

Abstract
There is an increased incidence of autism among the children of women who take the anti-epileptic, mood-stabilizing drug, valproic acid (VPA) during pregnancy; moreover, exposure to VPA in utero causes autistic-like symptoms in rodents and non-human primates. Analysis of RNA-seq data obtained from E12.5 fetal mouse brains 3 hours after VPA administration to the pregnant dam revealed that VPA rapidly and significantly increased or decreased the expression of approximately 7,300 genes. No significant sex differences in VPA-induced gene expression were observed. Expression of 399 autism risk genes was significantly altered by VPA as was expression of 258 genes that have been reported to modulate fetal brain development but are not otherwise linked to autism. Expression of genes associated with intracellular signaling pathways, neurogenesis, and excitation-inhibition balance as well as synaptogenesis, neuronal fate determination, axon and dendritic development, neuroinflammation, circadian rhythms, and epigenetic modulation of gene expression was dysregulated by VPA. Notably, at least 40 genes that are known to regulate embryonic neurogenesis were dysregulated by VPA. The goal of this study was to identify mouse genes that are: (a) significantly up- or downregulated by VPA in the fetal brain and (b) associated with autism and/or known to play a role in embryonic neurodevelopmental processes, perturbation of which has the potential to alter brain connectivity and, consequently behavior, in the adult. The genes meeting these criteria provide potential targets for future hypothesis-driven studies to elucidate the proximal causes of errors in brain connectivity underlying neurodevelopmental disorders such as autism.
Subject terms: Molecular neuroscience, Genomics
Introduction
Autism is a neurodevelopmental disorder (NDD) characterized by deficits in social interactions including language, and repetitive, stereotyped behavior with restricted interests [1]. Autistic individuals may also display an increased incidence of intellectual disability (ID), anxiety and seizures. The reported incidence of autism has increased dramatically over the last two decades [2], with some estimates now as high as 1:44 [3]. The severity of autism varies widely, ranging from high-functioning with minimal disability, to severely afflicted, where aggressive, self-destructive repetitive behaviors pose a threat to the safety of the patient, caregivers, and family. The term “autism spectrum disorder” (ASD) reflects this symptomatic variability. The neurological mechanisms underlying ASDs are not understood nor is it known whether autism is a single disorder or multiple disorders sharing common core features. It is generally assumed that dysregulation of one or more genes, due to either genetic variants or exposure to environmental stressors, underlies the abnormal development of the autistic brain.
The diagnosis of autism typically is made around two years of age when the child fails to meet normal milestones for social development and language. However, it has become increasingly evident that autism arises before birth [4, 5], e.g., infants destined for an autism diagnosis fail to show normal attention to faces [6] and machine learning analysis of gestational and perinatal biomarkers can predict ASD years later [7]. Some evidence indicates that neuronal hyperplasia in autism, possibly due to dysregulation of neurogenesis, arises during late gestation [8] or during delivery [9]. Epidemiological studies of folate supplementation also indicate that the onset of autism occurs during early fetal development [10].
Twin studies have revealed that 60 to 88% of autism cases are inherited [11, 12], however, many cases have been linked to in utero exposure to environmental factors such as pharmaceuticals, air pollution, insecticides, and maternal infection [13, 14]. For example, exposure to the anti-epileptic, mood-stabilizing drug, valproic acid (VPA; Depakote®) [15] or the organophosphate insecticide, chlorpyrifos [16], increases the incidence of autism and other NDDs in the offspring of women who are exposed during pregnancy. Other environmental causes of autism include maternal immune activation (MIA) [17–19], extremely pre-term birth and delivery by cesarean section [20, 21].
Studying the etiology of autism in humans is limited to epidemiological approaches implicating genetic variants (single nucleotide polymorphisms, copy number variants, and single-gene syndromic mutations). The Simons Foundation Autism Research Initiative (SFARI) has compiled a list of 1115 human genes (https://gene.sfari.org/database/human-gene/), referred to as the “SFARI List” below, for which there is evidence for association with autism based on genome-wide association studies (GWAS).
Hypothesis-driven experiments to determine cause and effect need to be done in animal models, which display behaviors that are remarkably similar to the core symptoms of autism [22–24]. These autistic-like behaviors have been reported in transgenic animals lacking ASD-linked genes such as Cntnap2 and Shank2 [25, 26], leading to the widespread use of these transgenic mice as animal models [27, 28]. Induction of MIA or systemic administration of VPA or chlorpyrifos in genetically normal pregnant rodents also leads to increased autistic-like behaviors in their offspring [18, 23, 24]. Unlike transgenic mouse models, these environmental toxicity models of autism provide the opportunity to control the timing of exposure of the fetus to the toxic stressor, as will be discussed below. Gene expression analyses of the brains of mice in the VPA and MIA models have been conducted using RNA microarray analysis or next-generation RNA sequencing (RNA-seq). In most of those animal studies, the fetus was exposed to the environmental stressor during mid-gestation, but RNA expression was analyzed in postnatal or adult animals (e.g., see refs. [29–33]).
There are two competing, but not mutually exclusive, theories about the role of differential gene expression in autism. In one, abnormal gene expression in the child or adult at the time of behavioral testing, is responsible for autistic-like behavior (c.f., refs. [34–36].). The second posits that abnormal gene expression, due to genomic variants or in utero environmental insults, interferes with one or more critical steps in the “program” controlling fetal brain development. Such errors could carry forward throughout life leading to a brain with subtle anatomical or connectivity defects that underlie the abnormal development of an autistic brain. This has been referred to as a “presymptomatic signature” [37]. The present study focused on the second hypothesis by examining altered gene expression in fetal brains when the environmental stressor (VPA) is still present. Meng et al. [38]. have taken an analogous approach, identifying genes that are dysregulated by long-term VPA treatment of human forebrain organoids in vitro and that are also linked to autism.
The goal of the present study was to address this question using the VPA mouse model in which pregnant mice receive a single i.p. injection of VPA at gestational day 12.5 (E12.5). Fetal brains were processed for RNA-seq three hours after VPA administration; male and female fetal brains were analyzed separately. Pharmacokinetic studies have demonstrated that injected VPA dissipates within 3−5 hours due to metabolism of the drug by the maternal liver [39, 40]; VPA levels in the fetal brain follow a similar trajectory [41, 42]. Consequently, with this protocol, the fetal brain receives a brief, transient exposure to VPA at a critical time for early brain development and VPA-induced molecular events occurring on E12.5 are both necessary and sufficient for the autistic-like behaviors in VPA-treated animals assessed 5 − 6 weeks later.
The reported incidence of autism is about 4-times higher in males than in females [43]. Sex differences in neural function are generally considered to be mediated by sex hormones acting from late prenatal brain development through to the adult. Since the time of VPA administration in the present study (E12.5) is just prior to the maturation of gonads and production of sex hormones [44], any sex differences in gene expression likely would be due to sexually-dimorphic gene expression rather than to hormonal effects.
Materials and methods
Timed-pregnant C57BL6 mice were generated by the University of Maryland School of Medicine Division of Veterinary Resources. VPA was obtained from Sigma.
One male was paired overnight with two females; the day of separation was designated E0.5. On E12.5 pregnant females received i.p injections of VPA (400 mg/kg) in sterile saline or saline alone. 7 pregnant females received VPA and 7 received saline. Three hours after VPA administration, pregnant females were euthanized by cervical dislocation and decapitation. Fetuses were transferred to ice-cold saline, decapitated and the entire brain was removed to Trizol and disrupted for 60 sec with a Bead Beater using 0.2 mm beads. The 105 individual fetal brain samples were frozen on dry ice and stored at −80 °C prior to RNA extraction and quality control (RIN = 10 for all samples).
Sex was determined by analyzing Sry and Gapdh RNA derived from the torso of each fetus by PCR. Males were identified by the presence of both Sry and Gapdh; Gapdh but not Sry RNA was expressed in females [45]. One male and one female brain from each of the 7 pregnancies at each condition were randomly selected and processed for next-generation RNA-seq by the University of Maryland Institute for Genome Sciences.
Libraries were prepared from 25 ng of RNA using the NEB Ultra II Directional RNA kit. Samples were sequenced on an Illumina NovaSeq 6000 with a 150 bp paired-end read configuration. The quality of sequences was evaluated by using FastQC [46]. The alignment was performed using HiSat (version HISAT2-2.0.4) [47] and Mus musculus GRCm38 as reference genome and annotation (version 102). Aligned bam files were used to determine the number of reads by gene using HTSeq [48]. On average, we sequenced 187,000,000 reads. 96.6% of them properly mapped the reference: 87% mapped exons, 2.4% mapped introns and the remaining 10.6% mapped intergenic regions.
Differential gene expression between mice treated with VPA and saline controls was conducted using DESeq2 [49], which models gene counts using the negative binomial distribution. P-values were corrected for false discovery rate (FDR) using the Benjamini-Hochberg procedure; pFDR ≤ 0.025 and log2-fold change ≥0.32 were used as the criteria for significance. Differentially expressed genes due to VPA treatment were tested for enrichment in autism risk genes (SFARI List) using Fisher’s Exact Test. Significance of sex differences in FC was analyzed by 2-way ANOVA using the Limma-Voom tool [50].
Results
RNA-seq
19,721 individual genes were analyzed in fetal mouse brain by RNA-seq. Male and female brains were analyzed separately. The raw data showing base counts for each gene in each of seven independent fetal brain samples of each sex, each from a different pregnancy, are shown in Supplemental Table S1; i.e., n = 7 fetal brains from 7 different pregnancies for each sex/condition. Only genes with mean base counts ≥100 were analyzed further. Genes that increased or decreased in response to VPA by <1.25-fold and >−1.25-fold and those with adjusted p-values (pFDR) > 0.025 were also filtered out; a gene was included if VPA increased or decreased its expression by ≤1.25-fold and ≥−1.25-fold in only one sex. Shown in Supplemental Table S2 are the 6516 genes significantly affected by VPA (pFDR ≤ 0.025) in both sexes, 498 in females only and 280 in males only (listed in three separate sections in Table S2). The apparent sex differences were due to variability in the replicates of one of the sexes (i.e., pFDR > 0.025).
Throughout this report, a positive fold-change (FC = ratio of VPA to control gene expression –V/C) indicates that VPA increased gene expression; a negative FC indicates that VPA decreased expression. For example, FC = −3.0 indicates that V/C = 0.33, a 67% reduction by VPA.
Curating the mouse genes dysregulated by VPA in the fetal brain
The 7294 genes that were significantly altered by VPA (Table S2) were further analyzed in two ways. First, they were merged with the genes on the SFARI List identifying 399 common genes (Table S3). It should be noted that inclusion on the SFARI List is based on GWAS; in many cases, a mechanistic role in neither brain development nor the etiology of autism is known and the validity of relying on such associations to explain the genetic underpinnings of human disease has been questioned [51, 52]. Importantly, in this report, genes on the SFARI List (based on GWAS) that are dysregulated by VPA are considered further if there is independent evidence in the literature that those genes play a role in embryonic brain development. Thus, only 67 genes on the SFARI List are discussed in this report.
Second, we identified 258 genes (Table S4) significantly altered by VPA, not on the list of SFARI risk genes but having a documented role in brain development including intracellular signaling, neurogenesis, and excitation-inhibition balance as well as 15 additional categories (c.f., Table 3 and Supplemental Discussion). As will be discussed below, although these 258 genes have not been associated with autism in GWAS, it is plausible that changes (either positive or negative) in the expression of their gene products in the fetal brain might adversely affect the trajectory of brain development leaving a permanent signature on brain structure and connectivity leading to autistic-like behavior in the adult.
Table 3.
Biological roles of 205 different genes that are dysregulated by VPA in the fetal brain and associated with autism or prenatal neurodevelopmental processes (from Tables S3 and S4).

Note that some genes are included in more than one category. The number in parentheses shows the FC induced by VPA (mean of males and females); red text indicates that VPA increased gene expression The basis for this categorization, with references, is given in the discussion above and in the Supplemental Discussion.
No significant sexually-dimorphic effects of VPA
An initial expectation was that one or more of these genes would exhibit sexually-dimorphic dysregulation by VPA, thereby suggesting a plausible underlying mechanism for the sex difference in the incidence of ASDs. Figure 1 is a plot of the log2 fold-change (FC) of males versus females for the 657 genes in Tables S3 and S4. Deviations from the regression line indicate possible sex differences; however, in no case was the effect of VPA found to be significantly different between males and females. 21 genes (magenta) were upregulated by >5-fold or downregulated >80% by VPA (average of males and females).
Fig. 1. Log2 fold-change (FC) induced by fetal VPA exposure plotted as females versus males for the 657 genes in Tables S3 and S4.
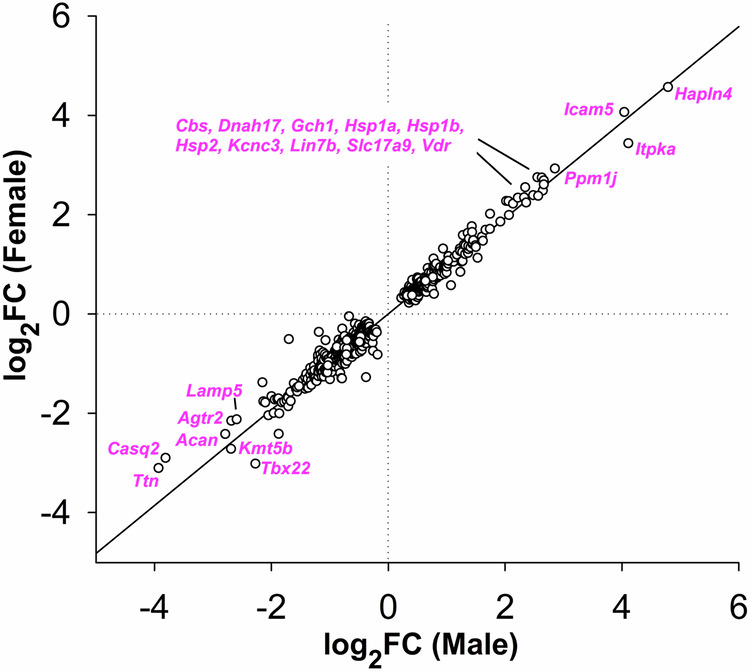
Deviations from the regression line (r2 = 0.973) indicate possible sex differences. The gap in the data reflects the ±1.25-fold FC cutoff. 21 genes (magenta) were upregulated by > 5-fold or downregulated >80% by VPA (average of males and females).
Limitations
The results reported here are restricted to VPA-induced changes in RNA levels; the extent to which these changes reflect commensurate changes in the expression of the proteins encoded by these genes is not known.
These results represent VPA-induced changes in gene expression at a single point in time (3 hr after VPA administration on E12.5). We do not know what genes would be dysregulated by VPA administered on other gestational days; however, earlier (E9) or later (E14.5) administration of VPA in mice produced no behavioral outcomes [53]. Whether the changes reported here are transient and reverse as the VPA levels dissipate, as has been reported for Bdnf [54], or are persistent, is not known.
There are many genes for which expression was increased or decreased by ≥1.25-fold or ≤−1.25-fold in response to VPA but the FDR-corrected p-value did not reach the ≤ 0.025 criterion applied in this study. Higher statistical power would be needed to determine whether these changes are significant.
The ±1.25 FC cutoff used here is arbitrary; it is possible that smaller changes in the expression of one or more critical genes could have profound effects on fetal brain development and these would not be identified in this analysis.
Only a fraction of the approximately 7300 genes whose expression is significantly affected by VPA were curated in this study. It is likely that other genes, in addition to those shown in Table 3, could play a role in mediating the effects of VPA on fetal brain development.
Caution is required before extrapolating these results from the rodent model to human autism. The greatly extended gestation and much higher complexity of the human brain provide many vulnerabilities to environmental insults not present in the mouse. Moreover, rodent behavior in the VPA model does not recapitulate the wide heterogeneity of behavioral symptoms in autistic individuals.
Reproducibility
The stimulation by VPA of Bdnf expression (Table S4) has been independently confirmed by quantitative RT-PCR [45, 54].
By separately analyzing gene expression in male and female brains, the results of this study were effectively replicated with independent biological samples (7 of each sex ± VPA, each from a different pregnancy). With very few exceptions, male and female gene expression levels were similar (c.f., Fig. 1), demonstrating reproducibility across independent data sets.
Discussion
The underlying hypothesis for this research is that abnormal connections within and among multiple brain regions contribute to NDDs such as ASDs. This abnormal connectivity is caused by dysregulation of one or more of the genes comprising the genetic program that establishes the complex neuronal networks underlying cognition and behavior [4, 55]. Although these networks are refined during late prenatal and postnatal brain development, it is likely that certain early prenatal developmental epochs are particularly vulnerable to alterations in the expression of genes controlling the neurodevelopmental program. Ben-Ari and Spitzer [56] have suggested that multiple “phenotypic checkpoints” in the fetal brain, which are necessary for normal circuit development, are vulnerable to environmental and genetic disruptions.
Although it is by no means clear that the fetal rodent brain accurately recapitulates the autistic human brain during prenatal development, the VPA model enables experimental control of the timing of exposure to an environmental factor that causes abnormal behavior resembling autism. In the present experimental paradigm, VPA is administered to the pregnant dam at E12.5, a time when multiple early neurodevelopmental events critical for brain organization and connectivity are occurring. These include proliferation of neural progenitors (NPs), specification of the neuronal fate of these NPs, migration of newborn neurons away from the proliferative ventricular zones, differentiation of NPs to establish a mature neuronal phenotype, extension and branching of axons and dendrites and establishment of synaptic connections. The goal of this study was to identify genes that are:
-
a. significantly up- or downregulated by VPA in the fetal mouse brain (Table S2)
AND
b. linked to autism in GWAS (Table S3) or known to play a role in embryonic neurodevelopmental processes, perturbation of which has the potential to alter brain connectivity in the postnatal and adult brain (Table S4).
The set of genes meeting these criteria would provide potential targets for future hypothesis-driven approaches to understanding the underlying proximal causes of defective brain connectivity in NDDs such as autism.
It was not practical to vet each of the nearly 7300 VPA-dysregulated genes for a potential role in abnormal brain development. However, as described below and in the Supplemental Discussion, the 258 genes in Table S4 and at least 67 of those in Table S3 have been reported to regulate specific aspects of fetal brain development. Consequently, it is plausible that the up- or downregulation of one or more of those genes could contribute to abnormal connectivity in subjects with NDDs. This could be tested in animals by determining the effect of manipulating the expression of individual (or combinations of) genes in the fetal brain on subsequent behavior.
A common subset of genes in GWAS and the present study
Several GWAS have identified genes linked to autism (see SFARI List). Here we tabulated the findings of four GWAS studies [57–60] and one report categorizing autism risk genes potentially involved in neurogenesis [61]. As shown in Table 1, many of the genes identified in these reports are also dysregulated by VPA in the fetal mouse brain (Tables S3 and S4). Genes discussed in the respective papers are listed in each column in Table 1; genes that are also dysregulated by VPA in the present study are shown in bold. Five VPA-regulated genes in Tables S3 and S4 that also appear in all five of the published reports are in red. (Arid1b, Dyrk1a, Pogz, Pten, Tbr1). Six genes in Tables S3 and S4 that also appear in four of the five other studies are in blue. (Adnp, Ash1l, Chd2, Kmt5b, Tcf7l2, Wac). There were four genes (yellow) identified in all five previously published studies that were not affected by VPA in the fetal mouse brain. Chd2, which is listed in three of the four published studies, and Chd3, (50% reduced by VPA), are both SFARI genes and have been reported to have similar functions to Chd8. Of the 11 red and blue genes in Table 1, three (Adnp, Dyrk1a, Pten) appear in Table 2 as VPA-regulated, autism-associated genes involved with the structural stability of neurons [62] (see below).
Table 1.

Genes dysregulated by VPA in fetal brain in the present study (Tables S2 and S3) are indicated in bold. The percentage of VPA-regulated genes is shown for each study. Red: genes identified in all five published papers and were affected by VPA the present study; blue: genes identified in four of the five published papers and were affected by VPA in the present study; yellow: autism-associated genes identified in all five published papers (Ank2, Chd8, Foxp1, Syngap1) but were not affected by VPA in the present study.
Table 2.
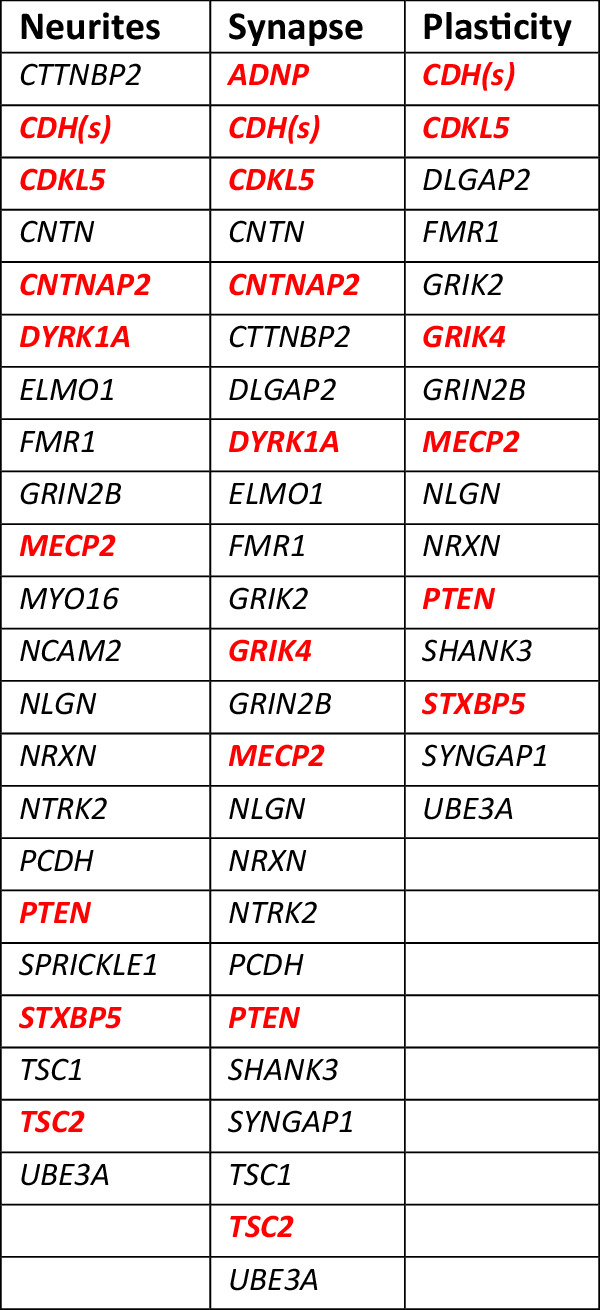
Genes linked to autism in GWAS that have also been reported to regulate “neurite outgrowth”, “synapse/spine formation”, or “synaptic plasticity” [62].
VPA dysregulation of high-confidence (hc)ASD genes in mid-fetal layer 5/6 cortical projection neurons
Willsey et al. [63]. identified 9 hcASD genes that are expressed in layer 5/6 projection neurons in the fetal human brain. Three of these genes (Dyrk1a, Pogz, Tbr1) were downregulated by VPA in this study (Table S3). The authors used the nine hcASD genes as seed genes for co-expression network analysis, which revealed 10 probable ASD genes, of which two (Bcl11a, Nfia) were downregulated and one (Aph1a) was upregulated by VPA (Table S4). All of these genes are on the SFARI List (Table S3) and the three seed genes were identified in all 5 reports analyzed in this study (Table 1).
Autism-associated genes and structural stability of neurons
Lin et al. [62]. tabulated genes linked to autism in GWAS that have also been reported to be involved in the structural stability of neurons. These genes were further sorted into three categories, viz., “neurite outgrowth”, “spine/synapse formation” and “synaptic plasticity”. 61 genes were assigned to these three categories although some genes appeared in two or all three, resulting in 29 different genes among the three categories (Table 2). Shown in red bold are 10 of these genes (35%) that were significantly up- or downregulated by VPA (i.e., in Table S2). Dysregulation of one or more of these genes (due to gene variants or fetal exposure to VPA) could alter the structure and function of synapses throughout brain development.
Throughout this report, the change in gene expression induced by VPA in the fetal brain at E12.5 (expressed as fold-change) is shown in parentheses (mean of males and females) when first mentioned.
Examination of Tables 1 and 2 reveals that Pten and Dyrk1a are dysregulated by VPA and are common to both the Lin et al. study [62] and to all five of the GWAS [57–61]. Pten (+1.4), a negative regulator of the PI3K-AKT-mTor signaling pathway, has a particularly strong association with autism, appearing in all five GWAS (Table 1) and is associated with the structural stability of neurons (Table 2). Dyrk1a (−1.5), is overexpressed in Down syndrome but, as explained in the Supplemental Discussion, haploinsufficiency of Dyrk1a is a cause of ID and ASD. Consequently, these two genes may be good candidates for investigating the molecular basis for the proximal causes of autism.
Biological roles of genes dysregulated by VPA
Table 3 lists 205 different genes, categorized by biological function, that were significantly dysregulated by VPA in the E12.5 fetal brain and have been reported to be involved in fetal brain development. Many of the genes affected by VPA are components of intracellular signaling pathways and, consequently, could mediate a wide variety of developmental processes. Some examples of these processes are discussed in the following sections (“Intracellular Signaling”, “Neurogenesis”, “Excitation-Inhibition”). The rationale for inclusion of the genes in the remaining 15 categories together with supporting references is provided in Supplemental Discussion.
Intracellular signaling
As summarized in Fig. 2, the genes encoding at least 10 members of the canonical signaling pathways downstream from receptor tyrosine kinase receptors are significantly upregulated by VPA in the fetal brain (green); four are downregulated (red).
Fig. 2. Schematic diagram of multiple signaling pathways downstream from receptor tyrosine kinases (RTKs), which mediate the action of ligands active in the developing brain.

The two subunits of the RTKs are trans-autophosphorylated upon activation by extracellular ligand, with the phosphorylated tyrosines serving as docking sites, which initiate downstream signaling. Components in red are significantly downregulated by VPA in the fetal brain; those in green are upregulated by VPA.
Fifteen additional genes associated with intracellular signaling are included in Table 3. Thus, abnormal signaling via these pathways is a potential mechanism by which VPA could interfere with normal brain development. Meta-analysis of GWAS and copy number variant studies of autism-related genes revealed that three signaling networks, regulating steroidogenesis, neurite outgrowth and excitatory synaptic function, were enriched [64]; A-kinase anchoring proteins (AKAPs) functionally integrate signaling cascades within and among these networks. VPA altered expression of both Akap8 (−1.3) and Akap8l (+1.6) in fetal mouse brains.
Dyrk1b (−2.2) a mediator of double-stranded DNA break repair [65], regulates Hedgehog signaling by activating the mTor/AKT pathway [66, 67]. Dyrk1b has been linked to metabolic syndrome and autism [68]. The 55% reduction in Dyrk1b expression induced by VPA could contribute to autistic-like behavior by dysregulating Hedgehog or mTor/AKT signaling.
Ppp1r1b (+3.7), encodes the dopamine- and cAMP-regulated neuronal phosphoprotein (DARPP-32) which amplifies and/or mediates many actions of cyclic AMP-dependent protein kinase at the plasma membrane and in the cytoplasm, and has a broad spectrum of potential targets and functions [69].
Intracellular Ca2+ signaling plays a critical role in generating neuronal diversity by determining neurotransmitter phenotype, dendritic morphology and axon growth and guidance in the embryonic brain [70]. The effects of fetal VPA exposure on multiple components of Ca2+ signaling are considered in the Supplemental Discussion.
Neurogenesis
Excitatory neurons in the mammalian cortex are generated from proliferating radial glia cells (RGCs), which undergo mitosis at the ventricular surface to expand the pool of neuroblasts and consequently determine the final number of neurons [71]. Neurogenesis proceeds according to a “program” that is tightly-regulated by intercellular signals from a variety of brain cells as well as the meninges. Disturbances in this regulation, for example by environmental factors such as VPA or gene mutations, have the potential to alter the neuronal population and configuration of the developing brain. An example of this disruption is the effect of VPA exposure at E12.5 on the number of neocortical neurons born on E14.5 measured one day before birth (E18.5) [72]. Thus, gene expression changes on E12.5 alter neurogenesis at E14.5, long after the VPA has dissipated. As discussed below, at least 40 genes associated with neurogenesis are dysregulated by VPA in the fetal brain.
Proteins encoded by the Runx genes regulate the transition from proliferation to differentiation in the developing nervous system [73]. Runx1 (−3.1) and Runx2 (−3.9) expression was reduced 68% and 74% by VPA in the fetal brain, potentially leading to a reduction in the generation of post-mitotic neurons.
Retinoic acid (RA) plays a number of roles in the regulation of cortical neurogenesis [74, 75]. RA is produced in the dorsal forebrain meninges and acts at receptors on the end-feet of RGCs [76]. The latter study used a transgenic mouse with reduced expression of Foxc1 (−1.9) leading to defects in forebrain meningeal formation and decreased neuron progenitor production due to failure of proliferating RGCs to exit the cell cycle. Decreased Foxc1 production would be predicted to lead to a delay in the generation of post-mitotic neurons and consequently, an expansion of the proliferating RGC neuroprogenitor pool.
Downregulation of Nr2f1 (−1.8), Sfrp2 (−1.8) and C3ar1 (−3.8) would be predicted to influence the balance between proliferation and neurogenesis [77, 78]. Multiple studies have implicated Eomes (Tbr2) (−1.8) in the regulation of cortical neurogenesis [79–84], while Neurog1 (−2.2) has been reported to play a role as a negative regulator of cortical neurogenesis [85].
Sulliman-Lavie et al. [86]. reported that Pogz (−2.2) is a negative regulator of transcription and Pogz deficiency upregulates expression of genes associated with ASD resulting in disruption of embryonic neurogenesis; Pogz was found to be associated with autism in all five GWAS (Table 1). The conversion from symmetrical to asymmetrical mitoses associated with the generation of post-mitotic neurons is regulated by the Plk1 (−1.7) – Lrrk1 (−2.9) – Cdk5rap2 (−1.3) cascade [87]. Several members of the heterogeneous nuclear ribonucleoprotein family have been linked to NDDs including Hnrnph1 (−1.6), Hnrnpk (−1.3), Hnrnpu (−1.6); expression of these genes in RGs has been reported to be necessary for normal neurodevelopment [88].
Comparison of the mechanisms controlling neurogenesis in rodents and humans has revealed that gyrification of the human brain is driven by changes in the expression of regulatory genes and growth factors [89]. Stahl et al. [90] reported that, over the period E12 to E16, decreased expression of the DNA-associated protein, Trnp1 (+1.5), leads to radial expansion of the cortex and the appearance of gyri-like folding of the mouse brain. In the present study, VPA induced a 50% increase in the expression of Trnp1 in the E12.5 fetal mouse brain which would be predicted to inhibit or delay the shift to radial growth of the cortex. Fibroblast growth factor 2 (FGF2) has also been reported to induce gyrification in the mouse brain, having an effect opposite to that of Trnp1 [91]. In the present study, VPA caused a 65% reduction on the expression of Fgf2 (−2.9). These findings together with the present results lead to a hypothesis that increased Trnp1 and decreased Fgf2 expression, induced by VPA in the fetal brain at E12.5, cause a delayed shift in radial growth.
WNT/β-catenin signaling plays several roles in regulating neurogenesis in the developing neocortex [92]. Wnt3a (−3.8) regulates the timing of differentiation of neocortical intermediate progenitors into neurons [93]. The 74% reduction in Wnt3a expression induced by VPA has the potential to disrupt the normal timing of cortical neurogenesis in the fetal brain. Zinc finger and BTB domain-containing 16 [Zbtb16 (−1.8)] has been reported to regulate cortical neurogenesis [94] and Zbtb16 knockout mice display a thinning of neocortical layer 6 and a reduction of TBR1-expressing neurons as well as increased dendritic spines and microglia [95]. Haploinsufficiency of Setbp1 (−3.2) reduces WNT/β-catenin signaling, impairs neurogenesis, reduces acquisition of ventral forebrain fate, and causes ID and ASD traits [96–98].
Cortical size is regulated, in part, by BAF-170, [encoded by Smarcc2 (−1.6)], a subunit of the chromatin remodeling complex, mSWI/SNF. Deletion of Smarcc2 in mouse increases the pool of intermediate progenitors resulting in an enlarged cortex [99]; de novo variants in Smarcc2 cause a syndrome with developmental delay and ID [100]. Mutation of a different BAF subunit, Arid1b (−2.2), alters the production of neuronal precursor cells in human cerebral organoids [84] and mutation of 36 high-risk ASD genes altered the fate of dorsal intermediate progenitors, ventral progenitors, and upper-layer excitatory neurons [84]. Expression of 18 of those genes (Adnp, Arid1b, Ash1l, Asxl3, Baz2b, Bcl11a, Chd2, Ddx3x, Irf2bpl, Kat2b, Kdm6b, Kmt5b, Mecp2, Pogz, Srcap, Tbl1xr1, Tbr1 and Wac) is dysregulated by VPA at E12.5 (c.f., Tables S3 and S4). Loss of Wdfy3 (−2.0) leads to overproduction of cortical neurons, a feature reported in autism [4], as well as neuronal migration defects [101].
Excitation-Inhibition
A widely discussed hypothesis is that autism is caused by an imbalance between neuronal excitation and inhibition due to increased excitation, reduced inhibition, or both [77, 102, 103]. Consequently, alterations in the developmental program in the fetal brain that result in too many excitatory (glutamatergic) or too few inhibitory (GABAergic) neurons (or synapses) could contribute to autistic behavior. Camk2a (+1.9), which is associated with excitatory synapses, is upregulated by 90% in response to VPA; CAMK2A is upregulated in the ASD superior temporal gyrus [104]. Two related genes, Maf (−1.6) and Mafb (−1.6), which are both downregulated by about 40% by VPA, have been reported to play redundant roles in the generation of interneurons from the fetal median ganglionic eminence; their deletion results in decreased numbers of cortical somatostatin-releasing, inhibitory interneurons [105]. Gad1 (−2.5) and Gad2 (−3.3) encode isoforms of the enzyme that synthesizes the inhibitory transmitter, GABA. VPA reduced Gad1 and Gad2 levels by 60% and 70%, respectively; GAD1 and GAD2 are downregulated in the superior temporal gyrus of ASD patients [104]. Insyn1 (−1.4) encodes a component of the dystroglycan complex at inhibitory synapses; loss of Insyn1 alters the composition of the GABAergic synapses, excitatory/inhibitory balance, and cognitive behavior [106, 107]. Using cortical assembloids together with CRISPR screening to analyze interneuron generation and migration, Meng et al. [108]. found that Csde1 (−2.0) is required for interneuron generation. Taken together, these findings are consistent with VPA increasing net brain excitation by decreasing the number of inhibitory interneurons generated during fetal brain development. These reductions in gene expression may contribute to a decrease in inhibition in mice exposed to VPA in utero [109] Nf1 (−1.3) encodes NF1, which is mutated in neurofibromatosis type 1, an inherited neurocutaneous disorder associated with NDDs including autism. Nf1 deletion results in the specific loss of parvalbumin-expressing inhibitory cortical interneurons [110]. In the present study, Nf1 was downregulated by about 25% in fetal mouse brains exposed to VPA. Semaphorin-4a [Sema4a (−1.6)] and Semaphorin-4d [Sema4d (+1.9)], promote inhibitory synapse development via the postsynaptic Plexin-B1 receptor encoded by Plxnb1 (−1.8) [111].
Hapln1 (−3.7) and Hapln4 (+25.7), encode extracellular matrix (ECM) proteins that are components of perineuronal nets (PNNs) [112, 113]. Ramsaran et al. [114]. reported that Hapln1 mediates the functional maturation of hippocampal parvalbumin interneurons through assembly of PNNs; this mechanism mediates the development of memory precision during early childhood. Hapln4 is a selective regulator of the formation of inhibitory GABAergic synapses between Purkinje and deep cerebellar nuclei neurons [115]; the cerebellum is one of many brain regions implicated in the etiology of autism [116]. The 70% decrease in Hapln1 expression and the massive, 26-fold increase in Hapln4 induced by VPA would be expected to alter PNN density, which could disrupt the developing connectivity of neuronal circuitry in the fetal brain. Aggrecan [Acan (−6.1)] is another major component of the ECM and PNNs that modulates the development of synaptic plasticity in the visual system (see “Synaptogenesis” in the Supplemental Discussion). Its role, if any, in the development of inhibitory neurons is not known.
Increased electrical activity has been shown to be an important regulator of neuroblast proliferation, neuronal migration and differentiation, and axon pathfinding [117]. While GABA is an inhibitory transmitter in the mature nervous system, it is excitatory early in development prior to a shift in the Cl− reversal potential during the late embryonic and perinatal periods [118]. This shift, which is mediated by oxytocin, is abolished in mice exposed to VPA in utero, leading to autistic-like behavior [119]; moreover, the diuretic bumetanide, which accelerates the shift, has been shown to be an effective treatment for autism [120] and to reverse the behavioral effects of embryonic VPA treatment in rodents [119]. While the gene encoding oxytocin (Oxt) was unaffected by VPA in the present study, genes encoding four isoforms of the GABAA receptor [Gabra1 (−1.7), Gabra2 (−1.7), Gabra4 (+2.5), Gabrq (+3.6)] are dysregulated by VPA in the fetal brain.
Summary
The approach for this study can be described as “hypothesis generating” in that we began without any preconceived idea (hypothesis) as to the mechanism by which a brief, transient dose of VPA, administered to the pregnant mouse at E12.5, can cause abnormal, autistic-like behavior in her offspring many weeks later. The analysis identified approximately 7300 genes, expression of which was significantly affected by VPA. Of those, we then identified genes that were a) also linked to autism by GWAS (SFARI List; Table S3) or b) not on the SFARI List but known to play a role in critical steps of prenatal brain development (Table S4). Interference with one or more of these steps in the fetal brain has the potential to interfere with the “program” directing brain development to create a persistent pathological signature that leads to abnormal neuronal circuitry in the adult, long after the increase in VPA has dissipated. Of those 657 genes, about half have known mechanisms of action associated with brain development or function, of which 205 (Table 3) are discussed below and in the Supplemental Discussion.
Genes affected by VPA in the fetal mouse brain appear in multiple GWAS studies (c.f., Tables 1 and 2) including Adnp, Arid1b, Ash1l, Cdh2, Cdk5l, Dyrk1a, Kmt5b, Mecp2, Nr2f1, Pogz, Pten, Tbr1, Tcf7l2, and Wac, moreover, nine of these genes were implicated in neurogenesis and cell fate determination in the embryonic brain [84]. This “short list” is a potential starting point for future hypothesis-driven studies to determine whether dysregulation of one or more of these genes by VPA is a proximal cause of the behavioral abnormalities identified in juvenile and adult animals. At least 20 genes encoding components of intracellular signaling pathways are dysregulated by VPA in the E12.5 brain (Fig. 2); disruption of these signaling pathways also has the potential to disrupt multiple developmental processes and may contribute to the autistic-like behavior induced by VPA. Considering that neurogenesis and the fate determination of excitatory and inhibitory neurons are occurring in the E12.5 mouse brain, genes involved in the regulation of these processes that are dysregulated by VPA (c.f., Table 3) are of particular interest. Nevertheless, it remains possible that dysregulation of one or more of the other genes altered by VPA contributes to the proximal cause of the autistic-like behavior.
Supplementary information
Acknowledgements
This research was supported by NIH grant R21ES130137 to BKK. We thank Tom Abrams, Edna Pereira Albuquerque, Brad Alger, Seth Ament, Tarik Haydar, and Alex Poulopoulos for helpful discussions and comments on the manuscript.
Author contributions
BKK and SGD conceived the research plan; BKK and MVL harvested fetal brains for analysis; MVL performed qPCR to determine fetal sex; EM performed bioinformatic analysis of the data; BKK performed literature searches to curate relevant genes; BKK, EM and SGD wrote and edited the manuscript.
Competing interests
The authors declare no competing interests.
Data deposition
The data analyzed in this study have been deposited in the NEMO Archive (RRID:SCR_002001) under identifier nemo:dat-nxjarjg accessible at https://assets.nemoarchive.org/dat-nxjarjg.
Ethics approval
All animal procedures were approved by the University of Maryland Baltimore, Institutional Animal Care and Use Committee (IACUC animal use protocol # 0419006).
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41398-024-03179-1.
References
- 1.American Psychiatric Association. Diagnostic and statistical manual of mental disorders (5th ed., text rev). 2022.
- 2.Zeidan J, Fombonne E, Scorah J, Ibrahim A, Durkin MS, Saxena S, et al. Global prevalence of autism: A systematic review update. Autism Res. 2022;15:778–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maenner MJ, Shaw KA. Bakian A v, Bilder DA, Durkin MS, Esler A et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years — autism and developmental disabilities monitoring network, 11 sites, United States, 2020. MMWR Surveill Summ. 2021;70:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Courchesne E, Gazestani VH, Lewis NE. Prenatal origins of ASD: the when, what, and how of ASD development. Trends Neurosci. 2020;43:326–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Packer A. Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci Biobehav Rev. 2016;64:185–95. [DOI] [PubMed] [Google Scholar]
- 6.Macari S, Milgramm A, Reed J, Shic F, Powell KK, Macris D, et al. Context-specific dyadic attention vulnerabilities during the first year in infants later developing autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. 2021;60:166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caly, H, Rabiei, H, Coste-Mazeau, P, Hantz, S, Alain, S, Eyraud, JL, et al. Machine learning analysis of pregnancy data enables early identification of a subpopulation of newborns with ASD. Sci Rep. 2021;11:10.1038/s41598-021-86320-0. [DOI] [PMC free article] [PubMed]
- 8.Bonnet-Brilhault F, Rajerison TA, Paillet C, Guimard-Brunault M, Saby A, Ponson L, et al. Autism is a prenatal disorder: evidence from late gestation brain overgrowth. Autism Res. 2018;11:1635–42. 10.1002/aur.2036. [DOI] [PubMed] [Google Scholar]
- 9.Cloarec, R, Riffault, B, Dufour, A, Rabiei, H, Gouty-Colomer, L-A, Dumon, C, et al. Pyramidal neuron growth and increased hippocampal volume during labor and birth in autism. Sci Adv. 2019;5:https://www.science.org. [DOI] [PMC free article] [PubMed]
- 10.Surén P, Roth C, Bresnahan M, Haugen M, Hornig M, Hirtz D, et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. J Am Med Assoc. 2013;309:570–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68:1095–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenberg RE, Law JK, Yenokyan G, Mcgready J, Kaufmann WE, Law PA. Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med. 2009;163:907–14. [DOI] [PubMed] [Google Scholar]
- 13.Landrigan PJ. What causes autism? Exploring the environmental contribution. Curr Opin Pediatr. 2010;22:219–25. [DOI] [PubMed] [Google Scholar]
- 14.Karimi P, Kamali E, Mousavi SM, Karahmadi M. Environmental factors influencing the risk of autism. J Res Med Sci. 2017;22:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. J Am Med Assoc. 2013;309:1696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burke RD, Todd SW, Lumsden E, Mullins RJ, Mamczarz J, Fawcett WP, et al. Developmental neurotoxicity of the organophosphorus insecticide chlorpyrifos: from clinical findings to preclinical models and potential mechanisms. J Neurochem. 2017;142:162–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bilbo SD, Block CL, Bolton JL, Hanamsagar R, Tran PK. Beyond infection - maternal immune activation by environmental factors, microglial development, and relevance for autism spectrum disorders. Exp Neurol. 2018;299:241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Careaga M, Murai T, Bauman MD. Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol Psychiatry. 2017;81:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zawadzka A, Cieślik M, Adamczyk A. The role of maternal immune activation in the pathogenesis of autism: a review of the evidence, proposed mechanisms and implications for treatment. Int J Mol Sci. 2021;22:11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padilla N, Eklöf E, Mårtensson GE, Bölte S, Lagercrantz H, Ådén U. Poor brain growth in extremely preterm neonates long before the onset of autism spectrum disorder symptoms. Cereb Cortex. 2017;27:1245–52. [DOI] [PubMed] [Google Scholar]
- 21.Curran EA, O’Neill SM, Cryan JF, Kenny LC, Dinan TG, Khashan AS, et al. Research review: birth by caesarean section and development of autism spectrum disorder and attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. J Child Psychol Psychiatry Allied Discip. 2015;56:500–8. [DOI] [PubMed] [Google Scholar]
- 22.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicolini C, Fahnestock M. The valproic acid-induced rodent model of autism. Exp Neurol. 2018;299:217–27. [DOI] [PubMed] [Google Scholar]
- 24.Lan A, Kalimian M, Amram B, Kofman O. Prenatal chlorpyrifos leads to autism-like deficits in C57Bl6/J mice. Environ Health. 2017;16:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peñagarikano O, Geschwind DH. What does CNTNAP2 reveal about autism spectrum disorder? Trends Mol Med. 2012;18:156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eltokhi A, Rappold G, Sprengel R. Distinct phenotypes of Shank2 mouse models reflect neuropsychiatric spectrum disorders of human patients with SHANK2 variants. Front Mol Neurosci. 2018;11:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bey AL, Jiang YH. Overview of mouse models of autism spectrum disorders. Curr Protoc Pharm. 2014;2014:5.66.1–5.66.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kazdoba TM, Leach PT, Yang M, Silverman JL, Solomon M, Crawley JN. Translational mouse models of autism: advancing toward pharmacological therapeutics. Curr Top Behav Neurosci. 2016;28:1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian Z, Zhang R, Zhou J, Sun S, Di Y, Ren W, et al. RNA-Seq data on prefrontal cortex in valproic acid model of autism and control rats. Data Brief. 2018;18:787–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao H, Wang Q, Yan T, Zhang Y. Xu H-j, Yu H-p et al. Maternal valproic acid exposure leads to neurogenesis defects and autism-like behaviors in non-human primates. Transl Psychiatry. 2019;9:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuo K, Shinoda Y, Abolhassani N, Nakabeppu Y, Fukunaga K. Transcriptome analysis in hippocampus of rats prenatally exposed to valproic acid and effects of intranasal treatment of oxytocin. Front Psychiatry. 2022;13:859198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang R, Zhou J, Ren J, Sun S, Di Y, Wang H, et al. Transcriptional and splicing dysregulation in the prefrontal cortex in valproic acid rat model of autism. Rep. Toxicol. 2018;77:53–61. [DOI] [PubMed] [Google Scholar]
- 33.Guerra M, Medici V, Weatheritt R, Corvino V, Palacios D, Geloso MC, et al. Fetal exposure to valproic acid dysregulates the expression of autism-linked genes in the developing cerebellum. Transl Psychiatry. 2023;13:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science. 2019;364:685–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wamsley B, Bicks L, Cheng Y, Kawaguchi R, Quintero D, Margolis M, et al. Molecular cascades and cell type-specific signatures in ASD revealed by single-cell genomics. Science (1979). 2024;384:adh2602. [DOI] [PubMed] [Google Scholar]
- 37.Ben-Ari Y. Neuro-archaeology: pre-symptomatic architecture and signature of neurological disorders. Trends Neurosci. 2008;31:626–36. [DOI] [PubMed] [Google Scholar]
- 38.Meng Q, Zhang W, Wang X, Jiao C, Xu S, Liu C et al. Human forebrain organoids reveal connections between valproic acid exposure and autism risk. Transl Psychiatry 2022;12:10.1038/s41398-022-01898-x. [DOI] [PMC free article] [PubMed]
- 39.Nau H, Löscher W. Valproic acid: brain and plasma levels of the drug and its metabolites, anticonvulsant effects and gamma-aminobutyric acid (GABA) metabolism in the mouse. J Pharm Exp Ther. 1982;220:654–9. [PubMed] [Google Scholar]
- 40.Nau H. Species differences in pharmacokinetics and drug teratogenesis. Environ Health Perspect. 1986;70:113–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nau H. Transfer of valproic acid and its main active unsaturated metabolite to the gestational tissue: correlation with neural tube defect formation in the mouse. Teratol. 1986;33:21–7. [DOI] [PubMed] [Google Scholar]
- 42.Dickinson RG, Lawyer CH, Kaufman SN, Lynn RK, Gerber N, Novy MJ, et al. Materno-fetal pharmacokinetics and fetal distribution of valproic acid in a pregnant rhesus monkey. Pediatr Pharm. 1980;1:71–83. [PubMed] [Google Scholar]
- 43.Ferri SL, Abel T, Brodkin ES. Sex differences in autism spectrum disorder: a review. Curr Psychiatry Rep. 2018;20:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross AJ, Capel B. Signaling at the crossroads of gonad development. Trends Endocrinol Metab. 2005;16:19–25. [DOI] [PubMed] [Google Scholar]
- 45.Konopko MA, Densmore AL, Krueger BK. Sexually-dimorphic epigenetic regulation of brain-derived neurotrophic factor in fetal brain in the valproic acid model of autism spectrum disorder. Dev Neurosci. 2017;39:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wingett SW, Andrews S. Fastq screen: A tool for multi-genome mapping and quality control. F1000Res. 2018;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bourgain C, Génin E, Cox N, Clerget-Darpoux F. Are genome-wide association studies all that we need to dissect the genetic component of complex human diseases? Eur J Hum Genet. 2007;15:260–3. [DOI] [PubMed]
- 52.Sundqvist J, Xu H, Vodolazkaia A, Fassbender A, Kyama C, Bokor A, et al. Replication of endometriosis-associated single-nucleotide polymorphisms from genome-wide association studies in a Caucasian population. Hum Reprod. 2013;28:835–9. [DOI] [PubMed] [Google Scholar]
- 53.Kataoka S, Takuma K, Hara Y, Maeda Y, Ago Y, Matsuda T. Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int J Neuropsychopharmacol. 2013;16:91–103. [DOI] [PubMed] [Google Scholar]
- 54.Almeida LEF, Roby CD, Krueger BK. Increased BDNF expression in fetal brain in the valproic acid model of autism. Mol Cell Neurosci. 2014;59:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gilbert J, Man HY Fundamental elements in autism: from neurogenesis and neurite growth to synaptic plasticity. Front Cell Neurosci. 2017;11:359. [DOI] [PMC free article] [PubMed]
- 56.Ben-Ari Y, Spitzer NC. Phenotypic checkpoints regulate neuronal development. Trends Neurosci. 2010;33:485–92. 10.1016/j.tins.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, de Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180:568–84.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yuen RKC, Merico D, Bookman M, Howe JL, Thiruvahindrapuram B, Patel RV, et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci. 2017;20:602–11. [DOI] [PMC free article] [PubMed]
- 59.Stessman HAF, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. 2017;49:515–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruzzo EK, Pérez-Cano L, Jung JY. Wang L kai, Kashef-Haghighi D, Hartl C, et al. Inherited and de novo genetic risk for autism impacts shared networks. Cell. 2019;178:850–66.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garcia-Forn M, Boitnott A, Akpinar Z, de Rubeis S. Linking autism risk genes to disruption of cortical development. Cells 2020;9:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin YC, Frei JA, Kilander MBC, Shen W, Blatt GJ. A subset of autism-associated genes regulate the structural stability of neurons. Front Cell Neurosci. 2016;10:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Poelmans G, Franke B, Pauls DL, Glennon JC, Buitelaar JK. AKAPs integrate genetic findings for autism spectrum disorders. Transl Psychiatry. 2013;3:e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong C, West KL, Tan XY, Li J, Ishibashi T, Yu C, et al. Screen identifies DYRK1B network as mediator of transcription repression on damaged chromatin. Proc Nat Acad Sci USA. 2020;117:17019–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Singh R, Kumar Dhanyamraju P, Lauth M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget. 2017;8:833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singh R, Lauth M. Emerging roles of DYRK kinases in embryogenesis and Hedgehog pathway control. J Dev Biol. 2017;5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orenstein N, Gofin Y, Shomron N, Ruhrman-Shahar N, Magal N, Hagari O, et al. DYRK1B haploinsufficiency in a family with metabolic syndrome and abnormal cognition. Clin Genet. 2022;101:265–6. [DOI] [PubMed] [Google Scholar]
- 69.Yger M, Girault JA. DARPP-32, jack of all trades…master of which? Front Behav Neurosci. 2011;5:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rosenberg SS, Spitzer NC. Calcium signaling in neuronal development. Cold Spring Harb Perspect Biol. 2011;3:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron 2016;89:248–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dufour A, Dumon C, Gouty-Colomer LA, Eftekhari S, Ferrari DC, Ben-Ari Y. Prenatal reduction of E14.5 embryonically fate-mapped pyramidal neurons in a mouse model of autism. Eur J Neurosci. 2022;56:3875–88. [DOI] [PubMed] [Google Scholar]
- 73.Zagami CJ, Zusso M, Stifani S. Runx transcription factors: lineage-specific regulators of neuronal precursor cell proliferation and post-mitotic neuron subtype development. J Cell Biochem. 2009;107:1063–72. [DOI] [PubMed]
- 74.Haushalter C, Asselin L, Fraulob V, Dollé P, Rhinn M. Retinoic acid controls early neurogenesis in the developing mouse cerebral cortex. Dev Biol. 2017;430:129–41. [DOI] [PubMed] [Google Scholar]
- 75.Shibata M, Pattabiraman K, Lorente-Galdos B, Andrijevic D, Kim SK, Kaur N, et al. Regulation of prefrontal patterning and connectivity by retinoic acid. Nature. 2021;598:483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 2009;139:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jourdon A, Wu F, Mariani J, Capauto D, Norton S, Tomasini L, et al. Modeling idiopathic autism in forebrain organoids reveals an imbalance of excitatory cortical neuron subtypes during early neurogenesis. Nat Neurosci. 2023;26:1505–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coulthard LG, Hawksworth OA, Conroy J, Lee JD, Woodruff TM. Complement C3a receptor modulates embryonic neural progenitor cell proliferation and cognitive performance. Mol Immunol. 2018;101:176–81. [DOI] [PubMed] [Google Scholar]
- 79.Sessa A, Ciabatti E, Drechsel D, Massimino L, Colasante G, Giannelli S, et al. The Tbr2 molecular network controls cortical neuronal differentiation through complementary genetic and epigenetic pathways. Cereb Cortex. 2017;27:3378–96. [DOI] [PubMed] [Google Scholar]
- 80.Elsen GE, Hodge RD, Bedogni F, Daza RAM, Nelson BR, Shiba N, et al. The protomap is propagated to cortical plate neurons through an Eomes-dependent intermediate map. Proc Natl Acad Sci USA. 2013;110:4081–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Englund C, Fink A, Lau C, Pham D, Daza RAM, Bulfone A, et al. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baala L, Briault S, Etchevers HC, Laumonnier F, Natiq A, Amiel J, et al. Homozygous silencing of T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat Genet. 2007;39:454–6. [DOI] [PubMed] [Google Scholar]
- 83.Arnold SJ, Huang GJ, Cheung AFP, Era T, Nishikawa SI, Bikoff EK, et al. The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev. 2008;22:2479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li C, Fleck JS, Martins-Costa C, Burkard TR, Themann J, Stuempflen M, et al. Single-cell brain organoid screening identifies developmental defects in autism. Nature. 2023;621:373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Han S, Dennis DJ, Balakrishnan A, Dixit R, Britz O, Zinyk D, et al. A non-canonical role for the proneural gene neurog1 as a negative regulator of neocortical neurogenesis. Development. 2018;145:dev157719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suliman-Lavie R, Title B, Cohen Y, Hamada N, Tal M, Tal N, et al. Pogz deficiency leads to transcription dysregulation and impaired cerebellar activity underlying autism-like behavior in mice. Nat Comm. 2020;11:5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hanafusa H, Kedashiro S, Tezuka M, Funatsu M, Usami S, Toyoshima F, et al. PLK1-dependent activation of LRRK1 regulates spindle orientation by phosphorylating CDK5RAP2. Nat Cell Biol. 2015;17:1024–35. [DOI] [PubMed] [Google Scholar]
- 88.Gillentine MA, Wang T, Hoekzema K, Rosenfeld J, Liu P, Guo H et al. Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders. Genome Med. 2021;13:10.1186/s13073-021-00870-6. [DOI] [PMC free article] [PubMed]
- 89.Borrell V, Götz M. Role of radial glial cells in cerebral cortex folding. Curr Opin Neurobiol. 2014;27:39–46. [DOI] [PubMed] [Google Scholar]
- 90.Stahl R, Walcher T, De Juan Romero C, Pilz GA, Cappello S, Irmler M, et al. Trnp1 regulates expansion and folding of the mammalian cerebral cortex by control of radial glial fate. Cell. 2013;153:535–49. [DOI] [PubMed] [Google Scholar]
- 91.Rash BG, Tomasi S, Lim HD, Suh CY, Vaccarino FM. Cortical gyrification induced by fibroblast growth factor 2 in the mouse brain. J Neurosci. 2013;33:10802–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Da Silva F, Zhang K, Pinson A, Fatti E, Wilsch‐Bräuninger M, Herbst J et al. Mitotic WNT signalling orchestrates neurogenesis in the developing neocortex. EMBO J. 2021;40:10.15252/embj.2021108041. [DOI] [PMC free article] [PubMed]
- 93.Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ. Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci. 2011;31:1676–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Usui, N, Berto, S, Konishi, A, Kondo, M, Konopka, G, Matsuzaki, H, et al. Zbtb16 regulates social cognitive behaviors and neocortical development. Transl Psychiatry. 2021;11:242. [DOI] [PMC free article] [PubMed]
- 95.Krontira AC, Cruceanu C, Dony L, Kyrousi C, Link M-H, Rek N, et al. Human cortical neurogenesis is altered via glucocorticoid-mediated regulation of ZBTB16 expression. Neuron. 2024;10.1016/j.neuron.2024.02.005. [DOI] [PubMed]
- 96.Antonyan L, Ernst C. Putative roles of SETBP1 dosage on the SET oncogene to affect brain development. Front Neurosci. 2022;16:813430. [DOI] [PMC free article] [PubMed]
- 97.Cardo LF, de la Fuente DC, Li M. Impaired neurogenesis and neural progenitor fate choice in a human stem cell model of SETBP1 disorder. Mol Autism. 2023;14:8. [DOI] [PMC free article] [PubMed]
- 98.Piazza R, Magistroni V, Redaelli S, Mauri M, Massimino L, Sessa A, et al. SETBP1 induces transcription of a network of development genes by acting as an epigenetic hub. Nat Commun. 2018;9:2192. [DOI] [PMC free article] [PubMed]
- 99.Tuoc TC, Boretius S, Sansom SN, Pitulescu ME, Frahm J, Livesey FJ, et al. Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev Cell. 2013;25:256–69. [DOI] [PubMed] [Google Scholar]
- 100.Machol K, Rousseau J, Ehresmann S, Garcia T, Nguyen TTM, Spillmann RC, et al. Expanding the spectrum of BAF-related disorders: de novo variants in SMARCC2 cause a syndrome with intellectual disability and developmental delay. Am J Hum Genet. 2019;104:164–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Orosco LA, Ross AP, Cates SL, Scott SE, Wu D, Sohn J et al. Loss of Wdfy3 in mice alters cerebral cortical neurogenesis reflecting aspects of the autism pathology. Nat Commun 2014;5:4692. [DOI] [PMC free article] [PubMed]
- 102.Rubenstein JLR, Merzenich MM. Model of autism: increased ratio of excitation/ inhibition in key neural systems. Genes Brain Behav. 2003;2:255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron. 2015;87:684–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang P, Omanska A, Ander BP, Gandal MJ, Stamova B, Schumann CM. Neuron-specific transcriptomic signatures indicate neuroinflammation and altered neuronal activity in ASD temporal cortex. Proc Natl Acad Sci USA. 2023;120:e2206758120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pai ELL, Vogt D, Clemente-Perez A, McKinsey GL, Cho FS, Hu JS, et al. Mafb and c-maf have prenatal compensatory and postnatal antagonistic roles in cortical interneuron fate and function. Cell Rep. 2019;26:1157–73.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Uezu A, Kanak DJ, Bradshaw TWA, Soderblom EJ, Catavero CM, Burette AC, et al. Identification of an elaborate complex mediating postsynaptic inhibition. Science. 2016;353:1123–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Uezu A, Hisey E, Kobayashi Y, Gao Y, Bradshaw TWA, Devlin P et al. Essential role for insyn1 in dystroglycan complex integrity and cognitive behaviors in mice. Elife. 2019;8:e50712. [DOI] [PMC free article] [PubMed]
- 108.Meng X, Yao D, Imaizumi K, Chen X, Kelley KW, Reis N et al. Assembloid CRISPR screens reveal impact of disease genes in human neurodevelopment. Nature. 2023;622:359–66. [DOI] [PMC free article] [PubMed]
- 109.Markram K, Rinaldi T, la Mendola D, Sandi C, Markram H. Abnormal fear conditioning and amygdala processing in an animal model of autism. Neuropsychopharmcol. 2008;33:901–12. [DOI] [PubMed] [Google Scholar]
- 110.Angara K, Ling-Lin Pai E, Bilinovich SM, Stafford AM, Nguyen JT, Li KX, et al. Nf1 deletion results in depletion of the Lhx6 transcription factor and a specific loss of parvalbumin+ cortical interneurons. Proc Natl Acad Sci USA. 2020;117:6189–95. [DOI] [PMC free article] [PubMed]
- 111.McDermott JE, Goldblatt D, Paradis S. Class 4 Semaphorins and Plexin-B receptors regulate GABAergic and glutamatergic synapse development in the mammalian hippocampus. Mol Cell Neurosci. 2018;92:50–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bosiacki M, Gąssowska-Dobrowolska M, Kojder K, Fabiańska M, Jeżewski D, Gutowska I, et al. Perineuronal nets and their role in synaptic homeostasis. Int J Mol Sci. 2019;20:4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nojima K, Miyazaki H, Hori T, Vargova L, Oohashi T. Assessment of possible contributions of hyaluronan and proteoglycan binding link protein 4 to differential perineuronal net formation at the calyx of held. Front Cell Dev Biol. 2021;9:730550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ramsaran AI, Wang Y, Golbabaei A, Stepan Aleshin S, de Snoo ML, Yeung BA, et al. A shift in the mechanisms controlling hippocampal engram formation during brain maturation. Science. 2023;380:543–51. [DOI] [PubMed] [Google Scholar]
- 115.Edamatsu M, Miyano R, Fujikawa A, Fujii F, Hori T, Sakaba T, et al. Hapln4/Bral2 is a selective regulator for formation and transmission of GABAergic synapses between Purkinje and deep cerebellar nuclei neurons. J Neurochem. 2018;147:748–63. [DOI] [PubMed] [Google Scholar]
- 116.Mapelli L, Soda T, D’Angelo E, Prestori F. The cerebellar involvement in autism spectrum disorders: from the social brain to mouse models. Int J Mol Sci. 2022;23:3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Spitzer NC. Electrical activity in early neuronal development. Nature. 2006;444:707–12. [DOI] [PubMed] [Google Scholar]
- 118.Watanabe M, Fukuda A. Development and regulation of chloride homeostasis in the central nervous system. Front Cell Neurosci. 2015;9:371. [DOI] [PMC free article] [PubMed]
- 119.Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 2014;343:675–9. [DOI] [PubMed] [Google Scholar]
- 120.Wang T, Shan L, Miao C, Xu Z, Jia F. Treatment effect of bumetanide in children with autism spectrum disorder: a systematic review and meta-analysis. Front Psychiatry. 2021;12:751575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.