

Summary
The depletion of disruptive variation caused by purifying natural selection (constraint) has been widely used to investigate protein-coding genes underlying human disorders1–4, but attempts to assess constraint for non-protein-coding regions have proven more difficult. Here we aggregate, process, and release a dataset of 76,156 human genomes from the Genome Aggregation Database (gnomAD), the largest public open-access human genome allele frequency reference dataset, and use this dataset to build a genomic constraint map for the whole genome (called Gnocchi). We present a refined mutational model that incorporates local sequence context and regional genomic features to detect depletions of variation. As expected, the average constraint for protein-coding sequences is stronger than for non-coding regions. Within the non-coding genome, constrained regions are enriched for known regulatory elements and variants implicated in complex human diseases and traits, facilitating the triangulation of biological annotation, disease association, and natural selection to non-coding DNA analysis. More constrained regulatory elements tend to regulate more constrained protein-coding genes, which in turn suggests that non-coding constraint can aid the identification of constrained genes that are as yet unrecognized by current gene constraint metrics. We demonstrate that this genome-wide constraint map improves the identification and interpretation of functional human genetic variation.
The expansion in the scale of human whole-genome or exome sequencing data has allowed characterization of the patterns of variation in human genes. With these data it is possible to directly assess the strength of negative selection on loss-of-function (LoF) and missense variation by modeling “constraint,” the depletion of variation in a gene compared to an expectation conditioned on that gene’s mutability. Using coding variant data from sequencing thousands to hundreds of thousands of humans5, we and others previously developed constraint metrics that classify each protein-coding gene along a spectrum of LoF/missense intolerance5–7, providing a valuable resource for studying the functional significance of human genes1–4. Although of outsized biological importance, protein-coding regions comprise less than 2% of the human genome, and the vast non-coding genome has been much less characterized, even though the importance of non-coding variation in human complex diseases has been long recognized8–12.
Several challenges arise when extending the gene constraint model to the non-coding space. First, the sample size of human whole-genome reference data has been relatively small compared to the exome, limiting the power of detecting depletions of variation at a fine scale. Second, in coding regions, the gene model enables accurate prediction of the effect of specific variants on amino acid translation; such nucleotide-specific models of the consequences of basepair changes are not available in non-coding regions. Third, there is a strong expectation from Mendelian genetics and existing constraint analyses that the coding regions, while a small fraction of the genome, are grossly overrepresented for rare and common disease mutations under selection. Fourth, the mutation rate in non-coding regions is highly heterogeneous and can be affected not only by local sequence context as commonly modeled in gene constraint metrics but also by a variety of genomic features at larger scales13,14.
Current methods attempting to evaluate non-coding constraint can be broadly divided into three categories: 1) context-dependent mutational models that assess the deviation of observed variation from an expectation based on the sequence composition of k-mers (e.g., Orion15, CDTS16, DR17); 2) machine-learning classifiers that are trained to differentiate between disease-associated variants and benign variants (e.g., CADD18, GWAVA19, JARVIS20); and 3) phylogenetic conservation scores that use comparative genomics data to infer evolutionary constraint (e.g., phastCons21, phyloP22). While all these methods aid in our understanding of the non-coding genome, each suffer from limitations/biases, respectively as 1) overlooking the influence of regional genomic features beyond the scale of flanking nucleotides on mutation rate; 2) a strong dependence on the availability of well-characterized functional mutations as training data; and 3) compromised power to detect regions that have only recently been under selection in the human lineage and may have a functional impact on human-specific traits or diseases.
Here we present a genome-wide map of human constraint (called Gnocchi: Genomic NOn-Coding Constraint of HaploInsufficient variation), generated from a high-quality set of variant calls from 76,156 whole-genome sequences (gnomAD v3.1.2 https://gnomad.broadinstitute.org). We describe an improved model of human mutation rates that jointly analyzes local sequence context and regional genomic features and quantifies the depletion of variation in tiled windows across the entire genome. Incorporating constraint evidence from functional elements linked to genes can enhance the identification of genes under strong constraint and aid in the functional interpretation of non-coding regions. Our study aims to depict a genome-wide view of how natural selection shapes patterns of human genetic variation and identify which functional genomic elements likely harbor variation with potential clinical significance.
Aggregating 76,156 whole genomes
We aggregated, reprocessed, and performed joint variant-calling on 153,030 whole genomes mapped to human genome reference build GRCh38, of which 76,156 samples were retained as high-quality sequences from unrelated individuals, without known severe pediatric disease, and with appropriate consent and data use permissions for the sharing of aggregate variant data (Supplementary Fig. 1–5 and Supplementary Table 1–3). Among these samples, 36,811 (48.3%) are of non-European ancestry, including 20,744 individuals with African ancestries and 7,647 individuals with admixed Amerindigineous ancestries. After stringent quality control, we discovered a set of 644,267,978 high-confidence short nuclear variants (single nucleotide/indel variants; gnomAD v3.1.2), of which 390,393,900 low-frequency (allele frequency [AF]≤0.1%), high-quality single nucleotide variants were used for building the genome-wide constraint map. These correspond to approximately one variant every 4.9 bp (one low-frequency variant every 8 bp) of the genome, providing a high density of variation.
Gnocchi quantifies genomic constraint
To construct a genome-wide mutational constraint map, we divided the genome into continuous non-overlapping 1kb windows, and quantified constraint for each window by comparing the expected and the observed variation in our gnomAD dataset. Here, we implemented a refined mutational model, which incorporates trinucleotide sequence context, base-level methylation, and regional genomic features to predict expected levels of variation under neutrality. In brief, we estimated the relative mutability for each single nucleotide substitution with one base of adjacent nucleotide context (e.g., ACG -> ATG), with adjustment for the effect of methylation on mutation rate at CpG sites, which become saturated for mutation at sample sizes above ~10K genomes23 (Extended Fig. 1a,b and Supplementary Fig. 6; Methods). Meanwhile, we adjusted the effects of regional genomic features for each trinucleotide mutation rate based on the occurrence of de novo mutations (N=413,304 previously detected in family-based whole-genome sequencing studies24,25; Extended Fig. 1c), and then applied it to establish the expected number of variants per 1kb across the entire genome (Methods).
We quantified the deviation from expectation for each 1kb window using a Z score7 - hereinafter referred to as “Gnocchi” (Methods; Extended Fig. 1d,e) - which was centered around zero for non-coding regions (median=0.08), and was significantly higher (more constrained) for windows containing any protein-coding sequences (median=1.47, Wilcoxon P<10−200; Fig. 1a). Gnocchi is positively correlated with the percentage of coding bases in a window and presented a substantial shift towards higher constraint for exonic sequences from directly concatenating coding exons into 1kb windows (median=3.17; Extended Fig. 2a–c). About 3.12% and 0.05% of the non-coding windows exhibited constraint as strong as the 50th and 90th percentile of exonic regions (Extended Fig. 2d). Comparing Gnocchi against the adjusted proportion of singletons (APS) score, a measure of constraint developed for structural variation (SV)26, we found a significant correlation (linear regression beta=0.01, P=4.3×10−65, Fig. 1b; Methods), providing an internal validation of our approach.
Fig. 1:
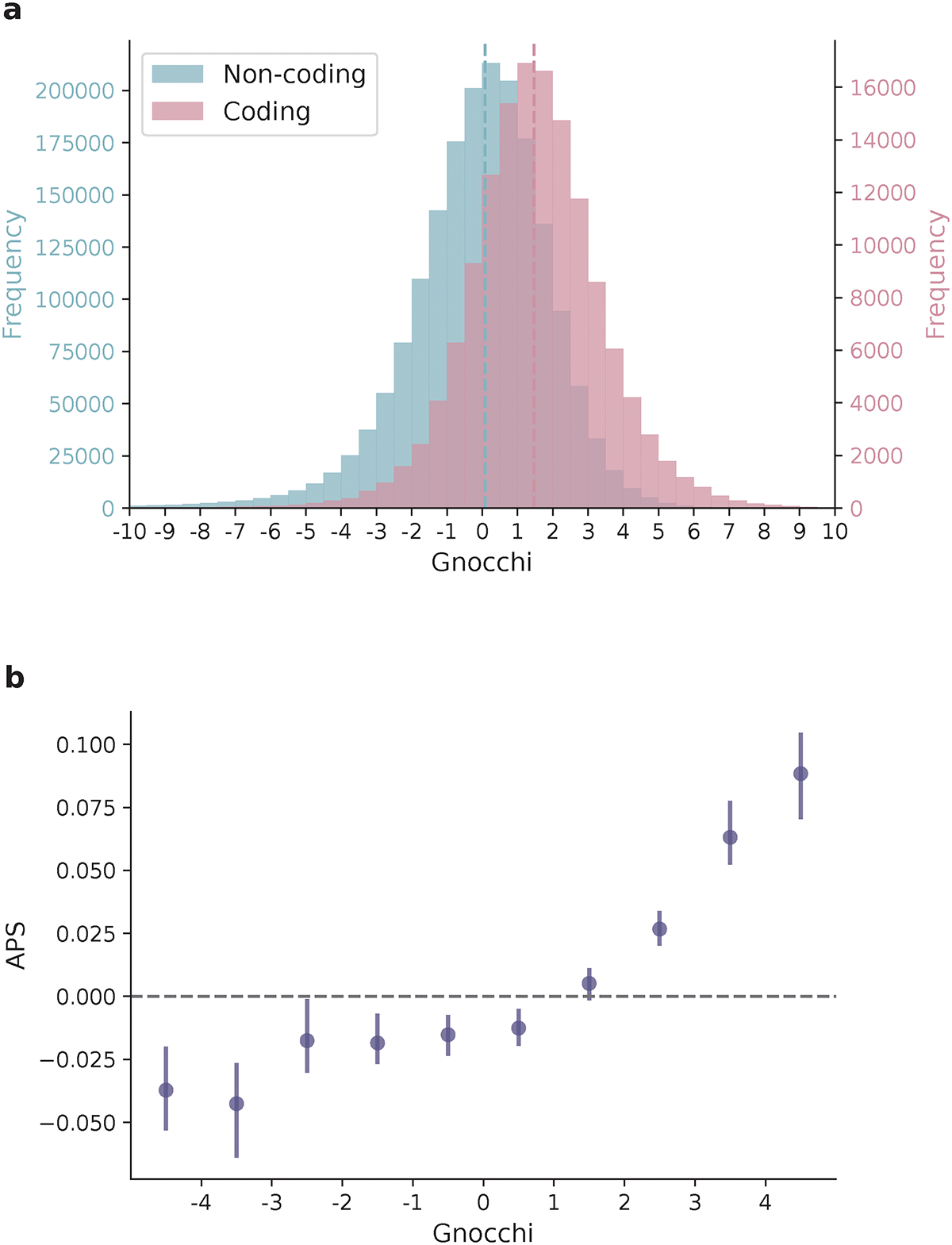
Distribution of Gnocchi scores across the genome. a, Histograms of Gnocchi scores for 1,984,900 1kb windows across the human autosomes. Windows overlapping coding regions (N=141,341 with ≥ 1bp coding sequence; red) overall exhibit a higher Gnocchi score (stronger negative selection) than windows that are exclusively non-coding (N=1,843,559; blue); dashed lines indicate the medians. b, The correlation between Gnocchi score and the adjusted proportion of singletons (APS) score developed for structural variation (SV) constraint. A collection of 116,184 autosomal SVs were assessed using Gnocchi by assigning each SV the highest Gnocchi score among all overlapping 1kb windows, which shows a significant positive correlation with the SV constraint metric APS. Error bars indicate 100-fold bootstrapped 95% confidence intervals of the mean values.
Gnocchi highlights non-coding function
To further validate the Gnocchi metric and investigate the functional relevance of non-coding regions under selection, we examined the correlation between Gnocchi and several annotations of functional non-coding sequences (Fig. 2a). First, we found that candidate cis-regulatory elements (cCREs, derived from ENCODE27 integrated DNase- and ChIP-seq data) are significantly enriched in the most constrained percentile of the genome (Gnocchi≥4, OR=2.77 compared to the genome-wide average, Fisher’s exact P<10−200); cCREs with a promoter-like signature (cCRE-PLS) presented the strongest enrichment (OR=7.28), followed by elements with a proximal/distal enhancer-like signature (pELS OR=4.35, dELS OR=2.14), and as a negative control, elements bound by CTCF but not associated with a regulatory signature showed no enrichment (CTCF-only OR=0.82). These patterns indicate that a large fraction of the constrained non-coding regions may serve a regulatory role, in line with previous findings15,16,20. Similarly, significant enrichment was found for an independent set of active, in vivo-transcribed enhancers (identified by FANTOM CAGE analyses28; OR=3.58) and super enhancers29 (OR=3.41), which are groups of enhancers in close genomic proximity regulating genes important for cell type specification30. By aggregating the regulatory annotations, we estimated that ~10.4% and ~6.3% of promoters and enhancers, respectively, are under selection as strong as the average constraint for coding exons (Extended Fig. 3a; Methods). A much higher proportion, 22.2%, was found for sequences encoding microRNAs (miRNAs), which are increasingly recognized as key mediators in various developmental and physiological processes31. In contrast, only 3.7% of long non-coding RNAs (lncRNAs) exhibited such strong constraint, similar to that of non-coding regions overall (3.1%; Extended Fig. 2d and 3b).
Fig. 2:
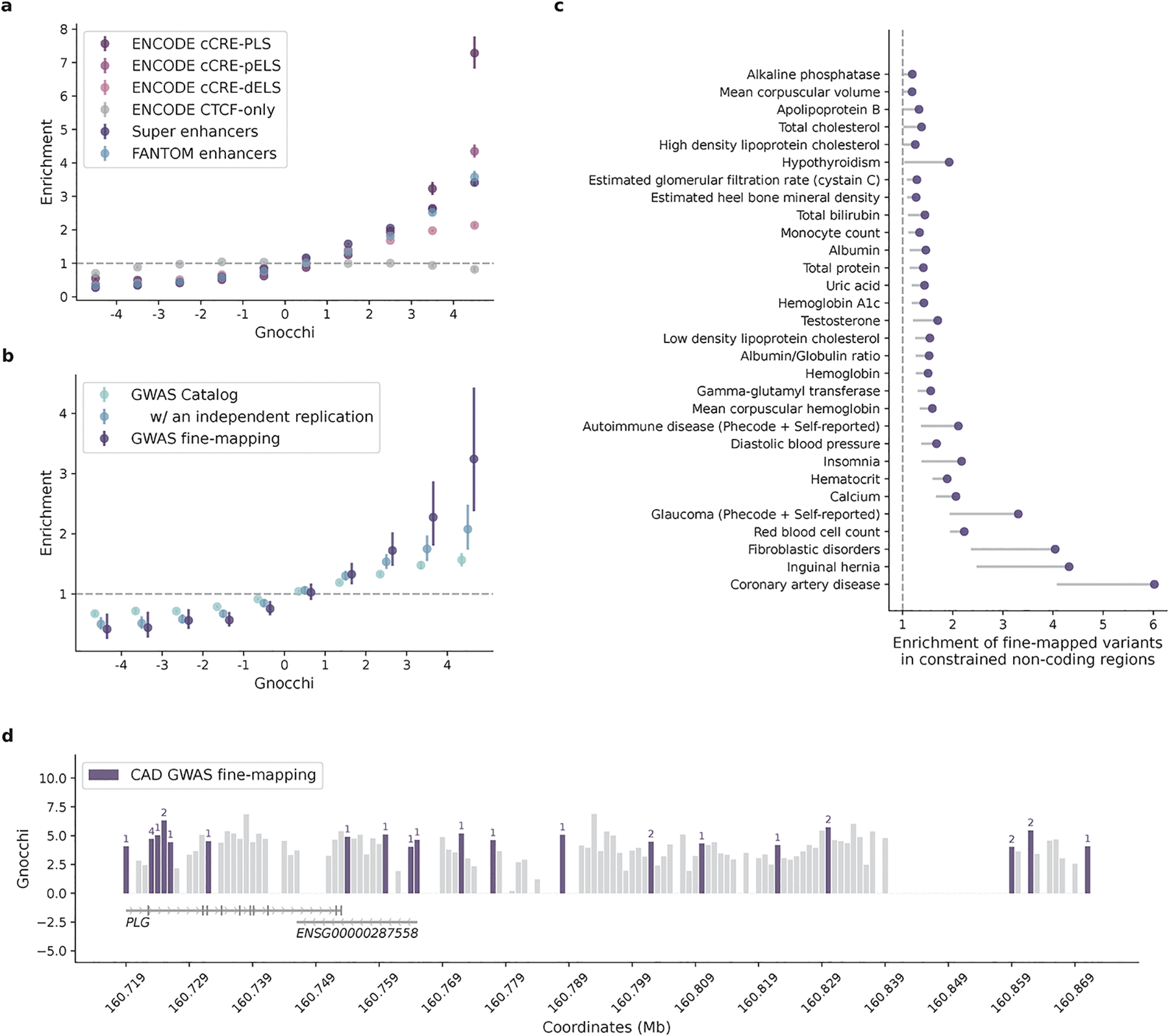
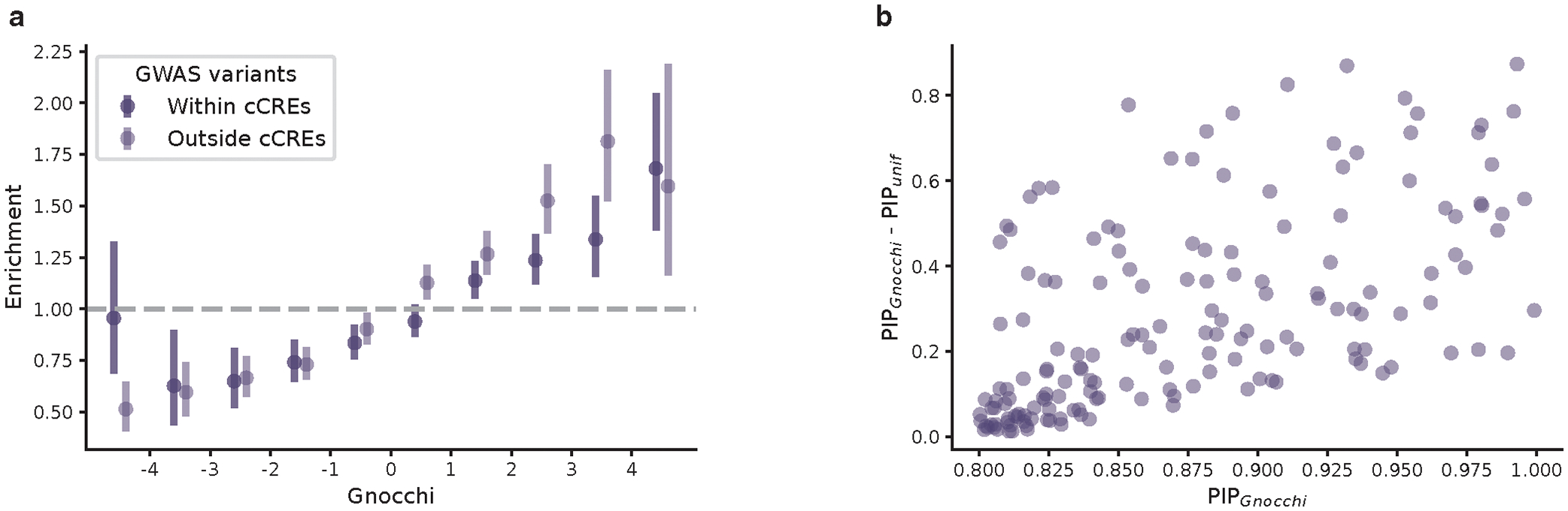
Correlation between Gnocchi and functional non-coding annotations. a,b, Distributions of candidate regulatory elements (a) and GWAS variants (b) along the spectrum of Gnocchi in non-coding regions. Enrichment was evaluated by comparing the proportion of non-coding 1kb windows, binned by Gnocchi, that overlap with a given functional annotation to the genome-wide average. Error bars indicate 95% confidence intervals of the odds ratios. cCRE, candidate cis-regulatory element: N=34,803 with a promoter-like signature (PLS), N=141,830 with a proximal enhancer-like signature (pELS), N=667,599 with a distal enhancer-like signature (dELS), N=56,766 bound by CTCF without a regulatory signature (CTCF-only); Super enhancers: N=331,601; FANTOM enhancers: N=63,285; GWAS Catalog: N=111,308 variants with an association P ≤5.0×10−8, N=9,229 with an independent replication; GWAS fine-mapping: N=2,191 variants fine-mapped with a posterior inclusion probability of causality≥0.9. See Methods for details on data collection. c, Enrichment of fine-mapped variants in constrained non-coding regions (Gnocchi≥4). Credible set (CS)-trat pairs with a significant enrichment are shown, ordered by the lower bound of 95% confidence interval; only lower bounds are shown for presentation purposes. d, The distribution of variants fine-mapped for coronary artery disease (CAD) in constrained regions (Gnocchi≥4) of PLG. Each bar shows the Gnocchi score of a 1kb window (gaps indicate windows removed by quality filters); windows containing fine-mapped variants are colored by purple, and the number of variants in each window is annotated on top of the bar correspondingly. Ten variants are located within PLG introns, four are mapped to the antisense gene of PLG (ENSG00000287558), and 14 reside in the downstream intergenic regions.
We next examined the distribution of putatively functional non-coding variants on the constraint spectrum. There was significant enrichment for non-coding variants implicated by genome-wide association studies (GWAS) in the constrained end of the genome: 837/19,471 constrained windows [Gnocchi≥4] overlapped with GWAS Catalog32 annotations (OR=1.57 compared to the genome-wide average of 51,430/1,843,559, Fisher’s exact P=2.5×10−32, Fig. 2b; Methods). The enrichment became stronger when restricted to the subset of variants that had been replicated by an independent study (OR=2.08, P=4.1×10−13). Moreover, further strong signals were found for likely causal GWAS variants fine-mapped for 148 complex diseases and traits in large-scale biobanks33 (OR=3.24, P=3.0×10−10; Methods). Across the 95% credible set (CS)-trait pairs, strong enrichment was predominantly seen in disease phenotypes, including coronary artery disease (CAD), inguinal hernia, fibroblastic disorders, and glaucoma (ORs 3.31–6.02, Fig. 2c; Methods). In the 95% CS of CAD, for instance, the highest Gnocchi score was found for rs1897107 and rs1897109 (both within the same genomic window chr6:160725000–160726000, Gnocchi=6.32); high constraint (Gnocchi≥4) was also found for 26 variants from the same CS (totaling 28/52), which together spanned a ~153 kb sequence downstream of the gene PLG (Fig. 2d). PLG encodes the plasminogen protein that circulates in blood plasma and is converted to plasmin to dissolve the fibrin of blood clots. While dysregulation of the PLG-plasmin system has been frequently associated with CAD34–39, no specific variants in PLG have been implicated. Our results prioritized a set of non-coding variants in highly constrained regions of PLG, which adds quantitative evidence to the implication of PLG in CAD and may help direct or prioritize follow-up functional experiments.
Collectively, these results demonstrated a significant positive correlation between constraint and functional non-coding annotations, illustrating the utility of Gnocchi in characterizing non-coding regions. Yet, we suggest that Gnocchi provides additional information to existing annotations For instance, prioritizing ENCODE cCREs by Gnocchi revealed increasingly stronger GWAS enrichment in the more constrained cCREs (Extended Fig. 4a), and constrained regions outside cCREs also captured significant signals, reflecting the value of Gnocchi independent of regulatory annotations. Moreover, besides prioritizing existing GWAS results, Gnocchi can be used as a prior for statistical fine-mapping. Using UK Biobank (UKBB) traits as examples, incorporating Gnocchi into the functionally informed fine-mapping model40 predicted ~13K variant-trait pairs to have an increased posterior inclusion probability of causality (ΔPIP≥0.01), in which 164 likely causal associations were newly identified at PIP≥0.8 (Extended Fig. 4b; Methods). While only functional tests can ultimately validate the underlying causality, our constraint map presents a valuable resource for expanding or refining the catalog of functional non-coding variants in the human genome.
Gnocchi versus other non-coding metrics
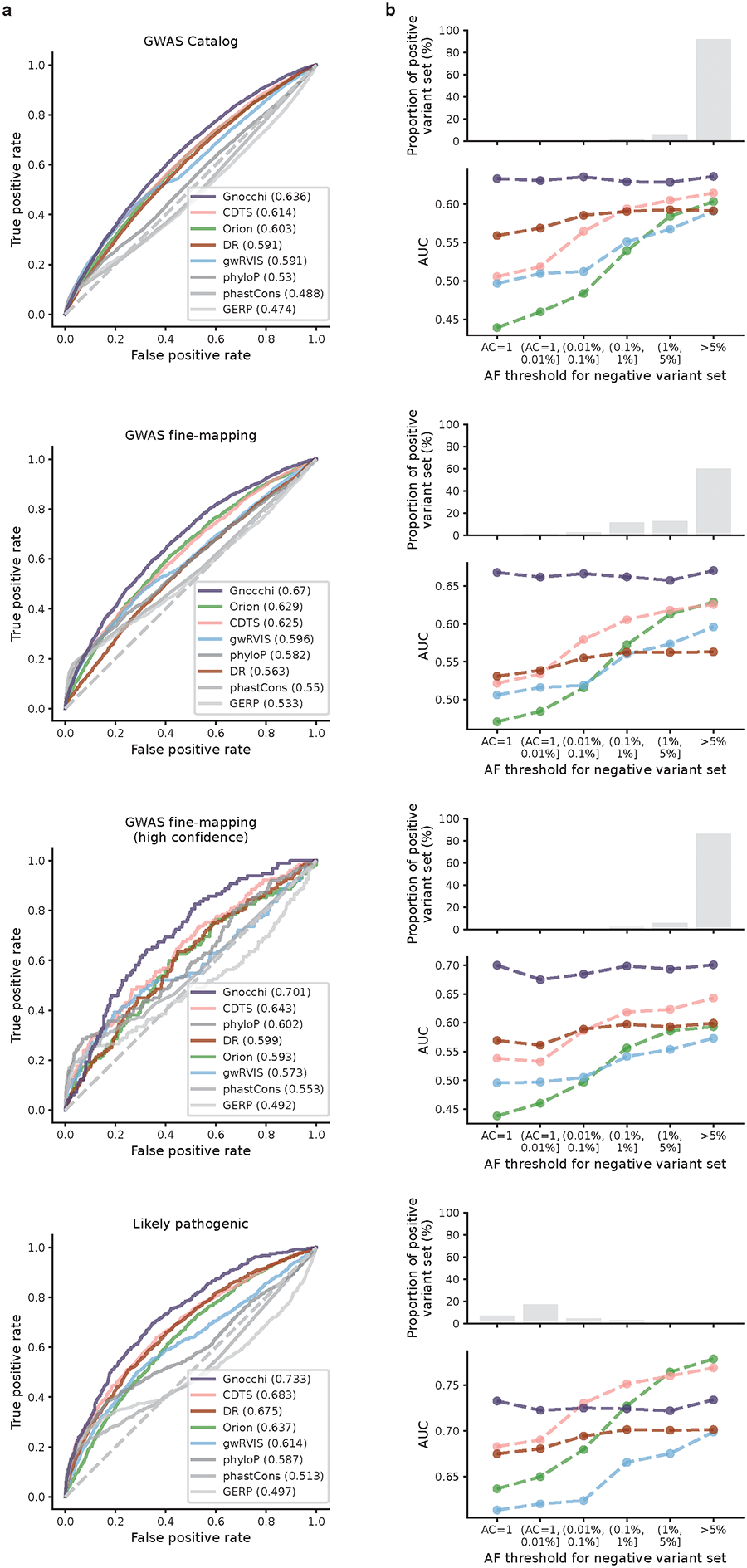
To benchmark the performance of Gnocchi in prioritizing non-coding variants, we extended the analyses of GWAS variants to compare it with other population genetics-based constraint metrics (Orion15, CDTS16, gwRVIS20, and DR17). Specifically, we assessed the performance of different metrics in identifying putative functional non-coding variants – as aforementioned, a) GWAS Catalog32 variants (N=9,229 with an independent replication); b) GWAS fine-mapping33 variants (N=2,191), and additionally, c) a subset of high-confidence causal variants from b (N=140); and d) likely pathogenic Mendelian variants (N=1,026 from ClinVar41) and the Human Gene Mutation Database (HGMD)42 – against background variants in the population with a similar allele frequency (hereafter referred to as “positive” and “negative” variant set, respectively; Methods). Overall, Gnocchi achieved the highest performance across all comparisons, as measured by the area under curve (AUC) statistic (Fig. 3a,b and Extended Fig. 5). The performance was also more stable than others when varying the allele frequency threshold for the negative variant set (Extended Fig. 5). This may be due to other metrics being informed by the site frequency spectrum, which made the classification performance sensitive to differences in allele frequency between the positive and negative variants. We also showed that our performance was robust to the artificial break of genomic windows (non-overlapping 1kb) by reconstructing Gnocchi scores in a sliding-window (1kb stepped by 100bp) approach as adopted by other metrics (Extended Fig. 6).
Fig. 3:
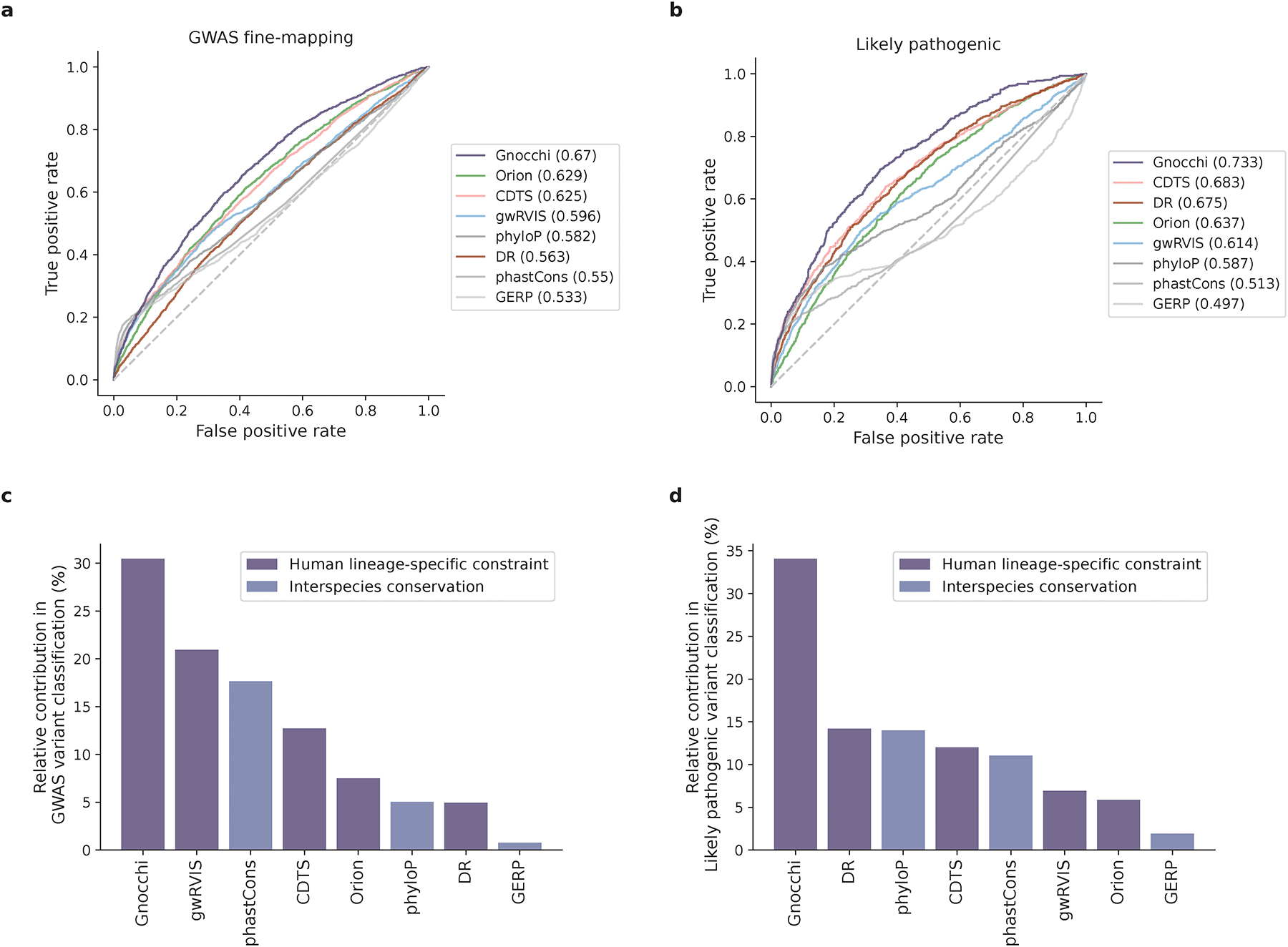
Performance of Gnocchi and other predictive metrics in prioritizing non-coding variants. a,b, Receiver operating characteristic (ROC) curves of Gnocchi and other seven metrics in classifying putative functional non-coding variants – 2,191 GWAS fine-mapping variants (a) and 1,026 likely pathogenic variants (b) – against background variants in the population. The performance of each metric was measured and ranked by the area under curve (AUC) statistic. c,d, The relative contribution of different metrics in classifying GWAS variants (b) and likely pathogenic variants (c). The eight metrics were modeled as eight independent predictors for the classification, and the relative contribution of one predictor over another was evaluated by estimating their additional contributions across all subset models.
Extending the comparison to include phylogeny-based conservation scores (phyloP22, phastCons21, and GERP43) revealed relatively low performance compared to the population genetics-based constraint metrics (Fig. 3a,b). The conservation scores were weakly correlated with constraint (Spearman’s rank correlation coefficient 0.017–0.19, Extended Fig. 7), suggesting that intraspecies (human lineage-specific) constrained regions complement, rather than reflect a subset of, regions that are conserved across species. Each individual metric also contributed to the classification when modeled as independent predictive variables (Fig. 3c,d; Methods), reinforcing the complementary nature of different approaches. Variants that were uniquely captured by Gnocchi, for instance, tended to be in regions with high recombination rates (3.45-fold the rest of the positive variant set) and high DNA methylation (2.74-fold; Methods), both associated with an increased mutation rate that had been adjusted in our refined mutational model. To further illustrate this improvement, we rebuilt our constraint model from solely the local sequence context, i.e., without adjustment on mutation rate by regional genomic features, and confirmed that Gnocchi outperformed such metrics (Extended Fig. 6). Altogether, we demonstrate that Gnocchi is an effective metric for identifying functional variants in the non-coding genome; at the same time, we suggest that a combination of different metrics is likely to provide the most informative results for prioritizing functional variation.
Gnocchi prioritizes copy number variants
Besides single nucleotide variants (SNVs) that have been extensively studied in GWAS, copy number variants (CNVs) causing dosage alterations (deletions/loss or duplications/gain) of DNA represent another important class of variation for contributing variability in risk for human disease44–49. Yet, unlike SNVs, CNVs can be large and determining the “minimal critical region”50 with a pathogenic effect has been a major challenge. Although CNVs primarily affect non-coding sequences, the most commonly studied mechanism is still the dosage alteration of overlapping protein-coding genes51. Using our genome-wide constraint map, we explored the possibility that constrained non-coding regions are also sensitive to a dosage effect, which may underlie the pathogenicity of corresponding CNVs.
We surveyed a collection of ~100K CNVs from a genome-wide CNV morbidity map of developmental delay and congenital birth defects52,53. There was a substantial excess of CNVs that affected constrained non-coding regions (Gnocchi≥4) among individuals with developmental disorders (DD cases) in comparison to healthy controls (42.6% versus 12.5%, OR=5.21, Fisher’s exact P<10−200, Fig. 4a; Methods). Moreover, of the 19 loci that had been previously identified as pathogenic52, all but one (94.7%) affected constrained non-coding regions; the high incidence was recapitulated in a curated set of ~4K putative pathogenic CNVs (85.5% in ClinVar41, Fig. 4a). Importantly, the case-control enrichment remained significant, albeit attenuated, after adjusting for the size and gene content of each CNV and when being tested in the subset of CNVs that are exclusively non-coding (Fig. 4b; Methods). Non-coding constraint presented high association with DD CNVs conditioning on gene constraint (log[OR]=1.06, logistic regression P<10−100), lending support to the possibility that dosage alteration of constrained non-coding regions may be an alternative explanation for the mechanism of CNVs underlying DDs.
Fig. 4:
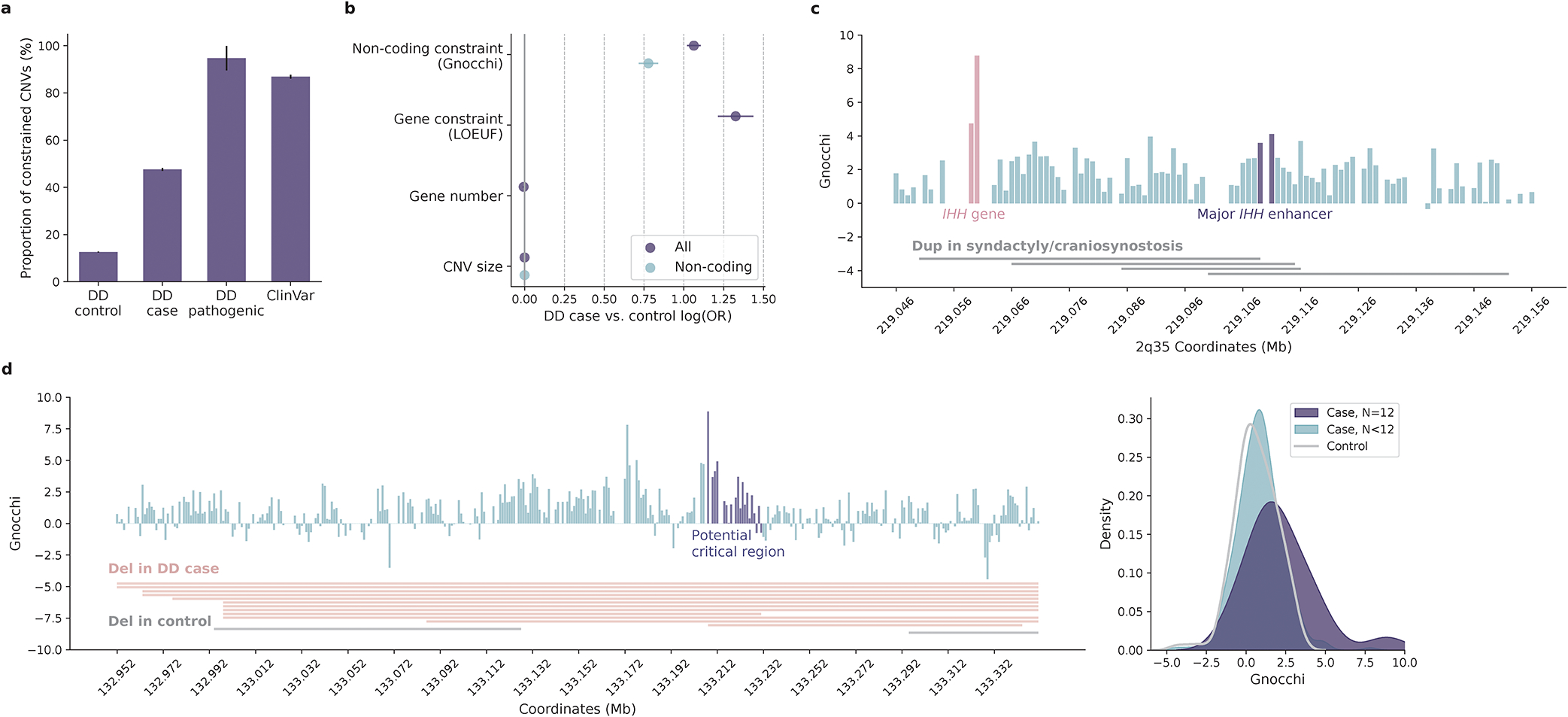
Contribution of non-coding constraint in evaluating copy number variants (CNVs). a, Proportions of constrained CNVs (Gnocchi≥4) identified in individuals with developmental delay (DD cases) versus healthy controls. Constrained CNVs are more common in DD cases than controls (7,239/17,004=42.6% versus 10,403/83,526=12.5%) and are most frequent for CNVs previously implicated as pathogenic (18/19=94.7% by DD and 3,433/4,014=85.5% by ClinVar). Error bars indicate standard errors of the proportions. b, Contribution of non-coding constraint to predicting CNVs in DD cases versus controls. Non-coding constraint remains a significant predictor for the case/control status of CNVs after adjusting for gene constraint (LOEUF score), gene number, and size of CNVs (Ncase=17,004, Ncontrol=83,526; purple), as well as being tested in the subset of non-coding CNVs (Ncase=8,702, Ncontrol=66,795; blue). Error bars indicate 95% confidence intervals of the log odds ratios. c, CNVs at the IHH locus associated with synpolydactyly and craniosynostosis. The four implicated duplications (grey horizontal bars) span a ~102kb sequence upstream of IHH. Each vertical bar shows the Gnocchi score of a 1kb window within the locus, with the highest score overlapping the IHH gene (red) and the highest non-coding score overlapping the major IHH enhancers (purple); gaps indicate windows removed by quality filters. d, Non-coding CNVs with the highest Gnocchi score identified in DD cases. The highest-scored window is located within the potential “critical region” (purple vertical bars) shared by 12 DD deletions (red horizontal bars; grey indicates two deletions observed in controls). The critical region overall, has a significantly higher Gnocchi score than the other regions affected by DD or control deletions, as shown in the kernel density estimate (KDE) plot on the right.
One known example of pathogenic non-coding dosage alteration is the duplication of IHH regulatory domain in synpolydactyly and craniosynostosis54–56. The four implicated duplications covered a ~102kb sequence upstream of IHH, with a ~10kb overlapping region (“critical region”50; Fig. 4c). The region contained no genes but exhibited high levels of constraint (median Gnocchi=2.52, Wilcoxon P=1.3×10−3 compared to the rest of the genome). The most constrained window (chr2:219111000–219112000, Gnocchi=4.12) overlapped with the major enhancer of IHH, the duplication of which has been shown to result in dosage-dependent IHH misexpression and consequently syndactyly and malformation of the skull56. This result highlights a potential use of the Gnocchi metric to prioritize non-coding regions within large CNVs. As a further illustration, we examined a set of non-coding CNVs that had the highest Gnocchi score among the DD cases. The most constrained genomic window (chr11:133208000–133209000, Gnocchi=8.87) was affected by 12 deletions spanning a ~400kb non-coding sequence (Fig. 4d). While of varying size, the deletions shared a common region of ~20kb (potential “critical region”), which encompassed the most constrained window and overall, showed a significantly higher constraint than the other affected regions (median Gnocchi=1.63 versus 0.84, Wilcoxon P=1.6×10−3; Fig. 4d). In addition, the ~400kb sequence also harbored two deletions from healthy controls, which interestingly, overlapped with the two lowest Gnocchi scores within the region and were significantly less constrained than those from DD cases (median Gnocchi=1.07 versus 0.62, Wilcoxon P=4.74×10−4). These findings suggest that Gnocchi can be a useful indicator of critical regions affected by large CNVs, facilitating the interpretation of non-coding risk factors in CNV disease association studies.
Gnocchi informs gene function
Given the significant role of non-coding regions in gene regulation, it is natural to expect that more constrained regulatory elements would regulate more constrained genes. To test this, we analyzed the constraint for enhancers that had been linked to specific genes57 (Methods). More constrained non-coding regions were more frequently linked to regulating a gene (Fig. 5a), and as expected, enhancers linked to constrained genes (predicted by loss-of-function observed/expected upper bound fraction [LOEUF]5, or curated disease genes from58–60; Methods) were significantly more constrained than those linked to presumably less constrained genes (median Gnocchi=2.71 versus 1.99, Wilcoxon P=1.3×10−26, Fig. 5b; Methods), thus supporting a correlated constraint between genes and their regulatory elements.
Fig. 5:
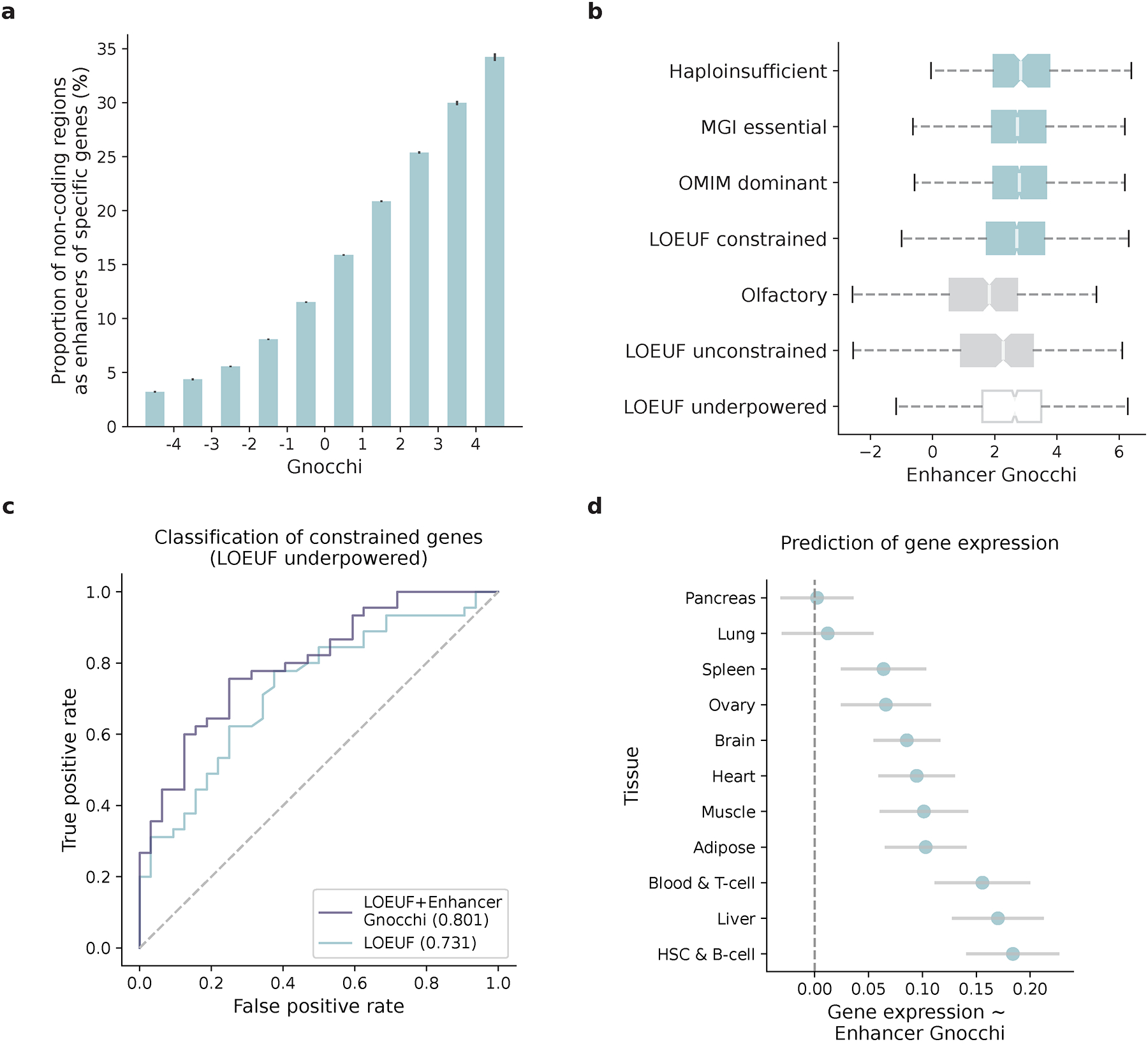
Correlation of constraint between non-coding regulatory elements and protein-coding genes. a, The proportion of non-coding 1kb windows overlapping with enhancers that were predicted to regulate specific genes, as a function of their Gnocchi scores. More constrained non-coding regions are more frequently linked to a gene (left to right: N=2,022/62,894, 2,743/62,653, 7,475/134,279, 20,383/252,354, 43,414/376,829, 66,343/417,743, 65,343/313,110, 38,785/152,787, 15,417/51,439, 6,663/19,471). Error bars indicate standard errors of the proportions. b, Comparison of the Gnocchi scores of enhancers linked to constrained and unconstrained genes. Enhancers of established sets of constrained genes (four blue boxes: N=189 haploinsufficient genes, N=2,454 essential genes, N=1,771 autosomal dominant disease genes, N=1,920 LOEUF-predicted constrained genes) are more constrained than enhancers of presumably less constrained genes (two grey boxes: N=356 olfactory receptor genes, N=189 LOEUF-predicted unconstrained genes). Enhancers of genes that are underpowered for gene constraint detection (“LOEUF underpowered”, N=1,117) present a higher constraint than those powered yet unconstrained genes (“LOUEF unconstrained”). The box plots show the distribution of Gnocchi scores of enhancers linked to different gene sets, denoting the median, quartiles and range (excepting outliers). c, Improvement of incorporating enhancer constraint into LOEUF in prioritizing underpowered genes. ROC curves and AUCs show the performance of two logistic regression models using LOEUF (blue) and LOEUF+enhancer Gnocchi score (purple) as independent predictive variables to classify constrained and unconstrained genes, tested on a set of 77 underpowered genes. d, Contribution of enhancer constraint to predicting gene expression in specific tissue types. The x-axis shows the linear regression coefficient of tissue-specific enhancer Gnocchi score predicting the expression level of target genes in matched tissue types (NHSC&B-cell=11,970, NBrain=11,555, NHeart=10,759, NPancreas=10,572, NBlood&T-cell=10,403, NMuscle=10,380, NAdipose=9,316, NLiver=8,838, NSpleen=8,308, NOvary=7,926, NLung=7,499), conditioning on gene constraint (LOEUF score). Error bars indicate 95% confidence intervals of the coefficient estimates.
On the other hand, a particularly interesting set of associations are the links between constrained enhancers and the “unconstrained” genes predicted by LOEUF, because these links may reflect functional significance of the “unconstrained” genes that had been previously unrecognized. The lack of predicted gene constraint can be explained by the design of LOEUF as a measure of intolerance to rare LoF variation, where small genes with few expected LoF variants are likely underpowered. Indeed, stratifying genes by the number of expected LoF variants showed a significantly higher enhancer constraint for genes that were underpowered (≤5 expected LoF variants)5 compared to genes that were sufficiently powered while scored as unconstrained (median Gnocchi=2.64 versus 2.27, Wilcoxon P=9.8×10−4, Fig. 5a). This suggests that certain underpowered genes may be functionally important but were not recognized in gene constraint evaluation. For instance, ASCL2, a basic helix-loop-helix (bHLH) transcription factor, had only 0.57 expected LoFs (versus 0 observed) across >125K exomes5; although being depleted for LoF variation, the absolute difference was too small to obtain a precise estimate of LoF intolerance. Yet, we found ASCL2 had a highly constrained enhancer (Gnocchi=5.58), located ~16kb upstream of the gene, where >40% of the expected variants were depleted (188.6 expected versus 112 observed, chr11:2286000–2287000). The same genomic window also contained an eQTL chr11:2286192:G>T that was predicted to be significantly associated with ASCL2 expression61; elevated ASCL2 expression has been implicated in the development and progression of several human cancers62–64. This example highlights the value of non-coding constraint – as a complementary metric to gene constraint – for identifying functionally important genes.
A practical implementation of this finding is to integrate the constraint of regulatory elements into the modeling of gene constraint, which essentially gains power from extending the functional unit of a gene to encompass its regulatory components. As a proof-of-principle, we tested whether adding the Gnocchi score of enhancer to LOEUF improves the prioritization of underpowered genes. The enhancer Gnocchi score was found a significant predictor of constrained genes (logistic regression P=7.4×10−11 conditioning on LOEUF) and improved the performance of LOEUF in identifying constrained genes that were underpowered (AUC = 0.80 versus 0.73, bootstrap P=0.03, Fig. 5b; Methods). Moreover, such approaches would allow incorporation of tissue/cell-type specific information into gene constraint modeling given the diverse range of epigenomic data. We explored this by testing whether the constraint of tissue-specific enhancers is predictive of tissue-specific gene expression (as a proxy for tissue-specific gene function). The enhancer Gnocchi score, again conditioning on LOEUF, was a significant predictor of the expression level of target genes in matched tissue types (Fig. 5c; Methods). These results further support the application of the Gnocchi metric for improving the characterization of gene function. While we acknowledge that the biological consequences of mutations in enhancers are not clearly understood and thus natural selection may differ in strength depending on mechanistic consequence, an extended model to incorporate non-coding variation information in a biologically-informed way holds promise to facilitate our understanding of the molecular mechanisms underlying selection.
Discussion
We have previously developed constraint metrics that leverage population-scale exome and genome sequencing data to evaluate genic intolerance to coding variation for each protein-coding gene5,23. Here, we adopted the same principle with an extended mutational model to assess constraint across the entire genome, using our latest release of gnomAD (v3.1.2), a dataset of harmonized high-quality whole-genome sequences from 76,156 individuals of diverse ancestries. Improvements to constraint modeling include unified fitting of the mutation rate for all substitution and trinucleotide contexts and inclusion of regional genomic features to refine the expected variation in non-coding regions (Methods). We validated our metric, called Gnocchi, using a series of external functional annotations, with a focus on the non-coding genome, and demonstrated the value of Gnocchi for prioritizing non-coding elements and identifying functionally important genes. We have made the Gnocchi scores publicly accessible via the gnomAD browser (https://gnomad.broadinstitute.org).
One key challenge in quantifying non-coding constraint is the estimation of the true base mutation rate, which can be affected by various genomic phenomena, potentially operating at different scales. To this end, we extended our previous mutational model, which computed the relative mutability of each substitution in a trinucleotide context, to include adjustments for regional genomic features that may index processes influencing mutagenesis. The adjustment was applied to each specific trinucleotide context and allowed a varying genomic scale for each specific feature (Methods). The added value of this adjustment was demonstrated by the improved performance of Gnocchi in identifying functional variants (Extended Fig. 6). Gnocchi also outperformed other genome-wide predictive scores, while each metric tended to provide complementary information. We note that all comparisons were restricted to non-coding regions for explicitly evaluating the metrics in prioritizing non-coding variants, and we further eliminated potential bias from nearby genes by recapitulating the results within regions >10kb away from any protein-coding exons (Supplementary Fig. 7). Overall, Gnocchi presented consistent, high performance in identifying functional non-coding variants in the human genome.
Despite the clear constraint signal identified for non-coding regions, many limitations exist. First, the lack of prior classification of the molecular consequences of non-coding variants, as analogous to “nonsynonymous” versus “synonymous” informed by the genetic code in coding regions, limits the resolution of non-coding constraint assessment (e.g., to measure constraint against “LoF” variation). While there are rich resources defining regulatory elements in the non-coding genome, no method is available for determining the impact of each possible variant on gene regulation and the distribution of their effect sizes genome-wide. Further, the interpretation of non-coding constraint, especially in the context of gene regulation, can only be informative when considered in a particular context, such as a tissue/cell type, developmental stage, or environment. Such information is not inherently built into our constraint metric nor in the mutational dataset; thus ad hoc integration of external annotations (e.g., tissue-specific enhancers as analyzed in this study) is often necessary for justifying specific biological implications. Also, since the detection of depletion of variation is immune to negative selection after reproductive age, genomic regions involved in late-onset phenotypes are likely to go underrecognized.
Finally, while this is among the largest datasets of human genomes examined to date for non-coding constraint, our method will substantially increase in power and resolution as sample sizes increase. Benchmarking on the depletion of variation seen in coding regions, we are currently well-powered to detect extreme non-coding constraint as strong as the 90th percentile of coding exons of similar size, and we estimate a sample size of ~340K genomes to detect constraint as to the 50th percentile (Extended Fig. 8a; Methods). Much larger sample sizes will be required for further increasing the resolution, for instance from 1kb to a 100bp scale, we would need ~5.3M samples (Extended Fig. 8b); under the current sample size, 1kb presented optimal performance when compared to varying window sizes tested from 100bp-3kb (Extended Fig. 8c). Meanwhile, we emphasize the importance of increasing genetic ancestral diversity in population-scale datasets like gnomAD. A more diverse population would identify a larger number of rare variants, thereby increasing the power of detecting depletions of variation. We explicitly demonstrated this by reconstructing Gnocchi from the subset of European population and comparing it to that from an equal-sized subset containing all diverse populations – the latter was proven to achieve a higher predictive power (Extended Fig. 8d). Future efforts towards a larger, more diverse human reference dataset would empower finer studies of the influence of human demography on constraint metrics, facilitating a fuller understanding of the distribution and effect of human genetic variation.
Overall, our study demonstrates the value of the genome-wide constraint map in characterizing both non-coding regions and protein-coding genes, providing a significant step towards a comprehensive catalog of functional genomic elements for humans.
Methods
Aggregation, variant-calling, and quality control of gnomAD genome data
We aggregated whole genome sequence data from 153,030 individuals spanning projects from case-control consortia and population cohorts, in a similar fashion to previous efforts65. Informed consent was obtained for the original studies that generated sequencing data and we keep a blank copy of those consents on file with our local Office of Research Subject Protection (ORSP). The Institutional Review Board (IRB) has approved our study protocol, and we confirm that we have complied with all relevant ethical regulations relating to human research subjects.
We harmonized the sequencing data using the GATK Best Practices pipeline and joint-called all samples using Hail66, and developed and utilized an updated pipeline of sample, variant, and genotype quality control to create a high-quality callset of 76,156 individuals, computing frequency information for several strata of this dataset based on attributes such as ancestry and sex for each of 644,267,978 short nuclear variants (see Supplementary Information).
Estimation of trinucleotide context-specific mutation rates
We estimated the probability of a given nucleotide mutating to one of the three other possible bases in a trinucleotide context (XY1Z -> XY2Z), by computing the proportion of all possible variants observed per context in the human genome. Since CpG transitions begin to saturate (proportion observed approaching 1) at a sample size of ~10K genomes, we downsampled the gnomAD dataset to 1,000 genomes for this calculation. The computed proportion observed values, which represent the relative mutability of each trinucleotide context, were further scaled so that the weighted genome-wide average is the human per-base, per-generation mutation rate (1.2×10−8) to obtain the absolute mutation rates . To estimate the proportion of variants expected to be observed in the full gnomAD dataset of 76,156 genomes, we fitted the actual proportion observed in the dataset against , using an exponential regression that caps at 1 for refining the estimates of (near-)saturated variant types (R2=0.999, Extended Fig. 1a,b; Supplementary Data 1).
A total of 390,393,900 high-quality, rare (AF≤0.1%) variants observed in 76,156 gnomAD genomes, a dataset of 6,079,733,538 possible variants at 2,026,577,846 autosomal sites (30–32X coverage), were used in the calculation of trinucleotide context-specific mutation rates. The estimates are well-correlated with the mutation rates reported in previous independent studies and are highly stable across different AF thresholds in gnomAD (Supplementary Fig. 6).
Adjustment of the effect of DNA methylation on CpG mutation rates
Given the strong effect of DNA methylation on increasing the mutation rate at CpG sites, we stratified all CpG sites by their methylation levels and computed the proportion observed within each context and methylation level. As an improvement to our previous methylation annotation (by averaging different tissues65), we analyzed methylation data from germ cells across 14 developmental stages, comprising eight from preimplantation embryos (sperm, oocyte, pronucleus, two-cell-, four-cell-, eight-cell-, morula-, and blastocyst-stage embryos)67 and six from primordial germ cells (7Wk, 10Wk, 11Wk, 13Wk, 17Wk, and 19Wk)68. For each stage, we computed methylation level at each CpG site as the proportion of whole-genome bisulfite sequencing reads corresponding to the methylated allele. To derive a composite score from the 14 stages, we regressed the observation of a CpG variant in gnomAD (0 or 1) on the methylation computed at the corresponding site (a vector of 14), and we used the coefficients from the regression model as weights to compute a composite methylation score for each CpG site. This metric was further discretized into 16 levels (by a minimum step of 0.05: [0,0.05], (0.05,0.1], (0.1,0.15], (0.15,0.2], (0.2,0.25], (0.25,0.3], (0.3,0.5], (0.5,0.55], (0.55,0.6], (0.6,0.65], (0.65,0.7], (0.7,0.75], (0.75,0.8], (0.8,0.85], (0.85,0.9], (0.9,1.0]) to stratify CpG variants in the mutation rate analysis.
Adjustment of the effects of regional genomic features on mutation rates
To estimate the effects of regional genomic features on mutation rates under neutrality, we uti3lized de novo mutations (DNMs), as a proxy of spontaneous mutations, and fitted logistic regression models using the genomic features as predictive variables. A set of 413,304 unique DNMs were compiled from two large-scale family-based whole-genome sequencing studies69,70, and an exclusive set of 4,104,879 genomic sites (~10× the DNMs) randomly drew from the genome was used as the “nonmutated” background. For each DNM or background site, we computed 13 genomic features (see Collection of genomic features) at four scales by taking the mean value of 1kb, 10kb, 100kb, and 1Mb windows centering at the site. This generated a feature matrix of 13×4=52 columns and 413,304+4,104,879 =4,518,183 rows. The matrix was further divided based on the trinucleotide context of each DNM or background site (by row) to assess the effects of genomic features on context-specific mutation rates. In particular, for CpG contexts, features that were correlated with DNA methylation (GC content, CpG_island, short interspersed nuclear element, and nucleosome density), which had been used for adjusting CpG mutation rates, were excluded from the analysis.
For each trinucleotide context, we first performed univariable logistic regression to select features that are significantly associated with an increased/decreased probability of observing a DNM. Features with a significant association surpassing the Bonferroni correction for 13×4=52 tests were selected; if a feature was significant at multiple genomic scales, the smallest window size was selected for the highest resolution (Extended Fig. 1c). Next, we fitted multivariable logistic regression using the selected features to predict DNMs from the background. To control for multicollinearity, we transformed the input feature matrix using principal components analysis (PCA71) to generate decorrelated predictive variables (i.e., the principal components or PCs). The regression coefficients were the primary output of interest, which represent the effects of genomic features on increasing (a positive coefficient) or decreasing (a negative coefficient) the mutation rate, and were used for adjusting the expected number of variants in a given region. The selected features, the PCs, and the coefficients are summarized in Extended Fig. 1c and are available as pickle files for implementation (see Code availability).
Prediction of expected number of variants per 1kb
Using the trinucleotide mutation rate estimates and the above adjustments, we computed the expected number of variants in a given 1kb genomic window as follow:
where denotes one of the 64 trinucleotide contexts; denotes one of the three bases substituting the central nucleotide; denotes one of the DNA methylation levels, where =16 for CpG sites (see Adjustment of the effect of DNA methylation on CpG mutation rates) and =1 for non-CpG sites (i.e., no stratification). Essentially, the expected value of variants in a genomic window is calculated by multiplying the number of possible variants () in by the probability of a variant () and summing across all trinucleotide contexts (), substitutions (), and methylation levels (); is the trinucleotide mutation rate estimated in this study (as described in Estimation of trinucleotide context-specific mutation rates).
Additionally, is adjusted by a factor , which represents the effect of regional genomic features of on mutation rate. For each , specific features have been pre-selected and their effects on mutation rate have been estimated using logistic regression models (see Adjustment of the effects of regional genomic features on mutation rates). Denote the feature values, computed centering and decorrelated by PCA, and the regression coefficients by and , respectively, where is the number of selected features for , the adjustment factor is defined as the ratio of logit given to that of the genome-wide average : ; since the adjustment is specific to each trinucleotide context, is further subscribed by .
Construction of Gnocchi
We created a signed score - called Gnocchi - to quantify the depletion of variation (constraint) at a 1kb scale by comparing the observed variation to an expectation:
The observed variant count () is the number of unique rare (AF≤0.1%) variants in a 1kb window identified in the gnomAD dataset of 76,156 genomes, and the expected number of variants () is established as described above based on the sequence context and the regional genomic features of the 1kb window.
Gnocchi scores were created for 2,689,987 non-overlapping 1kb windows across the human genome, comprising 2,561,056 on autosomes and 128,931 on chromosome X. Due to the lack of DNM data on chromosome X, the genomic feature adjustment factor was assessed using autosomal regions and extrapolated to chromosome X. We performed downstream analyses separately for autosomes and chromosome X and presented the former as primary, with the latter provided in Supplementary Fig. 8. For the analyses, we filtered the dataset to windows where 1) the sites contained at least 1,000 possible variants, 2) at least 80% of the observed variants passed all variant call filters (INFO/FILTER equals to “PASS”), and 3) the mean coverage in the gnomAD genomes was between 25–35X (or 20–25X for chromosome X). This resulted in 1,984,900 autosomal windows (77.5% of initial) for the primary analyses, of which 141,341 overlapped with coding regions and 1,843,559 were exclusively non-coding. The computed Gnocchi scores are available in Supplementary Data 2. We also computed the sores in a sliding window approach (1kb stepped by 100bp) and provided them in Supplementary Data 3.
Collection of genomic features
The 13 regional genomic features used for adjusting trinucleotide mutation rate are 1) GC content72, 2) low-complexity region73, 3) short and 4) long interspersed nuclear element72, distance from the 5) telomere and the 6) centromere72, 7) male and 8) female recombination rate69, 9) DNA methylation, 10) CpG island72, 11) nucleosome density74, 12) maternal and 13) paternal DNM cluster75. Data were downloaded from the referenced resources, lifted over to GRCh38 coordinates when needed using CrossMap76, and files in .bed or .BigWig format were processed using bedtools77 and bigWigAverageOverBed78 to obtain feature values within specific genomic windows.
Correlation between Gnocchi and APS
As an internal validation, we compared our Gnocchi score against the SV constraint score APS79. For each SV from the original study79, we assessed its constraint by assigning the highest Gnocchi score among all overlapping 1kb windows. The correlation between Gnocchi and APS was evaluated across 116,184 high-quality autosomal SVs scored by both metrics, using a linear regression test. In Fig. 1b, the correlation was presented by the mean value of APS across ascending constraint Gnocchi score bins, with 95% confidence intervals computed from 100-fold bootstrapping.
Correlation between Gnocchi and putative functional non-coding annotations
We validated the Gnocchi metric using a number of external functional annotations, including 926,535 ENCODE cCREs80 (34,803 promoter-like [PLS], 141,830 proximal enhancer-like [pELS], 667,599 distal enhancer-like [dELS), and 56,766 CTCF-only elements), 63,285 FANTOM581 enhancers, 331,601 super enhancers (SEdb82), 111,308 GWAS Catalog83 variants (with an association P ≤ 5.0×10−8; 9,229 with an independent replication), 2,191 GWAS variants fine-mapped across population biobanks with a posterior inclusion probability of causality≥0.984, and 100,530 CNVs from a CNV morbidity map of developmental delay85,86.
To assess the correlation between Gnocchi and the collected functional elements, we intersected each annotation with the scored 1kb windows binned by Gnocchi score (<-4, [-4,-3), [-3,-2), [-2,-1), [-1,-0), [0,1), [1,2), [2,3), [3,4), ≥4), and counted the frequency of overlapping windows within each bin. The enrichment of a given annotation (except CNVs) at a constraint level was evaluated by comparing the corresponding frequency to the genome-wide average using a Fisher’s exact test. In the analysis of CNVs, we assessed their enrichment in constrained regions by assigning each CNV the highest Gnocchi score among its overlapping windows and comparing the proportions of constrained CNVs (Gnocchi≥4) from cases of developmental delay and healthy controls (Supplementary Data 4). The enrichment was further examined using a logistic regression model to adjust for the size and gene content (gene constraint65 and gene number) of each CNV. We note that we performed all above analyses restricting to exclusively non-coding windows to evaluate the use of Gnocchi in characterizing the non-coding genome.
Estimation of constraint for aggregated regulatory annotations
We estimated how constrained the sequences encoding regulatory elements overall compared to coding exons by aggregating the regulatory annotations at a 1kb scale. These included 7,246 promoter-, 154,003 enhancer-, 117 microRNA (miRNA)-, and 414,084 long non-coding RNA (lncRNA)-1kb elements, created from concatenating ENCODE cCREs-PLS, cCREs-dELS, GENCODE87 miRNA, and FANTOM5 lncRNA88 annotations, respectively, into 1kb windows. Similarly, 27,875 exonic 1kb elements were created from aggregating all protein-coding exons. Gnocchi scores were computed for the created 1kb elements and the percentiles of each regulatory annotation were compared against the exonic region. Benchmarking on the 50th percentile (median) of exonic regions, we estimated the proportion of the regulatory elements that are under selection as strong as the coding exons.
Incorporation of Gnocchi into GWAS fine-mapping
To demonstrate the use of Gnocchi in statistical fine-mapping, we performed approximate functionally informed fine-mapping89 incorporating Gnocchi score and our previous fine-mapping results for 119 UK Biobank (UKBB) traits84. The Gnocchi scores were normalized and used as functional prior probabilities to update the posterior inclusion probabilities (PIPs; denoted as PIPZ) based on the previous UKBB fine-mapping (using a uniform prior, PIPunif) and SuSiE90. To exclude signals that potentially correspond to coding variants, we restricted our analysis to 60,121 non-coding variants in 6,592 SuSiE 95% credible set (CS)-trait pairs that do not contain variants within 1 kb of exonic regions. A total of 13,069 variant-trait pairs were predicted to have an increased PIP (ΔPIP≥0.01) of causality. The variants, associated traits, and PIP scores (PIPunif and PIPZ) are provided in Supplementary Data 5.
Comparison of Gnocchi and other predictive metrics
We compared the Gnocchi metric with other seven genome-wide predictive scores – Orion91, CDTS92, gwRVIS93, DR94, phyloP95, phastCons96, and GERP97. Each score was downloaded from the original study, lifted over to GRCh38 coordinates (for Orion) and multiplied by −1 (for CDTS, gwRVIS, and DR) when needed so that a higher value represents a higher constraint/conservation for all metrics. Pairwise correlation between the scores was assessed by comparing the mean value of each score on 1kb windows, using a Spearman’s rank correlation test.
We evaluated the predictive performance of each metric in distinguishing functional non-coding variants (“positive” variant set) from background variants (“negative” variant set). Four positive variant sets were compiled from public databases: 1) 9,229 variants from GWAS Catalog83 (with an independent replication), 2) 2,191 variants from a recent fine-mapping study84 (with a posterior inclusion probability of causality≥0.9), 3) 140 high-confidence variants from 2), and 4) 1,026 variants from ClinVar98 (annotated as “pathogenic” or “likely pathogenic”) and HGMD (annotated as ‘disease-causing mutation’ [DM] curated by92). All variants were filtered to non-coding regions; in particular, pathogenic variants were more strictly filtered to intergenic/intron variants given its strong predominance of variants close to protein-coding exons (>90% were splice site/region variants). A further stringent non-coding subset was generated by excluding variants within 10kb to any exons, which resulted in 1) 4,379, 2) 967, 3) 59, and 4) 45 variants. For each positive variant set, a negative variant set was created by randomly drawing variants from the Trans-Omics for Precision Medicine (TOPMed) whole-genome sequencing dataset (Freeze 8)99 to ~10× the size of corresponding positive variant set, of which the most severe molecular consequence is intergenic or intron and the AF approximates the positive variant set; AF>5% and allele count (AC)=1 were applied respectively for matching positive variant set 1)-3) and 4), based on their AF distributions in TOPMed (Fig. 3b). The selected variants were scored by each of the eight metrics, using bedtools77 (for .bed files) and bigWigAverageOverBed74 (for .BigWig files), and the performance of each metric in classifying positive and negative variants was assessed by the area under curve (AUC) statistic, as presented by the receiver operating characteristic (ROC) curve.
To investigate whether different metrics capture complementary information in the classification, we fitted logistic regression models using all eight metrics as independent variables. The relative contribution of each metric was evaluated by the dominance analysis100,101, which estimates the dominance of one predictor over another by comparing their additional contributions across all subset models. We further explored whether specific features were particularly captured by (and may have contributed to the performance of) our metric. We merged all positive variant sets and focused on a set of variants (N=204) that were uniquely prioritized by our metric, defined as being captured in the 99th percentile of Gnocchi score but not in that of any other scores. Specific features associated with these variants were evaluated by comparing values of the 13 genomic features of these variants to the rest of the positive variant set. The fold change was used to indicate the extent to which a feature is distinguished in variants captured by Gnocchi from others.
Correlation of constraint between non-coding regulatory elements and protein-coding genes
To examine whether constraint of non-coding regulatory elements informs the constraint of their target genes, we compared Gnocchi scores of enhancers linked to constrained genes and unconstrained genes. The former included well-established gene sets of 189 ClinGen102 haploinsufficient genes, 2,454 MGI103 essential genes mapped to human orthologs, 1, 771 OMIM104 autosomal dominant genes, and 1,920 LOEUF65 first-decile genes; and the latter included a curated list of 356 olfactory receptor genes and 189 LOEUF last-decile genes with at least 10 expected LoF variants (which are sufficiently powered to be classified into the most constrained decile65). The LOEUF underpowered list included 1,117 genes with ≤5 expected LoF variants. Enhancers linked to each gene were obtained from the Roadmap Epigenomics Enhancer-Gene Linking database, which used correlated patterns of activity between histone modifications and gene expression to predict enhancer-gene links105,106. For each gene, we aggregated and merged enhancers predicted from all 127 reference epigenomes and assigned the most constrained enhancer to each gene for the analysis of enhancer-gene constraint correlation (Supplementary Data 6).
In the analysis of correlation between tissue-specific enhancer constraint and tissue-specific gene expression, we processed the enhancer-gene links with the same principle as described above but within specific tissue types (as defined in the Roadmap Epigenomics metadata107). For each gene and tissue type, we searched for tissue-specific gene expression in the Genotype-Tissue Expression (GTEx108) database (RNASeQCv1.1.9) and computed a normalized median expression for each gene (log2(TPM+1)). Enhancer constraint and gene expression values were calculated for 11 matched tissue types, and the correlation within each tissue type was evaluated by regressing gene expression on enhancer constraint, including gene constraint (LOEUF score) as a covariate.
Incorporation of non-coding constraint of regulatory elements into gene constraint modeling
To demonstrate the practical value of non-coding constraint in improving gene constraint modeling, we compared two models – using 1) LOEUF and 2) LOEUF+enhancer Gnocchi score (as described in Correlation of constraint between non-coding regulatory elements and protein-coding genes) – in predicting constrained genes, with a particular focus on genes that were underpowered in LOEUF. A set of 3,220 unique constrained genes were curated from ClinGen102, MGI103, and OMIM104 (see Correlation of constraint between non-coding regulatory elements and protein-coding genes), and a set of 356 olfactory receptor genes was used as the unconstrained genes. We trained logistic regression models on 50% of the genes and tested the performance on 77 underpowered genes in the remaining 50%. The predictive performance of the two models were measured by AUC, and the significance of the difference in AUCs was assessed using a bootstrap test109.
Power of constraint detection
We estimated the power of our metric in detecting non-coding constraint as the percentage of the non-coding genome to obtain a high Gnocchi score (Gnocchi≥4) under a certain strength of negative selection, which was quantified by the level of depletion of variation (i.e., 1-observed/expected). For a given depletion of variation, the minimum number of expected variants to achieve a Gnocchi≥4 was determined, and the number of samples required to achieve the expected number of variants was estimated using a linear model of log(number of expected variants) ~ log(number of samples) from downsampling the gnomAD dataset. The power was estimated at two scales – 1kb (used in this study) and 100bp – and benchmarked by the depletion of variation observed in coding exons of similar size.
Extended Data
Extended Data Fig. 1:
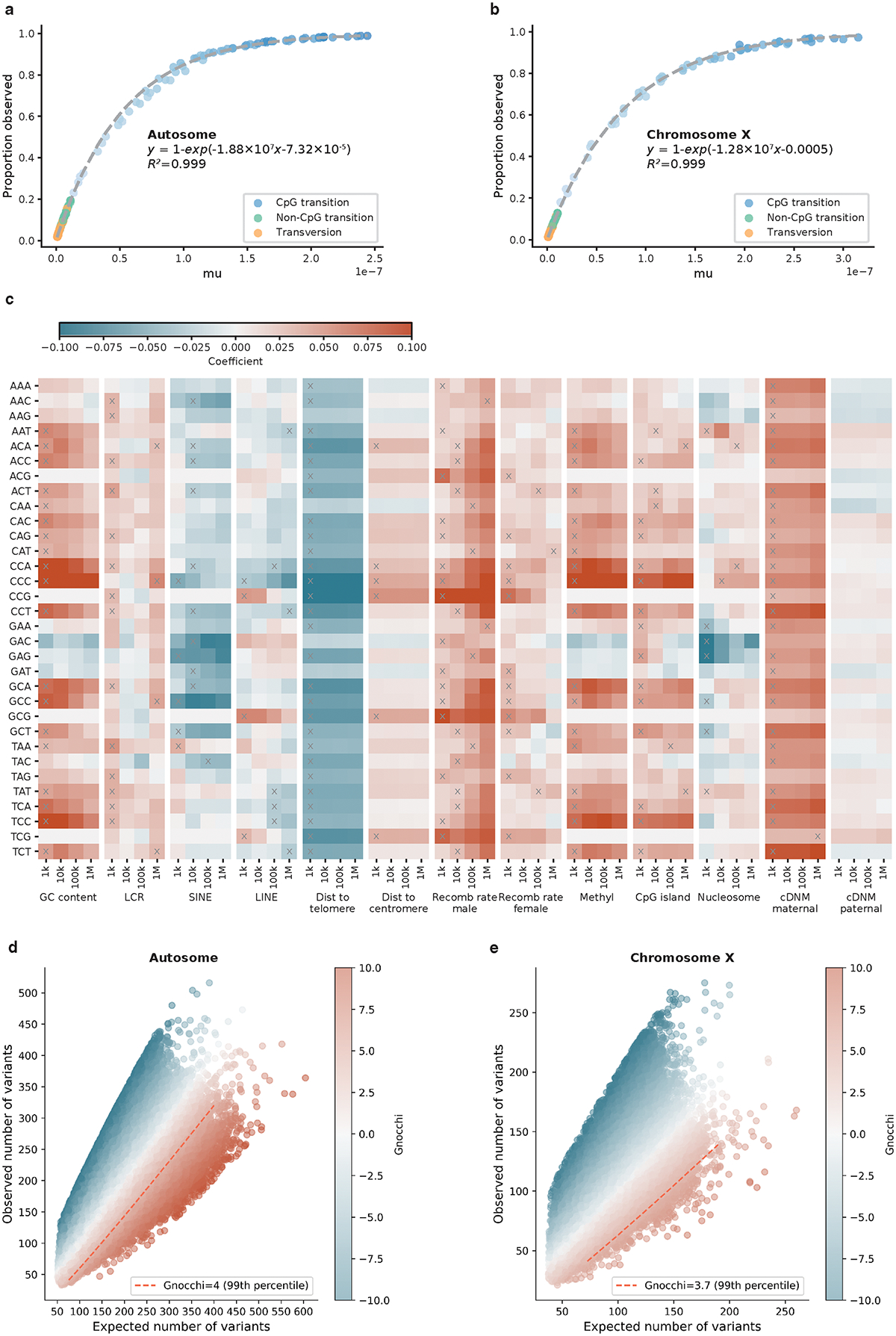
Construction of mutational model and Gnocchi score. a,b, Estimation of trinucleotide context-specific mutation rates. The proportion of possible variants observed for each substitution and context in 76,156 gnomAD genomes (y-axis) is exponentially correlated with the absolute mutation rate estimated from 1,000 downsampled genomes (x-axis). Fit lines were modeled separately for human autosomes (a) and chromosome X (b). c, Estimation of the effects of regional genomic features on mutation rates. The effects of 13 genomic features at four scales (window sizes 1kb-1Mb; x-axis) on the mutation rate of 32 trinucleotide contexts (y-axis) are shown, colored by the coefficient from regressing de novo mutations (DNMs) on each specific feature and window size. Red/Blue color indicates a positive/negative effect of increasing the feature value on mutation rates; grey crosses indicate significant features at the smallest possible window size after Bonferroni correction for 13×4=52 tests. Abbreviations: LCR=low-complexity region, SINE/LINE=short/long interspersed nuclear element, Dist=Distance, Recomb=Recombination, Methyl=Methylation. d,e, The distribution of Gnocchi score as a function of expected and observed variation. Each point represents the Gnocchi score of a 1kb window on the genome (N=1,984,900 on autosomes (d) and N=57,729 on chromosome X (e)), which quantifies the deviation of observed variation from expectation. A positive Gnocchi score (red) indicates depletion of variation (observed<expected) and the higher the score the stronger the depletion; the red dashed line indicates the 99th percentile of Gnocchi scores across the autosomes (d) or chromosome X (e).
Extended Data Fig. 2:
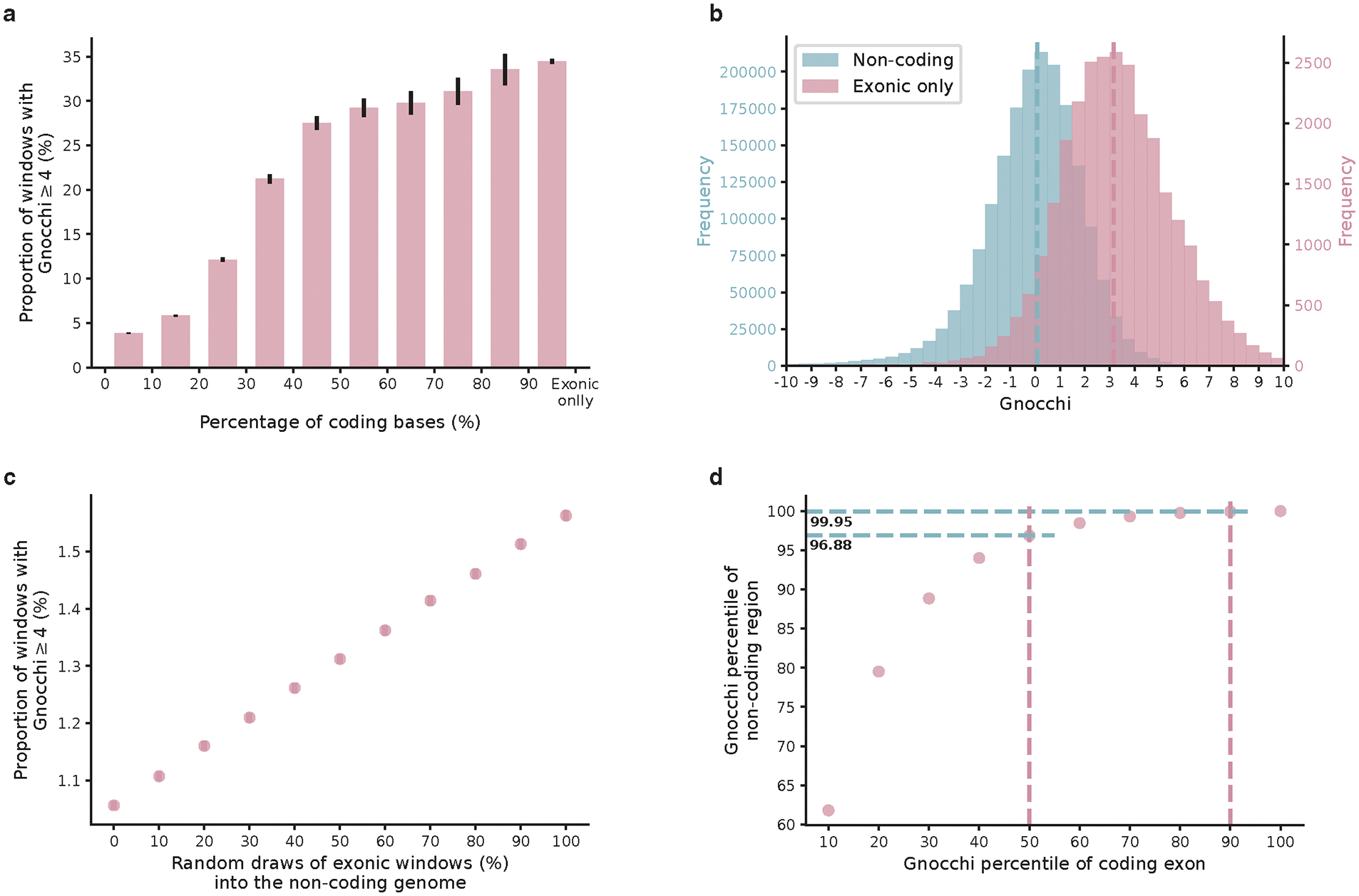
Comparison of Gnocchi score between coding and non-coding regions. a, The proportion of highly constrained windows (Gnocchi≥4) as a function of the percentage of coding sequences in a window (left to right: N=1,906/49,525, 3,244/55,676, 2,240/18,461, 1,506/7,094, 969/3,519, 569/1,946, 364/1,223, 283/910, 243/724, 10,392/30,138). The intervals (x-axis) are left exclusive and right inclusive. “Exonic only” refers to the 1kb windows created from directly concatenating coding exons into 1kb sequences. Error bars indicate standard errors of the proportions. b, The exonic-only regions (N=27,875; purple) present a significantly higher Gnocchi score than regions that are exclusively non-coding (N=1,843,559; blue). Dashed lines indicate the medians. c, The proportion of highly constrained windows (Gnocchi≥4) as a function of the proportion of exonic windows being added to the dataset of non-coding windows. d, Gnocchi score percentiles of non-coding versus exonic windows. About 0.05% (100–99.95%) and 3.12% (100–96.88%) of the non-coding windows exhibit similar constraint to the 90th and 50th of exonic regions, respectively.
Extended Data Fig. 3:
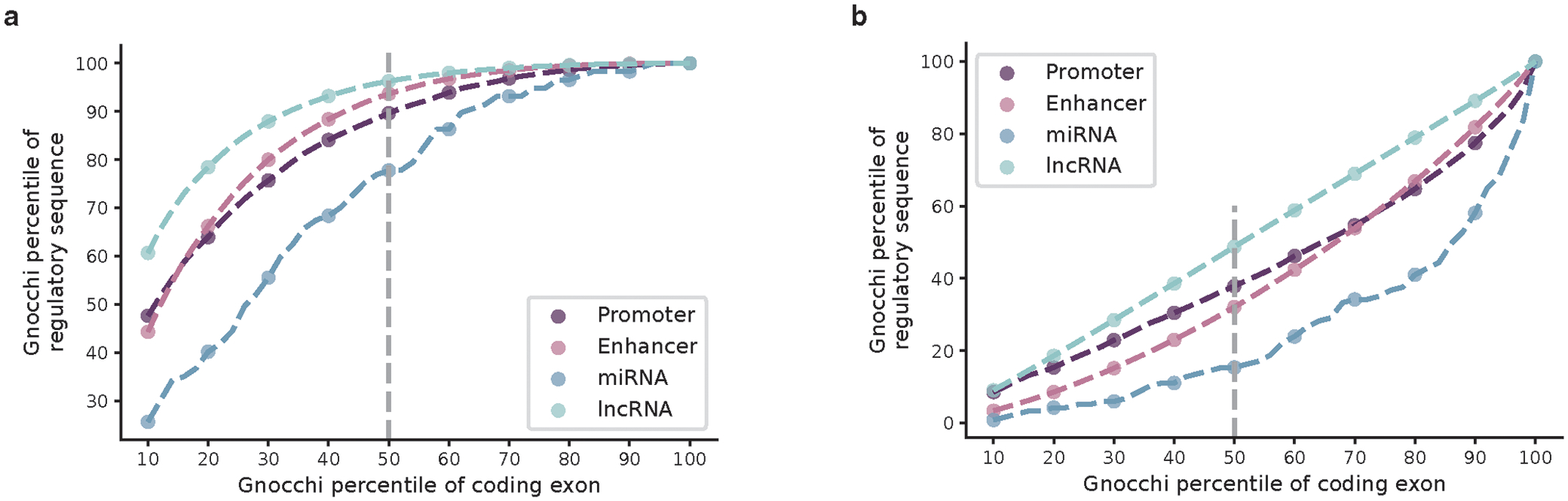
Estimation of constraint for aggregated regulatory annotations. a,b, Gnocchi scores of aggregated promoter (dark purple), enhancer (light purple), microRNA (miRNA; dark blue), and long non-coding RNA (lncRNA; light blue) annotations are compared against those of exonic (a) and non-coding (b) regions at a 1kb scale. The Gnocchi score percentiles of each annotation (y-axis) are benchmarked by the score deciles of exonic or non-coding regions (10–100 percentiles; x-axis); the grey dashed vertical line indicates the median (50th percentile).
Extended Data Fig. 4:
Applications of Gnocchi for characterizing non-coding regions in addition to existing functional annotations. a, Use of Gnocchi for prioritizing non-coding regions with or without a regulatory annotation (N=464,504 and 1,379,055, respectively). Constrained non-coding regions are enriched for GWAS variants, independent of the candidate cis-regulatory element (cCRE) annotation from ENCODE. Error bars indicate 95% confidence intervals of the odds ratios. b, Use of Gnocchi in statistical fine-mapping. The increase in posterior inclusion probability (PIP) when incorporating Gnocchi score as a functional prior into previous fine-mapping results (that used a uniform prior; denoted as PIPGnocchi and PIPunif, respectively) is shown for 164 new likely causal associations with a PIPGnocchi ≥0.8 as a function of PIPGnocchi.
Extended Data Fig. 5:
Comparison of Gnocchi and other predictive metrics in prioritizing non-coding variants. a, Receiver operating characteristic (ROC) curves of Gnocchi and other seven metrics in classifying putative functional non-coding variants (“positive” variant set) – left to right: 9,229 GWAS Catalog variants, 2,191 GWAS fine-mapping variants, a subset of 140 high-confidence fine-mapped variants, and 1,026 likely pathogenic variants – against “negative” variant set randomly drew from the population with a similar allele frequency (AF). AF>5% and allele count (AC)=1 were applied respectively for matching the three GWAS variant sets and the likely pathogenic variant set, based on their AF distributions in TOPMed (shown in b). b, AUCs of the classification with a varying AF threshold for the negative variant set. As most GWAS variants are common and most likely pathogenic variants are very rare (not seen in the population), AF>5% and AC=1 were applied respectively in the primary analyses shown in a.
Extended Data Fig. 6:
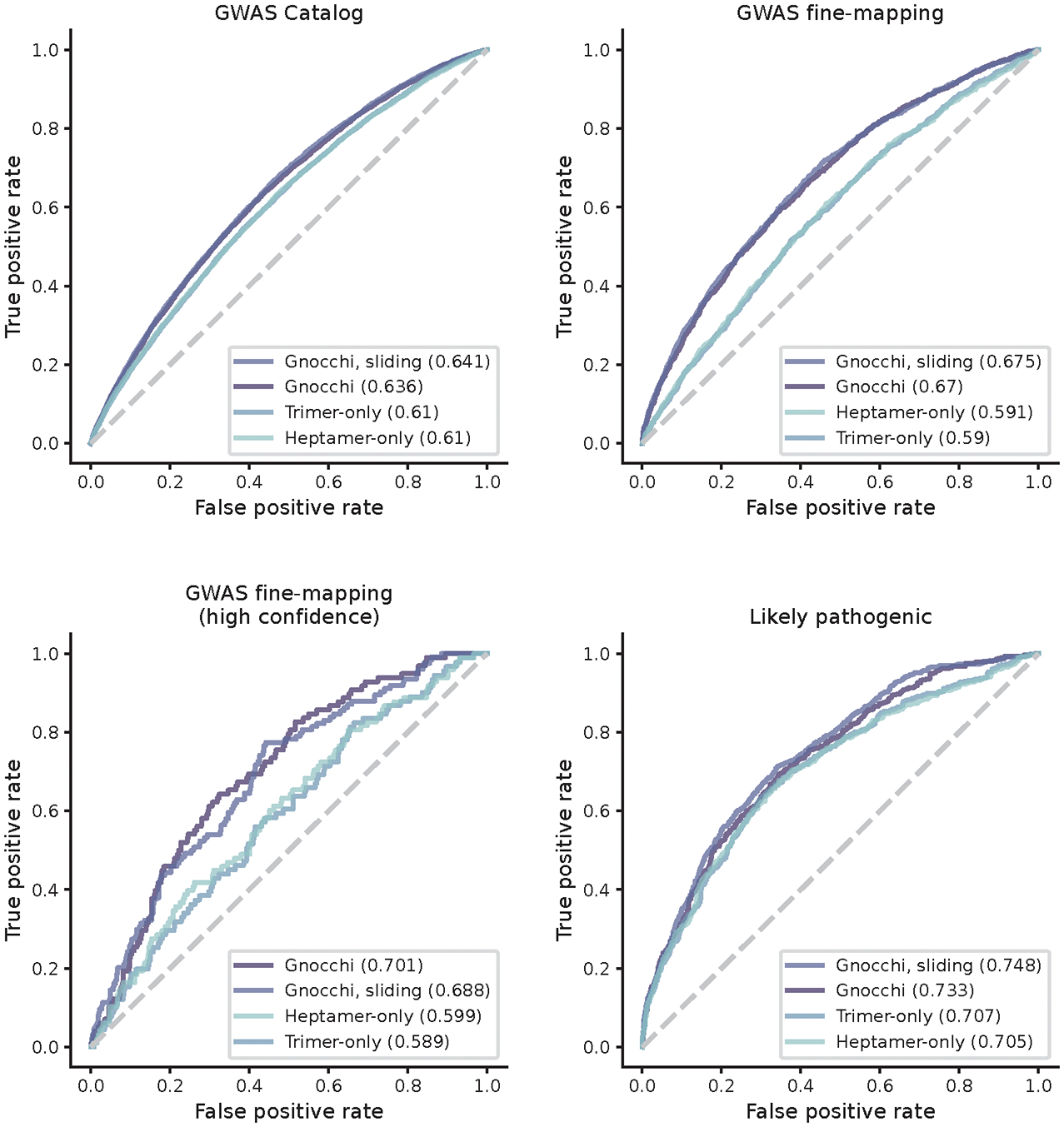
Comparison of constraint scores built from different mutational models and genomic windows. Gnocchi (presented in this study) outperforms the scores rebuilt from mutational models that only consider local sequence context – trinucleotide (trimer-only) or heptanucleotide (heptamer-only) – without adjustment on mutation rate by regional genomic features, and the performance is robust to the artificial break of genomic windows when computed at a 1kb sliding by 100bp scale.
Extended Data Fig. 7:
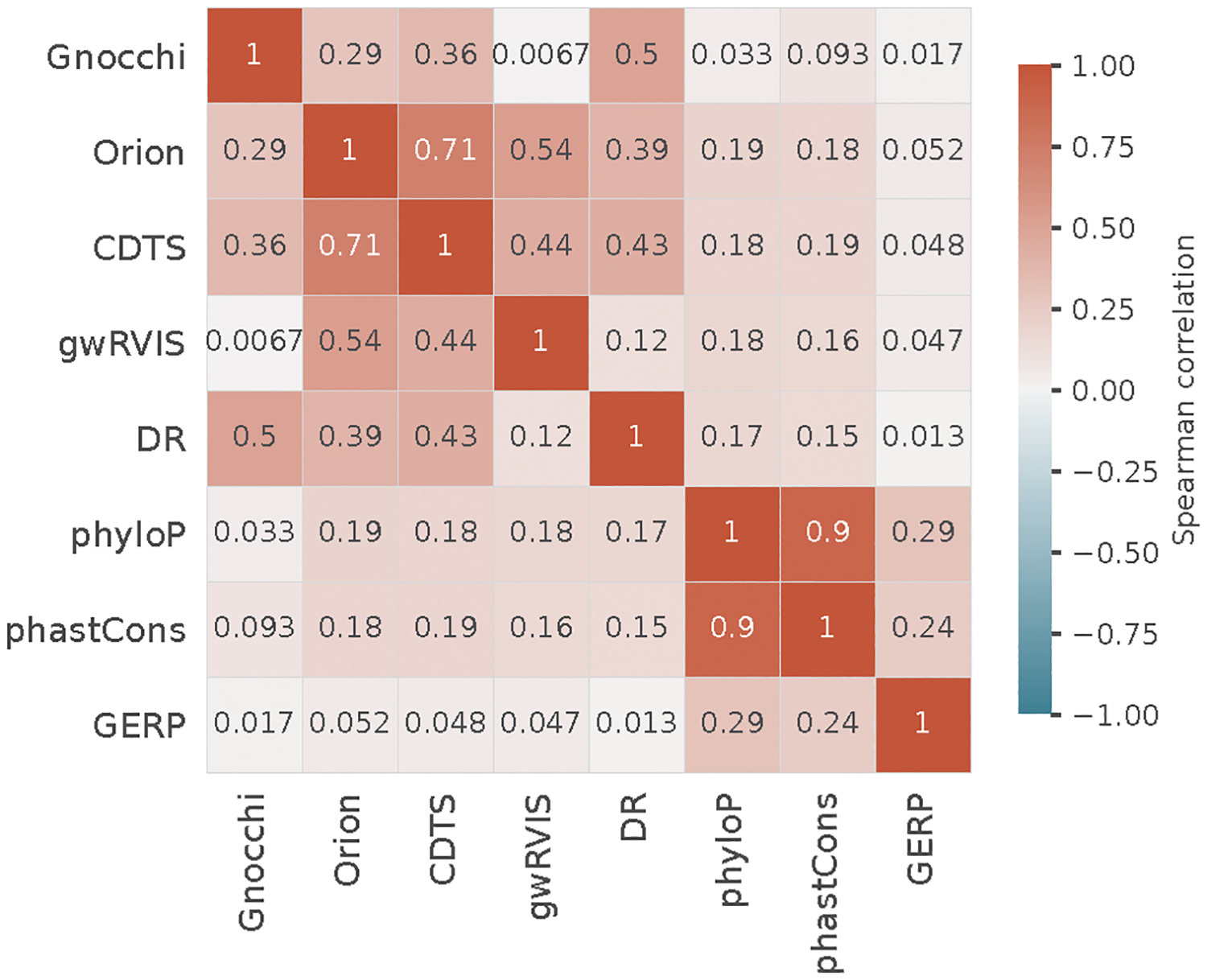
Pairwise correlations between different constraint/conservation metrics. The Spearman’s rank correlation between each pair of the eight metrics was computed based on the mean value of each score on 1kb windows across the genome.
Extended Data Fig. 8:
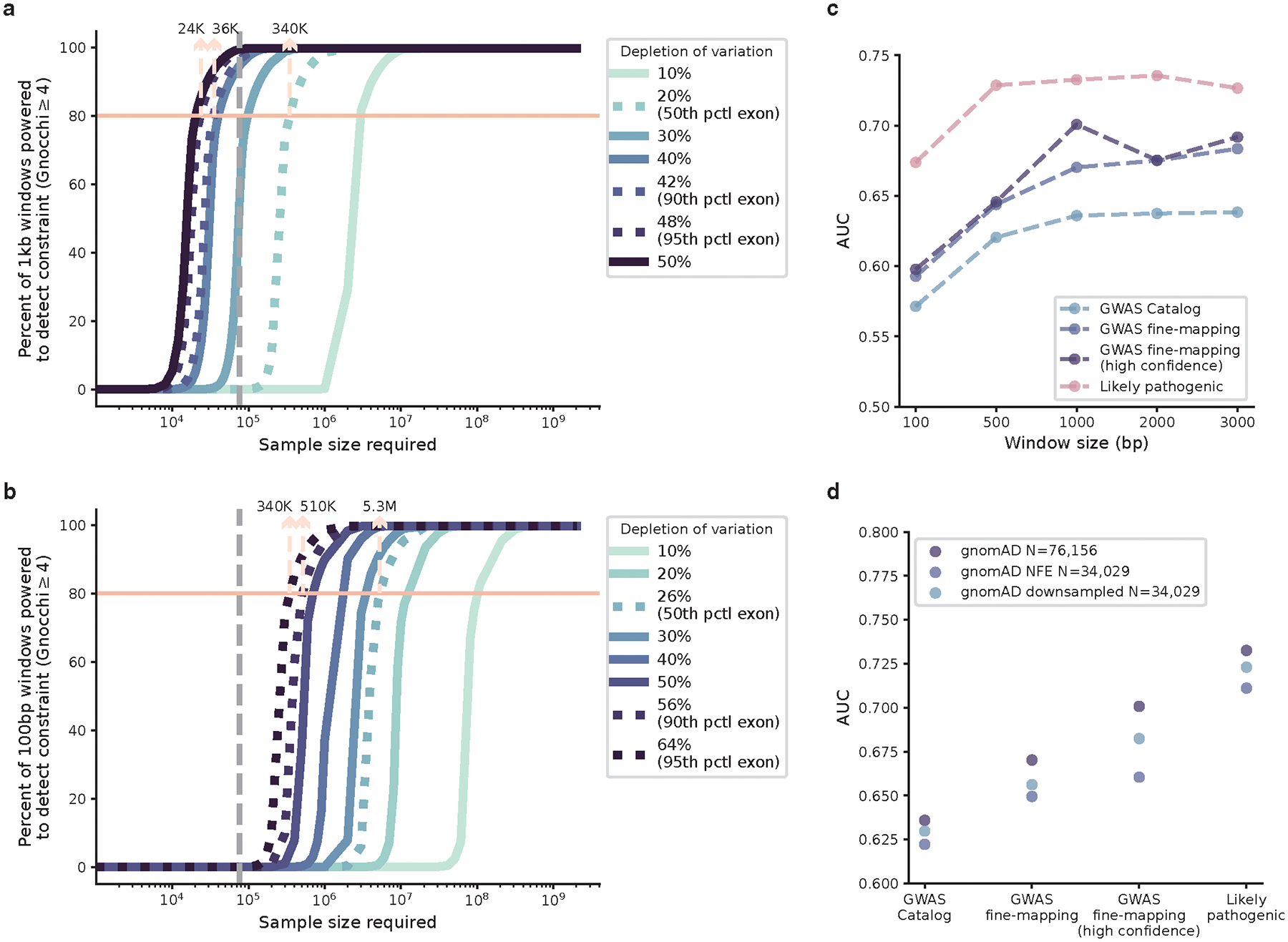
Power of constraint detection. a,b, The sample size required for well-powered non-coding constraint detection. The percentage of non-coding regions powered to detect constraint (Gnocchi≥4) at a 1kb (a) and 100bp (b) scale under varying levels of selection (depletion of variation) is shown as a function of log-scaled sample size. Lighter color indicates milder deletion of variation (weaker selection), which requires a larger sample size to detect constraint; the grey dashed vertical line indicates the current sample size of 76,156 genomes. Dotted curves (left to right) benchmark the 95th, 90th, and 50th percentile of depletion of variation observed in coding exons of similar size. The number of samples required to obtain an 80% detection power is labeled at corresponding benchmarks. c, AUCs of Gnocchi scores computed on different window sizes in identifying putative functional non-coding variants. 1kb (used in this study) presents the optimal window size with high performance while maintaining reasonable resolution. d, AUCs of Gnocchi scores computed from different subsets of gnomAD in identifying putative functional non-coding variants. While with an equal sample size, the downsampled dataset with diverse ancestries presents higher performance than the Non-Finnish European (NFE)-only dataset.
Supplementary Material
Acknowledgements
We thank the individuals whose data is in gnomAD for their contributions to research. Development of the Genome Aggregation Database was supported by NIDDK U54DK105566 and the NHGRI of the National Institutes of Health under award number U24HG011450. Additional funding for Genome Aggregation Database Consortium members is listed in the Supplementary Information. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing Interest
KJK is a consultant for Vor Biopharma, Tome Biosciences, and is on the Scientific Advisory Board of Nurture Genomics. DGM is a paid advisor to GSK, Insitro, Variant Bio and Overtone Therapeutics, and has previously received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Merck, Pfizer and Sanofi-Genzyme.
Genome Aggregation Database Consortium
Maria Abreu15, Carlos A. Aguilar Salinas16, Tariq Ahmad17, Christine M. Albert18,19, Jessica Alföldi1,2, Diego Ardissino20, Irina M. Armean1,2, Elizabeth G. Atkinson21,22, Gil Atzmon23,24, Eric Banks6, John Barnard25, Samantha M. Baxter1, Laurent Beaugerie26, Emelia J. Benjamin27,28,29, David Benjamin6, Louis Bergelson6, Michael Boehnke30, Lori L. Bonnycastle31, Erwin P. Bottinger32, Donald W. Bowden33,34,35, Matthew J. Bown36,37, Harrison Brand3,38, Steven Brant39,40,41, Ted Brookings6,42, Sam Bryant2,22, Sarah E. Calvo1,3, Hannia Campos43,44, John C. Chambers45,46,47, Juliana C. Chan48, Katherine R. Chao1,2, Sinéad Chapman1,2,7, Daniel I. Chasman18,49, Siwei Chen1,2, Rex Chisholm50, Judy Cho32, Rajiv Chowdhury51, Mina K. Chung52, Wendy K. Chung53,54,55, Kristian Cibulskis6, Bruce Cohen56,57, Ryan L. Collins1,3,4, Kristen M. Connolly58, Adolfo Correa59, Miguel Covarrubias6, Beryl B. Cummings1,4, Dana Dabelea60, Mark J. Daly1,2,12, John Danesh51, Dawood Darbar61, Phil Darnowsky1, Joshua Denny62, Stacey Donnelly10, Ravindranath Duggirala63, Josée Dupuis64,65, Patrick T. Ellinor1,66, Roberto Elosua67,68,69, James Emery6, Eleina England1,70, Jeanette Erdmann71,72,73, Tõnu Esko1,74, Emily Evangelista1, Yossi Farjoun9, Diane Fatkin75,76,77, Steven Ferriera11, Jose Florez49,78,79, Laurent C. Francioli1,2, Andre Franke80,81, Jack Fu1,3,38, Martti Färkkilä82,83,84, Stacey Gabriel11, Kiran Garimella6, Laura D. Gauthier6, Jeff Gentry6, Gad Getz49,85,86, David C. Glahn87,88, Benjamin Glaser89, Stephen J. Glatt90, David Goldstein91,92, Clicerio Gonzalez93, Julia K. Goodrich1, Riley Grant1, Leif Groop94,95, Sanna Gudmundsson1,2,8, Namrata Gupta1,11, Andrea Haessly6, Christopher Haiman96, Ira Hall97, Craig L. Hanis98, Matthew Harms99,100, Mikko Hiltunen101, Matti M. Holi102, Christina M. Hultman103,104, Chaim Jalas105, Thibault Jeandet6, Mikko Kallela106, Masahiro Kanai1,2, Diane Kaplan6, Jaakko Kaprio95, Konrad J. Karczewski1,2,7, Sekar Kathiresan3,49,107, Eimear E. Kenny108, Bong-Jo Kim109, Young Jin Kim109, Daniel King1, George Kirov110, Zan Koenig2,7, Jaspal Kooner46,111,112, Seppo Koskinen113, Harlan M. Krumholz114, Subra Kugathasan115, Soo Heon Kwak116, Markku Laakso117,118, Nicole Lake119, Trevyn Langsford6, Kristen M. Laricchia1,2, Terho Lehtimäki120, Monkol Lek119, Emily Lipscomb1, Christopher Llanwarne6, Ruth J.F. Loos32,121,122, Wenhan Lu1, Steven A. Lubitz1,66, Teresa Tusie Luna123,124, Ronald C.W. Ma48,125,126, Daniel G. MacArthur1,13,14, Gregory M. Marcus127, Jaume Marrugat128,129, Alicia R. Martin1,2,7, Kari M. Mattila120, Steven McCarroll7,130, Mark I. McCarthy131,132,133, Jacob L. McCauley134,135, Dermot McGovern136, Ruth McPherson137, James B. Meigs1,49,138, Olle Melander139, Andres Metspalu140, Deborah Meyers141, Eric V. Minikel1, Braxton D. Mitchell142, Vamsi K. Mootha1,143, Ruchi Munshi6, Aliya Naheed144, Saman Nazarian145,146, Benjamin M. Neale1,2, Peter M. Nilsson147, Sam Novod6, Anne O’Donnell-Luria1,3,8, Michael C. O’Donovan148, Yukinori Okada5,149,150, Dost Ongur49,56, Lorena Orozco151,152, Michael J. Owen148, Colin Palmer153, Nicholette D. Palmer33, Aarno Palotie2,7,95, Kyong Soo Park116,154, Carlos Pato155, Nikelle Petrillo6, William Phu1,8, Timothy Poterba1,2,7, Ann E. Pulver156, Dan Rader145,157, Nazneen Rahman158, Heidi L. Rehm1,3, Alex Reiner159,160, Anne M. Remes161, Dan Rhodes1, Stephen Rich162,163, John D. Rioux164,165, Samuli Ripatti10,95,166, David Roazen6, Dan M. Roden167,168, Jerome I. Rotter169, Valentin Ruano-Rubio6, Nareh Sahakian6, Danish Saleheen170,171,172, Veikko Salomaa173, Andrea Saltzman1, Nilesh J. Samani37,174, Kaitlin E. Samocha1,3, Alba Sanchis-Juan3, Jeremiah Scharf1,3,7, Molly Schleicher1, Heribert Schunkert175,176, Sebastian Schönherr177, Eleanor G. Seaby1,178, Cotton Seed2,7, Svati H. Shah179,180, Megan Shand6, Ted Sharpe6, Moore B. Shoemaker181, Tai Shyong182,183, Edwin K. Silverman184,185, Moriel Singer-Berk1, Pamela Sklar186,187,188, Jonathan T. Smith6, J. Gustav Smith189,190, Hilkka Soininen191, Harry Sokol192,193,194, Matthew Solomonson1,2, Rachel G. Son1, Jose Soto6, Tim Spector195, Christine Stevens1,2,7, Nathan O. Stitziel196,197,198, Patrick F. Sullivan103,199, Jaana Suvisaari173, E. Shyong Tai200,201,202, Michael E. Talkowski1,3,7, Yekaterina Tarasova1, Kent D. Taylor169, Yik Ying Teo200,203,204, Grace Tiao1,2, Kathleen Tibbetts6, Charlotte Tolonen6, Ming Tsuang205,206, Tiinamaija Tuomi95,207,208, Dan Turner209, Teresa Tusie-Luna210,211, Erkki Vartiainen212, Marquis Vawter213, Christopher Vittal1,2, Gordon Wade6, Lily Wang214, Qingbo Wang1,5, Arcturus Wang1,2,7, James S. Ware1,215,216, Hugh Watkins217, Nicholas A. Watts1,2, Rinse K. Weersma218, Ben Weisburd6, Maija Wessman95,219, Nicola Whiffin1,220,221, Michael W. Wilson1,2, James G. Wilson222, Ramnik J. Xavier223,224, Mary T. Yohannes1
15University of Miami Miller School of Medicine, Gastroenterology, Miami, USA
16Unidad de Investigacion de Enfermedades Metabolicas, Instituto Nacional de Ciencias Medicas y Nutricion, Mexico City, Mexico
17Peninsula College of Medicine and Dentistry, Exeter, UK
18Division of Preventive Medicine, Brigham and Women’s Hospital, Boston, MA, USA
19Division of Cardiovascular Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA
20Department of Cardiology University Hospital, Parma, Italy
21Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA
22Stanley Center for Psychiatric Research, The Broad Intitute of MIT and Harvard, Cambridge MA, USA
23Department of Biology Faculty of Natural Sciences, University of Haifa, Haifa, Israel
24Departments of Medicine and Genetics, Albert Einstein College of Medicine, Bronx, NY, USA
25Department of Quantitative Health Sciences, Lerner Research Institute Cleveland Clinic, Cleveland, OH, USA
26Sorbonne Université, APHP, Gastroenterology Department Saint Antoine Hospital, Paris, France
27NHLBI and Boston University’s Framingham Heart Study, Framingham, MA, USA
28Department of Medicine, Boston University Chobanian & Avedisian School of Medicine, Boston, MA, USA
29Department of Epidemiology, Boston University School of Public Health, Boston, MA, USA
30Department of Biostatistics and Center for Statistical Genetics, University of Michigan, Ann Arbor, MI, USA
31National Human Genome Research Institute, National Institutes of Health Bethesda, MD, USA
32The Charles Bronfman Institute for Personalized Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, USA
33Department of Biochemistry, Wake Forest School of Medicine, Winston-Salem, NC, USA
34Center for Genomics and Personalized Medicine Research, Wake Forest School of Medicine, Winston-Salem, NC, USA
35Center for Diabetes Research, Wake Forest School of Medicine, Winston-Salem, NC, USA
36Department of Cardiovascular Sciences and NIHR Leicester Biomedical Research Centre, University of Leicester, Leicester, UK
37NIHR Leicester Biomedical Research Centre, Glenfield Hospital, Leicester, UK
38Department of Neurology, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA
39Department of Medicine, Rutgers Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, New Brunswick, NJ, USA
40Department of Genetics and the Human Genetics Institute of New Jersey, School of Arts and Sciences, Rutgers, The State University of New Jersey, Piscataway, NJ, USA
41Meyerhoff Inflammatory Bowel Disease Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
42Fulcrum Genomics, Boulder, CO, USA
43Harvard School of Public Health, Boston, MA, USA
44Central American Population Center, San Pedro, Costa Rica
45Department of Epidemiology and Biostatistics, Imperial College London, London, UK
46Department of Cardiology, Ealing Hospital, NHS Trust, Southall, UK
47Imperial College, Healthcare NHS Trust Imperial College London, London, UK
48Department of Medicine and Therapeutics, The Chinese University of Hong Kong, Hong Kong, China
49Department of Medicine, Harvard Medical School, Boston, MA, USA
50Northwestern University, Evanston, IL, USA
51University of Cambridge, Cambridge, England
52Departments of Cardiovascular, Medicine Cellular and Molecular Medicine Molecular Cardiology, Quantitative Health Sciences, Cleveland Clinic, Cleveland, OH, USA
53Department of Pediatrics, Columbia University Irving Medical Center, New York, NY, USA
54Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, NY, USA
55Department of Medicine, Columbia University Medical Center, New York, NY, USA
56McLean Hospital, Belmont, MA, USA
57Department of Psychiatry, Harvard Medical School, Boston, MA, USA
58Genomics Platform, Broad Institute of MIT and Harvard, Cambridge, MA, USA
59Department of Medicine, University of Mississippi Medical Center, Jackson, MI, USA
60Department of Epidemiology Colorado School of Public Health Aurora, CO, USA
61Department of Medicine and Pharmacology, University of Illinois at Chicago, Chicago, IL, USA
62Vanderbilt University Medical Center, Nashville, TN, USA
63Department of Life Sciences, College of Arts and Scienecs, Texas A&M University-San Antonio, San Antonio, TX, USA
64Department of Biostatistics, Boston University School of Public Health, Boston, MA, USA
65Department of Epidemiology, Biostatistics and Occupational Health, McGill University, Montreal, QC, Canada
66Cardiac Arrhythmia Service and Cardiovascular Research Center, Massachusetts General Hospital, Boston, MA, USA
67Cardiovascular Epidemiology and Genetics, Hospital del Mar Medical Research Institute (IMIM), Barcelona, Catalonia, Spain
68CIBER CV, Spain
69Departament of Medicine, Faculty of Medicine, University of Vic-Central University of Catalonia, Vic Catalonia, Spain
70Clalit Genomics Center, Israel
71Institute for Cardiogenetics, University of Lübeck, Lübeck, Germany
72German Research Centre for Cardiovascular Research, Hamburg/Lübeck/Kiel, Lübeck, Germany
73University Heart Center Lübeck, Lübeck, Germany
74Estonian Genome Center, Institute of Genomics University of Tartu, Tartu, Estonia
75Victor Chang Cardiac Research Institute, Darlinghurst, NSW, Australia
76Faculty of Medicine, UNSW Sydney, Kensington, NSW, Australia
77Cardiology Department, St Vincent’s Hospital, Darlinghurst, NSW, Australia
78Diabetes Unit and Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA, USA
79Programs in Metabolism and Medical & Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA
80Institute of Clinical Molecular Biology, Christian-Albrechts-University of Kiel, Kiel, Germany
81University Hospital Schleswig-Holstein, Kiel, Germany
82Helsinki University and Helsinki University Hospital Clinic of Gastroenterology, Helsinki, Finland
83Helsinki University and Helsinki University Hospital, Helsinki, Finland
84Abdominal Center
85Bioinformatics Program MGH Cancer Center and Department of Pathology, Boston, MA, USA
86Cancer Genome Computational Analysis, Broad Institute of MIT and Harvard, Cambridge, MA, USA
87Department of Psychiatry and Behavioral Sciences, Boston Children’s Hospitaland Harvard Medical School, Boston, MA, USA
88Harvard Medical School Teaching Hospital, Boston, MA, USA
89Department of Endocrinology and Metabolism, Hadassah Medical Center and Faculty of Medicine, Hebrew University of Jerusalem, Israel
90Department of Psychiatry and Behavioral Sciences, SUNY Upstate Medical University, Syracuse, NY, USA
91Institute for Genomic Medicine, Columbia University Medical Center Hammer Health Sciences, New York, NY, USA
92Department of Genetics & Development Columbia University Medical Center, Hammer Health Sciences, New York, NY, USA
93Centro de Investigacion en Salud Poblacional, Instituto Nacional de Salud Publica, Mexico
94Lund University Sweden, Sweden
95Institute for Molecular Medicine Finland, (FIMM) HiLIFE University of Helsinki, Helsinki, Finland
96Center for Genetic Epidemiology, Department of Population and Public Health Sciences, University of Southern California, Los Angeles, CA, USA
97Washington School of Medicine, St Louis, MI, USA
98Human Genetics Center, University of Texas Health Science Center at Houston, Houston, TX, USA
99Department of Neurology Columbia University, New York City, NY, USA
100Institute of Genomic Medicine, Columbia University, New York City, NY, USA
101Institute of Biomedicine, University of Eastern Finland, Kuopio, Finland
102Department of Psychiatry, Helsinki University Central Hospital Lapinlahdentie, Helsinki, Finland
103Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden
104Icahn School of Medicine at Mount Sinai, New York, NY, USA
105Bonei Olam, Center for Rare Jewish Genetic Diseases, Brooklyn, NY, USA
106Department of Neurology, Helsinki University, Central Hospital, Helsinki, Finland
107Cardiovascular Disease Initiative and Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA
108Institute for Genomic Health, Icahn School of Medicine at Mount Sinai, New York, NY, USA
109Division of Genome Science, Department of Precision Medicine, National Institute of Health, Republic of Korea
110MRC Centre for Neuropsychiatric Genetics & Genomics, Cardiff University School of Medicine, Cardiff, Wales
111Imperial College, Healthcare NHS Trust, London, UK
112National Heart and Lung Institute Cardiovascular Sciences, Hammersmith Campus, Imperial College London, London, UK
113Department of Health THL-National Institute for Health and Welfare, Helsinki, Finland
114Section of Cardiovascular Medicine, Department of Internal Medicine, Yale School of Medicine, Center for Outcomes Research and Evaluation Yale-New Haven Hospital, New Haven, CT, USA
115Division of Pediatric Gastroenterology, Emory University School of Medicine, Atlanta, GA, USA
116Department of Internal Medicine, Seoul National University Hospital, Seoul, Republic of Korea
117The University of Eastern Finland, Institute of Clinical Medicine, Kuopio, Finland
118Kuopio University Hospital, Kuopio, Finland
119Department of Genetics, Yale School of Medicine, New Haven, CT, USA
120Department of Clinical Chemistry Fimlab Laboratories and Finnish Cardiovascular Research Center-Tampere Faculty of Medicine and Health Technology, Tampere University, Finland
121The Mindich Child Health and Development, Institute Icahn School of Medicine at Mount Sinai, New York, NY, USA
122The Novo Nordisk Foundation Center for Basic Metabolic Research, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark
123National Autonomous University of Mexico, Mexico City, Mexico
124Salvador Zubirán National Institute of Health Sciences and Nutrition, Mexico City, Mexico
125Li Ka Shing Institute of Health Sciences, The Chinese University of Hong Kong, Hong Kong, China
126Hong Kong Institute of Diabetes and Obesity, The Chinese University of Hong Kong, Hong Kong, China
127Division of Cardiology, University of California San Francisco, San Francisco, CA, USA
128Hospital del Mar Medical Research Institute (IMIM), Barcelona, Spain
129CIBERCV, Madrid, Spain
130Department of Genetics, Harvard Medical School, Boston, MA, USA
131Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Churchill Hospital Old Road Headington, Oxford, OX, LJ, UK
132Welcome Centre for Human Genetics, University of Oxford, Oxford, OX, BN, UK
133Oxford NIHR Biomedical Research Centre, Oxford University Hospitals, NHS Foundation Trust, John Radcliffe Hospital, Oxford, OX, DU, UK
134John P. Hussman Institute for Human Genomics, Leonard M. Miller School of Medicine, University of Miami, Miami, FL, USA
135The Dr. John T. Macdonald Foundation Department of Human Genetics, Leonard M. Miller School of Medicine, University of Miami, Miami, FL, USA
136F. Widjaja Foundation Inflammatory Bowel and Immunobiology Research Institute Cedars-Sinai Medical Center, Los Angeles, CA, USA
137Atherogenomics Laboratory University of Ottawa, Heart Institute, Ottawa, Canada
138Division of General Internal Medicine, Massachusetts General Hospital, Boston, MA, USA
139Department of Clinical Sciences University, Hospital Malmo Clinical Research Center, Lund University, Malmö, Sweden
140Estonian Genome Center, Institute of Genomics, University of Tartu, Tartu, Estonia
141University of Arizona Health Science, Tuscon, AZ, USA
142University of Maryland School of Medicine, Baltimore, MD, USA
143Howard Hughes Medical Institute and Department of Molecular Biology, Massachusetts General Hospital, Boston, MA, USA
144International Centre for Diarrhoeal Disease Research, Bangladesh
145Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
146Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA
147Lund University, Dept. Clinical Sciences, Skåne University Hospital, Malmö, Sweden
148Centre for Neuropsychiatric Genetics & Genomics, Cardiff University School of Medicine, Cardiff, Wales
149Laboratory of Statistical Immunology, Immunology Frontier Research Center (WPI-IFReC), Osaka University, Suita, Japan
150Integrated Frontier Research for Medical Science Division, Institute for Open and Transdisciplinary Research Initiatives, Osaka University, Suita, Japan
151Instituto Nacional de Medicina Genómica, (INMEGEN) Mexico City, Mexico
152Laboratory of Immunogenomics and Metabolic Diseases, INMEGEN,Mexico City, Mexico
153Medical Research Institute, Ninewells Hospital and Medical School University of Dundee, Dundee, UK
154Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science and Technology, Seoul National University, Seoul, Republic of Korea
155Department of Psychiatry Keck School of Medicine at the University of Southern California, Los Angeles, CA, USA
156Department of Psychiatry and Behavioral Sciences, Johns Hopkins University School of Medicine, Baltimore, MD, USA
157Children’s Hospital of Philadelphia, Philadelphia, PA, USA
158Division of Genetics and Epidemiology, Institute of Cancer Research, London, UK
159University of Washington, Seattle, WA, USA
160Fred Hutchinson Cancer Research Center, Seattle, WA, USA
161Medical Research Center, Oulu University Hospital, Oulu Finland and Research Unit of Clinical Neuroscience Neurology University of Oulu, Oulu, Finland
162Center for Public Health Genomics, University of Virginia, Charlottesville, VA, USA
163Department of Public Health Sciences, University of Virginia, Charlottesville, VA, USA
164Research Center Montreal Heart Institute, Montreal, Quebec, Canada
165Department of Medicine, Faculty of Medicine Université de Montréal, Québec, Canada
166Department of Public Health Faculty of Medicine, University of Helsinki, Helsinki, Finland
167Departments of Medicine, Pharmacology, Biomedical Informatics Vanderbilt, University Medical Center, Nashville, TN, USA
168Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA
169The Institute for Translational Genomics and Population Sciences, Department of Pediatrics, The Lundquist Institute for Biomedical Innovation at Harbor-UCLA Medical Center, Torrance, CA, USA
170Department of Biostatistics and Epidemiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
171Department of Medicine, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
172Center for Non-Communicable Diseases, Karachi, Pakistan
173National Institute for Health and Welfare, Helsinki, Finland
174Department of Cardiovascular Sciences, University of Leicester, Leicester, UK
175Department of Cardiology, Deutsches Herzzentrum München, Technical University of Munich, DZHK Munich Heart Alliance, Germany
176Technische Universität München, Germany
177Institute of Genetic Epidemiology, Department of Genetics, Medical University of Innsbruck, 6020 Innsbruck, Austria
178Faculty of Medicine, University of Southampton, Southampton, SO16 6YD, UK
179Duke Molecular Physiology Institute, Durham, NC
180Division of Cardiology, Department of Medicine, Duke University School of Medicine, Durham, NC, USA
181Division of Cardiovascular Medicine, Nashville VA Medical Center, Vanderbilt University School of Medicine, Nashville, TN, USA
182Division of Endocrinology, National University Hospital, Singapore
183NUS Saw Swee Hock School of Public Health, Singapore
184Channing Division of Network Medicine, Brigham and Women’s Hospital, Boston, MA, USA
185Harvard Medical School, Boston, MA, USA
186Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY, USA
187Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, USA
188Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
189The Wallenberg Laboratory/Department of Molecular and Clinical Medicine, Institute of Medicine, Gothenburg University and the Department of Cardiology, Sahlgrenska University Hospital, Gothenburg, Sweden
190Department of Cardiology, Wallenberg Center for Molecular Medicine and Lund University Diabetes Center, Clinical Sciences, Lund University and Skåne University Hospital, Lund, Sweden
191Institute of Clinical Medicine Neurology, University of Eastern Finad, Kuopio, Finland
192Sorbonne Université, INSERM, Centre de Recherche Saint-Antoine, CRSA, AP-HP, Saint Antoine Hospital, Gastroenterology department, F-75012 Paris, France
193INRA, UMR1319 Micalis & AgroParisTech, Jouy en Josas, France
194Paris Center for Microbiome Medicine, (PaCeMM) FHU, Paris, France
195Department of Twin Research and Genetic Epidemiology King’s College London, London, UK
196Department of Medicine, Washington University School of Medicine, Saint Louis, MO, USA
197Department of Genetics, Washington University School of Medicine, Saint Louis, MO, USA
198The McDonnell Genome Institute at Washington University, Saint Louis, MO, USA
199Departments of Genetics and Psychiatry, University of North Carolina, Chapel Hill, NC, USA
200Saw Swee Hock School of Public Health National University of Singapore, National University Health System, Singapore
201Department of Medicine, Yong Loo Lin School of Medicine National University of Singapore, Singapore
202Duke-NUS Graduate Medical School, Singapore
203Life Sciences Institute, National University of Singapore, Singapore
204Department of Statistics and Applied Probability, National University of Singapore, Singapore
205Center for Behavioral Genomics, Department of Psychiatry, University of California, San Diego, CA, USA
206Institute of Genomic Medicine, University of California San Diego, San Diego, CA, USA
207Endocrinology, Abdominal Center, Helsinki University Hospital, Helsinki, Finland
208Institute of Genetics, Folkhalsan Research Center, Helsinki, Finland
209Juliet Keidan Institute of Pediatric Gastroenterology Shaare Zedek Medical Center, The Hebrew University of Jerusalem, Jerusalem, Israel
210Instituto de Investigaciones Biomédicas, UNAM, Mexico City, Mexico
211Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, Mexico
212Department of Public Health Faculty of Medicine University of Helsinki, Helsinki, Finland
213Department of Psychiatry and Human Behavior, University of California Irvine, Irvine, CA, USA
214Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Bioinformatics and Integrative Genomics Program, Harvard Medical School, Boston, MA, USA
215National Heart & Lung Institute & MRC London Institute of Medical Sciences, Imperial College London, London, UK
216Royal Brompton & Harefield Hospitals, Guy’s and St. Thomas’ NHS Foundation Trust, London, UK
217Radcliffe Department of Medicine, University of Oxford, Oxford, UK
218Department of Gastroenterology and Hepatology, University of Groningen and University Medical Center Groningen, Groningen, Netherlands
219Folkhälsan Institute of Genetics, Folkhälsan Research Center, Helsinki, Finland
220Big Data Institute, University of Oxford, UK
221Wellcome Centre for Human Genetics, University of Oxford, UK
222Division of Cardiology, Beth Israel Deaconess Medical Center, Boston, MA USA
223Program in Infectious Disease and Microbiome, Broad Institute of MIT and Harvard, Cambridge, MA, USA
224Center for Computational and Integrative Biology, Massachusetts General Hospital, Boston, MA, USA
Footnotes
Code availability
All code to perform quality control of the resource is publicly available at https://github.com/broadinstitute/gnomad_qc, and many of the functions are documented in a Python package (gnomad) at https://broadinstitute.github.io/gnomad_methods/index.html. The code to compute the constraint statistics is available at https://github.com/atgu/gnomad_nc_constraint.
Lists of authors and their affiliations appear at the end of the paper
Data availability
We release the aggregated allele frequency dataset at https://gnomad.broadinstitute.org, in a browser and bulk downloads for VCFs and Hail Tables, as well as all constraint statistics described in this manuscript. Additionally, we provide a subset of the dataset that includes individual level data for the HGDP110 and the 1000 Genomes projects111: the generation and use of this dataset is described in a companion manuscript112. There are no restrictions on the aggregate data released. External datasets used in this study are available in the following public resources:
ENCODE cCREs https://screen-v2.wenglab.org/, super enhancers http://www.licpathway.net/sedb/download.php, FANTOM5 enhancers https://fantom.gsc.riken.jp/5/datafiles/reprocessed/hg38_latest/extra/enhancer/, miRNA https://genome.ucsc.edu/cgi-bin/hgTables (All GENCODE V32 track), FANTOM5 lncRNA https://fantom.gsc.riken.jp/cat/v1/#/genes, GWAS Catalog https://genome.ucsc.edu/cgi-bin/hgTables (GWAS Catalog track), GWAS fine-mapping https://www.finucanelab.org/data, CNV morbidity map of developmental delay https://genome.ucsc.edu/cgi-bin/hgTables (Development Delay track), ClinVar https://genome.ucsc.edu/cgi-bin/hgTables (ClinVar Variants track), TOPMed https://bravo.sph.umich.edu/freeze8/hg38/downloads, ClinGen https://genome.ucsc.edu/cgi-bin/hgTables (ClinGen track), MGI https://www.informatics.jax.org/, OMIM https://www.omim.org/, Roadmap Epigenomics Enhancer-Gene Linking https://ernstlab.biolchem.ucla.edu/roadmaplinking/, GTEx https://gtexportal.org/home/datasets.
References
- 1.Short PJ et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 555, 611–616, doi: 10.1038/nature25983 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Satterstrom FK et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584 e523, doi: 10.1016/j.cell.2019.12.036 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh T et al. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat Genet 49, 1167–1173, doi: 10.1038/ng.3903 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganna A et al. Quantifying the Impact of Rare and Ultra-rare Coding Variation across the Phenotypic Spectrum. Am J Hum Genet 102, 1204–1211, doi: 10.1016/j.ajhg.2018.05.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443, doi: 10.1038/s41586-020-2308-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrovski S, Wang Q, Heinzen EL, Allen AS & Goldstein DB Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9, e1003709, doi: 10.1371/journal.pgen.1003709 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet 46, 944–950, doi: 10.1038/ng.3050 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hindorff LA et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106, 9362–9367, doi: 10.1073/pnas.0903103106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanyi JK Photochromism of halorhodopsin. cis/trans isomerization of the retinal around the 13–14 double bond. J Biol Chem 261, 14025–14030 (1986). [PubMed] [Google Scholar]
- 10.Mathelier A, Shi W & Wasserman WW Identification of altered cis-regulatory elements in human disease. Trends Genet 31, 67–76, doi: 10.1016/j.tig.2014.12.003 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Spielmann M & Mundlos S Looking beyond the genes: the role of non-coding variants in human disease. Hum Mol Genet 25, R157–R165, doi: 10.1093/hmg/ddw205 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Zhang F & Lupski JR Non-coding genetic variants in human disease. Hum Mol Genet 24, R102–110, doi: 10.1093/hmg/ddv259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seplyarskiy VB & Sunyaev S The origin of human mutation in light of genomic data. Nat Rev Genet 22, 672–686, doi: 10.1038/s41576-021-00376-2 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Seplyarskiy VB et al. Population sequencing data reveal a compendium of mutational processes in the human germ line. Science 373, 1030–1035, doi: 10.1126/science.aba7408 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gussow AB et al. Orion: Detecting regions of the human non-coding genome that are intolerant to variation using population genetics. PLoS One 12, e0181604, doi: 10.1371/journal.pone.0181604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.di Iulio J et al. The human noncoding genome defined by genetic diversity. Nat Genet 50, 333–337, doi: 10.1038/s41588-018-0062-7 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Halldorsson BV et al. The sequences of 150,119 genomes in the UK Biobank. Nature 607, 732–740, doi: 10.1038/s41586-022-04965-x (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kircher M et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46, 310–315, doi: 10.1038/ng.2892 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yousefian-Jazi A, Jung J, Choi JK & Choi J Functional annotation of noncoding causal variants in autoimmune diseases. Genomics 112, 1208–1213, doi: 10.1016/j.ygeno.2019.07.006 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Vitsios D, Dhindsa RS, Middleton L, Gussow AB & Petrovski S Prioritizing non-coding regions based on human genomic constraint and sequence context with deep learning. Nat Commun 12, 1504, doi: 10.1038/s41467-021-21790-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siepel A et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050, doi: 10.1101/gr.3715005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pollard KS, Hubisz MJ, Rosenbloom KR & Siepel A Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20, 110–121, doi: 10.1101/gr.097857.109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291, doi: 10.1038/nature19057 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halldorsson BV et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 363, doi: 10.1126/science.aau1043 (2019). [DOI] [PubMed] [Google Scholar]
- 25.An JY et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362, doi: 10.1126/science.aat6576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins RL et al. A structural variation reference for medical and population genetics. Nature 581, 444–451, doi: 10.1038/s41586-020-2287-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Consortium, E. P. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710, doi: 10.1038/s41586-020-2493-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersson R et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461, doi: 10.1038/nature12787 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang Y et al. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res 47, D235–D243, doi: 10.1093/nar/gky1025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pott S & Lieb JD What are super-enhancers? Nat Genet 47, 8–12, doi: 10.1038/ng.3167 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Bartel DP Metazoan MicroRNAs. Cell 173, 20–51, doi: 10.1016/j.cell.2018.03.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welter D et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42, D1001–1006, doi: 10.1093/nar/gkt1229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanai M et al. Insights from complex trait fine-mapping across diverse populations. medRxiv, 2021.2009.2003.21262975, doi: 10.1101/2021.09.03.21262975 (2021). [DOI] [Google Scholar]
- 34.Jung RG et al. Association between plasminogen activator inhibitor-1 and cardiovascular events: a systematic review and meta-analysis. Thromb J 16, 12, doi: 10.1186/s12959-018-0166-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song C, Burgess S, Eicher JD, O’Donnell CJ & Johnson AD Causal Effect of Plasminogen Activator Inhibitor Type 1 on Coronary Heart Disease. J Am Heart Assoc 6, doi: 10.1161/JAHA.116.004918 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaefer AS et al. Genetic evidence for PLASMINOGEN as a shared genetic risk factor of coronary artery disease and periodontitis. Circ Cardiovasc Genet 8, 159–167, doi: 10.1161/CIRCGENETICS.114.000554 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Li YY Plasminogen activator inhibitor-1 4G/5G gene polymorphism and coronary artery disease in the Chinese Han population: a meta-analysis. PLoS One 7, e33511, doi: 10.1371/journal.pone.0033511 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drinane MC, Sherman JA, Hall AE, Simons M & Mulligan-Kehoe MJ Plasminogen and plasmin activity in patients with coronary artery disease. J Thromb Haemost 4, 1288–1295, doi: 10.1111/j.1538-7836.2006.01979.x (2006). [DOI] [PubMed] [Google Scholar]
- 39.Lowe GD et al. Tissue plasminogen activator antigen and coronary heart disease. Prospective study and meta-analysis. Eur Heart J 25, 252–259, doi: 10.1016/j.ehj.2003.11.004 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Wang QS et al. Leveraging supervised learning for functionally informed fine-mapping of cis-eQTLs identifies an additional 20,913 putative causal eQTLs. Nat Commun 12, 3394, doi: 10.1038/s41467-021-23134-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landrum MJ et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46, D1062–D1067, doi: 10.1093/nar/gkx1153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stenson PD et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21, 577–581, doi: 10.1002/humu.10212 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Davydov EV et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6, e1001025, doi: 10.1371/journal.pcbi.1001025 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenway SC et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet 41, 931–935, doi: 10.1038/ng.415 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mefford HC et al. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet 81, 1057–1069, doi: 10.1086/522591 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sebat J et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449, doi: 10.1126/science.1138659 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stefansson H et al. Large recurrent microdeletions associated with schizophrenia. Nature 455, 232–236, doi: 10.1038/nature07229 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walsh T et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320, 539–543, doi: 10.1126/science.1155174 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Wright CF et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 385, 1305–1314, doi: 10.1016/S0140-6736(14)61705-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spielmann M, Lupianez DG & Mundlos S Structural variation in the 3D genome. Nat Rev Genet 19, 453–467, doi: 10.1038/s41576-018-0007-0 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Spielmann M & Mundlos S Structural variations, the regulatory landscape of the genome and their alteration in human disease. Bioessays 35, 533–543, doi: 10.1002/bies.201200178 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Coe BP et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46, 1063–1071, doi: 10.1038/ng.3092 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cooper GM et al. A copy number variation morbidity map of developmental delay. Nat Genet 43, 838–846, doi: 10.1038/ng.909 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klopocki E et al. Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am J Hum Genet 88, 70–75, doi: 10.1016/j.ajhg.2010.11.006 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barroso E et al. Identification of the fourth duplication of upstream IHH regulatory elements, in a family with craniosynostosis Philadelphia type, helps to define the phenotypic characterization of these regulatory elements. Am J Med Genet A 167A, 902–906, doi: 10.1002/ajmg.a.36811 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Will AJ et al. Composition and dosage of a multipartite enhancer cluster control developmental expression of Ihh (Indian hedgehog). Nat Genet 49, 1539–1545, doi: 10.1038/ng.3939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330, doi: 10.1038/nature14248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rehm HL et al. ClinGen--the Clinical Genome Resource. N Engl J Med 372, 2235–2242, doi: 10.1056/NEJMsr1406261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blake JA et al. The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res 39, D842–848, doi: 10.1093/nar/gkq1008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKusick VA Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet 80, 588–604, doi: 10.1086/514346 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585, doi: 10.1038/ng.2653 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu H et al. Elevated ASCL2 expression in breast cancer is associated with the poor prognosis of patients. Am J Cancer Res 7, 955–961 (2017). [PMC free article] [PubMed] [Google Scholar]
- 63.Jubb AM et al. Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene 25, 3445–3457, doi: 10.1038/sj.onc.1209382 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Tian Y et al. MicroRNA-200 (miR-200) cluster regulation by achaete scute-like 2 (Ascl2): impact on the epithelial-mesenchymal transition in colon cancer cells. J Biol Chem 289, 36101–36115, doi: 10.1074/jbc.M114.598383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443, doi: 10.1038/s41586-020-2308-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hail v. 0.2.62–84fa81b9ea3d. https://github.com/hail-is/hail/commit/84fa81b9ea3d.
- 67.Zhu P et al. Single-cell DNA methylome sequencing of human preimplantation embryos. Nat Genet 50, 12–19, doi: 10.1038/s41588-017-0007-6 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Tang WW et al. A Unique Gene Regulatory Network Resets the Human Germline Epigenome for Development. Cell 161, 1453–1467, doi: 10.1016/j.cell.2015.04.053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Halldorsson BV et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 363, doi: 10.1126/science.aau1043 (2019). [DOI] [PubMed] [Google Scholar]
- 70.An JY et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362, doi: 10.1126/science.aat6576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ross DA, Lim J, Lin R-S & Yang M-H Incremental learning for robust visual tracking. International journal of computer vision 77, 125–141 (2008). [Google Scholar]
- 72.Karolchik D et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32, D493–496, doi: 10.1093/nar/gkh103 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 30, 2843–2851, doi: 10.1093/bioinformatics/btu356 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davis CA et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 46, D794–D801, doi: 10.1093/nar/gkx1081 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goldmann JM et al. Germline de novo mutation clusters arise during oocyte aging in genomic regions with high double-strand-break incidence. Nat Genet 50, 487–492, doi: 10.1038/s41588-018-0071-6 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Zhao H et al. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007, doi: 10.1093/bioinformatics/btt730 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, doi: 10.1093/bioinformatics/btq033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kent WJ, Zweig AS, Barber G, Hinrichs AS & Karolchik D BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207, doi: 10.1093/bioinformatics/btq351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Collins RL et al. A structural variation reference for medical and population genetics. Nature 581, 444–451, doi: 10.1038/s41586-020-2287-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Consortium EP et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710, doi: 10.1038/s41586-020-2493-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andersson R et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461, doi: 10.1038/nature12787 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang Y et al. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res 47, D235–D243, doi: 10.1093/nar/gky1025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Welter D et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42, D1001–1006, doi: 10.1093/nar/gkt1229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kanai M et al. Insights from complex trait fine-mapping across diverse populations. medRxiv, 2021.2009.2003.21262975, doi: 10.1101/2021.09.03.21262975 (2021). [DOI] [Google Scholar]
- 85.Coe BP et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46, 1063–1071, doi: 10.1038/ng.3092 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cooper GM et al. A copy number variation morbidity map of developmental delay. Nat Genet 43, 838–846, doi: 10.1038/ng.909 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harrow J et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774, doi: 10.1101/gr.135350.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hon CC et al. An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 543, 199–204, doi: 10.1038/nature21374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang QS et al. Leveraging supervised learning for functionally informed fine-mapping of cis-eQTLs identifies an additional 20,913 putative causal eQTLs. Nat Commun 12, 3394, doi: 10.1038/s41467-021-23134-8 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang G, Sarkar A, Carbonetto P & Stephens M A simple new approach to variable selection in regression, with application to genetic fine-mapping. BioRxiv, 501114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gussow AB et al. Orion: Detecting regions of the human non-coding genome that are intolerant to variation using population genetics. PLoS One 12, e0181604, doi: 10.1371/journal.pone.0181604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.di Iulio J et al. The human noncoding genome defined by genetic diversity. Nat Genet 50, 333–337, doi: 10.1038/s41588-018-0062-7 (2018). [DOI] [PubMed] [Google Scholar]
- 93.Vitsios D, Dhindsa RS, Middleton L, Gussow AB & Petrovski S Prioritizing non-coding regions based on human genomic constraint and sequence context with deep learning. Nat Commun 12, 1504, doi: 10.1038/s41467-021-21790-4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Halldorsson BV et al. The sequences of 150,119 genomes in the UK Biobank. Nature 607, 732–740, doi: 10.1038/s41586-022-04965-x (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pollard KS, Hubisz MJ, Rosenbloom KR & Siepel A Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20, 110–121, doi: 10.1101/gr.097857.109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Siepel A et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050, doi: 10.1101/gr.3715005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Davydov EV et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 6, e1001025, doi: 10.1371/journal.pcbi.1001025 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Landrum MJ et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46, D1062–D1067, doi: 10.1093/nar/gkx1153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Taliun D et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299, doi: 10.1038/s41586-021-03205-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Budescu DV Dominance analysis: a new approach to the problem of relative importance of predictors in multiple regression. Psychological bulletin 114, 542 (1993). [Google Scholar]
- 101.Azen R & Budescu DV The dominance analysis approach for comparing predictors in multiple regression. Psychological methods 8, 129 (2003). [DOI] [PubMed] [Google Scholar]
- 102.Rehm HL et al. ClinGen--the Clinical Genome Resource. N Engl J Med 372, 2235–2242, doi: 10.1056/NEJMsr1406261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Blake JA et al. The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res 39, D842–848, doi: 10.1093/nar/gkq1008 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McKusick VA Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet 80, 588–604, doi: 10.1086/514346 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ernst J et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49, doi: 10.1038/nature09906 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu Y, Sarkar A, Kheradpour P, Ernst J & Kellis M Evidence of reduced recombination rate in human regulatory domains. Genome Biol 18, 193, doi: 10.1186/s13059-017-1308-x (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330, doi: 10.1038/nature14248 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Consortium, G. T. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585, doi: 10.1038/ng.2653 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Robin X et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC bioinformatics 12, 1–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bergstrom A et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 367, doi: 10.1126/science.aay5012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74, doi: 10.1038/nature15393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koenig Z et al. A harmonized public resource of deeply sequenced diverse human genomes. bioRxiv, 2023.2001. 2023.525248 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We release the aggregated allele frequency dataset at https://gnomad.broadinstitute.org, in a browser and bulk downloads for VCFs and Hail Tables, as well as all constraint statistics described in this manuscript. Additionally, we provide a subset of the dataset that includes individual level data for the HGDP110 and the 1000 Genomes projects111: the generation and use of this dataset is described in a companion manuscript112. There are no restrictions on the aggregate data released. External datasets used in this study are available in the following public resources:
ENCODE cCREs https://screen-v2.wenglab.org/, super enhancers http://www.licpathway.net/sedb/download.php, FANTOM5 enhancers https://fantom.gsc.riken.jp/5/datafiles/reprocessed/hg38_latest/extra/enhancer/, miRNA https://genome.ucsc.edu/cgi-bin/hgTables (All GENCODE V32 track), FANTOM5 lncRNA https://fantom.gsc.riken.jp/cat/v1/#/genes, GWAS Catalog https://genome.ucsc.edu/cgi-bin/hgTables (GWAS Catalog track), GWAS fine-mapping https://www.finucanelab.org/data, CNV morbidity map of developmental delay https://genome.ucsc.edu/cgi-bin/hgTables (Development Delay track), ClinVar https://genome.ucsc.edu/cgi-bin/hgTables (ClinVar Variants track), TOPMed https://bravo.sph.umich.edu/freeze8/hg38/downloads, ClinGen https://genome.ucsc.edu/cgi-bin/hgTables (ClinGen track), MGI https://www.informatics.jax.org/, OMIM https://www.omim.org/, Roadmap Epigenomics Enhancer-Gene Linking https://ernstlab.biolchem.ucla.edu/roadmaplinking/, GTEx https://gtexportal.org/home/datasets.