

Abstract
In this study, we establish that conjugated enynes undergo selective 1,4-hydroamination under Pd catalysis to deliver chiral allenes with pendant allylic amines. Several primary and secondary aliphatic and aryl-substituted amines couple with a wide range of mono- and disubstituted enynes in a nonenantioselective reaction where DPEphos serves as the ligand for Pd. Benzophenone imine acts as an ammonia surrogate to afford primary amines in a two-step/one-pot process. Examination of chiral catalysts revealed a high degree of reversibility in the C–N bond formation that negatively impacted enantioselectivity. Consequently, an electron-poor ferrocenyl-PHOX ligand was developed to enable efficient and enantioselective enyne hydroamination.
Graphical Abstract
1. INTRODUCTION
The development of new transformations for the installation of amine functionality is critically important for the preparation of new medicines, agrochemicals, and natural products. Intermolecular hydroamination1 provides an atom economical way of generating chiral amines from readily available unsaturated hydrocarbons via C–N bond formation. Catalytic enantioselective reactions involving alkynes,2 allenes,3 cyclic4 and acyclic5 1,3-dienes, vinylarenes,6 cyclopropenes,7 and simple α-olefins8 have been disclosed.9
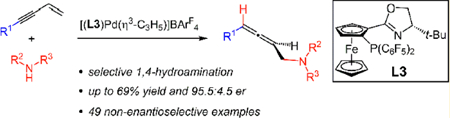
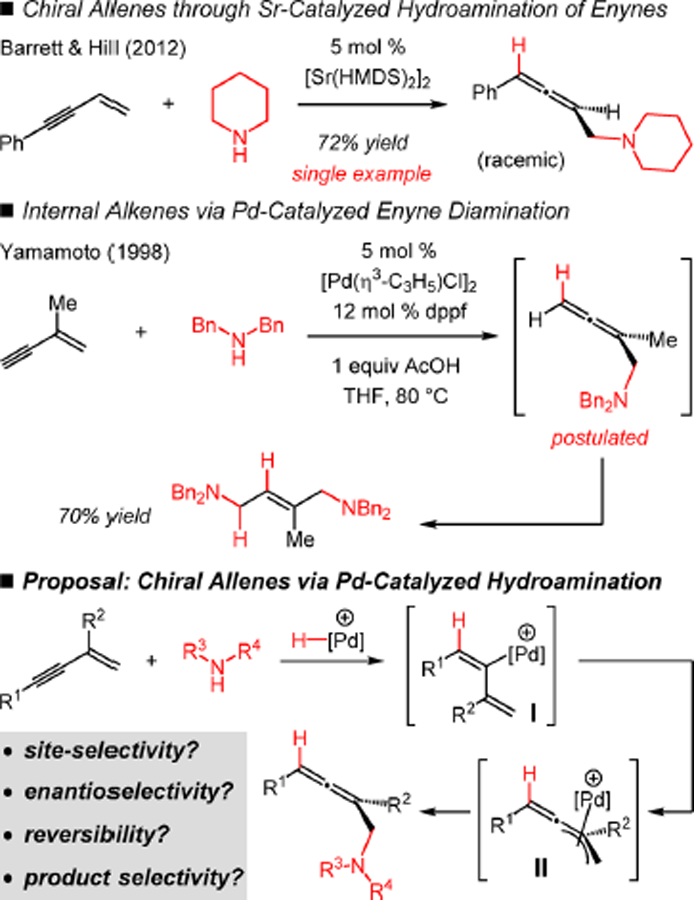
In contrast to these numerous examples are the paucity of reports detailing hydroamination of conjugated enynes. Recently, Barrett, Hill, and co-workers have demonstrated a single, nonenantioselective Sr-catalyzed hydroamination of an enyne that delivers an aminomethyl-substituted allene (Scheme 1).10 The Yamamoto laboratory has shown that 3-substituted enynes undergo Pd–bis(phosphine)-catalyzed double addition of aliphatic amines to yield 2-butenyl-1,4-diamines.11 These reactions were suggested to take place by the intermediacy of an aminomethyl-substituted allene. To our knowledge, there are no reported examples of enantioselective hydroamination of nonpolarized enynes.12,13 In general, intermolecular couplings of unactivated enynes with nucleophiles are rare, especially in a catalytic enantioselective fashion.14
Scheme 1.
Hydroaminations of Enynes
In fact, the majority of late transition-metal-catalyzed enyne hydrofunctionalizations have concerned reductive couplings with aldehydes or ketones as electrophiles.15 Interestingly, reactions that take place by initial metal–hydride insertion16 (Ru-, Ir-, or Cu-based catalysts) generally do so at the alkene, forming a propargyl metal species that is in equilibrium with an allenyl metal. It should be noted that Cu-catalyzed enyne hydroboration17 delivers chiral allenes through the direct σ-bond metathesis of the metal for boron in the allenyl metal intermediate.18
In part inspired by Yamamoto’s proposal that enyne diaminations occur by initial formation of an aminomethyl allene,11 we hypothesized that under milder reaction conditions and with the appropriate catalyst, Pd-catalyzed hydroamination of enynes might lead to allene products (Scheme 1). Although Pd–H insertion could occur at the olefin, this cannot lead to a stable π-allyl complex for amine addition. Instead, alkyne insertion would lead to η1-butadienyl–Pd I, which could then lead to the η3-butadienyl–Pd II. Nucleophilic attack at the least hindered electrophilic carbon would then afford the allene product.19 Such a π-allyl complex has been invoked as an intermediate in several Pd-catalyzed allylic substitution and related reactions.20
Yet even if this site-selectivity could be achieved for hydroamination, several questions remained with regard to the enantio- and chemoselectivity of the Pd-catalyzed process. (1) To achieve high enantioselectivity, a chiral catalyst would have to control the direction of the 90° single bond rotation of dienyl I in forming π-allyl II or, alternatively, nucleophilic attack upon II or its diastereomer would have to be under Curtin–Hammett control.20 Although there was some precedent in allylic substitution with malonate,20a,b,d amine,20b,c and amide nucleophiles,20c–e would this be possible in hydroamination? (2) Even if kinetic control of enantioselectivity could be achieved, would reaction reversibility via C–N bond ionization and reformation of I erode it over the course of the reaction? (3) Would the optimal catalyst for controlling site-selectivity and enantioselectivity also selectively lead to allene formation without further reaction to produce a diamine?11 In this work, we illustrate the successful realization of an enyne 1,4-hydroamination process to prepare myriad racemic aminomethyl-substituted allenes with DPEphos-ligated Pd as the catalyst. Through the design and development of a new PHOX ligand, we demonstrate the principles needed for enantioselectivity control in this transformation.
2. RESULTS AND DISCUSSION
2.1. Development of Nonenantioselective Enyne Hydroamination with Alkyl Amines, Aryl-Substituted Amines, and Benzophenone Imine.
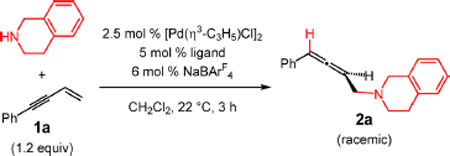
We began our study by investigating the nonenantioselective addition of tetrahydroisoquinoline (THIQ) to phenyl-substituted enyne 1a (Table 1). Catalyst was generated in situ from 2.5 mol % [(η3-C3H5)PdCl]2 and 6 mol % NaBArF4 with 5 mol % of several achiral bis(phosphine) ligands. No matter the phosphine identity, allene 2a was obtained selectively after 3 h at ambient temperature in CH2Cl2. Other product regioisomers and diamination of the enyne were not observed. The efficiency of the reaction was correlated with the natural bite angle of the phosphine (entries 1–6).21,22 This phenomenon could perhaps be attributed to an increased rate of nucleophilic attack upon the Pd-π-allyl with wider bite angle ligands.22 As DPEphos and Xantphos gave roughly equivalent results, we opted to continue with the former. NaBF4 in place of NaBArF4 gave nearly the same result as well (entry 7) although having a noncoordinating counterion was important for reaction efficiency.23 The isolated Pd complex (Pd-1) behaved similarly to that formed in situ (entry 8), and so for exploration of reaction scope, we employed isolated Pd-1. Under the optimized conditions, allene 2a was obtained in 88% yield.
Table 1.
Role of Ligand Bite Angle on Efficiency of the Nonenantioselective Pd-Catalyzed Processa
 | |||
|---|---|---|---|
| entry | ligand | bite angle | yield of 2a (%)b |
| 1 | dppe | 86° | 14 |
| 2 | dppp | 91° | 44 |
| 3 | dppb | 94° | 62 |
| 4 | dppf | 99° | 63 |
| 5 | DPEphos | 104° | 84 |
| 6 | Xantphos | 108° | 83 |
| 7c | DPEphos | 104° | 86 |
| 8d | DPEphos | 104° | 88 |
Reactions under N2 with 0.2 mmol of tetrahydroisoquinoline (0.8 M).
Isolated yield of 2a after purification.
NaBF4 instead of NaBArF4.
Reaction with isolated [(DPEPhos)Pd(η3-C3H5)]BF4 (Pd-1).
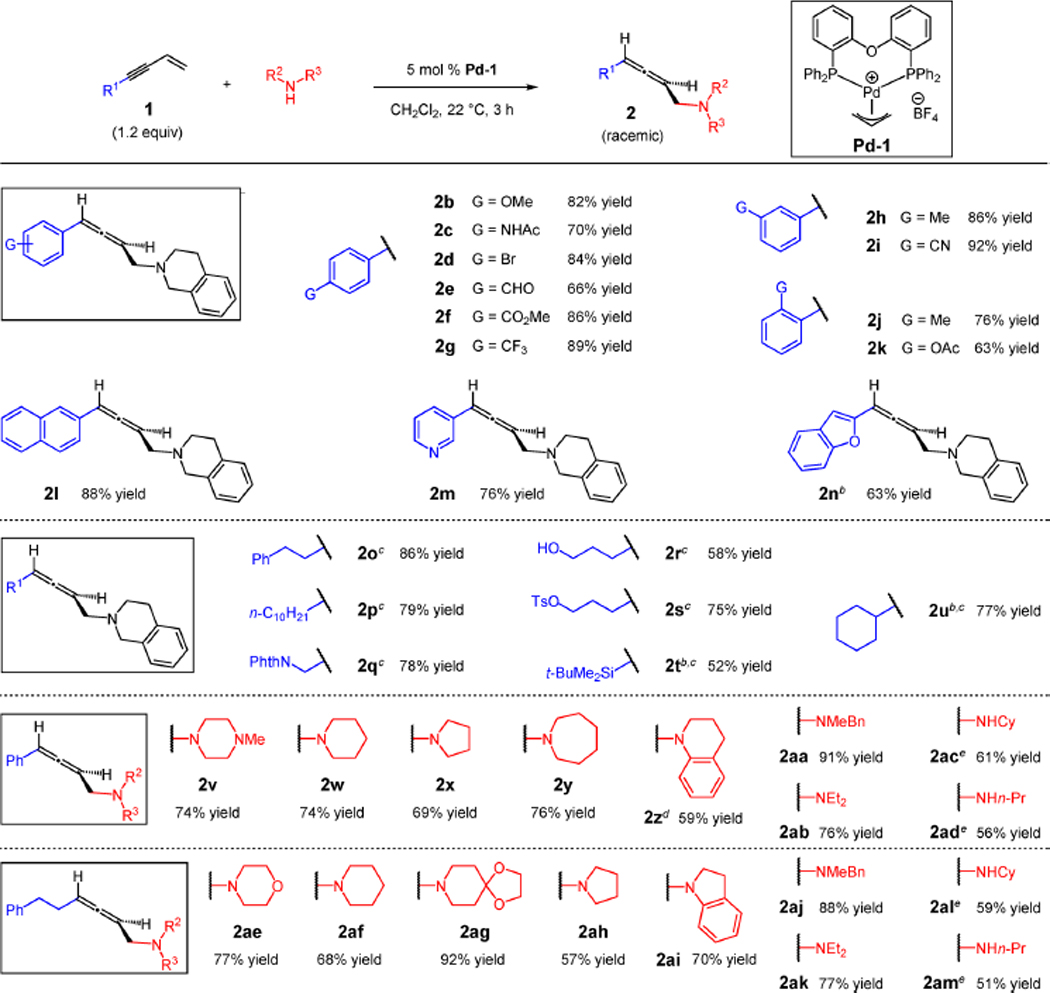
Numerous readily accessible enynes couple with a wide range of commercially available amines for 1,4-hydroamination with Pd-1 (Table 2). A variety of aryl-substituted enynes are effective partners in transformations with THIQ (2b–k, 63–92% yield). Substrate electronics have little obvious effect on reaction efficiency, and several aryl substituents are compatible with the hydroamination process, including functionality with acidic protons (2c), aryl bromides (2d), and aldehydes (2e). ortho-Substituted arenes afford products with only slightly diminished yields (2j–k). A naphthyl group (2l) and heteroaromatic rings (2m–n), including a Lewis basic pyridyl substituent, are tolerated.
Table 2.
Scope of Nonenantioselective Enyne Hydroamination To Prepare Disubstituted Allenesa
 |
Reactions under N2 with 0.2 mmol of amine (0.8 M) and 5 mol % Pd-1 for 3 h unless otherwise noted. Isolated yields of purified products.
15 h reaction.
Catalyst prepared in situ with NaBArF4.
20 h reaction.
10 mol % Pd-1.
Alkyl-substituted enynes and a silyl-substituted enyne undergo efficient hydroamination with THIQ24 as well (Table 2, 2o–u, 52–86% yield). The functional group tolerance is again impressive, a testament to the mild reaction conditions for this catalytic process. A propargylic phthalimido group (2q) and a primary tosylate (2s) remain intact. A free hydroxyl group (2r) does not interfere in the process. With extended reaction times, steric hindrance imposed at the alkyne by a TBS (2t) or cyclohexyl (2u) group can also be overcome by the Pd-based catalyst.
The amine scope is similarly broad (Table 2), with both phenyl-substituted enyne 1a and phenethyl-containing enyne 1o; the atom economic reactions generate allenes 2v–am in 51–92% yield. Several amines, including the more Lewis basic piperidine (2w and 2af) and pyrrolidine (2x and 2ah) that may at times prove challenging to late transition metal catalysts, couple in good yields. Additionally, azepane (2y) and a ketal-containing piperidine (2ag), which may serve as an ammonia surrogate,25 are also effective partners. Aryl-substituted amines, such as tetrahydroquinoline and indoline, afford allenes 2z and 2ai in 59% and 70% yields, respectively. Acyclic secondary amines such as N-methylbenzylamine (2aa and 2aj) and sterically hindered diethylamine (2ab and 2ak) add smoothly to enynes. Primary amines are also capable of participating in the enyne hydroamination (2ac–ad and 2al–am) although the transformations require 10 mol % Pd-1 and catalyst turnover is low (51–61% yield). The 1,4-hydroamination of enyne 1a is readily scalable (5.0 mmol scale) and can be carried out with as little as 2 mol % Pd catalyst, prepared in situ with NaBArF4 (Scheme 2). Under these conditions, 1.2 g of the THIQ adduct 2a was obtained in 92% yield.
Scheme 2.
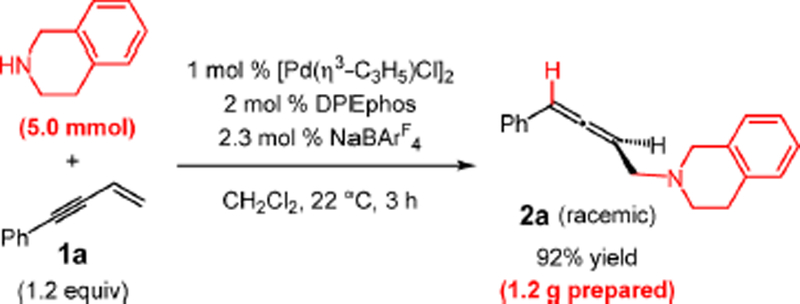
Scalability of Nonenantioselective Hydroamination with Tetrahydroisoquinoline
We were also pleased to find that benzophenone imine,3i which serves as a surrogate for ammonia, undergoes facile addition to enynes 1 with 5 mol % Pd-1 in the presence of Et3N as a proton shuttle (Table 3). Selective 1,4-addition occurs to afford 3 after 24 h. The imine may be isolated or hydrolyzed in the workup under mildly acidic conditions to form the corresponding primary amine 4. For example, reaction with enyne 1a leads to allene 3 in 86% yield, but addition to naphthyl-substituted enyne 1l and in situ hydrolysis of the resulting imine delivers primary amine 4a in 65% yield. Alkyl-substituted enynes also react with benzophenone imine to form allenes 4b–c after hydrolysis. Thus, through the 1,4-hydroamination process, myriad chiral allenes that contain tertiary, secondary, and primary amines may be obtained in racemic form.
Table 3.
Enyne Hydroamination with Benzophenone Imine as an Ammonia Surrogatea
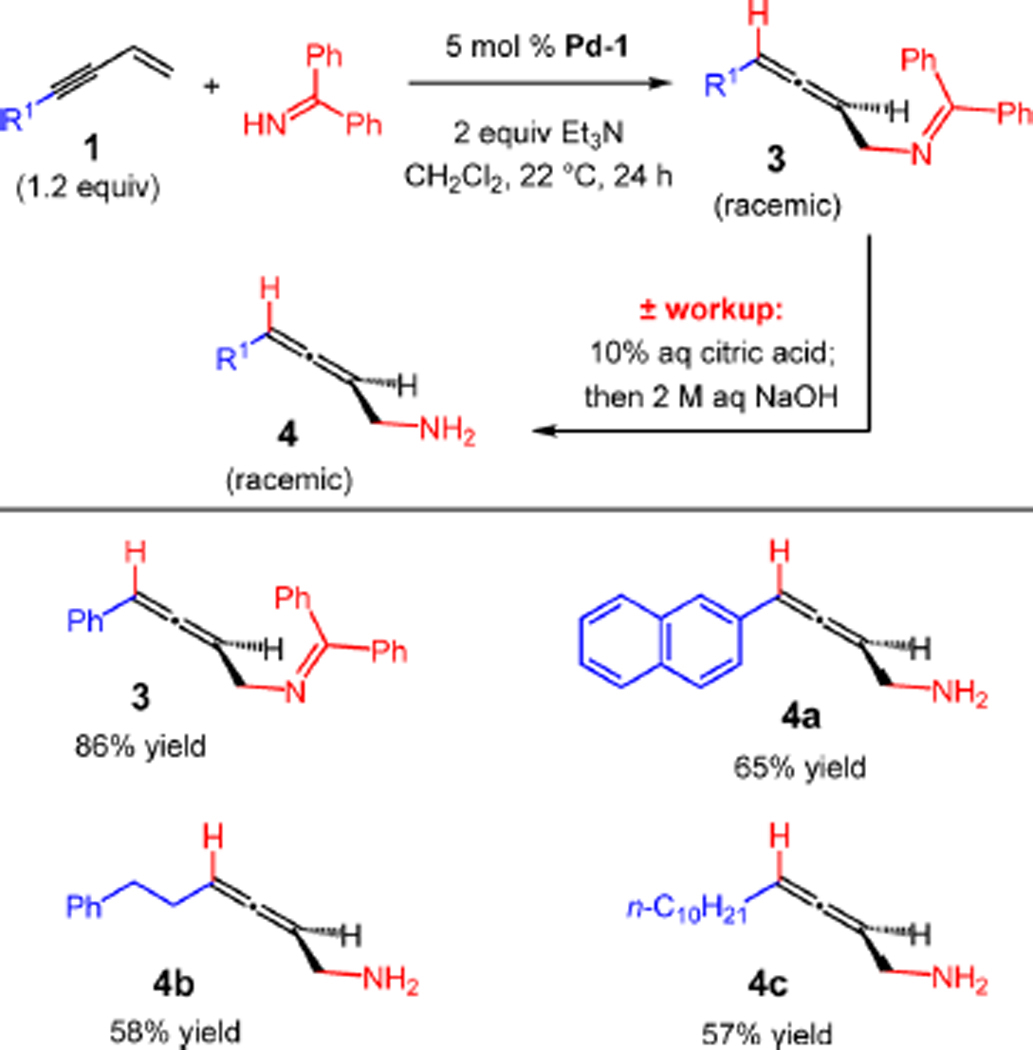 |
Reactions under N2 with 1.0 mmol benzophenone imine (0.8 M).
Furthermore, we have investigated the addition of THIQ to a handful of 1,3-disubstituted enynes 5 (Table 4). Selective 1,4-addition, catalyzed by Pd-1, enables the isolation of trisubstituted allenes 6 in 51–82% yield. With a phenyl substituent at C1, several groups are tolerated at the 3-position, including methyl (6a), linear alkyl (6b), α-branched cyclohexyl (6c), and a trifluoromethyl group (6d). An enyne bearing alkyl groups at both the 1- and 3-positions furnishes trialkyl-containing allene 6e in 82% yield; however, with aryl substitution at both positions, the enyne proved too unstable to allow for study.
Table 4.
1,3-Disubstituted Enyne Scope for Trisubstituted Allene Synthesisa
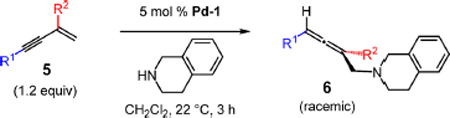 | ||
|---|---|---|
| entry | product, R1, R2 | yield (%)b |
| 1 | 6a, Ph, Me | 60 |
| 2 | 6b, Ph, n-C6H13 | 66 |
| 3 | 6c, Ph, Cy | 51 |
| 4 | 6d, Ph, CF3 | 60 |
| 5 | 6e, Cy, Me | 82 |
Reactions under N2 with 0.2 mmol of tetrahydroisoquinoline (0.8 M).
Isolated yields of purified products.
We additionally examined the addition of THIQ to 1,4-disubstituted enyne (Z)-7 (Scheme 3). Compared to reactions with the 1,3-disubstituted congeners, the transformation is markedly less efficient in the absence of Et3N. After 20 h with [Pd(DPEphos)]BArF4, aminoethyl allene 8 is isolated in 25% yield as a 2:1 mixture of diastereomers. The stereochemical configuration of the major isomer was not determined. In the presence of Et3N, allene 8 is isolated in 59% yield (1:1 dr).
Scheme 3.
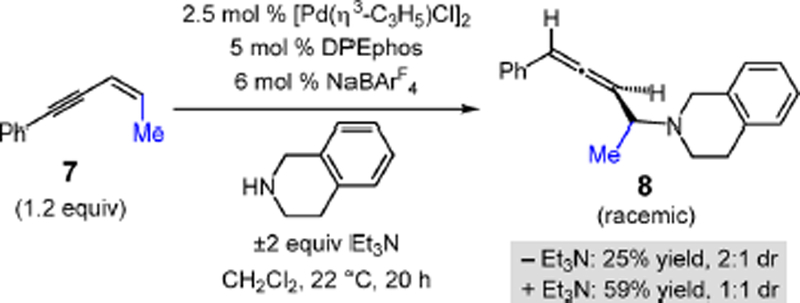
THIQ Addition to a 1,4-Disubstituted Enyne
2.2. Development of Enantioselective Enyne Hydroamination with Pd(PHOX) Catalysts.
Having established the feasibility of transforming conjugated enynes to chiral allenes by site-selective 1,4-addition of amines with an achiral Pd catalyst, we next sought to develop an enantioselective variant. With our previous successes in 1,3-diene hydro-functionalization reactions with Pd(PHOX) catalysts,5b,c,26 we once again turned to ligand L1, containing an electron-deficient phosphine (Table 5), for enyne hydroamination.
Table 5.
Initial Examination of Conditions for Enantioselective Enyne Hydroaminationa
 | ||||||
|---|---|---|---|---|---|---|
 | ||||||
| entry | additive | Et3N (Y/N) | solvent | yield of 2a (%)b | 2a:9c | er of 2ad |
| 1 | NaBF4 | N | CH2Cl2 | <2 | – | – |
| 2e | NaBF4 | N | CH2Cl2 | 56 | 4:1 | 53.5:46.5 |
| 3 | NaBF4 | Y | CH2Cl2 | <2 | – | – |
| 4e | NaBF4 | Y | CH2Cl2 | 71 | 19:1 | 63:37 |
| 5e | NaBArF4 | N | Et2O | 73 | 13:1 | 69.5:30.5 |
| 6 | NaBArF4 | Y | Et2O | 48–73f | >20:1 | <52:48–74:26f |
| 7 | NaBArF4 | Y | CH2Cl2 | 65–70f | >20:1 | 54:46–82:18f |
See Table 1.
Isolated yield of 2a (single data point unless otherwise noted).
Determined by 400 MHz 1H NMR analysis of the unpurified mixture.
Determined by HPLC analysis (single data point unless otherwise noted).
Performed with isolated catalyst; see the Supporting Information.
Range for three experiments.
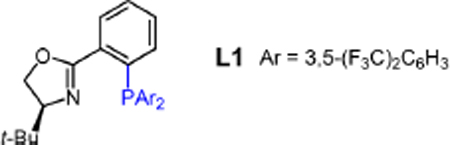
However, we quickly discovered numerous differences between the Pd(DPEphos)- and Pd(PHOX)-catalyzed processes. First, in situ generation of the catalyst derived from L1 and NaBF4 fails to generate allene 2a from enyne 1a and THIQ (entries 1 and 3). Contrastingly, 5 mol % isolated [(η3-C3H5)Pd(L1)]BF4 delivers a 4:1 mixture of allene 2a and diamine 9 in 56% yield (entry 2). The observation of diamine 9, which likely arises from hydroamination of allene 2a,11 is another departure from reactions promoted by DPEphos, which result in complete selectivity for the allene. The second amine addition can be largely suppressed by the inclusion of 2.0 equiv of Et3N (entry 4, 19:1 2a:9), but enantioselectivity remains poor. With the catalyst bearing L1 and a BArF4 counterion, the allene could also be selectively obtained even without Et3N (entry 5, 13:1 2a:9). The collective data in Table 4 (entries 2 and 4–5) suggest that the identity of the ammonium salt that serves as the acid source during the reaction is critical to suppressing overamination. Triethylammonium with either BF4 or BArF4 as the counterion selectively leads to proton transfer when Pd is bound to the alkyne of 1a rather than the allene of product 2a. Comparatively, the ammonium salt of THIQ shows significantly different selectivity profiles depending on its counterion. Thus, the inclusion of Et3N in the reaction mixture seemed likely to be beneficial for product selectivity in the eventual expansion of the reaction to other amine nucleophiles beyond THIQ.
Switching to NaBArF4 as the additive also allows for the in situ preparation of an active catalyst (entries 6–7) perhaps due to its faster formation of NaCl. With the NaBArF4 additive and Et3N, only the desired allene was observed; however, the product yield and especially the enantioselectivity of the transformation proved highly variable under several conditions. In a number of identical experiments, the enantiopurity of 2a ranged from 82:18 er to nearly racemic.23
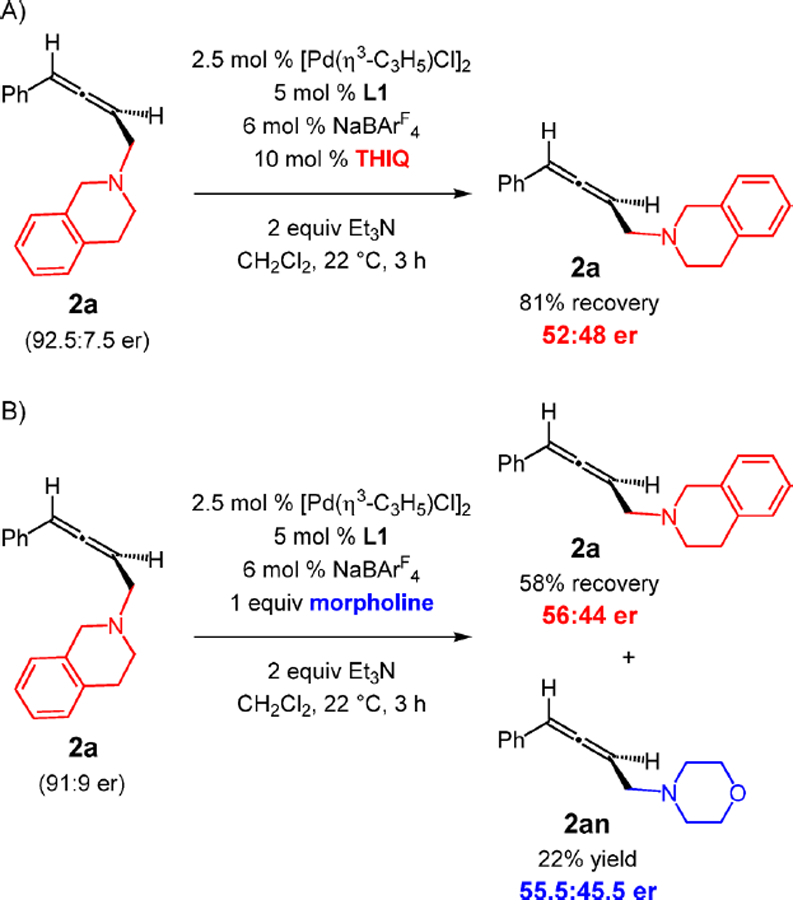
We thought that the variability in enantioselectivity might be due to reaction reversibility. Erosion of enantiopurity over time has been previously observed in Pd-catalyzed 1,3-diene hydroamination by such a process,27 including Pd(PHOX) catalysts.5c Indeed, subjection of highly enantioenriched 2a (92.5:7.5 er) to the Pd(L1) catalyst with 10 mol % THIQ leads to rapid racemization of the allene (Scheme 4A). Futhermore, the addition of 1 equiv of morpholine to enantioenriched 2a (91:9 er) and the Pd(L1) catalyst results in 58% recovery of 2a in only 56:44 er and 22% yield of morpholine adduct 2an in equally low enantiopurity (Scheme 4B). Although both 2a and 2an were nearly racemic after 3 h, determination of the equilibrium ratio of the two allenes by employing the Pd(PHOX) catalyst was thwarted by formation of diamine products from 2a after extended reaction times. However, the same process with achiral Pd(DPEphos) affords a 1.3:1 2a:2an mixture within 3 h.23
Scheme 4.
Reaction Reversibility and Transamination Studies
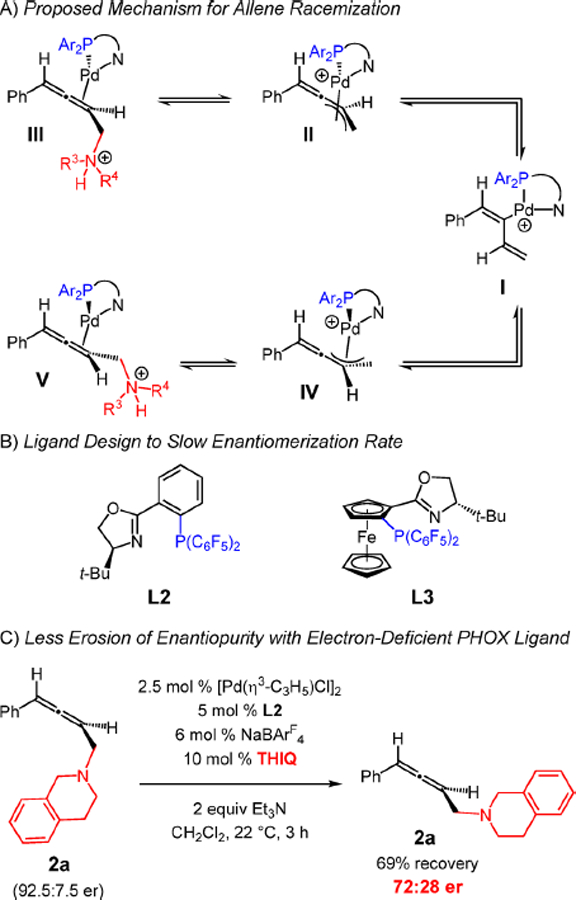
Although we cannot rule out other mechanisms28 at this time, the racemization pathway likely proceeds by ionization of the C–N bond in Pd(0)–allene complex III (Scheme 5A) to regenerate π-allyl–Pd II. Isomerization to the diastereomeric complex IV via alkenyl–Pd I then leads to V after C–N bond formation. Dissociation of the Pd(PHOX) catalyst from III and V leads to allene enantiomers.
Scheme 5.
Design of a Chiral PHOX Ligand To Minimize Erosion of Allene Enantiopurity
Since the ionization of the ammonium group in III or V is at the heart of the racemization process, we rationalized that redesigning the catalyst to slow this event by minimizing the trans effect of the phosphine ligand within complex III/V could lead to more reproducible results and potentially higher enantioselectivity. Reasoning that an even more electron-deficient phosphine might accomplish this goal, perhaps also disfavoring allene coordination to the catalyst compared to an enyne, led us to prepare ligands L2 and L3 (Scheme 5B) containing a bis(perfluorophenyl)phosphino group. We were heartened to find that, consistent with our hypothesis, the enantiomerization rate was significantly retarded with L2 (Scheme 5C): the combination of 2a (91:9 er), Pd(L2) catalyst, and 10 mol % THIQ allows for allene 2a to be recovered in 72:28 er after 3 h (cf., Scheme 4A).
Importantly, addition of THIQ to 1a with the Pd(L2) catalyst allows for a high degree of reproducibility (Table 6, entry 1) with 2a formed in ca. 70% yield and 82:18 er at room temperature in CH2Cl2 after 3 h. Ferrocenyl-PHOX L3 gave slightly higher enantioselectivity (entry 2), and so we chose to optimize further with this ligand.23 Impressively, even in CH2Cl2 as a polar solvent, which we have observed in diene hydroamination to increase the enantiomerization rate compared to reactions in Et2O,5c enantioselectivity only decreases to 82.5:17.5 er after 19 h at room temperature (entry 3). After the reaction was cooled to 4 °C, allene 2a was obtained in 63% yield and 92.5:7.5 er (entry 4).
Table 6.
Improved Enantioselectivity with Electron-Deficient PHOX Ligandsa
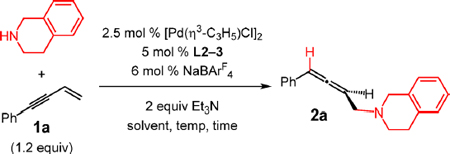 | ||||||
|---|---|---|---|---|---|---|
| entry | ligand | solvent | temp (°C) | time (h) | yield of 2a (%)b | er of 2ac |
| 1 | L2 | CH2Cl2 | 22 | 3 | 67–73d | 82:18–82.5:17.5d |
| 2 | L3 | CH2Cl2 | 22 | 3 | 66 | 86.5:13.5 |
| 3 | L3 | CH2Cl2 | 22 | 19 | 78 | 82.5:17.5 |
| 4 | L3 | CH2Cl2 | 4 | 19 | 63 | 92.5:7.5 |
See Table 1.
Isolated yield of 2a (average of 2–3 experiments unless otherwise noted).
Determined by HPLC analysis (average of 2–3 experiments unless otherwise noted).
Range for three experiments.
The addition of THIQ to several aryl-substituted enynes 1 proceeds with moderate reaction efficiency but good levels of enantioselectivity in the presence of 5 mol % Pd catalyst formed from L3 (Table 7, Condition A: CH2Cl2, 4 °C, 20 h). Both electron-rich and electron-poor enynes react with roughly equal efficiency: anisole 2b is isolated in 64% yield (91:9 er), and benzoate 2f, in 57% yield (94:6 er). An aryl bromide is tolerated in the coupling, with 2d formed in 52% yield and 94.5:5.5 er. An ortho-tolyl group leads to lower enantioselectivity (86:14 er), but 2j is still generated in 57% yield. Although a pyridyl substituent had little impact on hydroamination with the Pd(DPEphos) catalyst, the heterocycle significantly impedes the L3-derived catalyst: allene 2m is obtained in 91:9 er but only 32% yield.
Table 7.
Scope of Enantioselective Enyne Hydroaminationa
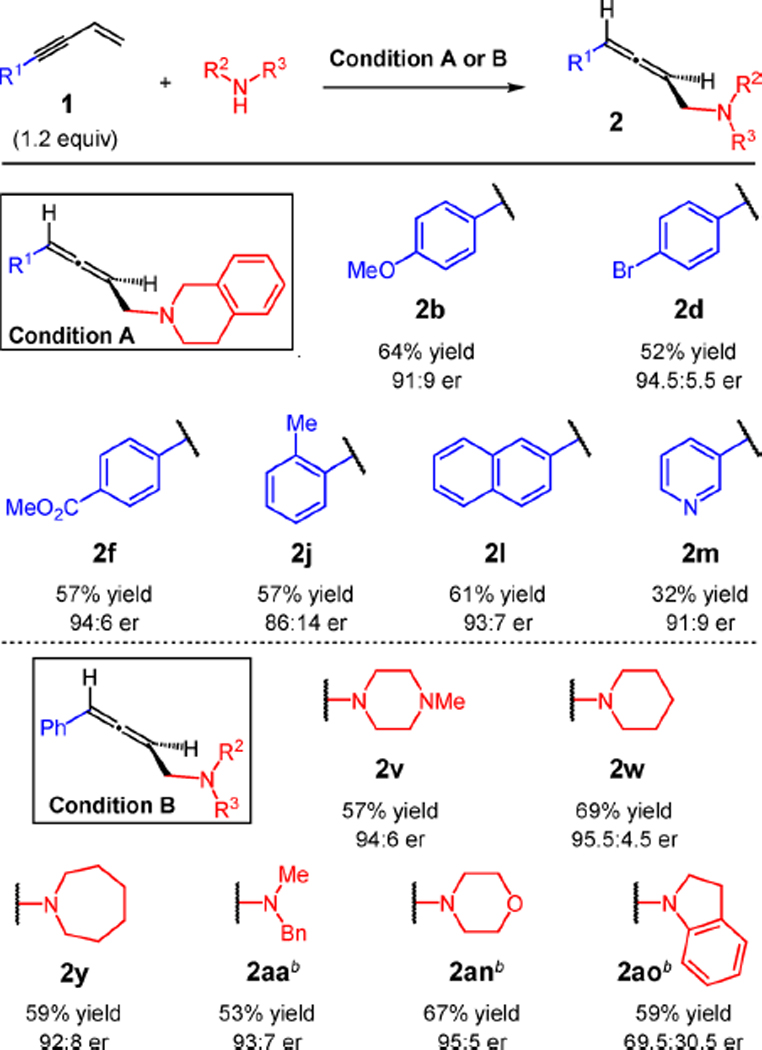 |
Reactions run under N2 with 0.2 mmol of amine (0.8 M). Condition A: 2.5 mol % [Pd(η3-C3H5)Cl]2, 5 mol % L3, 6 mol % NaBArF4, 2 equiv of Et3N, CH2Cl2, 4 °C, 20 h. Condition B: 1 mol % [Pd(η3-C3H5)Cl]2, 2 mol % L3, 2.5 mol % NaBArF4, 2 equiv of Et3N, Et2O, 4 °C, 3 h.
5 h reaction.
Whereas addition of THIQ to enynes required a 5 mol % catalyst loading to obtain allenes in good yields,23 other amines couple efficiently with enyne 1a with only 2 mol % of the catalyst formed from L3 (Table 7, Condition B: Et2O, 4 °C, 3 h). These optimal conditions enable allene products to be isolated with greater enantiopurity. As a result, cyclic amines (2v, 2w, 2y, and 2an) are isolated in 92:8 to 95.5:4.5 er (57–69% yield). Acyclic alkyl-substituted amines are also tolerated: 2aa is obtained in 53% yield and 93:7 er. Despite displaying similar reaction efficiency as aliphatic amines, indoline addition to 1a leads to allene 2ao with only 69.5:30.5 er (59% yield). The lower enantioselectivity is not due to more rapid product enantiomerization but rather to lower kinetic selectivity; in fact, the enantiopurity of 2ao remains constant throughout the course of the reaction with the L3-derived catalyst.23
The addition of THIQ to alkyl-substituted enynes as promoted by the Pd(L3) catalyst, however, occurs with low enantioselectivity,23 perhaps suggesting that ionization of the C–N bond of the product is competitive with ligand exchange of the allene for another alkyl-substituted enyne substrate at the Pd(0) center (displacement of the allene within III, Scheme 5A, for enyne). The more substituted enynes 5 and 7 (Table 4 and Scheme 3, respectively) are relatively unreactive with the Pd(PHOX) catalyst; benzophenone imine also does not add to enyne 1a.23
2.3. Reaction Mechanism Investigations.
Our proposed mechanism for the enyne 1,4-hydroamination proceeds through η3-butadienyl–Pd II (Scheme 6), itself formed from η1-butadienyl–Pd I, the product of alkyne insertion to a palladium hydride species VI.29 This pathway mirrors that suggested by Yamamoto11 in a related process and is also based on prior work in Pd–bis(phosphine)-catalyzed hydroamination of dienes and styrene,22 which have been shown to proceed through outer-sphere addition of the amine to a π-allyl–Pd or π-benzyl–Pd complex, respectively. Alkene insertion to the palladium hydride in complex VII might also occur to generate propargylic Pd species VIII. Although VIII cannot collapse to a stable π-allyl–Pd complex, we wondered if alkene insertion were a competitive process or if alkyne insertion (VI to I) were the exclusive reaction pathway. We also questioned whether DPEphos- and L3-derived Pd catalysts might show different kinetic site-selectivity profiles for migratory insertion.
Scheme 6.
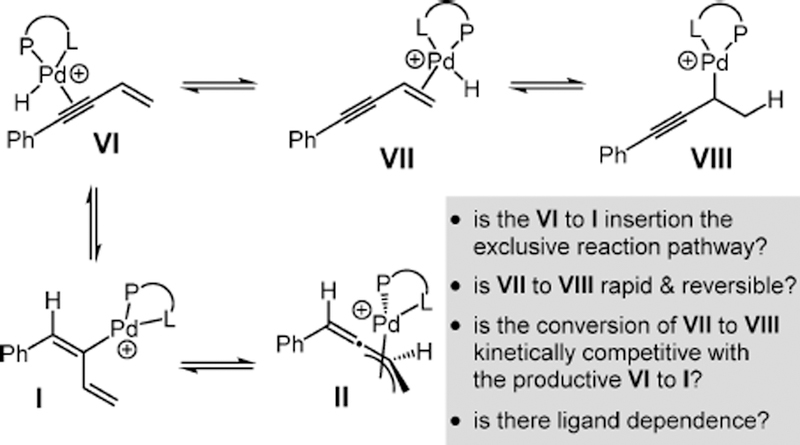
Possible Reaction Pathways in the Course of Pd-Catalyzed Enyne Hydroamination
To investigate these site-selectivity questions, we employed N-deuterated THIQ (90% labeled) in a reaction with enyne 1a (Scheme 7). With either bis(phosphine) or PHOX ligand, the deuterium label is confined to C1 in d-2a (80% labeled) with <5% incorporation at the allylic C4 position. No deuterium label was detected in the recovered enyne 1a. Therefore, we can conclude that insertion of the alkyne to the palladium hydride is significantly faster than olefin insertion.
Scheme 7.
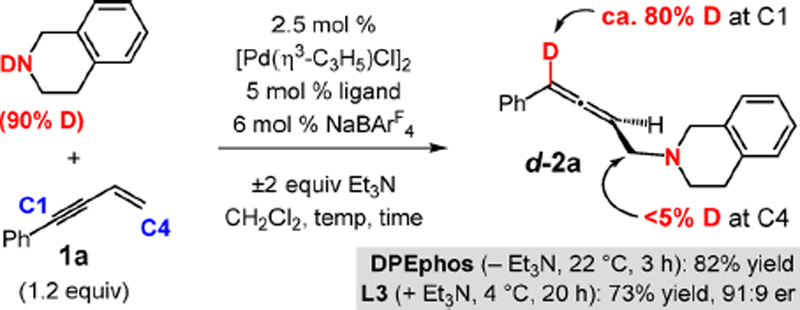
Deuterium Labeling Reveals Kinetic Selectivity for Alkyne Insertion to Pd–H
Having determined that Pd–H migratory insertion occurs kinetically at the alkyne to furnish η1-butadienyl–Pd I, we next examined the origin of high enantioselectivity in enyne hydroamination. Two possibilities seemed likely, the first being selective collapse of I to η3-butadienyl–Pd II followed by faster attack of the amine (Scheme 8). The second option is that II might be in rapid equilibrium with diastereomeric complex IV, with amine attack upon II being faster than addition to IV (Curtin–Hammett kinetics). Recognizing that the Trost laboratory20b has observed a Curtin–Hammett situation in allylic aminations involving allene substrates that share common intermediates II and IV, we investigated the allylic amination of racemic allylic acetate 10. Addition of morpholine to rac-10 with the Pd(L3) catalyst under the conditions for enyne hydroamination affords aminomethyl-substituted allene 2an in 44% yield and 91:9 er (cf. 95:5 er from enyne hydroamination) with the (R)-enantiomer still as the major isomer (Scheme 8). Allene 10 is recovered as the racemate, illustrating enantioconvergence in the reaction (i.e., not kinetic resolution). The data indicate that allylic amination with Pd(L3) is under Curtin–Hammett control and strongly suggests that enyne hydroaminations are as well.
Scheme 8.
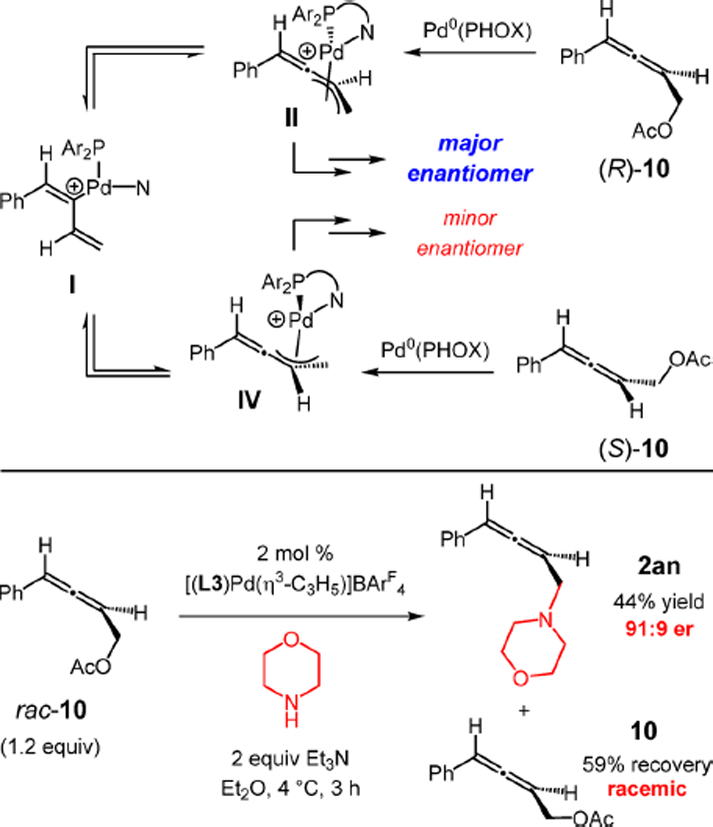
Allylic Substitution Suggests Curtin–Hammett Kinetics Operative in Enyne Hydroamination
The preferential attack of the amine upon η3-butadienyl II compared to IV can be rationalized in terms of the transition states leading to allene–Pd complexes III and V, respectively (Scheme 9). In both π-allyl–Pd complexes II and IV, the phosphine lies trans to the allyl ligand’s methylene carbon, which undergoes attack by the amine (trans effect). The C–N bond formation causes rehybridization at this carbon, which leads to steric clash with the tert-butyl group of the oxazoline en route to V. Comparatively there is less interaction between these substituents in the amine addition to II that leads to III.
Scheme 9.
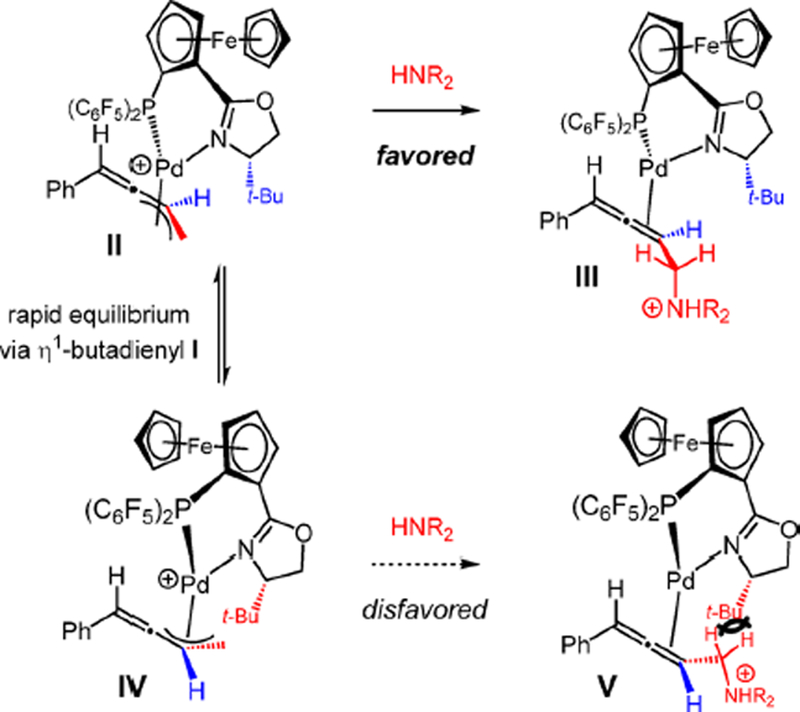
Proposed Stereochemical Model for Enyne Hydroamination
3. CONCLUSION
In this study, we have demonstrated the selective 1,4-addition of aliphatic amines, aryl-substituted amines, and benzophenone imine to deliver chiral aminomethyl-substituted allenes. Several tertiary, secondary, and primary amines can be obtained in racemic form with a Pd(DPEphos) catalyst. The enyne scope is equally broad with several 1-substituted and 1,3-disubstituted enynes amenable to the reaction.
Furthermore, we have demonstrated the first examples of catalytic enantioselective intermolecular addition of nucleophiles to nonpolarized 1,3-enynes. Transformations take place in good yield and enantioselectivity with a Pd(PHOX) catalyst that bears an electron-poor bis(perfluorophenyl)phosphino group. With the electron-deficient Pd catalyst, reaction reversibility is slowed significantly, thereby preserving the stereochemistry set in the initial C–N bond-forming step. Still, these studies highlight future areas for improvement, including hydroamination with alkyl-substituted enynes, disubstituted substrates, and reactions of benzophenone imine. Likely success in these objectives will require further catalyst development.
Initial mechanistic investigations indicate that, unlike enyne reductive couplings with electrophiles that proceed via Rh–H, Ir–H, or Cu–H insertion at the olefin, Pd–H insertion in enyne hydroamination takes place exclusively at the alkyne with both Pd(DPEphos) and Pd(PHOX) catalysts. Additionally, comparison with known allylic substitutions indicates that enantioselectivity in the Pd(PHOX)-catalyzed enyne hydroaminations likely takes place under Curtin–Hammett kinetics involving rapid equilibration of diastereomeric π-allyl–Pd complexes.
1,4-Addition of nucleophiles to conjugated enynes provides a new avenue for the synthesis of multisubstituted chiral allenes. Studies directed toward the catalytic enantioselective addition of other nucleophiles to 1,3-enynes are underway in this laboratory.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Science Foundation (CHE-1800012) and Duke University for supporting this research. N.J.A. is grateful for support from NIGMS (T32GM007105) and for a Burroughs-Welcome fellowship from the Duke Chemistry Department.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b02637.
Experimental procedures, analytical data for new compounds (PDF)
NMR spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) For reviews, see: (a)Reznichenko AL; Nawara-Hultzsch AJ; Hultzsch KC Asymmetric Hydroamination. Top. Curr. Chem 2013, 343, 191–260. [DOI] [PubMed] [Google Scholar]; (b) Huang L; Arndt M; Gooßen K; Heydt H; Gooßen LJ Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev 2015, 115, 2596–2697. [DOI] [PubMed] [Google Scholar]
- (2).Chen Q-A; Chen Z; Dong VM Rhodium-Catalyzed Enantioselective Hydroamination of Alkynes with Indolines. J. Am. Chem. Soc 2015, 137, 8392–8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) For a recent review, see: Koschker P; Breit B Branching Out: Rhodium-Catalyzed Allylation with Alkynes and Allenes. Acc. Chem. Res 2016, 49, 1524–1536. [DOI] [PubMed] [Google Scholar]; For examples, see: (b) Butler KL; Tragni M; Widenhoefer RA Gold(I)-Catalyzed Stereo-convergent, Intermolecular Enantioselective Hydroamination of Allenes. Angew. Chem., Int. Ed 2012, 51, 5175–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cooke ML; Xu K; Breit B Enantioselective Rhodium-Catalyzed Synthesis of Branched Allylic Amines by Intermolecular Hydroamination of Terminal Allenes. Angew. Chem., Int. Ed 2012, 51, 10876–10879. [DOI] [PubMed] [Google Scholar]; (d) Xu K; Gilles T; Breit B Asymmetric Synthesis of N-Allylic Indoles via Regio- and Enantioselective Allylation of Aryl Hydrazines. Nat. Commun 2015, 6, 7616–7622. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xu K; Thieme N; Breit B Atom-Economic, Regiodivergent, and Stereoselective Coupling of Imidazole Derivatives with Terminal Allenes. Angew. Chem., Int. Ed 2014, 53, 2162–2165. [DOI] [PubMed] [Google Scholar]; (f) Li C; Kähny M; Breit B Rhodium-Catalyzed Chemo-, Regio-, and Enantioselective Addition of 2-Pyridones to Terminal Allenes. Angew. Chem., Int. Ed 2014, 53, 13780–13784. [DOI] [PubMed] [Google Scholar]; (g) Haydl AM; Xu K; Breit B Regio- and Enantioselective Synthesis of N-Substituted Pyrazoles by Rhodium-Catalyzed Asymmetric Addition to Allenes. Angew. Chem., Int. Ed 2015, 54, 7149–7153. [DOI] [PubMed] [Google Scholar]; (h) Xu K; Raimondi W; Bury T; Breit B Enantioselective Formation of Tertiary and Quaternary Allylic C–N Bonds via Allylation of Tetrazoles. Chem. Commun 2015, 51, 10861–10863. [DOI] [PubMed] [Google Scholar]; (i) Xu K; Wang Y-H; Khakyzadeh V; Breit B Asymmetric Synthesis of Allylic Amines via Hydroamination of Allenes with Benzophenone Imine. Chem. Sci 2016, 7, 3313–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Löber O; Kawatsura M; Hartwig JF Palladium-Catalyzed Hydroamination of 1,3-Dienes: A Colorimetric Assay and Enantio-selective Additions. J. Am. Chem. Soc 2001, 123, 4366–4367. [DOI] [PubMed] [Google Scholar]
- (5).(a) Yang X-H; Dong VM Rhodium-Catalyzed Hydrofunctionalization: Enantioselective Coupling of Indolines and 1,3-Dienes. J. Am. Chem. Soc 2017, 139, 1774–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Adamson NJ; Hull E; Malcolmson SJ Enantioselective Intermolecular Addition of Aliphatic Amines to Acyclic Dienes with a Pd–PHOX Catalyst. J. Am. Chem. Soc 2017, 139, 7180–7183. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Park S; Malcolmson SJ Development and Mechanistic Investigations of Enantioselective Pd-Catalyzed Intermolecular Hydroaminations of Internal Dienes. ACS Catal 2018, 8, 8468–8476. [Google Scholar]
- (6).(a) Kawatsura M; Hartwig JF Palladium-Catalyzed Intermolecular Hydroamination of Vinylarenes Using Arylamines. J. Am. Chem. Soc 2000, 122, 9546–9547. [Google Scholar]; (b) Utsunomiya M; Hartwig JF Intermolecular, Markovnikov Hydroamination of Vinylarenes with Alkylamines. J. Am. Chem. Soc 2003, 125, 14286–14287. [DOI] [PubMed] [Google Scholar]
- (7).Teng H-L; Luo Y; Wang B; Zhang L; Nishiura M; Hou Z Synthesis of Chiral Aminocyclopropanes by Rare-Earth-Metal-Catalyzed Cyclopropene Hydroamination. Angew. Chem., Int. Ed 2016, 55, 15406–15410. [DOI] [PubMed] [Google Scholar]
- (8).Reznichenko AL; Nguyen HN; Hultzsch KC Asymmetric Intermolecular Hydroamination of Unactivated Alkenes with Simple Amines. Angew. Chem., Int. Ed 2010, 49, 8984. [DOI] [PubMed] [Google Scholar]
- (9).(a) For umpolung enantioselective hydroamination reactions, see: Miki Y; Hirano K; Satoh T; Miura M Copper-Catalyzed Intermolecular Regioselective Hydroamination of Styrenes with Polymethylhydrosiloxane and Hydroxylamines. Angew. Chem., Int. Ed 2013, 52, 10830–10834. [DOI] [PubMed] [Google Scholar]; (b) Zhu S; Niljianskul N; Buchwald SL Enantio- and Regioselective CuH-Catalyzed Hydroamination of Alkenes. J. Am. Chem. Soc 2013, 135, 15746–15749. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Niljianskul N; Zhu S; Buchwald SL Enantioselective Synthesis of α-Aminosilanes by Copper-Catalyzed Hydroamination of Vinylsilanes. Angew. Chem., Int. Ed 2015, 54, 1638–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yang Y; Shi S-L; Niu D; Liu P; Buchwald SL Catalytic Asymmetric Hydroamination of Unactivated Internal Olefins to Aliphatic Amines. Science 2015, 349, 62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nishikawa D; Hirano K; Miura M Asymmetric Synthesis of α-Aminoboronic Acid Derivatives by Copper-Catalyzed Enantioselective Hydroamination. J. Am. Chem. Soc 2015, 137, 15620–15623. [DOI] [PubMed] [Google Scholar]; (f) Xi Y; Butcher TW; Zhang J; Hartwig JF Regioselective, Asymmetric Formal Hydroamination of Unactivated Internal Alkenes. Angew. Chem., Int. Ed 2016, 55, 776–780. [DOI] [PubMed] [Google Scholar]; (g) Ichikawa S; Zhu S; Buchwald SL A Modified System for the Synthesis of Enantioenriched N-Arylamines through Copper-Catalyzed Hydroamination. Angew. Chem., Int. Ed 2018, 57, 8714–8718. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhou Y; Engl OD; Bandar JS; Chant ED; Buchwald SL CuH-Catalyzed Asymmetric Hydroamidation of Vinylarenes. Angew. Chem., Int. Ed 2018, 57, 6672–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Guo S; Yang JC; Buchwald SL A Practical Electrophilic Nitrogen Source for the Synthesis of Chiral Primary Amines by Copper-Catalyzed Hydroamination. J. Am. Chem. Soc 2018, 140, 15976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Brinkmann C; Barrett AGM; Hill MS; Procopiou PA Heavier Alkaline Earth Catalysts for the Intermolecular Hydroamination of Vinylarenes, Dienes, and Alkynes. J. Am. Chem. Soc 2012, 134, 2193–2207. [DOI] [PubMed] [Google Scholar]
- (11).(a) Radhakrishnan U; Al-Masum M; Yamamoto Y Palladium Catalyzedz Hydroamination of Conjugated Enynes. Tetrahedron Lett 1998, 39, 1037–1040. [Google Scholar]; For a related process that affords achiral allenes by C–C bond formation, see: (b) Salter MM; Gevorgyan V; Saito S; Yamamoto Y Synthesis of Allenes via Palladium Catalysed Addition of Certain Activated Methynes to Conjugated Enynes. Chem. Commun 1996, 32, 17–18. [Google Scholar]
- (12).(a) For conjugate additions to polarized enynes, see: Hayashi T; Tokunaga N; Inoue K Rhodium-Catalyzed Asymmetric 1,6-Addition of Aryltitanates to Enynones Giving Axially Chiral Allenes. Org. Lett 2004, 6, 305–307. [DOI] [PubMed] [Google Scholar]; (b) Nishimura T; Makino H; Nagaosa M; Hayashi T Rhodium-Catalyzed Enantioselective 1,6-Addition of Arylboronic Acids to Enynamides: Asymmetric Synthesis of Axially Chiral Allenylsilanes. J. Am. Chem. Soc 2010, 132, 12865–12867. [DOI] [PubMed] [Google Scholar]; (c) Qian H; Yu X; Zhang J; Sun J Organocatalytic Enantioselective Synthesis of 2,3-Allenoates by Intermolecular Addition of Nitroalkanes to Activated Enynes. J. Am. Chem. Soc 2013, 135, 18020–18023. [DOI] [PubMed] [Google Scholar]; (d) Wang M; Liu Z-L; Zhang X; Tian P-P; Xu Y-H; Loh T-P Synthesis of Highly Substituted Racemic and Enantioenriched Allenylsilanes via Copper-Catalyzed Hydrosilylation of (Z)-Alken-4-ynoates with Silylboronate. J. Am. Chem. Soc 2015, 137, 14830–14833. [DOI] [PubMed] [Google Scholar]; (e) Yao Q; Liao Y; Lin L; Lin X; Ji J; Liu X; Feng X Efficient Synthesis of Chiral Trisubstituted 1,2-Allenyl Ketones by Catalytic Asymmetric Conjugate Addition of Malonic Esters to Enynes. Angew. Chem., Int. Ed 2016, 55, 1859–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For a nonenantioselective intramolecular enyne hydroamination, see: Zhang W; Werness JB; Tang W Base-Catalyzed Intramolecular Hydroamination of Conjugated Enynes. Org. Lett 2008, 10, 2023–2026. [DOI] [PubMed] [Google Scholar]
- (14).(a) For catalytic enantioselective Cu–boryl addition to enynes followed by carbonyl coupling, see: Meng F; Haeffner F; Hoveyda AH Diastereo- and Enantioselective Reactions of Bis(pinacolato)diboron, 1,3-Enynes, and Aldehydes Catalyzed by an Easily Accessible Bisphosphine–Cu Complex. J. Am. Chem. Soc 2014, 136, 11304–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gan X-C; Zhang Q; Jia X-S; Yin L Asymmetric Construction of Fluoroalkyl Tertiary Alcohols through a Three-Component Reaction of (Bpin)2, 1,3-Enynes and Fluoroalkyl Ketones Catalyzed by a Copper(I) Complex. Org. Lett 2018, 20, 1070–1073. [DOI] [PubMed] [Google Scholar]; For other nucleophile additions, see: (c) Todo H; Terao J; Watanabe H; Kuniyasu H; Kambe N Cu-Catalyzed Regioselective Carbomagnesiation of Dienes and Enynes with sec- and tert-Alkyl Grignard Reagents. Chem. Commun 2008, 44, 1332–1334. [DOI] [PubMed] [Google Scholar]; (d) Tomida Y; Nagaki A; Yoshida J.-i. Asymmetric Carbolithiation of Conjugated Enynes: A Flow Microreactor Enables the Use of Configurationally Unstable Intermediates before They Epimerize. J. Am. Chem. Soc 2011, 133, 3744–3747. [DOI] [PubMed] [Google Scholar]; (e) Mori Y; Kawabata T; Onodera G; Kimura M Remarkably Selective Formation of Allenyl and Dienyl Alcohols via Ni-Catalyzed Coupling Reaction of Conjugated Enyne, Aldehyde, and Organozinc Reagents. Synthesis 2016, 48, 2385–2395. [Google Scholar]; (f) Zhu X; Deng W; Chiou M-F; Ye C; Jian W; Zeng Y; Jiao Y; Ge L; Li Y; Zhang X; Bao H Copper-Catalyzed Radical 1,4-Difunctionalization of 1,3-Enynes with Alkyl Diacyl Peroxides and N-Fluorobenzenesulfonamide. J. Am. Chem. Soc 2019, 141, 548–559. [DOI] [PubMed] [Google Scholar]
- (15).For a recent review, see: Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) For examples with Ru, see: Patman RL; Williams VM; Bower JF; Krische MJ Carbonyl Propargylation from the Alcohol or Aldehyde Oxidation Level Employing 1,3-Enynes as Surrogates to Preformed Allenylmetal Reagents: A Ruthenium Catalyzed C-C Bond Forming Transfer Hydrogenation. Angew. Chem., Int. Ed 2008, 47, 5220–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Geary LM; Leung JC; Krische MJ Ruthenium Catalyzed Reductive Coupling of 1,3-Enynes and Aldehydes via Transfer Hydrogenation: anti-Diastereoselective Carbonyl Propargylation. Chem. - Eur. J 2012, 18, 16823–16827. [DOI] [PubMed] [Google Scholar]; (c) Nguyen KD; Herkommer D; Krische MJ Ruthenium-BINAP Catalyzed Alcohol C-H tert-Prenylation via 1,3-Enyne Transfer Hydrogenation: Beyond Stoichiometric Carbanions in Enantioselective Carbonyl Propargylation. J. Am. Chem. Soc 2016, 138, 5238–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an example with Ir, see: (d) Geary LM; Woo SK; Leung JC; Krische MJ Diastereo- and Enantioselective Iridium Catalyzed Carbonyl Propargylation from the Alcohol or Aldehyde Oxidation Level: 1,3-Enynes as Allenylmetal Equivalents. Angew. Chem., Int. Ed 2012, 51, 2972–2976. [DOI] [PubMed] [Google Scholar]; For an example with Cu, see: (e) Yang Y; Perry IB; Lu G; Liu P; Buchwald SL Copper-Catalyzed Asymmetric Addition of Olefin-Derived Nucleophiles to Ketones. Science 2016, 353, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Huang Y; del Pozo J; Torker S; Hoveyda AH Enantioselective Synthesis of Trisubstituted Allenyl-B(pin) Compounds by Phosphine-Cu-Catalyzed 1,3-Enyne Hydroboration. In-sights Regarding Stereochemical Integrity of Cu-Allenyl Intermediates. J. Am. Chem. Soc 2018, 140, 2643–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sang HL; Yu S; Ge S Copper-Catalyzed Asymmetric Hydroboration of 1,3-Enynes with Pinacolborane to Access Chiral Allenylboronates. Org. Chem. Front 2018, 5, 1284–1287. [Google Scholar]; (c) Gao D-W; Xiao Y; Liu M; Liu Z; Karunananda MK; Chen JS; Engle KM Catalytic, Enantioselective Synthesis of Allenyl Boronates. ACS Catal 2018, 8, 3650–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) For a related enantioselective Pd-catalyzed hydrosilylation, see: Han JW; Tokunaga N; Hayashi T Palladium-Catalyzed Asymmetric Hydrosilylation of 4-Substituted 1-Buten-3-ynes. Catalytic Asymmetric Synthesis of Axially Chiral Allenylsilanes. J. Am. Chem. Soc 2001, 123, 12915–12916. [DOI] [PubMed] [Google Scholar]; For a related non-enantioselective Pd-catalyzed hydroboration, see: (b) Matsumoto Y; Naito M; Hayashi T Palladium(o)-Catalyzed Hydroboration of 1-Buten-3-ynes: Preparation of Allenylboranes. Organometallics 1992, 11, 2732–2734. [Google Scholar]
- (19).(a) For reviews on catalytic enantioselective synthesis of allenes, see: Ogasawara M Catalytic Enantioselective Synthesis of Axially Chiral Allenes. Tetrahedron: Asymmetry 2009, 20, 259–271. [Google Scholar]; (b) Chu W-D; Zhang Y; Wang J Recent Advances in Catalytic Asymmetric Synthesis of Allenes. Catal. Sci. Technol 2017, 7, 4570–4579. [Google Scholar]
- (20).(a) Ogasawara M; Ueyama K; Nagano T; Mizuhata Y; Hayashi T Palladium-Catalyzed Asymmetric Synthesis of Axially Chiral (Allenylmethyl)silanes and Chirality Transfer to Stereogenic Carbon Centers in SE′ Reactions. Org. Lett 2003, 5, 217–219. [DOI] [PubMed] [Google Scholar]; (b) Trost BM; Fandrick DR; Dinh DC Dynamic Kinetic Asymmetric Allylic Alkylations of Allenes. J. Am. Chem. Soc 2005, 127, 14186–14187. [DOI] [PubMed] [Google Scholar]; (c) Imada Y; Nishida M; Kutsuwa K; Murahashi S-I; Naota T Palladium-Catalyzed Asymmetric Amination and Imidation of 2,3-Allenyl Phosphates. Org. Lett 2005, 7, 5837–5839. [DOI] [PubMed] [Google Scholar]; (d) Li Q; Fu C; Ma S Catalytic Asymmetric Allenylation of Malonates with the Generation of Central Chirality. Angew. Chem., Int. Ed 2012, 51, 11783–11786. [DOI] [PubMed] [Google Scholar]; (e) Li Q; Fu C; Ma S Palladium-Catalyzed Asymmetric Amination of Allenyl Phosphates: Enantioselective Synthesis of Allenes with an Additional Unsaturated Unit. Angew. Chem., Int. Ed 2014, 53, 6511–6514. [DOI] [PubMed] [Google Scholar]
- (21).Birkholz M-N; Freixa Z; van Leeuwen PWNM Bite Angle Effects of Diphosphines in C–C and C–X Bond Forming Cross Coupling Reactions. Chem. Soc. Rev 2009, 38, 1099–1118. [DOI] [PubMed] [Google Scholar]
- (22).For similar observations in other Pd-catalyzed hydroamination reactions, see: Johns AM; Utsunomiya M; Incarvito CD; Hartwig JF A Highly Active Palladium Catalyst for Intermolecular Hydroamination. Factors that Control Reactivity and Additions of Functionalized Anilines to Dienes and Vinylarenes. J. Am. Chem. Soc 2006, 128, 1828–1839. [DOI] [PubMed] [Google Scholar]
- (23). For additional details, see the Supporting Information.
- (24). Transformations involving THIQ addition to alkyl-substituted enynes lead to small inseparable quantities of a byproduct, tentatively assigned as benzylic oxidation of the N-heterocycle of the desired product, affording a lactam. This process is unique to reactions of THIQ with alkyl-substituted enynes and was discovered to be mitigated by in situ generation of the catalyst.
- (25).Shimano M; Meyers AI Asymmetric Diastereoselective Conjugate Additions of Lithium Amides to Chiral Naphthyloxazolines Leading to Novel β-Amino Acids. J. Org. Chem 1995, 60, 7445–7455. [Google Scholar]
- (26).Adamson NJ; Wilbur KCE; Malcolmson SJ Enantioselective Intermolecular Pd-Catalyzed Hydroalkylation of Acyclic 1,3-Dienes with Activated Pronucleophiles. J. Am. Chem. Soc 2018, 140, 2761–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pawlas J; Nakao Y; Kawatsura M; Hartwig JF A General Nickel-Catalyzed Hydroamination of 1,3-Dienes by Alkylamines: Catalyst Selection, Scope, and Mechanism. J. Am. Chem. Soc 2002, 124, 3669–3679. [DOI] [PubMed] [Google Scholar]
- (28).Horvéth A; Bäckvall J-E Mild and Efficient Palladium(II)-Catalyzed Racemization of Allenes. Chem. Commun 2004, 40, 964–965. [DOI] [PubMed] [Google Scholar]
- (29). The Pd–H is initially formed by a sequence involving amine attack upon the π-allyl ligand in the starting Pd complex, which generates a Pd(0) species and an N-allyl ammonium salt, followed by oxidative protonation of the Pd(0) by this ammonium acid source.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.