

CONSPECTUS:
Catalytic intermolecular hydroamination of alkenes is an atom- and step-economical method for the synthesis of amines, which have important applications as pharmaceuticals, agrochemicals, catalysts, and materials. However, hydroaminations of alkenes in high yield with high selectivity are challenging to achieve because these reactions often lack a thermodynamic driving force and often are accompanied by side reactions, such as alkene isomerization, telomerization, and oxidative amination. Consequently, early examples of hydroamination were generally limited to the additions of N–H bonds to conjugated alkenes or strained alkenes, and the catalytic hydroamination of unactivated alkenes with late transition metals has only been disclosed recently. Many classes of catalysts, including early transition metals, late transition metals, rare-earth metals, acids, and photocatalysts, have been reported for catalytic hydroamination. Among them, late transition-metal complexes possess several advantages, including their relative ease of handling and their high compatibility of substrates containing polar or sensitive functional groups.
This Account describes the progression in our laboratory of hydroaminations catalyzed by late transition-metal complexes from the initial additions of N–H bonds to activated alkenes to the more recent additions to unactivated alkenes. Our developments include the Markovnikov and anti-Markovnikov hydroamination of vinylarenes with palladium, rhodium, and ruthenium, the hydroamination of dienes and trienes with nickel and palladium, the hydroanimation of bicyclic strained alkenes with neutral iridium, and the hydroamination of unactivated terminal and internal alkenes with cationic iridium and ruthenium. Enantioselective hydroaminations of these classes of alkenes to form enantioenriched, chiral amines also have been developed.
Mechanistic studies have elucidated the elementary steps and the turnover-limiting steps of these catalytic reactions. The hydroamination of conjugated alkenes catalyzed by palladium, rhodium, nickel, and ruthenium occurs by turnover-limiting nucleophilic attack of the amine on a coordinated benzyl, allyl, alkene, or arene ligand. On the other hand, the hydroamination of unconjugated alkenes catalyzed by ruthenium and iridium occurs by turnover-limiting migratory insertion of the alkene into a metal–nitrogen bond. In addition, pathways for the formation of side products, including isomeric alkenes and enamines, have been identified during our studies. During studies on enantioselective hydroamination, the reversibility of the hydroamination has been shown to erode the enantiopurity of the products. Based on our mechanistic understandings, new generations of catalysts that promote catalytic hydroaminations with higher rates, chemoselectivity, and enantioselectivity have been developed. We hope that our discoveries and mechanistic insights will facilitate the further development of catalysts that promote selective, practical, and efficient hydroamination of alkenes.
Graphical Abstract
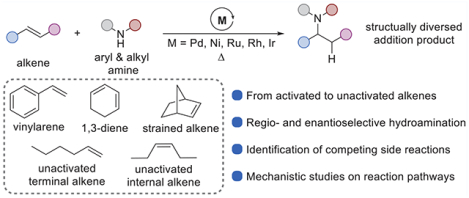
1. INTRODUCTION
The intermolecular hydroamination of alkenes is the addition of an N−H bond across a C=C double bond and is an atom- and step-economical method for the synthesis of alkylamines.5 Due to their Lewis basicity and hydrogen-bond acceptor properties, alkylamines are common substructures in pharmaceuticals, agrochemicals, catalysts, and materials. Because amines and alkenes are both nucleophilic, the hydroamination of alkenes generally does not occur without a catalyst.5l Since the first observation of the catalytic hydroamination of ethylene with ammonia in the presence of metallic sodium or lithium in 1954,6 a series of catalysts, including alkali metals, early transition-metal complexes, and late transition-metal complexes, have been reported for the hydroamination of multiple classes of alkenes, such as vinylarenes, dienes, allenes, and strained alkenes,5 but only recently have catalysts for the thermal and photocatalytic7 intermolecular addition of N–H bonds to unactivated alkenes been disclosed.
Several challenges needed to be overcome to create highly selective and active catalysts for hydroamination. First, the reaction is close to thermoneutral or even endergonic,8 as shown by experimental measurements of equilibria, thereby limiting the conversion of certain combinations of N–H donors and alkenes and rendering enantioselective intermolecular hydroaminations difficult to achive.2,3,9 Second, systems that catalyze hydroamination often promote accompanying side reactions, such as the isomerization and telomerization of alkenes, as well as oxidative amination,1–3 making precise control of the chemoselectivity during hydroamination essential. Third, the hydroamination of alkenes can form constitutional isomers, and the challenge confronting regioselective hydroamination is especially pronounced for the intermolecular addition of unsymmetrical internal alkenes.3,10 Finally, for metal-catalyzed hydroaminations, poisoning of the catalyst by amines is common because they are stronger σ donors and usually bind more strongly to metals than alkenes do.5l
Since 2000, our group has extensively studied the intermolecular hydroamination of alkenes and has discovered a variety of catalysts based on late transition metals for the hydroamination of vinylarenes, dienes, trienes, bicyclic strained alkenes, and unactivated terminal and internal alkenes. We focused on creating catalysts based on late transition metals for these reactions because such catalysts are easier to handle and are more tolerant of polar functional groups than those based on alkali metals, early transition metals, or rare-earth metals. As part of this work, we reported a series of asymmetric hydroaminations to form highly enantioenriched amines from both feedstock alkenes and complex alkenes. Detailed mechanistic studies have elucidated the steps of the catalytic cycles, which included novel complexes and elementary reactions. This information has been used to develop more active and selective catalysts for a wide range of hydroaminations. This Account describes our development of intermolecular hydroamination reactions, from our initial examples with activated alkenes, such as vinylarenes and dienes, to our more recent reactions with unstrained and unconjugated terminal and internal alkenes. Interleaved with these studies on catalyst development are mechanistic information and accompanying features of our design that led to selective catalysis.
2. OVERVIEW OF HYDROAMINATION OF VINYLARENES CATALYZED BY COMPLEXES OF LATE TRANSITION METALS
The hydroamination of vinylarenes is a valuable reaction for the synthesis of phenethylamines as building blocks for pharmaceuticals.11 The intermolecular hydroamination of vinylarenes is widely studied, and both Markovnikov and anti-Markovnikov hydroaminations of vinylarenes have been reported to proceed in the presence of transition metals,12 rare-earth metals,5c alkali metals,13 and acids.10,14
2.1. Markovnikov Hydroamination of Vinylarenes Catalyzed by Complexes of Late Transition Metals
In 2000, Dr. Motoi Kawatsura discovered the Pd-catalyzed Markovnikov hydroamination of vinylarenes with arylamines (Figure 1a).11 The combination of Pd(TFA)2, DPPF, and TfOH was found to catalyze the hydroamination of both electron-poor and electron-neutral vinylarenes to afford sec-phenethylamines in high yield. This reaction was compatible with primary anilines containing methoxy and trifluoromethyl substituents, as well as secondary N-methylanilines. The addition of aniline to para-trifluoromethylstyrene was rendered enantioselective to afford the corresponding product in 81% ee when [(R)-BINAP]Pd(OTf)2 was used as the catalyst (Figure 1b). The acid cocatalyst and the counteranion of the acid were important to achieve a high rate of reaction. The scope of amine that underwent Pd-catalyzed Markovnikov hydroamination of vinylarenes was extended to secondary aliphatic amines and benzylic amines by the replacement of toluene with dioxane as the solvent (Figure 1c).15
Figure 1.
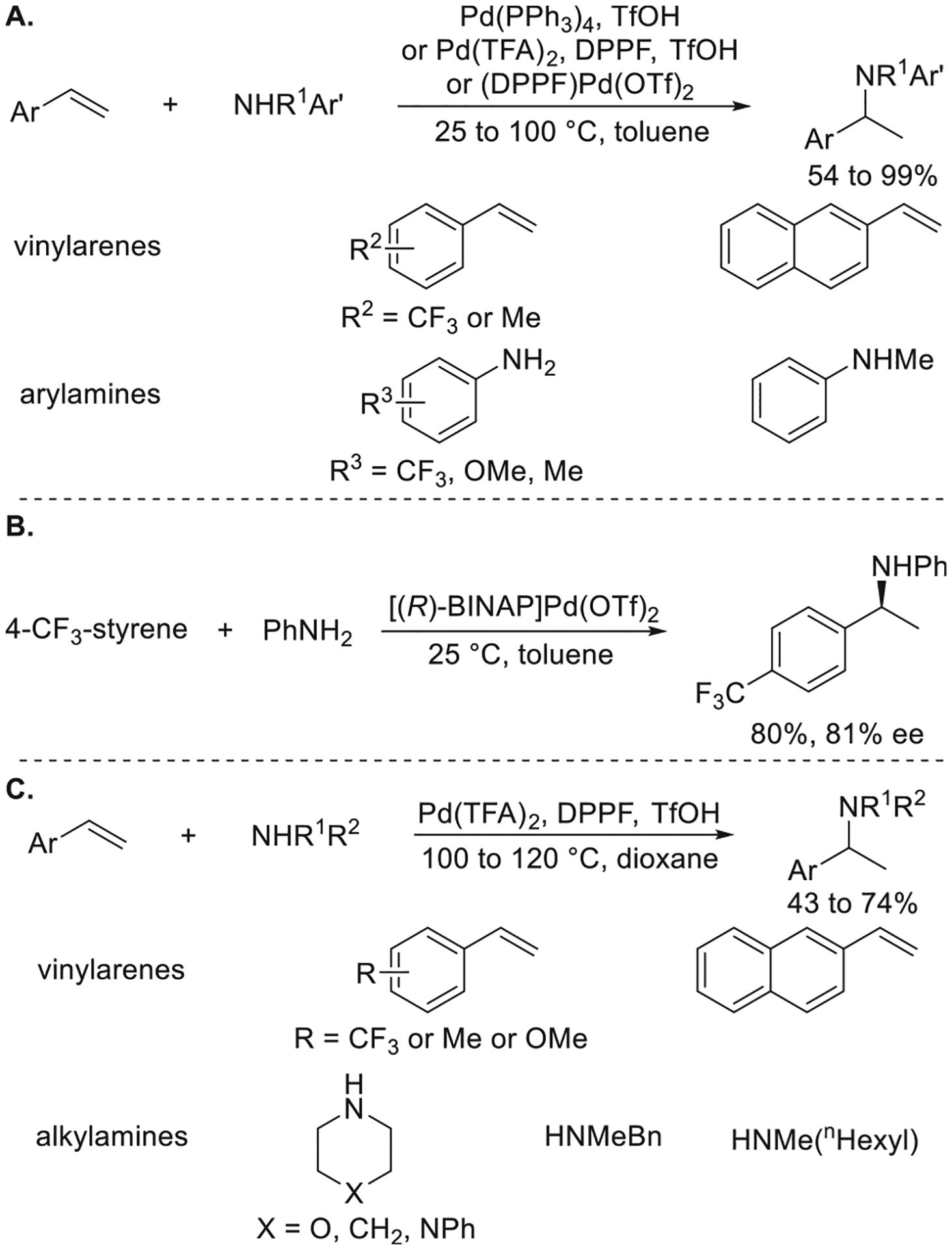
(A) Pd-catalyzed Markovnikov hydroamination of vinylarenes with arylamines. (B) Enantioselective hydroamination of vinylarenes with arylamines. (C) Pd-catalyzed hydroamination of vinylarenes with alkylamines. Yields and enantioselectivities are from refs 11 and 15.
Dr. Ulrike Nettekoven conducted mechanistic studies to understand the possible pathway for this Pd-catalyzed hydroamination of vinylarenes (Figure 2).16 The resting state of the catalyst for the reaction of vinylnaphthalene with aniline in the presence of [(R)-Tol-BINAP]Pd(OTf)2 was determined, by NMR spectroscopy, to be a mixture of diastereomeric forms of [(R)-Tol-BINAP][1-(2-naphthyl-ethyl)]Pd(OTf) (Figure 2a). The major diastereomer was characterized crystallographically, and the hydroamination of vinylnaphthalene with aniline catalyzed by the isolated diastereomeric mixture afforded product in a comparable yield and at a comparable rate to those catalyzed by the species generated from [(R)-Tol-BINAP]Pd(OTf)2. Treatment of [(R)-Tol-BINAP][(S)-1-(2-naphthyl-ethyl)]Pd(OTf) with aniline afforded the (R)-N-1-(2-naphthyl)ethylamine product in 71% ee. This result suggested that the C–N bond in the product is primarily formed by the external nucleophilic attack of aniline at the benzylic position, rather than by a concerted reductive elimination from a (phenethyl)Pd anilide complex (Figure 2a). Consequently, the catalytic cycle shown in Figure 2b was proposed for the hydroamination of vinylarenes with arylamines. L2Pd(OTf)2 enters the catalytic cycle by reacting with the vinylarene and the amine to form L2Pd(H)(OTf). Coordination of the vinylarene to the Pd center, followed by migratory insertion of the alkene into the Pd–H bond, affords the Pd(η3-benzyl) complex as the catalyst resting state. Turnover-limiting nucleophilic attack of aniline on the Pd(η3-benzyl) complex, followed by proton transfer, generates the hydroamination product and regenerates L2Pd(H)(OTf) to complete the catalytic cycle.
Figure 2.
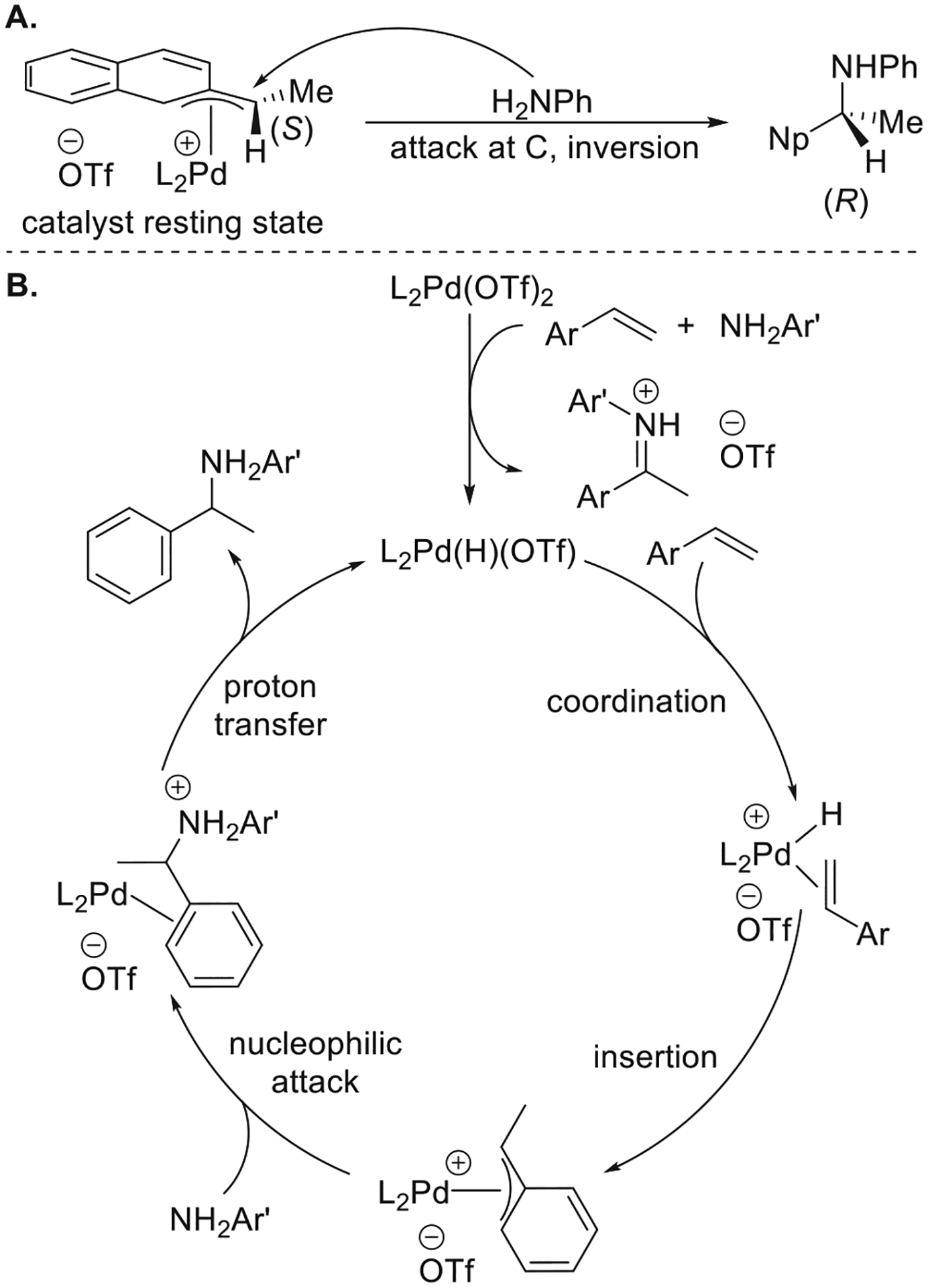
(A) Catalyst resting state for the Pd-catalyzed Markovnikov hydroamination and possible pathway for the formation of the C–N bond. (B) Proposed mechanism for the Pd-catalyzed hydroamination of vinylarenes with arylamines.
A further increase in the rate of hydroamination of vinylarenes with arylamines was achieved by accelerating the turnover-limiting, C–N bond-forming step occurring by nucleophilic attack of the arylamine on the Pd(η3-benzyl) complex. Stoichiometric reactions of isolated L2Pd(η3-CH2Ph)(OTf) complexes with aniline occurred with a positive relationship between the rate of the nucleophilic attack and the bite angle of the bidentate ligand. Consequently, (Xantphos)Pd(OTf)2 was identified as a more active catalyst for the hydroamination of vinylarenes because the bite angle of Xantphos (108°) is larger than that of DPPF (102°). As a result, the reaction between para-cyanoaniline and styrene catalyzed by a combination of (Xantphos)Pd(OTf)2 and TfOH occurred to form the product in 93% yield, which was 7 times greater than the yield obtained with (DPPF)Pd(OTf)2 and TfOH as the catalyst. With the newly developed (Xantphos)Pd(OTf)2 as the catalyst in the presence of a catalytic amount of TfOH, the hydroamination of styrene occurred with anilines bearing alcohol, amine, ester, acid, and amide groups (Figure 3).9a
Figure 3.
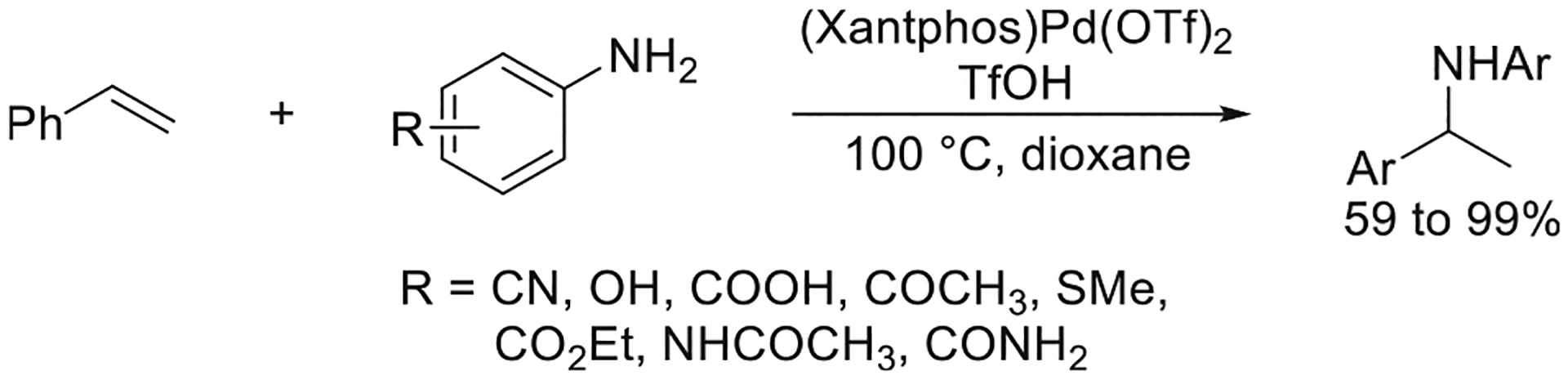
Improved scope of arylamines for the Pd-catalyzed hydroamination of vinylarenes. Yields are from ref 9a.
The development of an efficient catalytic hydroamination of vinylarenes enabled the direct measurement of the thermodynamic parameters of this reaction (Figure 4). Measurements of the equilibrium constants showed that the hydroaminations of vinylarenes with anisidine and N-methylaniline were nearly ergoneutral, and van’t Hoff analysis showed that a favorable enthalpy was counterbalanced by the unfavorable entropy of an intermolecular reaction.8 Consequently, the use of excess alkene was required to achieve a high yield for the hydroamination of certain vinylarenes, such as indene and dihydronaphthalene, due to the unfavorable thermodynamics of the reaction.
Figure 4.
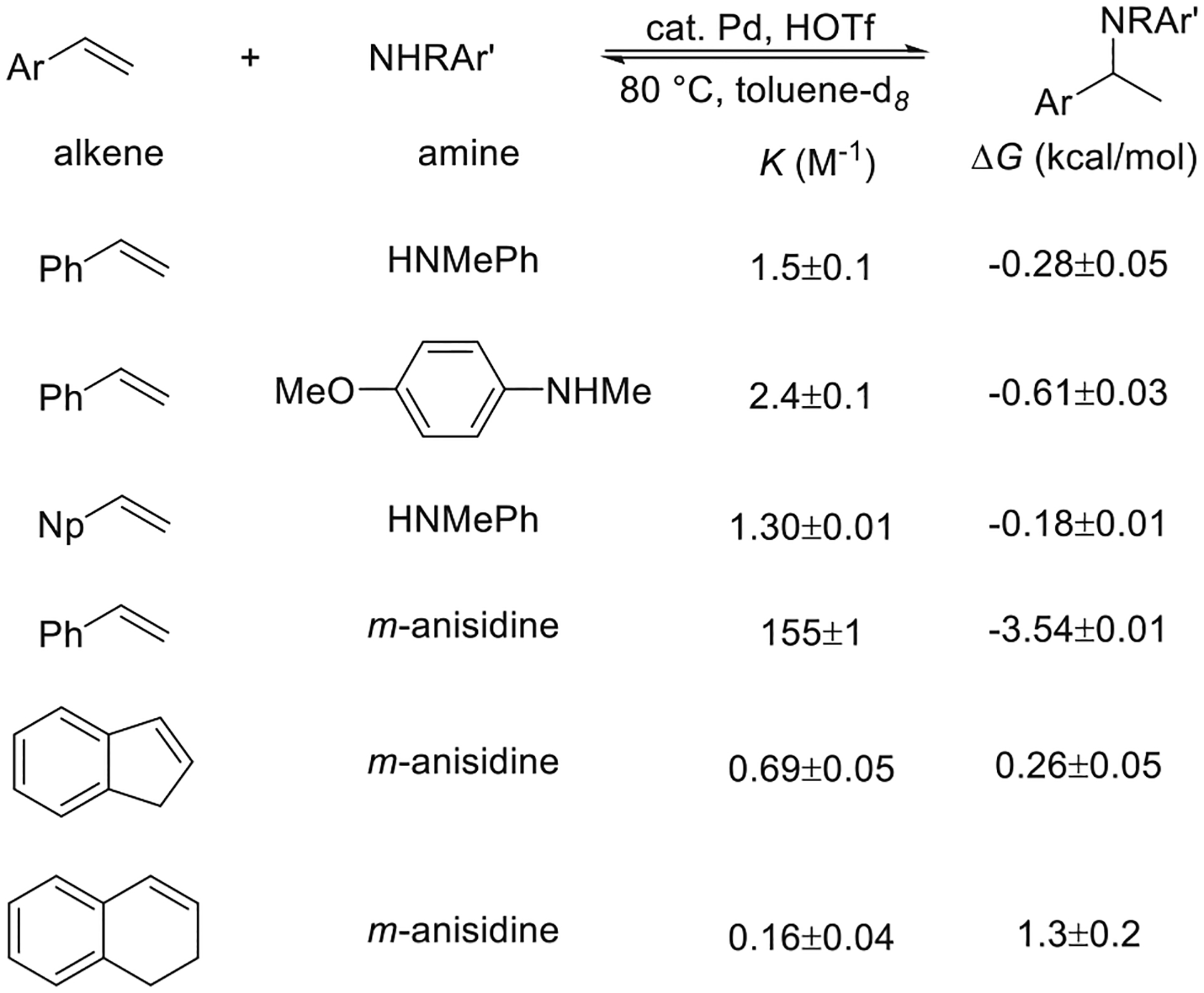
Measurements of the thermodynamics of the Markovnikov hydroamination of vinylarenes with arylamines. Equilibrium constants and Gibbs free energies are from ref 8.
2.2. Anti-Markovnikov Hydroamination of Vinylarenes Catalyzed by Complexes of Late Transition Metals
Vinylarenes also react with amines through organometallic intermediates to form products from anti-Markovnikov aminations. Beller reported the oxidative amination of vinylarenes with alkylamines with anti-Markovnikov selectivity to form enamines as the major product catalyzed by [Rh(cod)2]-BF4 and PPh3.17 Masaru Utsunomiya in our group later showed that vinylarenes reacted with secondary alkylamines catalyzed by [Rh(cod)(DPEphos)]BF4 with the same anti-Markovnikov selectivity, but to form amines by hydroamination.18 The scope of the reaction catalyzed by [Rh(cod)(DPEphos)]BF4 encompassed cyclic and acyclic secondary alkylamines as well as both electron-poor and electron-rich vinylarenes (Figure 5a). Enamines formed by oxidative amination were the major side product. The selectivity for hydroamination over the oxidative amination depended on the alkene and was higher with more electron-rich vinylarenes and at a lower concentration of vinylarene. These results suggested that the vinylarene served as both a reactant and an ancillary ligand, and a possible mechanism that accounted for the observations was proposed (Figure 5b). This mechanism involves the formation of a β-amino, α-alkyl rhodacycle from either the migratory insertion of the vinylarene into a Rh–N bond or the nucleophilic attack of aniline on a Rh-bound vinylarene. Due to the rigidity of the fourmembered rhodacycle, reductive elimination from the rhodacycle to form the hydroamination product was preferred over the β-hydrogen elimination from this rhodacycle to form the oxidative amination product. If this rhodacycle equilibrated with a Rh-alkyl complex containing vinylarene as a dative ligand, then β-hydrogen elimination could occur, thereby accounting for the lower selectivity for hydroamination with more electron-poor vinylarenes (which would bind more tightly to the Rh(III) species) and at higher concentrations of the vinylarene.
Figure 5.
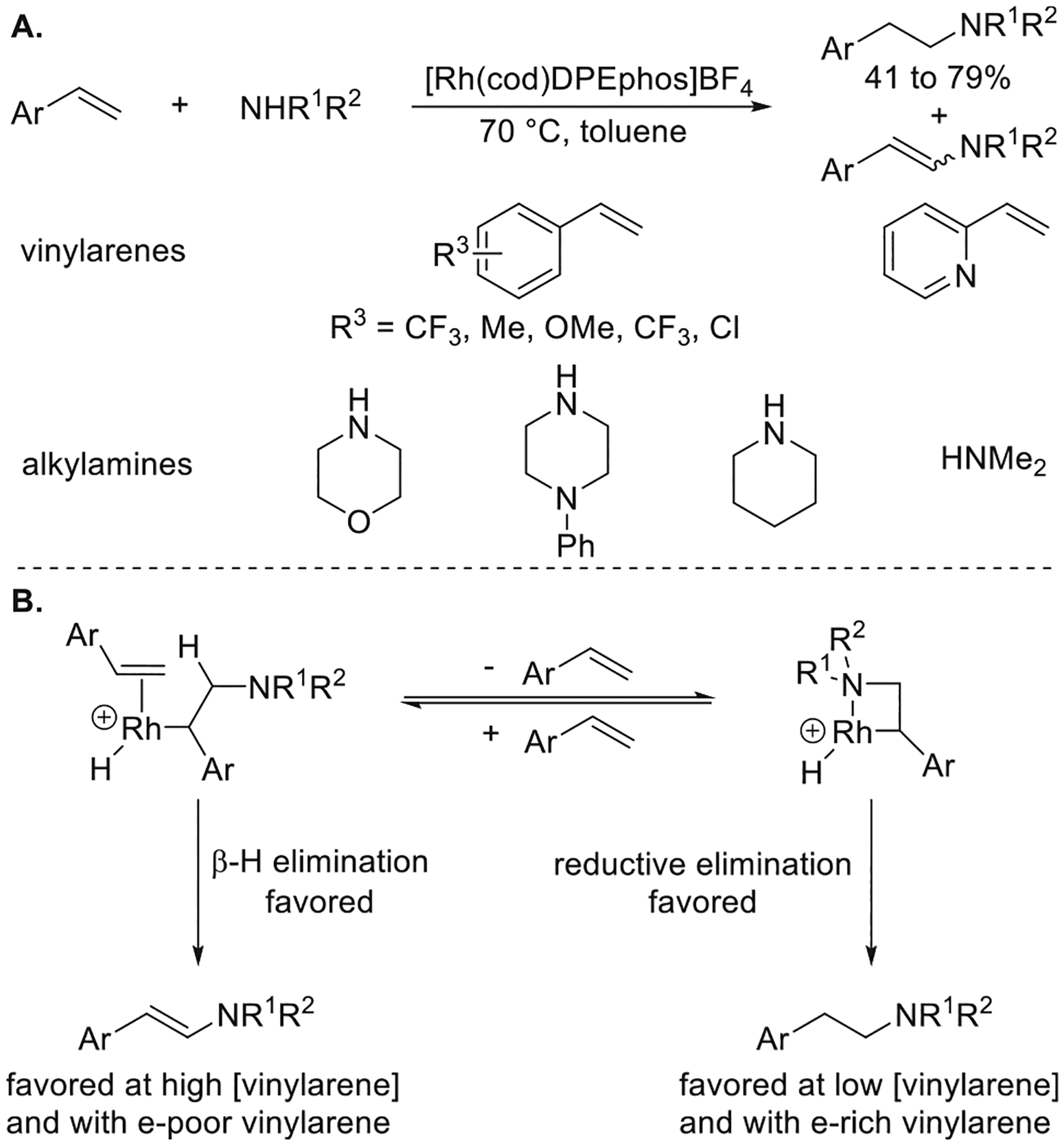
(A) Rh-catalyzed anti-Markovnikov hydroamination of vinylarenes with alkylamines. (B) Possible explanation for the low selectivity for hydroamination with more electron-poor vinylarenes and at higher concentrations of vinylarenes. Yields are from ref 18.
Seeking to suppress the formation of enamine side products, Utsunomiya discovered a ruthenium catalyst for the anti-Markovnikov hydroamination of vinylarenes with secondary alkylamines to form amines without competing oxidative amination.19 A combination of Ru(cod)(methylallyl)2, DPPPent, and TfOH catalyzes the anti-Markovnikov hydroamination of electron-poor and electron-rich vinylarenes with secondary alkylamines (Figure 6a) as well as α-methylstyrene for the first time.
Figure 6.
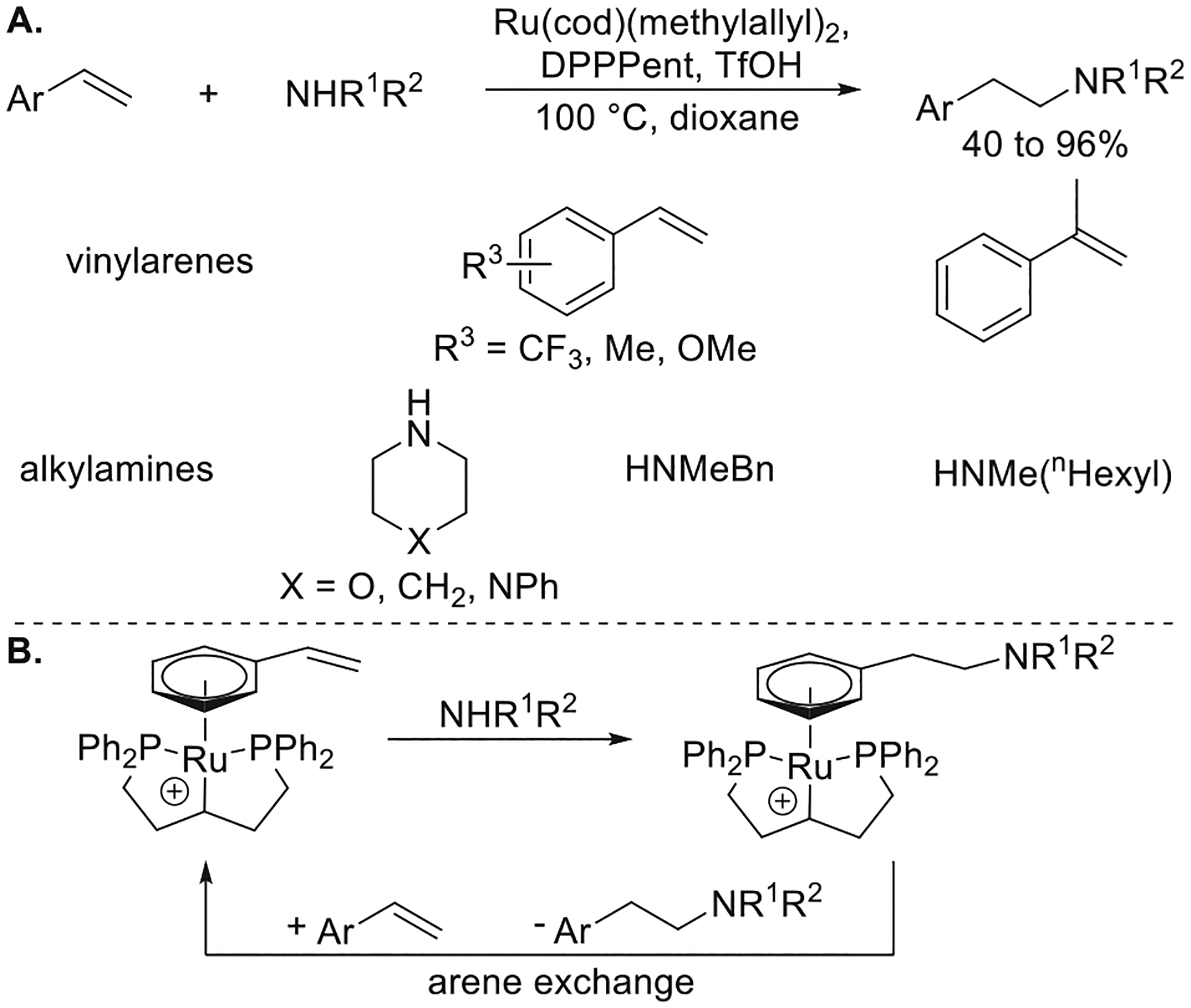
(A) Ru-catalyzed anti-Markovnikov hydroamination of vinylarenes with alkylamines. (B) Proposed mechanism. Yields are from ref 19.
Detailed mechanistic studies revealed a new pathway for hydroamination and an unusual example of catalysis through π-arene metal complexes. The combination of Ru(cod)-(methylallyl)2, DPPPent, and TfOH generates a PCP-pincer ligand in the active form of the catalyst by cleavage of the C–H bond on the central carbon, and the remaining coordination sites are occupied by the arene of the vinylarene (Figure 6b).20 [Ru(DPPPent)(styrene)](OTf) synthesized independently was found to be chemically and kinetically competent to be the active catalyst. Furthermore, treatment of this complex with excess morpholine formed a new Ru π-arene complex by nucleophilic attack of morpholine on the coordinated styrene at the terminal position. The π-bound hydroamination product in the resulting complex was subsequently released by ligand exchange with excess styrene. While this system was not designed to react through π-arene complexes, it was one of the first examples of a catalytic process that occurred through this class of complex.21
3. HYDROAMINATION OF CONJUGATED DIENES AND TRIENES CATALYZED BY COMPLEXES OF LATE TRANSITION METALS
The hydroamination of dienes and trienes forms allylamines and has been reported to occur with transition metals,22 rare-earth metals,23 and acids.24 Having observed the hydroamination of vinylarenes and considering the literature on the telomerization of butadiene with amines,25 the 1:1 addition of amines to dienes was a logical target, and Dr. Oliver Löber in our laboratory developed conditions for the Pd-catalyzed hydroamination of dienes with arylamines. A combination of Pd(PPh3)4 and TFA was shown by a colorimetric high-throughput screening method to catalyze the 1,4-addition of aniline to cyclohexadiene in high yield (Figure 7a).26 The scope of the reaction encompassed the addition of primary and N-methyl anilines bearing electron-donating and electron-withdrawing substituents to cyclic and acyclic 1,3-dienes. However, reactions of acyclic dienes formed N,N-diallylamines as side products, along with the primary arylamine products. Enantioselective hydroamination of cyclohexadiene with aniline was achieved with a combination of [Pd(allyl)Cl]2 and a Trost ligand as the catalyst (Figure 7b). This reaction likely proceeds through a mechanism similar to that followed by the Pd-catalyzed hydroamination of vinylarenes. A migratory insertion of the diene into the Pd–H bond would generate a Pd η3-allyl intermediate. The allylamine product would then form by turnover-limiting nucleophilic attack of the arylamine on the Pd η3-allyl complex, followed by a proton transfer to regenerate the palladium hydride (Figure 8).9a,26
Figure 7.
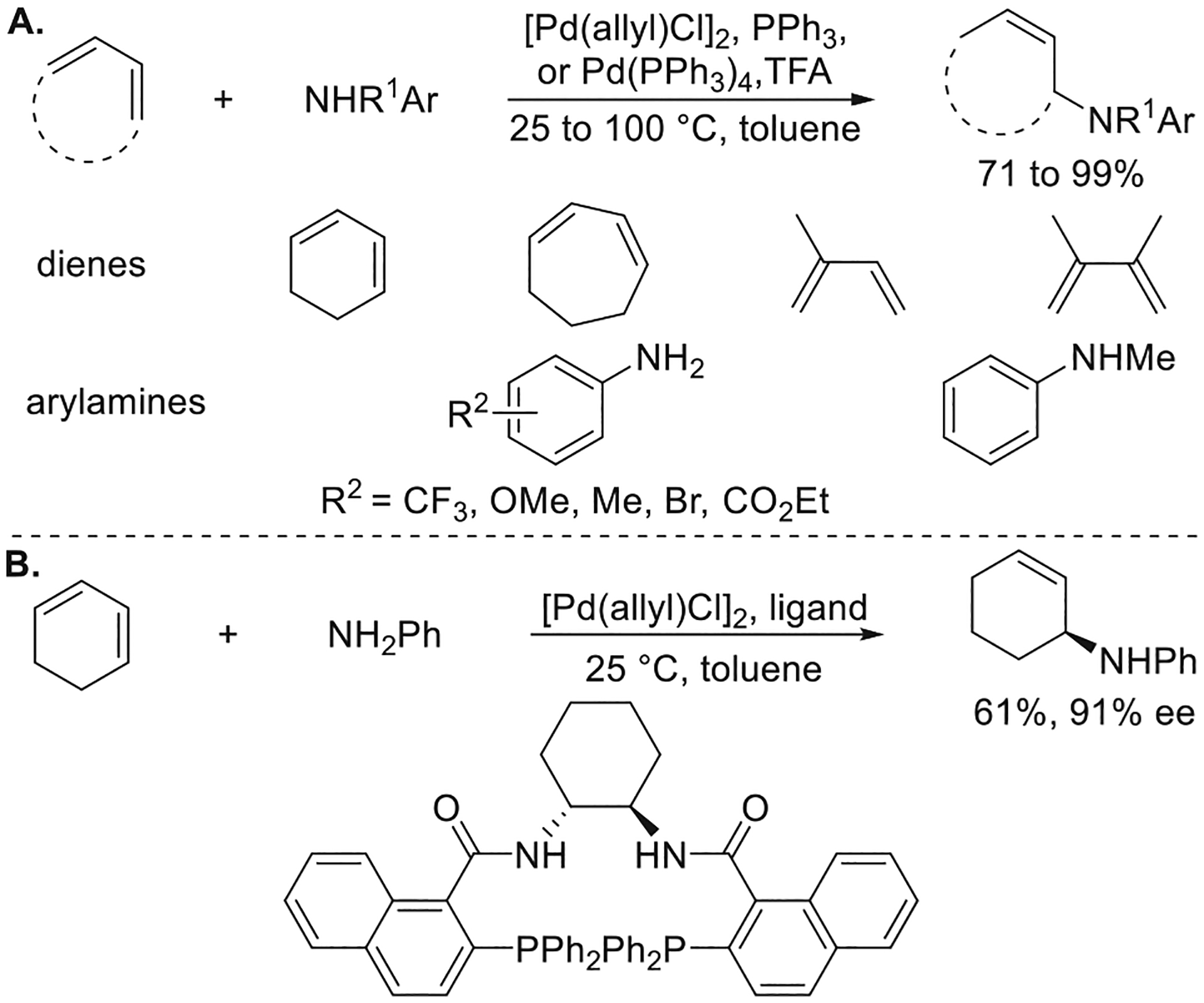
(A) Pd-catalyzed hydroamination of 1,3-dienes with arylamines. (B) Pd-catalyzed enantioselective hydroamination of 1,3-dienes with arylamines. Yields and enantioselectivities are from ref 26.
Figure 8.
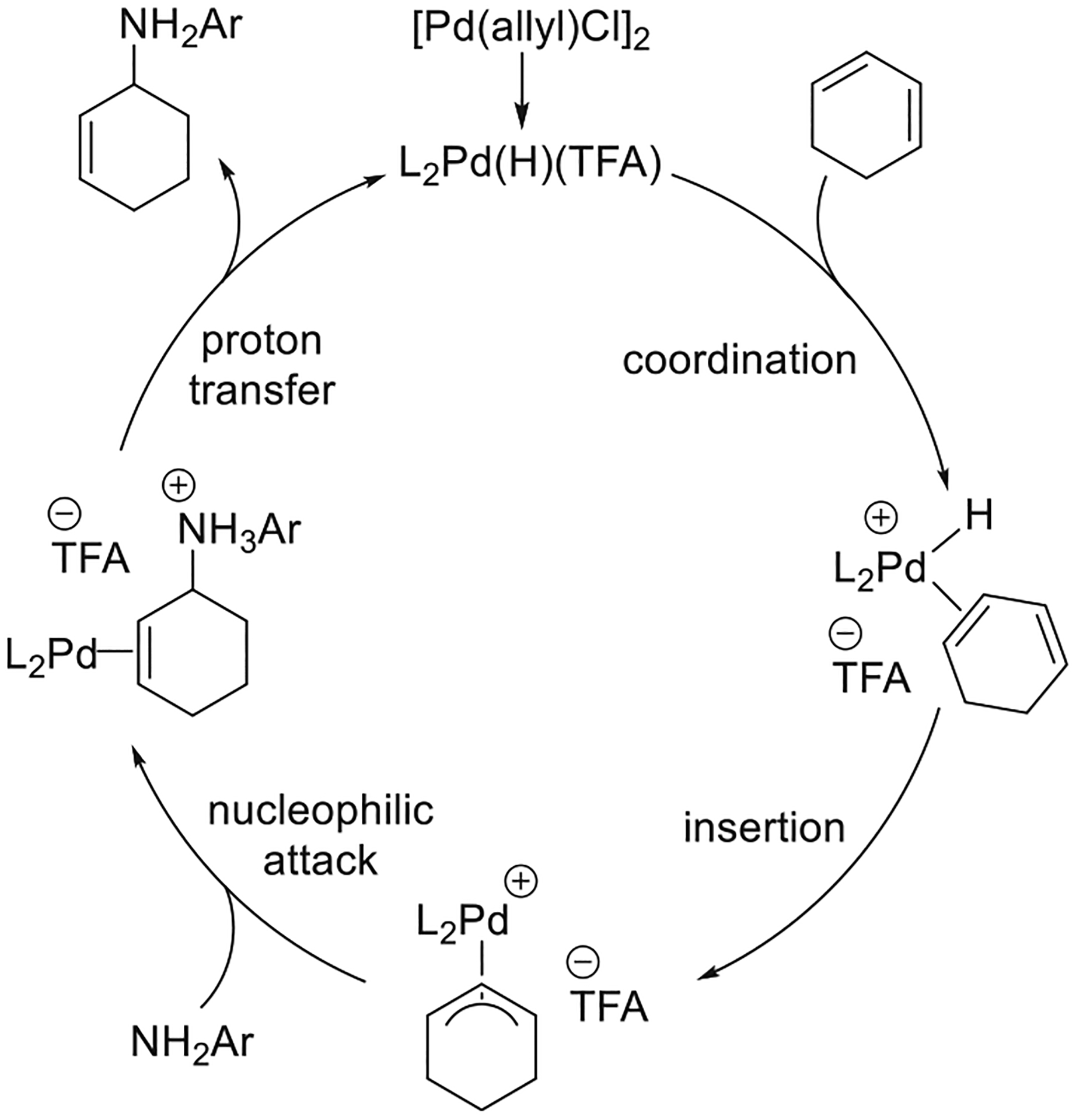
Proposed mechanism for the Pd-catalyzed hydroamination of 1,3-dienes.
Like the hydroamination of vinylarenes, the hydroamination of dienes was improved with a Pd catalyst containing a bidentate ligand possessing a large bite angle (Figure 9a).9a For example, a 5-fold increase in the rate of the reaction between para-cyanoaniline and cyclohexadiene was observed when [Pd(allyl)-Cl]2 and Xantphos were used instead of [Pd(allyl)Cl]2 and PPh3 as the catalyst. Moreover, the combination of [Pd(allyl)Cl]2 and Xantphos catalyzes the reactions of additional N–H donors, such as hydrazones, hydrazines, and hydroxylamines, with a variety of cyclic and acyclic dienes (Figure 9b).27 The synthesis of tropene derivatives from a sequential intermolecular and subsequent transannular, intramolecular hydroamination of cycloheptatriene was also reported with a combination of Pd(TFA)2, Xantphos, and benzoic acid as the catalyst (Figure 9c)28.
Figure 9.
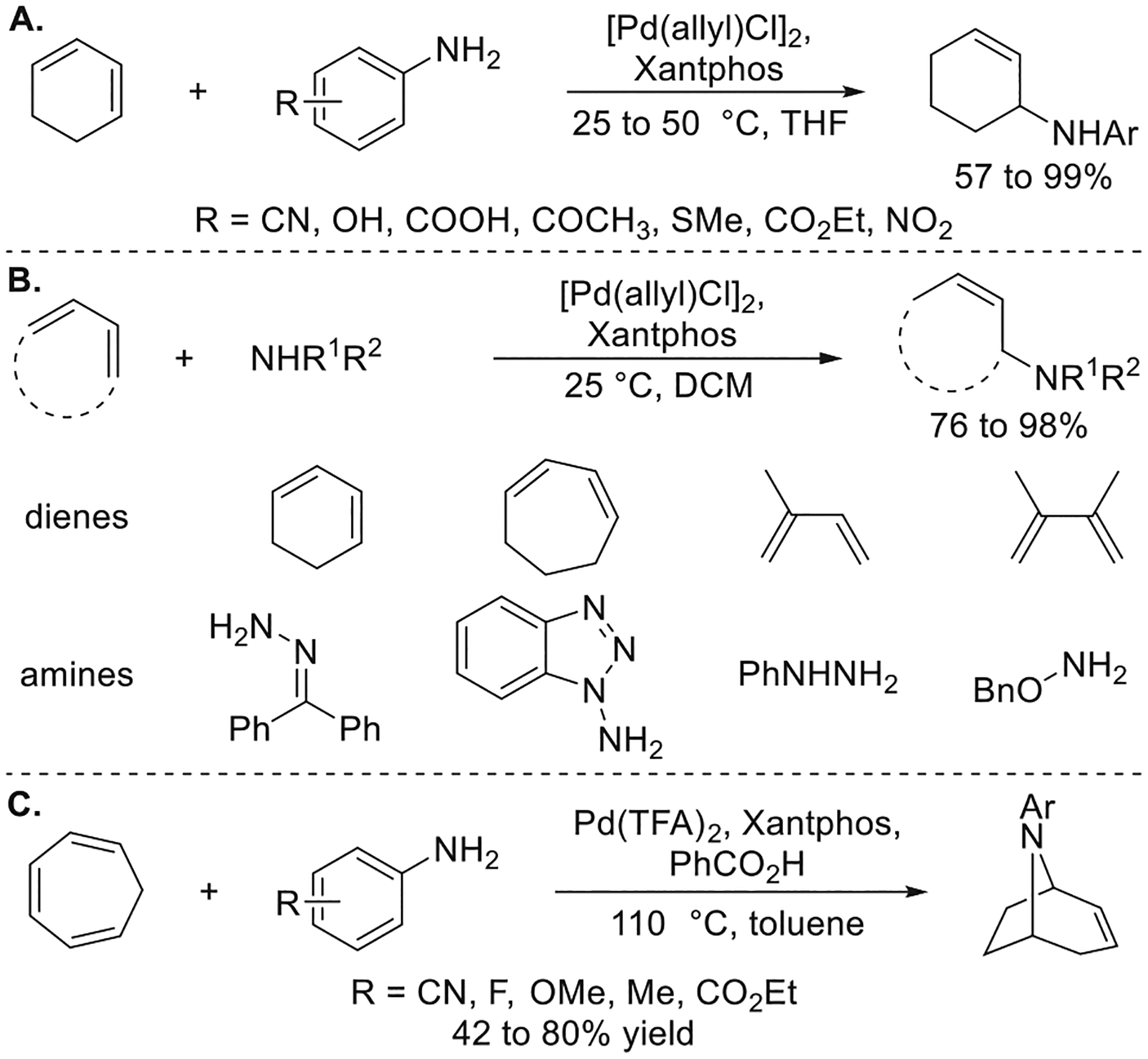
(A) Improved scope of arylamines for the Pd-catalyzed hydroamination of 1,3-dienes. (B) Pd-catalyzed hydroamination of 1,3-dienes with hydrazones, hydrazines, and hydroxylamines. (C) Synthesis of tropene derivatives by Pd-catalyzed Markovnikov hydroamination of cycloheptatrienes. Yields are from refs 9a, 27, and 28.
During the Pd-catalyzed hydroamination of dienes, the population of catalytically active [(bisphosphine)Pd(η3-allyl)-(Cl)] was higher than that of the inactive (bisphosphine)PdCl2 when the bisphosphine possessed a larger bite angle.9a This difference in the speciation of the catalyst, together with the faster turnover-limiting nucleophilic attack when the metal bears the large bite angle of Xantphos, contributed to the observed high rate of catalysis.9a
A nickel-catalyzed hydroamination of 1,3-dienes with alkylamines was discovered by Motoi Kawatsura using high-throughput screening methods and was developed and studied mechanistically by Jan Pawlas and visiting scholar Yoshiaki Nakao.9b A combination of Ni(cod)2, DPPF, and TFA was found to catalyze the hydroamination of cyclic dienes with alkyl and benzyl amines (Figure 10a). For reactions of butadiene, the product from 1,2-addition of the amine was favored kinetically, and the product from 1,4-addition was favored thermodynamically.
Figure 10.
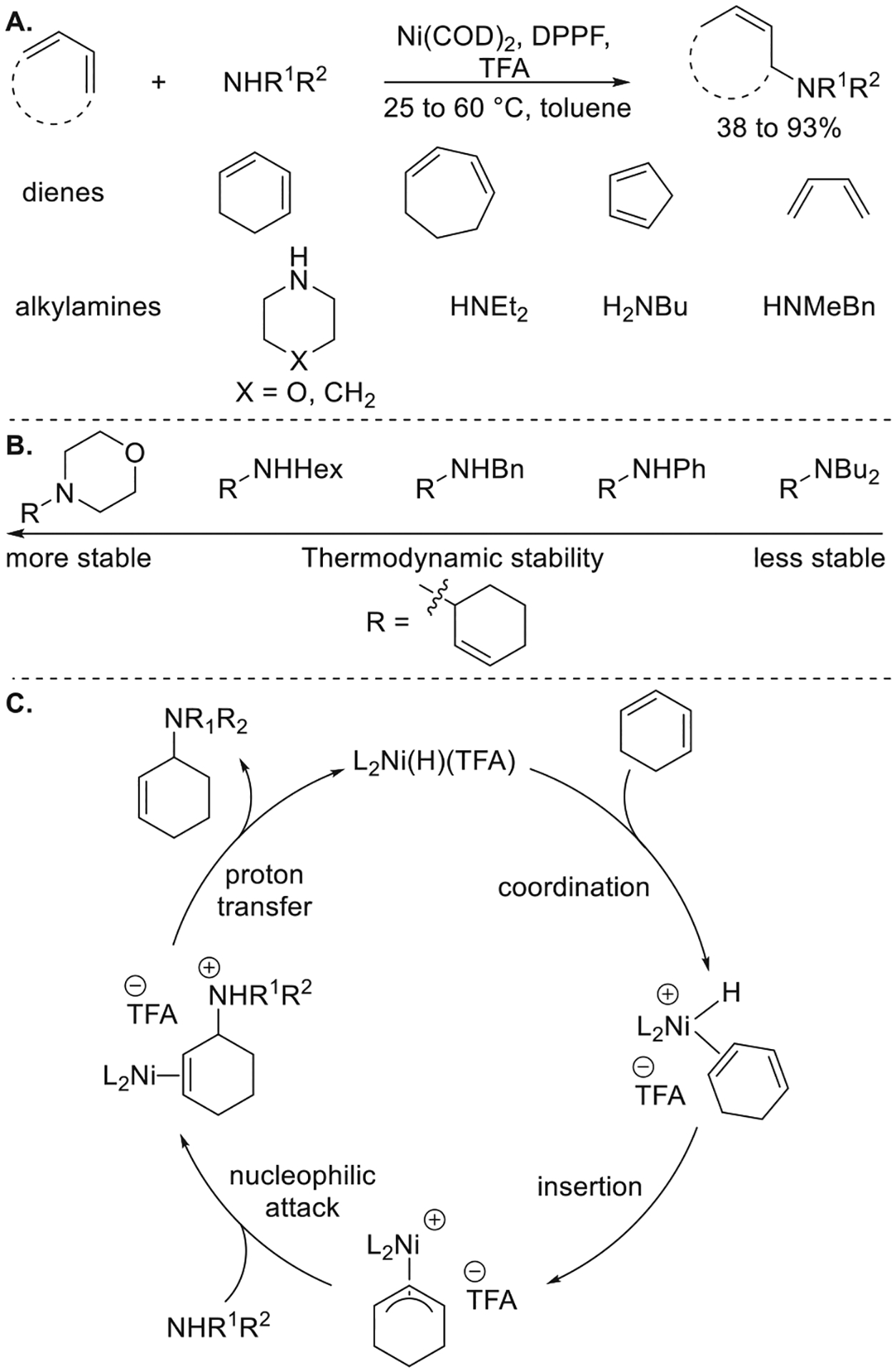
(A) Ni-catalyzed hydroamination of 1,3-dienes with alkylamines. (B) Evaluation of the thermodynamic stability of allylamines. (C) Proposed mechanism. Yields are from ref 9b.
This result, together with the observation that allylic amines reacted with primary or secondary alkyl or arylamines in the presence of this catalyst to generate mixtures of allylic amines, suggested that the hydroamination of 1,3-dienes with alkylamines was reversible. The thermodynamic stability of a variety of allylamines derived from a series of amines was determined by the exchange process, and the order of stability was cyclic secondary amines > primary alkylamines > primary benzylamines > primary arylamines > acyclic secondary amines (Figure 10b).
Mechanistic studies revealed the pathway for this Ni-catalyzed hydroamination of dienes (Figure 10c).9b The catalyst resting state in the reaction between cyclohexadiene and methylbenzylamine was determined to be [(DPPF)Ni(η3-cyclohexenyl)]-(TFA) by 31P NMR spectroscopy and independent synthesis. The reaction between a 1:1 ratio of [(DPPF)Ni(η3-cyclohexenyl)](TFA) and methylbenzylamine afforded the allylamine in only 15% yield, but the same reaction in the presence of excess cyclooctadiene afforded the allylamine in 90% yield, indicating that the nucleophilic attack of the allylnickel complex by amines was feasible but endergenic and that the diene was needed to make the reaction exergonic by trapping the Ni–H intermediate as the allyl complex. These studies on the catalytic hydroamination of conjugated dienes seeded the further development of enantioselective hydroamination of dienes22e,29 and hydroamination of internal dienes30 by groups including those of Mazet, Malcolmson, and Dong.
4. HYDROAMINATION OF BICYCLIC STRAINED ALKENES CATALYZED BY COMPLEXES OF LATE TRANSITION METALS
The hydroamination of strained alkenes, such as cyclopropene,5f,h cyclobutene,31 and bicyclic alkenes,32 is an attractive method for the synthesis of amine-substituted carbocyclic compounds. Milstein reported the first hydroaminations of strained, bicyclic alkenes, which occurred with six turnovers in the presence of an iridium catalyst containing PEt3 as the ligand.32e Togni reported the first enantioselective examples in high yield but with low enantioselectivity or in low yield but with high enantioselectivity in the presence of iridium and chiral bisphosphine.32a
An iridium system that catalyzes the enantioselective hydroamination of strained bicyclic alkenes and dienes in both high yield and high enantioselectivity was discovered in our laboratory by Jianrong (Steve) Zhou (Figure 11a).33 A combination of [Ir(coe)2Cl]2, DTBM-SEGPHOS, and KHMDS catalyzed the enantioselective addition of arylamines to norbornenes in high yield and selectivity. While fluoride was important for catalyst activity in prior work by Togni,32a the key to achieving activity in Steve Zhou’s systems was the replacement of the chloride ligand with an anilido ligand by aniline and KHMDS. Bisphosphines possessing biaryl backbones with smaller dihedral angles and bulky, electron-donating aryl groups on phosphorus increased the yield and enantioselectivity of the reaction over those with larger dihedral angles and less bulky aryl groups on phosphorus. The scope of the reaction encompassed arylamines with both electron-donating and electron-withdrawing substituents and the bicyclic alkenes including norbornene, norbornadiene, and benzo- as well as imide-fused norbornenes.
Figure 11.
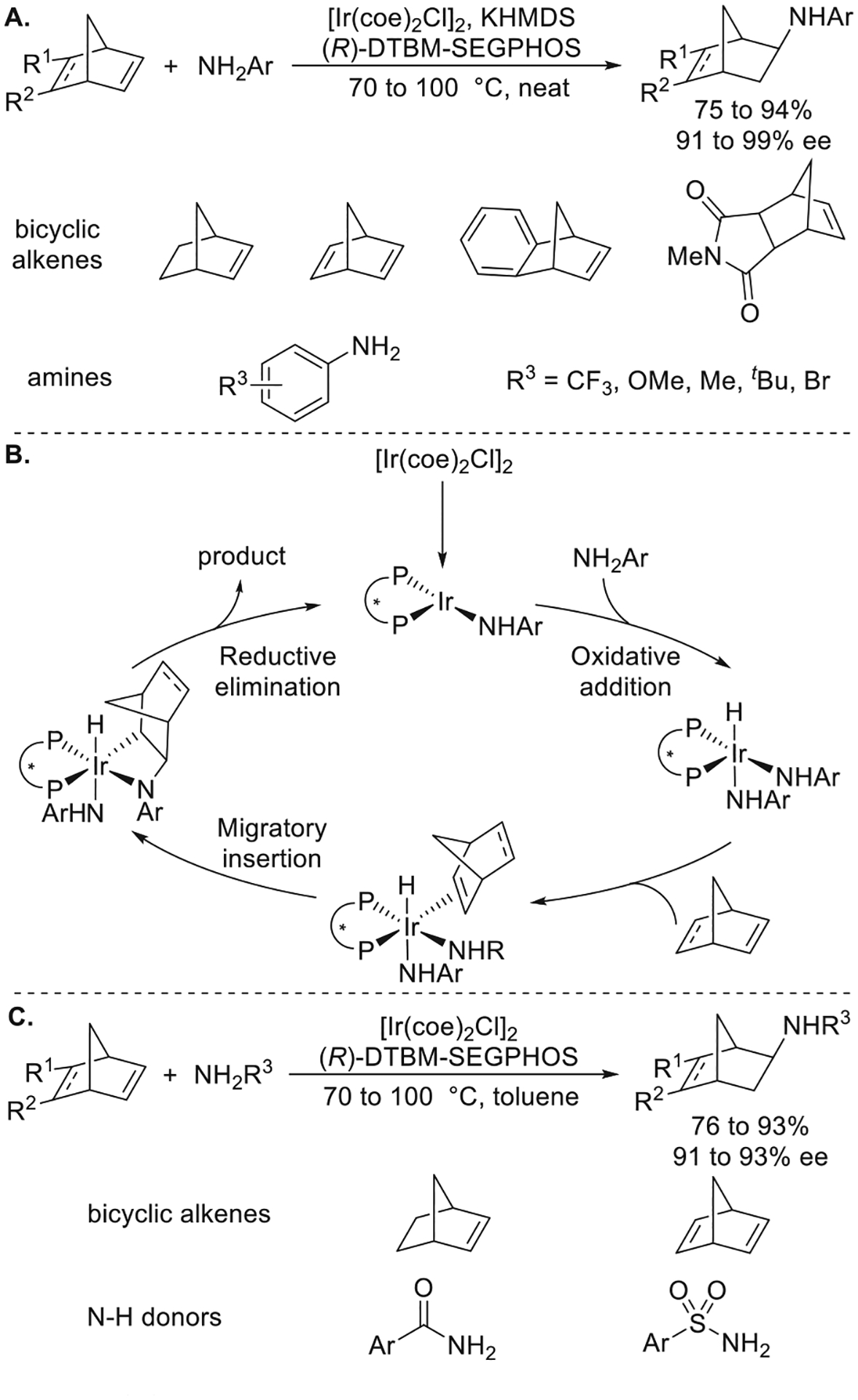
(A) Ir-catalyzed enantioselective hydroamination of bicyclic alkenes with arylamines. (B) Proposed mechanism. (C) Ir-catalyzed enantioselective hydroamination of bicyclic alkenes with arylamides and arylsulfonamides. Yields and enantioselectivities are from refs 1 and 33.
In addition to prior work, our studies with iridium catalysts were motivated by our direct observation of the oxidative addition of N–H bonds in arylamines and ammonia to welldefined Ir(I) complexes to form hydrido amido complexes.34 Consistent with these studies on N–H activation, mechanistic studies implied that the catalytic reactions occurred by the oxidative addition of an N–H bond, followed by alkene insertion into the Ir–N bond. The reaction between N-deuterated m-xylylamine and norbornene formed the product from syn addition of the deuterium and the amino group across the alkene. The anionic bis-anilide complex K[Ir(DM-SEGPHOS)(NHm-Xylyl)2] catalyzed the reaction of m-xylylamine with norbornene in high yield only in the presence of an equimolar amount of m-XylylNH3+, which would generate the neutral five-coordinate complex [Ir(DM-SEGPHOS)(H)-(NHm-Xylyl)2]. The neutral dimeric complex [Ir(DM-SEGPHOS)(NHm-Xylyl)]2 did not catalyze the hydroamination, implying that the monomeric unsaturated complex, [Ir(DM-SEGPHOS)(H)(NHm-Xylyl)2], was the active catalyst for the reaction and that the anilide dimer resisted dissociation to a monomer. [Ir(DM-SEGPHOS)(H)(NHm-Xylyl)2] was presumably generated in the catalytic reaction by the oxidative addition of aniline to the species generated from [Ir(coe)2Cl]2, DTBM-SEGPHOS, and potassium anilide. Norbornene would then coordinate to [Ir(DM-SEGPHOS)(H)(NHm-Xylyl)2], as depicted in the catalytic cycle of Figure 11b,33 and undergo migratory insertion into the Ir–N bond. Reductive elimination would form the C–H bond. Christo Sevov in our group showed that a similar system comprising [Ir(coe)2Cl]2 and DTBM-SEGPHOS catalyzed the addition of benzamides and sulfonamides to norbornene and norbornadiene in high yield and enantioselectivity (Figure 11c).1 These studies set the stage for reactions of N–H bonds with unactivated alkenes catalyzed by iridium complexes.
5. MARKOVNIKOV HYDROAMINATION OF UNACTIVATED ALKENES CATALYZED BY COMPLEXES OF LATE TRANSITION METALS
The hydroamination of unactivated alkenes could be a straightforward, atom-economical method for the preparation of aliphatic amines. However, unlike conjugated alkenes, for which insertion can generate η3-allyl or benzyl intermediates, and strained alkenes, for which migratory insertion is promoted by the release of ring strain, unactivated aliphatic alkenes had rarely been shown to undergo migratory insertion into late transition-metal–nitrogen bonds.35 Consequently, reports of direct N–H additions to unactivated alkenes are uncommon, and these reactions often required a large excess of alkene and occurred with limited tolerance of functional groups.36 Several alternative strategies for the hydroamination of unactivated alkenes also have been developed, including the reactions of alkenes bearing a directing group,37 formal hydroaminations with silanes and esters of hydroxylamines38 or dioxazolones as the source of an amino group,39 and hydroamination under photocatalytic conditions.7 Our group has sought to achieve the hydroamination of unactivated alkenes by the oxidative addition or deprotonation of the N–H bond to generate metal amido complexes that can form the C–N bond by insertion of the alkene.
Christo Sevov built upon the initial results of Dr. Steve Zhou in our group to achieve the Ir-catalyzed hydroamination of unactivated terminal alkenes with arylamides and arylsulfonamides catalyzed by a combination of [Ir(coe)2Cl]2 and DTBM-SEGPHOS in neat alkene (Figure 12a).1 Enamides from oxidative amination were the major side product, but a high yield of the N-alkylamide was achieved by the addition of H2 or isopropanol to the reaction mixture after the amination process.
Figure 12.
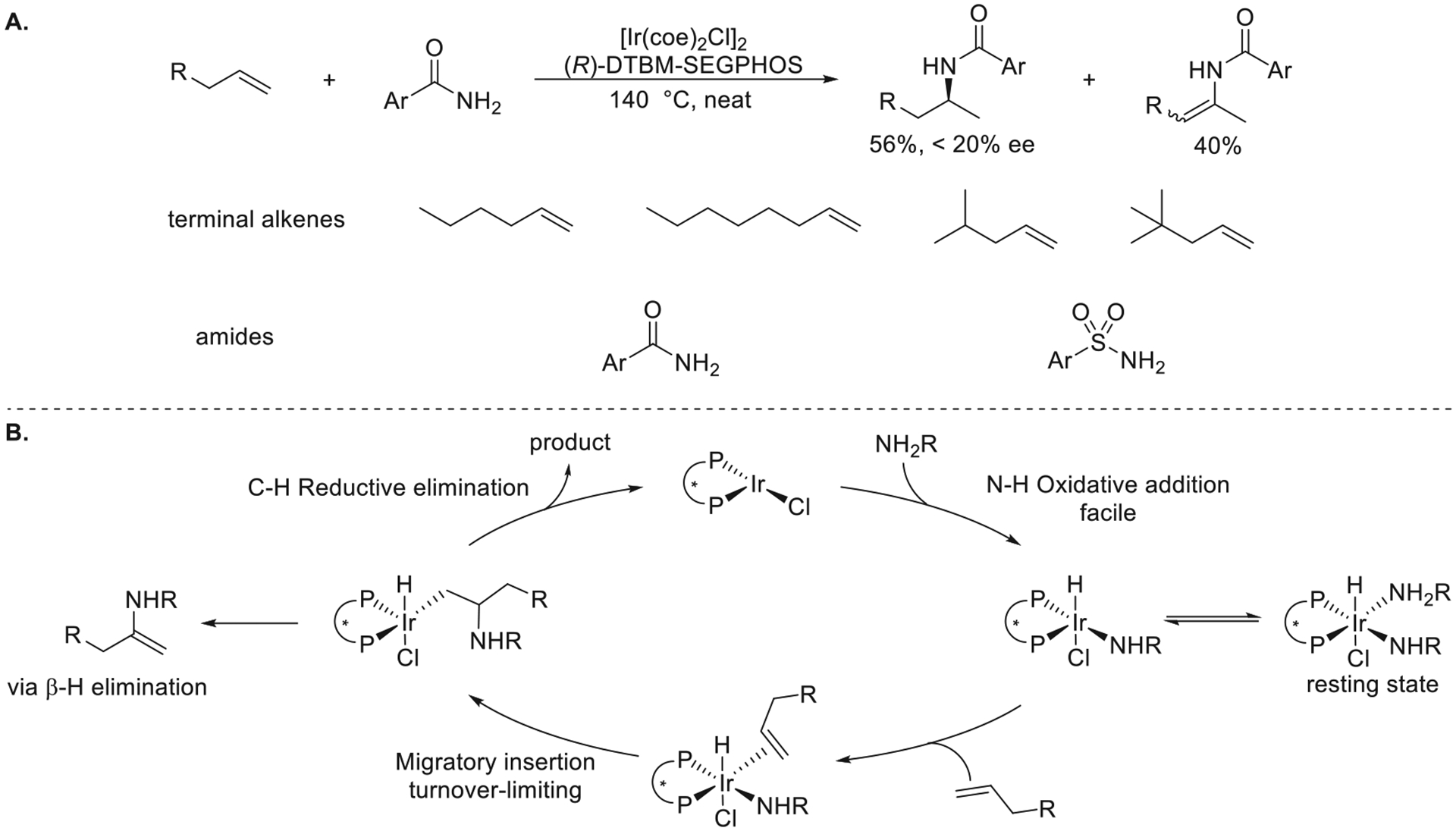
(A) Ir-catalyzed hydroamidation of unactivated terminal alkenes with arylamides and arylsulfonamides. (B) Proposed mechanism. Yields and enantioselectivities are from ref 1.
The resting state of the catalyst during the reaction was determined to be (L2)Ir(H)(Cl)(NHCOAr)(NH2COAr), which was formed by the oxidative addition of one molecule of the arylamide to the iridium center and the dative coordination of a second arylamide. This complex was determined to be chemically and kinetically competent for catalyst hydroamination. Kinetic measurements showed that the reaction was first order in the concentration of the Ir catalyst, inverse first order in the concentration of the arylamide, and first order in the concentration of the terminal alkene. Consequently, this reaction was proposed to occur by the dissociation of the dative arylamide from (L2)Ir(H)(Cl)(NHCOAr)(NH2COAr), followed by sequential migratory insertion of the terminal alkene into the Ir–N bond and reductive elimination to form the alkylamide product (Figure 12b).
Sevov and Zhou also showed that the combination of [Ir(cod)Cl]2 and DTBM-SEGPHOS catalyzed the hydroamination of unactivated alkenes with indoles (Figure 13a).40 The reaction occurred through selective addition of the N–H bond of the indole to the alkene, and side products from the addition of the indoyl C–H bond to the alkene were not observed. With ethyl acetate as a cosolvent, the reaction occurred with as few as 1.5 equiv of alkene versus indole.
Figure 13.
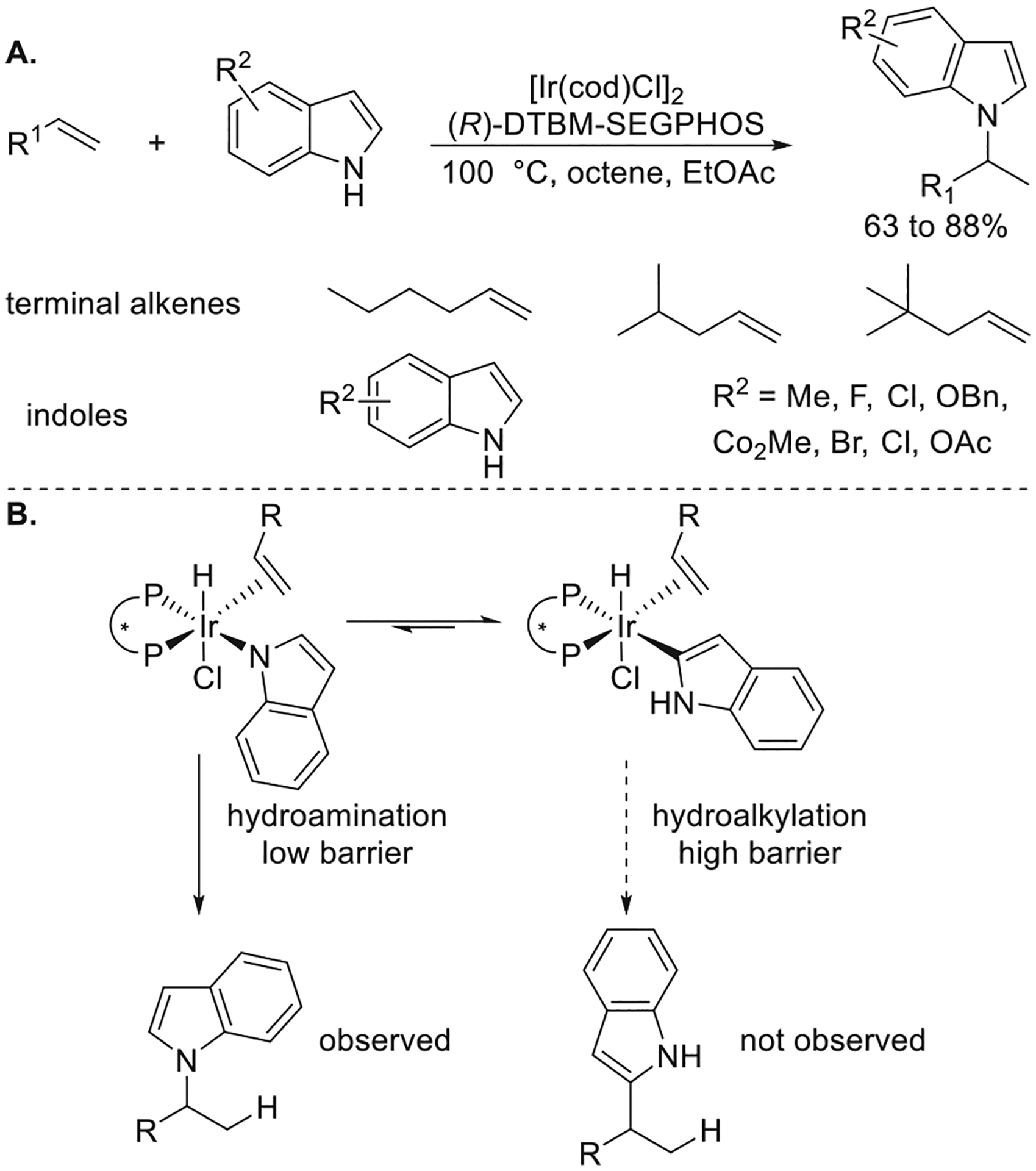
(A) Ir-catalyzed hydroamination of unactivated terminal alkenes with indoles. (B) Explanation of the selectivity of hydroamination over hydroalkylation. Yields are from ref 40.
Mechanistic analysis showed that the resting state of the reaction, unexpectedly, was a C-bound iridium indolyl alkene complex. This observed complex isomerized to its reactive N-bound isomer before undergoing migratory insertion and reductive elimination to generate the product from hydroamination. Computational studies by DFT indicated that the addition of the N–H bond over the C–H bond to the alkene occurred because the transition state for migratory insertion of the alkene into the Ir–N bond was lower in energy than that for the migratory insertion of the alkene into the Ir–C bond of the Ir-indolyl isomers (Figure 13b). These relative barriers would not be predicted by the vast prior literature on insertions of alkenes into metal–carbon bonds and scarce literature on insertions into metal–nitrogen bonds. However, they are consistent with the growing body of literature on the insertions of alkenes into metal–heteroatom bonds and analyses of the factors controlling these relative rates.41
While this work showed that the intermolecular hydroamination of unactivated alkenes catalyzed by late transition metals was possible, the reactions required excess alkene and did not occur with internal alkenes. Thus, Yumeng Xi and Chris Hill in our group sought to redesign the catalysts and amines to address these shortcomings.
Achieving enantioselective intermolecular hydroamination of unactivated internal alkenes is particularly challenging because internal alkenes tend to bind more weakly to transition metals than do terminal alkenes, undergo migratory insertion less readily, and undergo isomerization and oxidation by β-hydrogen elimination after the insertion event. To tackle these challenges, Yumeng focused on systems comprising a cationic Ir catalyst and a judiciously designed amine that collectively promoted the key alkene coordination, migratory insertion, and reductive elimination steps of the catalytic cycle for hydroamination (Figure 14a).3 Cationic iridium catalysts were used because they readily bind and insert internal alkenes, as shown by Crabtree for his hydrogenation systems many years ago,42 and an amine possessing a second coordinating group was used to favor the thermodynamics of oxidative addition.
Figure 14.
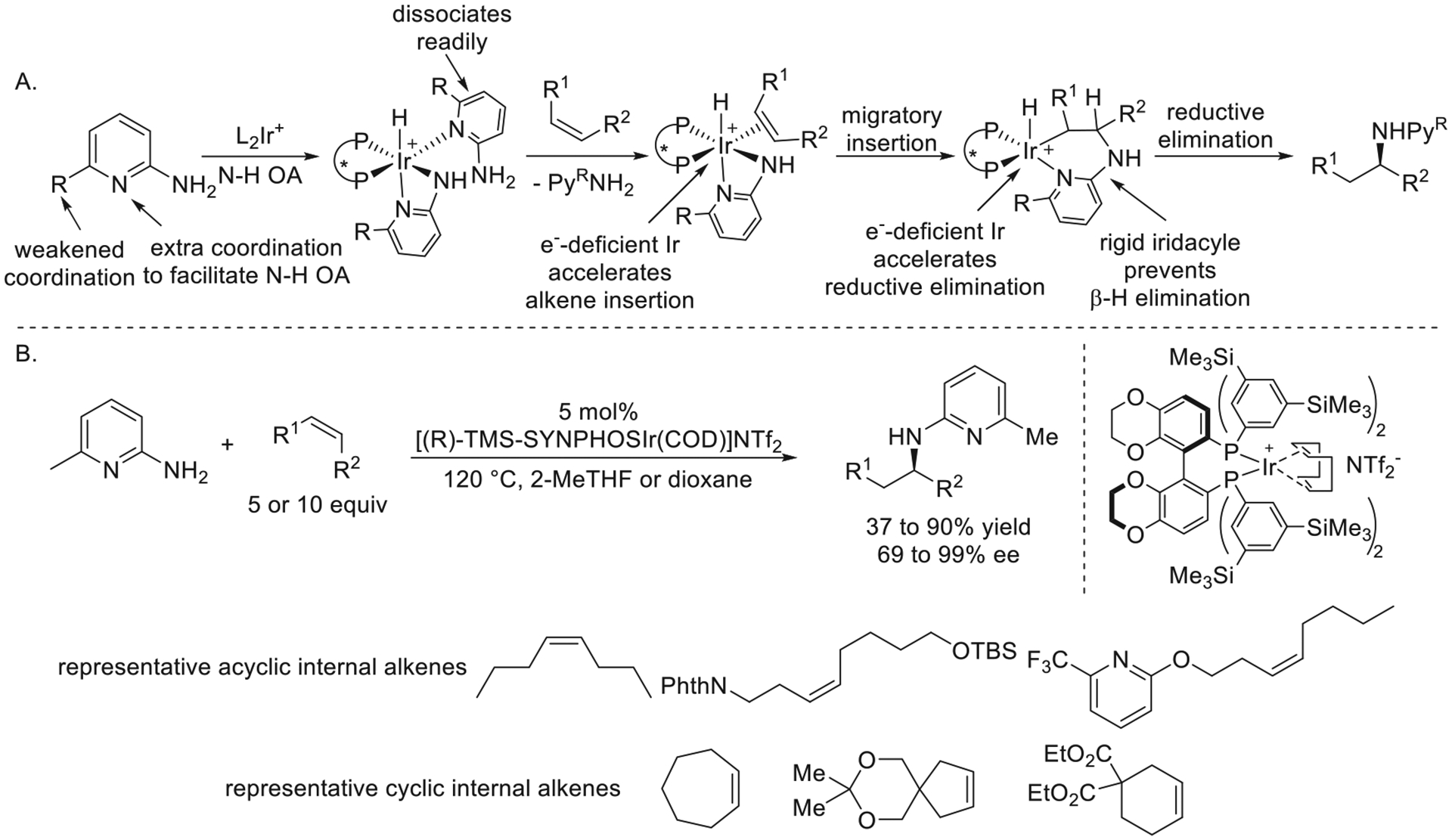
(A) Design elements for the catalytic hydroamination of unactivated internal alkenes. (B) Ir-catalyzed enantioselective hydroamination of unactivated internal alkenes. Adapted with permission from ref 3. Copyright 2020 Springer Nature.
Following this design, the enantioselective hydroamination of unactivated symmetrical and unsymmetrical linear Z-alkenes, as well as cyclic internal alkenes, occurred with 6-methyl-2-aminopyridine catalyzed by [(R)-TMS-SYNPHOSIr(COD)]-NTf2 (Figure 14b). The products of this hydroamination were converted into primary amines with little to no erosion of enantiopurity by sequential hydrogenation and reduction. Due to competing alkene isomerization, excess alkene was required for this reaction to occur with high regioselectivity, and further development was needed to address this limitation. Mechanistic experiments showed that this reaction occurred by turnoverlimiting migratory insertion of the alkene into the Ir–N bond, and DFT computations suggested that both steric and electronic properties of the ligand contributed to the high enantioselectivity of this transformation.
This design was also applicable to the hydroamination of terminal alkenes, but further modifications of the catalysts by Senjie Ma were needed to increase the functional-group tolerance and to enable reactions to be conducted without excess alkene.2 Initial mechanistic experiments on the reactions of terminal alkenes, including deuterium-labeling and 1H NMR spectroscopy analysis of the reaction, revealed three undesired pathways: (1) the retrohydroamination of the product amine (Figure 15a, path (i)), (2) the irreversible isomerization of allylbenzene to β-methylstyrene (path (ii)), and (3) the reversible dehydrogenation of the product amine (path (iii)). A catalytic system comprising [Ir[(S)-DTBM-SEGPHOS]-(ethylene)(Cl)], which contains a monodentate, unstrained, and volatile alkene, and NaBArF to generate the cation was discovered to catalyze the hydroamination of allylbenzene with 6-methyl-2-aminopyridine at 60 °C with no detectable side products.2 The hydroamination occurred with equimolar 6-methyl-2-aminopyridine and >40 structurally diverse terminal alkenes (Figure 15b). This reaction occurred equally well on gram scale and even with 0.2 mol % catalyst for selected substrates.
Figure 15.
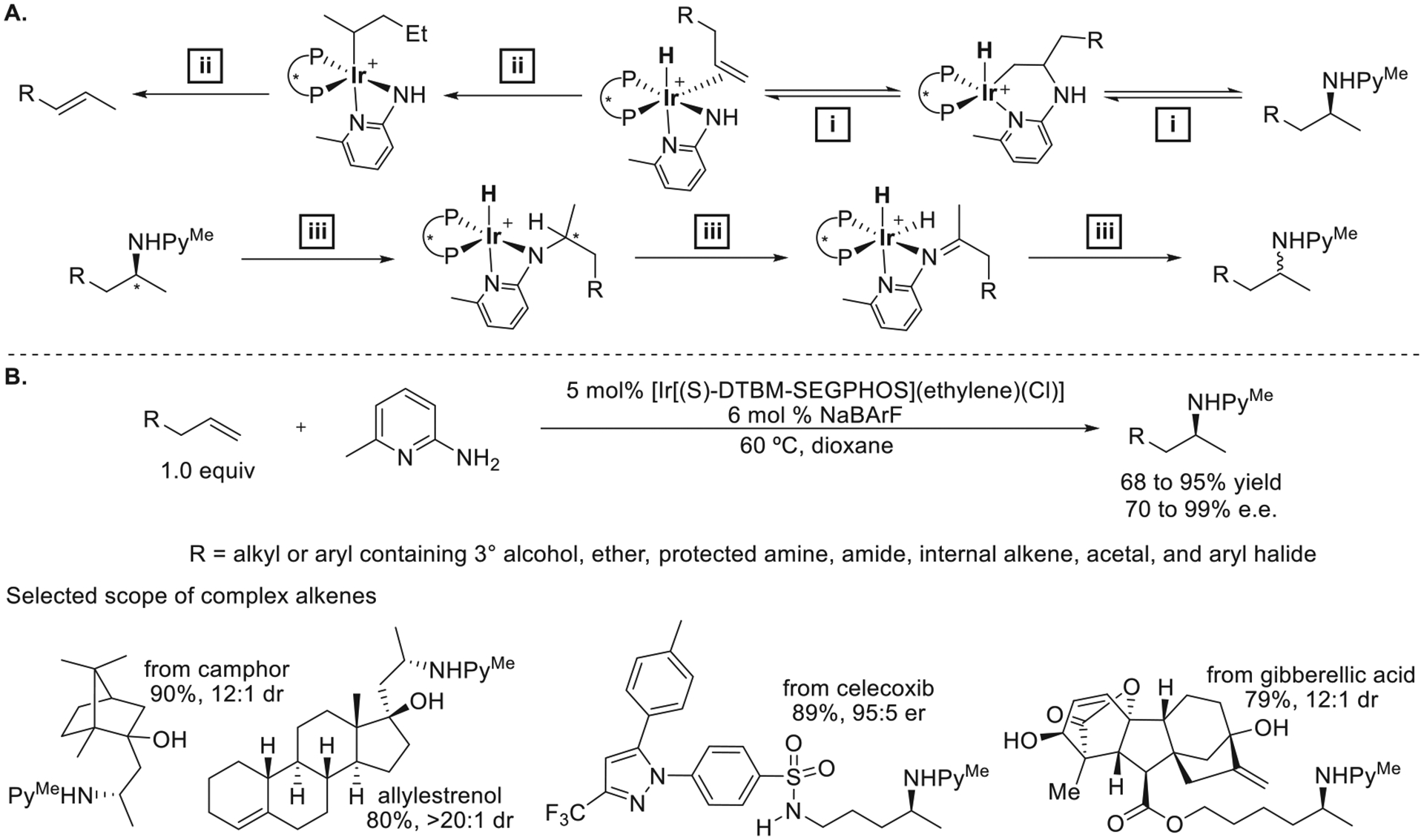
(A) Undesired pathways for the isomerization of the alkene and the racemization of the product. (B) Ir-catalyzed enantioselective hydroamination of unactivated terminal alkenes. Adapted with permission from ref 2. Copyright 2022 Elsevier.
An iridium-catalyzed remote hydroamination of unactivated disubstituted alkenes was developed by Senjie Ma.43 This remote hydroamination was achieved by isomerization of a disubstituted alkene, followed by selective hydroamination of the terminal alkene that installs the amino group at the subterminal, unactivated, methylene position. A combination of electron-deficient 2-aminopyridines and an iridium catalyst bearing a new DIP-Ad-SEGPHOS ligand was identified by modular changes to the system to form the most active and selective reagent and catalyst for this remote hydroamination. This reaction empassed alkenes and aminopyridines bearing electron-donating and electron-withdrawing substituents and even occurred with mixtures of isomeric alkenes to form a single product (Figure 16). Subsequent functionalization of the products from hydroamination was achieved by nucleophilic aromatic substitution with a variety of nucleophiles.
Figure 16.
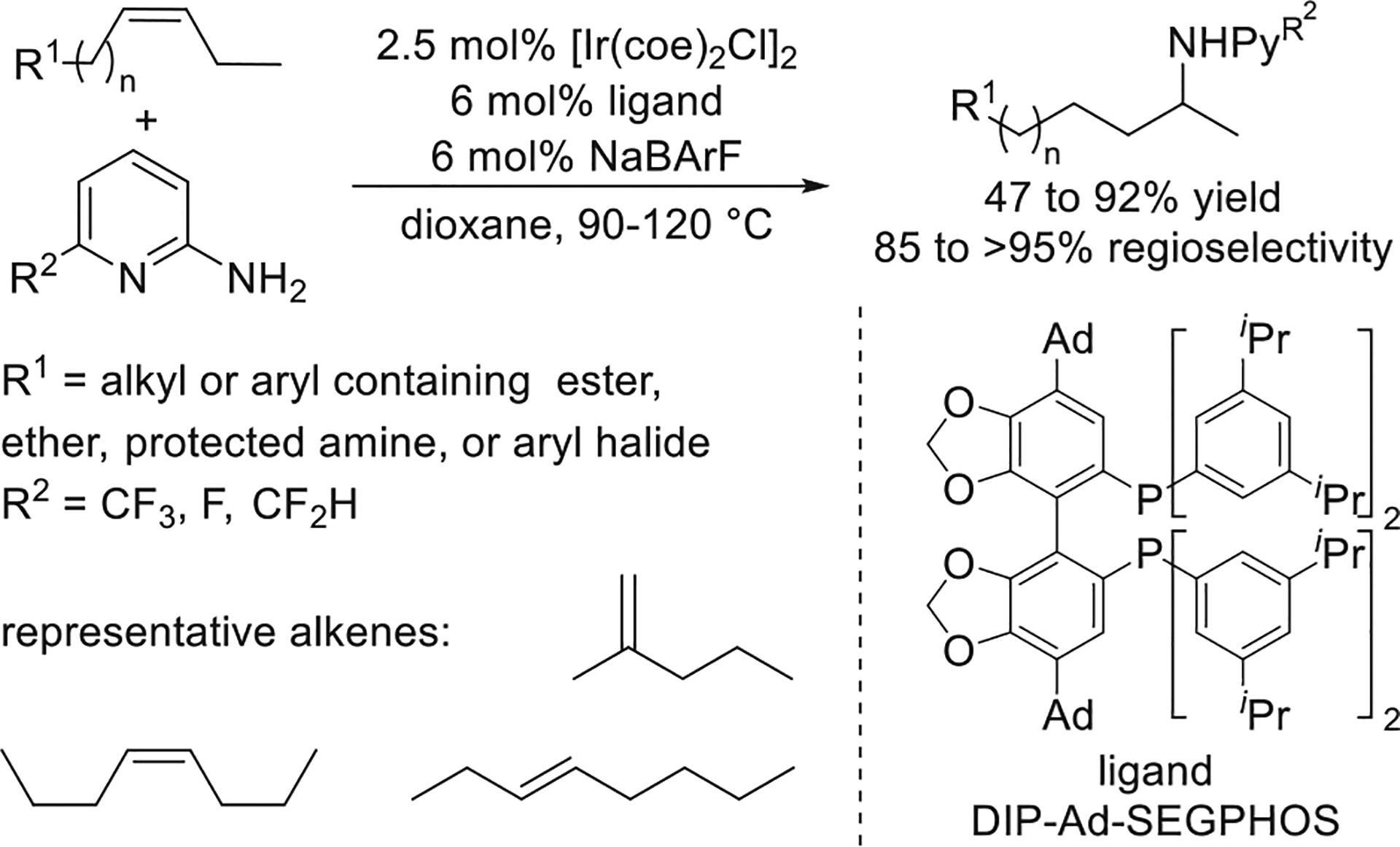
Ir-catalyzed remote hydroamination of unactivated disubstituted alkenes. Yields are from ref 43.
Prior to the discovery of this iridium-catalyzed hydroamination of terminal alkenes, Chris Hill in our group discovered a ruthenium-catalyzed, intermolecular Markovnikov hydroamination of unactivated terminal alkenes with equimolar 2-aminopyridines, and this reaction occurred via an unusual mechanism involving sequential oxidative amination and transfer hydrogenation (Figure 17a).4 Studies on the mechanism by Senjie Ma showed that the catalyst resting state was the Ru-amido complex [Ru(PEt3)3(amido)](NTf2) formed by coordination of the 2-aminopyridine to ruthenium, followed by deprotonation of the acidic N–H bond of the aminopyridine. Kinetic measurements and DFT computations revealed that this reaction occurred by a turnover-limiting migratory insertion of the alkene into the Ru–N bond, and deuterium labeling studies implied that the product forms by initial oxidative amination, followed by reduction of the resulting imine (Figure 17b).
Figure 17.
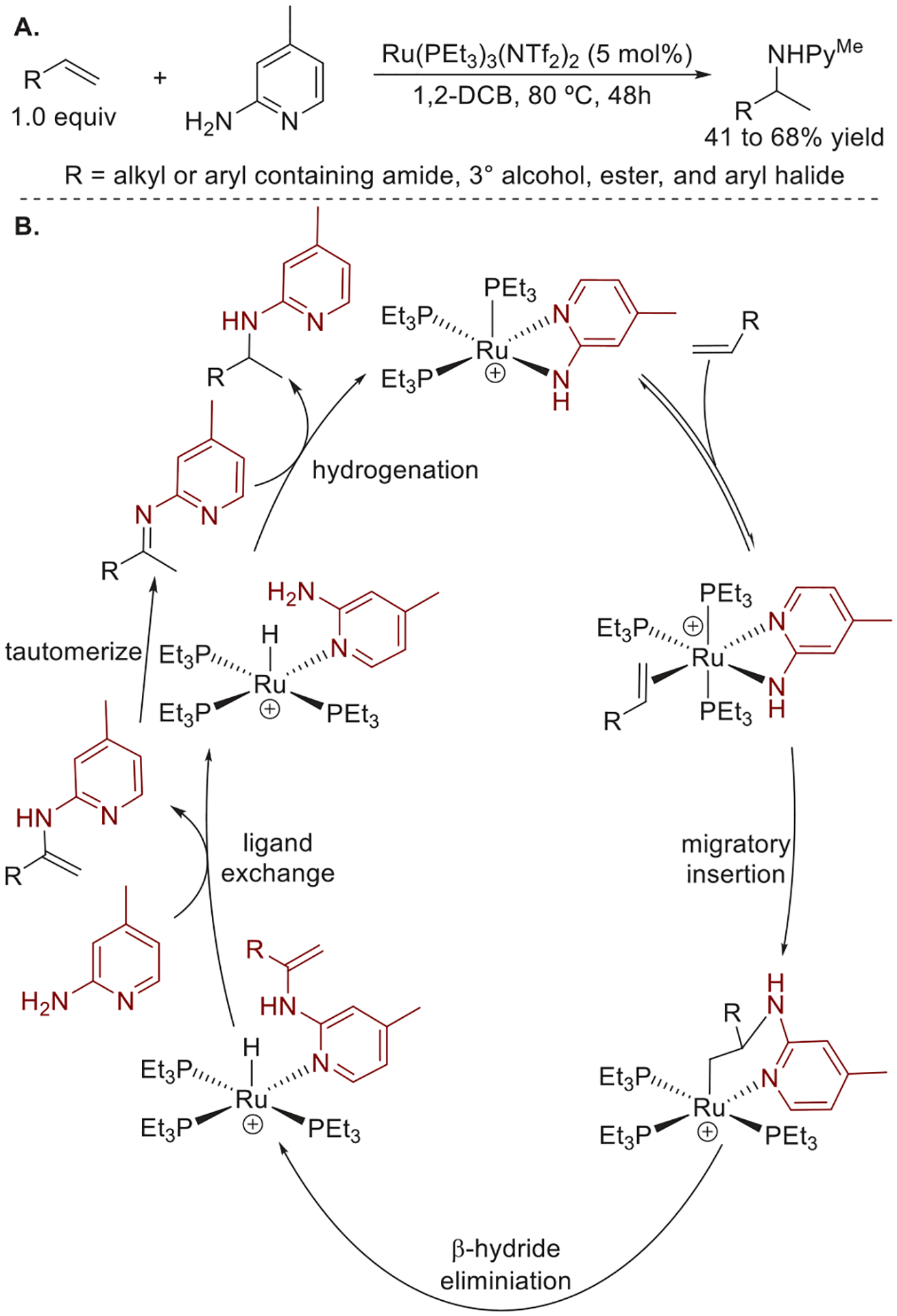
(A) Ru-catalyzed hydroamination of unactivated terminal alkenes. (B) Proposed mechanism for the Ru-catalyzed hydroamination of unactivated terminal alkenes. Yields are from ref 4.
6. CONCLUSIONS AND OUTLOOK
This Account described the progression of hydroamination catalyzed by late transition metal complexes in our laboratory from the addition of N–H bonds to conjugated alkenes to the addition to unconjugated and unstrained internal and terminal alkenes. While reactions of the conjugated alkenes catalyzed by palladium, rhodium, and ruthenium occur by nucleophilic attack of the amine on a coordinated benzyl, allyl, alkene, or arene ligand, the reactions of unconjugated alkenes occur by migratory insertion into metal–nitrogen bonds in complexes formed by oxidative addition or deprotonation of the N–H bond. Fundamental studies have shown that the oxidative addition of N–H bonds to Ir(I) is close to thermoneutral and is influenced by subtle properties of the ancillary ligand,41 causing reactions to be more favorable with substrates containing more acidic N–H bonds or secondary binding groups.44 The hydroamination of alkenes also was shown experimentally to be close to thermoneutral, and common side products were found to result from oxidative amination and alkene isomerization. The facility of alkene insertion into metal–nitrogen bonds during these reaction runs counter to the rarity of this reaction in the prior literature and presages multiple possible future alkene amination processes. On the basis of these principles, future catalytic systems for the addition of N–H donors that are more directly relevant to synthetic applications than aminopyridines to unactivated alkenes, for the regioselective hydroamination of unsymmetrical internal alkenes, and for the hydroamination of hindered, multisubstituted alkenes will be sought. In parallel, the application of these principles to create intramolecular reactions to form a variety of saturated nitrogen heterocycles that avoid the translational, entropic penalty of intermolecular processes will be sought.
ACKNOWLEDGMENTS
The authors thank the National Institutes of Health under grant R35GM130387 and the Director, Office of Science of the U.S. Department of Energy under contract number DE-AC02-05CH11231 for the support of our current work on catalytic hydroamination discussed in this Account.
Biographies
Senjie Ma obtained his B.A. degree from Carleton College with Prof. Matthew T. Whited in 2018 and his Ph.D. degree from the University of California, Berkeley with Prof. John F. Hartwig in 2023. His research focused on the development of new transition-metal catalysts for the hydroamination of unactivated alkenes and the understanding of the mechanism of those catalytic reactions. He is currently a scientist at Bristol Myers Squibb.
John F. Hartwig is the Henry Rapoport Professor of Chemistry at the University of California, Berkeley. The work in his laboratory has focused on revealing the fundamental principles that control metalcentered reactivity and creating catalysts and new catalytic reactions using this information. He is the author of the textbook “Organotransition Metal Chemistry – From Bonding to Catalysis.” Outside the laboratory, he enjoys cycling, hiking, skiing, music, theater, and cooking.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.3c00141
The authors declare no competing financial interest.
Contributor Information
Senjie Ma, Department of Chemistry, University of California, Berkeley, California 94720, United States; Division of Chemical Sciences, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States.
John F. Hartwig, Department of Chemistry, University of California, Berkeley, California 94720, United States; Division of Chemical Sciences, Lawrence Berkeley National Laboratory, Berkeley, California 94720, United States
REFERENCES
- (1).Sevov CS; Zhou J; Hartwig JF Iridium-Catalyzed Intermolecular Hydroamination of Unactivated Aliphatic Alkenes with Amides and Sulfonamides. J. Am. Chem. Soc 2012, 134 (29), 11960–11963. [DOI] [PubMed] [Google Scholar]; 1Iridium-catalyzed intermolecular amination of unactivated terminal alkenes and bicycloalkenes with arylamides and sulfonamides generates synthetically useful protected amine products in high yields.
- (2).Ma S; Xi Y; Fan H; Roediger S; Hartwig JF Enantioselective hydroamination of unactivated terminal alkenes. Chem. 2022, 8 (2), 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]; 2A system comprising a specific iridium precatalyst catalyzes highly enantioselective hydroamination of structurally varied unactivated alkenes containing a wide range of functional groups with a 1:1 ratio of the amine and the alkene.
- (3).Xi Y; Ma S; Hartwig JF Catalytic asymmetric addition of an amine N–H bond across internal alkenes. Nature 2020, 588 (7837), 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]; 3A cationic iridium system bearing a new TMS-SYNPHOS ligand catalyzed the intermolecular hydroamination of a range of unactivated, internal alkenes, including those in both acyclic and cyclic alkenes, to afford chiral amines with high enantioselectivity.
- (4).Ma S; Hill CK; Olen CL; Hartwig JF Ruthenium-Catalyzed Hydroamination of Unactivated Terminal Alkenes with Stoichiometric Amounts of Alkene and an Ammonia Surrogate by Sequential Oxidation and Reduction. J. Am. Chem. Soc 2021, 143 (1), 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]; 4Ruthenium-catalyzed intermolecular hydroaminations of a variety of unactivated terminal alkenes occurred through oxidative amination in concert with reduction of the resulting imine.
- (5).(a) Bytschkov I; Doye S Group-IV Metal Complexes as Hydroamination Catalysts. Eur. J. Org. Chem 2003, 2003 (6), 935–946. [Google Scholar]; (b) Pohlki F; Doye S The catalytic hydroamination of alkynes. Chem. Soc. Rev 2003, 32 (2), 104–114. [DOI] [PubMed] [Google Scholar]; (c) Hong S; Marks TJ Organolanthanide-Catalyzed Hydroamination. Acc. Chem. Res 2004, 37 (9), 673–686. [DOI] [PubMed] [Google Scholar]; (d) Hultzsch KC Transition Metal-Catalyzed Asymmetric Hydroamination of Alkenes (AHA). Adv. Synth. Catal 2005, 347 (2–3), 367–391. [Google Scholar]; (e) Koschker P; Breit B Branching Out: Rhodium-Catalyzed Allylation with J. Am. Chem. Soc.Alkynes and Allenes. Acc. Chem. Res 2016, 49 (8), 1524–1536. [DOI] [PubMed] [Google Scholar]; (f) Teng H-L; Luo Y; Wang B; Zhang L; Nishiura M; Hou Z Synthesis of Chiral Aminocyclopropanes by Rare-Earth-Metal-Catalyzed Cyclopropene Hydroamination. Angew. Chem., Int. Ed 2016, 55 (49), 15406–15410. [DOI] [PubMed] [Google Scholar]; (g) Xu K; Wang Y-H; Khakyzadeh V; Breit B Asymmetric synthesis of allylic amines via hydroamination of allenes with benzophenone imine. Chem. Sci 2016, 7 (5), 3313–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Teng H-L; Luo Y; Nishiura M; Hou Z Diastereodivergent Asymmetric Carboamination/Annulation of Cyclopropenes with Aminoalkenes by Chiral Lanthanum Catalysts. J. Am. Chem. Soc 2017, 139 (46), 16506–16509. [DOI] [PubMed] [Google Scholar]; (i) Adamson NJ; Jeddi H; Malcolmson SJ Preparation of Chiral Allenes through Pd-Catalyzed Intermolecular Hydroamination of Conjugated Enynes: Enantioselective Synthesis Enabled by Catalyst Design. J. Am. Chem. Soc 2019, 141 (21), 8574–8583. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Hilpert LJ; Sieger SV; Haydl AM; Breit B Palladium- and Rhodium-Catalyzed Dynamic Kinetic Resolution of Racemic Internal Allenes Towards Chiral Pyrazoles. Angew. Chem., Int. Ed 2019, 58 (11), 3378–3381. [DOI] [PubMed] [Google Scholar]; (k) Müller TE; Beller M Metal-Initiated Amination of Alkenes and Alkynes. Chem. Rev 1998, 98 (2), 675–704. [DOI] [PubMed] [Google Scholar]; (l) Huang L; Arndt M; Gooßen K; Heydt H; Gooßen LJ Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev 2015, 115 (7), 2596–2697. [DOI] [PubMed] [Google Scholar]
- (6).Howk BW; Little EL; Scott SL; Whitman GM Alkali Metal-catalyzed Amination of Olefins. J. Am. Chem. Soc 1954, 76 (7), 1899–1902. [Google Scholar]
- (7).(a) Musacchio AJ; Lainhart BC; Zhang X; Naguib SG; Sherwood TC; Knowles RR Catalytic intermolecular hydroaminations of unactivated olefins with secondary alkyl amines. Science 2017, 355 (6326), 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chinn AJ; Sedillo K; Doyle AG Phosphine/Photoredox Catalyzed Anti-Markovnikov Hydroamination of Olefins with Primary Sulfonamides via α-Scission from Phosphoranyl Radicals. J. Am. Chem. Soc 2021, 143 (43), 18331–18338. [DOI] [PubMed] [Google Scholar]
- (8).Johns AM; Sakai N; Ridder A; Hartwig JF Direct Measurement of the Thermodynamics of Vinylarene Hydroamination. J. Am. Chem. Soc 2006, 128 (29), 9306–9307. [DOI] [PubMed] [Google Scholar]
- (9).(a) Johns AM; Utsunomiya M; Incarvito CD; Hartwig JF A Highly Active Palladium Catalyst for Intermolecular Hydroamination. Factors that Control Reactivity and Additions of Functionalized Anilines to Dienes and Vinylarenes. J. Am. Chem. Soc 2006, 128 (6), 1828–1839. [DOI] [PubMed] [Google Scholar]; (b) Pawlas J; Nakao Y; Kawatsura M; Hartwig JF A General Nickel-Catalyzed Hydroamination of 1,3-Dienes by Alkylamines: Catalyst Selection, Scope, and Mechanism. J. Am. Chem. Soc 2002, 124 (14), 3669–3679. [DOI] [PubMed] [Google Scholar]
- (10).Rosenfeld DC; Shekhar S; Takemiya A; Utsunomiya M; Hartwig JF Hydroamination and Hydroalkoxylation Catalyzed by Triflic Acid. Parallels to Reactions Initiated with Metal Triflates. Org. Lett 2006, 8 (19), 4179–4182. [DOI] [PubMed] [Google Scholar]
- (11).Kawatsura M; Hartwig JF Palladium-Catalyzed Intermolecular Hydroamination of Vinylarenes Using Arylamines. J. Am. Chem. Soc 2000, 122 (39), 9546–9547. [Google Scholar]
- (12).(a) Qian H; Widenhoefer RA Platinum-Catalyzed Intermolecular Hydroamination of Vinyl Arenes with Carboxamides. Org. Lett 2005, 7 (13), 2635–2638. [DOI] [PubMed] [Google Scholar]; (b) Hu A; Ogasawara M; Sakamoto T; Okada A; Nakajima K; Takahashi T; Lin W Palladium-Catalyzed Intermolecular Asymmetric Hydroamination with 4,4′-Disubstituted BINAP and SEGPHOS. Adv. Synth. Catal 2006, 348 (15), 2051–2056. [Google Scholar]; (c) Kaspar LT; Fingerhut B; Ackermann L Titanium-Catalyzed Intermolecular Hydroamination of Vinylarenes. Angew. Chem., Int. Ed 2005, 44 (37), 5972–5974. [DOI] [PubMed] [Google Scholar]
- (13).Horrillo-Martínez P; Hultzsch KC; Gil A; Branchadell V Base-Catalyzed Anti-Markovnikov Hydroamination of Vinylarenes – Scope, Limitations and Computational Studies. Eur. J. Org. Chem 2007, 2007 (20), 3311–3325. [Google Scholar]
- (14).Seshu Babu N; Mohan Reddy K; Sai Prasad PS; Suryanarayana I; Lingaiah N Intermolecular hydroamination of vinyl arenes using tungstophosphoric acid as a simple and efficient catalyst. Tetrahedron Lett. 2007, 48 (43), 7642–7645. [Google Scholar]
- (15).Utsunomiya M; Hartwig JF Intermolecular, Markovnikov Hydroamination of Vinylarenes with Alkylamines. J. Am. Chem. Soc 2003, 125 (47), 14286–14287. [DOI] [PubMed] [Google Scholar]
- (16).Nettekoven U; Hartwig JF A New Pathway for Hydroamination. Mechanism of Palladium-Catalyzed Addition of Anilines to Vinylarenes. J. Am. Chem. Soc 2002, 124 (7), 1166–1167. [DOI] [PubMed] [Google Scholar]
- (17).Beller M; Eichberger M; Trauthwein H Anti-Markovnikov Functionalization of Olefins: Rhodium-Catalyzed Oxidative Aminations of Styrenes. Angew. Chem., Int. Ed 1997, 36 (20), 2225–2227. [Google Scholar]
- (18).Utsunomiya M; Kuwano R; Kawatsura M; Hartwig JF Rhodium-Catalyzed Anti-Markovnikov Hydroamination of Vinylarenes. J. Am. Chem. Soc 2003, 125 (19), 5608–5609. [DOI] [PubMed] [Google Scholar]
- (19).Utsunomiya M; Hartwig JF Ruthenium-Catalyzed Anti-Markovnikov Hydroamination of Vinylarenes. J. Am. Chem. Soc 2004, 126 (9), 2702–2703. [DOI] [PubMed] [Google Scholar]
- (20).Takaya J; Hartwig JF Mechanistic Studies of Ruthenium-Catalyzed Anti-Markovnikov Hydroamination of Vinylarenes: Intermediates and Evidence for Catalysis through π-Arene Complexes. J. Am. Chem. Soc 2005, 127 (16), 5756–5757. [DOI] [PubMed] [Google Scholar]
- (21).(a) Otsuka M; Yokoyama H; Endo K; Shibata T Rucatalyzed β-selective and enantioselective addition of amines to styrenes initiated by direct arene-exchange. Org. Biomol. Chem 2012, 10 (19), 3815–3818. [DOI] [PubMed] [Google Scholar]; (b) Takemoto S; Matsuzaka H Recent topics on catalytic transformations of aromatic molecules via η6-arene transition metal complexes. Tetrahedron Lett. 2018, 59 (8), 697–703. [Google Scholar]
- (22).(a) Minami T; Okamoto H; Ikeda S; Tanaka R; Ozawa F; Yoshifuji M (η3-Allyl)palladium Complexes Bearing Diphosphinidenecyclobutene Ligands: Highly Active Catalysts for the Hydroamination of 1,3-Dienes. Angew. Chem., Int. Ed 2001, 40 (23), 4501–4503. [DOI] [PubMed] [Google Scholar]; (b) Brouwer C; He C Efficient Gold-Catalyzed Hydroamination of 1,3-Dienes. Angew. Chem., Int. Ed 2006, 45 (11), 1744–1747. [DOI] [PubMed] [Google Scholar]; (c) Goldfogel MJ; Roberts CC; Meek SJ Intermolecular Hydroamination of 1,3-Dienes Catalyzed by Bis-(phosphine)carbodicarbene–Rhodium Complexes. J. Am. Chem. Soc 2014, 136 (17), 6227–6230. [DOI] [PubMed] [Google Scholar]; (d) Yang X-H; Lu A; Dong VM Intermolecular Hydroamination of 1,3-Dienes To Generate Homoallylic Amines. J. Am. Chem. Soc 2017, 139 (40), 14049–14052. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tran G; Shao W; Mazet C Ni-Catalyzed Enantioselective Intermolecular Hydroamination of Branched 1,3-Dienes Using Primary Aliphatic Amines. J. Am. Chem. Soc 2019, 141 (37), 14814–14822. [DOI] [PubMed] [Google Scholar]
- (23).Ryu J-S; Li GY; Marks TJ Organolathanide-Catalyzed Regioselective Intermolecular Hydroamination of Alkenes, Alkynes, Vinylarenes, Di- and Trivinylarenes, and Methylenecyclopropanes. Scope and Mechanistic Comparison to Intramolecular Cyclohydroaminations. J. Am. Chem. Soc 2003, 125 (41), 12584–12605. [DOI] [PubMed] [Google Scholar]
- (24).Pradhan S; Das S; Kumar G; Chatterjee I Transition-MetalFree Regioselective Intermolecular Hydroamination of Conjugated 1,3Dienes with Heterocyclic Amines. Org. Lett 2022, 24 (12), 2452–2456. [DOI] [PubMed] [Google Scholar]
- (25).Takahashi S; Shibano T; Hagihara N The Dimerization of Butadiene by Palladium Complex Catalysts. Bull. Chem. Soc. Jpn 1968, 41 (2), 454–460. [Google Scholar]
- (26).Löber O; Kawatsura M; Hartwig JF Palladium-Catalyzed Hydroamination of 1,3-Dienes: A Colorimetric Assay and Enantioselective Additions. J. Am. Chem. Soc 2001, 123 (18), 4366–4367. [DOI] [PubMed] [Google Scholar]
- (27).Johns AM; Liu Z; Hartwig JF Primary tert- and sec-Allylamines via Palladium-Catalyzed Hydroamination and Allylic Substitution with Hydrazine and Hydroxylamine Derivatives. Angew. Chem., Int. Ed 2007, 46 (38), 7259–7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Sakai N; Ridder A; Hartwig JF Tropene Derivatives by Sequential Intermolecular and Transannular, Intramolecular Palladium-Catalyzed Hydroamination of Cycloheptatriene. J. Am. Chem. Soc 2006, 128 (25), 8134–8135. [DOI] [PubMed] [Google Scholar]
- (29).(a) Adamson NJ; Hull E; Malcolmson SJ Enantioselective Intermolecular Addition of Aliphatic Amines to Acyclic Dienes with a Pd–PHOX Catalyst. J. Am. Chem. Soc 2017, 139 (21), 7180–7183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jiu AY; Slocumb HS; Yeung CS; Yang X-H; Dong VM Enantioselective Addition of Pyrazoles to Dienes. Angew. Chem., Int. Ed 2021, 60 (36), 19660–19664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Park S; Malcolmson SJ Development and Mechanistic Investigations of Enantioselective Pd-Catalyzed Intermolecular Hydroaminations of Internal Dienes. ACS Catal. 2018, 8 (9), 8468–8476. [Google Scholar]
- (31).Feng S; Hao H; Liu P; Buchwald SL Diastereo- and Enantioselective CuH-Catalyzed Hydroamination of Strained Trisubstituted Alkenes. ACS Catal. 2020, 10 (1), 282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).(a) Dorta R; Egli P; rcher F; Togni A The [IrCl-(Diphosphine)]2/Fluoride System. Developing Catalytic Asymmetric Olefin Hydroamination. J. Am. Chem. Soc 1997, 119 (44), 10857–10858. [Google Scholar]; (b) Ackermann L; Kaspar LT; Gschrei CJ TiCl4-Catalyzed Intermolecular Hydroamination Reactions of Norbornene. Org. Lett 2004, 6 (15), 2515–2518. [DOI] [PubMed] [Google Scholar]; (c) Kemper J; Studer A Stable Reagents for the Generation of N-Centered Radicals: Hydroamination of Norbornene. Angew. Chem., Int. Ed 2005, 44 (31), 4914–4917. [DOI] [PubMed] [Google Scholar]; (d) McBee JL; Bell AT; Tilley TD Mechanistic Studies of the Hydroamination of Norbornene with Electrophilic Platinum Complexes: The Role of Proton Transfer. J. Am. Chem. Soc 2008, 130 (49), 16562–16571. [DOI] [PubMed] [Google Scholar]; (e) Casalnuovo AL; Calabrese JC; Milstein D Rational design in homogeneous catalysis. Iridium(I)-catalyzed addition of aniline to norbornylene via nitrogen-hydrogen activation. J. Am. Chem. Soc 1988, 110 (20), 6738–6744. [Google Scholar]
- (33).Zhou J; Hartwig JF Intermolecular, Catalytic Asymmetric Hydroamination of Bicyclic Alkenes and Dienes in High Yield and Enantioselectivity. J. Am. Chem. Soc 2008, 130 (37), 12220–12221. [DOI] [PubMed] [Google Scholar]
- (34).(a) Zhao J; Goldman AS; Hartwig JF Oxidative Addition of Ammonia to Form a Stable Monomeric Amido Hydride Complex. Science 2005, 307 (5712), 1080–1082. [DOI] [PubMed] [Google Scholar]; (b) Kanzelberger M; Zhang X; Emge TJ; Goldman AS; Zhao J; Incarvito C; Hartwig JF Distinct Thermodynamics for the Formation and Cleavage of N–H Bonds in Aniline and Ammonia. Directly-Observed Reductive Elimination of Ammonia from an Isolated Amido Hydride Complex. J. Am. Chem. Soc 2003, 125 (45), 13644–13645. [DOI] [PubMed] [Google Scholar]
- (35).(a) Zhao P; Krug C; Hartwig JF Transfer of Amido Groups from Isolated Rhodium(I) Amides to Alkenes and Vinylarenes. J. Am. Chem. Soc 2005, 127 (34), 12066–12073. [DOI] [PubMed] [Google Scholar]; (b) Hanley PS; Marković D; Hartwig JF Intermolecular Insertion of Ethylene and Octene into a Palladium–Amide Bond. Spectroscopic Evidence for an Ethylene Amido Intermediate. J. Am. Chem. Soc 2010, 132 (18), 6302–6303. [DOI] [PubMed] [Google Scholar]; (c) Hanley PS; Hartwig JF Intermolecular Migratory Insertion of Unactivated Olefins into Palladium–Nitrogen Bonds. Steric and Electronic Effects on the Rate of Migratory Insertion. J. Am. Chem. Soc 2011, 133 (39), 15661–15673. [DOI] [PubMed] [Google Scholar]; (d) Hanley PS; Hartwig JF Migratory Insertion of Alkenes into Metal–Oxygen and Metal–Nitrogen Bonds. Angew. Chem., Int. Ed 2013, 52 (33), 8510–8525. [DOI] [PubMed] [Google Scholar]
- (36).(a) Pan S; Endo K; Shibata T Ir(I)-Catalyzed Intermolecular Regio- and Enantioselective Hydroamination of Alkenes with Heteroaromatic Amines. Org. Lett 2012, 14 (3), 780–783. [DOI] [PubMed] [Google Scholar]; (b) Reznichenko AL; Nguyen HN; Hultzsch KC Asymmetric Intermolecular Hydroamination of Unactivated Alkenes with Simple Amines. Angew. Chem., Int. Ed 2010, 49 (47), 8984–8987. [DOI] [PubMed] [Google Scholar]; (c) Zhang Z; Lee SD; Widenhoefer RA Intermolecular Hydroamination of Ethylene and 1-Alkenes with Cyclic Ureas Catalyzed by Achiral and Chiral Gold(I) Complexes. J. Am. Chem. Soc 2009, 131 (15), 5372–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).(a) Gurak JA Jr.; Yang KS; Liu Z; Engle KM Directed, Regiocontrolled Hydroamination of Unactivated Alkenes via Protodepalladation. J. Am. Chem. Soc 2016, 138 (18), 5805–5808. [DOI] [PubMed] [Google Scholar]; (b) Vanable EP; Kennemur JL; Joyce LA; Ruck RT; Schultz DM; Hull KL Rhodium-Catalyzed Asymmetric Hydroamination of Allyl Amines. J. Am. Chem. Soc 2019, 141 (2), 739–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).(a) Rucker RP; Whittaker AM; Dang H; Lalic G Synthesis of Tertiary Alkyl Amines from Terminal Alkenes: Copper-Catalyzed Amination of Alkyl Boranes. J. Am. Chem. Soc 2012, 134 (15), 6571–6574. [DOI] [PubMed] [Google Scholar]; (b) Miki Y; Hirano K; Satoh T; Miura M Copper-Catalyzed Intermolecular Regioselective Hydroamination of Styrenes with Polymethylhydrosiloxane and Hydroxylamines. Angew. Chem., Int. Ed 2013, 52 (41), 10830–10834. [DOI] [PubMed] [Google Scholar]; (c) Gao Y; Cui Y; Huo Y; Chen J; She M; Li X; Chen Q; Hu X-Q Nickel-Catalyzed Hydroamination of Olefins with Anthranils. J. Org. Chem 2021, 86 (17), 12107–12118. [DOI] [PubMed] [Google Scholar]; (d) Lee C; Kang H-J; Seo H; Hong S Nickel-Catalyzed Regio- and Enantioselective Hydroamination of Unactivated Alkenes Using Carbonyl Directing Groups. J. Am. Chem. Soc 2022, 144 (20), 9091–9100. [DOI] [PubMed] [Google Scholar]; (e) Yang Y; Shi S-L; Niu D; Liu P; Buchwald SL Catalytic asymmetric hydroamination of unactivated internal olefins to aliphatic amines. Science 2015, 349 (6243), 62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wagner-Carlberg N; Rovis T Rhodium(III)-Catalyzed Anti-Markovnikov Hydroamidation of Unactivated Alkenes Using Dioxazolones as Amidating Reagents. J. Am. Chem. Soc 2022, 144 (49), 22426–22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sevov CS; Zhou J; Hartwig JF Iridium-Catalyzed, Intermolecular Hydroamination of Unactivated Alkenes with Indoles. J. Am. Chem. Soc 2014, 136 (8), 3200–3207. [DOI] [PubMed] [Google Scholar]
- (41).Tye JW; Hartwig JF Computational Studies of the Relative Rates for Migratory Insertions of Alkenes into Square-Planar, Methyl, – Amido, and – Hydroxo Complexes of Rhodium. J. Am. Chem. Soc 2009, 131 (41), 14703–14712. [DOI] [PubMed] [Google Scholar]
- (42).Verendel JJ; Pàmies O; Diéguez M; Andersson PG Asymmetric Hydrogenation of Olefins Using Chiral Crabtree-type Catalysts: Scope and Limitations. Chem. Rev 2014, 114 (4), 2130–2169. [DOI] [PubMed] [Google Scholar]
- (43).Ma S; Fan H; Day CS; Xi Y; Hartwig JF Remote Hydroamination of Disubstituted Alkenes by a Combination of Isomerization and Regioselective N–H Addition. J. Am. Chem. Soc 2023, 145 (7), 3875–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wang DY; Choliy Y; Haibach MC; Hartwig JF; Krogh-Jespersen K; Goldman AS Assessment of the Electronic Factors Determining the Thermodynamics of “Oxidative Addition” of C–H and N–H Bonds to Ir(I) Complexes. J. Am. Chem. Soc 2016, 138 (1), 149–163. [DOI] [PubMed] [Google Scholar]