

Abstract
Ketotherapeutics is a potential metabolic intervention for mitigating dementias; however, its mechanisms and optimal methods of application are not well understood. Our previous in vitro study showed that β‐hydroxybutyrate (BHB), a major ketone body, reverses pathological features of amyloid‐β oligomer (AβO)‐activated microglia. Here we tested the in vivo effects of BHB on microglia and synaptic plasticity in the 5xFAD Alzheimer's disease (AD) mouse model. A short 1‐week regimen of daily intraperitoneal injection of BHB (250 mg/kg), which induced brief and mild daily episodic ketosis, was sufficient to mitigate pro‐inflammatory microglia activation and reduce brain amyloid‐β deposition by enhancing phagocytosis. Remarkably, it mitigated the deficits of hippocampal long‐term depression but not long‐term potentiation, and this effect was linked to suppression of NLRP3 inflammasome‐generated IL‐1β. As ketogenic diets are known for poor compliance, our study opens the possibility for alternative approaches such as short‐term BHB injections or dietary ketone esters that are less restrictive, potentially safer, and easier for compliance.
Keywords: Alzheimer's, ketosis, metabolism, microglia, synapse
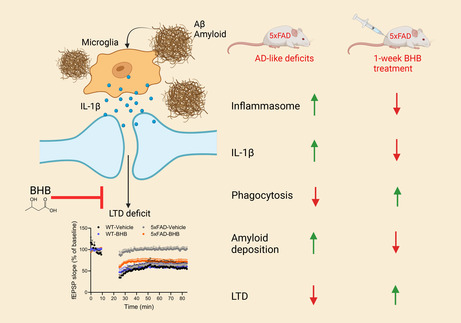
A short 1‐week treatment of the ketone body BHB is able to rectify impairment of microglia‐regulated synaptic plasticity in 5xFAD by reducing NLRP3 inflammasome‐generated IL‐1β and enhancing Aβ phagocytosis.
1. INTRODUCTION
Alzheimer's disease is the most common form of dementia in the elderly, affecting more than 6.7 million Americans in 2023. 1 Targeting risk factors associated with metabolic health is an important part of a precision medicine approach. 2 In Alzheimer's disease brains, microglia lose their homeostatic molecular signature and show profound functional impairments. 3 Recently, the pivotal roles of microglia‐orchestrated neuroinflammation in Alzheimer's disease have been established. 4 , 5 , 6 , 7 Microglia, the innate immune cells of the brain, continuously survey their microenvironment and rapidly respond to major and minor physiological alterations and pathological insults. 8 , 9 The constant motility, interaction with neighboring cells, phagocytosis of debris, release of mediators, and numerous other activities require versatile and flexible metabolic programs. A generally accepted hypothesis is that dysfunctional microglia with impaired immunometabolic states fail to do the above normal functions and release cytotoxic substances and pro‐inflammatory cytokines that cause neuronal/synaptic damage and aggravate Alzheimer's disease pathology. 4 , 10 , 11 From an interventional point of view, a critical question is how to reverse or fine‐tune the Alzheimer's disease‐associated microglial metabolic states toward beneficial immunometabolic outcomes to improve brain/synaptic function.
A major metabolic interventional approach is to induce ketosis, which can be achieved by fasting, strenuous exercise, or consumption of ketogenic diet (KD, a low‐carbohydrate, high‐fat, and moderate protein diet). 12 , 13 , 14 Common to these approaches is a metabolic shift toward fatty acid metabolism, mainly by hepatocytes, to generate the ketone bodies acetoacetate and β‐hydroxybutyrate (BHB). The consequent ketosis is a well‐known physiological condition with elevated blood BHB levels. In humans, serum levels of BHB rise from the basal levels of low μM to a few hundred μM after 12–16 h of fasting, and 1–2 mM after 2 days of fasting. Serum levels of 1–2 mM BHB can also be reached after 90 min of intense exercise. 12 , 15 Our previous mouse studies also showed similar levels of ketosis following KD consumption. 16 , 17 Ketosis has been linked to better memory and cognitive performance, prompting a great deal of interest in the KD as a potential intervention to mitigate dementias. 18 , 19 , 20 Our recent demonstration that KD rescues the impaired synaptic plasticity, manifested by suppressed hippocampal long‐term potentiation (hLTP), in a mouse model of Alzheimer's disease provides a physical basis for such an improvement. 21 Because BHB, the active principle of KD and other induced ketosis, penetrates the blood–brain barrier and signals downstream pathways through its hydroxycarboxylic acid receptor (HCAR2) and can also be used as a neural energy source, 15 it is important to understand how BHB affects brain cell functions.
In a previous study, we investigated how BHB may benefit Alzheimer's disease via microglia. We used Aβ oligomer (AβO)‐treated human iPS cell‐derived microglia (HiMG) as a model of early Alzheimer's disease‐like microglia impairments and directly tested the effects of BHB on microglia. 22 We found that HiMG responded to AβO with pro‐inflammatory activation, which was mitigated by BHB. AβO stimulated glycolytic transcripts, suppressed genes in the β‐oxidation pathway and triggered mitochondrial abnormalities. BHB potently ameliorated all the above mitochondrial changes, resulting in reduced inflammasome activation and recovery of the phagocytotic function impaired by AβO. In the current study, we tested the effects of BHB on microglia in vivo using 5xFAD mice, which harbor five familial mutations of APP and PSEN1 genes and show robust Aβ production and Aβ‐associated microglia activation and neuroinflammation. 23 In this model, AβO is a major species of Aβ aggregates and could stimulate microglial action in vivo as in the above microglia cultures. 24 Notably, we employed a “subacute” 1‐week regimen of daily injection of BHB, which induces episodic ketosis similar to that induced by daily strenuous exercise, exercise or fasting. The subacute course was expected to allow a metabolic shift to develop. This regimen resulted in significant impact on microglia function and microglia‐regulated synaptic plasticity in 5xFAD mice, indicating that microglia are one of the primary cellular targets of ketosis and supporting the principle that metabolic shift induced by BHB regulates microglial function in vivo.
2. MATERIALS AND METHODS
2.1. Mouse model
Tg6799 5xFAD mice on the C57BL/6 background were purchased from Jackson Laboratory. This line co‐expresses human APP695 with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) mutations and human PS1 harboring M146L and L286V mutations. Mice at 12 months of age were randomly assigned to treatment groups. Roughly equal numbers of male and female mice were used. All mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC) and we have complied with all relevant ethical regulations for animal use. Mice for this study were housed in the same facility under the same husbandry conditions.
2.2. Intraperitoneal (IP) injection and measurement of blood BHB
Sodium 3‐hydroxybutyrate (BHB) (Sigma‐Aldrich, St. Louis, MO) was dissolved in sterile saline solution for a final concentration of 75 mg/mL. Mice were injected daily intraperitoneally at 5 pm with either 250 mg/kg BHB or PBS as control. Mice were treated for seven consecutive days. Prior to termination, blood BHB was measured with a Precision Xtra glucose and ketone monitoring system (Abbott Diabetes Care Inc., Alameda, CA) using blood from a tail nick at 0, 5, 10‐, 30‐, 60‐, and 120‐min post‐injection, as well as immediately before sacrifice (15 h post‐injection).
2.3. Induction of hippocampal long‐term potentiation and hippocampal long‐term depression (hLTD)
The preparation of mouse hippocampal slices and the induction of hLTP and hLTD by stimulation of the Schaffer collateral afferents were conducted on a subset of mice. Coronal slices (300 μm) of mouse hippocampus were prepared from wild‐type (WT) and 5xFAD mice. The animals were subjected to deep anesthesia with isoflurane and decapitated. The brain was rapidly removed and transferred to a modified artificial cerebrospinal fluid (HI‐ACSF) containing (in mM): 220 sucrose, 2 KCl, 0.2 CaCl2, 6 MgSO2, 26 NaHCO3, 1.3 NaH2PO4, and 10 D‐glucose (pH 7.4, set by aeration with 95% O2 and 5% CO2). Coronal brain slices were cut in ice‐cold modified artificial cerebrospinal fluid (ACSF) with the use of a DTK‐1000 D.S.K Microslicer (TedPella, Inc., Redding, CA, USA). The slices were immediately transferred into an ACSF solution containing (in mM): 126 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 10 D‐glucose (pH 7.4, set by aeration with 95% O2 and 5% CO2) for at least 30 min at controlled temperature of 37°C. After subsequent incubation for at least 1 h at room temperature, hemi‐slices were transferred to the recording chamber, which was perfused with standard ACSF at a constant flow rate of ~2 mL/min. Recordings of field excitatory postsynaptic potentials (fEPSPs) were obtained from the stratum radiatum of the CA1 region of the hippocampus after stimulation of the Schaffer collateral afferents. Extracellular recording electrodes were prepared from borosilicate capillaries with an outer diameter of 1.5 mm (Sutter Instruments) and were filled with 3 M NaCl (resistance, 1–2 MΩ). Baseline stimulation rate was 0.05 Hz. fEPSPs were filtered at 2 kHz and digitized at 10 kHz with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA). Data were collected and analyzed with pClamp 10.3 software (Molecular Devices). Slope values of fEPSPs were considered for quantitation of the responses. A 10 min of stable baseline was recorded of fEPSPs evoked every 20 s by application of a constant current pulse of 0.2–0.4 mA with a duration of 60 ms at the current intensity set to evoke 50%–60% of the maximal response. LTP was elicited by high frequency stimulation (HFS), consisting of two trains of 100‐Hz (1 s) stimulation with the same intensity and pulse duration used in sampling of baseline fEPSPs. For induction of LTD, low frequency stimulation 25 was applied, consisting of 900 stimuli at 1 Hz. Recording was then continued for 60 min with stimulation of fEPSPs every 20 s. For antibody experiments, brains slices were incubated with different concentrations of IL‐1β antibody (R&D systems) for 1 h. Antibody was also present during recordings.
2.4. Tissue homogenate preparation and Western blot analysis
Brain tissue samples (mid‐coronal cryosections including hippocampus) were homogenized in lysis buffer (150 mM NaCl, 10 mM NaH2PO4, 1 mM EDTA, 1% Triton X‐100, 0.5% SDS) with protease inhibitor cocktail and phosphatase inhibitor (Sigma). Equivalent amounts of protein were analyzed by 4%–20% Tris‐Glycine gel electrophoresis (Invitrogen). Proteins were transferred to polyvinylidene difluoride membranes and probed with antibodies. Visualization was enabled using enhanced chemiluminescence (GE Healthcare Pharmacia). The following primary antibodies (dilutions) were used: anti‐CD11b (1:1000, Invitrogen, #MA5‐17857), anti‐NF‐kB (1:1000, Cell Signaling, #8242), anti‐Cleaved IL‐1β (1:1000, Cell Signaling, #63124), anti‐Pro‐Caspase1 (1:1000, Cell Signaling, #24232), and β‐actin (1:5000, Cell Signaling, #3700).
For β‐Amyloid detection, brain homogenates were separated on 16% Tricine gel (Invitrogen) and then immunoblotted on PVDF membrane. The blots were incubated with 6E10 anti‐Aβ antibody (1:1000, BioLegend, #803001).
Secondary antibodies were HRP‐conjugated anti‐rabbit or anti‐mouse antibody (1:1000, Cell Signaling, #7074 and #7076).
2.5. ELISA quantification
Brain tissue samples (mid‐coronal cryosections including hippocampus) were homogenized in lysis buffer (100 mM TRIS, pH 7.4; 150 mM NaCl; 1 mM EGTA; 1 mM EDTA; 1% Triton X‐100; 0.5% Sodium deoxycholate; proteinase inhibitor mix), and centrifuged for 20 min at 15,000 rpm at 4°C. The supernatants were directly used for measuring total cytokine (IL‐1β, TNF‐α, and IL‐6). Concentrations of IL‐1β, TNF‐α, and IL‐6 were measured using the Quantikine sandwich ELISA kit (R&D systems, Minneapolis, MN).
2.6. Acute isolation of microglia
Microglia were acutely isolated from a forebrain hemisphere without culturing as we described. 26 Briefly, brains were dissociated enzymatically with a Neural Tissue Dissociation Kit (Miltenyi Biotec). Microglia were subsequently purified by magnetic‐activated cell sorting (MACS) using anti‐CD11b magnetic beads (Miltenyi Biotec). The whole procedure took about 90 min.
2.7. Quantitative PCR
Total RNA from acutely isolated microglia and tissue samples were extracted using RNeasy Plus Universal Mini Kit (Qiagen). RNA samples from acutely isolated microglia were further reverse‐transcribed and pre‐amplified as we previously described. 27 T The primer sequences used are as followed: il1b (5′‐CCCCAAGCAATACCCAAAGA‐3′; 5‐TACCAGTTGGGGAACTCTG‐3′); tnfa (5′‐ GACGTGGAACTGGCAGAAGAG‐3′; 5‐TGCCACAAGCAGGAATGAGA‐3′); il6 (5′‐ GTTCTCTGGGAAATCGTGGA‐3′; 5‐TTCTGCAAGTGCATCATCGT‐3′); arg1 (5′‐ CCAACTCTTGGGAAGACAGC‐3′; 5‐TATGGTTACCCTCCCGTTGA‐3′); mcr1 (5′‐ TCATCCCTGTCTTGTTCAGC‐3′; 5‐ATGGCACTTAGAGCGTCCAC‐3′); chil3 (5′‐ AGGAAGCCCTCCTAAGGACA‐3′; 5‐TGAGTAGCAGCCTTGGAATG‐3′); nlrp3 (5′‐ ATTACCCGCCCGAGAAAGG‐3′; 5‐TCGCAGCAAAGATCCACACAG‐3′); asc (5′‐ CTTGTCAGGGGATGAACTCAAAA‐3′; 5‐GCCATACGACTCCAGATAGTAGC‐3′); casp1 (5′‐ ACAAGGCACGGGACCTATG‐3′; 5‐ TCCCAGTCAGTCCTGGAAATG‐3′); actin (5′‐TCAAGATCATTGCTCCTCCTGAG‐3′; 5‐ ACATCTGCTGGAAGGTGGACA‐3′). Relative cDNA levels for the target genes were analyzed by the 2−∆∆Ct method using Actb as the internal control for normalization.
2.8. Immunofluorescence staining
Cryostat brain sections (16 μm) were fixed with 4% PFA and blocked with 10% normal goat serum with 0.3% Triton‐X. Sections were incubated with 80% formic acid to expose β‐amyloid epitopes. Sections were incubated with primary antibody, anti‐Iba1 (1:400, Biocare Medical, #290), and anti‐Amyloid Beta 4G8 (BioLegend, #800701) overnight at 4°C. Sections were then incubated with Alexa Fluor‐conjugated secondary antibody (1:700, Invitrogen). For LAMP1 and Amyloid Beta colocalization, brain sections were incubated with anti‐LAMP1‐Alexa594. Amyloid plaques were detected by 400 nmol/L FSB (Sigma). Immunostained slides were imaged under a Nikon Eclipse E600 microscope and photographed by a digital camera (SPOT RTsCMOS, SPOT Diagnostics). The images were randomly taken in a blind fashion within a defined anatomic region. The images were transformed to 8‐bit grayscale and analyzed by ImageJ program.
2.9. Flow cytometry analysis of inflammasome proteins
Dissociated brain‐cell suspensions were prepared from a forebrain hemisphere using a Neural Tissue Dissociation kit (P) (Miltenyi Biotec) according to the manufacturer's instructions. Cell suspensions were incubated with anti‐Iba1‐Alexa488 (Cell Signaling, #20825), anti‐NLRP3‐Alexa594 (R&D Systems, #IC7578T) or anti‐ASC‐Alexa549 (Cell Signaling, #29956) for 30 min at 37°C. Cells were then washed with phosphate‐buffered saline (PBS; 0.1 M). Fluorescence intensities were assessed by a flow cytometer (Accuri C6 Plus, BD Biosciences). Colocalization of Iba1‐positive cells with NLRP3 or ASC‐positive cells was assessed with FlowJo 10.
2.10. RNA sequencing and analysis
Total RNA was extracted from mid‐coronal sections (including hippocampus) of brains using RNeasy Plus Universal Mini Kit (Qiagen). RNA quality was assessed based on RNA Integrity Number (RIN). RNA library and paired‐end RNA sequencing was performed using NovaSeq X Plus system (Illumina) with 6 Gb throughout by NovoGene (Sacramento, CA, USA). All RNA‐seq reads were aligned to the mouse reference genome (mm10) using Hisat2 version 2.1.0. FeatureCounts version 1.4.6 was used to quantify reads counts. Differentially expressed genes (DEGs) were analized using DESeq2 package using the open source, web‐based platform Galaxy (usegalaxy.org). To control false‐positive rates, we applied the Benjamini–Hochberg false discovery rate (FDR) approach. Gene Ontology (GO) and KEGG pathway enrichment analysis was performed using DAVID Bioinformatics Resources. KEGG pathway visualization was performed using Pathview. To identify biological pathways enriched with differentially expressed genes, we performed a gene set enrichment analysis (GSEA). Datasets were analyzed in reference to the h.all.v.2023.0.2.symbols.gmt gene set database.
2.11. Statistics
The mouse phenotype and cellular assays were conducted in a blinding manner. Statistical analyses were performed using GraphPad Prism 10 (GraphPad Software). All data are presented as means ± SEM. Exact sample sizes and statistical test used for each comparison were provided in corresponding figure legends. p < 0.05 was considered to be statistically significant.
3. RESULTS
3.1. One‐week BHB mitigates the deficits of hLTD but not hLTP in 5xFAD mice
We administered a 1‐week regimen of daily IP injection of BHB at a dose of 250 mg/kg to 12 month‐old 5xFAD mice and their WT littermates. The injection was given daily at 5 pm before the beginning of the dark cycle. This injection produced a spike of blood BHB level above the baseline (~0.75–0.8 mM) level within at least the first hour post‐injection. The BHB concentrations reached the highest level at 5 min (means = 1.73 mM for WT and 1.6 mM for 5xFAD), maintained slightly above 1 mM at 1 h (means = 1.05 mM for WT and 1.06 mM for 5xFAD), and fell below 1 mM at 2 h (Figure 1A). This regimen, therefore, generated episodic ketosis with blood levels similar to or slightly lower than those achieved by daily strenuous exercise, intermittent fasting, or KD consumption in either humans or mice. 15 , 21 , 28 There was no significant difference in blood levels of BHB between 5xFAD and WT mice at any time points following injection. We sacrificed the mice at 8 am the day after the completion of the regimen, that is, 15 h after the last dose of BHB, when the injected BHB had long been washed out, and the brain was then used for various studies. Figure 1B shows blood BHB levels at the time of sacrifice. All through the course of BHB injections, the mice were provided ad libitum access to a regular chow diet to avoid the complicating effect of additional BHB production from fasting. The rationale for a 7‐day course was that it would generate “subacute” effects of BHB that allow the metabolic shift of microglia to evolve and potentially the recovery of their normal functions. The fact that the blood BHB had long resumed the baseline level when we obtained the brain tissue excludes the likelihood that we were measuring the immediate effects of BHB.
FIGURE 1.
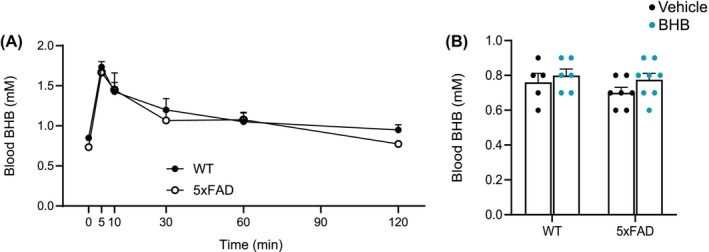
Blood levels of BHB in WT and 5xFAD mice after 1‐week injections. (A) Blood BHB levels at 0, 5, 10, 30, 60, 90, and 120 min post injection. BHB concentrations reached highest level at 5 min. N = 4 (B) Blood BHB levels after a week of treatment with either vehicle or BHB. These measurements were completed 15 h after the last BHB dose. Data are presented ad mean ± SEM.
We used hippocampal slices to evaluate both hLTP and hLTD at the Schaffer collateral‐CA1 synapses, induced by standard high‐frequency stimulation 29 and low‐frequency stimulation, 30 respectively. The hippocampus of 5xFAD mice showed stunted hLTP and hLTD. Different from the effect of 7‐month KD we reported before, 21 the 1‐week BHB regimen showed no effects on hLTP (Figure 2A, blue arrows). In contrast, this regimen significantly improved the hLTD deficits in both early induction and late maintenance phases (Figure 2B, red arrows). As stated above, this BHB effect on hLTD does not likely reflect a direct action of BHB on the synapse as the recordings were done at least 15 h after the last BHB dose when the exogenous BHB was long washed out and no increased BHB was detected in the blood (Figure 1B).
FIGURE 2.
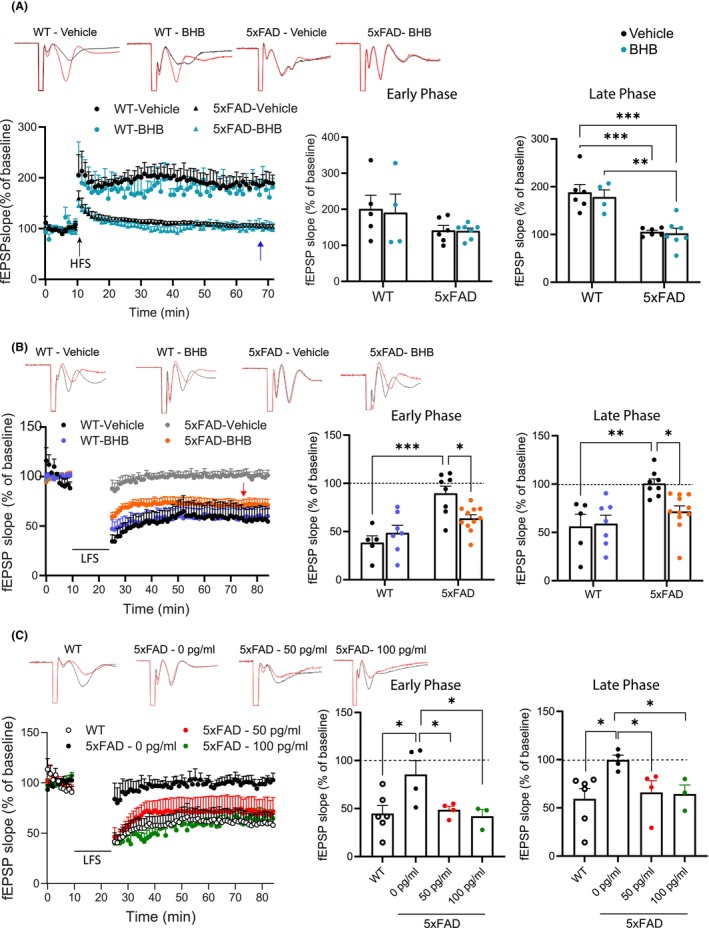
BHB treatment rescues hLTD, but not hLTD, impairment in 5xFAD mice. Shown are representative traces, time course chart, and summary bar graphs of hLTP induced with high frequency stimulation (HFS) (A) or hLTD induced with low frequency stimulation (B, C). Representative traces show the fEPSP of baseline (black trace) and at 60 min (red trace) after stimulation. Summary bar graphs show the average fEPSP slope during the early phase (first 5 min after induction) and the late phase (between 50 and 60 min after induction). (A) BHB does not rescue hLTP impairment in 5xFAD mice. Group size: WT‐Vehicle = 5; WT‐BHB = 4; 5xFAD‐Vehicle = 6; 5xFAD‐BHB = 7. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between genotypes in late phase (F(1,13) = 20.81, p < 0.001). (B) BHB mitigates hLTD impairment in 5xFAD mice. Group size: WT‐Vehicle = 5; WT‐BHB = 7; 5xFAD‐Vehicle = 8; 5xFAD‐BHB = 11. The blue arrow points to no improvement of 5xFAD hLTP by BHB. The red arrow points to the recovery of hLTD in 5xFAD mice by BHB. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between genotypes in early phase (F(1,27) = 24.83, p < 0.001) and late phase F(1,27) = 13.79, p < 0.001. (C) Pre‐incubation with IL‐1β antibody mitigates hLTD impairment in 5xFAD mice. Group size: WT = 6; 5xFAD‐0 pg/mL (anti‐IL‐1β) = 4; 5xFAD‐50 pg/mL = 4: 5xFAD‐100 pg/mL = 3. Statistical analysis was performed by one‐way ANOVA with Newman–Keuls correction for multiple comparisons. Data are presented as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
3.2. Microglia may contribute to hLTD deficits in 5xFAD mice via IL‐1β production
We hypothesize that this subacute influence of BHB on synaptic plasticity is an indirect effect related to the ability of BHB to induce microglia immunometabolic shift. 22 Several mediators released by microglia such as IL‐1βwere reported to influence hLTD. 31 We previously found that BHB dose‐dependently inhibited amyloid‐β oligomer (AβO)‐induced IL‐1β expression in human iPSC‐derived microglia. 22 To determine the role of IL‐1β in abrogating hLTD in 5xFAD hippocampus, we pre‐incubated hippocampal slices with 0, 50, and 100 pg/mL of a neutralizing IL‐1β antibody for 1 h prior to recording. Anti‐IL‐1β at both 50 and 100 pg/mL recovered the hLTD in 5xFAD hippocampus to the WT levels (Figure 2C). This result suggests that the synaptic structure and function underlying hLTD is intact in 5xFAD mice; the hLTD impairment could be attributed to microglia releasing excessive IL‐1β, which can be corrected by BHB.
3.3. One‐week BHB reduces microglial pro‐inflammatory activation in 5xFAD mice
In our previous in vitro study using human iPSC‐derived microglia, we demonstrated that BHB was able to block AβO‐induced expression of pro‐inflammatory mediators (IL‐1β, TNF‐α, and IL‐6), suppress NLRP3 (NOD‐, LRR‐ and pyrin domain‐containing protein 3)‐inflammasome formation, and improve phagocytotic activities. 22 We therefore examined whether the 1‐week BHB regimen could regulate microglia‐orchestrated neuroinflammation in the brain, including IL‐1β production. One‐week BHB reduced the number of Iba‐1 positive microglia in the 5xFAD brain to the WT level (Figure 3A), attesting to its potent effect on microglia. Western blot performed on homogenates made from mid‐coronal cryosections of forebrain showed that 1‐week BHB reduced the levels of CD11b and NF‐κB (Figure 3B,C) in 5xFAD mice. ELISA performed on the same sets of homogenates showed reductions of three major pro‐inflammatory cytokines that were increased in 5xFAD brains, TNF‐α, IL‐1β, and IL‐6 (Figure 3D). We further acutely isolated microglia from the forebrain and quantitated RNA expression by qPCR. The 1‐week BHB regimen reduced the upregulated pro‐inflammatory cytokines TNF‐α and IL‐1β in 5xFAD (Figure 3E). It increased the expression of anti‐inflammatory markers ARG1 and MRC1 in 5xFAD microglia while Chil3 expression was induced by BHB treatment in both WT and 5xFAD mice but did not reach statistical significance between groups (Figure 3F). Interestingly, the level of IL‐6 transcript was not increased in 5xFAD microglia and was not affected by BHB treatment (Figure 3E).
FIGURE 3.
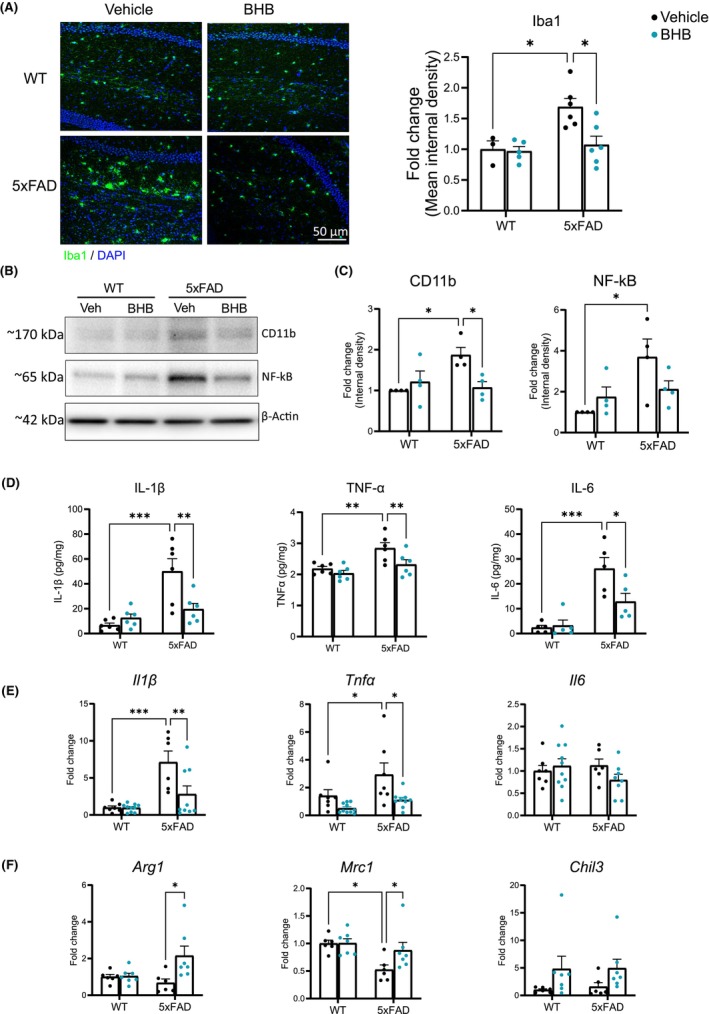
BHB treatment reduces microglial pro‐inflammatory activation in 5xFAD mice. (A) Representative fluorescent images from hippocampus co‐stained for Iba‐1 (green) and DAPI (blue). Bar graph shows quantification of Iba‐1 immunoreactivity. Group size: WT‐Vehicle = 3; WT‐BHB = 5; 5xFAD‐Vehicle = 6; 5xFAD‐BHB = 6. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between treatments (F(1,16) = 5.578, p = 0.031) and between genotypes F(1,16) = 8.446, p = 0.010. (B, C) Representative images of Western blots for CD11b and NF‐kB and quantification of band intensity expressed as fold change. n = 4 per group. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between genotypes in NF‐kB (F(1,13) = 7.996, p = 0.015). (D) ELISA quantification of pro‐inflammatory cytokines IL‐1β, TNF‐α, and IL‐6 levels in brain homogenates. n = 6 per group. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between treatments in IL‐1β (F(1,20) = 4.401, p = 0.049); and TNF‐α (F(1,20) = 6.718, p = 0.017); and significant differences between genotypes in IL‐1β (F(1,20) = 18.60, p < 0.001); TNF‐α (F(1,20) = 13.03, p = 0.002); and IL‐6 (F(1,16) = 30.27, p < 0.001); (E, F) qPCR quantification of pro‐inflammatory (E) and anti‐inflammatory (F) cytokines transcript expression in acutely isolated microglia. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between treatments in il1β (F(1,28) = 6.334, p = 0.018); tnfα (F(1,29) = 6.180, p = 0.019); arg1 (F(1,22) = 5.758, p = 0.025); and chil3 (F(1,22) = 5.170, p = 0.033); and significant differences between genotypes in IL‐1β (F(1,28) = 21.21, p < 0.001); tnfα (F(1,29) = 10.31, p = 0.003); and mrc1 (F(1,22) = 8.911, p = 0.007). Data are presented as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
3.4. One‐week BHB reduces microglial NLRP3‐inflammasome formation in the 5xFAD brain
Because of the significance of IL‐1β in mediating microglial effects on hLTD and the fact that BHB inhibited microglial inflammasome formation in vitro, 22 we tested whether BHB could block NLRP3‐inflammasome to reduce IL‐1β release in vivo. As expected, in microglia acutely isolated from 5xFAD mice, the levels of transcripts for NLRP3, apoptosis‐associated speck‐like protein containing a CARD (ASC), and Caspase‐1 that are signals and components required for inflammasome formation, were upregulated compared to those from WT microglia (Figure 4A). These increases were suppressed by the 1‐week BHB regimen. We next analyzed the levels of two core inflammasome assembly proteins, NLRP3 and ASC, in microglia by flow cytometry and found that the levels of both in IBA1‐positive cells were increased in 5xFAD microglia compared to WT microglia. One‐week BHB treatment significantly reduced the level of microglial NLRP3 protein (Figure 4B); it also induced a trend of reduction in ASC but this did not reach statistical significance (Figure 4C p value = 0.112). Because we were not able to optimize the flow cytometry method for pro‐caspase 1, the other inflammasome assembly protein, and because of the limited quantity of extractable material from isolated microglia, we analyzed it by Western blot using brain homogenates. Pro‐caspase 1 was significantly increased in 5xFAD mice and was decreased by BHB treatment (Figure 4D,E). At the effector side, Western blot showed increased cleaved IL‐1β levels in 5xFAD mice that were suppressed to the WT levels by BHB treatment, consistent with the above qPCR and ELISA results (Figure 4D,E). Taken together, these results indicate that 1‐week BHB treatment suppressed the elevated NLRP3 inflammasome activity in 5xFAD microglia and reduced their IL‐1β release.
FIGURE 4.
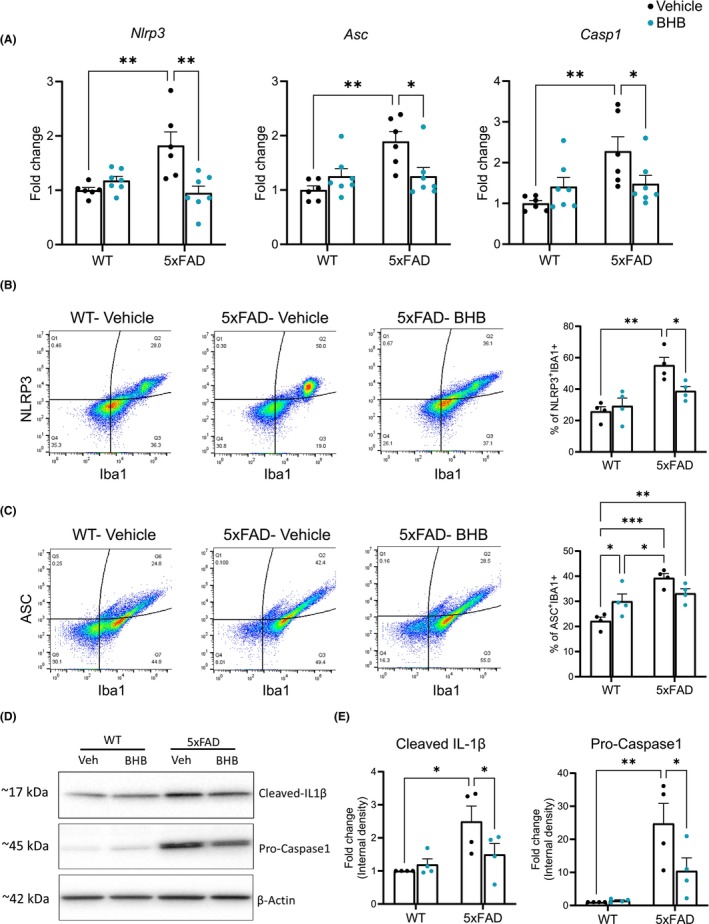
BHB treatment reduces microglial NLRP3‐inflammasome formation in 5xFAD mice. (A) qPCR quantification of nlrp3, asc, and caspase1 RNA expression in acutely isolated microglia. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between treatments in nlrp3 (F(1,22) = 6.019, p = 0.023); and significant differences between genotypes in nlrp3 (F(1,22) = 4.413, p = 0.047); asc (F(1,22) = 9.329, p = 0.006); and caspase1 (F(1,22) = 8.240, p = 0.009). (B) Flow cytometry quantification of NLRP3 in Iba1‐positive microglia. Shown are representative density plots and quantification of co‐localization for NLRP3 and Iba1 in dissociated brain cells. n = 4 per group. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between genotype (F(1,12) = 23.20, p < 0.001). (C) Flow cytometry quantification of ASC in Iba1‐positive microglia. Shown are representative density plots and quantification of co‐localization for ASC and Iba1 in dissociated brain cells. n = 4 per group. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between genotype (F(1,12) = 23.98, p < 0.001) (D, E) Representative images of Western blots for Cleaved IL‐1β and Caspase1 (D) and quantification of band intensity expressed as fold change (E). n = 4 per group. Two‐way ANOVA with Newman–Keuls's post‐hoc tests shows significant differences between genotype in cleaved IL‐1β (F(1,12) = 9.115, p = 0.011); and caspase1 (F(1,12) = 20.40, p < 0.001). Data are presented as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
3.5. One‐week BHB treatment regulates gene sets related to inflammation and microglia function
To determine how the 1‐week BHB injection alters the gene expression of 5xFAD mice, we conducted next‐generation RNA‐sequencing (RNA‐seq)‐based transcriptome analysis on comparable mid‐coronal sections (including hippocampus) of brains from vehicle‐treated and BHB‐treated 5xFAD mice. Unsupervised analysis of the transcript count data using principal component analysis (PCA) identified a distinction in clustering between vehicle and BHB‐treated 5xFAD (Figure S1). Differentially expressed genes (DEGs) analysis showed that a total of 452 genes were differentially expressed in 5xFAD+BHB compared to 5xFAD+Vehicle group, with 213 upregulated genes and 239 downregulated genes (Figure 5A). Functional enrichment analyses of the DEGs revealed multiple cellular and molecular pathways that were altered by BHB treatment in 5xFAD mice. Gene ontology (GO) analysis showed that BHB treatment enriched the biological processes (BP) of terms related to synaptic plasticity, such as “phosphorylation” (p = 7.30E‐04), “response to electrical stimulus” (p = 2.00E‐02), and “receptor internalization” (p = 5.00E‐02) (Figure 5B). GO analysis together with KEGG analysis (Figure S2) also found that BHB treatment upregulated pathways related to insulin signaling 28 (Figure 5B) involving the JNK, PI3K, and RAS pathways (Figures S3 and S4), consistent with the overall neuronal changes following KD in a previous report. 28 To test which pathways are likely to be involved in 5xFAD responses to BHB treatment, we performed gene set enrichment analysis (GSEA) of normalized counts of all genes using the Hallmark database. The top results by normalized score (NES), presented in Figure 5C, identified insulin and other metabolism related modules such as “glycolysis,” “p53 pathway,” “PI3K AKT mTOR signaling,” and “bile acid metabolism,” which have been considered to be altered in ketogenic conditions or with changes in dietary fat. 22 , 32 , 33 Among the five gene sets significantly down‐regulated by BHB (FDR < 25%) were “inflammatory response” (NES = −1.490), “complement” (NES = −1.380), and “cholesterol homeostasis” (NES = −1.520) which are relevant to microglia function. Degrees of individual transcript changes within the gene sets are presented in Figure S5. An examination of enriched genes in these categories show decreased expression of gene linked to pro‐inflammatory state of microglia and inflammasome formation (Nlrp3, Clec5a, Mmp8, Nfkb1, Il6, Il1a, Ifngr2, Il18rap, Tlr1, and Kcna3) and upregulation of anti‐inflammatory genes (Nfkbie, Rchy1, Slc16a6, and Tob1) (Figure S5) by BHB. Interestingly, the two genes most robustly upregulated by BHB, Nfkbie encodes the NF‐κB inhibitor protein called I‐kappa‐B‐epsilon (IκBε) 34 and Rchy1 encodes the ring finger and CHY zinc finger domain‐containing protein 1, which is an E3 ligase shown to negatively regulate histone deacetylase 2 (HDAC2) (Figure S5, red circles). 35 The upregulation of these two genes is consistent with BHB's ability to inhibit NF‐κB and HDACs and provides potential novel transcriptional mechanisms for BHB signaling. 15 , 36 , 37
FIGURE 5.
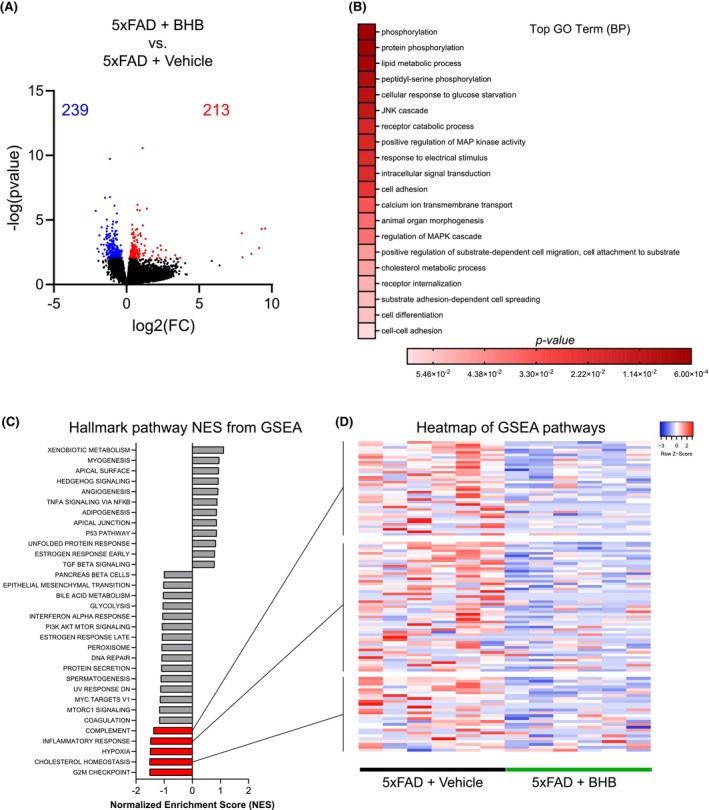
BHB treatment regulates gene sets related to inflammation and microglia function. (A) Volcano plot showing the top 20,000 genes. Red dots show the significantly up‐regulated genes, while blue dots the significantly down‐regulated genes in 5xFAD+BHB versus 5xFAD+vehicle. (B) Top gene ontology terms of DEGs in 5xFAD+BHB compared to 5xFAD+vehicle. (C) Results of GSEA Hallmark analysis showing enriched gene sets. Bars in red indicate significant enrichment at FDR < 25%; bars in gray represent gene sets with FDR > 25% and a nominal p value <5%. A positive Normalized Enrichment Score (NES) value indicates enrichment in the BHB phenotype, a negative NES indicates enrichment in the vehicle phenotype. (D) Heatmap of the three categories of interest from GSEA analysis.
3.6. One‐week BHB enhances amyloid phagocytosis and reduces cerebral amyloid load in the 5xFAD brain
We reported that BHB corrects AβO‐induced deficits in microglial phagocytic activity in vitro. 22 To test whether this is the case in vivo, we used an established in vivo phagocytosis assay 38 , 39 to co‐stain brain sections with lysosomal marker LAMP1 and amyloid dye FSB ((E,E)‐1‐fluoro‐2,5‐bis(3‐hydroxycarbonyl‐4‐hydroxy) styrylbenzene). BHB treatment resulted in a remarkable accentuation of colocalization of LAMP1 and FSB (yellow color in Figure 6A) surrounding each amyloid plaque, indicating enhanced amyloid phagocytosis. Enhanced amyloid phagocytosis may result in reduced cerebral amyloid load. Immunohistochemistry using 4G8, an antibody against Aβ, showed that BHB treatment resulted in smaller and less numerous amyloid plaques in 5xFAD mice with a ~ 25% reduction of cerebral amyloid deposit (p = 0.029) (Figure 6B). We previously identified that 5xFAD brain solubilized by RIPA buffer contained mainly SDS‐stable Aβ dimers. 40 Western blot of such prepared brain extracts showed that BHB treatment significantly reduced the levels of SDS‐stable dimer and trimer (Figure 6C). These results indicate that the 1‐week BHB regimen was able to reduce cerebral amyloid load in 5xFAD mice via enhancing amyloid phagocytosis.
FIGURE 6.
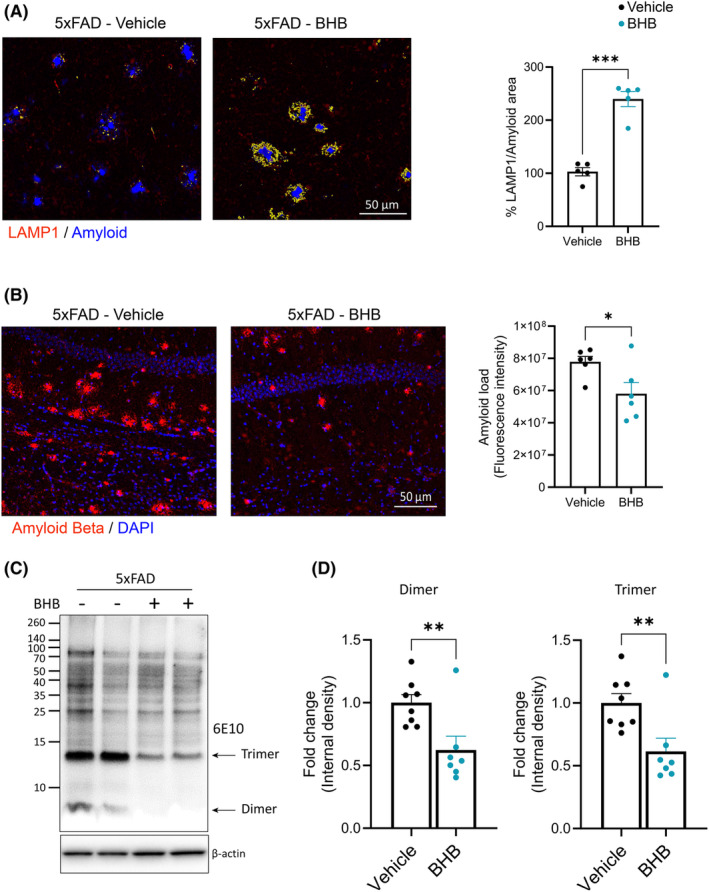
BHB treatment enhances amyloid phagocytosis and reduces cerebral amyloid load in the 5xFAD brain. (A) Representative fluorescent images and quantification of the colocalization of LAMP1 (red) and amyloid plaques (FSB, Blue) staining in cerebral cortex. Co‐localization of LAMP1 and amyloid is highlighted in yellow. n = 4. (B) Representative fluorescent images from hippocampus co‐stained for Amyloid Beta (4G8, red) and DAPI (blue). Bar graph shows quantification of the amyloid load indicated by 4G8 immunoreactivity. n = 6 (C, D) Representative images of Western blots for β‐amyloid (clone 6E10) (C) and bar graphs showing quantification of the band intensity for Aβ dimer trimer, expressed as fold change (D). n = 8. Data are presented as mean ± SEM *p < 0.05, **p < 0.01, ***p < 0.001, unpaired t‐test.
4. DISCUSSION
Our 1‐week regimen of BHB injection generated a daily brief spike of low‐level ketosis. Although the ketosis was brief and mild, this regimen was able to mitigate synaptic deficits, pro‐inflammatory microglia activation, phagocytotic deficits, and amyloid deposition, key pathological features in 5xFAD mice. These effects replicated those effected by BHB in vitro on human iPS cell‐derived microglia, 22 and we linked improved hLTD to reduced NLRP3 inflammasome‐generated IL‐1β and reduced amyloid load to enhanced Aβ phagocytosis.
Perhaps the most intriguing finding in our study is that the 1‐week BHB regimen mitigated hLTD but not hLTP deficits in 5xFAD mice. Synaptic strengthening through LTP and weakening through LTD, less studied and less understood, are considered to shape learning and memory. 41 , 42 , 43 , 44 Recent studies demonstrated distinct roles of hLTD in specific aspects of hippocampus‐dependent associative learning and long‐term memory, 41 , 44 and that hLTD strengthens the robustness of stored experience by pruning away synapses that are weakly integrated into a synaptic network. 44 , 45 Thus, together with hLTP, hLTD is required for the integrity of hippocampal function. In Alzheimer's disease, alterations of hippocampal synaptic efficacy occur prior to frank neuronal degeneration, and AβO mediates major synaptotoxic offenses. 46 Both hippocampal hLTP and hLTD have been shown to be impaired in several Alzheimer's disease mouse models. 47 However, we are not aware of any previous reports demonstrating hLTD deficits in the widely used 5xFD model. Here we showed for the first time that 12‐month‐old 5xFAD mice had significant hLTD deficits in the Schaffer collateral‐CA1 synapse. Because impairments of hLTD are implicated in human Alzheimer's disease, 47 5xFAD mice could serve as a model of early events leading to Alzheimer's disease‐like hippocampal synaptic failure. 46
We recently reported that a 7‐month course of daily meal‐fed ketogenic diet 19 recovered the deficits of hLTP in APP‐PS1 mice, but hLTD was not investigated. 21 In this study, the blood level of BHB remained chronically elevated compared to the control diet as the mice were maintained on a KD diet throughout the study. In contrast, the present study produced daily, episodic ketosis generated by the 1‐week course of BHB administration. Although the paradigms used in these two studies have several clear differences, both achieved comparable maximum daily BHB levels. Our results, therefore, may suggest a higher sensitivity of hLTD to ketogenic metabolic regulations or microglia influences than hLTP. This result provides important clues to the differential mechanisms regulating hLTP and hLTD.
Microglia were shown to influence hLTD by releasing mediators such as ATP, 48 BDNF, 49 and IL‐1β, 31 and evidence also suggests the involvement of microglia pruning of weak synapses. 44 , 45 However, whether microglia are involved in hLTD deficits in Alzheimer's disease has not been studied. We designed the “subacute” 1‐week protocol with the goal to study BHB effects on microglia in vivo, rather than the immediate effects of BHB on synaptic neurotransmission or on energy metabolism. Here we show that the hLTD deficit in 5xFAD mice requires IL‐1β for its expression and is not related to intrinsic synaptic impairments because blockage of IL‐1β fully recovered hLTD. BHB treatment inhibited the 5xFAD upregulation of IL‐1β via inhibition of NLRP3 inflammasome formation, a mechanism that may contribute to its hLTD rescuing effect and explain the high sensitivity of hLTD deficits to the effect of BHB.
The other intriguing finding is that, even with this relatively short course, BHB treatment was able to significantly reduce the Aβ‐amyloid load, including amyloid plaques recognized by a Congo‐red derivative dye in brain sections and the soluble but SDS‐stable Aβ dimer and trimer shown in Western blot. These reductions were linked to enhanced amyloid phagocytosis, which was robust around all large and small amyloid plaques following BHB treatment (Figure 6). While Aβ deposition due to imbalance in its production and clearance has been hypothesized as a “very early, often initiating factor in Alzheimer's disease” in the amyloid hypothesis, 50 other factors such as chronic inflammation and impaired mitochondria bioenergetics may also be among the early initiating events, with Aβ deposition as a consequence. 51 It is intriguing to speculate that BHB may rapidly reduce the Aβ‐amyloid load via reducing microglia‐orchestrated chronic inflammation and correcting overall mitochondrial dysfunction, leading to enhanced clearance and reduced production of Aβ. Our prior in vitro data using human microglia indeed supports this hypothesis. 22
BHB has been increasingly recognized as an active signaling molecule with a wide range of direct actions and indirect actions via its metabolites. 15 The known direct actions include its activation of cell surface receptors HCAR2 (hydroxycarboxylic acid receptor 2) and FFAR3 (free fatty acid receptor 3), inhibition of histone deacetylases (HDACs) to affect epigenetics, modulation of potassium channel function, and direct β‐hydroxybutyrylation of proteins as a form of post‐translational modification. 15 Inhibition of potassium efflux through potassium channels may explain BHB's effects on NLRP3 inflammasome. 52 Notably, in the brain, HCAR2 is expressed selectively by microglia, and niacin, a high‐affinity ligand for HCAR2, was shown to modulate microglial response and limit disease progression in 5xFAD mice. 53 It is likely that BHB, also a ligand for HCAR2, induces a mechanism parallel to that of niacin. Furthermore, our RNA‐seq data showed that BHB treatment upregulated Nfkbie and Rchy1 genes encoding negative regulators of NF‐κB and HDAC2, respectively. This result provides possible molecular mechanisms via which BHB suppresses NF‐κB (and thereby the downstream NLRP3) 54 and HDAC2.
Further investigations are warranted to identify signaling pathways required for the pleotropic BHB effects on microglia. Our previous study showed that BHB was able to induce a metabolic shift to enhance the endogenous mitochondrial ketogenic pathway (MKP) of microglia under pro‐inflammatory conditions. 22 Here we propose that microglia respond to episodic ketosis with such a metabolic shift to propagate the effects of BHB perhaps via promoting endogenous MKP. Many cells have the ability to generate ketone bodies by MPK. In several published instances, ketogenesis was shown to upregulate gene expression of components of the MKP, 55 , 56 similar to what we have shown in human iPS cell‐derived microglia 22 and therefore further promoting ketosis. Leucine, an essential branched‐chain amino acid metabolized to generate BHB, 57 potently increases lipid content to further enhance ketogenesis and shifts microglia toward anti‐inflammatory states. 58 , 59 BHB generated by cells may play important functional roles in a cell autonomous or non‐autonomous manner. 37 , 60 , 61 , 62 Focally produced BHB may act as an endogenous inhibitor of histone deacetylases to regulate gene expression. 37 It may also act as a paracrine chemical mediator similar to those among the growing list of metabolic signaling molecules such as ATP, lactate, and epoxyeicosatrienoic acids. 63 , 64 In addition, the possibility of an “astrocyte‐neuron ketone shuttle” was raised. 13 , 65 Our observation of the significant impact by a short regimen of episodic ketosis provides strong rationale to investigate this MKP activation mechanism in vivo and how BHB may affect not only microglia or astrocytes but also neuron–glia interactions to ultimately influence synaptic plasticity.
Because beneficial results were achieved with mild episodic BHB elevation alone without diet restriction (mice had ad libitum access to a regular chow) and without the need of feeding a KD, our results have significant implications to human ketotherapeutics. KDs are known for their restrictive nature, challenging long‐term compliance, and, due to their high amount of fat, potential harmful effects in some individuals, for example, elevated LDL cholesterol. These limitations particularly hinder the compliance of elderly individuals in risk of Alzheimer's disease. Our results open the possibility for approaches to manipulate blood BHB levels that are less restrictive, potentially safer, and easier to follow than a KD. Future translational and mechanistic studies using approaches such as short‐term BHB injections or dietary ketone esters, a translatable form of induced ketosis compared favorably among other ketogenic interventions, 66 are warranted.
AUTHOR CONTRIBUTIONS
L.‐W. Jin and I. Maezawa conceived and designed the research; J. Di Lucente performed the research, acquired, analyzed and interpreted the data; All authors were involved in drafting and revising the manuscript.
FUNDING INFORMATION
This work was supported by a grant from the U.S. NIH/NIA grants P01 AG025532 to J.D‐L., L.‐W.J., J.L. and I.M., and in part by RF1 AG071665 to I.M. and P30 AG072972 to L.‐W.J. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICT OF INTEREST STATEMENT
The authors report no competing interests.
Supporting information
Data S1: Supporting information.
ACKNOWLEDGMENTS
We thank Dr. Jennifer Rutkowsky and Dr. Zeyu Zhou for reviewing the manuscript and providing helpful recommendations.
Di Lucente J, Ramsey JJ, Jin L‐W, Maezawa I. The impact of mild episodic ketosis on microglia and hippocampal long‐term depression in 5xFAD mice. FASEB BioAdvances. 2024;6:581‐596. doi: 10.1096/fba.2024-00123
Contributor Information
Lee‐Way Jin, Email: lwjin@ucdavis.edu.
Izumi Maezawa, Email: imaezawa@ucdavis.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. Uncropped blots are provided in Figure S6. RNAseq data are deposited on the GEO repository and are accessible with number GSE269488.
REFERENCES
- 1. 2023 Alzheimer's disease facts and figures. Alzheimers Dement. 2023;19:1598‐1695. [DOI] [PubMed] [Google Scholar]
- 2. Niotis K, Saperia C, Saif N, Carlton C, Isaacson RS. Alzheimer's disease risk reduction in clinical practice: a priority in the emerging field of preventive neurology. Nat Mental Health. 2024;2:25‐40. [Google Scholar]
- 3. Marschallinger J, Iram T, Zardeneta M, et al. Lipid‐droplet‐accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci. 2020;23:194‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnson ECB, Dammer EB, Duong DM, et al. Large‐scale proteomic analysis of Alzheimer's disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020;26:769‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bellenguez C, Kucukali F, Jansen IE, et al. New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat Genet. 2022;54:412‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314‐1318. [DOI] [PubMed] [Google Scholar]
- 9. Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752‐758. [DOI] [PubMed] [Google Scholar]
- 10. Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110:3458‐3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bernier LP, York EM, MacVicar BA. Immunometabolism in the brain: how metabolism shapes microglial function. Trends Neurosci. 2020;43:854‐869. [DOI] [PubMed] [Google Scholar]
- 12. Paoli A, Tinsley GM, Mattson MP, De Vivo I, Dhawan R, Moro T. Common and divergent molecular mechanisms of fasting and ketogenic diets. Trends Endocrinol Metab. 2024;35:125‐141. [DOI] [PubMed] [Google Scholar]
- 13. Koppel SJ, Swerdlow RH. Neuroketotherapeutics: a modern review of a century‐old therapy. Neurochem Int. 2018;117:114‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Taylor MK, Swerdlow RH, Sullivan DK. Dietary neuroketotherapeutics for Alzheimer's disease: an evidence update and the potential role for diet quality. Nutrients. 2019;11:1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Newman JC, Verdin E. β‐Hydroxybutyrate: a signaling metabolite. Annu Rev Nutr. 2017;37:51‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pathak SJ, Zhou Z, Steffen D, et al. 2‐month ketogenic diet preferentially alters skeletal muscle and augments cognitive function in middle aged female mice. Aging Cell. 2022;21:e13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roberts MN, Wallace MA, Tomilov AA, et al. A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 2017;26:539‐546. e535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fortier M, Castellano CA, Croteau E, et al. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 2019;15:625‐634. [DOI] [PubMed] [Google Scholar]
- 19. Dilmore AH, Martino C, Neth BJ, et al. Effects of a ketogenic and low‐fat diet on the human metabolome, microbiome, and foodome in adults at risk for Alzheimer's disease. Alzheimers Dement. 2023;19:4805‐4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davis JJ, Fournakis N, Ellison J. Ketogenic diet for the treatment and prevention of dementia: a review. J Geriatr Psychiatry Neurol. 2021;34:3‐10. [DOI] [PubMed] [Google Scholar]
- 21. Di Lucente J, Persico G, Zhou Z, et al. Ketogenic diet and BHB rescue the fall of long‐term potentiation in an Alzheimer's mouse model and stimulates synaptic plasticity pathway enzymes. Commun Biol. 2024;7:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jin LW, Di Lucente J, Ruiz Mendiola U, et al. The ketone body β‐hydroxybutyrate shifts microglial metabolism and suppresses amyloid‐β oligomer‐induced inflammation in human microglia. FASEB J. 2023;37:e23261. [DOI] [PubMed] [Google Scholar]
- 23. Oakley H, Cole SL, Logan S, et al. Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129‐10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin LW, Lucente JD, Nguyen HM, et al. Repurposing the KCa3.1 inhibitor senicapoc for Alzheimer's disease. Ann Clin Transl Neurol. 2019;6:723‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martinello KA, Meehan C, Avdic‐Belltheus A, et al. Acute LPS sensitization and continuous infusion exacerbates hypoxic brain injury in a piglet model of neonatal encephalopathy. Sci Rep. 2019;9:10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin LW, Horiuchi M, Wulff H, et al. Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: a mechanism for mitochondrial dysfunction and neurotoxicity. J Neurosci. 2015;35:2516‐2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Di Lucente J, Nguyen HM, Wulff H, Jin LW, Maezawa I. The voltage‐gated potassium channel Kv1.3 is required for microglial pro‐inflammatory activation in vivo. Glia. 2018;66:1881‐1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koppel SJ, Pei D, Wilkins HM, et al. A ketogenic diet differentially affects neuron and astrocyte transcription. J Neurochem. 2021;157:1930‐1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maezawa I, Nguyen HM, Di Lucente J, et al. Kv1.3 inhibition as a potential microglia‐targeted therapy for Alzheimer's disease: preclinical proof of concept. Brain. 2017;141:596‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dudek SM, Bear MF. Homosynaptic long‐term depression in area CA1 of hippocampus and effects of N‐methyl‐D‐aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363‐4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ikegaya Y, Delcroix I, Iwakura Y, Matsuki N, Nishiyama N. Interleukin‐1beta abrogates long‐term depression of hippocampal CA1 synaptic transmission. Synapse. 2003;47:54‐57. [DOI] [PubMed] [Google Scholar]
- 32. Liu K, Li F, Sun Q, et al. p53 β‐hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019;10:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McDaniel SS, Rensing NR, Thio LL, Yamada KA, Wong M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52:e7‐e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hayden MS, Ghosh S. Shared principles in NF‐kappaB signaling. Cell. 2008;132:344‐362. [DOI] [PubMed] [Google Scholar]
- 35. Choi M, Choi YM, An IS, Bae S, Jung JH, An S. E3 ligase RCHY1 negatively regulates HDAC2. Biochem Biophys Res Commun. 2020;521:37‐41. [DOI] [PubMed] [Google Scholar]
- 36. Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite β‐hydroxybutyrate blocks NLRP3 inflammasome‐mediated inflammatory disease. Nat Med. 2015;21:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shimazu T, Hirschey MD, Newman J, et al. Suppression of oxidative stress by β‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kiyota T, Machhi J, Lu Y, et al. URMC‐099 facilitates amyloid‐β clearance in a murine model of Alzheimer's disease. J Neuroinflammation. 2018;15:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bolmont T, Haiss F, Eicke D, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283‐4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hong HS, Maezawa I, Budamagunta M, et al. Candidate anti‐Abeta fluorene compounds selected from analogs of amyloid imaging agents. Neurobiol Aging. 2008;31:1690‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Compans B, Camus C, Kallergi E, et al. NMDAR‐dependent long‐term depression is associated with increased short term plasticity through autophagy mediated loss of PSD‐95. Nat Commun. 2021;12:2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lüscher C, Malenka RC. NMDA Receptor‐Dependent Long‐Term Potentiation and Long‐Term Depression (LTP/LTD). Cold Spring Harb Perspect Biol. 2012;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu X, Gu QH, Duan K, Li Z. NMDA receptor‐dependent LTD is required for consolidation but not acquisition of fear memory. J Neurosci. 2014;34:8741‐8748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stacho M, Manahan‐Vaughan D. The intriguing contribution of hippocampal long‐term depression to spatial learning and long‐term memory. Front Behav Neurosci. 2022;16:806356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Piochon C, Kano M, Hansel C. LTD‐like molecular pathways in developmental synaptic pruning. Nat Neurosci. 2016;19:1299‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789‐791. [DOI] [PubMed] [Google Scholar]
- 47. Mango D, Saidi A, Cisale GY, Feligioni M, Corbo M, Nisticò R. Targeting synaptic plasticity in experimental models of Alzheimer's disease. Front Pharmacol. 2019;10:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pougnet JT, Toulme E, Martinez A, Choquet D, Hosy E, Boué‐Grabot E. ATP P2X receptors downregulate AMPA receptor trafficking and postsynaptic efficacy in hippocampal neurons. Neuron. 2014;83:417‐430. [DOI] [PubMed] [Google Scholar]
- 49. Mizui T, Ishikawa Y, Kumanogoh H, et al. BDNF pro‐peptide actions facilitate hippocampal LTD and are altered by the common BDNF polymorphism Val66Met. Proc Natl Acad Sci USA. 2015;112:E3067‐E3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taylor MK, Sullivan DK, Keller JE, Burns JM, Swerdlow RH. Potential for ketotherapies as amyloid‐regulating treatment in individuals at risk for Alzheimer's disease. Front Neurosci. 2022;16:899612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite beta‐hydroxybutyrate blocks NLRP3 inflammasome‐mediated inflammatory disease. Nat Med. 2015;21:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Moutinho M, Puntambekar SS, Tsai AP, et al. The niacin receptor HCAR2 modulates microglial response and limits disease progression in a mouse model of Alzheimer's disease. Sci Transl Med. 2022;14:eabl7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cullingford TE, Eagles DA, Sato H. The ketogenic diet upregulates expression of the gene encoding the key ketogenic enzyme mitochondrial 3‐hydroxy‐3‐methylglutaryl‐CoA synthase in rat brain. Epilepsy Res. 2002;49:99‐107. [DOI] [PubMed] [Google Scholar]
- 56. Seira O, Kolehmainen K, Liu J, et al. Ketogenesis controls mitochondrial gene expression and rescues mitochondrial bioenergetics after cervical spinal cord injury in rats. Sci Rep. 2021;11:16359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bixel MG, Hamprecht B. Generation of ketone bodies from leucine by cultured astroglial cells. J Neurochem. 1995;65:2450‐2461. [DOI] [PubMed] [Google Scholar]
- 58. De Simone R, Vissicchio F, Mingarelli C, et al. Branched‐chain amino acids influence the immune properties of microglial cells and their responsiveness to pro‐inflammatory signals. Biochim Biophys Acta. 2013;1832:650‐659. [DOI] [PubMed] [Google Scholar]
- 59. Rivera ME, Lyon ES, Johnson MA, Vaughan RA. Leucine increases mitochondrial metabolism and lipid content without altering insulin signaling in myotubes. Biochimie. 2020;168:124‐133. [DOI] [PubMed] [Google Scholar]
- 60. Adijanto J, Du J, Moffat C, Seifert EL, Hurle JB, Philp NJ. The retinal pigment epithelium utilizes fatty acids for ketogenesis. J Biol Chem. 2014;289:20570‐20582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang Q, Zhou Y, Rychahou P, et al. Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ. 2017;24:458‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Royo T, Pedragosa MJ, Ayté J, Gil‐Gómez G, Vilaró S, Hegardt FG. Testis and ovary express the gene for the ketogenic mitochondrial 3‐hydroxy‐3‐methylglutaryl‐CoA synthase. J Lipid Res. 1993;34:867‐874. [PubMed] [Google Scholar]
- 63. Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end‐product to signalling molecule. Nat Rev Neurosci. 2018;19:235‐249. [DOI] [PubMed] [Google Scholar]
- 65. Guzmán M, Blázquez C. Is there an astrocyte‐neuron ketone body shuttle? Trends Endocrinol Metab. 2001;12:169‐173. [DOI] [PubMed] [Google Scholar]
- 66. Soto‐Mota A, Norwitz NG, Clarke K. Why a d‐β‐hydroxybutyrate monoester? Biochem Soc Trans. 2020;48:51‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supporting information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Uncropped blots are provided in Figure S6. RNAseq data are deposited on the GEO repository and are accessible with number GSE269488.