

Abstract
Hydrogen atom transfer (HAT) processes provide an important strategy for selective C–H functionalization. Compared with the popularity of 1,5-HAT processes, however, net-1,2-HAT reactions have been reported less frequently. Herein, we report a unique visible-light-mediated net-1,2-HAT of amidyl radicals for the synthesis of β-amido ketone derivatives. Single-electron transfer (SET) to N-aryloxy amides generates nitrogen-centered radicals (N˙), which undergo a rare net-1,2-HAT to form carbon-centered radicals (C˙). The C-centered radicals are then captured by silyl enol ethers on the way to β-amido ketones. A series of β-amido ketone derivatives (33 examples, up to 97% yield) were prepared with good functional group tolerance demonstrating the synthetic utility of this method. Mechanistic studies, including EPR, radical trapping experiments, deuterium labeling and KIE measurements, suggest an intramolecular radical net-1,2-HAT pathway.
Visible-light-driven N-centered radicals lead to C-centered α-amino radicals through rare net-1,2-HAT processes, with trapping by silyl enol ethers to access β-amido ketone derivatives.
Introduction
The selective functionalization of C–H bonds has received tremendous attention over the last few decades with many milestones achieved.1–10 Despite significant advances, the C–H activation and chemical elaboration at specific sites of organic compounds is in its infancy and remains a challenge due to the similar reactivity of inequivalent C–H bonds.11,12 Efforts to circumvent the selectivity problem have employed directing groups to steer reactive metal catalysts to specific C–H bonds, engendering selectivity.13–19 A classic strategy that has recently witnessed a resurgence is the generation of reactive O- and N-centered radicals that undergo energetically favorable 1,5-hydrogen atom transfer (1,5-HAT) reactions (Scheme 1, path A).20–27 The resulting translocated radicals can be readily functionalized through formation of C–C, C–N, C–O and C–S bonds, adding great synthetic value to this approach.28–39
Scheme 1. Reactions of heteroatom-centered radicals. (Path A) The dominant course of heteroatom-centered radicals, the 1,5-HAT. (Path B) The unfavorable 1,2-HAT. (Path C) An alternative reaction pathway, the base mediated net-1,2-HAT.

The fidelity of the 1,5-HAT reaction stems from its low energy 6-membered ring transition state compared to HAT from other positions.40 Recent reports, however, hint that the facile intramolecular 1,5-HAT process can be energetically undercut by an apparent 1,2-HAT under certain conditions.41–48 These results were quite surprising, given that the classical 1,2-HAT proceeds through a strained, high energy 3-membered transition state (Scheme 1, path B). Thus, alternative mechanistic hypotheses were necessary to explain the observed reactivity,49 on which the foundation of a general 1,2-HAT method can be built (Scheme 1, path C).
Our entry into this area arose from our generation of radicals using the organic super electron donors (SEDs), 2-azaallyl anions.50–53 2-Azaallyl anions are strong reductants,54 and upon donation of an electron they form stabilized 2-azaallyl radicals55 that participate in a variety of radical–radical coupling reactions to form C–C and C–X coupled products.56–65 Of note, we found that 2-azaallyl anions react with N-aryloxy amides to afford amidyl radicals (N˙).66,67 Interestingly, the amidyl radical did not undergo the expected 1,5-HAT but instead underwent a base-promoted deprotonation/reprotonation leading to net-1,2-HAT. This isomerization converts the N-centered radicals into α-amino C-centered radicals (Scheme 2a).66 Radical–radical coupling then furnished diamine derivatives. This mechanism was supported experimentally and computationally.
Scheme 2. Reaction development. (a) Base-assisted 1,2-HAT of amidyl radicals. (b) Visible-light-mediated base-assisted 1,2-HAT. (c) Proton-coupled electron transfer (PCET) followed by 1,2-HAT.

Shortly after our publication, Chen and Wang observed a similar base-promoted net-1,2-HAT of an amidyl radical under photoredox catalysis conditions (Scheme 2b).68 Their mechanism was also supported by DFT calculations. They further developed the net-1,2-HAT with this Boc-protected substrate with capture by trifluoromethyl-substituted alkenes.69 They focused on the N-trifluoroethylamide substrates to synthesize α-trifluoromethylamine and N-trifluoroethylamine derivatives. Along these lines, the Besset group recently developed a photoredox catalyzed trifluoromethylthiolation of amides. They proposed proton-coupled electron transfer (PCET) from an amide N–H to generate an N-centered radical. This amidyl radical was envisioned to undergo a 1,2-HAT to form an α-amino C-centered radical that underwent thiolation with in situ generated CF3S–SCF3 to furnish trifluoromethylthiolated amides (Scheme 2c).70
Inspired by the high synthetic value and predominance of the 1,5-HAT as a method to selectively functionalize C–H bonds,71–74 the goal of the current study was to evaluate the generality of the base-mediated net-1,2-HAT with the aim of establishing it as a viable synthetic strategy to perform chemoselective C–H functionalizations located next to the amino group of amides. Using photoredox catalysis and N-aryloxy amides, we envisioned generation of amidyl radicals (Scheme 3). We anticipated that the amidyl radicals could be deprotonated by the aryloxide to reveal α-amino C-centered radicals. We hypothesized that the generated α-amino radicals could be captured by radical acceptors for the construction of new C–C bonds.75–80 For the trapping of the formed radicals, we chose silyl enol ethers because they are known to readily react with radicals to form ketones.81–83 Herein we describe the fruition of this plan. This study represents another step toward demonstrating the potential utility of the base-mediated net-1,2-HAT as a synthetic tool for mild and selective C–H functionalizations.
Scheme 3. This work. Net-1,2-Hydrogen Atom Transfer (HAT) of amidyl radicals.

Results and discussion
We initiated this study by using N-benzyl N-aryloxy amide 1a and phenylmethylsilyl ether 2a as model reactants. We initially used 2 mol% fac-Ir(ppy)3 as the photoredox catalyst to investigate the effect of solvents (DMF, DMSO, CH3CN, DCM, DCE, THF, PhCl, PhCF3, and EtOH, Table 1, entries 1–9). Among the solvents tested, reactions in DMSO showed the best conversions to the coupled product 3aa (68% assay yield, AY, as determined by 1H NMR integration against an internal standard, Table 1, entry 2). Given that the generation of amidyl radicals requires a strong reductant (1a, E1/2 = −0.935 V vs. SCE in DMSO, see the ESI† for details),71,84 we evaluated appropriate photoredox catalysts. Switching photoredox catalysts to 4CzIPN, [Ir(dtbbpy)(ppy)2][PF6], [Ir(ppy)2(bpy)]PF6, and Eosin Y, however, led to lower AY (entries 10–13). Based on the results, fac-Ir(ppy)3 was chosen for continued optimization. Considering that some of the N-aryloxy amide 1a was reduced to the amide 1a′ and aryloxide (Table S6, see the ESI† for details),85,86 we next examined the 1a : 2a ratio. We were pleased to find that increasing the ratio of 1a and 2a from 1 : 1.5 to 1.5 : 1 under otherwise identical conditions resulted in 94% AY and 90% isolated yield of the amido ketone (entry 14). Furthermore, varying the concentration from 0.1 to 0.2 or 0.05 M, led to a reduction in the AY to 86% or 77% (entries 15 and 16). Control experiments showed that the photoredox catalyst and light irradiation were both necessary for the success of this transformation (entries 17 and 18).
Table 1. Optimization of coupling of amide 1a and silyl enol ether 2aa,b.
| ||||
|---|---|---|---|---|
| Entry | Solvent | Photocatalyst | Conc. [M] | Assay yield (%) |
| 1 | DMF | fac-Ir(ppy)3 | 0.1 | 40 |
| 2 | DMSO | fac-Ir(ppy)3 | 0.1 | 68 |
| 3 | CH3CN | fac-Ir(ppy)3 | 0.1 | 27 |
| 4 | DCM | fac-Ir(ppy)3 | 0.1 | 0 |
| 5 | DCE | fac-Ir(ppy)3 | 0.1 | 12 |
| 6 | THF | fac-Ir(ppy)3 | 0.1 | 18 |
| 7 | PhCl | fac-Ir(ppy)3 | 0.1 | 20 |
| 8 | PhCF3 | fac-Ir(ppy)3 | 0.1 | Trace |
| 9 | EtOH | fac-Ir(ppy)3 | 0.1 | 0 |
| 10 | DMSO | 4CzIPN | 0.1 | 13 |
| 11 | DMSO | [Ir(dtbbpy)(ppy)2][PF6] | 0.1 | 31 |
| 12 | DMSO | [Ir(ppy)2(bpy)]PF6 | 0.1 | 26 |
| 13 | DMSO | Eosin Y | 0.1 | 18 |
| 14c | DMSO | fac-Ir(ppy)3 | 0.1 | 94 (90)d |
| 15c | DMSO | fac-Ir(ppy)3 | 0.2 | 86 |
| 16c | DMSO | fac-Ir(ppy)3 | 0.05 | 77 |
| 17c | DMSO | — | 0.1 | 0 |
| 18e | DMSO | fac-Ir(ppy)3 | 0.1 | 0 |
Reaction conditions: 1a (0.1 mmol, 1.0 equiv.), 2a (0.15 mmol, 1.5 equiv.), PC (2 mol%), rt, 6 h.
Assay yields determined by 1H NMR spectroscopy of the crude reaction mixtures using CH2Br2 as an internal standard.
1.5 : 1 ratio of 1a to 2a.
Isolated yield after chromatographic purification.
Without blue LEDs.
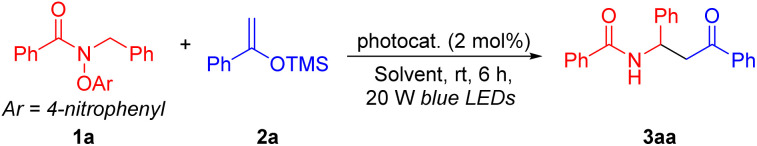
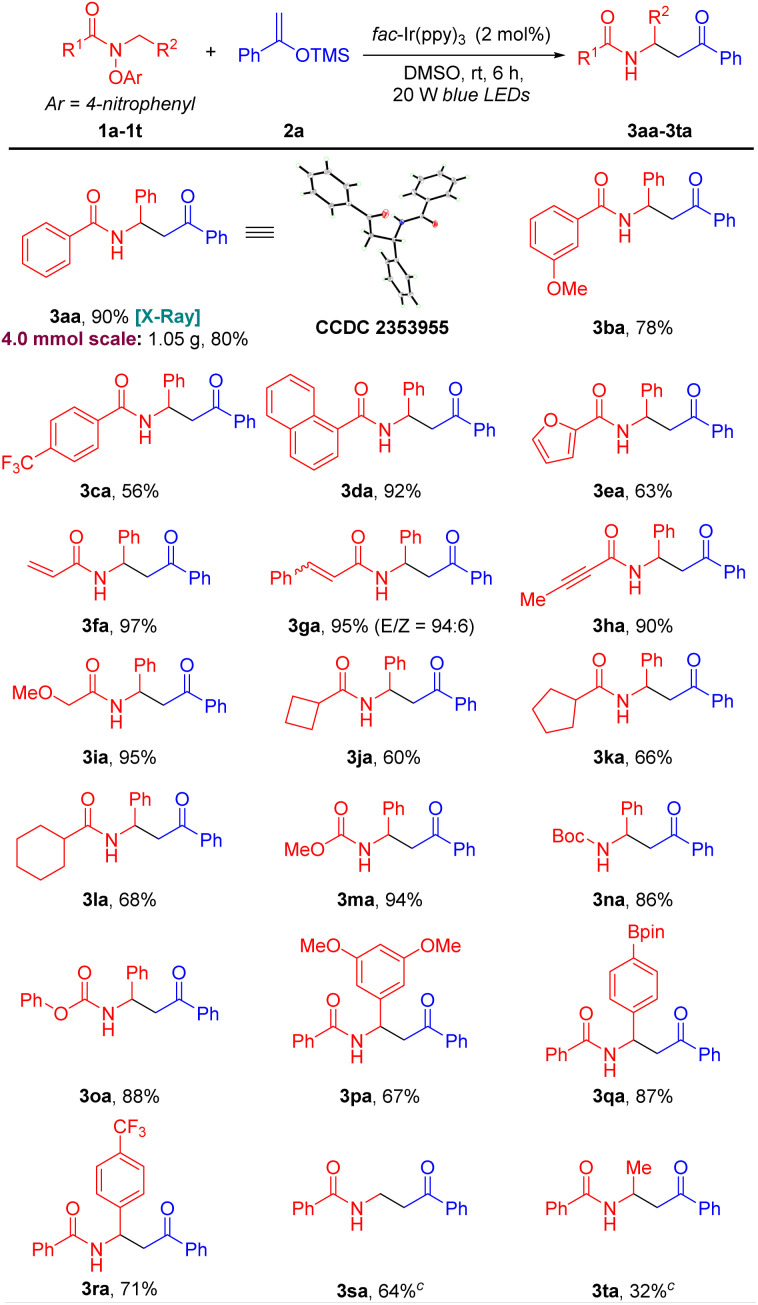
With the optimized conditions in hand (Table 1, entry 14), we initiated exploration of the scope of amides 1. As shown in Table 2, in general, amides bearing various substituted aryl, alkyl and alkoxy groups delivered products in moderate to excellent yields under the optimized conditions. Aryl-substituted amides with electron-withdrawing 3-OMe (1b) or 4-CF3 (1c), reacted with silyl enol ether 2a to furnish coupling products 3ba and 3ca in 78 and 56% yields, respectively. Sterically hindered 1-naphthyl-substituted amide 1d provided coupling product 3da in 92% yield. Notably, a medicinally relevant heterocyclic amide possessing a 2-furyl group (1e) delivered coupling product 3ea in 63% yield. It is noteworthy that amides bearing alkenes (vinyl 1f; E-styryl 1g) and alkyne (1h) were also successfully converted to the desired C(sp3)–C(sp3) cross-coupling products (3fa–3ha) in 97, 95, and 90% yields, respectively. These results suggest that the intermolecular coupling with the silyl enol ether is faster than intramolecular cyclization onto the unsaturated moiety of the substrate. The alkyl ether (1i) was an excellent substrate, providing the desired coupled product in 95% yield. Furthermore, alkyl-substituted amides bearing cyclobutyl (1j), cyclopentyl (1k), and cyclohexyl (1l) groups were also suitable coupling partners, offering products 3ja, 3ka and 3la, respectively, in 60–68% yields. We were pleased to find that carbamates bearing methyl (1m), t-butyl (1n) and benzyl (1o), performed well, furnishing the desired products 3ma, 3na and 3oa in 94, 86 and 88% yields, respectively.
Table 2. Scope of amides 1a,b.
|
Reactions were conducted on a 0.2 mmol scale using 1.5 equiv. (1a–1t), 1.0 equiv. 2a, and fac-Ir(ppy)3 (2 mol%) at 0.1 M.
Isolated yields after chromatographic purification.
NaHCO3 added (4.5 equiv.).
Different N-benzyl groups were briefly surveyed. For example, N-benzyl groups bearing 3,5-di-OMe (1p), 4-Bpin (1q) and 4-CF3 (1r) were determined to be competent coupling partners, leading to the expected products 3pa, 3qa and 3ra in 67, 87 and 71% yields, respectively. Meanwhile, the scope of amide substrates could be expanded by switching from N-benzyl amides to N-alkyl amides. Interestingly, the addition of base (NaHCO3) was necessary when using N-alkyl amides for coupling reactions, likely due to the decreased acidity of the amidyl radical's α-C–H bonds. In the absence of NaHCO3, products derived from nitrogen radical addition to the silyl enol ether were obtained (see the ESI† for details).87–89 Under the NaHCO3 modified conditions, N-methyl (1s) and N-ethyl (1t) amides gave the corresponding products 3sa and 3ta in 64 and 32% yields, respectively. Unfortunately, the net-1,2-HAT reaction did not occur when an amide derived from phenethyl amine was employed, which would form a secondary α-amino radical, probably due to the steric hindrance to the deprotonation of the amidyl radical.
Finally, a gram-scale synthesis was conducted to demonstrate the utility and scalability of this coupling process. Reaction of N-benzyl amide 1a with the silyl enol ether 2a under the standard conditions provided 1.05 g of 3aa in 80% yield. The structure of 3aa was confirmed by X-ray crystallography (CCDC 2353955).90
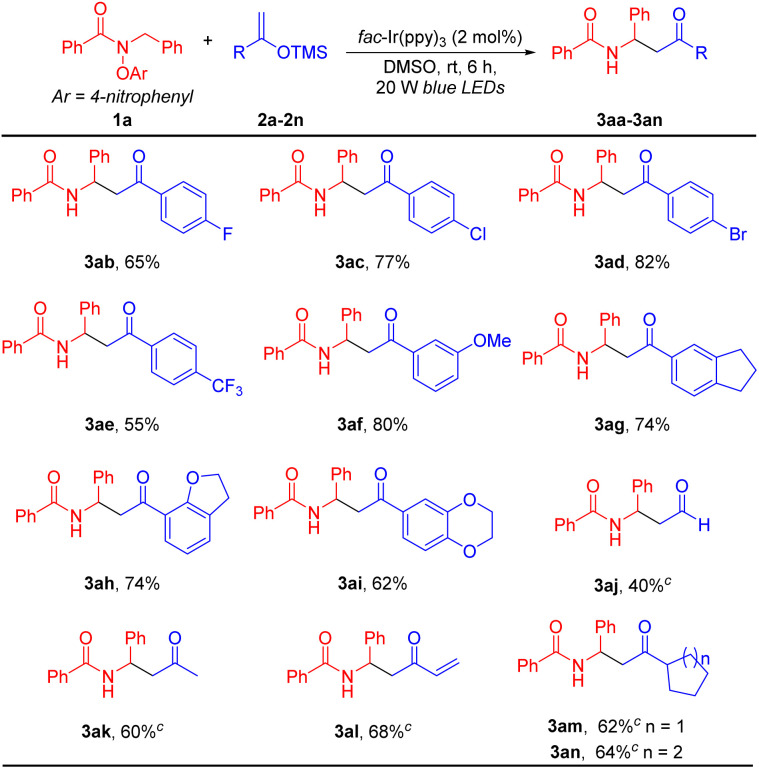
Encouraged by the results with various amides, we evaluated the scope of substituted silyl enol ethers (2), which were readily synthesized from the corresponding ketones.91 As shown in Table 3, we selected amide 1a as the coupling partner. A wide range of silyl enol ethers bearing various groups were compatible with the standard conditions, generating the β-amido ketone products in moderate to good yields (40–82%). For instance, aryl-substituted silyl enol ethers with electronegative or electron-withdrawing groups, such as 4-F (2b), 4-Cl (2c), 4-Br (2d), 4-CF3 (2e) and 3-OMe (2f), or electron-donating groups, such as dihydrindene (2g), 2,3-dihydrofuran (2h), and 1,4-dioxane (2i) reacted with N-benzyl amide 1a to furnish coupling products 3ab–3ai, respectively, in 55–82% yields. Interestingly, silyl enol ethers derived from aldehydes (2j), such as acetaldehyde, from aliphatic ketones (2k), such as acetone, and from methyl acrylate (2l) were viable reaction partners. These substrates gave the net-1,2-HAT coupling products 3aj, 3ak and 3al in 40, 60 and 68% yields, respectively. Finally, alkyl-substituted silyl enol ethers bearing cyclopentyl (1m), and cyclohexyl (1n) groups were also suitable partners, affording the coupling products 3am and 3an in 62 and 64% yields, respectively.
Table 3. Scope of silyl enol ethersa,b.
|
Reactions were conducted on a 0.2 mmol scale using 1.5 equiv. 1a, 1.0 equiv. (2a–2n), and fac-Ir(ppy)3 (2 mol%) at 0.1 M.
Isolated yields after chromatographic purification.
fac-Ir(ppy)3 (10% mmol).
Next, we used amide 1v with hydrogen atoms positioned at the 1,2-, 1,5- and 1,6-positions relative to the amide nitrogen to explore the HAT site-selectivity under the reaction conditions. As shown in Scheme 4, treatment of 1v with NaHCO3 (4.5 equiv.) under standard conditions gave the net-1,2-HAT product 3va in 9% yield, and no 1,5- or 1,6-HAT coupling product 3va′ or 3va′′ was detected. Most of the amide 1v was converted to the reduced product 1v′ in 60% yield (see the ESI† for details).
Scheme 4. Site-selectivity studies. Net-1,2-HAT vs. 1,5- and 1,6-HAT.

To explore the reaction mechanism, a series of experiments were conducted. First, Stern–Volmer quenching studies suggested that amide 1a quenches the excited state of *IrIII(ppy)3 more efficiently than silyl enol ether 2a (Scheme 5a, see the ESI† for details). To probe for the presence of radical species, EPR experiments employing phenyl N-t-butylnitrone (PBN) as the radical spin trap were conducted under the standard conditions. Thus, treatment of N-benzyl aryl amide 1a with silyl enol ether 2a in the presence of PBN resulted in the generation of a PBN-trapped carbon-centered radical, as detected by EPR spectroscopy (Scheme 5b and 5c). The generated EPR signals (g = 1.9995, AN = 14.7 G, AH = 3.3 G) are in agreement with previously reported literature for trapping C-centered radicals.92,93 The cationic intermediate originating from radical 4 can be detected in the reaction mixtures by high-resolution mass spectrometry (HRMS calculated for C25H27N2O2+ 387.2067, found 387.2068 [M]+).
Scheme 5. Mechanistic studies. (a) Stern–Volmer fluorescence quenching experiments. (b) The PBN-trapped carbon-centered radical. (c) EPR spectrum of the PBN-trapped radical assigned as 4.

Experiments with the addition of 2.5 equiv. of radical scavenger 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) were conducted under otherwise standard conditions. The coupling product 3sa was not detected, despite 1s being consumed and converted to the amide PhCONHMe. The TEMPO trapping products 5sa and 6aa were isolated in 18% and 80% yields, respectively (Scheme 6a). Compound 5sa is the product from trapping the C-centered α-amino radical formed from the net-1,2-HAT pathway. 6aa has previously been observed in other photoredox reactions with enol silanes when radical trapping experiments were performed with TEMPO.94,95 In our case, it is likely the aryloxide byproduct binds to the Si of the TMS group of the silyl enol ether and generates an enolate. The enolate is oxidized by the Ir4+ photoredox catalyst to furnish the α-carbonyl radical that is trapped by TEMPO (see Fig. S11† for a proposed mechanism).
Scheme 6. Mechanistic experiments. (a) Radical trapping experiment with TEMPO. (b) Radical inhibitor experiment with BHT. (c) Control experiment with imine 1a′′.

Meanwhile, the addition of butylated hydroxytoluene (BHT) inhibited the reaction and resulted in a low yield of 3aa (7%) (Scheme 6b). It is also conceivable that the reaction could proceed by formation of the amidyl radical with oxidation to the imine 1a′′ by the photoredox catalyst (IrIV(ppy)3)+, which could then react with the silyl enol ether via a Mannich type reaction to afford the observed product. Thus, N-benzoyl-benzaldimine 1a′′ was prepared and reacted with silyl ether 2a under standard conditions (Scheme 6c, see the ESI† for details). The coupling product 3aa, however, was not detected, which suggested that such a pathway was not operative. These results lead us to favor a radical pathway, as outlined in Scheme 3.
Additional experiments were performed to probe the formation of the proposed α-amidyl radical. N-Allyl-substituted amide 1u was prepared and reacted with 2a under standard conditions (Scheme 7a). The net-1,2-HAT coupling product 3ua was obtained, but only in 10% yield. The more thermodynamically favored product 3ua′ was afforded in 55% yield. Interestingly, the direct coupling of the nitrogen-centered radical with the silyl enol ether gave 3ua′′ in 28% yield.89 These results trace the intermediates in this process from the N-centered radical to the resonance stabilized C-centered radical and coupling products.
Scheme 7. Mechanistic experiments. (a) Radical trapping experiment with the N-allyl amide 1u. (b) Intramolecular deuterium isotope effect experiment. (c) Parallel KIE experiments.

Next, a deuterium-substituted amide 1a-d1 with D : H = 1.0 : 1.0 at the amide nitrogen α-position was prepared (Scheme 7b, see the ESI† for details). Subjecting labeled amide 1a-d1 and enol silane 2a to the standard conditions delivered the coupling products 3aa-d1 and 3aa in 84% yield with 3aa-d1 : 3aa = 0.76 : 0.24 (kH/kD = 3.2, Scheme 7b). The different rates of C–H and C–D isotopes are consistent with deprotonation. Furthermore, the parallel kinetic isotope effect (KIE) was measured, giving KIE kH/kD = 1.8, which indicates that the deprotonation of the amidyl radical may be the rate-determining step (Scheme 7c, see the ESI† for details). It is interesting to compare this result to parallel KIEs measured in 1,5-HAT of amidyl radicals, which show values of 1.6–2.6.96–99
Finally, the dehydrogenative coupling of 1a was chosen for kinetic studies. Initial rate experiments disclosed that the reaction was zero-order in silyl enol ether, first-order in amide and saturation behavior in [fac-Ir(ppy)3] (first-order at 0 < [fac-Ir(ppy)3] < 2 mol% and zero-order at [fac-Ir(ppy)3] > 2 mol%).100–103 (see the ESI† for full initial-rate kinetics).
According to the above mechanistic investigations and relevant literature,66 a plausible mechanism for the formation of amido ketones was proposed. As outlined in Scheme 8, the catalytic cycle was envisioned to start with the light absorption by the photoredox catalyst IrIII(ppy)3 to its excited state IrIII(ppy)3*. Oxidative quenching of IrIII(ppy)3* via reduction of N-benzyl aryl amide 1a would generate the N-centered radical I, which undergoes base-assisted-1,2-HAT to produce α-amino C-centered radical II.66 At this stage, radical II is envisioned to add to the silyl enol ether 2a to generate α-oxygen radical III, which undergoes SET and desilylation to furnish product 3aa while simultaneously regenerating the catalyst IrIII(ppy)3.
Scheme 8. Proposed mechanism.

Conclusions
In summary, we have developed a unique visible-light-driven SET to form amidyl radicals. The electron deficient amidyl radical acidifies the neighboring C–H bonds such that the relatively weak base, ArO−, can deprotonate the N-α-C–H to trigger a net-1,2-HAT. The resulting C-centered radical adds to the silyl enol ether to form a C–C bond toward the generation of β-amido ketone derivatives. This method is complementary to Shang, Fu and co-workers' use of redox active esters to generate α-amino amide radicals.104 A variety of functionalized amides are suitable for this transformation, and a range of aromatic or aliphatic silyl enol ethers were used as radical acceptors. A gram-scale synthesis exhibits the potential synthetic utility of this approach. A series of mechanistic and EPR experiments demonstrate an intramolecular radical net-1,2-HAT pathway. Notably, this method establishes a new synthetic strategy for the preparation of β-amido ketones under mild conditions. Further investigation of net-1,2-HAT processes with amidyl radicals is underway in our laboratory.
Data availability
All experimental data, procedures for data analysis, and pertinent data sets are provided in the ESI.†
Author contributions
X. Y., Y. J. and P. J. W. conceived of the project. X. Y., H. Z. and C. Z. designed the experiments. Y. J., H. L., H. T., Q. Z., and H. Y. performed the research. X. Y. and P. J. W. wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by grants from the NSFC (22361050), National Key R&D Program of China (2019YFE0109200), Central Government Guides Local Science and Technology Development Fund (202207AA110007), Ling-Jun Scholars Yunnan Province (202005AB160003), Program for Xingdian Talents (Yun-Ling Scholars), Postdoctoral Fellowship Program of CPSF (GZC20232214) and Cai-Yun Postdoctoral Innovation Project (Y. G. J.). P. J. W. thanks the US National Science Foundation (CHE-2154593) for financial support. The authors thank Prof. Chengfeng Xia for help with the EPR experiments.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04997g
Notes and references
- He J. Wasa M. Chan K. S. L. Shao Q. Yu J.-Q. Chem. Rev. 2016;117:8754–8786. doi: 10.1021/acs.chemrev.6b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moselage M. Li J. Ackermann L. ACS Catal. 2015;6:498–525. doi: 10.1021/acscatal.5b02344. [DOI] [Google Scholar]
- Du B. Chan C.-M. Au C.-M. Yu W.-Y. Acc. Chem. Res. 2022;55:2123–2137. doi: 10.1021/acs.accounts.2c00283. [DOI] [PubMed] [Google Scholar]
- Dutta U. Maiti S. Bhattacharya T. Maiti D. Science. 2021;372:701. doi: 10.1126/science.abd5992. [DOI] [PubMed] [Google Scholar]
- Lam N. Y. S. Wu K. Yu J. Q. Angew. Chem., Int. Ed. 2021;60:15767–15790. doi: 10.1002/anie.202011901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. Kim B.-S. Li M. Walsh P. J. Org. Lett. 2016;18:2371–2374. doi: 10.1021/acs.orglett.6b00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Sha S.-C. Bellomo A. Trongsiriwat N. Gao F. Tomson N. C. Walsh P. J. J. Am. Chem. Soc. 2016;138:4260–4266. doi: 10.1021/jacs.6b01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S. Li M. Ma X. Chen W. Li L. Zhang H. Yang X. Walsh P. J. Adv. Synth. Catal. 2018;360:4837–4842. doi: 10.1002/adsc.201801045. [DOI] [Google Scholar]
- Liu Z. Li M. Wang B. Deng G. Chen W. Kim B.-S. Zhang H. Yang X. Walsh P. J. Org. Chem. Front. 2018;5:1870–1876. doi: 10.1039/C8QO00207J. [DOI] [Google Scholar]
- Tian X. Li X. Duan S. Du Y. Liu T. Fang Y. Chen W. Zhang H. Li M. Yang X. Adv. Synth. Catal. 2020;363:1050–1058. doi: 10.1002/adsc.202001254. [DOI] [Google Scholar]
- Newton C. G. Wang S.-G. Oliveira C. C. Cramer N. Chem. Rev. 2017;117:8908–8976. doi: 10.1021/acs.chemrev.6b00692. [DOI] [PubMed] [Google Scholar]
- Shao Q. Wu K. Zhuang Z. Qian S. Yu J.-Q. Acc. Chem. Res. 2020;53:833–851. doi: 10.1021/acs.accounts.9b00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.-H. Ouyang Y. Chekshin N. Yu J.-Q. ACS Catal. 2022;12:10581–10586. doi: 10.1021/acscatal.2c03400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J. F. Li J. J. Wang P. Yu J. Q. Angew. Chem., Int. Ed. 2019;58:18141–18145. doi: 10.1002/anie.201910395. [DOI] [PubMed] [Google Scholar]
- Zhao H. Xu X. Luo Z. Cao L. Li B. Li H. Xu L. Fan Q. Walsh P. J. Chem. Sci. 2019;10:10089–10096. doi: 10.1039/C9SC03672E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rej S. Chatani N. Angew. Chem., Int. Ed. 2019;58:8304–8329. doi: 10.1002/anie.201808159. [DOI] [PubMed] [Google Scholar]
- Shi B.-F. Zhou T. Zhou Y.-B. Li J.-Y. Qian P.-F. SynOpen. 2023;07:466–485. doi: 10.1055/a-2167-8298. [DOI] [Google Scholar]
- Sunny S. Karvembu R. Adv. Synth. Catal. 2021;363:4309–4331. doi: 10.1002/adsc.202100558. [DOI] [Google Scholar]
- Tian X. Pan A. Du Y. Zhang L. Qin C. Duan S. Yang L. Yang X. Adv. Synth. Catal. 2022;364:4421–4426. doi: 10.1002/adsc.202200965. [DOI] [Google Scholar]
- Wang C. Harms K. Meggers E. Angew. Chem., Int. Ed. 2016;55:13495–13498. doi: 10.1002/anie.201607305. [DOI] [PubMed] [Google Scholar]
- Kärkäs M. D. ACS Catal. 2017;7:4999–5022. doi: 10.1021/acscatal.7b01385. [DOI] [Google Scholar]
- Davies J. Morcillo S. P. Douglas J. J. Leonori D. Chem.–Eur. J. 2018;24:12154–12163. doi: 10.1002/chem.201801655. [DOI] [PubMed] [Google Scholar]
- Li Z. Wang Q. Zhu J. Angew. Chem., Int. Ed. 2018;57:13288–13292. doi: 10.1002/anie.201807623. [DOI] [PubMed] [Google Scholar]
- Nagib D. Stateman L. Nakafuku K. Synthesis. 2018;50:1569–1586. doi: 10.1055/s-0036-1591930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X. Wang Q. Zhu J. Angew. Chem., Int. Ed. 2019;58:2139–2143. doi: 10.1002/anie.201813356. [DOI] [PubMed] [Google Scholar]
- Guo W. Wang Q. Zhu J. Chem. Soc. Rev. 2021;50:7359–7377. doi: 10.1039/D0CS00774A. [DOI] [PubMed] [Google Scholar]
- Murray P. R. D. Cox J. H. Chiappini N. D. Roos C. B. McLoughlin E. A. Hejna B. G. Nguyen S. T. Ripberger H. H. Ganley J. M. Tsui E. Shin N. Y. Koronkiewicz B. Qiu G. Knowles R. R. Chem. Rev. 2021;122:2017–2291. doi: 10.1021/acs.chemrev.1c00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J. C. K. Rovis T. Nature. 2016;539:272–275. doi: 10.1038/nature19810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.-F. Chu J. C. K. Rovis T. J. Am. Chem. Soc. 2017;139:14897–14900. doi: 10.1021/jacs.7b09306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. Guo L. Yu S. Org. Lett. 2018;20:6255–6259. doi: 10.1021/acs.orglett.8b02737. [DOI] [PubMed] [Google Scholar]
- Chen H. Fan W. Yuan X.-A. Yu S. Nat. Commun. 2019;10:4743. doi: 10.1038/s41467-019-12722-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. Xiao H. Zhang B. Shen H. Zhu L. Li C. Angew. Chem., Int. Ed. 2019;58:2510–2513. doi: 10.1002/anie.201813425. [DOI] [PubMed] [Google Scholar]
- Thullen S. M. Treacy S. M. Rovis T. J. Am. Chem. Soc. 2019;141:14062–14067. doi: 10.1021/jacs.9b07014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B. Tambar U. K. ACS Catal. 2019;9:4627–4631. doi: 10.1021/acscatal.9b00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Stateman L. M. Nagib D. A. Chem. Sci. 2019;10:1207–1211. doi: 10.1039/C8SC04366C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.-H. Dong X.-Y. Du X.-Y. Gu Q.-S. Li Z.-L. Liu X.-Y. Nat. Commun. 2019;10:5689. doi: 10.1038/s41467-019-13705-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q. Peng Q. Chai H. Huo Y. Wang S. Xu Z. Nat. Commun. 2020;11:1463. doi: 10.1038/s41467-020-15167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L.-J. Xiong Z.-Q. Ouyang X.-H. Li Y. Song R.-J. Sun Q. Lu X. Li J.-H. J. Am. Chem. Soc. 2021;144:339–348. doi: 10.1021/jacs.1c10053. [DOI] [PubMed] [Google Scholar]
- Wang Y. Meng H. Li S. Shu W. ACS Catal. 2024;14:2402–2408. doi: 10.1021/acscatal.3c05701. [DOI] [Google Scholar]
- Moran D. Jacob R. Wood G. P. F. Coote M. L. Davies M. J. O'Hair R. A. J. Easton C. J. Radom L. Helv. Chim. Acta. 2006;89:2254–2272. doi: 10.1002/hlca.200690210. [DOI] [Google Scholar]
- Che C. Huang Q. Zheng H. Zhu G. Chem. Sci. 2016;7:4134–4139. doi: 10.1039/C5SC04980F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che C. Qian Z. Wu M. Zhao Y. Zhu G. J. Org. Chem. 2018;83:5665–5673. doi: 10.1021/acs.joc.8b00666. [DOI] [PubMed] [Google Scholar]
- Chen Y. Liu D. Zhang J. Synlett. 2020;32:356–361. doi: 10.1055/a-1300-3453. [DOI] [Google Scholar]
- Zhang J. Liu D. Liu S. Ge Y. Lan Y. Chen Y. iScience. 2020;23:100755. doi: 10.1016/j.isci.2019.100755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand A. W. Yin H. Xu L. Giacoboni J. Martin-Montero R. Romano C. Montgomery J. Martin R. ACS Catal. 2020;10:4671–4676. doi: 10.1021/acscatal.0c01318. [DOI] [Google Scholar]
- Zhong L.-J. Wang H.-Y. Ouyang X.-H. Li J.-H. An D.-L. Chem. Commun. 2020;56:8671–8674. doi: 10.1039/D0CC03619F. [DOI] [PubMed] [Google Scholar]
- Yue W.-J. Day C. S. Martin R. J. Am. Chem. Soc. 2021;143:6395–6400. doi: 10.1021/jacs.1c03126. [DOI] [PubMed] [Google Scholar]
- Lombardi L. Cerveri A. Giovanelli R. Castiñeira Reis M. Silva López C. Bertuzzi G. Bandini M. Angew. Chem., Int. Ed. 2022;61:e202211732. doi: 10.1002/anie.202211732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechab M. Mondal S. Bertrand M. P. Chem.–Eur. J. 2014;20:16034–16059. doi: 10.1002/chem.201403951. [DOI] [PubMed] [Google Scholar]
- Li M. Berritt S. Matuszewski L. Deng G. Pascual-Escudero A. Panetti G. B. Poznik M. Yang X. Chruma J. J. Walsh P. J. J. Am. Chem. Soc. 2017;139:16327–16333. doi: 10.1021/jacs.7b09394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. Gutierrez O. Berritt S. Pascual-Escudero A. Yeşilçimen A. Yang X. Adrio J. Huang G. Nakamaru-Ogiso E. Kozlowski M. C. Walsh P. J. Nat. Chem. 2017;9:997–1004. doi: 10.1038/nchem.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. Li M. Deng G. Wei W. Feng P. Zi Q. Li T. Zhang H. Yang X. Walsh P. J. Chem. Sci. 2020;11:7619–7625. doi: 10.1039/D0SC00031K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Liu Z. Tian X. Zi Y. Duan S. Fang Y. Chen W. Jing H. Yang L. Yang X. Org. Lett. 2021;23:1714–1719. doi: 10.1021/acs.orglett.1c00135. [DOI] [PubMed] [Google Scholar]
- Panetti G. B. Carroll P. J. Gau M. R. Manor B. C. Schelter E. J. Walsh P. J. Chem. Sci. 2021;12:4405–4410. doi: 10.1039/D0SC04822D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifert D. Studer A. Angew. Chem., Int. Ed. 2019;59:74–108. doi: 10.1002/anie.201903726. [DOI] [PubMed] [Google Scholar]
- Deng G. Duan S. Wang J. Chen Z. Liu T. Chen W. Zhang H. Yang X. Walsh P. J. Nat. Commun. 2021;12:3860. doi: 10.1038/s41467-021-24027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G. Li M. Yu K. Liu C. Liu Z. Duan S. Chen W. Yang X. Zhang H. Walsh P. J. Angew. Chem., Int. Ed. 2019;58:2826–2830. doi: 10.1002/anie.201812369. [DOI] [PubMed] [Google Scholar]
- Yu K. Li M. Deng G. Liu C. Wang J. Liu Z. Zhang H. Yang X. Walsh P. J. Adv. Synth. Catal. 2019;361:4354–4359. doi: 10.1002/adsc.201900497. [DOI] [Google Scholar]
- Wang J. Deng G. Liu C. Chen Z. Yu K. Chen W. Zhang H. Yang X. Adv. Synth. Catal. 2020;362:2268–2273. doi: 10.1002/adsc.201901553. [DOI] [Google Scholar]
- Duan S. Deng G. Zi Y. Wu X. Tian X. Liu Z. Li M. Zhang H. Yang X. Walsh P. J. Chem. Sci. 2021;12:6406–6412. doi: 10.1039/D1SC00972A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S. Zi Y. Wang L. Cong J. Chen W. Li M. Zhang H. Yang X. Walsh P. J. Chem. Sci. 2022;13:3740–3747. doi: 10.1039/D2SC00500J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. Li M. Gao G. Sanz-Vidal A. Zheng B. Walsh P. J. J. Org. Chem. 2022;87:8099–8103. doi: 10.1021/acs.joc.2c00767. [DOI] [PubMed] [Google Scholar]
- Duan S. Zi Y. Du Y. Cong J. Sun X. Jing H. Zhao J. Chen W. Yang X. Org. Lett. 2023;25:3687–3692. doi: 10.1021/acs.orglett.3c01121. [DOI] [PubMed] [Google Scholar]
- Deng G. Chen Z. Bai Y. Zhang L. Xia D. Duan S. Chen W. Zhang H. Walsh P. J. Yang X. Org. Lett. 2024;26:3855–3860. doi: 10.1021/acs.orglett.4c01014. [DOI] [PubMed] [Google Scholar]
- Zi Q. Li M. Cong J. Deng G. Duan S. Yin M. Chen W. Jing H. Yang X. Walsh P. J. Org. Lett. 2022;24:1786–1790. doi: 10.1021/acs.orglett.2c00140. [DOI] [PubMed] [Google Scholar]
- Jiang Y. Liu D. Rotella M. E. Deng G. Liu Z. Chen W. Zhang H. Kozlowski M. C. Walsh P. J. Yang X. J. Am. Chem. Soc. 2023;145:16045–16057. doi: 10.1021/jacs.3c04376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. Liu D. Zhang L. Qin C. Li H. Yang H. Walsh P. J. Yang X. Chem. Sci. 2024;15:2205–2210. doi: 10.1039/D3SC05210A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Zhou T. Xuan L. Lin Y. Li F. Wang H. Lyu J. Yan Q. Zhou H. Wang W. Chen F.-E. Org. Lett. 2023;25:8693–8699. doi: 10.1021/acs.orglett.3c03594. [DOI] [PubMed] [Google Scholar]
- Chen B. Chen Q. Liu Y. Chen J. Zhou X. Wang H. Yan Q. Wang W. Cai Z. Chen F.-E. Org. Lett. 2023;25:9124–9129. doi: 10.1021/acs.orglett.3c03523. [DOI] [PubMed] [Google Scholar]
- Doche F. Poisson T. Besset T. ACS Catal. 2023;13:14112–14120. doi: 10.1021/acscatal.3c03249. [DOI] [Google Scholar]
- Wu K. Wang L. Colón-Rodríguez S. Flechsig G. U. Wang T. Angew. Chem., Int. Ed. 2019;58:1774–1778. doi: 10.1002/anie.201811004. [DOI] [PubMed] [Google Scholar]
- Schmidt V. A. Quinn R. K. Brusoe A. T. Alexanian E. J. J. Am. Chem. Soc. 2014;136:14389–14392. doi: 10.1021/ja508469u. [DOI] [PubMed] [Google Scholar]
- Choi G. J. Zhu Q. Miller D. C. Gu C. J. Knowles R. R. Nature. 2016;539:268–271. doi: 10.1038/nature19811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morcillo S. P. Dauncey E. M. Kim J. H. Douglas J. J. Sheikh N. S. Leonori D. Angew. Chem., Int. Ed. 2018;57:12945–12949. doi: 10.1002/anie.201807941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L. Chen Y. Angew. Chem., Int. Ed. 2016;55:13312–13315. doi: 10.1002/anie.201607813. [DOI] [PubMed] [Google Scholar]
- Chu L. Ohta C. Zuo Z. MacMillan D. W. C. J. Am. Chem. Soc. 2014;136:10886–10889. doi: 10.1021/ja505964r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa K. Koike T. Akita M. Adv. Synth. Catal. 2014;356:2749–2755. doi: 10.1002/adsc.201400556. [DOI] [Google Scholar]
- Cheng W.-M. Shang R. Fu Y. ACS Catal. 2016;7:907–911. doi: 10.1021/acscatal.6b03215. [DOI] [Google Scholar]
- Qin T. Cornella J. Li C. Malins L. R. Edwards J. T. Kawamura S. Maxwell B. D. Eastgate M. D. Baran P. S. Science. 2016;6287:801–805. doi: 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmatsu K. Suzuki R. Furukawa Y. Sato M. Ooi T. ACS Catal. 2019;10:2627–2632. doi: 10.1021/acscatal.9b04491. [DOI] [Google Scholar]
- Yasu Y. Koike T. Akita M. Chem. Commun. 2012;48:5355–5357. doi: 10.1039/C2CC31748F. [DOI] [PubMed] [Google Scholar]
- Cai S.-H. Xie J.-H. Song S. Ye L. Feng C. Loh T.-P. ACS Catal. 2016;6:5571–5574. doi: 10.1021/acscatal.6b01230. [DOI] [Google Scholar]
- Zhao G. Mukherjee U. Zhou L. Wu Y. Yao W. Mauro J. N. Liu P. Ngai M.-Y. Chem. Sci. 2022;13:6276–6282. doi: 10.1039/D2SC01042A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svejstrup T. D. Ruffoni A. Juliá F. Aubert V. M. Leonori D. Angew. Chem., Int. Ed. 2017;56:14948–14952. doi: 10.1002/anie.201708693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. Svejstrup T. D. Fernandez Reina D. Sheikh N. S. Leonori D. J. Am. Chem. Soc. 2016;138:8092–8095. doi: 10.1021/jacs.6b04920. [DOI] [PubMed] [Google Scholar]
- Reina D. F. Dauncey E. M. Morcillo S. P. Svejstrup T. D. Popescu M. V. Douglas J. J. Sheikh N. S. Leonori D. Eur. J. Org Chem. 2017;2017:2108–2111. doi: 10.1002/ejoc.201601607. [DOI] [Google Scholar]
- Pandey G. Laha R. Angew. Chem., Int. Ed. 2015;54:14875–14879. doi: 10.1002/anie.201506990. [DOI] [PubMed] [Google Scholar]
- Zhang M. Duan Y. Li W. Xu P. Cheng J. Yu S. Zhu C. Org. Lett. 2016;18:5356–5359. doi: 10.1021/acs.orglett.6b02711. [DOI] [PubMed] [Google Scholar]
- Kim J. H. Ruffoni A. Al-Faiyz Y. S. S. Sheikh N. S. Leonori D. Angew. Chem., Int. Ed. 2020;59:8225–8231. doi: 10.1002/anie.202000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. CSD Commun. 2024 doi: 10.5517/ccdc.csd.cc2k0h14. [DOI] [Google Scholar]
- Khan I. Reed-Berendt B. G. Melen R. L. Morrill L. C. Angew. Chem., Int. Ed. 2018;57:12356–12359. doi: 10.1002/anie.201808800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner G. R. Free Radical Biol. Med. 1987;3:259–303. doi: 10.1016/S0891-5849(87)80033-3. [DOI] [PubMed] [Google Scholar]
- Das B. G. Chirila A. Tromp M. Reek J. N. H. de Bruin B. J. Am. Chem. Soc. 2016;138:8968–8975. doi: 10.1021/jacs.6b05434. [DOI] [PubMed] [Google Scholar]
- Cao H. Ma S. Feng Y. Guo Y. Jiao P. Chem. Commun. 2022;58:1780–1783. doi: 10.1039/D1CC06529G. [DOI] [PubMed] [Google Scholar]
- Seefeldt P. Villinger A. Brasholz M. Adv. Synth. Catal. 2024;366:24–30. doi: 10.1002/adsc.202300967. [DOI] [Google Scholar]
- Groendyke B. J. AbuSalim D. I. Cook S. P. J. Am. Chem. Soc. 2016;138:12771–12774. doi: 10.1021/jacs.6b08171. [DOI] [PubMed] [Google Scholar]
- Bao X. Wang Q. Zhu J. Nat. Commun. 2019;10:769. doi: 10.1038/s41467-019-08741-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Xuan L. Chen Q. Fan R. Zhao F. Dong J. Wang H. Yan Q. Zhou H. Chen F.-E. J. Am. Chem. Soc. 2024;146:6307–6316. doi: 10.1021/jacs.4c00023. [DOI] [PubMed] [Google Scholar]
- Becker P. Duhamel T. Stein C. J. Reiher M. Muñiz K. Angew. Chem., Int. Ed. 2017;56:8004–8008. doi: 10.1002/anie.201703611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L. Ren J. Zhou J. Wu X. Shao Z. Yang X. Qian D. Nat. Commun. 2022;13:6861. doi: 10.1038/s41467-022-34615-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avidan-Shlomovich S. Ghosh H. Szpilman A. M. ACS Catal. 2014;5:336–342. doi: 10.1021/cs501744e. [DOI] [Google Scholar]
- Davies J. Booth S. G. Essafi S. Dryfe R. A. W. Leonori D. Angew. Chem., Int. Ed. 2015;54:14017–14021. doi: 10.1002/anie.201507641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandar J. S. Pirnot M. T. Buchwald S. L. J. Am. Chem. Soc. 2015;137:14812–14818. doi: 10.1021/jacs.5b10219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M.-C. Shang R. Zhao B. Wang B. Fu Y. Science. 2019;363:1429–1434. doi: 10.1126/science.aav3200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental data, procedures for data analysis, and pertinent data sets are provided in the ESI.†