

Abstract
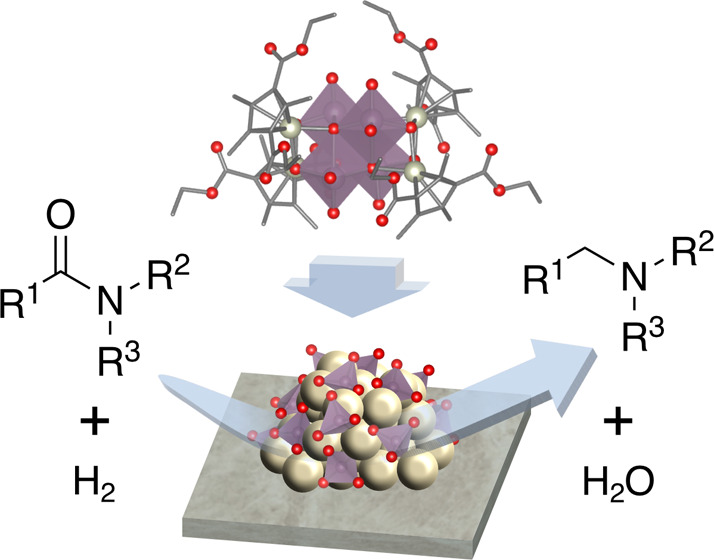
Amide hydrogenation is an important process for producing amines, with the development of efficient heterogeneous catalysts relying on the creation of bimetallic active sites where the two components interact synergistically. In this study, we develop a method for preparing catalysts using ligand-functionalized organometallic polyoxometalates by synthesizing a Rh–Mo organometallic polyoxometalate, [(RhCpE)4Mo4O16] (CpE = C5(CH3)3(COOC2H5)2), with Rh–O–Mo interfacial structures and ethoxycarbonyl-functionalized ligands as a catalyst precursor. The activity of supported Rh–Mo catalysts for amide hydrogenation depend on the precursor used, with [(RhCpE)4Mo4O16] showing the highest activity, followed by [(RhCp*)4Mo4O16] (Cp* = C5(CH3)5), and then RhCl3 combined with (NH4)6[Mo7O24]·4H2O. The catalyst prepared from [(RhCpE)4Mo4O16] effectively hydrogenates tertiary, secondary, and primary amides under mild conditions (0.8 MPa H2, 353–393 K), demonstrating a high activity and selectivity (conversion: 97%, selectivity: 76%) for primary amide hydrogenation under NH3-free conditions. Furthermore, we determine that carbonyl oxygen atoms in CpE ligands contribute to the electrostatic interaction with Al2O3, leading to the high dispersibility of [(RhCpE)4Mo4O16] on the support. We conclude that the high efficiency of [(RhCpE)4Mo4O16] as a catalyst precursor originates from the effective formation of Rh/Mo interfacial active sites, which is assisted by the electrostatic interaction between the CpE ligands and support.
Keywords: hydrogenation, nanoparticles, organometallic polyoxometalates, primary amide, supported catalysts
1. Introduction
Amines are one of the most important classes of organic compounds that are used for agrochemicals, drugs, detergents, lubricants, food additives, and polymers.1 Although amides are considered among the most difficult compounds to hydrogenate compared with other carbonyl compounds, their hydrogenation is considered as the most efficient method for producing amines. Amide hydrogenation is typically performed using stoichiometric amounts of hydride reagents, such as LiAlH4 and NaBH4.2 To achieve greater atom efficiency, these processes should be replaced by catalytic hydrogenation, which uses molecular H2 and produces water as the only byproduct.3
Recently, numerous studies have focused on developing heterogeneous catalysts for amide hydrogenation.4,5 Interestingly, these catalysts are predominantly composed of bimetallic species, as reported in pioneering studies using Rh–Mo6,7 and for recently developed catalysts, such as Rh–V,8 Pt–V,9 Ru–Mo,10−12 Rh–Mo,13 Ir–Mo,14 Ru–W,15,16 Pd–Re,17 and Pt–Re18 catalysts. These synergistic activities are a result of the oxophilicity of partially reduced secondary components, such as V, Mo, W, and Re, which form before or during the reaction. These components contribute to the activation of amide C=O bonds. Therefore, the key to improving the activity is to efficiently form active sites where two components are in close proximity.
To form interfacial active sites composed of two components, we previously focused on the possibility of catalyst preparation by hybrid clustering.19−21 In this method, organometallic polyoxometalates,22 formulated as [(M1L)xM2yOn] (M1: metal ions; M2yOn: metal oxide clusters; L: organic ligands), were used as catalyst precursors. Because the surface oxygen atoms of the metal oxide clusters were coordinated to the metal ions, they could be viewed as building blocks with M1–O–M2 interfaces. Therefore, we predicted that catalysts prepared from organometallic polyoxometalates would comprise a high density of interfacial sites, which would contribute to efficient amide hydrogenation.
In this study, we focused on ligand-functionalizing organometallic polyoxometalates to use them as catalyst precursors for amide hydrogenation. Rh–Mo-based catalysts, which are among the most promising bimetallic combinations for amide hydrogenation, were prepared using Rh–Mo organometallic polyoxometalates as catalyst precursors. We synthesized a Rh–Mo organometallic polyoxometalate, [(RhCpE)4Mo4O16] (CpE = C5(CH3)3(COOC2H5)2), with ethoxycarbonyl groups (−COOC2H5) in the ligands. The catalyst prepared from [(RhCpE)4Mo4O16] was used to hydrogenate tertiary, secondary, and primary amides under mild reaction conditions (0.8 MPa H2, 353–393 K). The effect of the introduction of ethoxycarbonyl groups on the activity was studied by comparing the activity of [(RhCpE)4Mo4O16] with that of [(RhCp*)4Mo4O16] (Cp* = C5(CH3)5).
2. Results and Discussion
2.1. Catalyst Preparation and Characterization
The Rh–Mo organometallic polyoxometalate with functionalized ligands, [(RhCpE)4Mo4O16], was synthesized by reacting [RhCpECl2]223 with [MoO4]2– under MeOH/H2O. The procedure was similar to that reported for the synthesis of [(RhCp*)4Mo4O16], where water was used as the solvent.24 Methanol was required to facilitate the reaction because of the insolubility of [RhCpECl2]2 in water. The product was characterized by single crystal X-ray diffraction, positive-ion electrospray ionization mass spectrometry, and Fourier transform infrared spectroscopy. The single crystal X-ray diffraction analysis revealed that the molecular structure of [(RhCpE)4Mo4O16] was similar to that reported for [(RhCp*)4Mo4O16],24 which contained a triple-cubane framework consisting of Rh4Mo4O16 (Figures 1 and S3 and Table S1). The monovalent cations observed in the positive-ion electrospray ionization mass spectra were assigned to the proton adduct [H(RhCpE)4Mo4O16]+ (Figure S1). The Fourier transform infrared spectrum displayed characteristic absorption patterns assigned to the terminal (η1) Mo=O stretching and bridging (μ2) Mo–O–Mo vibration bands, which were similar to those of [(RhCp*)4Mo4O16] (Figure S2).
Figure 1.

[(RhCpE)4Mo4O16] as catalyst precursor with Rh–O–Mo interfacial structure and functionalized ligands. Hydrogen atoms are omitted for clarity.
[(RhCpE)4Mo4O16] was applied as a precursor to prepare a supported Rh–Mo catalyst, Rh4Mo4(CpE)/Al2O3. After the precursor was adsorbed on γ-Al2O3 by impregnation with methanol, the catalyst was prepared by calcination under air at 573 K, followed by reduction under H2 flow at 573 K. To elucidate the effect of the functionalized ligand on the activity of the catalyst, [(RhCp*)4Mo4O16] was used to prepare a reference catalyst, Rh4Mo4(Cp*)/Al2O3. The coimpregnated Rh–Mo catalyst, Rh–Mo/Al2O3, and a pristine Rh catalyst, Rh/Al2O3, were also prepared from RhCl3 and (NH4)6[Mo7O24]·4H2O under identical thermal treatment conditions to act as references.
According to the results of a high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis, the average diameters of Rh4Mo4(CpE)/Al2O3, Rh4Mo4(Cp*)/Al2O3, and Rh–Mo/Al2O3 were estimated to be 0.8 ± 0.2, 0.8 ± 0.2, and 1.2 ± 0.3 nm, respectively. This suggested that small nanoparticles were formed by using the clusters as precursors, regardless of the ligand structures (Figure 2).
Figure 2.
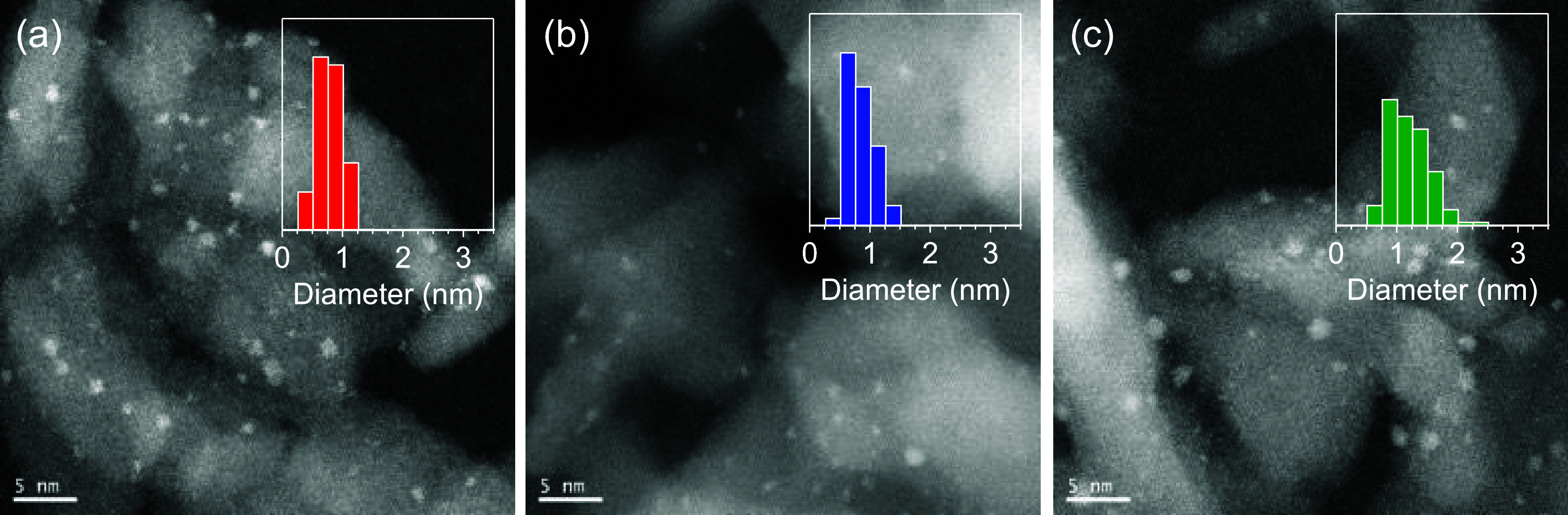
High-angle annular dark-field scanning transmission electron microscopy images of (a) Rh4Mo4(CpE)/Al2O3, (b) Rh4Mo4(Cp*)/Al2O3, and (c) Rh–Mo/Al2O3 (scale bar = 5 nm).
The electronic and local structures of Rh4Mo4(CpE)/Al2O3, Rh4Mo4(Cp*)/Al2O3, and Rh–Mo/Al2O3 were studied by Rh and Mo K-edge X-ray absorption spectroscopy. The Rh K-edge X-ray absorption near edge structure (XANES) spectra revealed that the Rh species in the catalysts were Rh3+ (Figure 3a). The Fourier transforms of extended X-ray absorption fine structure spectra of the catalysts showed a peak derived from the first coordination sphere (Rh–O bond) (Figure S4 and Table S2). The weak intensity of the peaks derived from the second coordination sphere suggested the presence of small nanoparticles in the catalysts, which was consistent with the results of the HAADF-STEM analysis. Because the pre-edge peaks in the Mo K-edge XANES spectra were assigned to weak quadrupole-allowed (1s → 4d) and strong dipole-allowed (1s → 5p) transitions, the local structural disorder of the molybdenum oxide species reflected the intensity. The Mo K-edge XANES spectra of the three catalysts were similar, implying that the local structure of Mo was independent of the catalyst preparation method (Figure 3b).
Figure 3.

(a) Rh and (b) Mo K-edge X-ray absorption near edge structure spectra of (i) Rh4Mo4(CpE)/Al2O3, (ii) Rh4Mo4(Cp*)/Al2O3, and (iii) Rh–Mo/Al2O3.
2.2. Catalytic Application to Amide Hydrogenation
Catalytic activities were tested through the hydrogenation of 4-acetylmorpholine (1a) to 4-ethylmorpholine (2a). The catalyst prepared from [(RhCpE)4Mo4O16], Rh4Mo4(CpE)/Al2O3, showed a higher catalytic activity than that of the catalyst derived from [(RhCp*)4Mo4O16], Rh4Mo4(Cp*)/Al2O3. This suggested that the choice of ligand in the precursors affected the activity of the supported catalysts (Table 1). The catalytic activity depended on the catalyst preparation method, with Rh4Mo4(CpE)/Al2O3 achieving the highest activity, followed by Rh4Mo4(Cp*)/Al2O3 and Rh–Mo/Al2O3. The pristine Rh/Al2O3 exhibited a low activity, implying that the formation of Rh/Mo interfaces was crucial for improving the activity. The catalysts supported on Al2O3, TiO2, and ZrO2 showed comparable activities, implying that the activity mainly originated from Rh/Mo interfaces (Table S3). The recycle tests suggested that Rh4Mo4(CpE)/Al2O3 could be reused at least four times without significant loss of activity (Figure S5). According to the Rh and Mo K-edge X-ray absorption spectroscopy analyses, the electronic and local structures of Rh and Mo were independent of the catalyst preparation method (Figure 3). Therefore, the high activity of Rh4Mo4(CpE)/Al2O3 was explained by the amount of Rh/Mo interfaces, rather than the chemical properties of Rh and Mo alone. Since the average diameters of Rh4Mo4(CpE)/Al2O3 and Rh4Mo4(Cp*)/Al2O3 were comparable (Figure 2), the formation efficiency of such interfaces could be affected by the choice of ligand in the precursors.
Table 1. Hydrogenation of 1a Catalyzed by Rh-Based Catalystsa.

| entry | catalyst | yield (%) |
|---|---|---|
| 1 | Rh4Mo4(CpE)/Al2O3 | 89 |
| 2 | Rh4Mo4(Cp*)/Al2O3 | 53 |
| 3 | Rh–Mo/Al2O3 | 24 |
| 4 | Rh/Al2O3 | 2 |
Reaction conditions: 1a (0.1 mmol), H2 (0.8 MPa), n-octane (0.5 mL), catalyst (10 mg, Rh: 0.97 mol %), molecular sieves (3A, 20 mg), 353 K, and 4 h.
Rh4Mo4(CpE)/Al2O3 also demonstrated activity toward various amides, including tertiary, secondary, and primary amides. The hydrogenation of 1-acetylpiperidine (1b) proceeded under the same H2 pressure (0.8 MPa) as that of 1a. However, a higher reaction temperature (393 K) was required to achieve a good yield (Table 2, entries 1 and 2). The hydrogenation of a secondary amide, N-butylpropionamide (1c), also proceeded under the same conditions (Table 2, entry 3). In particular, Rh4Mo4(CpE)/Al2O3 was used to successfully hydrogenate a primary amide, n-octanamide (1d) (Table 2, entry 4). In this reaction, a desired primary amine (2d) was mainly formed, and a secondary amine was obtained as a major side product. The formation of a primary alcohol and a tertiary amine was also confirmed as minor side products (Table S4, entry 1). The catalyst with a high loading (Rh: 5 wt %) was used because it showed a better selectivity than that of the catalyst with a typical (Rh: 1 wt %) loading (Table S4, entry 2). The addition of Al2O3 decreased the selectivity to 2d and increased it to secondary amines (Table S4, entry 3). Therefore, the minimal use of supports in highly loaded catalysts prevented undesired side reactions. The absence of molecular sieves decreased the conversion and selectivity to 2d, implying that the removal of water generated as a byproduct was important to achieve the desired C–O cleavage selectively (Table S4, entry 4).
Table 2. Hydrogenation of Amides to Amines Catalyzed by Rh4Mo4(CpE)/Al2O3.

Reaction conditions: 1 (0.1 mmol), H2 (0.8 MPa), n-octane (0.5 mL), catalyst (10 mg, Rh: 1 wt %, 0.97 mol %), molecular sieves (3A, 20 mg), and 6 h.
1 (0.025 mmol), H2 (0.8 MPa), n-octane (2 mL), catalyst (4 mg, Rh: 5 wt %, 7.8 mol %), molecular sieves (3A, 20 mg), and 10 h.
The hydrogenation of primary amides to their corresponding primary amines is usually difficult because of the formation of secondary (and tertiary) amines and primary alcohols as byproducts.4 The former is generated by the reaction of imine intermediates with primary amines, whereas the latter forms because of C–N bond cleavage. Although the hydrogenation of 1d to 2d showed a relatively low selectivity (76%) compared with those of the tertiary and secondary amides, it was considerably higher than those of other reported catalysts (Table S5).
As previously described, various catalysts, including bimetallic ones, are effective for amide hydrogenation. However, their use has mainly been limited to tertiary and secondary amides, and bimetallic catalysts have rarely been applied to primary amides. In particular, the reported catalysts required an excess of additives such as NH3 to hydrogenate acyclic primary amides (CH3(CH2)nC(O)NH2).6,15,25,26 Amide hydrogenation without the addition of NH3 is preferable as an environmentally friendly and atom-economical transformation. To the best of our knowledge, Rh4Mo4(CpE)/Al2O3 is the first example of a catalyst that can induce the hydrogenation of primary amides to amines with a high selectivity under mild (0.8 MPa H2, 393 K) and NH3-free conditions (Table S5). This emphasizes the utility of ligand-functionalized organometallic polyoxometalates as precursors for supported bimetallic catalysts.
2.3. Origin of the High Activity of Rh4Mo4(CpE)/Al2O3
We predicted that the presence of ethoxycarbonyl groups in CpE ligands would contribute to the adsorption behavior of the catalyst precursor onto the support. Cp* ligands are typically used for organometallic polyoxometalates because Cp* complexes are stable and accessible, making them suitable for starting materials. However, the hydrophobic nature of Cp* ligands leads to minimal interaction with supports such as Al2O3, the surface of which is covered with polarized functional groups, such as −OH. Therefore, the ligand-functionalization of organometallic polyoxometalates is necessary to enhance the interaction. To evaluate the degree of interaction, an adsorption experiment of the precursor organometallic polyoxometalate on the Al2O3 support was conducted. After the mixture of the precursor and Al2O3 in methanol was dried up, the amount of precursor redissolved in methanol was evaluated by UV–vis absorption spectroscopy. The amount of redissolved [(RhCpE)4Mo4O16] was 22%, whereas nearly all (92%) [(RhCp*)4Mo4O16] was redissolved (Figure 4). This implied that the presence of the ethoxycarbonyl group significantly enhanced the interaction of the precursor with Al2O3. When the adsorption experiment was conducted without a drying process (i.e., the mixture of the precursor and Al2O3 was simply filtrated), no adsorption was observed for either precursor (Figure S6). This suggested that [(RhCpE)4Mo4O16] showed minimal interaction while solvated and was adsorbed onto Al2O3 during the drying process.
Figure 4.

UV–vis spectra of pristine precursor (dotted) and extract from dried mixture with support (solid): (a) [(RhCpE)4Mo4O16] and (b) [(RhCp*)4Mo4O16].
The interaction of [(RhCpE)4Mo4O16] with Al2O3 was also confirmed by the dependence of the catalytic activity on the loading amount. The loading amount did not have a significant influence on the catalytic activity of Rh4Mo4(CpE)/Al2O3 for hydrogenating 1a, whereas the catalytic activities of Rh4Mo4(Cp*)/Al2O3 and Rh–Mo/Al2O3 significantly decreased with an increase in loading amount (Figure 5). This result was consistent with those obtained during the hydrogenation of 1d, where a highly loaded catalyst showed an improved selectivity without a decrease in conversion (Table S4, entries 1 and 2). These results implied that the interaction of the precursor with the support contributed to the high dispersibility of the active species.
Figure 5.

Initial reaction rate of hydrogenation of 1a over Rh–Mo-based catalysts with different Rh loadings. Reaction conditions were identical to those cited in Table 1, except for the amount of catalysts used (1–10 mg, Rh: 0.97 mol %).
DFT calculations were conducted to elucidate the interaction site of [(RhCpE)4Mo4O16]. The electrostatic potential was mapped onto the total electron density surfaces of [(RhCpE)4Mo4O16] and [(RhCp*)4Mo4O16] (Figures 6 and S7). The negative electrostatic potential represented the nucleophilic region, where the clusters likely interacted with the Al2O3 support. Despite the presence of negative areas on the terminal oxygen atoms in the Rh4Mo4O16 triple-cubane structure for both clusters, they were structurally hindered, making them unsuitable as interaction sites. Unlike [(RhCp*)4Mo4O16], [(RhCpE)4Mo4O16] presented exposed negative areas on the carbonyl oxygen atoms in the CpE ligands. Therefore, we concluded that the introduction of ethoxycarbonyl groups to the ligands contributed to the interaction with the support with negatively charged carbonyl oxygen atoms. This interaction prevented catalyst precursors from aggregating on the support and promoted the formation of a highly dispersed Rh–Mo catalyst (Figure 7).
Figure 6.
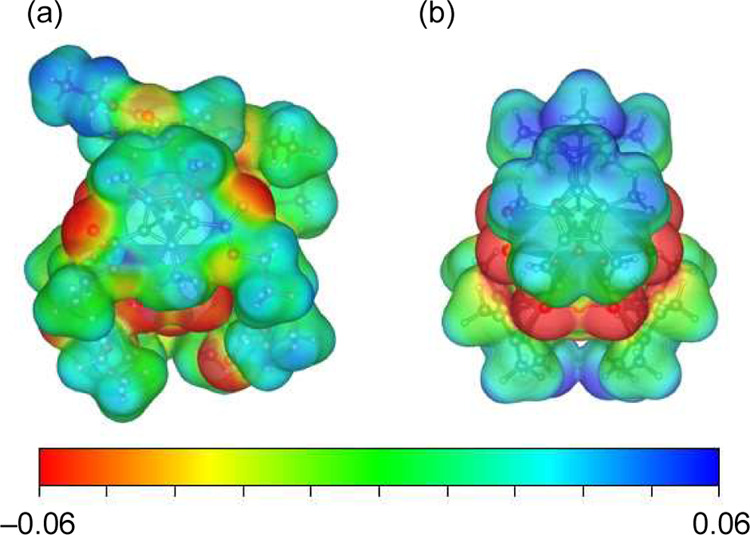
Electrostatic potentials mapped on electron density surfaces of (a) [(RhCpE)4Mo4O16] and (b) [(RhCp*)4Mo4O16].
Figure 7.

Role of precursors on structures of supported catalysts: (a) [(RhCpE)4Mo4O16], (b) [(RhCp*)4Mo4O16], and (c) RhCl3 + (NH4)6[Mo7O24]·4H2O. Precursors in solution (left) are adsorbed onto support during drying process (middle) and thermally treated to form supported catalysts (right).
3. Conclusions
Toward the efficient formation of interfacial catalytic active sites, a ligand-functionalized Rh–Mo organometallic polyoxometalate, [(RhCpE)4Mo4O16] (CpE = C5(CH3)3(COOC2H5)2), was newly synthesized. A Rh–Mo catalyst, Rh4Mo4(CpE)/Al2O3, was prepared from [(RhCpE)4Mo4O16] and applied for amide hydrogenation. Rh4Mo4(CpE)/Al2O3 showed a higher activity than those of Rh4Mo4(Cp*)/Al2O3 prepared from [(RhCp*)4Mo4O16] and Rh–Mo/Al2O3 prepared by coimpregnation. Rh4Mo4(CpE)/Al2O3 was applicable to the hydrogenation of tertiary, secondary, and primary amides under mild conditions (0.8 MPa H2, 353–393 K). In particular, it showed good activity and selectivity for the hydrogenation of primary amide under NH3-free conditions. Adsorption experiments of the precursors to Al2O3 suggested that CpE ligands interacted with the Al2O3 surface. Rh4Mo4(CpE)/Al2O3 showed a comparable activity with increased loadings, whereas a significant decrease in the activities was observed for Rh4Mo4(Cp*)/Al2O3 and Rh–Mo/Al2O3. DFT calculations revealed that the carbonyl oxygen atoms in the CpE ligands were negatively charged, leading to electrostatic interaction with the Al2O3 surface. Therefore, the high efficiency of [(RhCpE)4Mo4O16] as a catalyst precursor originated from the efficient formation of Rh/Mo interfacial active sites owing to the electrostatic interaction of CpE ligands with the support. These findings suggest a simple catalyst design strategy: catalyst precursors that possess both local M1/M2 interfacial structures and interaction sites with supports are advantageous for preparing supported bimetallic catalysts with highly dispersed M1/M2 interfacial active sites.
4. Experimental Section
4.1. Synthesis of Catalyst Precursors
[(RhCp*)4Mo4O16] was synthesized according to a previously described procedure.24 [(RhCpE)4Mo4O16] was synthesized according to the following procedure: [RhCpECl2]223 (170 mg, 0.20 mmol) was mixed with methanol (1 mL), to which an aqueous solution of Na2MoO4·2H2O (484 mg, 2.0 mmol, 4 mL) was added. This mixture was stirred for 4 h at 353 K using a heating block. After the reaction, black precipitate was collected by filtration and washed with water. The precipitate was dried well, and methanol was added to extract the desired product. Then, the mixture was filtrated to remove insoluble black precipitate, and the crude product was obtained by evaporating the solvent by using a rotary evaporator. The purified product was obtained via recrystallization from toluene/hexane (75% yield). Orange plate-like crystals suitable for single crystal X-ray diffraction analysis were obtained by slow vapor diffusion of hexane into toluene solution.
IR (KBr pellet): 1096, 1052, 1016, 937, 914, 854, 775, 683, 618, and 560 cm–1. HRMS (ESI+) Calcd for C56H77O32Rh4Mo4 ([M + H]+): 2064.6834, Found: 2064.6895.
4.2. Catalyst Preparation
The supported [(RhCpE)4Mo4O16] catalyst, namely Rh4Mo4(CpE)/Al2O3, was prepared as follows: [(RhCpE)4Mo4O16] (25.0 mg, 12.2 μmol) was dissolved in toluene (10 mL) and diluted with methanol (10 mL). This solution was added dropwise to a methanolic dispersion of the γ-Al2O3 support (490 mg in 40 mL). The mixture was stirred for 2 h before being slowly dried at 373 K using a heating block. Then, the solid was dried overnight at 353 K. The catalyst was obtained by performing calcination at 573 K for 1 h under static air using a muffle furnace, followed by reduction at 573 K for 1 h under H2 flow (10 mL/min) using a tube furnace. Rh4Mo4(Cp*)/Al2O3 was prepared using [(RhCp*)4Mo4O16] as the precursor. Methanol was used as the solvent. Rh–Mo/Al2O3 and Rh/Al2O3 were prepared using RhCl3 and (NH4)6[Mo7O24]·4H2O as precursors. Water was used as the solvent. The calculated loading amounts of Rh and Mo were 1.0 and 0.93 wt %, respectively (Mo/Rh = 1).
4.3. Characterization
Single-crystal X-ray diffraction studies were conducted using a Rigaku XtaLAB Synergy custom with Mo Kα radiation from the rotating-anode target combined with a confocal mirror. The CrysAlisPro software package (version 171.40.60a) was used for processing the diffraction data, including the application of numerical absorption correction. The structure was solved by the dual space method using SHELXT-2018/2.27 SHELXL-2019/3,28 with neutral-atom scattering factors, was used to refine the crystal structure. Details of the data collection, refinement, and results of the refinement are summarized in Table S1. Positive-ion electrospray ionization mass spectrometry was conducted with a Shimadzu LCMS-9050. The sample was dissolved in dichloromethane/methanol and electrosprayed at a bias voltage of +4 kV. FT–IR analysis was conducted using a JASCO FT/IR-6100 in the transmission mode. The sample was mixed with KBr and pressed to form a pellet, which was used for analysis. UV–vis absorption spectroscopy was conducted using a JASCO V-550 in the transmission mode. High-angle annular dark-field scanning transmission electron microscopy analysis was conducted using a JEOL JEM-ARM200F. The sample was prepared by dropping an ethanolic dispersion of the catalyst onto a copper grid with a support membrane and evaporating the solvent. The Mo and Rh K-edge X-ray absorption spectroscopy measurements were conducted using a BL14B2 beamline at the SPring-8 of the Japan Synchrotron Radiation Research Institute. The incident X-ray beam was monochromatized using a Si(311) double-crystal monochromator. The measurements were conducted in the transmission mode at room temperature. The samples were ground with boron nitride and pressed to form pellets. The X-ray absorption spectroscopy data was analyzed using the Rigaku REX2000 program. After background subtraction, the k3-weighted χ spectrum within a k range of 3–14 Å–1 was Fourier transformed into an r space. Curve-fitting analysis was performed for the first (Rh–O) and second (Rh–Rh) coordination spheres in the r ranges of 1.3–1.9 and 2.0–2.8 Å, respectively. The phase shifts and backscattering amplitude functions for the Rh–O and Rh–Rh bonds were extracted from [(RhCp*)4Mo4O16]24 using the FEFF8 program29 by setting the Debye–Waller Factor (σ2) to 0.0036 Å2. Gas chromatography analysis was conducted with a Shimadzu GC-2025 gas chromatograph comprising a flame ionization detector equipped with an InertCap 5 capillary column (internal diameter = 0.25 mm, length = 30 m).
4.4. Catalytic Test
The typical procedure for amide hydrogenation is as follows. The catalyst (10 mg, 0.97 mol % Rh) was placed in a glass tube, to which 4-acetylmorpholine (0.1 mmol), n-octane (0.5 mL), and molecular sieves (3A, 20 mg) were subsequently added. n-Decane (0.05 mmol) was also added as an internal standard. The glass tube was placed in an autoclave to which H2 was introduced. The reaction mixture was stirred at 353 K for 6 h using a heating block. After the reaction, acetone or 2-propanol was added, and the catalyst was removed by filtration. The conversions and yields were estimated based on the initial amounts of reactants used. The typical procedure for recycle tests is as follows. After the reaction, the catalyst and molecular sieves were collected by filtration, washed three times with 2-propanol (0.5 mL), and dried at 333 K. The used catalyst and molecular sieves were placed in a glass tube without separation, and the reactant, solvent, and fresh molecular sieves were added subsequently. For the hydrogenation of n-octanamide, the selectivities to dimerized and trimerized products were estimated based on the amounts of the reactants that were consumed. A representative gas chromatography chart of the reaction mixture is shown in Figure S8.
4.5. Adsorption Experiment
The amount of the organometallic polyoxometalate adsorbed on Al2O3 was estimated according to the following procedure: Al2O3 (50 mg) was added to a solution of [(RhCpE)4Mo4O16] in methanol (50 μM, 10 mL). This mixture was stirred for 1 h. Then, the solvent was evaporated using a rotary evaporator, and methanol (10 mL) was added. After stirring for 1 h, Al2O3 was removed by filtration. The filtrate was analyzed by UV–vis adsorption spectroscopy.
4.6. Theoretical Calculation
DFT calculations were conducted using the B3LYP density functional method with LanL2DZ (Mo, Rh) and 6-31++G(d,p) (H, C, O) as the basis sets, utilizing the Gaussian 16 program.30 The structures of [(RhCpE)4Mo4O16] and [(RhCp*)4Mo4O16] were optimized with C2 and S4 point-group symmetries, respectively. The absence of an imaginary frequency was confirmed by vibrational analysis. Structural models were prepared using VESTA programs.31
Acknowledgments
This research was financially supported by a Grant-in-Aid for Scientific Research (24K17563) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT). The HAADF-STEM analysis was conducted at the Advanced Research Infrastructure for Materials and Nanotechnology in Japan of the University of Tokyo, which was supported by the MEXT (JPMXP1224UT0115). The synchrotron radiation experiments were performed with the approval of the Japan Synchrotron Radiation Research Institute (2024A1763 and 2024A1918). The DFT calculations were performed at the Research Center for Computational Science, Okazaki, Japan (23-IMS-C263).
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.4c00071.
Results of characterization and catalytic tests (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Froidevaux V.; Negrell C.; Caillol S.; Pascault J. P.; Boutevin B. Biobased Amines: From Synthesis to Polymers; Present and Future. Chem. Rev. 2016, 116, 14181–14224. 10.1021/acs.chemrev.6b00486. [DOI] [PubMed] [Google Scholar]
- Volkov A.; Tinnis F.; Slagbrand T.; Trillo P.; Adolfsson H. Chemoselective Reduction of Carboxamides. Chem. Soc. Rev. 2016, 45, 6685–6697. 10.1039/C6CS00244G. [DOI] [PubMed] [Google Scholar]
- Constable D. J. C.; Dunn P. J.; Hayler J. D.; Humphrey G. R.; Leazer J. L.; Linderman R. J.; Lorenz K.; Manley J.; Pearlman B. A.; Wells A.; Zaks A.; Zhang T. Y. Key Green Chemistry Research Areas—a Perspective from Pharmaceutical Manufacturers. Green Chem. 2007, 9, 411–442. 10.1039/B703488C. [DOI] [Google Scholar]
- Smith A. M.; Whyman R. Review of Methods for the Catalytic Hydrogenation of Carboxamides. Chem. Rev. 2014, 114, 5477–5510. 10.1021/cr400609m. [DOI] [PubMed] [Google Scholar]
- Yang H.; Garcia H.; Hu C. Hydrogenation of Amides to Amines by Heterogeneous Catalysis: A Review. Green Chem. 2024, 26, 2341–2364. 10.1039/D3GC04175A. [DOI] [Google Scholar]
- Hirosawa C.; Wakasa N.; Fuchikami T. Hydrogenation of Amides by the Use of Bimetallic Catalysts. Tetrahedron Lett. 1996, 37, 6749–6752. 10.1016/S0040-4039(96)01458-X. [DOI] [Google Scholar]
- Beamson G.; Papworth A. J.; Philipps C.; Smith A. M.; Whyman R. Selective Hydrogenation of Amides Using Rh/Mo Catalysts. J. Catal. 2010, 269, 93–102. 10.1016/j.jcat.2009.10.020. [DOI] [Google Scholar]
- Mitsudome T.; Miyagawa K.; Maeno Z.; Mizugaki T.; Jitsukawa K.; Yamasaki J.; Kitagawa Y.; Kaneda K. Mild Hydrogenation of Amides to Amines over a Platinum-Vanadium Bimetallic Catalyst. Angew. Chem., Int. Ed. 2017, 56, 9381–9385. 10.1002/anie.201704199. [DOI] [PubMed] [Google Scholar]
- Pennetier A.; Hernandez W. Y.; Kusema B. T.; Streiff S. Efficient Hydrogenation of Aliphatic Amides to Amines over Vanadium-Modified Rhodium Supported Catalyst. Appl. Catal., A 2021, 624, 118301 10.1016/j.apcata.2021.118301. [DOI] [Google Scholar]
- Beamson G.; Papworth A. J.; Philipps C.; Smith A. M.; Whyman R. Selective Hydrogenation of Amides Using Ruthenium/ Molybdenum Catalysts. Adv. Synth. Catal. 2010, 352, 869–883. 10.1002/adsc.200900824. [DOI] [Google Scholar]
- Zhang Y.; Zhang F.; Li L.; Liu F.; Wang A. Highly Chemoselective Reduction of Amides to Amines over a Ruthenium-Molybdenum Bimetallic Catalyst. ChemistrySelect 2022, 7, e202203030 10.1002/slct.202203030. [DOI] [Google Scholar]
- Zhang Y.; Zhang F.; Li L.; Qi H.; Yu Z.; Liu X.; Cao C.; Liu F.; Wang A.; Zhang T. Decoration of Ru Nanoparticles with Mononuclear MoOx Boosts the Hydrodeoxygenation of Amides to Amines. J. Catal. 2023, 417, 301–313. 10.1016/j.jcat.2022.12.012. [DOI] [Google Scholar]
- Nakagawa Y.; Tamura R.; Tamura M.; Tomishige K. Combination of Supported Bimetallic Rhodium–Molybdenum Catalyst and Cerium Oxide for Hydrogenation of Amide. Sci. Technol. Adv. Mater. 2015, 16, 014901 10.1088/1468-6996/16/1/014901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T.; Shi Z.; Zhang G.; Chan H. C.; Shu Y.; Gao Q.; Tang Y. Molybdenum-Incorporated Mesoporous Silica: Surface Engineering toward Enhanced Metal-Support Interactions and Efficient Hydrogenation. ACS Appl. Mater. Interfaces 2018, 10, 42475–42483. 10.1021/acsami.8b16496. [DOI] [PubMed] [Google Scholar]
- Coeck R.; Berden S.; De Vos D. E. Sustainable Hydrogenation of Aliphatic Acyclic Primary Amides to Primary Amines with Recyclable Heterogeneous Ruthenium-Tungsten Catalysts. Green Chem. 2019, 21, 5326–5335. 10.1039/C9GC01310E. [DOI] [Google Scholar]
- Zhang Y.; Li L.; Liu F.; Qi H.; Zhang L.; Guan W.; Liu Y.; Wang A.; Zhang T. Synergy between Ru and WOX Enables Efficient Hydrodeoxygenation of Primary Amides to Amines. ACS Catal. 2022, 12, 6302–6312. 10.1021/acscatal.2c01264. [DOI] [Google Scholar]
- Stein M.; Breit B. Catalytic Hydrogenation of Amides to Amines under Mild Conditions. Angew. Chem., Int. Ed. 2013, 52, 2231–2234. 10.1002/anie.201207803. [DOI] [PubMed] [Google Scholar]
- Burch R.; Paun C.; Cao X. M.; Crawford P.; Goodrich P.; Hardacre C.; Hu P.; McLaughlin L.; Sá J.; Thompson J. M. Catalytic Hydrogenation of Tertiary Amides at Low Temperatures and Pressures Using Bimetallic Pt/Re-Based Catalysts. J. Catal. 2011, 283, 89–97. 10.1016/j.jcat.2011.07.007. [DOI] [Google Scholar]
- Hayashi S.; Shishido T. High-Density Formation of Metal/Oxide Interfacial Catalytic Active Sites through Hybrid Clustering. ACS Appl. Mater. Interfaces 2021, 13, 22332–22340. 10.1021/acsami.1c02240. [DOI] [PubMed] [Google Scholar]
- Hayashi S.; Endo S.; Miura H.; Shishido T. Highly Active and Durable Rh–Mo-Based Catalyst for the NO–CO–C3H6–O2 Reaction Prepared by Using Hybrid Clustering. ACS Mater. Au 2023, 3, 456–463. 10.1021/acsmaterialsau.3c00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S.; Shishido T. High-Density Formation of Ir/MoOx Interface through Hybrid Clustering for Chemoselective Nitrostyrene Hydrogenation. ACS Org. Inorg. Au 2023, 3, 283–290. 10.1021/acsorginorgau.3c00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putaj P.; Lefebvre F. Polyoxometalates Containing Late Transition and Noble Metal Atoms. Coord. Chem. Rev. 2011, 255, 1642–1685. 10.1016/j.ccr.2011.01.030. [DOI] [Google Scholar]
- Shibata Y.; Tanaka K. Catalytic [2 + 2 + 1] Cross-Cyclotrimerization of Silylacetylenes and Two Alkynyl Esters to Produce Substituted Silylfulvenes. Angew. Chem., Int. Ed. 2011, 50, 10917–10921. 10.1002/anie.201105517. [DOI] [PubMed] [Google Scholar]
- Hayashi Y.; Toriumi K.; Isobe K. Novel Triple Cubane-Type Organometallic Oxide Clusters: [MCp*MoO4]4•nH2O (M = Rh and Ir; Cp* = C5Me5; n = 2 for Rh and 0 for Ir). J. Am. Chem. Soc. 1988, 110, 3666–3668. 10.1021/ja00219a056. [DOI] [Google Scholar]
- Guo W.; Xia Q.; Jia H.; Guo Y.; Liu X.; Pan H.; Wang Y.; Wang Y. Highly Selective Synthesis of Primary Amines from Amide over Ru-Nb2O5 Catalysts. Chem. - Asian J. 2022, 17, e202101256 10.1002/asia.202101256. [DOI] [PubMed] [Google Scholar]
- Yang H.; Zhou L.; Chen H.; Zeng Y.; Li D.; Hu C. Efficient Hydrogenation of Aliphatic Acyclic Amides to Amines by Bimetallic NiMo Nitrides via Heterogeneous Catalysis. Chem. Eng. J. 2023, 473, 145374 10.1016/j.cej.2023.145374. [DOI] [Google Scholar]
- Sheldrick G. M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankudinov A. L.; Ravel B.; Rehr J. J.; Conradson S. D. Real-Space Multiple-Scattering Calculation and Interpretation of X-ray-Absorption near-Edge Structure. Phys. Rev. B 1998, 58, 7565 10.1103/PhysRevB.58.7565. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.. et al. Gaussian 16, Revision C.02; Gaussian, Inc.: Wallingford CT, 2019.
- Momma K.; Izumi F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. 10.1107/S0021889811038970. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.