

Abstract
Haematopoietic stem cell (HSC) gene therapy (GT) may provide lifelong reconstitution of the haematopoietic system with gene-corrected cells1. However, the effects of underlying genetic diseases, replication stress and ageing on haematopoietic reconstitution and lineage specification remain unclear. In this study, we analysed haematopoietic reconstitution in 53 patients treated with lentiviral-HSC-GT for diverse conditions such as metachromatic leukodystrophy2,3 (MLD), Wiskott–Aldrich syndrome4,5 (WAS) and β-thalassaemia6 (β-Thal) over a follow-up period of up to 8 years, using vector integration sites as markers of clonal identity. We found that long-term haematopoietic reconstitution was supported by 770 to 35,000 active HSCs. Whereas 50% of transplanted clones demonstrated multi-lineage potential across all conditions, the remaining clones showed a disease-specific preferential lineage output and long-term commitment: myeloid for MLD, lymphoid for WAS and erythroid for β-Thal, particularly in adult patients. Our results indicate that HSC clonogenic activity, lineage output, long-term lineage commitment and rates of somatic mutations are influenced by the underlying disease, patient age at the time of therapy, the extent of genetic defect correction and the haematopoietic stress imposed by the inherited disease. This suggests that HSCs adapt to the pathological condition during haematopoietic reconstitution.
Subject terms: Haematopoietic stem cells, Stem-cell research, Gene therapy, Genomics
Haematopoietic stem cell (HSC) clonogenic activity, lineage output, lineage commitment and somatic mutation rates are influenced by the underlying disease, patient age, extent of genetic defect correction and hematopoietic stress imposed by the inherited disease, suggesting HSC adaptation.
Main
In haematopoietic stem cell (HSC) gene therapy (GT) (HSC-GT) integrating viral vectors are used to deliver therapeutic genetic material into haematopoietic stem and progenitor cells (HSPCs) from patients affected by inherited disorders and restore the defective function. Autologous gene-corrected HSPCs engraft, self-renew and give rise to all cells of the blood and the immune system providing long-term therapeutic benefits to patients. HSC-GT is an effective treatment option for several monogenic diseases including blood disorders, such as primary immune deficiencies and hemoglobinopathies, in which the administered HSPCs restore the functionality of the defective lineages, and non-haematopoietic diseases such as lysosomal storage disorders, in which the transduced HSPC progeny such as monocytes and macrophages perform the clearance of the stored material and deliver the therapeutic enzyme to cross-correct other haematopoietic and non-haematopoietic cells in tissues1,7,8. To assess the safety and effectiveness of the treatment and to shed light on the biology of haematopoiesis in various disease conditions, it is important to understand how haematopoietic reconstitution occurs in terms of lineage output and commitment, kinetics, clonal composition and succession.
In patients receiving GT, the process of haematopoietic reconstitution has been analysed by tracking viral vector integration sites (ISs) retrieved from whole peripheral blood (PB) and bone marrow (BM) and purified cell lineages gathered at sequential time points after reinfusion2,4,6,9–12. Indeed, because vector integrations occur semi-randomly in the host cells, each IS represents a distinct marker of clonal identity that is stably maintained over time and inherited by all its progeny. High-throughput sequencing and mapping of the junctions between the integrated vector and the host cellular genome13,14 allowed retrieval and identification of hundreds of thousands of ISs that are used to monitor the clonal composition during the haematopoietic reconstitution, the multi-lineage potential of HSPC subsets and the hierarchical relationships among haematopoietic lineages.
Previous clonal tracking studies on patients receiving HSC-GT affected by primary immune deficiencies, including adenosine-deaminase severe combined immune-deficiency (SCID), X-linked SCID type 1 (XSCID) and Wiskott–Aldrich syndrome (WAS), in which B and T cells are profoundly impaired, have clearly shown a selective advantage of genetically modified lymphoid cells after transplantation indicating that the disease background influences the proportion and the type of lineage-committed cells over long periods of time4,10,11,15–19. On the other hand, in other diseases such as chronic granulomatous disease, a progressive phagocytic disorder or leukodystrophies and lysosomal storage disorders2,3,9,20–22 no selective advantage in growth or survival was observed in any cell lineage. Comparative analyses of HSPC lineage commitment were performed in a few studies with a limited number of patients, mainly in primary immune deficiencies such as WAS and hemoglobinopathies (β-thalassaemia (β-Thal) and sickle cell disease)15,16.
It is also relevant to understand how the replicative stress imposed by haematopoietic reconstitution would affect HSPCs’ multi-lineage potential, dynamics of lineage commitment and, ultimately, long-term safety, and how this may vary in different disease conditions. Indeed, patients with sickle cell disease have an increased risk of developing haematologic malignancies, and an increased frequency of haematopoietic clones with driver mutations associated with myeloid cancer or clonal haematopoiesis, as observed in untreated and treated patients receiving GT23–26.
A comprehensive comparison of the haematopoietic reconstitution in different disease backgrounds with large cohorts of patients, including both haematopoietic and non-haematopoietic disorders and with or without selective advantage and different amounts of haematopoietic stress, is still missing.
Here we present a comparative analysis of the haematopoietic reconstitution at the clonal level in 53 patients enrolled in three distinct lentiviral vector-based HSC-GT for metachromatic leukodystrophy (MLD, a neurodegenerative lysosomal storage disorder), WAS and β-Thal carried out by our institution2–6, and, to further extend and validate some findings, of ten patients with XSCID treated with lentiviral vector-based HSC-GT in another institution and previously published19.
Our results uncover that the long-term output, lineage commitment of HSPCs, and accumulation of somatic mutations, are strongly modulated by the patients’ genetic backgrounds, conceivably to better compensate for the demands posed by the specific clinical condition.
Clonal dynamics of vector-marked cells
We studied the clonal reconstitution and multi-lineage potential over time (up to 8 years of follow-up) in 53 patients receiving HSC-GT affected by three different diseases: 29 with MLD2,3,22, 15 with WAS4,5,15,17 and 9 with β-Thal6 (Supplementary Table 1). The lentiviral vector constructs used in these clinical programmes have the same backbone but different promoters and transgenes: MLD vector contains the ubiquitous human phosphoglycerate kinase promoter driving the expression of the human arylsulfatase A complementary DNA (cDNA)2; WAS vector contains a portion of the human WAS gene promoter driving the expression of WASP cDNA4 and β-Thal vector contains in addition to the human β-globin promoter two hypersensitive sites from the β-globin locus control region driving the expression of β-globin gene. In each of the three clinical programmes, different conditioning regimens have been adopted (Methods). Most of the patients analysed in this study were paediatric (age range from 6 months to 15 years) at the time of treatment, except for three adult patients with β-Thal(age range 31–35 years).
Clonal tracking was performed on total BM and PB cells, as well as on purified myeloid cells (CD13+, CD14+, CD15+), B cells (CD19+), T cells (CD3+, CD4+, CD8+), erythroid cells and precursor (GpA+ for MLD and WAS and GpA+, CD36+ for β-Thal) and CD34+ progenitor cells (Fig. 1a, Methods and Supplementary Table 2), with purity levels in line with the previous reports. Samples were collected at 1, 3, 6, 9 and 12 months posttreatment during the first year, and once a year thereafter. By this strategy, we were able to analyse more than 6,700 samples overall, processed by PCR methods13,27 and sequenced with Illumina paired-end reads (Methods). After all the quality controls, we obtained 1,516,818 unique ISs for MLD, 1,647,351 ISs for WAS and 1,180,274 ISs for β-Thal, totalling more than 4.3 million univocally mapped ISs. ISAnalytics28 was used also to streamline clonal analyses such as clonal abundance, clonal population diversity, population size estimate of active stem cells, common insertion site identification and the analyses of shared ISs across lineages and time points (Methods).
Fig. 1. HSPC clonal complexity, size and source time point of long-lasting clones.

a, Clonal tracking through ISs begins with a patient’s autologous transplantation of vector-marked cells. Periodic sampling and DNA processing isolate markers for distinct lineages (myeloid, erythroid, B and T cells). ISs are retrieved by means of custom PCRs and deep sequencing, allowing clonal population diversity, abundance, lineage tracking and vector integration frequency to be assessed for treatment safety and efficacy. b, The cumulative number of ISs retrieved for each patient by disease (MLD, WAS and β-Thal) increases over time after gene therapy, with a log-based regression curve showing the progression (confidence interval (CI) 0.75). c, Clonal population diversity index (Shannon index, y axis) by clinical trial, tissue (BM and PB) and lineage (different colours), over time (months, x axis); spline CI 0.75. d, The analytical process of HSPC estimate, starting from ISs derived by short-lived myeloid cells in PB, filtering ISs by low sequencing reads (removing ISs with n < 3), and estimating HSPCs over time by triplets of consecutive time points using the Chao1 model. e, Results of estimated HSPCs (y axis) over time (x axis) for each clinical trial, normalized by in vivo VCN for samples with VCN > 1. f, Percentage of the estimated active HSPCs on the total number of infused CD34+ BM cells, stratified by clinical trial; statistical results obtained with Fisher’s exact test. g, Long-term clones are identified by tracking ISs back to their first observed time points, categorized into early (1–6 months), mid (12–18 months) and steady (24–30 months) phases. Statistical comparisons were made using the Kruskal–Wallis test corrected by FDR. h, Percentage of long-lasting clones, for each lineage (in colours) and clinical study, backtracked by the first observed time point grouped by the haematopoietic reconstitution phase (early, mid and steady). RBC, red blood cell; MK, megakaryocyte; NK, natural killer cell; DCs, dendritic cells; Mφ, macrophage. Graphics in a were created using BioRender (https://biorender.com).
The analysis of ISs, which serve as a marker for clonal identity, showed that more than 75% of ISs were captured within the first 12 months posttreatment in most patients, with fewer new ISs appearing over time (Fig. 1b). This indicates that most active clones contributing to haematopoiesis were identified early and remained stable over nearly a decade. ISs tended to integrate into gene-dense regions (Extended Data Fig. 1a,b), forming hotspots and gene ontology (GO) analysis confirmed a preference for genes involved in chromatin and histone modification, as already observed in previous studies2,4 (Extended Data Fig. 1c), with comparable targeting specific gene classes among the three clinical trials (correlation greater than 0.96, Extended Data Fig. 1d), and well-known hotspots of lentiviral vector integration (targeting genes such as KDM2A, PACS1, HLA, TNRC6C, SETD2; Extended Data Fig. 1e) without significant differences in gene targeting frequencies (Supplementary Table 3) across studies. Clonal abundance analysis (Methods) did not find any persisting dominant clone (Extended Data Fig. 1f). Moreover, specific ISs were identified at several time points and across different lineages, highlighting the persistence of long-term repopulating clones and multi-lineage marking.
Extended Data Fig. 1. Genome wide distribution of IS.

(A) Circular representation of the genomic distribution of the ISs for the three clinical trials (MLD in the blue track, WAS in the green track, β-Thal in the red track). (B) UpSet plot displaying IS distribution around targeted genes for a sample patient in each trial (Pt16 for MLD, Pt52 for WAS, Pt45 for β-Thal). The rows list gene features, with combinations represented by connecting lines and histograms showing element counts. (C) Gene ontology analysis for the three trials, showing the results for biological processes (BP), first 15 classes. (D) GO similarity plot comparing ontological enrichments among the trials. Pairwise similarity scores (“MLD-WAS,” “MLD-β-Thal,” “WAS-β-Thal”) are presented on the y-axis, covering all GO categories cellular components (CC), biological processes (BP), and molecular functions (MF). (E) CIS results displayed as volcano plots for each trial. Each gene is represented as a dot, with integration frequency on the x-axis (log2 scale) and the associated p-value on the y-axis (−log10). The orange dashed line indicates the alpha value of 0.05. Genes are colored gray if never significant or violet if significant in at least one patient. (F) Stacked bar plot tracking top clones (≥ 1% in MLD/WAS, ≥0.1% in β-Thal) over time for the first patient in each trial (MLD Pt42, WAS Pt52, β-Thal Pt45). Each colored bar represents a clone, with height proportional to abundance. Colored ribbons connect neighboring time points for recaptured clones. BM-derived clones are on the left, PB-derived IS on the right, with the number of observed IS reported above each bar.
The diversity of clones in each patient, measured by the Shannon diversity index (H) and corrected by a Bayesian multivariate linear regression algorithm to model remove biases induced by different technical confounding factors as well as their interactions (Methods), showed that all patients had a polyclonal repertoire (Fig. 1c), with some lineages showing higher complexity depending on the underlying disease. Specifically, to compare the diversity index of the lineages against all the other lineages within the same clinical programme and the same lineage across the different clinical programmes, we first selected the interval of 24–60 months after therapy (at stability) and normalized the H index of each lineage by Z-score (Extended Data Fig. 2a), and then compared by the Kruskal–Wallis non-parametric test (Extended Data Fig. 2b,c). Patients with MLD had more complex myeloid lineages, patients with WAS had more complex B and T lymphocyte lineages and patients with β-Thal had more complex erythroid lineages. These differences suggest that the underlying disease biology significantly affects haematopoietic reconstitution.
Extended Data Fig. 2. Clonal complexity and diversity.

(A) Population diversity index (Shannon entropy, H) over time, normalized with Z-score by patient and time point, divided by disease condition and tissue. Colors refer to lineages (CD34+ cells, myeloid, erythroid, B and T); spline with CI 0.75. (B) Statistical comparisons of population diversity index among the different diseases grouping values by lineages and tissues (Kruskal–Wallis test). The bars represent the median, the whiskers extend to 1.5 times the IQR, and the p-value threshold is set at 0.05. (C) Similar to (B), statistical comparisons of the population diversity index among the different lineages, grouping values by disease and tissues.
HSPC population size
We compared the estimated number of active HSPCs and the contribution of CD34+ cells towards myeloid, B, T and erythroid precursor cells over time in the three disease conditions. Using the Chao1 model29 and short-living cells as surrogates of stemness (Methods), active HSPC populations were estimated during early haematopoietic reconstitution and found to decrease significantly after 24 months posttreatment (Fig. 1d). Active HSPCs were on average in patients with MLD 8,984 (range 3,583–19,498), in WAS 55,495 (range 5,378–363,649) and in β-Thal 54,297 (range 6,470–196,773) (Fig. 1e). After 24 months from treatment, the number of active HSPCs decreased significantly to an average of 6,497 (range 1,865–16,302, roughly 1.3-fold) in MLD, 6,151 (range 1,882–23,774, roughly ninefold) in WAS and 10,446 (range 3,475–18,480, roughly 5.2-fold) in β-Thal (Extended Data Fig. 3a). The decrease was more rapid in patients with β-Thal due to faster recovery from mobilized CD34+ cells (cut off to 12 and 24 months, Extended Data Fig. 3a). These results are compatible with published models, showing early phases of haematopoietic reconstitution sustained by many progenitors and short-term HSCs that were exhausted by 24 months and progressively replaced by a smaller, yet substantial number of long-term HSCs that stably sustained steady-state haematopoiesis2,4,15,16. The fraction of engrafted transduced HSCs actively contributing to the haematopoiesis at steady state, estimated as the recaptured population size divided by the total CD34+ cell number infused, was similar across the three trials, regardless as to whether HSPCs were collected from the BM or mobilized PB (MPB): MLD 0.007% (range 0.001–0.027%), WAS 0.003% (range 0.001–0.005%) and β-Thal 0.003% (range 0.001–0.004%) (Fig. 1f and Supplementary Table 4). These percentages corresponded to 15–270 active long-term HSCs per million infused CD34+ cells in MLD, 10–48 in WAS and 9–44 in β-Thal, and from two- to tenfold more short-term engrafting progenitors.
Extended Data Fig. 3. HSPC population size.

(A) Estimated number of HSPCs (y-axis) by disease comparing early time points (<24 months post GT) and late time points (>24 months post GT) using Wilcoxon matched pairs signed rank test. β-Thal patients showed not statistically significant decrease when comparing <24 Vs. >24 months, while showed a significant decrease when comparing <12 Vs. >24 months. (B) Statistical analysis of the composition of long-lasting clones analyzed by the time of origin (first observed time point). For each lineage (in columns) and clinical trial (in rows), we plot the percentages of shared ISs (in dots, distributions with violin plots) within each time point grouped by the three phases (“early” from 1 to 6 months after GT, “mid” from 12 to 18 months, and “steady” from 24 to 30 months). P-values obtained by Kruskal Wallis test. (C) Percentages of the composition of long-lasting clones analyzed by the time of origin expressed as fold change (y-axis) of the “mid” and “steady” phase on the “early” phase (x-axis) for the three diseases (MLD in blue, WAS in green, β-Thal in red), separated by lineage.
We then investigated when long-term clones originated during the haematopoietic reconstitution. To this aim, we selected the ISs retrieved from 36 months after infusion until the latest time point (long-term ISs, long-term clones) and tracked each long-term clone backward to the time point when it appeared for the first time, and we determined the percentage of long-term clones collected within the following three time intervals: from 1 to 6 months (‘early’ phase of haematopoietic reconstitution), from 9 to 18 months (‘mid’) and from 24 to 30 months (‘steady’) (Fig. 1g). We found that the long-term clones were poorly retrieved (between 0.5 and 4.5%) during the early time points but increased during the mid and steady phases between 1.3- and 18-fold for the early phase for all lineages and disease conditions (Fig. 1h and Extended Data Fig. 3b,c). We calculated the percentage of ISs detected at long-term (more than 24 months after therapy) for the three patients that showed the greatest number of ISs. In the patient with WAS with roughly 450,000 ISs (Pt17) the proportion of ISs detected long term was 4.22%, whereas in the two patients with β-Thal with roughly 432,000 and 220,000 ISs (respectively, Pt36 and Pt41), the proportions were roughly 3.5 and 1.74%, respectively. These data further confirmed that the early phase of the haematopoietic reconstitution is sustained by short-lived progenitors and probably, at least in part, by HSPCs clones that have undergone exhaustion.
Given the observed inter-patient variability in clonality and HSPC size among patients and clinical programmes, we investigated whether the number of estimated active HSPCs and the clonal complexity (diversity index) calculated in patients at steady state would correlate with treatment variables such as vector copy number (VCN) (average VCN in PB-derived myeloid cells), transduction efficiency in vitro in the clonogenic outgrowth of the infused cell product, age at treatment and the dose of infused CD34+ cells per kg (Extended Data Fig. 4a). Our results showed that the patient’s age at treatment had a negative correlation with transduction efficiency (Pearson correlation −0.51 with P = 0.01, meaning that younger patients had a higher transduction efficiency) and a positive correlation with active HSPC size (Pearson correlation 0.53, P = 0.006). Active HSPC size also negatively correlated with transduction efficiency (Pearson correlation −0.56, P = 0.003), but positively correlated with cell dose (Pearson correlation 0.45, P = 0.046). The Shannon population diversity index correlated with VCN in vivo (Pearson correlation 0.53, P = 0.006) and with cell dose (Pearson correlation 0.46, P = 0.035). Multivariate analyses with principal component analysis (PCA) confirmed pair-wise correlations and combined the variables by reducing the results in two main dimensions (with a percentage of explained data greater than 74%). Moreover, clustering results showed that patients treated with MPB CD34+ cells were well separated from all the remaining patients transplanted with BM CD34+ cells or a mixed BM-MPB dose (Extended Data Fig. 4b).
Extended Data Fig. 4. Integrated and unsupervised analysis of patients’ variables, combining clinical and molecular data.

(A) Pair-wise correlation results among all variables (represented in the diagonal). The distribution of each variable is shown within each box in the diagonal. On the lower triangular below the diagonal, the bivariate scatter plots with a fitted line. On the upper triangular side of the matrix above the diagonal, the value of the correlation plus the significance level (p-values, adjusted Benjamini). (B) PCA analysis with biplot representation showing on the axes the first 2 dimensions explaining >74% of the data; dots are all patients labeled with patient ID; blue arrows are the eigenvectors of the PCA; in colors the three GT types (BM = bone marrow, MPB = mobilized peripheral blood) with centroids of clustering as elliptic shapes.
Lineage output of CD34+ cells
We then analysed the HSPC contribution to each lineage over time in the different trials. To this end, we calculated the percentage of ISs retrieved from CD34+ cells shared with lineage-specific cells (sharing ratio, Methods) from PB and BM (Fig. 2a). We selected ISs represented by three or more sequencing reads and excluded those with fewer than 10% of the sequence reads to eliminate confounding effects. To address dataset size differences and prevent biases, we used the Good–Turing model and Bayesian multivariate linear regression (Methods) (Fig. 2b).
Fig. 2. Lineage output of CD34+ cells.

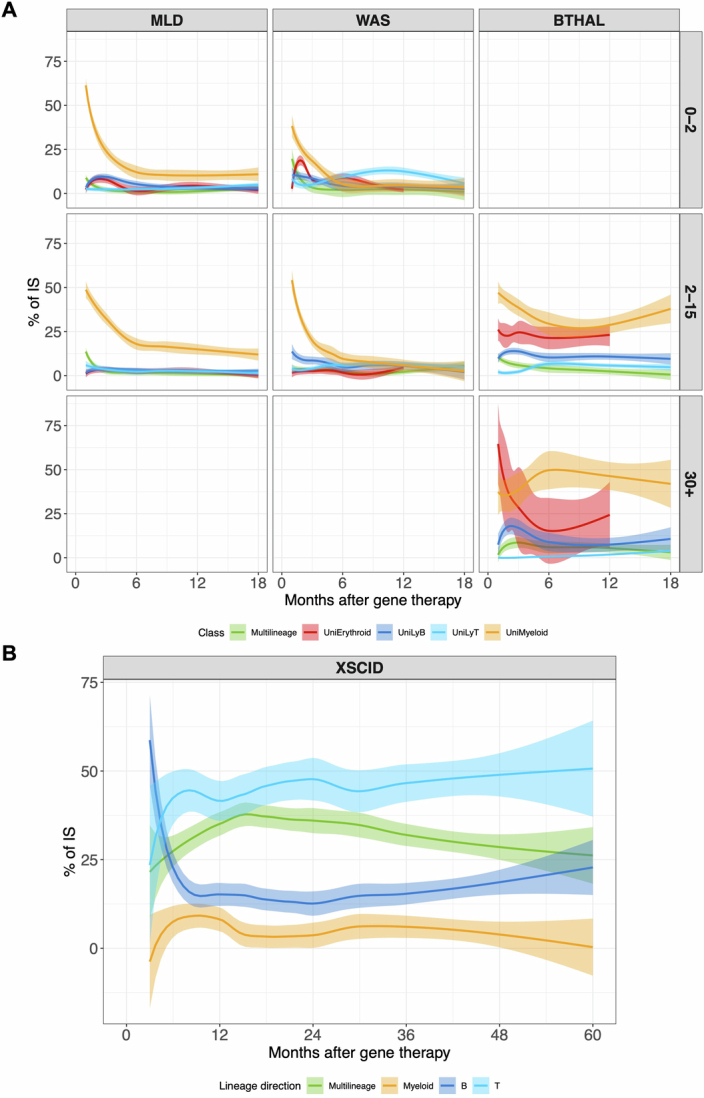
a, Strategy for the analysis of CD34+ cells and lineage output: for each time point, we computed the proportion of the shared ISs retrieved for each lineage (among myeloid, erythroid, B and T cells) and CD34+ ISs (sharing ratio). b, Dynamics of the sharing ratio (y axis) as lineage output (with different colours) of CD34+ cells over time (x axis) for the three clinical trials. Spline curves with CI 0.75. c, Box plots of sharing ratio (bar indicates median, whisker 25–75 percentiles), normalized by Z-score (y axis) isolated in all patients from 24 months and averaged, in BM and PB tissues (circle or triangular dot shapes), grouping cell markers (colours) by cell lineages. Statistical tests are performed between pairs of clinical studies (Kruskal–Wallis test). The bars represent the median, the whiskers extend to 1.5 times the interquartile range (IQR) and the P value threshold is set at 0.05. d, Similar to b, CD34+ BM lineage output over time stratified by age groups (0–2, 2–15 and older than 30 years old).
In patients with MLD, the ISs shared between CD34+ cells and myeloid lineages increased from 10 to 12% initially to 20% at 24 months, stabilizing thereafter (Fig. 2b). B, T and erythroid lineage sharing was initially minimal but increased to around 10% for B and T cells and 5% for erythroid cells after 24 months. In patients with WAS, T cells had the highest contribution from CD34+ cells, reaching 20% within 30 months, whereas B cells reached roughly 12.5% at 18 months and slowly increased to roughly 20% at 60 months. Myeloid and erythroid lineages were slower, reaching roughly 14 and 8%, respectively. In patients with β-Thal, sharing between CD34+ cells and myeloid, B and T lineages plateaued at 12 months at around 4, 3 and 2%, respectively, but the contribution to erythroid lineage increased to 40% at the latest time points.
We compared the contribution of CD34+ cells to each lineage in different disease conditions using Z-score transformation (Extended Data Fig. 5a) and the non-parametric Kruskal–Wallis test (Methods). Patients with MLD showed higher sharing with myeloid cells and lower with lymphoid cells compared to patients with WAS, who had higher T cell lineage sharing (in line with the selective advantage of gene-corrected T cell precursors). In patients with β-Thal, the erythroid lineage showed the highest sharing (Fig. 2c).
Extended Data Fig. 5. CD34+ cell output towards mature lineages.

(A) Dynamics of the lineage output over time (x-axis) calculated with the sharing ratio (y-axis) between CD34+ cells and each lineage (different colors) expressed as percentage normalized with Z-score by patient and time point, reported by clinical study. Spline curves with CI 0.75. (B) Similar to (A), dynamics of the lineage output (y-axis) over time (x-axis) stratified by patient age (0–2 years, 2–15 years, >30 years). (C) Lineage output for XSCID patients (N = 10) under LV-HSPC gene therapy treatment (De Ravin S., et al. Nature Communications 2022). (D) Box-plots of sharing ratio (bar indicates median, whisker 25–75 percentiles), normalized by Z-score (y-axis) isolated in all patients from 24 months and averaged, in BM and PB tissues (circle or triangular dot shapes), grouping cell markers (colors) by cell lineages, and stratified by patient’s age within each clinical trial. Statistical tests are performed comparing the different lineages (Kruskal Wallis test). The bars represent the median, the whiskers extend to 1.5 times the IQR, and the p-value threshold is set at 0.05. (E) Similar to (D), box-plot representation of normalized sharing ratio (y-axis) stratified by lineage within each clinical trial, comparing the different groups of treatment age (Kruskal Wallis test).
We stratified the patients by treatment age into three groups: from 0 to 2 years, represented by 18 patients with MLD and five patients with WAS, from more than 2 to 15 years, represented by 11 MLD, ten WAS and six patients with β-Thal, and adults with more than 30 years of age represented by only three patients with β-Thal, and performed the analyses of the output of CD34+ cells into mature lineages (Fig. 2d). In the adult patients with β-Thal (above 30 years), the lineage sharing of CD34+ cells with the erythroid lineage were roughly 5% until 24 months and then progressively increased over time and reached up to roughly 40% at 60 months after therapy. The erythroid output in the six paediatric (4–13 years) patients with β-Thal was 5% until 12 months, increasing progressively to roughly 15% at 36 months. The output towards B cell and especially the T cell lineages was delayed in adult and paediatric patients with β-Thal compared to patients with WAS. The contribution towards B and T cell linages was superior in the paediatric patients with β-Thal (15%) compared to adults (5% for B cells and roughly 2% for T cells), as expected by the higher number of BM B-lymphoid committed progenitors in children versus adults30 and the specific myeloablative conditioning that results in a delayed T cell reconstitution similar to that observed in patients with MLD.
The contribution of CD34+ cells to mature lineages across different diseases and age groups was analysed using Z-score transformation and the Kruskal–Wallis test (Methods and Extended Data Fig. 5b). In patients with MLD aged 0–2 years, CD34+ cells showed significantly higher sharing with myeloid cells, with T cells having the lowest sharing. Similar patterns were observed in the 2–15 years age group, except T cell sharing was lower than B cells. Patients with WAS aged 0–2 and 2–15 years showed predominant T cell sharing, with B cell sharing being lower, reflecting a less pronounced selective advantage5,31. Both paediatric and adult patients with β-Thal showed higher sharing with erythroid cells than myeloid, T and B cells (Fig. 2e). The only significant age-related difference was in patients with β-Thal over the age of 30, who had higher erythroid lineage sharing compared to younger patients (Fig. 2f).
To confirm that the increased lineage output for the lymphoid B and T cell lineages in patients with WAS was not specific to the disease but a general characteristic of the lymphoid impairment observed also in other immunodeficiencies, we analysed previously published19 IS datasets of ten patients receiving XSCID treated with lentiviral vector-HSC-GT. The output of CD34+ cells towards the T and B cell lineages was initially low (less than 2%) but increased progressively up to 25% at 60 months, the last time point of the analysis (Extended Data Fig. 5c). Therefore, our data show that the preferential output towards T and B cells is common between these two lymphoid immunodeficiencies. B cell output was similar to or faster than T cell output in patients receiving XSCID, contrary to expectations.
Longitudinal analysis of HSPC commitment
We then further investigated whether the disease condition could also affect the multi-lineage potential of HSPC by promoting long-term commitment to a specific lineage. We defined clones with multi-lineage potential when the ISs, represented by 3 or more sequencing reads, were shared by at least two mature lineages (among myeloid, B, T or erythroid cells) at any time point. We defined lineage-committed clones when their ISs were retrieved consistently in a single lineage at least during two distinct time points, whereas clones observed in one time point only were defined as singletons (Fig. 3a). We addressed biases arising from confounding variables with a Bayesian multivariate linear regression model and biases from the comparison of datasets with varying sizes using the Good–Turing model (Methods); then we calculated the contribution of each multi- and uni-lineage-committed class over time (Fig. 3b). We compared multi-lineage and lineage-committed clones across different diseases using Z-score scaling and the Kruskal–Wallis test (Methods and Extended Data Fig. 6a). Multi-lineage clones in BM and PB reached up to 75% at early time points (less than 12 months) and stabilized at 50–60% in all clinical programmes, with MLD showing fewer multi-lineage clones compared to patients with WAS and β-Thal. Myeloid-committed clones were higher in MLD than in WAS but similar to β-Thal. T-committed clones were higher in WAS, whereas erythroid-committed clones were highest in β-Thal. Overall, our results showed that the disease background affected haematopoietic reconstitution, determining a preferential and specific lineage commitment of HSPC clones (Fig. 3c). Multi-lineage clones amounted to roughly 90–75% at the earliest time points and decreased progressively to 50–60% in all clinical programmes. But age affected lineage commitment, with older patients with MLD and β-Thal showing fewer multi-lineage clones (Extended Data Fig. 6b–d). In patients with MLD, myeloid-committed clones increased over time, particularly in the 2–15 years age group. In patients over the age of 30 with β-Thal, myeloid commitment was higher but did not increase over time as in MLD. Erythroid commitment was low in older patients with β-Thal, peaking at 12.5% before decreasing. By contrast, erythroid clones remained below 1% in patients with MLD and WAS across all age groups.
Fig. 3. HSC lineage commitment.

a, The workflow for analysing HSC commitment involved computing IS sharing among myeloid, erythroid, B and T cell markers at each time point. Shared ISs were categorized as multi-lineage (if found in several lineages, ‘Multi’) or uni-lineage (if found in one lineage, ‘Uni-myeloid’ for uni-lineage myeloid and ‘Uni-LyT’ or ‘Uni-LyB’ for uni-lineage lymphoid B or T cell, respectively). We then calculated the percentage of each lineage and analysed the profiles over time. b, HSC lineage commitment was tracked for different diseases and tissues, showing the relative percentage of shared ISs over time for multi-lineage and mature uni-lineage clones, using spline regression with a 0.75 CI. c,The box plot compares normalized HSC lineage commitment (Z-score) across clinical trials, tissues and lineages, with statistical significance indicated by Kruskal–Wallis test P values. The bars represent the median, the whiskers extend to 1.5 times the IQR, and the P value threshold is set at 0.05. d–g, Box plots represent lineage commitment (myeloid (d), T cell (e), B cell (f), erythroid (g)) during early and late phases of haematopoietic reconstitution in BM and PB, focusing on multi-lineage clones transitioning to uni-lineage and those remaining committed. Statistical comparisons used one-way analysis of variance (ANOVA). The central line represents the median, and the whiskers indicate the range, showing the minimum and maximum values. h, Box plots for multi-lineage clones remaining multi-lineage in BM and PB, with statistical analysis as above. The bar represents the median and the whiskers the range.
Extended Data Fig. 6. HSC lineage commitment.

(A) Z-score normalization of the HSC lineage commitment over time expressed as percentage on the recaptured clones. The plot represents the scaled relative percentage of the shared IS (y-axis) over time (x-axis) for recurrent clones (IS observed in at least two time points for each patient) on the overall number of cells observed in multilineage (green line) or mature uni-lineages (erythroid in red, myeloid in yellow, B in blue, T in light blue). Lines are spline regression curves using a log curve with 0.75 CI. (B) HSC lineage commitment as the percentage of shared ISs of recurrent clones (y-axis) over time (x-axis) for PB samples stratified by clinical trials (in columns) and patient’s age (in rows the three classes of age: 0–2 years, 2–15 years, and >30 years). Colors are associated with multilineage clones (green line) or mature uni-lineages (erythroid in red, myeloid in yellow, B in blue, T in light blue). (C) HSC lineage commitment in BM, similar to (B). (D) Similar to (A), Z-score normalization of the HSC lineage commitment over time expressed as percentage on the recaptured clones and stratified by age. (E) Boxplot representation of the HSC lineage commitment normalized (Z-score) and averaged to compare the different clinical studies (in columns) for multi-lineage or mature committed lineages and tissues (rows); patients are grouped by their age of treatment (groups “0–2” years, “2–15” years, and “>30” years). Statistical results are expressed with lines between pairs of age groups (Kruskal Wallis test). The bars represent the median, the whiskers extend to 1.5 times the IQR, and the p-value threshold is set at 0.05.
To analyse lineage commitment dynamics over time, clones were classified as transitioning from multi- to uni-lineage or remaining consistently committed between early and late phases of haematopoietic reconstitution (less than 24 months and more than 24 months, respectively). This single clone level analysis allowed us to compare if and how the different disease conditions affected the rate of lineage commitment over time as well as the relative contribution of long-lived clones already committed since the early phases of haematopoietic reconstitution. Patients with MLD showed higher myeloid commitment (10–12%) compared to patients with WAS and β-Thal (less than 5%) (Fig. 3d). Persistently myeloid-committed clones (uni-myeloid) were more prevalent in patients with MLD and β-Thal (20–30%) than in patients with WAS (less than 5%). T cell commitment was higher in WAS (Fig. 3e,f), whereas erythroid commitment was notably higher in patients with β-Thal (Fig. 3g). In agreement with our previous findings, multi-lineage clones remained abundant across all programmes, exceeding 50% (Fig. 3h). Stratifying by age, older patients with MLD and β-Thal had more uni-myeloid clones, whereas younger patients with MLD had more T cell commitment. In patients with β-Thal over the age of 30, persisting multi-lineage clones decreased significantly compared to younger cohorts (Extended Data Fig. 7a–e). Age did not significantly affect other lineages within the studied age ranges.
Extended Data Fig. 7. Unilineage or multilineage commitment.

(A) The first six panels show the percentage of multilineage clones (identified <24 months post-transplant) in BM and PB that transition into unimyeloid clones in the late phase (>24 months). The remaining six panels show the percentage of unimyeloid clones that remain committed in the late phase. Each panel compares lineage commitment across clinical programs by age at treatment: 0–2 years and 2–15 years for MLD and WAS, and 2–15 and >30 years for β-Thal. Statistical comparisons used Student’s T-test. (B-E) Box plots representing T-cell, B-cell, erythroid, and multilineage commitment during early and late hematopoietic reconstitution. Panel order and statistical analysis are consistent with (A).
A recent study in nonhuman primates suggested that clonal abundance might bias vector integration studies32, encompassing lineage output and commitment. To investigate this, we compared clonal abundances between uni-lineage (erythroid, B, T, myeloid) and multi-lineage clones during early and late phases (Extended Data Fig. 8a). The abundance distributions for multi-lineage, uni-lineage and transitioning clones were similar, with no significant differences found between early and late datasets (Extended Data Fig. 8b,c). A bootstrap approach added a confidence interval to each IS generated through using incremental percentage of reads’ sampling (50, 70, 80, 90%) and ten randomizations (Methods) and further confirmed the robustness of our findings, showing high accuracy (more than 0.9 from 80% subsampling) across lineage classes (Extended Data Fig. 9). These results indicate that clonal abundance did not significantly affect lineage output or commitment in our dataset.
Extended Data Fig. 8. Transitioning clones in HSPC commitment.

(A) Boxplot representation of single IS clonal abundances per class (multi-lineage or specific uni-linage) in early time points (<24 months) or late time points (>24 months). In all boxplots, the bars represent the median, the whiskers extend to 1.5 times the IQR. (B) Lineage commitment per clone comparing clonal abundances over time (early versus late, <24 months and >24 months respectively) in BM and PB across the different classes (multi-lineage or uni-lineage) and transitions (multi-multi, multi-uni, or uni-uni). Statistical results are expressed with lines between pairs of age groups (Kruskal Wallis test). Patients in each clinical program are represented with dots and colored in blue, green, or red if enrolled in MLD, WAS, or β-Thal study. (C) Similar to (A), we compared lineage commitment clonal abundances per class (multi-lineage or uni-linage) within early time points (<24 months) or late time points (>24 months).
Extended Data Fig. 9. Reliability of HSPC commitment through IS analysis.

(A) The bootstrap strategy. At each timepoint (T = 1 and T = 60 in this case), each IS is observed in different lineages (B, T, and Myeloid here), with a specific number of reads. An IS is given a label (Multi-lineage “Multi” or unilineage “UniT”/”My”) based on the lineages and the number of reads observed. During the bootstrap procedure, each IS undergoes read subsampling, which reduces the number of reads and may affect its labeling. For example, due to low observed abundances, IS1 might not be detected in B cells, causing its final label at the last time point to change to “UniMy”. After 10 randomizations, we evaluate each IS by assessing the accuracy of classifications in early and late phases across bootstrap samples. ISs with fluctuating labels, like IS1, will show low accuracy, whereas ISs with consistent results and a 100% label, like IS2 and IS3, will yield more robust classifications. (B) Mean accuracy with standard error of all IS Confidence Intervals (CI), y-axis, across randomizations, by sampling percentage (x-axis), per class (multi-lineage or uni-linage), for each clinical study. The bars represent the median, the whiskers while the whiskers indicate the range showing the minimum and maximum values.
Clones captured at only one time point (singletons), specifically in the early phase of haematopoietic reconstitution (less than 9 months), were used as surrogates of progenitors and low-abundant HSPCs. Analysis of these early-phase singletons revealed that myeloid cells were dominant across all clinical studies, with patients with β-Thal also showing erythroid lineage singletons, suggesting erythroid precursor expansion (Extended Data Fig. 10a). This indicates early haematopoietic reconstitution is driven by myeloid-committed clones regardless of the disease.
Extended Data Fig. 10. Singleton clones, exhausting HSPCs, Lineage commitment in XSCID patients.
(A) HSC commitment analysis of ISs observed only at one time point (singletons), representing committed clones that exhaust after GT. Results were stratified by clinical trials and patient age, with trends shown for multilineage and mature committed lineages in BM samples, using log spline curves with a 0.75 CI. (B) HSC lineage commitment over time expressed as percentage on the recaptured clones in XSCID patients. The plot represents the scaled relative percentage of the shared IS (y-axis) over time (x-axis) for recurrent clones (IS observed in at least two time points for each patient) on the overall number of cells observed in multilineage (green line) or mature uni-lineages (erythroid in red, myeloid in yellow, B in blue, T in light blue). Lines are spline regression curves using a log curve with 0.75 CI.
Lineage commitment analysis, performed for the ten patients receiving XSCID described above, showed that the T cell committed clones were already 25% at the early time points and increased up to 50% at 60 months (Extended Data Fig. 10b). Multi-lineage clones initially rose but later declined, whereas B cell clones started high (more than 50%) but decreased to roughly 25%. Myeloid commitment was roughly 6%, similar to patients with WAS, indicating a strong uni-lineage T cell commitment in XSCID, with a lesser extent for B cells.
TPO and EPO in patients with WAS and β-Thal
Given that haematopoietic cytokines influence HSPC lineage fate and are regulated by disease states, we examined whether elevated thrombopoietin (TPO) concentrations in patients with WAS indicated haematopoietic recovery by promoting lymphoid and platelet maturation, and if elevated erythropoietin (EPO) concentrations in patients with β-Thal—the classic hallmark of anaemic patients with β-thalassaemia and patients receiving GT33–35—correlate with increased erythroid output.
In patients with WAS the TPO concentrations before transplant were all near or within the normal TPO concentrations ranging around 99 pg ml−1. Elevated TPO concentrations were noted at 1 year post-GT, especially in those treated at 0–2 years, but decreased to near-normal concentrations over time. The highest TPO concentrations were significantly higher in younger patients (0–2 years) compared to others (Fig. 4a).
Fig. 4. TPO and EPO concentrations in patients with WAS and β-Thal, and somatic mutations in patients with MLD and β-Thal.

a, TPO concentration s in patients with WAS before GT (Pre-GT), at 1 year follow-up (1 yr FU) and 3–4 years post-GT (3–4 yr FU), stratified by age at treatment: 0–2 years (0–2 yr) and 2–15 years (2–15 yr). b, EPO concentrations in patients with β-Thal at 1 year (1 yr FU), 2 year (2 yr FU) and 3 year (3 yr FU) follow-ups, stratified by age at treatment: 2–15 years (2–15 yr) and more than 30 years (>30 yr). One-way ANOVA. c,d, Somatic mutations in patients with β-Thal (n = 9) (c) and MLD (n = 16) (d) over time (T0, before infusion; TP1, 2 years posttransplant; TP2, max 5–7 years posttransplant). No significant differences by Friedman’s test. e, Comparison of somatic mutation frequencies between patients with β-Thal and MLD by age group. One-way ANOVA. f, Percentage of VAF over time for somatic mutations found in several time points in patients with MLD and β-Thal, with mutated genes and clinical programmes indicated. In all box plots, the central line represents the median and the whiskers indicate the range, showing the minimum and maximum values.
In patients with β-Thal, EPO concentrations were significantly elevated 1 year post-GT, particularly in adults (older than 30 years) where concentrations were 17-fold higher than normal (reported at 24 mU ml−1). Although EPO concentrations slightly decreased over the following years, they remained elevated, with adult patients showing 3.5-fold higher concentrations than younger patients (Fig. 4b).
Somatic mutation in MLD and β-Thal
We conducted a comprehensive analysis of somatic mutations in 40 genes linked to clonal haematopoiesis and myeloid cancer using the Illumina AmpliSeq Myeloid Panel. We examined peripheral blood mononuclear cells before treatment and at roughly 2 years posttreatment and the last available time point (2.5 to 7.5 years) in nine patients with β-Thal and 23 with MLD. From 20 ng of genomic DNA per time point, we obtained more than 100 million high-quality sequence reads, with an average depth of 4,400 reads per base for β-Thal and 4,300 for patients with MLD (Methods and Supplementary Table 5). Variant analysis, with custom filters to remove false positives, revealed 96 somatic mutations (85 with a variant allele frequency (VAF) less than 2%). The most abundant mutation discovered in a patient with MLD, involving the p53 gene with an average VAF of 15%, was annotated as benign (Supplementary Table 6).
In patients with β-Thal, we found 68 somatic mutations (67 single nucleotide variants and one single nucleotide deletion), from 2 to 24 mutations per patient. The average number of mutations in patients with β-Thal remained consistent across all time points, showing no statistically significant variations (P > 0.9 by Friedman test) (Fig. 4c). Considering that the sequenced genomic interval corresponds to 76,715 bp and that we analysed a total of 8,100 equivalent genomes per patient, the resulting mutation rate in patients with β-Thal was 1.21 × 10−8 mutations per bp. In patients with MLD, we identified 26 somatic mutations (25 single nucleotide variants and 1 deletion), from one to three mutations per patient resulting in a mutation rate of 2.6 × 10−9 mutations per bp. The average number of mutations in patients with MLD was similar at all time points without any statistically significant variations (P > 0.9 by Friedman test) (Fig. 4d). Adult patients with β-Thal had a significantly higher mutation rate than paediatric patients, and both groups had higher rates than patients with MLD (P < 0.05) (Fig. 4e). Five out of the 96 mutations (four in β-Thal and one in MLD) were detected at several time points, none showing a progressive increase in abundance, indicating no selective advantage (Fig. 4f).
Discussion
In this work, we carried out a detailed analysis of lentiviral vector integrations in terminally differentiated myeloid, lymphoid and erythroid cell lineages, as well as CD34+ HSPCs purified from BM or collected in MPB of patients subjected to HSC-GT for MLD, WAS and β-Thal. Thanks to the large number of patients and clones tracked over extended periods of time (up to 8 years from therapy), our work constitutes a large and highly detailed dataset addressing the safety and graft dynamics and, consequently, the overall safety outlook of lentiviral vector-HSC-GT. Exploiting ISs as unique clonal markers we confirm and provide further evidence of long-term efficacy and safety of GT in patients with MLD, WAS and β-Thal and provide biological insights into haematopoietic reconstitution and lineage commitment in the different disease contexts, further modulated by the specific conditioning protocols and the age of the patients at the time of treatment (see Supplementary Discussion for IS sensitivity).
Studies by us15 and others36–38 have highlighted different cell division rates and latency characteristics of distinct HSPC subsets, explaining the plateau we observed in most patients with the switch from short- to long-term HSCs during the 12 to 24 months’ time window. One patient with WAS (Pt17) and two with β-Thal (Pt36 and Pt41) showed continuous retrieval of new ISs beyond 24 months and until the latest follow-up, reaching up to 450,000 ISs. Of note, these increases in new ISs may not raise per se safety concerns, contrary to the increasing abundance of a single IS. The biological reasons behind these exceptions remain to be explained but suggest a particularly high number of genetically engineered HSCs that were engrafted and continue to be recruited for blood cell production. Potential explanations include higher baseline HSC frequency within the transduced CD34+ cells, higher permissiveness of these HSCs to the ex vivo engineering process or more efficient engraftment, for example, by prolonged in vivo persistence after infusion and/or availability of extra niche space in these patients.
In this study, the number of active HSPCs during steady-state haematopoiesis in patients ranged from 1,800 to 74,000, representing roughly 0.0007 to 0.03% of the total infused CD34+ cells. A positive correlation was observed between the number of estimated HSCs and the total infused CD34+ cells16, with no evidence of saturation even at high doses. Patients with β-Thal, who received the highest CD34+ cell doses, showed significantly higher HSC estimates than patients with MLD and WAS. However, when normalized to the total number of infused CD34+ cells, HSC frequency was similar across the three diseases, regardless of whether HSPCs were collected from BM or MPB. This suggests that higher CD34+ cell doses are beneficial for gene therapy, as they do not saturate stem cell niches and may protect against clonal haematopoiesis and malignancy39. This study also highlights the advantages of using MPB-derived40 CD34+ cells in gene therapy, as they yield more transplantable cells and may allow faster blood cell recovery. Furthermore, the higher number of active HSPCs in patients with β-Thal may be due to more efficient engraftment from intrabone infusion compared to intravenous infusion40,41, although the superiority of this method in long-term engraftment remains debated42–44.
The estimated number of HSPCs and their frequency in the infused product align with previous findings in patients with MLD, WAS and β-Thal, and patients receiving sickle cell anaemia GT2,15,16. However, studies of native haematopoiesis in healthy donors45–47 suggest higher HSPC numbers, ranging from 20,000 to 200,000: two to ten times more than in patients receiving HSC-GT. The lower HSPC estimates in patients receiving HSC-GT may be due to underestimation in young patients, competition for engraftment in damaged niches or the detrimental effects of ex vivo culture and lentiviral vector transduction on stemness. Furthermore, the number of persisting clones after haematopoietic reconstitution exceeded initial HSPC estimates, suggesting that capture–recapture methods may underestimate true HSC numbers.
We found that for all the diseases analysed, the output of HSPCs with multi-lineage potential represented only 50% of the clones, whereas the remaining 50% was constituted by clones committed towards a specific lineage and with specific differences across the clinical programmes. In patients with MLD, WAS and β-Thal, the haematopoiesis showed a preferential output and lineage commitment towards myeloid, lymphoid and erythroid lineages respectively, even after years from treatment. It is important to note that we classify HSPC clones as multi-lineage if they are detected in at least two mature lineages, rather than in all four lineages analysed. Our choice is driven by the high polyclonality of our patients, which reduces of recapture in all four linages, whereas barcoding tracking studies in animal models showed higher sensitivity48.
These persistent biases might be influenced by the disease type, the age at which treatment occurs and incomplete correction of the disease posttherapy. In patients with MLD, a significant number of long-lived myeloid clones were present early after transplant, in agreement with previous reports on mouse models49,50, with older patients (aged 2–15 years) showing higher amounts of these myeloid cells compared to younger patients (0–2 years). This difference may be attributed to the accumulation of disease burden with age, causing chronic inflammation that primes HSPCs towards myeloid differentiation. Despite this, the overall balance between multi- and uni-lineage clones remains stable over time, indicating a long-term homeostatic equilibrium. By contrast, patients with WAS and those receiving XSCID, treated with reduced-intensity conditioning, showed rapid lymphoid repopulation, particularly in T cells, due to the selective advantage of gene-corrected cells and the effects of lymphodepleting agents4,15,16. Patients from the β-Thal study showed the most complex scenario in which lineage output and commitment are influenced by factors such as age at treatment and therapeutic outcomes. Adult patients with β-Thal, who remained transfusion dependent, showed a significant increase in erythroid lineage output over time, whereas younger patients (aged 4–13 years) had a lower but stable erythroid output, suggesting a correlation with the residual presence of ineffective erythropoiesis6. Despite normal B cell lineage output, patients with β-Thal showed higher amounts of long-lived B cell committed clones, in agreement with other reports on animal models51, indicating that the disease affects both erythroid and B cell lineages. Furthermore, older patients with β-Thal demonstrated increased uni-myeloid commitment and reduced multi-lineage contributions compared to younger patients, suggesting that age at treatment or therapeutic outcomes play a role in lineage commitment. The stable yet altered HSPC output and commitment observed in both patients with WAS and β-Thal over time indicates that the BM microenvironment and specific cytokines may drive long-term commitment to the most needed lineages, reflecting incomplete normalization of haematopoiesis posttherapy52–57. In agreement with these hypotheses, in patients with WAS, TPO concentrations were initially high after GT but decreased over time, suggesting partial therapeutic success. In patients with β-Thal, EPO concentrations varied, being normal in some transfusion-independent paediatric patients but abnormally high in others, particularly in adults and transfusion-dependent individuals. This indicates a partial correlation between EPO concentrations and therapeutic outcomes, with other factors probably influencing HSPC behaviour in β-Thal58–60. Environmental signals, such as mitogens and cytokines, together with intrinsic or epigenetic factors, may lead to a biased but not exclusive production of deficient mature lineages. The increased abundance of these lineages suggests higher detection rates, whereas contributions to other lineages might fall below the detection threshold. Future experiments aimed at analysing the expression signatures and epigenetic states of HSPCs subsets in bulk and at the single cell level will allow us to better explain the biological mechanisms underlying the biological differences in each disease condition.
No harmful somatic mutations were found from GT in patients with β-Thal and MLD, but patients with β-Thal had significantly more somatic mutations than those with MLD, probably due to haematopoietic stress from oxidative damage related to iron overload, which could result in increased amounts of genomic and mitochondrial DNA damage and telomere erosion, previously reported only in leukocytes, and the altered BM niche25,60–64.
Despite these disease-specific differences, lentiviral vector-based genetic modification of HSPCs and their transplantation were shown to preserve the cells’ ability to respond to disease demands and environmental stressors. An important aspect of this study is that despite the observed differences in these disease conditions, lentiviral vector-based genetic modification of HSPCs and their transplantation do not harm their remarkable capability to respond to the demands imposed by genetic diseases and the ability to respond to infections and environmental damage. Indeed, genetically modified HSPCs with multi-lineage potential once reached stability, contributed to about 50% of the haematopoiesis while the remaining clones were committed. The concept that LT-HSCs are the sole contributors to haematopoiesis has already been challenged65 and our observations reinforce the hypothesis that at late time points after transplantation haematopoiesis is maintained by many lineage-committed HSPCs able to persist for several years with no evidence of clonal dominance after several years from GT.
Methods
Characteristics of patients in the study
Patients receiving GT who were included in the study were treated in the context of clinical trials or early access programmes approved by ethical committee and competent regulatory authorities. The treatment was administered at the Bone Marrow Transplantation Unit at the San Raffaele Scientific Institute in Milan, Italy. We have complied with all the ethical regulations for retrieving biological materials from patients receiving GT. Parents signed informed consent for research protocols approved by the San Raffaele Scientific Institute’s Ethics Committee (TIGET06 and TIGET09). All patients received autologous HSPC transduced with transgene encoding lentiviral vectors under the same transduction protocol, as previously described2,4–6. Patients with MLD received full myeloablative conditioning2,3,22 with busulfan at doses ranging from 10 to 14 mg kg−1. Patients with WAS received a reduced-intensity conditioning regimen consisting of the combined administration of a monoclonal antibody against CD20, busulfan (7.6 to 10.1 mg kg−1) and fludarabine (60 mg m−2)4,5,15,17. In patients with β-Thal, the conditioning consisted in thiotepa (6–8 mg kg) and dose- adjusted treosulfan (14 g m−2) administration6. Patients with MLD were enrolled in a phase 1 or 2 clinical trial (NCT01560182) or treated with a hospital exemption programme or compassionate use programme. A total of 29 patients had been treated with the HSPC gene therapy clinical protocol for arylsulfatase A deficiency). Sixteen of the treated patients (Pt16, Pt32, Pt47, Pt02, Pt20, Pt34, Pt31, Pt03, Pt37, Pt33, Pt08, Pt53, Pt23, Pt40, Pt25, Pt28) were affected by late infantile MLD in a presymptomatic stage and have been identified by molecular and biochemical tests in the presence of at least an affected older sibling, whereas 13 patients (Pt38, Pt01, Pt10, Pt43, Pt42, Pt04, Pt30, Pt51, Pt44, Pt18, Pt50, Pt14, Pt07) were affected by early juvenile MLD in a pre- or early-symptomatic stage. Patients were treated with a myeloablative busulfan conditioning regimen administered before reinfusion of the engineered HSPCs3,22. Fourteen male patients with WAS for whom no human leukocyte antigen–identical sibling donor or suitable matched unrelated donor was available underwent lentiviral GT after a reduced conditioning regimen protocol. Patients Pt52, Pt21, Pt48, Pt13, Pt29, Pt11, Pt39 and Pt17 were enrolled in an open-label, non-randomized, phase 1 or 2 clinical study5 registered with ClinicalTrial.gov (number NCT01515462) and EudraCT (number 2009-017346-32). The other patients with WAS were treated under an early access programme, compassionate use programme or hospital exemption. Among patients with β-Thal, three adults and six children with β0 or severe β+ mutations were enrolled in a phase 1 or 2 trial (NCT02453477) for intrabone administration of GLOBE lentiviral vector–modified HSPCs after myeloablative conditioning with treosulfan–thiotepa6.
Sample collection
PB and BM samples were obtained from patients before and after gene therapy at different times. From each patient, 12 ml of blood was collected at time points of 30 days, 60 days (only for PB sample), 90 days, 6 months during the first year and once every year and half year thereafter. BM cells were isolated by aspirate of 12 ml and specific cell lineages purified as previously described2,4,6. Briefly, PB and BM mononuclear cells from patients receiving GT were isolated using Ficoll-Hypaque gradient separation (Lymphoprep, Fresenius). Mature PB lineages and BM progenitors were purified using positive selection with immunomagnetic beads according to the manufacturer’s specifications (Miltenyi Biotec) and genomic DNA was extracted with the QIAamp DNA Blood Mini or Micro Kit (Qiagen). CD34+ cells were isolated using the CD34+ MicroBead Kit UltraPure, human (lyophilized), MILTENYI BIOTEC CD15+ were isolated from granulocytes cells using CD15 Micro Beads Human, MILTENYI BIOTEC. CD13+ (CD13 Antibody, Antihuman, Biotin and Antibiotin Micro Beads UltraPure), CD3+ (CD3 Micro Beads Human, MILTENYI BIOTEC), CD56+ (CD56 Micro Beads Human, MILTENYI BIOTEC), GLYA+ (Glycophorin A CD235a Micro Beads, human, MILTENYI BIOTEC S.R.L), CD19+ (CD19 Micro Beads Human, MILTENYI BIOTEC) were isolated in sequence from mononuclear cells after CD34+ purification. PB mononuclear cells were isolated by aspirate of 12 ml and specific cell lineages were purified. From granulocytes, CD15+ were isolated using CD15 Micro Beads Human, MILTENYI BIOTEC). From peripheral mononuclear cells CD14+ (CD14 Micro Beads Human, MILTENYI BIOTEC), CD19+ (CD19 Micro Beads Human, MILTENYI BIOTEC), CD3+ (CD3 Micro Beads Human, MILTENYI BIOTEC) and CD56+ (CD56 Micro Beads Human, MILTENYI BIOTEC) were isolated in sequence.
Genomic DNA was extracted using the QIAamp DNA blood mini kit (Qiagen) or QIAamp DNA Micro Kit (Qiagen), based on a cell pellet. The QIAamp DNA Micro Kit was used with fewer than 1 × 106 cells, whereas the QIAamp DNA Mini Kit was used with a dry pellet from 0.5 × 106 to 5 × 106 cells.
Retrieval of vector ISs
The genomic DNA from the patients’ cells was extracted and split in three technical replicates that were subjected to custom PCR amplification to retrieve vector ISs, initially the linear amplification-mediated (LAM)-PCR27 and in more recent samples the sonication linker-mediated (SLiM)-PCR13. Briefly, the SLiM-PCR procedure consists of the following steps: (1) fragmentation by sonication of the DNA, (2) ligation of the fragments to a linker cassette and (3) two consecutive rounds of PCR, to specifically amplify vector to cellular-genome junctions, by using primers annealing to the vector genome end (long terminal repeats) and the linker cassette. The list of primers and sequences used for SLiM-PCR procedure is reported in Supplementary Tables 7–9. Primers contain DNA barcodes, which allow univocal barcoding of all the SLiM-PCR replicates, and sequencing adaptors that allow multiplexed sequencing on Illumina sequencers. The list of samples with the details on the applied PCR procedure and the number of reads retrieved is reported in Supplementary Table 2.
The resulting PCR products were sequenced using Illumina platforms (Mi-seq, Hi-seq, Next-seq and Nova-seq), for a total amount of 194 sequencing pools for more than 17,000 PCR reactions and more than 14.5 × 109 raw reads.
Identification of vector ISs
We used VISPA2 (ref. 66) to identify ISs from PCR samples sequenced with Illumina paired-end reads. Briefly, for each Illumina sequencing library, paired-end reads are filtered for quality standards, barcodes are identified for sample demultiplexing, vector sequences are trimmed from each read and the remaining cellular genomic sequence is mapped on the reference Human Genome (Human Genome_GRCh37/hg19 February 2019). For the quantification of the number of genomes representing each clone, we adopted an estimation method based on the number of distinct fragments for each IS, SonicLength67, such that each IS abundance will be proportional to the initial number of contributing cells, allowing to estimate the clonal abundance in the starting sample. The final list of unique ISs is composed of univocally mapped loci annotated with the nearest RefSeq gene.
We then used a new R package, ISAnalytics28 to integrate the output files of VISPA2 and perform downstream analyses of ISs, from quality controls to the analyses of shared ISs among samples. A detailed report and code of ISAnalytics is available in the GitHub repository (https://github.com/calabrialab/Code_HSPCdynamics). We first removed the same IS present in different independent samples, named collisions, using the same approach previously described2. Briefly, given two patients and the list of shared ISs between the two patients, we assign to one patient the IS if it was observed and retrieved for the first time in that patient or if the relative abundance of that read was at least ten times higher than the other patient. The analysis and removal of collisions in independent samples was realized using ISAnalytics. We then performed data quality analysis of sequencing samples by analysing the number of reads of each PCR sample in each sequencing pool, and we removed the samples containing a number of raw reads highly under-represented (threefold less) than the average number of reads of the other samples in the pool. The quality control analyses included (1) the removal of sequencing samples with a number of raw reads threefold less than the average number of reads per sample in the pool, (2) the removal of contaminations identified as identical ISs retrieved in independent samples as previously described2. Through these filtering approaches, 99 samples (0.7%) were removed because they had a number of raw reads below the acceptance threshold (less than threefold difference on raw reads average per pool), whereas the amount of removed contaminant ISs was less than 1%. For common insertion site analysis, we used Grubbs test for outliers68 (implemented in ISAnalytics). Briefly, for each patient, we computed the targeting frequency of each gene using the number of ISs landing in the gene body ±100 kbp and then normalized by the gene length. After the log2 transformation of the gene distribution frequency, we computed the Grubbs test for outliers to identify genes with a targeting frequency significantly higher than the average observed frequency. The analysis of the cumulative ISs over time allowed us to quantify how many new ISs we observed at each collection or sample compared to all previous samplings. To this aim, we used all ISs observed at each time point. We computed the cumulative number of ISs using ISAnalytics with function ‘cumulative_is’.
Clonal population diversity
An ecological system is maintained stable if the populating species are in equilibrium, suggesting a healthy environment. Population dynamics can be characterized in terms of species diversity, using for example a quantitative measure such as the Shannon diversity index (H index). The H index accounts for the number of distinct species (richness) and their relative abundance with the following formula:
where i is a clone (an IS), pi is the clonal abundance and R is the set of clones.
Several clonal studies already approached the analysis of heterogeneity and complexity of the vector-marked cells over time, tissues and differentiated lineages, using IS as surrogate of different species and the IS size as species abundance, and then measuring richness and evenness over time to quantify long-term efficacy (when the H index is maintained at high values) or observe malignant occurrences (when the H index drastically decreases over time). The diversity index has been computed using the R package Vegan and is integrated in ISAnalytics. Z-score normalization has been performed on the H index by patient and time point using the R function ‘scale’.
As different PCR methods (LAM-PCR and SLiM-PCR) were used to retrieve ISs, which have different retrieval efficiencies, we performed a comparative analysis of clonal diversity only on samples in which the same technology within the same clinical programme was used. For MLD and β-Thal, we selected only the samples processed by SLiM-PCR whereas for patients with WAS only LAM-PCR samples were available. We used the Bayesian multivariate linear regression algorithm to model and correct biases induced by different technical confounding factors as well as their interactions. These factors included PCR method, amount of DNA used, dose of CD34+ infused per kg, VCN, sequencing depth, patient gender and age.
Estimate of active repopulating HSPCs
Similar to what was done in our previous studies2,4, we used short-living cells as the readout of HSC output. Specifically, from our data, we selected myeloid-derived cells from PB samples (cell markers CD14+ and CD15+) from each time point and we then filtered ISs from these cell lineages by sequence count sum less than three across time. When estimating HSPC from several samples, we used mark-recapture statistics that leverage on the number of recaptured ISs across the different observations to estimate the size of the source population. Population size estimate has been performed in R using the package Rcapture with Chao1 (ref. 69) model considering a closed population. For the estimate of HSPCs over time, we used triplets of consecutive time points using a sliding window from the first month in vivo to the last follow-up month. For the estimate of the size of HSPCs per patient, we selected used all stable time points (from months 9 or 12 after gene therapy). Then, we corrected the number of estimated HSPCs by the VCN in in vivo cells if VCN > 1. Moreover, when we correlated the HSPC size to the cell dose, we corrected the dose by the engraftment using the in vitro VCN (if VCN < 1) as reported16. HSPC estimate has been implemented in ISAnalytics, here run with the following prototype function ‘HSC_population_size_estimate’.
Clonal composition through IS sharing
To analyse the composition of long-lasting clones by tracking the originating time points (that is, the first observed time point of each specific IS) for each patient, sample and tissue, we processed the matrix of ISs to label each clone with the first observed time point (among the previous ones) such that if the clones were recaptured in a subsequent time point we could backtrack from which month it has been initially found. We then computed the percentage of the recaptured clones on the total observed clones at each time point. For this purpose, we used the function ‘iss_source’ in ISAnalytics.
To quantify the composition of long-lasting clones, we isolated ISs retrieved from 36 months (both PB and BM) and averaged the percentages for each source time point. Source time points were then grouped by haematopoietic phases: 1–6 months as early, 12–18 months as mid and 24–30 months as steady. Once collected data from each patient and calculated the percentage in each lineage, we then performed the non-parametric Kruskal–Wallis statistical test on the means between pairs of groups (early versus mid, early versus steady, mid versus steady) using the R function compare_means and returned P value after false discovery rate (FDR) correction.
Sharing ratio and Z-score
A single HSPC clone can proliferate and differentiate in distinct mature lineages. Using ISs, we can account for how many lineages have been retrieved for every single clone, thus studying the output of HSPCs. Specifically, given two distinct lineages (namely A and B), we can calculate the number of shared ISs or not shared ISs (as exclusive of A or B) on the overall number of clones per set (A or B). This ratio (or relative percentage) is called the ‘sharing ratio’.
The sharing ratio for CD34+ BM cells is calculated as the number of shared IS retrieved in both CD34+ BM cells and each mature cell marker on the total number of CD34+ BM IS. From each patient’s matrix of IS, we applied the filter for intra-marker contamination (as reported in previous works2,4, here using ISAnalytics with the function named ‘purity_filter’ with parameters impurity_threshold = 10, min_value = 3) and we then computed the sharing ratio. The parameter ‘input_threshold’ is the fold difference, set to 10 as already configured in past analyses, whether ‘min_value’ is the minimum number of reads to accept and IS as true.
Profiles of the sharing ratio over time were generated using the log-spline function in R. The Z-score was computed within each patient, time point and tissue by the R function ‘scale’. The statistical test to compare means was the non-parametric Kruskal–Wallis test with FDR correction. For data selection for time course analysis, repeated measurements were obtained as the average of ISs between 24 and 60 months, corresponding to the overlapping time points among the three clinical studies, with a minimum number of three observations per patient.
The sharing ratio for whole populations (in BM and PB separately) is calculated as the number of shared IS retrieved in both whole populations and each mature cell marker on the total number of IS retrieved in the whole population. Once computed the sharing ratio, we then compared these values among the lineages to observe any skewing or imbalance within each clinical trial and performed the non-parametric Kruskal–Wallis test (with FDR correction) on the means to quantify the differences. When we compared the different clinical trials, we needed to apply Z-score statistics and transform the data before data comparison.
HSC lineage commitment
If we considered pairs of sets in the sharing ratio, allowing a clone to belong to different mature lineages, to study lineage commitment, we required a method that accounted for sharing across all lineages together, assigning each IS unambiguously to a specific intersection (Fig. 3a). In our study, we defined the following sets to intersect for each patient and time point: CD34+, erythroid, myeloid, B and T cells. A single IS (with a minimum of three reads) exclusively belongs to either a single lineage or to an intersection of several lineages. For each time point, we classified an IS as ‘multi-lineage’ if it was shared among at least two mature lineages (excluding CD34+ cells). Otherwise, it was labelled as ‘uni-lineage’ if the IS was found exclusively within one mature lineage (or shared with CD34+ cells).
Computationally, we addressed this by generating a binary table that included all possible combinations of intersections for each time point (see Supplementary Methods for more details).
If a clone has been found in several time points, then it is considered ‘recurrent’, otherwise it is a ‘singleton’. The union of recurrent and singletons sum to all patient IS. For the analysis of lineage commitment in a percentage, the sharing ratio was then computed for each time point placing at the numerator the number of IS observed in a specific group (among multi-lineage of one of the uni-lineage) and at the denominator the number of clones captured at that time point (belonging to one of the groups, recurrent or singleton, if the ratio was within the group). For single clone lineage commitment, we tracked the classification dynamics of each clone over time and then we selected recurrent clones in early and late phases (less than 24 months and more than 24 months, respectively). For the analyses of the abundance of committed clones over time, we then joined each IS with the relative abundance (quantified with the number of genomes) and averaged the clonal abundance within each assemblage.
Gene Ontology and feature annotation
Gene Ontology has been realized using R packages for Gene Ontology clusterProfiler, the annotation DB org.Hs.eg.db and msigdbr. Semantic similarity has been done with the R package GOSemSim70. Feature annotations have been realized with the R packages ChIPseeker and database TxDb.Hsapiens.UCSC.hg19.knownGene (up-set plot) and the closest genes have been annotated with RefGene table (UCSC database hg19). The circos plot was generated by the R package ‘circlize’.
Statistical analysis
For pair-wise correlation and PCA analysis we used the R packages PerformanceAnalytics and factoextra, respectively. Kruskal–Wallis test performed in the R package ggpubr and stat_compare_means function. The R library ‘stats’ has been used for all P value corrections. The following libraries and software version have been used for IS analysis and statistics: Prism (v.10); R v.4.0.3 (package dependencies linked to this version include ISAnalytics, scales, dplyr, rstatix for wilcox_test, splines, Rcapture, vegan, psych, cluster, DescTools, fpc, pca, factorextra, ggplot). The following packages have been used for Good–Turing and Bayesian regression: R v.4.2.2 (2022-10-31), plyr_1.8.9, tools_4.2.2, jsonlite_1.8.8, grid_4.2.2, tidyselect_1.2.0; Python v.3.8.15, packaged by conda-forge, sklearn v.0.2, joblib v.1.2.0, numpy v.1.24.1, scipy v.1.10.1 and threadpoolctl v.3.1.0.
Multivariate Bayesian linear regression
Accurate determination of species counts and their respective abundances may face challenges due to technical or patient-specific confounders, potentially hindering the identification of true variability within the data. To overcome this, in our analyses focusing on clonal diversity, the assessment of long-lasting clones and HSC lineage commitment, we included these factors as covariates in a multivariate Bayesian linear regression model. The set of covariates includes positive real values and categorical values: time after gene therapy (positive real value), amount of sequenced DNA (positive real value), next-generation sequencing technology (categorical value), PCR methodology (categorical value), average VCN (positive real value), dose of cells (CD34 pro per kg) (positive real value) and patient gender and age (positive real value). Further details are reported in Supplementary Methods.
To model the nonlinearity among the response and the predictors, a second-order spline transformation on the input data X is performed before fitting the model and categorical predictors are included in the model by means of one-hot encoding. The preprocessing has been carried out through the functions SplineTransformer() and OneHotEncoder(), whereas for the model fitting, the ARDRegression() function was used; all functions are implemented in the scikit-learn Python package. We carried out a Bayesian linear regression to predict the Shannon diversity index (H index), the Sharing ratio (S ratio) and the CD34 output, taking into account confounding covariates to reduce their effect on the observed quantity.
Good–Turing estimator
The Good–Turing model has been used to estimate the number of undetected species in an assemblage. This estimation can be extended to evaluate the number of shared species between two assemblages and it is particularly useful in situations in which rare species, often not detected, make up a significant portion of the total. This method has been used in ecology to correct the bias between the observed and true number of species in a given area. By treating each IS as a species and cell markers as distinct assemblages, we can apply this approach to better estimate the richness of ISs within specific cell markers and the number of shared ISs between CD34 and each marker, accounting for undetected species. Two assemblage models exist and have been used: single assemblage and two assemblages. In the single assemblage model, the true number of species includes both observed and undetected species. The undetected species count is estimated based on the rarest observed species by using singletons and doubletons. This model is extended to two assemblages when estimating shared species between two sets. The true count of shared species accounts for those undetected in one or both assemblages. Estimations are based on observed species frequencies and population sizes, with specific formulas for undetected species in each case. A detailed formulation is reported in Supplementary Methods.
We applied the Good–Turing estimators to account for undetected ISs in lineage analyses. In single assemblage cases, we calculated the adjusted richness of CD34+ cells. For two assemblages, we estimated shared ISs between CD34+ cells and mature cell markers. The adjusted sharing ratio was then used in a multivariate Bayesian linear regression model to analyse lineage commitment and patient heterogeneity. We categorized each IS as multi- or uni-lineage and calculated sharing ratios, correcting them for confounding factors using the Good–Turing method and Bayesian analysis.
IS confidence interval by bootstrap
Each IS at each time point has been classified as either multi- or uni-lineage if shared across distinct cell lineages or a single cell lineage, respectively. To test whether the potential subsampling bias for poorly abundant clones could result in misclassification of an IS as uni-lineage instead of multi-lineage, we further evaluated the robustness of the commitment results by performing a parametric bootstrap with incremental subsampling percentages of the number of reads per IS and replicating this procedure ten times (ten is the number of randomizations). By this approach, we can evaluate the robustness and stability of the assigned label class (multi- or uni-lineage) for each IS during early, less than 24 months, versus late, greater than or equal to 24 months, phases of haematopoietic reconstitution by assessing how many times each single IS maintains the same commitment or multi-lineage state (in percent) generating the confidence interval. The standard error of the confidence interval is then computed across all IS.
In more detail, for each experiment i, let Xi be the observed read counts matrix. In this matrix, the rows represent the distinct ISs and the columns represent the distinct time points and cell markers, with the column-wise sums yielding the total number of observed reads for the given sample. We assumed to collect a subsample of the total number of reads, testing the robustness using 50, 70, 80 and 90% of the observed read counts. For each subsampling percentage p, we generated a matrix X(p)i. To generate several stochastic bootstrap replicates, we then sampled each matrix entry from a negative binomial distribution, using as mean and overdispersion the subsampled number of reads X(p)i. For each replicate, we performed the classification of the ISs as multi- or uni-lineage. We then compared each IS’s classifications with those obtained from the observed matrix (without subsampling, 100%) using a measure of accuracy, defined as accuracy = (number of correct predictions)/(number of total predictions).
Somatic mutations with the myeloid panel
DNA was extracted from PB mononuclear cells of 71 MLD (23 patients) and 27 β-Thal samples (nine patients) using the QIAamp DNA Blood Mini Kit (Qiagen). Twenty nanograms of genomic DNA were subjected to the AmpliSeq for Illumina Myeloid Panel (Illumina) procedure, following the manufacturer’s instructions. Special control DNA samples to assess the performance of next-generation sequencing assays for the detection of somatic mutations (Horizon Discovery HD701 and HD729) were processed simultaneously. PCR products were barcoded with AmpliSeq for Illumina CD Indexes Set A and B and pooled in one library that was paired-end sequenced on 1 Nova-Seq SP500 flow cells.
Sequences were aligned to the human reference genome (hg19/GRChg37) using BWA-MEM. Pileup files were generated using Samtools (options -B -q 1), followed by variant calling with VarScan2 (options --min-coverage 100, --min-var-freq 0.01). To filter out false positives, we removed mutations that: (1) appeared in several samples, (2) were in low-covered amplicons (fewer than 200 reads), (3) were germline (49 < VAF < 51 or VAF > 99), (4) occurred in the last 3 bp of reads or (5) were in poly-T or poly-A regions. Most somatic mutations had a VAF of less than 2%. Detected mutations were annotated using gnomAD, dbSNP and ClinVar databases. Further details are reported in Supplementary Methods.
TPO and EPO assays
To measure TPO in patients with WAS, we used the Quantikine ELISA—Human Thrombopoietin Immunoassay kit on the frozen plasma of patients according to the manufacturer’s instructions.
EPO was dosed from each patient’s serum by chemiluminescence with the Immulite 2000Xpi. Normal values are 3–24 mU ml−1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41586-024-08250-x.
Supplementary information
This file contains Supplementary Methods, legends for Tables 1–9, Discussion and References.
Patients’ summary. Summary table of treated patients under analysis, showing details of the disease, the ID of the patient (Patient ID), the body weight at treatment, the cell dose expressed as millions of CD34+ cells 10 per kg, the transduction efficiency, the GT source (expressed as BM, MPB for mobilized PB or BM and MPB), the VCN at treatment, the age of the patient at treatment and the maximum follow-up available with molecular data, and the gender.
Patients’ samples and details with ISs. The table presents all details of each patient sample used in this work, using the following columns: Disease, Patient ID, Tissue of sample origin (PB and BM), Lineage, Cell Marker, Time point (expressed in months after GT), VCN, amount of genomic material (reported as ng of DNA), PCR Method for IS retrieval (between LAM-PCR and SLiM-PCR), sequencing technology and platform, Number of reads supporting the ISs and Number of ISs, population diversity index (using Shannon H index).
Normalized integration frequency by gene. Pair-wise comparison of gene normalized integration frequency for patients with MLD, WAS and β-Thal, after homogeneous subsampling and randomization. For each gene (column GeneName) and pair-wise comparison (column Comparison) we reported the average number of ISs for study and the final corrected P value (column corrected p-value avg).
Estimated number of active HSPC. For each available patient, the table reports the number of estimated HSPCs observed at early time points (less than 24 months) and from 24 months (column Pop. Size >24 m) calculated with the Chao1 model and corrected with the VCN (if VCN > 1). We also reported the number of IS retrieved less than 24 months and more than 24 months, and the number of shared IS between the two sets. The last column reports the fraction of the estimated HSPCs greater than 12 months on the total number of observed IS more than 24 months.
Mean depth coverage of somatic mutations data. The table shows the mean depth coverage reached for each patient with MLD and β-Thal coming from the Illumina’s AmpliSeq Myeloid Panel Targeted exome sequencing used for the somatic mutations analysis. Columns report: Disease, Patient ID, time point in days of the sample and mean depth coverage.
Somatic mutations. The table presents all the somatic mutations for each patient with MLD and β-Thal coming from the Illumina’s AmpliSeq Myeloid Panel Targeted exome sequencing. Columns report: Disease, Patient ID, time point in days of the sample, cell type, total number of reads in the position of the mutation, number of reads supporting the reference variant, number of reads supporting the mutated variance, VAF, chromosome, starting position, ending position, nucleotide reported by the reference, nucleotide reported by the mutation, functional annotation of the position (intron, exon), gene, exon modification, aminoacidic change, effect of the mutation reported by ClinVar, frequency of the mutation in the population reported by gnomAD and ID of the mutation reported by avsnp147.
Primers used for the first (Exponential) PCR. A list of primers used for the first exponential PCR with details about name, type and sequence.
Primers used for the second (Fusion) PCR. List of primers used for the second fusion PCR with details about name, type, sequence and barcode (eight nucleotides) of each primer.
Linker cassette sequences. The linker cassette sequences used in the SLiM-PCR procedure. Each linker cassette was generated by the annealing of the short oligo with a long oligo. The sequence of each oligo and its barcode (eight nucleotides) is reported.
Source data
Acknowledgements
We acknowledge the physicians and nursing team of the Pediatric Immunohematology Unit, Stem Cell Transplant Program of the IRCCS San Raffaele Scientific Institute, for their professional care of patients during hospitalization and visits; Fondazione Telethon ‘Just Like Home’ Program, S. Zancan and all in the SR-TIGET Clinical Trial Office, M. Soncini and all at the SR-TIGET clinical laboratory for their support; the team of AGC Biologics (formerly MolMed) for manufacturing the vector and medicinal product. We thank E. Zonari for the support with Fig. 1, C. Sartirana for technical support on TPO measurements and C. Cipriani for his support with R. We thank A. Biffi as former PI of the MLD GT study. This work has been supported by grants to A.C. and D. Cesana from Italian Ministry of Health (grant no. Giovani Ricercatori GR-2016-02363681) and to E.M. from the Telethon Foundation (grant nos. TGT11D1, TGT16B01 and TGT16B03) and from the European Leukodystrophies Association (ELA, grant no. 2021-019I1).
Extended data figures and tables
Author contributions
A.C. performed most of the research, interpreted data, designed the study, developed the computational biology analyses and wrote the manuscript. G.S., G.P., F.G., E.B. and M.O. performed bioinformatics analyses under the supervision of A.C., G.C. and E.M. D. Cesana supported the design and the interpretation of lineage commitment analyses. B.G., S. Scala, M.R.L. and S. Scaramuzza critically interpreted the results of the analyses and supported the revision of the manuscript. F.B., A. Albertini and S.E. performed most of the experiments. F.T. and D. Canarutto contributed to patient follow-up and data collection. F.D.M., F.D., S.C. and S.G. managed clinical samples. S.M., V.C., F. Fumagalli and F. Ferrua performed clinical follow-up to patients receiving G.T. G.C. supervised the Good–Turing and Bayesian data corrections. F.C., L.N., G.F. and A.A. provided interpretation of all data and critically reviewed the manuscript. E.M. contributed to the study design, data collection and interpretation of all data, manuscript writing and provided overall supervision.
Peer review
Peer review information
Nature thanks Vijay Sankaran, Stefan Cordes and Mark Walters for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
Sequencing data of mutations have been deposited into the Sequencing Read Archive (NCBI SRA) with the BioProject PRJNA1150995. IS data have been deposited in GitHub at the following project archive: https://github.com/calabrialab/Code_HSPCdynamics. Source data are provided with this paper.
Code availability
We released our source code in GitHub (https://github.com/calabrialab/Code_HSPCdynamics).
Competing interests
The San Raffaele Telethon Institute for GT (SR-Tiget) is a joint venture between the Telethon Foundation and Ospedale San Raffaele. Lentiviral vector-based gene therapy for metachromatic leukodystrophy (MLD), developed at SR-Tiget, was licensed to Orchard Therapeutics in 2018. Lentiviral vector-based gene therapy for Wiskott–Aldrich (WAS) syndrome was developed by Fondazione Telethon. Lentiviral vector-based gene therapy for β-thalassemia was developed at SR-Tiget. Gene therapy for MLD is approved in the EU (Libmeldy) and in the US (Lenmeldy). A.A. was the principal investigator of the pilot and pivotal SR-Tiget clinical trial of GT for MLD, WAS and β-Thal. The other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Andrea Calabria, Email: calabria.andrea@hsr.it.
Eugenio Montini, Email: montini.eugenio@hsr.it.
Extended data
is available for this paper at 10.1038/s41586-024-08250-x.
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-024-08250-x.
References
- 1.Ferrari, G., Thrasher, A. J. & Aiuti, A. Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet.22, 216–234 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Biffi, A. et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science341, 1233158–1233158 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Sessa, M. et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet10.1016/s0140-6736(16)30374-9 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Aiuti, A. et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science341, 1233151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrua, F. et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol.6, e239–e253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marktel, S. et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ss-thalassemia. Nat. Med.25, 234–241 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Tucci, F. et al. Bone marrow harvesting from paediatric patients undergoing haematopoietic stem cell gene therapy. Bone Marrow Transplant54, 1995–2003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tucci, F., Galimberti, S., Naldini, L., Valsecchi, M. G. & Aiuti, A. A systematic review and meta-analysis of gene therapy with hematopoietic stem and progenitor cells for monogenic disorders. Nat. Commun.13, 1315 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eichler, F. et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N. Engl. J. Med.377, 1630–1638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hacein-Bey Abina, S. et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA313, 1550–1563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hacein-Bey-Abina, S. et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. New Engl. J. Med.371, 1407–1417 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohn, D. B. et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat. Med.10.1038/s41591-019-0735-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cesana, D. et al. Retrieval of vector integration sites from cell-free DNA. Nat. Med.10.1038/s41591-021-01389-4 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Firouzi, S. et al. Development and validation of a new high-throughput method to investigate the clonality of HTLV-1-infected cells based on provirus integration sites. Genome Med.6, 46–46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scala, S. et al. Dynamics of genetically engineered hematopoietic stem and progenitor cells after autologous transplantation in humans. Nat. Med.24, 1683–1690 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Six, E. et al. Clonal tracking in gene therapy patients reveals a diversity of human hematopoietic differentiation programs. Blood135, 1219–1231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biasco, L. et al. In vivo tracking of human hematopoiesis reveals patterns of clonal dynamics during early and steady-state reconstitution phases. Cell Stem Cell19, 107–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aiuti, A. et al. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J. Clin. Invest.117, 2233–2240 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Ravin, S. S. et al. Lentivector cryptic splicing mediates increase in CD34+ clones expressing truncated HMGA2 in human X-linked severe combined immunodeficiency. Nat. Commun.13, 3710 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cartier, N. et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science326, 818–823 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Gentner, B. et al. Hematopoietic stem- and progenitor-cell gene therapy for Hurler syndrome. N. Engl. J. Med.385, 1929–1940 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Fumagalli, F. et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet399, 372–383 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goyal, S. et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N. Engl. J. Med.386, 138–147 (2022). [DOI] [PubMed] [Google Scholar]
- 24.Jones, R. J. & DeBaun, M. R. Leukemia after gene therapy for sickle cell disease: insertional mutagenesis, busulfan, both, or neither. Blood138, 942–947 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Spencer Chapman, M. et al. Clonal selection of hematopoietic stem cells after gene therapy for sickle cell disease. Nat. Med.10.1038/s41591-023-02636-6 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanter, J. et al. Lovo-cel gene therapy for sickle cell disease: treatment process evolution and outcomes in the initial groups of the HGB-206 study. Am. J. Hematol.98, 11–22 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt, M. et al. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR). Nat. Methods4, 1051–1057 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Pais, G. et al. ISAnalytics enables longitudinal and high-throughput clonal tracking studies in hematopoietic stem cell gene therapy applications. Brief Bioinform.24, bbac551 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chao, A., Chiu, C. H. & Jost, L. Phylogenetic diversity measures based on Hill numbers. Phil. Trans. R. Soc. B: Biol. Sci.365, 3599–3609 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Lochem, E. G. et al. Immunophenotypic differentiation patterns of normal hematopoiesis in human bone marrow: reference patterns for age-related changes and disease-induced shifts. Cytometry B Clin. Cytom.60, 1–13 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Magnani, A. et al. Long-term safety and efficacy of lentiviral hematopoietic stem/progenitor cell gene therapy for Wiskott-Aldrich syndrome. Nat. Med.28, 71–80 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radtke, S. et al. Stochastic fate decisions of HSCs after transplantation: early contribution, symmetric expansion, and pool formation. Blood142, 33–43 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Locatelli, F. et al. Betibeglogene autotemcel gene therapy for non-beta(0)/beta(0) genotype beta-thalassemia. N. Engl. J. Med.386, 415–427 (2022). [DOI] [PubMed] [Google Scholar]
- 34.Thompson, A. A. et al. Gene therapy in patients with transfusion-dependent beta-thalassemia. N. Engl. J. Med.378, 1479–1493 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Cazzola, M. et al. Red blood cell precursor mass as an independent determinant of serum erythropoietin level. Blood91, 2139–2145 (1998). [PubMed] [Google Scholar]
- 36.Laurenti, E. et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell16, 302–313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaufmann, K. B. et al. A latent subset of human hematopoietic stem cells resists regenerative stress to preserve stemness. Nat. Immunol.22, 723–734 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Bernitz, J. M., Kim, H. S., MacArthur, B., Sieburg, H. & Moore, K. Hematopoietic stem cells count and remember self-renewal divisions. Cell167, 1296–1309 e1210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glait-Santar, C. et al. Functional niche competition between normal hematopoietic stem and progenitor cells and myeloid leukemia cells. Stem Cells33, 3635–3642 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lidonnici, M. R. et al. Plerixafor and G-CSF combination mobilizes hematopoietic stem and progenitors cells with a distinct transcriptional profile and a reduced in vivo homing capacity compared to plerixafor alone. Haematologica102, e120–e124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larochelle, A. et al. AMD3100 mobilizes hematopoietic stem cells with long-term repopulating capacity in nonhuman primates. Blood107, 3772–3778 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felker, S. et al. Differential CXCR4 expression on hematopoietic progenitor cells versus stem cells directs homing and engraftment. JCI Insight7, e151847 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng, Q. et al. Nonhuman primate allogeneic hematopoietic stem cell transplantation by intraosseus vs intravenous injection: engraftment, donor cell distribution, and mechanistic basis. Exp. Hematol.36, 1556–1566 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Yahata, T. et al. A highly sensitive strategy for SCID-repopulating cell assay by direct injection of primitive human hematopoietic cells into NOD/SCID mice bone marrow. Blood101, 2905–2913 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Lee-Six, H. et al. Population dynamics of normal human blood inferred from somatic mutations. Nature561, 473–478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Werner, B. et al. Reconstructing the in vivo dynamics of hematopoietic stem cells from telomere length distributions. eLife4, e08687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitchell, E. et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature606, 343–350 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adair, J. E. et al. DNA barcoding in nonhuman primates reveals important limitations in retrovirus integration site analysis. Mol. Ther. Methods Clin. Dev.17, 796–809 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pei, W. et al. Resolving fates and single-cell transcriptomes of hematopoietic stem cell clones by PolyloxExpress barcoding. Cell Stem Cell27, 383–395 e388 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Pietras, E. M. et al. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell17, 35–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eisele, A. S. et al. Erythropoietin directly remodels the clonal composition of murine hematopoietic multipotent progenitor cells. eLife11, e66922 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mossadegh-Keller, N. et al. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature497, 239–243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grover, A. et al. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J. Exp. Med.211, 181–188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Endele, M. et al. CSF-1-induced Src signaling can instruct monocytic lineage choice. Blood129, 1691–1701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brown, G. The social norm of hematopoietic stem cells and dysregulation in leukemia. Int. J. Mol. Sci.23, 5063 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ding, Y., Liu, Z. & Liu, F. Transcriptional and epigenetic control of hematopoietic stem cell fate decisions in vertebrates. Dev. Biol.475, 156–164 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Meng, Y. et al. Epigenetic programming defines haematopoietic stem cell fate restriction. Nat. Cell Biol.25, 812–822 (2023). [DOI] [PubMed] [Google Scholar]
- 58.Takizawa, H., Boettcher, S. & Manz, M. G. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood119, 2991–3002 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Batsivari, A. et al. Dynamic responses of the haematopoietic stem cell niche to diverse stresses. Nat. Cell Biol.22, 7–17 (2020). [DOI] [PubMed] [Google Scholar]
- 60.Aprile, A. et al. Hematopoietic stem cell function in beta-thalassemia is impaired and is rescued by targeting the bone marrow niche. Blood136, 610–622 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nanthatanti, N. et al. Leukocyte telomere length in patients with transfusion-dependent thalassemia. BMC Med. Genomics13, 73 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aprile, A. et al. Inhibition of FGF23 is a therapeutic strategy to target hematopoietic stem cell niche defects in beta-thalassemia. Sci. Transl. Med.15, eabq3679 (2023). [DOI] [PubMed] [Google Scholar]
- 63.Crippa, S. et al. Bone marrow stromal cells from beta-thalassemia patients have impaired hematopoietic supportive capacity. J. Clin. Invest.129, 1566–1580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gorur, V., Kranc, K. R., Ganuza, M. & Telfer, P. Haematopoietic stem cell health in sickle cell disease and its implications for stem cell therapies and secondary haematological disorders. Blood Rev.63, 101137 (2024). [DOI] [PubMed] [Google Scholar]
- 65.Cavazzana-Calvo, M. et al. Is normal hematopoiesis maintained solely by long-term multipotent stem cells. Blood117, 4420–4424 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spinozzi, G. et al. VISPA2: a scalable pipeline for high-throughput identification and annotation of vector integration sites. BMC Bioinf.18, 520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berry, C. C. et al. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics10.1093/bioinformatics/bts004 (2012). [DOI] [PMC free article] [PubMed]
- 68.Biffi, A. et al. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood117, 5332–5339 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Chao, A. et al. Rarefaction and extrapolation with Hill numbers: a framework for sampling and estimation in species diversity studies. Ecol. Monogr.84, 45–67 (2014). [Google Scholar]
- 70.Yu, G. Gene ontology semantic similarity analysis using GOSemSim. Methods Mol. Biol.2117, 207–215 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains Supplementary Methods, legends for Tables 1–9, Discussion and References.
Patients’ summary. Summary table of treated patients under analysis, showing details of the disease, the ID of the patient (Patient ID), the body weight at treatment, the cell dose expressed as millions of CD34+ cells 10 per kg, the transduction efficiency, the GT source (expressed as BM, MPB for mobilized PB or BM and MPB), the VCN at treatment, the age of the patient at treatment and the maximum follow-up available with molecular data, and the gender.
Patients’ samples and details with ISs. The table presents all details of each patient sample used in this work, using the following columns: Disease, Patient ID, Tissue of sample origin (PB and BM), Lineage, Cell Marker, Time point (expressed in months after GT), VCN, amount of genomic material (reported as ng of DNA), PCR Method for IS retrieval (between LAM-PCR and SLiM-PCR), sequencing technology and platform, Number of reads supporting the ISs and Number of ISs, population diversity index (using Shannon H index).
Normalized integration frequency by gene. Pair-wise comparison of gene normalized integration frequency for patients with MLD, WAS and β-Thal, after homogeneous subsampling and randomization. For each gene (column GeneName) and pair-wise comparison (column Comparison) we reported the average number of ISs for study and the final corrected P value (column corrected p-value avg).
Estimated number of active HSPC. For each available patient, the table reports the number of estimated HSPCs observed at early time points (less than 24 months) and from 24 months (column Pop. Size >24 m) calculated with the Chao1 model and corrected with the VCN (if VCN > 1). We also reported the number of IS retrieved less than 24 months and more than 24 months, and the number of shared IS between the two sets. The last column reports the fraction of the estimated HSPCs greater than 12 months on the total number of observed IS more than 24 months.
Mean depth coverage of somatic mutations data. The table shows the mean depth coverage reached for each patient with MLD and β-Thal coming from the Illumina’s AmpliSeq Myeloid Panel Targeted exome sequencing used for the somatic mutations analysis. Columns report: Disease, Patient ID, time point in days of the sample and mean depth coverage.
Somatic mutations. The table presents all the somatic mutations for each patient with MLD and β-Thal coming from the Illumina’s AmpliSeq Myeloid Panel Targeted exome sequencing. Columns report: Disease, Patient ID, time point in days of the sample, cell type, total number of reads in the position of the mutation, number of reads supporting the reference variant, number of reads supporting the mutated variance, VAF, chromosome, starting position, ending position, nucleotide reported by the reference, nucleotide reported by the mutation, functional annotation of the position (intron, exon), gene, exon modification, aminoacidic change, effect of the mutation reported by ClinVar, frequency of the mutation in the population reported by gnomAD and ID of the mutation reported by avsnp147.
Primers used for the first (Exponential) PCR. A list of primers used for the first exponential PCR with details about name, type and sequence.
Primers used for the second (Fusion) PCR. List of primers used for the second fusion PCR with details about name, type, sequence and barcode (eight nucleotides) of each primer.
Linker cassette sequences. The linker cassette sequences used in the SLiM-PCR procedure. Each linker cassette was generated by the annealing of the short oligo with a long oligo. The sequence of each oligo and its barcode (eight nucleotides) is reported.
Data Availability Statement
Sequencing data of mutations have been deposited into the Sequencing Read Archive (NCBI SRA) with the BioProject PRJNA1150995. IS data have been deposited in GitHub at the following project archive: https://github.com/calabrialab/Code_HSPCdynamics. Source data are provided with this paper.
We released our source code in GitHub (https://github.com/calabrialab/Code_HSPCdynamics).