using the reduced isoprene oxidation mechanism provided in ref. 35. The results are presented in Extended Data Fig. 1b. The simulation is carried out by the F0AM model43. The model requires input parameters such as isoprene, OH, HO2 and O3 concentrations measured by our instruments.
Another important parameter is the wall-loss rate of IP-OOM. We present an experiment in which we manipulate the loss rate of IP-OOM by turning off the light source and increasing the mixing fan spinning rate from 12% to 100% from the equilibrium conditions in Extended Data Fig. 1a. By turning off the light source, the production of IP-OOM stops. Furthermore, by increasing the fan speed, we increase the maximum wall-loss rate from approximately 1.6 × 10−3 s−1 to 8.5 × 10−3 s−1. The decay rates of C5H10O3 and C5H12O6, with lifetimes of 137 s and 112 s, respectively, are similar to the decay rate of HIO3 (129 s) and also, from previous measurements, H2SO4. Because HIO3 has an accommodation coefficient of unity to the chamber wall, we conclude that C5H10O3 and other species with lower volatilities have similar wall-loss rates. In this study, we apply a general wall-loss rate for these species of 1.6 × 10−3 s−1. This wall-loss rate is calculated from the measured H2SO4 wall-loss rate by correcting the diffusivity of C5H12O6 at −30 °C using the method described by our earlier study58.
We further conduct simulations for all of our experiments using the same procedure, and the ratio of IEPOX in C5H10O3 versus OH concentration is presented in Extended Data Fig. 1b. As anticipated, the IEPOX ratio is positively correlated with OH concentrations. For further analysis, a fit with an expression of ratio of is plotted.
Gas-phase oxidized isoprene products
The gas-phase measurement of IP-OOM was achieved by using a combination of NO3-CIMS, Br-MION2-CIMS and NH4-PTR3-CIMS. As defined in this study, only the species with carbon and oxygen numbers equal to or larger than 4 are considered in the IP0-2N, which are primarily produced from OH oxidation of ISOPOOH and IEPOX with and without involving nitrogen oxides. Furthermore, the particle-phase IP0-2N were monitored by a FIGAERO69, which operates with the bromide chemical-ionization method60 in CLOUD15 (Br-FIGAERO-CIMS) and with the iodide chemical-ionization method in CLOUD16 (I-FIGAERO-CIMS). These chemical-ionization methods exhibit varying preferences for analytes. For example, the NO3-CIMS is renowned for detecting highly oxygenated organic molecules70 that contain more than 5 oxygen atoms. The H3O-PTR-MS is the only one that can detect isoprene, whereas both the NH4-PTR3-CIMS and the Br-MION2-CIMS are capable of detecting semi-volatile organic compounds. Consequently, the combination of these CIMS methods enables the measurement of IP-OOM at different oxidation states.
It is worth mentioning our specialized approach to measuring IP-OOM using Br-MION2-CIMS during experiments involving excess HONO and/or H2O2, as described previously. In these experiments, the primary ions (Br− and H2OBr−) were substantially transformed into product ions such as HONOBr−, H2O2Br− and (H2O2)2Br−. Consequently, the measurement of IP-OOM could be compromised if HONO and H2O2 strongly bind with Br−, thereby impeding the ligand exchange with IP-OOM. Therefore, we extensively compared the Br-MION2-CIMS measurements with those of NO3-CIMS and NH4-PTR3-CIMS during experiments with and without such primary ion saturation to ensure reliable measurements. We found that the Br-MION2-CIMS measurement remained uncompromised when we included HONOBr−, H2O2Br− and (H2O2)2Br− as the primary ions. This is probably because of the relatively weak bonding of HONO and H2O2 with Br−, which enables effective charging of IP-OOM by allowing ligand exchange reaction. Quantum-chemical calculations further suggest that the formation free enthalpies of HONOBr− and H2O2Br− are −23.6 and −21.2 kcal mol−1, respectively. These numbers are sufficiently lower than other molecules that are detected at the collision limit by Br-MION2-CIMS55.
To produce IP0-2N, we conducted a set of experiments in which we varied the concentrations of isoprene (ranging from 1.4 × 109 to 4.2 × 1010 cm−3) and OH (ranging from 0.1 to 6.9 × 107 cm−3) to alter the distribution of oxidation products35. To analyse the results of these experiments, we present a generic algorithm to calculate the total sum of gaseous IP0-2N produced, with a focus on those with carbon and oxygen numbers greater than 3:
IP-OOM are independently identified by each of the CIMS instruments. Their responses to the isoprene oxidation in the chamber are observed to distinguish them from any background contaminations originating from either the chamber or the ion sources. If an individual IP0-2N is affected by contaminants of the same molecular formula, its background, derived from the nearest cleaning stage, is subtracted from its concentrations.
If an IP0-2N is detected by only one of the three CIMS, it is added to the total sum directly.
If several CIMS detect species with the same molecular formula, their measured signals are compared in pairs to derive a correlation coefficient. A pair is considered to measure identical molecules if the correlation coefficient is greater than 0.5. However, owing to the transfer of the H2SO4 calibration factors to the measured IP0-2N (NO3-CIMS and Br-MION2-CIMS), the concentration of any molecule with a lower detection efficiency than H2SO4 may be underestimated. The extent of this underestimation depends on the chemical-ionization method used, as the binding enthalpies of the analyte-Br−, analyte-NO3− and analyte-NH4+ may differ. To address this, we add the highest measured concentration of the three CIMS to the IP0-2N and discard the rest, as the highest concentration is probably the closest to the actual concentration.
If the correlation coefficient is less than 0.5, we consider that this pair represents two different molecular structures, that is, two isomers or conformers. In this case, both will be added to the IP0-2N.
However, maintaining all three instruments to be operational throughout all experiments presents a challenge, for instance, the Br-MION2-CIMS operated in the APi-TOF mode to measure charged clusters. Therefore, we excluded data collected during periods in which any one of the instruments was not available.
Charged clusters
Naturally charged clusters were measured with two APi-TOF mass spectrometers (Aerodyne Research Inc.) operating at negative and positive ion mode71. The first instrument was equipped with a MION2 operating in the APi-TOF mode (MION2-APi-TOF)55,72 by deactivating the inlet voltages responsible for directing charged reagent ions into the sample flow. The second device was coupled with an ion-molecule reaction chamber (APi-TOF). Overall, the APi-TOF was less sensitive than the MION2-APi-TOF. The charged clusters reported in Fig. 3 were measured with the MION2-APi-TOF, which was validated by the APi-TOF. Because the MION2 inlet was operated in bromide chemical-ionization mode in some experiments, part of the data reported in Extended Data Fig. 7 was measured by the APi-TOF.
Particle-phase measurements
We measured the chemical composition of small particles using a FIGAERO coupled to a chemical-ionization mass spectrometer69. Particles were sampled from the CLOUD chamber onto a 5-µm-pore polytetrafluoroethylene (PTFE) filter (MilliporeSigma). Filter mass loading is dependent on particle distribution in the chamber, collection flow rate (typically 7–8 l min−1) and total collection time (1–2 h in this study). After particle collection, the filter was automatically moved to in front of the ion molecule reactor. The filter aligned with a sealed port that constantly flushes pure N2. In CLOUD15, the flow rate during chemical measurement was 3 l min−1 and it was increased to 5 l min−1 in CLOUD16 for more efficient heat transfer in a longer port. Pure N2 was heated from room temperature up to 180 °C using programmed thermal desorption controlled by eyeon software v2.1.4.5. As the filter temperature increased, we detected lower-volatility molecules partitioning back into the gas phase. For the particle filter loadings in this study, we observed that all signals decreased back to the baseline by the end of the heating cycle, indicating no notable remaining mass.
Typically, FIGAERO-CIMS is operated using I− chemical-ionization in a reduced-pressure ion molecule reactor (about 120–150 mbar). Pure N2 is flowed around a CH3I permeation tube (Vici) and through a 210Po ionizer (NRD LLC) to produce iodide ions. These polarizable ions effectively form adducts with oxygenated organic compounds, with a small fraction of interactions leading to charge transfer between the ion and neutral compound. In CLOUD15, we used Br− chemical-ionization to distinguish between our chemical-ionization reagent and iodine species inside the CLOUD chamber. The set-up is the same as iodide ionization mode except we exchange a CH2Br2 permeation tube and heat it to 40 °C to increase permeation rates. These chemical-ionization techniques are both sensitive to oxygenated organic compounds, organics with nitrate and sulfate functional groups and inorganic acids60,69. Compounds chemically transformed through deprotonation or thermal decomposition have been excluded, as their parent molecule is unknown.
Particle number size distribution
The Neutral cluster and Air Ion Spectrometer73,74 (NAIS) was used to measure the naturally charged particle number size distribution from 0.8 to 41 nm and the particle number size distribution (naturally charged + neutral) from 2 to 42 nm in both negative and positive polarities. The nano-condensation nucleus counter was used to measure the particle number size distribution between 1 and 3 nm. It consists of a particle size magnifier75 (PSM, Airmodus Oy). The PSM, which is an aerosol pre-conditioner, uses diethylene glycol to grow aerosol particles as small as 1 nm to sizes that can be easily detected by a CPC75. Furthermore, a butanol CPC (TSI 3776) was used to measure the total number concentration of particles with diameters greater than 2.5 nm. A nano-scanning mobility particle sizer (TSI 3938)76 coupled to a butanol CPC (TSI 3776), was used to measure the particle-size distribution within the range 6–65 nm, whereas particles larger than 65 nm were measured using a commercially available long-SMPS (TSI 3082) coupled to a water butanol CPC (TSI 3775).
Yield of IP0N from ISOPOOH
As shown in a previous section, the IP0N in this study is defined as species with C,O ≥ 4. Therefore, ISOPOOH and IEPOX are not included in the IP0N. ISOPOOH and IEPOX are treated as the direct precursors of IP0N, which in turn contribute to isoprene new particle formation. It is worth noting that both ISOPOOH and IEPOX undergo oxidation, producing compounds with C,O ≥ 4. However, the reaction rate of ISOPOOH is approximately ten times larger than IEPOX35. To account for the difference in reaction-rate coefficients, we predict the ratio of IEPOX in C5H10O3 using the data shown in Extended Data Fig. 1b based on the OH concentrations. Assuming that the concentration of IP0N is at equilibrium, the primary mechanism for IP0N loss is wall deposition, which is approximately equal to the production of IP0N from ISOPOOH and IEPOX. Therefore,
in which kOH-ISOPOOH and kOH-IEPOX are the reaction-rate coefficients of ISOPOOH (10−10 cm3 s−1) and IEPOX (10−11 cm3 s−1) with OH (ref. 35), respectively; [IP0N], [OH], [ISOPOOH] and [IEPOX] show concentrations and kwall is the wall-loss rate of C5H12O6; R represents the yield of IP0N from C5H10O3.
We then define the reacted C5H10O3 (cm−3) as:
The yield of IP0N from reacted C5H10O3 is depicted in Extended Data Fig. 2. We find that the yields of IP0N are approximately 46% at −30 °C and 55% at −50 °C. However, it is essential to note that the detection of C5H10O3, IP0N and OH has various uncertainties. We estimate that the derived yield has an uncertainty of at least a factor of two, with the quantification of IP0N being the main source of uncertainty.
One further source of error in determining the yield is the contribution of highly oxygenated molecule production from the first-generation isoprene hydroxy peroxy radical (ISOPOO, C5H9O3) through auto-oxidation or dimer formation. For example, the reaction between two ISOPOO radicals can generate C10H18O4, and intramolecular H-shift followed by HO2 termination of ISOPOO produces C5H10O5. Although these two molecules only contribute to a small fraction of IP0N in this study, other similar channels may contribute to a greater extent to IP0N, thereby reducing the yield of IP0N from C5H10O3. As disentangling first-generation and second-generation highly oxygenated molecules from isoprene oxidation is not the objective of this study, future research is necessary to investigate this direction.
Quantum-chemical calculations
Quantum-chemical methods are used to compute cluster formation free enthalpies and proton affinities. Initially, the Spartan’18 program is used for the conformational sampling with the MMFF method. Subsequently, density function theory (DFT) methods are used to optimize the molecules first at the B3LYP/6-31+G(d) level of theory, followed by optimization and frequency calculations at the ωB97X-D/aug-cc-pVTZ-PP level of theory77,78 on conformers within 2 kcal mol−1 in relative electronic energies. Bromine pseudopotential definitions are obtained from the Environmental Molecular Sciences Laboratory (EMSL) basis set library79,80. The DFT calculations are carried out using the Gaussian 16 program81. To refine the DFT-calculated enthalpies, an extra coupled-cluster single-point energy correction is performed at the DLPNO-CCSD(T)/def2-QZVPP level of theory on the lowest-energy conformers. This coupled-cluster calculation is conducted using the ORCA program version 5.0.3 (ref. 82).
Calculation of the nucleation and growth rates
The nucleation rate, J1.7, is calculated on the basis of PSM measurement of particles at a mobility diameter of 1.7 nm (1.4 nm in physical diameter83), which are generally considered to be larger than their critical cluster sizes and thus stable.
To determine the nucleation rates, the time evolution of the particle concentration is analysed, taking into account various loss processes that also affect the concentration. However, because loss processes in a chamber setting differ from those in the atmosphere, the calculation method must be adjusted for chamber experiments84. Specifically, the nucleation rate (J1.7) is calculated by factoring in losses specific to the CLOUD chamber, such as dilution, wall and coagulation losses. We calculated Jdp as follows:
in which dN/dt is the time derivative of the total particle concentration above a certain particle size (here >1.7 nm for J1.7) and Sdil, Swall and Scoag are the particle losses owing to dilution, wall and coagulation. The details can be found in ref. 84. To calculate the coagulation sink, we used the combined particle-size distribution from three instruments (NAIS, nano-SMPS and long-SMPS).
Furthermore, the nucleation rate at 2.5 nm, J2.5, derived from the butanol CPC and corrected by the same method described above, is calculated. The results are presented in Extended Data Fig. 6 in the same format as in Fig. 2. Because the CPC was not affected by the systematic upgrade in the cooling system between CLOUD15 and CLOUD16, it serves to distinguish subtle changes in our data. For example, the nucleation rates from experiments with NOx (filled squares in Fig. 2) seem to be similar to the experiments without NOx (filled circles in Fig. 2). This is probably a result of systematic errors introduced by changing either the cooling system or the instrument-calibration experiments. On the other hand, Extended Data Fig. 6 shows that experiments with NOx have nucleation rates higher than the experiments without NOx, therefore, isoprene nitrates (IP1-2N) do contribute, despite to a lesser extent compared with IP0N, to particle nucleation.
To calculate particle growth rates, we use the 50% appearance-time method, as outlined in previous studies58,84,85. It is worth noting that the appearance-time method can overestimate growth rates when the impacts of coagulation (coagulation sink, coagulation source and particle coagulation growth) are non-negligible compared with the condensation growth, but the coagulation impact is rather small in the CLOUD experiments. For a deeper understanding of the molecular-level theory behind the method, we refer to the theoretical derivation presented in ref. 58. The particle number size distribution data used to calculate growth rates between 3.2 and 8.0 nm are measured by the NAIS. During previous experiments with α-pinene and sulfuric acid, we have confirmed that the growth rates measured with the NAIS in total mode are similar to those measured with the DMA-train86.
Comparison of experimental and ambient conditions
To compare the CLOUD experimental conditions with ambient conditions in the tropical upper troposphere, we summarize in Extended Data Table 1 the key chemical and physical parameters of the CLOUD experiments and the CAFE-Brazil (CB) flight campaign13. The CLOUD statistics are summarized from all experiments presented in this study, separated into two temperature conditions at −30 °C and −50 °C, respectively. Statistics of the CB flight campaign are derived from a single research flight, RF 19, samples T4 and T9, shown in Fig. 1 of ref. 13. All vapour concentrations are presented in units of molecules per cm−3—the quantities as measured—for both CLOUD and CB. The values from the CB campaign are not corrected to their values at standard temperature and pressure, to allow for a direct comparison of the chemical and aerosol formation kinetics between the CLOUD experiments and the flight measurements. We elaborate below on three aspects of this comparison: (1) isoprene non-nitrates (IP0N) and nitrates (IP1-2N); (2) atmospheric acids; and (3) impact of atmospheric pressure on particle nucleation.
Distribution of IP0N and IP1-2N
In general, the CLOUD experiments were designed to mimic ambient conditions as closely as possible. Key parameters such as temperature, relative humidity (RH), isoprene, O3, NO and HO2/OH ratios are directly comparable between the CLOUD experiments and the CB measurements. The largest differences between CLOUD and CB are higher atmospheric pressure in CLOUD and higher OH/HO2 concentrations, resulting in a higher HO2/NO ratio in CLOUD. The higher OH concentration in the CLOUD chamber is required to reproduce ambient IP-OOM concentrations at a chamber-wall-loss rate of approximately 2 × 10−3 s−1. In the upper troposphere, the condensation sink for low-volatility gaseous species could be several times or even up to one order of magnitude lower than the chamber-wall-loss rate.
Because this study aims to investigate the contribution of IP0N and IP1-2N to particle nucleation and growth, these parameters are critical for CLOUD to reproduce at atmospheric concentrations. A consequence of the relatively higher HO2/NO ratio in CLOUD is that the IP0N to IP1-2N ratio is elevated compared with CB. However, because the higher operating pressure in CLOUD favours the formation of IP1-2N by accelerating the reaction of organic peroxy radical (RO2) with NO as well as enhancing the organic nitrate formation branching ratio35 (see Extended Data Table 1), this effect largely compensates for the higher HO2/NO ratio in CLOUD. Nevertheless, regardless of the chemical details, which will be presented by follow-up studies, CLOUD successfully reproduces isoprene oxidation products IP0N and IP1-2N, in terms of both absolute values and relative ratios (Extended Data Table 1). The wide range of IP0N and IP1-2N concentrations and IP0N/IP1-2N ratios covered by CLOUD experiments enables CLOUD to reasonably simulate particle nucleation and growth dynamics, consistent with CB measurements during dawn hours (T4 period in Extended Data Table 1) and morning (T9 period in Extended Data Table 1). As the daylight hours proceed beyond the period measured by CB, both OH and HO2 concentrations will increase, whereas NOx concentrations decrease, favouring the formation of IP0N over IP1-2N. Thus, the importance of IP0N may be further enhanced after noon. It is also noteworthy that chemical distribution and nucleation dynamics might differ in other seasons and locations from those covered by CB measurements, such as the cases presented in Fig. S11 of ref. 5. Therefore, we believe that the wide range of conditions explored by CLOUD provides valuable data to enable global models to evaluate the impact of isoprene on new particle formation in other upper-tropospheric environments in which NOx concentrations may differ from those measured by CB.
Impact of sulfuric acid and iodine oxoacids
In this study, we observe a large enhancement of IP-OOM nucleation rates from trace amounts of atmospheric acids (specifically, H2SO4 and HIOx). The enhancement starts at acid concentrations of 105 cm−3 and, at an acid concentration of 2 × 106 cm−3, the particle-nucleation rate is approximately 100-fold faster than without added acids.
The CB measurements are unable to comment on this synergistic role of acids for IP-OOM particle nucleation owing to their H2SO4 detection limit of several times 106 cm−3. Nevertheless, acid enhancement of IP-OOM nucleation can be expected to occur in the atmosphere. Aircraft measurements indicate that approximately 10 pptv SO2 is ubiquitous in the global atmosphere between the marine boundary layer and the upper troposphere87,88. Global simulations also suggest that H2SO4 concentrations greater than 105 cm−3 are widespread throughout the troposphere44. The global distribution of HIOx is less well known and global simulations and aircraft measurements are needed to quantify its concentrations in the upper troposphere. However, recent measurements of iodine oxide and particle-phase iodine in the upper troposphere89 suggest that HIOx may also play a role in enhancing IP-OOM particle nucleation.
Impact of atmospheric pressure on particle nucleation
Cluster-forming interactions (such as nucleation) are of the form:
in which A and B are two molecules (or small clusters) forming the cluster C and C* is a vibrationally excited state of the cluster containing the cluster energy EAB as part of its internal vibrational energy. The excited cluster C* will lose energy to the bath gas, M, with a concentration given by pressure through the ideal gas law. Cluster-forming interactions can therefore depend on ambient pressure90. When pressure is relatively ‘low’, reaction R3 will be the rate-limiting step for cluster formation and the overall rate will be of the third order (in A, B and M). However, when pressure is relatively high, reaction R1 will be the rate-limiting step and the rate will be of the second order (in A and B) and be independent of pressure, that is, at the so-called ‘high-pressure limit’. The critical pressure occurs when the rates of R2 and R3 are equal and therefore depends on the lifetime (evaporation rate) of C* through reaction R2, as well as the ambient pressure through reaction R3.
In practice, only systems with very few heavy atoms (four or fewer heavy atoms, in which ‘heavy’ excludes hydrogen) would show any meaningful pressure dependence in the atmosphere91, as quantified below. Because IP-OOM nucleation typically involves more than 15 heavy atoms, measurements in the CLOUD chamber at near 1 bar are directly applicable to the upper troposphere near 0.2 bar, provided the results are interpreted in terms of number concentrations (thus accounting for the dilution effect of reduced pressure) and not mixing ratios.
The (microcanonical) decomposition rates (inverse lifetimes) of the cluster are given by Rice–Ramsperger–Kassel–Marcus theory90. It depends strongly on the number of internal vibrational modes in C*. As a rule, it can be estimated (by Rice, Ramsperger and Kassel theory)90 roughly as the ‘fractional excess free energy’, , in which υ is a typical frequency, 100 THz or so, e is the cluster energy above the ground vibrational state and e0 is the critical energy for decomposition (the cluster energy EAB) and s is an effective number of vibrational modes in the cluster. Approximately, e − e0 will be on the order kT (200 cm−1), whereas for the systems nucleating under atmospheric conditions, e0 will be on the order 800 cm−1 or more. Thus, e will be on the order 1,000 cm−1 and (e − e0)/e will be on the order 0.2. Again, approximately and conservatively, s is 3N − 7, in which N is the number of heavy (non-H) atoms in C*. The ‘7’ excludes external modes as well as the reaction coordinate. A system with five heavy atoms would have a decay coefficient of roughly 2.6 × 108 s−1, whereas the collision frequency at 1 atm is near 1010 s−1. Such a system would (barely) show some pressure dependence. By contrast, for isoprene oxidation products and H2SO4, N is probably 15, so s = 3N − 7 = 38. Given this, the microcanonical decay coefficients for these clusters will be on the order 3 × 10−13 s−1. This is extremely slow. In practice, it means that the energy distribution in cluster C will be entirely thermal, that is, given by a Boltzmann term at the ambient temperature, and the rates of cluster formation (and decomposition or evaporation) will be unaffected by pressure.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-024-08196-0.
Acknowledgements
We thank CERN for supporting CLOUD with important technical and financial resources and for providing a particle beam from the CERN Proton Synchrotron. Funding: this work was supported by Research Council of Finland ACCC Flagship projects 337550, 337549 and 337552; Research Council of Finland professorship (302958); Research Council of Finland’s Centre of Excellence 346371; Research Council of Finland projects 349659, 346371, 345982, 1325656, 311932, 334792, 316114, 325647, 325681, 339489, 347782 and 355966; the Strategic Research Council (SRC) at the Research Council of Finland (352431); European Research Council project ATM-GTP (742206); European Union through Non-CO2 Forcers and their Climate, Weather, Air Quality and Health Impacts (FOCI); European Research Council Consolidator Grant INTEGRATE grant 865799; EU MSCA Doctoral Network CLOUD-DOC 101073026; German Federal Ministry of Education and Research project CLOUD-22 01LK2201A; US National Science Foundation grants AGS-2132089, AGS-2215489, CHE-2336463, AGS-1602086, AGS-1801329, AGS-1951514, AGS-1801280, AGS-2027252 and AGS-2215489; NASA ROSES programme grant 80NSSC19K0949; Swiss National Science Foundation grants 200021_213071 and 216181; Knut and Alice Wallenberg Foundation projects 2021.0169 and 2021.0298; Federal Ministry of Education and Research (BMBF) financed project CLOUD-22 (01LK2201B); Untersuchung von Aerosolnukleation, Aerosolwachstum und Wolkenaktivierung an der CLOUD-Kammer am CERN zur Erforschung des Einflusses auf das Klima; Estonian Research Council project RVTT3 – ‘CERN Science Consortium of Estonia’ and project PRG714; Horizon 2020 research and innovation programme under grant 856612. We thank L. Yang for her contribution to Fig. 5. We thank the Jane and Aatos Erkko Foundation and the ‘Gigacity’ project financed by Wihuri foundation. A.M. and P.M.W. acknowledge financial support by the Vienna Doctoral School in Physics (VDSP).
Extended data figures and tables
Author contributions
X.-C.H., J.S., D.M.R., J.K., J.C. and M.H. planned the experiments. J.S., D.M.R., J.De., F.K., R.B., B.M., W.S., W.Y., L.C.-P., E.S., E.A., D.A., J.A., A.A., L.J.B., H.B., M.B., N.B., M.R.C., A.C., L.D., J.D., M.G., L.G.S., M.H., H.K., T.K., A.K., L.L., M.M., A.M., A.O., M.P., P.R., M.R., B.R., M.K.S., M.Si., M.Su., K.T., R.C.T., A.T., Y.T., J.T., N.S.U., J.W., B.Y., M.Z.-W., J.Z., Z.Z., T.C., I.E.H., R.C.F., H.J., U.R., S.S., R.V., P.M.W., A.H., K.L., N.M.D., J.L., H.H., M.K., D.R.W., J.K., J.C. and X.-C.H. prepared the CLOUD facility or measuring instruments. J.S., D.M.R., J.De., F.K., R.B., B.M., W.S., W.Y., L.C.-P., E.S., E.A., J.A., A.A., L.J.B., H.B., N.B., A.C., R.C.-S., J.D., M.G., L.G.S., M.H., H.K., T.K., M.L., L.L., B.L., M.M., A.M., M.P., P.R., M.R., S.R., B.R., M.Si., M.Su., K.T., R.C.T., A.T., Y.T., J.T., G.U., L.V., J.W., C.X., B.Y., M.Z.-W., J.Z., Z.Z., I.E.H., O.M., R.V., P.M.W., A.H., K.L., H.H., J.K., J.C. and X.-C.H. collected the data. S.I. carried out quantum-chemical calculations. J.S., X.-C.H., D.M.R., J.De., F.K., R.B., B.M., W.S., W.Y., L.C.-P., E.S., H.K., T.K., L.L., P.R., M.Si., M.Su., Y.T., J.W., B.Y., M.Z.-W., J.Z., Z.Z., H.H. and J.K. analysed the data. J.K. and X.-C.H. wrote the manuscript, with contributions from J.S., D.M.R., J.De. and N.M.D. Finally, J.S., D.M.R., J.De., F.K., R.B., B.M., W.S., E.S., R.C.-S., L.D., H.G., B.R., M.Si., Y.T., L.V., C.X., B.Y., U.B., I.E.H., R.C.F., I.R., S.S., R.V., A.H., N.M.D., J.L., M.K., D.R.W., J.K., J.C. and X.-C.H. commented on and edited the manuscript.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Data availability
The full dataset shown in the figures and tables is available open access at 10.5281/zenodo.13736557 (ref.92).
Code availability
Codes conducting the analysis presented here can be obtained on reasonable request to the corresponding authors.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jiali Shen, Douglas M. Russell
Contributor Information
Jasper Kirkby, Email: Jasper.Kirkby@cern.ch.
Joachim Curtius, Email: curtius@iau.uni-frankfurt.de.
Xu-Cheng He, Email: xucheng.he@helsinki.fi.
Extended data
is available for this paper at 10.1038/s41586-024-08196-0.
References
- 1.Krejci, R. Evolution of aerosol properties over the rain forest in Surinam, South America, observed from aircraft during the LBA-CLAIRE 98 experiment. J. Geophys. Res. Atmos.108, 4561 (2003). [Google Scholar]
- 2.Andreae, M. O. et al. Aerosol characteristics and particle production in the upper troposphere over the Amazon Basin. Atmos. Chem. Phys.18, 921–961 (2018). [Google Scholar]
- 3.Williamson, C. J. et al. A large source of cloud condensation nuclei from new particle formation in the tropics. Nature574, 399–403 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Clarke, A. D. et al. Particle production in the remote marine atmosphere: cloud outflow and subsidence during ACE 1. J. Geophys. Res. Atmos.103, 16397–16409 (1998). [Google Scholar]
- 5.Palmer, P. I., Marvin, M. R., Siddans, R., Kerridge, B. J. & Moore, D. P. Nocturnal survival of isoprene linked to formation of upper tropospheric organic aerosol. Science375, 562–566 (2022). [DOI] [PubMed] [Google Scholar]
- 6.Gordon, H. et al. Causes and importance of new particle formation in the present-day and preindustrial atmospheres. J. Geophys. Res. Atmos.122, 8739–8760 (2017). [Google Scholar]
- 7.Kerminen, V.-M. et al. Atmospheric new particle formation and growth: review of field observations. Environ. Res. Lett.13, 103003 (2018). [Google Scholar]
- 8.Wang, X., Gordon, H., Grosvenor, D. P., Andreae, M. O. & Carslaw, K. S. Contribution of regional aerosol nucleation to low-level CCN in an Amazonian deep convective environment: results from a regionally nested global model. Atmos. Chem. Phys.23, 4431–4461 (2023). [Google Scholar]
- 9.Kulmala, M. et al. Deep convective clouds as aerosol production engines: role of insoluble organics. J. Geophys. Res. Atmos.111, 2005JD006963 (2006). [Google Scholar]
- 10.Ekman, A. M. L. et al. Do organics contribute to small particle formation in the Amazonian upper troposphere? Geophys. Res. Lett.35, 2008GL034970 (2008). [Google Scholar]
- 11.Zhu, J. et al. Decrease in radiative forcing by organic aerosol nucleation, climate, and land use change. Nat. Commun.10, 423 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao, B. et al. Global variability in atmospheric new particle formation mechanisms. Nature631, 98–105 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Curtius, J. et al. Isoprene nitrates drive new particle formation in Amazon’s upper troposphere. Nature10.1038/s41586-024-08192-4 (2024). [DOI] [PMC free article] [PubMed]
- 14.Zha, Q. et al. Oxidized organic molecules in the tropical free troposphere over Amazonia. Natl Sci. Rev.11, nwad138 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guenther, A. et al. A global model of natural volatile organic compound emissions. J. Geophys. Res. Atmos.100, 8873–8892 (1995). [Google Scholar]
- 16.Andreae, M. O. et al. The Amazon Tall Tower Observatory (ATTO): overview of pilot measurements on ecosystem ecology, meteorology, trace gases, and aerosols. Atmos. Chem. Phys.15, 10723–10776 (2015). [Google Scholar]
- 17.Bardakov, R., Thornton, J. A., Riipinen, I., Krejci, R. & Ekman, A. M. L. Transport and chemistry of isoprene and its oxidation products in deep convective clouds. Tellus B Chem. Phys. Meteorol.73, 1–21 (2021). [Google Scholar]
- 18.Lelieveld, J. et al. Atmospheric oxidation capacity sustained by a tropical forest. Nature452, 737–740 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Squire, O. J. et al. Influence of isoprene chemical mechanism on modelled changes in tropospheric ozone due to climate and land use over the 21st century. Atmos. Chem. Phys.15, 5123–5143 (2015). [Google Scholar]
- 20.Claeys, M. Formation of secondary organic aerosols through photooxidation of isoprene. Science303, 1173–1176 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Paulot, F. et al. Unexpected epoxide formation in the gas-phase photooxidation of isoprene. Science325, 730–733 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C. & Seinfeld, J. H. Secondary organic aerosol formation from isoprene photooxidation. Environ. Sci. Technol.40, 1869–1877 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Massoli, P. et al. Ambient measurements of highly oxidized gas-phase molecules during the Southern Oxidant and Aerosol Study (SOAS) 2013. ACS Earth Space Chem.2, 653–672 (2018). [Google Scholar]
- 24.Pandis, S. N., Paulson, S. E., Seinfeld, J. H. & Flagan, R. C. Aerosol formation in the photooxidation of isoprene and β-pinene. Atmos. Environ. Part A Gen. Top.25, 997–1008 (1991). [Google Scholar]
- 25.Weber, J. et al. Chemistry-driven changes strongly influence climate forcing from vegetation emissions. Nat. Commun.13, 7202 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanakidou, M. et al. Organic aerosol and global climate modelling: a review. Atmos. Chem. Phys.5, 1053–1123 (2005). [Google Scholar]
- 27.Budisulistiorini, S. H. et al. Real-time continuous characterization of secondary organic aerosol derived from isoprene epoxydiols in downtown Atlanta, Georgia, using the Aerodyne Aerosol Chemical Speciation Monitor. Environ. Sci. Technol.47, 5686–5694 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Lamkaddam, H. et al. Large contribution to secondary organic aerosol from isoprene cloud chemistry. Sci. Adv.7, eabe2952 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.St. Clair, J. M. et al. Kinetics and products of the reaction of the first-generation isoprene hydroxy hydroperoxide (ISOPOOH) with OH. J. Phys. Chem. A120, 1441–1451 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Krechmer, J. E. et al. Formation of low volatility organic compounds and secondary organic aerosol from isoprene hydroxyhydroperoxide low-NO oxidation. Environ. Sci. Technol.49, 10330–10339 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Liu, J. et al. Efficient isoprene secondary organic aerosol formation from a non-IEPOX pathway. Environ. Sci. Technol.50, 9872–9880 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Kiendler-Scharr, A. et al. New particle formation in forests inhibited by isoprene emissions. Nature461, 381–384 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Heinritzi, M. et al. Molecular understanding of the suppression of new-particle formation by isoprene. Atmos. Chem. Phys.20, 11809–11821 (2020). [Google Scholar]
- 34.McFiggans, G. et al. Secondary organic aerosol reduced by mixture of atmospheric vapours. Nature565, 587–593 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Wennberg, P. O. et al. Gas-phase reactions of isoprene and its major oxidation products. Chem. Rev.118, 3337–3390 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Kirkby, J. et al. Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature476, 429–433 (2011). [DOI] [PubMed] [Google Scholar]
- 37.He, X.-C. et al. Role of iodine oxoacids in atmospheric aerosol nucleation. Science371, 589–595 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Crutzen, P. J. et al. High spatial and temporal resolution measurements of primary organics and their oxidation products over the tropical forests of Surinam. Atmos. Environ.34, 1161–1165 (2000). [Google Scholar]
- 39.Bates, K. H. et al. Gas phase production and loss of isoprene epoxydiols. J. Phys. Chem. A118, 1237–1246 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Jacobs, M. I., Darer, A. I. & Elrod, M. J. Rate constants and products of the OH reaction with isoprene-derived epoxides. Environ. Sci. Technol.47, 12868–12876 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Kroll, J. H. & Seinfeld, J. H. Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ.42, 3593–3624 (2008). [Google Scholar]
- 42.Liu, Y. J., Herdlinger-Blatt, I., McKinney, K. A. & Martin, S. T. Production of methyl vinyl ketone and methacrolein via the hydroperoxyl pathway of isoprene oxidation. Atmos. Chem. Phys.13, 5715–5730 (2013). [Google Scholar]
- 43.Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R. & Liao, J. The Framework for 0-D Atmospheric Modeling (F0AM) v3.1. Geosci. Model Dev.9, 3309–3319 (2016). [Google Scholar]
- 44.Dunne, E. M. et al. Global atmospheric particle formation from CERN CLOUD measurements. Science354, 1119–1124 (2016). [DOI] [PubMed] [Google Scholar]
- 45.He, X.-C. et al. Iodine oxoacids enhance nucleation of sulfuric acid particles in the atmosphere. Science382, 1308–1314 (2023). [DOI] [PubMed] [Google Scholar]
- 46.Stolzenburg, D. et al. Enhanced growth rate of atmospheric particles from sulfuric acid. Atmos. Chem. Phys.20, 7359–7372 (2020). [Google Scholar]
- 47.Lucas, D. D. & Prinn, R. G. Tropospheric distributions of sulfuric acid-water vapor aerosol nucleation rates from dimethylsulfide oxidation. Geophys. Res. Lett.30, 2136 (2003). [Google Scholar]
- 48.Zauner-Wieczorek, M. et al. Mass spectrometric measurements of ambient ions and estimation of gaseous sulfuric acid in the free troposphere and lowermost stratosphere during the CAFE-EU/BLUESKY campaign. Atmos. Chem. Phys.22, 11781–11794 (2022). [Google Scholar]
- 49.Stolzenburg, D. et al. Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range. Proc. Natl Acad. Sci.115, 9122–9127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clark, C. H. et al. Temperature effects on secondary organic aerosol (SOA) from the dark ozonolysis and photo-oxidation of isoprene. Environ. Sci. Technol.50, 5564–5571 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Dias, A. et al. Temperature uniformity in the CERN CLOUD chamber. Atmos. Meas. Tech.10, 5075–5088 (2017). [Google Scholar]
- 52.Taira, M. & Kanda, Y. Continuous generation system for low-concentration gaseous nitrous acid. Anal. Chem.62, 630–633 (1990). [Google Scholar]
- 53.Xiao, M. et al. The driving factors of new particle formation and growth in the polluted boundary layer. Atmos. Chem. Phys.21, 14275–14291 (2021). [Google Scholar]
- 54.Martinez, M. et al. Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: airborne measurements. Atmos. Chem. Phys.10, 3759–3773 (2010). [Google Scholar]
- 55.He, X.-C. et al. Characterisation of gaseous iodine species detection using the multi-scheme chemical ionisation inlet 2 with bromide and nitrate chemical ionisation methods. Atmos. Meas. Tech.16, 4461–4487 (2023). [Google Scholar]
- 56.Thalman, R. & Volkamer, R. Inherent calibration of a blue LED-CE-DOAS instrument to measure iodine oxide, glyoxal, methyl glyoxal, nitrogen dioxide, water vapour and aerosol extinction in open cavity mode. Atmos. Meas. Tech.3, 1797–1814 (2010). [Google Scholar]
- 57.Kürten, A., Rondo, L., Ehrhart, S. & Curtius, J. Calibration of a chemical ionization mass spectrometer for the measurement of gaseous sulfuric acid. J. Phys. Chem. A116, 6375–6386 (2012). [DOI] [PubMed] [Google Scholar]
- 58.He, X.-C. et al. Determination of the collision rate coefficient between charged iodic acid clusters and iodic acid using the appearance time method. Aerosol Sci. Technol.55, 231–242 (2021). [Google Scholar]
- 59.Finkenzeller, H. et al. The gas-phase formation mechanism of iodic acid as an atmospheric aerosol source. Nat. Chem.15, 129–135 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang, M. et al. Measurement of iodine species and sulfuric acid using bromide chemical ionization mass spectrometers. Atmos. Meas. Tech.14, 4187–4202 (2021). [Google Scholar]
- 61.Graus, M., Müller, M. & Hansel, A. High resolution PTR-TOF: quantification and formula confirmation of VOC in real time. J. Am. Soc. Mass. Spectrom.21, 1037–1044 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Canaval, E., Hyttinen, N., Schmidbauer, B., Fischer, L. & Hansel, A. NH4+ association and proton transfer reactions with a series of organic molecules. Front. Chem.7, 191 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Breitenlechner, M. et al. PTR3: an instrument for studying the lifecycle of reactive organic carbon in the atmosphere. Anal. Chem.89, 5824–5831 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Hansel, A., Scholz, W., Mentler, B., Fischer, L. & Berndt, T. Detection of RO2 radicals and other products from cyclohexene ozonolysis with NH4+ and acetate chemical ionization mass spectrometry. Atmos. Environ.186, 248–255 (2018). [Google Scholar]
- 65.Bernhammer, A.-K., Breitenlechner, M., Keutsch, F. N. & Hansel, A. Technical note: Conversion of isoprene hydroxy hydroperoxides (ISOPOOHs) on metal environmental simulation chamber walls. Atmos. Chem. Phys.17, 4053–4062 (2017). [Google Scholar]
- 66.Rivera‐Rios, J. C. et al. Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry. Geophys. Res. Lett.41, 8645–8651 (2014). [Google Scholar]
- 67.Mentler, B. Measuring Isoprene Hydroxy Hydroperoxides and Isoprene Epoxydiols Using a Novel Funnel SRI-TOF-MS. Thesis, Univ. Innsbruck (2017).
- 68.Liu, Y. et al. Isoprene photochemistry over the Amazon rainforest. Proc. Natl Acad. Sci.113, 6125–6130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lopez-Hilfiker, F. D. et al. A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO). Atmos. Meas. Tech.7, 983–1001 (2014). [Google Scholar]
- 70.Ehn, M. et al. A large source of low-volatility secondary organic aerosol. Nature506, 476–479 (2014). [DOI] [PubMed] [Google Scholar]
- 71.Junninen, H. et al. A high-resolution mass spectrometer to measure atmospheric ion composition. Atmos. Meas. Tech.3, 1039–1053 (2010). [Google Scholar]
- 72.Rissanen, M. P., Mikkilä, J., Iyer, S. & Hakala, J. Multi-scheme chemical ionization inlet (MION) for fast switching of reagent ion chemistry in atmospheric pressure chemical ionization mass spectrometry (CIMS) applications. Atmos. Meas. Tech.12, 6635–6646 (2019). [Google Scholar]
- 73.Manninen, H. E., Mirme, S., Mirme, A., Petäjä, T. & Kulmala, M. How to reliably detect molecular clusters and nucleation mode particles with Neutral cluster and Air Ion Spectrometer (NAIS). Atmos. Meas. Tech.9, 3577–3605 (2016). [Google Scholar]
- 74.Mirme, S. & Mirme, A. The mathematical principles and design of the NAIS – a spectrometer for the measurement of cluster ion and nanometer aerosol size distributions. Atmos. Meas. Tech.6, 1061–1071 (2013). [Google Scholar]
- 75.Vanhanen, J. et al. Particle size magnifier for nano-CN detection. Aerosol Sci. Technol.45, 533–542 (2011). [Google Scholar]
- 76.Tröstl, J. et al. Fast and precise measurement in the sub-20 nm size range using a Scanning Mobility Particle Sizer. J. Aerosol Sci.87, 75–87 (2015). [Google Scholar]
- 77.Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys.10, 6615 (2008). [DOI] [PubMed] [Google Scholar]
- 78.Kendall, R. A., Dunning, T. H. & Harrison, R. J. Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys.96, 6796–6806 (1992). [Google Scholar]
- 79.Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem.17, 1571–1586 (1996). [Google Scholar]
- 80.Peterson, K. A., Figgen, D., Goll, E., Stoll, H. & Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys.119, 11113–11123 (2003). [Google Scholar]
- 81.Frisch, M. J. et al. Gaussian 16, Revision C.01 (Gaussian, Inc., 2016).
- 82.Neese, F. The ORCA program system: the ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci.2, 73–78 (2012). [Google Scholar]
- 83.Ku, B. K. & de la Mora, J. F. Relation between electrical mobility, mass, and size for nanodrops 1–6.5 nm in diameter in air. Aerosol Sci. Technol.43, 241–249 (2009). [Google Scholar]
- 84.Dada, L. et al. Formation and growth of sub-3-nm aerosol particles in experimental chambers. Nat. Protoc.15, 1013–1040 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Lehtipalo, K. et al. Methods for determining particle size distribution and growth rates between 1 and 3 nm using the Particle Size Magnifier. Boreal Environ. Res.19, 215–236 (2014). [Google Scholar]
- 86.Stolzenburg, D., Steiner, G. & Winkler, P. M. A DMA-train for precision measurement of sub-10 nm aerosol dynamics. Atmos. Meas. Tech.10, 1639–1651 (2017). [Google Scholar]
- 87.Thompson, C. R. et al. The NASA Atmospheric Tomography (ATom) mission: imaging the chemistry of the global atmosphere. Bull. Am. Meteorol. Soc.103, E761–E790 (2022). [Google Scholar]
- 88.Ranjithkumar, A. et al. Constraints on global aerosol number concentration, SO2 and condensation sink in UKESM1 using ATom measurements. Atmos. Chem. Phys.21, 4979–5014 (2021). [Google Scholar]
- 89.Koenig, T. K. et al. Quantitative detection of iodine in the stratosphere. Proc. Natl Acad. Sci.117, 1860–1866 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marcus, R. A. Unimolecular dissociations and free radical recombination reactions. J. Chem. Phys.20, 359–364 (1952). [Google Scholar]
- 91.Atkinson, R. et al. Evaluated kinetic and photochemical data for atmospheric chemistry: supplement III. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry. J. Phys. Chem. Ref. Data18, 881–1097 (1989). [Google Scholar]
- 92.Shen, J. et al. New particle formation from isoprene under upper- tropospheric conditions: data sources. Zenodo10.5281/zenodo.13736557 (2024). [DOI] [PMC free article] [PubMed]
- 93.Pfeifer, J. et al. Measurement of ammonia, amines and iodine compounds using protonated water cluster chemical ionization mass spectrometry. Atmos. Meas. Tech.13, 2501–2522 (2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The full dataset shown in the figures and tables is available open access at 10.5281/zenodo.13736557 (ref.92).
Codes conducting the analysis presented here can be obtained on reasonable request to the corresponding authors.