

Abstract
This study examines the impact of cytogenetic abnormalities and their co‐segregation on the prognosis of newly diagnosed multiple myeloma patients. The analysis included 1304 patients from four different GEM‐PETHEMA clinical trials. Genetic alterations, such as t(4;14), t(14;16), del(17p), +1q, and del(1p), were investigated using FISH on CD38 purified plasma cells. The frequency of genetic alterations detected were as follows: del(17p) in 8%, t(4;14) in 12%, t(14;16) in 3%, +1q in 43%, and del(1p) in 8%. The median follow‐up was 61 months, and the median progression‐free survival (PFS) and overall survival (OS) were 44 months and not reached, respectively. Consistent with previous reports, the presence of t(4;14) was associated with shorter PFS and OS. In our series, the presence of t(14;16) did not impact survival, maybe due to limitations in sample size. Del(17p) was linked to poor prognosis using a cut‐off level of ≥20% positive cells, without any impact of higher cut‐off in prognosis, except for patients with clonal fraction ≥80% who had a dismal outcome. Cosegregation of cytogenetic abnormalities patients worsened the prognosis in t(4;14) patients but not in patients with del(17p), which retained its adverse prognosis even as a solitary abnormality. Gain(1q) was associated with significantly shorter PFS and OS, while del(1p) affected PFS but not OS. Nevertheless, when co‐segregation was eliminated, the detrimental effect of +1q or del(1p) was no longer observed. In conclusion, this study confirms the prognostic significance of high‐risk cytogenetic abnormalities in MM and highlights the importance of considering co‐occurrence for accurate prognosis assessment.
INTRODUCTION
Multiple myeloma (MM) is a genetically complex neoplasm because of the multiple chromosomal and genetic lesions present in the clonal plasma cells. 1 Some of these abnormalities have been consistently associated with poor prognosis, which has led to the definition of high‐risk cytogenetic alterations encompassing t(4;14), t(14;16), deletion of 17p13 [del(17p)] and, more recently, chromosome 1 abnormalities. 1 Prognosis has dramatically changed in the last decades due to the advent of new treatment strategies that included proteasome inhibitors, immunomodulatory agents, and monoclonal antibodies. 2 , 3 Still, patients harboring high‐risk features continue to do poorly, and, so far, no treatment or combination has been able to completely abrogate the poor prognosis of high‐risk cytogenetic abnormalities, and they continue to be one of the more relevant baseline prognostic features. 4 The diagnostic approach for MM genetic alterations in the clinical setting is based on fluorescence in situ hybridization (FISH) in separated plasma cells because it is the most standardized, time and cost‐optimized approach to obtain a reliable result and to individualize risk assessment. 5 , 6 Indeed, high‐risk cytogenetic abnormalities have been incorporated into the revised international scoring system (R‐ISS) recommended by the International Myeloma Working Group (IMWG). 6 , 7 Whether each high‐risk alteration can condition an unfavorable prognosis on its own or whether, on the contrary, some of them need the cooperation of other alterations to negatively impact patient survival is not fully elucidated. In this regard, it has been shown that the poor prognosis associated with chromosome 13 monosomy was caused by its close association with other high‐risk alterations, such as t(4;14). 7 Additionally, other studies have demonstrated that the co‐segregation of more than one of the high‐risk lesions significantly impairs the prognosis. 5 , 8 , 9
In this study, we aimed to analyze the prognostic impact of cytogenetic abnormalities detected by FISH in a large series of newly diagnosed MM patients included in four clinical trials conducted by the Spanish myeloma group, with particular interest in assessing the role of additional chromosomal changes in the patient outcome.
METHODS
Patients from four large prospective Spanish trials who had been studied for the presence of high‐risk cytogenetic abnormalities were included in this study. Two of the trials evaluated the frontline treatment in newly diagnosed transplant‐eligible (NDTE) (GEM05MENOS65 and GEM2012) 10 , 11 , 12 , and the other two were conducted in newly diagnosed nontransplant eligible MM patients (GEM05MAS65 and GEM2010 trials). 13 , 14
The institutional review board or independent ethics committee at each participating center approved the study. In accordance with the Declaration of Helsinki, all patients provided written informed consent before screening.
FISH studies
FISH studies included the detection of t(4;14), t(14;16), del(17p), 1q gain [gain(1q)], and 1p loss [del(1p)]. All analyses were carried out in CD138‐selected bone marrow plasma cells as described previously. 7 , 15 According to the recommendations made by the European Myeloma Network (EMN) FISH workshop, 5 the cut‐off level for considering a positive result was set at 10% for IGH translocations (fusion/break‐apart probes), and 20% for del(17p), gain(1q) and del(1p). Additionally, for patients with gain(1q), 1q amplification [amp(1q)] was defined as four or more copies of 1q.
Statistical analyses
To explore differences between comparison groups, chi‐square, student t, and Mann–Whitney U tests were used, as appropriate. p < 0.05 was considered statistically significant.
Progression‐free survival (PFS) was measured as the time from the date of diagnosis to disease progression or death, regardless of cause, and overall survival (OS) as the time from diagnosis to death from any cause. If progression or death did not occur at the last follow‐up date, they were censored for PFS or OS. Kaplan–Meier method was used to estimate time‐to‐event, and significance was determined with a two‐sided long‐rank test.
The prognostic impact of the high‐risk cytogenetic abnormalities was analyzed by comparing the outcomes in terms of complete response (CR) rate, PFS, and OS of patients between high‐risk versus standard‐risk cytogenetic abnormalities. Disease response was assessed according to the IMWG Response criteria 2014. 12 Cox proportional hazards regression model was used to estimate hazard ratio (HR) and 95% confidence interval (CI). All statistical analyses were performed by using IBM© SPSS© Version 24.0 software and STATA v15.0.
RESULTS
Patient characteristics
A total of 1304 patients were enrolled in the Spanish Myeloma Trials, namely GEM05MAS65, GEM05MENOS65, GEM2010, and GEM2012 (Supporting information S1: Table S1). Baseline characteristics are listed in Table 1.
Table 1.
Basal characteristics of 1304 MM patients included in the Spanish Myeloma upfront trials (GEM05MAS65, GEM05MENOS65, GEM2010, and GEM2012).
| Basal characteristics | No. (%) |
|---|---|
| Gender | |
| Male | 684 (52.5) |
| Female | 620 (47.6) |
| Age at diagnosis | |
| ≥75 years | 194 (14.9) |
| <75 years | 1109 (85.1) |
| Type of MM | |
| Ig G, IG A or Ig D MM | 1118 (85.7) |
| Light chain MM | 175 (13.4) |
| Light chain subtype | |
| Kappa | 805 (61.7) |
| Lambda | 488 (37.4) |
| Serum M‐protein | |
| <1 g/dL | 224 (17.2) |
| ≥1 g/dL | 1065 (81.7) |
| Serum M‐protein | |
| ≥3 g/dL | 706 (54.14) |
| <3 g/dL | 576 (44.17) |
| BM PC by morphology | |
| ≥60% | 270 (20.71) |
| 10%–59% | 960 (73.62) |
| Hypercalcemia (>11 g/dL) | 112 (8.59) |
| Creatinine | |
| ≥1.5 g/dL | 133 (10.2) |
| <1.5 g/dL | 1165 (89.3) |
| Hemoglobin | |
| ≤10 g/dL | 486 (37.3) |
| >10 g/dL | 813 (62.4) |
| ISS stage | |
| I | 432 (33.5) |
| II | 545 (42.3) |
| III | 312 (24.2) |
| High LDH | 193 (14.8) |
| High‐risk cytogenetic abnormalities | 234 (17.9) |
Abbreviations: BM PC, bone marrow plasma cells; ISS, International Staging System; LDH, lactate dehydrogenase; MM, multiple myeloma; No., number of patients; NS, not statistically significant; yrs, years old.
Although most of the patients were simultaneously evaluated for the presence of the five aforementioned cytogenetic abnormalities by FISH, it was not possible to study all of them in all patients, due to the lack of samples. In particular, del(17p) was studied in 1157 out of 1304 patients (89%); t(4;14) in 1165 (89%); t(14;16) in 1150 (88%); gain(1q) in 906 (69%) and del(1p) in 902 (69%) (Table 2). In the early trials, the investigation of chromosome 1 abnormalities was not mandatory, which explains the lower number of cases analyzed.
Table 2.
Frequency of cytogenetic abnormalities and distribution across newly diagnosed MM patients included in the Spanish Myeloma upfront (GEM05MAS65, GEM05MENOS65, GEM2010, and GEM2012).
| Spanish myeloma trials | del(17p) | t(4;14) | t(14;16) | gain(1q) | del(1p) |
|---|---|---|---|---|---|
| All | |||||
| Patients with genetic alteration, n (%) | 89 (7.7) | 142 (12.2) | 31 (2.7) | 358 (39.5) | 47 (5.2) |
| Total of patients included, n | 1304 | 1304 | 1304 | 1304 | 1304 |
| Pts analyzed, n | 1157 | 1165 | 1150 | 906 | 902 |
| Missing data, n | 147 | 139 | 154 | 398 | 402 |
| GEM05MAS65*1 | |||||
| Patients with genetic alteration, n (%) | 24 (10.4) | 20 (8.7) | 3 (1.3) | 59 (38.1) | 7 (4.5) |
| Total of patients included, n | 257 | 257 | 257 | 257 | 257 |
| Pts analyzed, n | 230 | 231 | 229 | 155 | 154 |
| Missing data, n | 27 | 26 | 28 | 102 | 103 |
| GEM2010*1 | |||||
| Patients with genetic alteration, n (%) | 10 (5.0) | 23 (11.5) | 2 (1.0) | 70 (46.4) | 5 (3.3) |
| Total of patients included, n | 236 | 236 | 236 | 236 | 236 |
| Pts analyzed, n | 200 | 200 | 194 | 151 | 150 |
| Missing data, n | 36 | 36 | 42 | 85 | 86 |
| GEM05MENOS65*2 | |||||
| Patients with genetic alteration, n (%) | 20 (6.1) | 42 (12.6) | 9 (2.7) | 74 (33.9) | 7 (3.2) |
| Total of patients included, n | 354 | 354 | 354 | 354 | 354 |
| Pts analyzed, n | 328 | 333 | 332 | 218 | 218 |
| Missing data, n | 26 | 21 | 22 | 136 | 136 |
| GEM2012*2 | |||||
| Patients with genetic alteration, n (%) | 35 (8.8) | 57 (14.2) | 17 (4.3) | 155 (40.5) | 28 (7.4) |
| Total of patients included, n | 457 | 457 | 457 | 457 | 457 |
| Pts analyzed, n | 399 | 401 | 395 | 382 | 380 |
| Missing data, n | 58 | 56 | 62 | 75 | 77 |
Abbreviations: *1, trials for nontransplant eligible patients; *2, trials for transplant‐eligible patients; n, number of patients; Pts, patients.
As expected, the most frequent cytogenetic abnormalities were those affecting chromosome 1, present in half of these newly diagnosed MM patients. Gain(1q) was observed in 43% of patients, while del(1p) in 8% of the cases. t(4;14) was the most frequent IGH translocation, observed in 12%. Finally, del(17p) was detected in 8% of patients (Table 2). The distribution of genetic abnormalities according to the clinical trial is also shown in Table 2. No differences in the CR rate after induction were observed between patients with or without cytogenetic abnormalities (Supporting information S1: Table S2). Besides, we analyzed the impact of cytogenetic abnormalities in the outcome of transplant‐eligible and nontransplant‐eligible patients, and no differences were observed for del(17p) and gain(1q), only we did see a difference in the impact of t(4;14) in the two groups with an HR of 1.73 (95% confidence interval [CI]: 1.17–2.57; p 0.006) with a median PFS in younger patients of 41.4 versus 23.7 m in patients older than 65 years old. This comparison was not analyzed for the t(14;16) and del(1p) due to sample size limitations. In addition, we analyzed the distribution of the different hits among the two age groups and found no differences in the univariate analysis (p 0.09).
The median follow‐up was 60.9 months (interquartile range [IQR]: 51.1–74.3 months) for the whole population. Overall, 816 (62.6%) progressed or died at the last follow‐up, and the estimated median PFS was 43.8 months (95% CI: 40.2–47.4 months). So far, 483 (37%) patients have died, and the median OS was not yet reached during this follow‐up, with a 3‐year OS probability of 77.6% and a 5‐year OS of 63.4%.
t(4;14)
This translocation was found in 142 (12.2%) patients (Table 2). This group was characterized by more frequent anemia at presentation (47% vs. 36%, p 0.01), higher amount of M‐protein (>3 g/dL in 67% vs. 54.7% of patients, respectively, p 0.006), and a lower proportion of light chain MM as compared to those without t(4;14) (7.7% vs. 13.9%, p 0.04) (Supporting information S1: Table S3).
With regards to outcome, patients with t(4;14) had shorter PFS (median PFS 27.7 vs. 45.3 months, HR: 1.5 [95% CI: 1.2–1.8], p < 0.001). Furthermore, t(4;14) was associated with twice the risk of death (HR: 2.0 [95% CI: 1.5–2.5]; p < 0.001), and, consequently, the 3‐year OS probability was significantly worse for patients with t(4;14) than for those without it (61.0% vs. 80.2%, respectively) (Supporting information S1: Table S3).
This cytogenetic abnormality was presented more frequently in association with other genetic alterations or hits (del(17p), gain(1q), or (1p‐) than alone (66.0% vs. 44.0%). Among 106 patients with t(4;14) studied for all abnormalities, co‐occurrence with another hit was observed in 56.6% of cases, with 2 hits in 9.4% and with 3 hits in one patient (Table 3). The coexistence of t(4;14) with other high‐risk abnormalities had a negative impact on outcome. Thus, in the group t(4;14) with one or more additional hits, PFS was significantly shorter than in those with t(4;14) alone (median PFS 23.8 vs. 63.3 months HR: 2.8 (1.6–5.1); p < 0.001), and OS as well with a median of 38.5 vs. 80.7 months, respectively, HR: 3.2 (1.5–7.0); p = 0.003. (Figure 1A–D).
Table 3.
Summary of genetic abnormalities, including description of additional genetic hits in the 898 patients studied for all abnormalities.
| Genetic abnormality | Co‐segregation (n, %) | +1 hit (n, %) | 2 or more hits (n, %) | Partner (n) |
|---|---|---|---|---|
|
del(17p) n = 73 |
41 (56.1) | 26 (35.6) | 15 (20.5) |
t(4;14): 14 t(14;16): 8 gain(1q): 28 del(1p): 9 |
|
t(4;14) n = 106 |
70 (66.0) | 60 (56.6) | 10 (9.4) |
del(17p): 12 t(14;16): ‐ gain(1q): 58 del(1p): 2 |
|
t(14;16) n = 28 |
20 (71.4) | 14 (50.0) | 6 (21.4) |
del(17p): 8 t(4;14): ‐ gain(1q): 17 del(1p): 3 |
|
gain(1q) n = 356 |
111 (31.1) | 95 (26.7) | 16 (4.5) |
del(17p): 28 t(4;14): 61 t(14;16): 17 del(1p): 24 |
|
del(1p) n = 47 |
32 (68.1) | 28 (59.6) | 4 (8.5) |
del(17p): 9 t(4;14): 5 t(14;16): 3 gain(1q): 24 |
Figure 1.
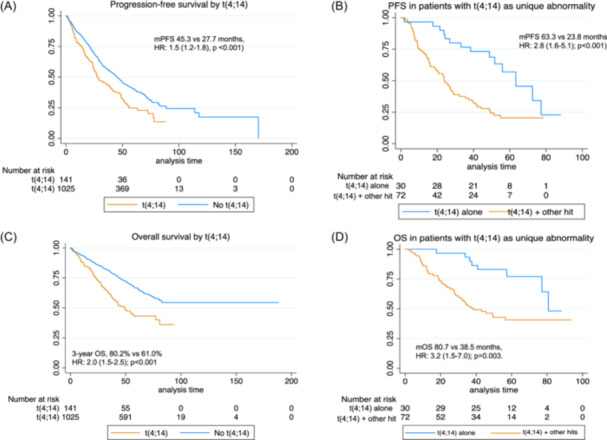
Survival curves by status of t(4;14). (A) Progression‐free survival (PFS) by overall presence or absence of t(4;14). (B) PFS in patients with t(4;14) alone or with other hits. (C) Overall survival by t(4;14). (D) Overall survival in patients with t(4;14) alone or with other hits. mPFS, median progression‐free survival; mOS, median overall survival; OS, overall survival.
t(14;16)
In our series, only 31 (2.7%) patients harbored t(14;16) at the moment of diagnosis (Table 2). This translocation was associated with ISS 3 (43% vs. 24%, p 0.05) (Supporting information S1: Table S4). In fact, higher mean ₂‐microglobulin values were found in the group with t(14;16) than in the group without it (6.2 vs. 4.5 mg/L, respectively, p = 0.005).
The presence of t(4;16) was not associated with statistically significant differences in terms of PFS or OS (Table 4). Interestingly, 8 patients with t(14;16) co‐harbored del(17p) (25.8%) and displayed a very short OS (18.1 months vs. NR for patients without del(17p); HR: 3.0 [95% CI: 1.01–8.73]; p = 0.05).
Table 4.
Outcomes of newly diagnosed MM patients included in the Spanish Myeloma trials (GEM05MAS65, GEM05MENOS65, GEM2010, GEM2012) by cytogenetic abnormalities.
| Frec, % (No. of patients studied) | Median PFS (m) | 3‐yr PFS (%) | HR (95% CI) | p‐value | Median OS (m) | 3‐yr OS (%) | HR (95% CI) | p‐value | |
|---|---|---|---|---|---|---|---|---|---|
| t(4;14) | 12.2 (1165) | 27.7 | 44.3 | 1.5 (1.2–1.8) | <0.001 | 49.3 | 61.0 | 2.0 (1.5–2.5) | <0.001 |
| Non t(4;14) | 45.3 | 56.7 | NR | 80.2 | |||||
| t(14;16) | 2.7 (1150) | 27.5 | 45.2 | 1.2 (0.8–1.9) | NS | NR | 58.1 | 1.4 (0.8–2.4) | NS |
| Non t(14;16) | 43.6 | 55.3 | NR | 78.4 | |||||
| del17p | 7.7 (1157) | 23.9 | 29.5 | 1.9 (1.5–2.4) | <0.001 | 36.9 | 50.9 | 2.5 (1.9–3.3) | <0.001 |
| Non del17p | 46.0 | 57.5 | NR | 80.4 | |||||
| Gain (1q) | 40.1 (906) | 36.5 | 50.1 | 1.4 (1.2–1.7) | <0.001 | 80.6 | 73.9 | 1.5 (1.2–1.9) | <0.001 |
| Non gain (1q) | 53.2 | 62.2 | NR | 83.4 | |||||
| del(1p) | 5.2 (902) | 41.1 | 51.0 | 0.7 (0.5–0.97) | 0.04 | NR | 80.6 | 0.7 (0.4–1.1) | NS |
| Non del(1p) | 48 | 57.0 | NR | 67.6 |
Abbreviations: CI, confidence interval; frec, frequency; HR, hazard ratio; MM, multiple myeloma; mo, months; No., number of patients; NR, not reached; NS, not statistically significant; OS, overall survival; PFS, progression‐free survival; yr, years.
del(17p)
There were 89 (7.7%) MM patients harboring del(17p) (Table 2). This abnormality was slightly more frequent in light chain MM than in IgG, IgA, or IgD MM (22 vs. 13%, respectively, p = 0.01) as well as in patients with oligosecretory disease (26 vs. 16%, p = 0.02). Interestingly, other characteristics of poor prognosis were also more frequent in the del(17p) group. In fact, one‐third of these patients had high LDH at the moment of diagnosis (30.3% vs. 13.7%, p < 0.001), advanced clinical stage (ISS III) (33.7% vs. 23.8%, p 0.03), or high bone marrow infiltration (≥60% BMPC) (30.5% vs. 20.8%, p 0.04) (Supporting information S1: Table S5).
To evaluate the impact on survival of the clonal fraction, different cut‐off points were analyzed based on the percentage of cells harboring del(17p). In our cohort, there were no differences in the outcome among the different subgroups analyzed (≥20% up to ≥70%) (Supporting information S1: Table S6). The median PFS estimated in those with ≥80% cells with del(17p) was just over a year, although this group of 22 patients was especially enriched in other high‐risk cytogenetic abnormalities, 17 out of 22 (77%) had another hit (8 patients with gain(1q), 6 with 1p‐, 2 with (4;14) and 1 with t(14;16)).
Among 73 patients with del(17p) who had been studied for coexistence with other genetic abnormalities (translocations IGH [(t4;14) and t(14;16)], gain(1q) or del(1p)), co‐occurrence of another hit was observed in 26 (35.6%) patients and with 2 hits or more in 15 (20.5%). In the remaining 43.8% of cases, del(17p) occurred alone. No statistically significant differences were found in PFS or OS when patients with del(17p) alone were compared with those with co‐occurrence of one or more hits, including del(17p) (Figure 2A–D).
Figure 2.
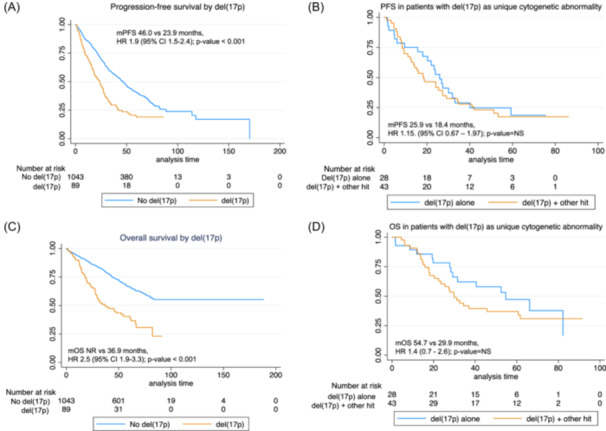
Survival curves by status of del(17p). (A) Progression‐free survival (PFS) by overall presence or absence of del(17p). (B) PFS in patients with del(17p) as a single cytogenetic abnormality. (C) Overall survival (OS) by del(17p). (D) OS in patients with del(17p) as a single cytogenetic abnormality. NS, nonsignificant.
Gain(1q)
Gain(1q) was analyzed in 906 out of the total of 1304 patients. A total of 358 out of 906 patients tested (39.5%) were positive for this abnormality (Table 2). In 346 positive patients, the 1q gains were differentiated from the amplifications so that gain(1q) was present in 217 patients (62.7% of positive cases), and amp(1q) (4 or more copies) was detected in 129 patients (37.2%). The presence of gain(1q) was evenly distributed between the 4 studies (Table 2). Patients with gain(1q) were more frequently associated with ISS 2 or 3, anemia, and t(4;14) or t(14;16) as compared to patients without gain(1q).
Patients harboring gain(1q) showed a shorter PFS as compared to patients without gain(1q) (36.5 vs. 53.2 months, HR 1.40 [95% CI: 1.2–1.7], p = 0.0001) as well as decreased OS (80.6 vs. NR; HR 1.5 [95% CI: 1.2–1.9], p = 0.0002) (Table 4 and Figure 3A,B).
Figure 3.
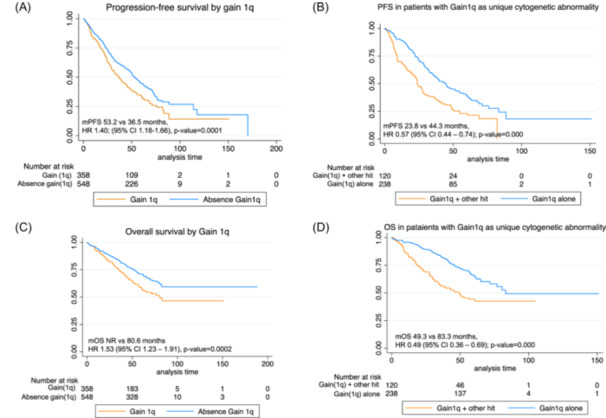
Survival curves by status of gain(1q). (A) Progression‐free survival by overall presence or absence of gain(1q). (B) Progression‐free survival in patients with gain(1q) as a single cytogenetic abnormality. (C) Overall survival by gain(1q). (D) Overall survival in patients with gain(1q) as a single cytogenetic abnormality.
Gain(1q) was also associated with other high‐risk cytogenetic abnormalities. Thus, 28 patients had also del(17p), 61 patients t(4;14), 17 patients t(14;16), and 24 patients del(1p). Thereby, when we analyzed patients harboring gain(1q) as an isolated cytogenetic aberration (excluding patients with co‐segregation of other high‐risk cytogenetic abnormalities), no negative impact in PFS was found, and median PFS was comparable between patients with gain(1q) as single alteration and those without gain(1q) (median PFS 44.3 vs. 53.2 months; HR: 1.17 [95% CI: 0.96–1.43], p = 0.110). Similar results were found for OS (median OS 83.2 months vs. not reached; HR: 1.2 [95% CI: 0.92–1.56], p = 0.173), suggesting that co‐segregation of other cytogenetic abnormalities is critical in the prognosis of gain(1q). Thus, patients with gain(1q) harboring other cytogenetic abnormalities showed an inferior PFS (23.8 vs. 44.3 months, HR: 0.57 [95% CI: 0.44–0.74]; p = 0.0001) and OS (49.3 vs 83.3 months, HR: 0.49 [95% CI: 0.36–0.69]; p = 0.0001), as compared to patients with gain(1q) alone (Figure 3C,D). We also analyzed the impact of different cut‐off values for gain(1q); however, no differences in PFS were found between the cut‐offs analyzed.
Amp(1q) was also associated with other high‐risk genetic abnormalities. Among 129 patients with amp(1q) in this cohort, 32 and 9 patients presented 1 and 2 additional genetic hits, respectively. Patients with amp(1q) only had a longer PFS (median PFS 46.0 m [95% CI: 35.7–60.5]) compared to patients with 1 additional hit (median PFS 21.9 m [95% CI: 9.2–44.2 m]) or two additional hits (median PFS 15.4 m [7.9–NE]); HR: 1.62 (95% CI: 1.15–2.25); p 0.0081. The median PFS for patients with amp(1q) only was similar to that of patients without amp(1q) (median PFS 46 m [95% CI: 35.7–60.5] vs. 44.1 m [95% CI: 39.9–47.8 m]). Likewise, the median OS for patients with amp(1q) alone was longer (median NE, [95% CI: 65.1–NE] as compared to two patients with 1 (median NE [95% CI: 18.6–NE]) or 2 additional hits, respectively (median 22.4 m [95% CI: 15.9–NR].
In our series, we were not able to find significant differences in outcome between 1q gains and amplifications (median PFS gain(1q) 33.3 vs. 38.2 months for amp(1q); HR: 0.86 [95% CI: 0.66−1.12]; p = 0.25).
del(1p)
Among all included patients, deletions of 1p (del(1p)) were found in 47 patients out of the 902 analyzed (5.2%) (Table 2). A negative impact in PFS was observed with a median PFS in patients with del(1p) of 41.1 m as compared to 48 m for patients without del(1p) (HR: 0.7 [95% CI: (0.48–0.97)]; p = 0.04). However, OS was not affected by the presence of del(1p) abnormality (Table 4 and Figure 4A). Co‐segregation with other cytogenetic abnormalities was less common except for gain(1q), which was present in 24 out of the 47 patients (51%) with del(1p). Other abnormalities observed were t(4;14) in five patients, t(14;16) in three patients, and del(17p) in nine patients. Interestingly, upon analyzing patients with del(1p) as the unique cytogenetic abnormality, the negative impact of del(1p) in PFS was abrogated (HR: 1.3, [95% CI: 0.84–1.97], p 0.24). Finally, upon analyzing the role of co‐segregation in patients with del(1p), median PFS was numerically shorter in patients with del(1p) plus other hits, although differences were not statistically different (Figure 4B,D).
Figure 4.
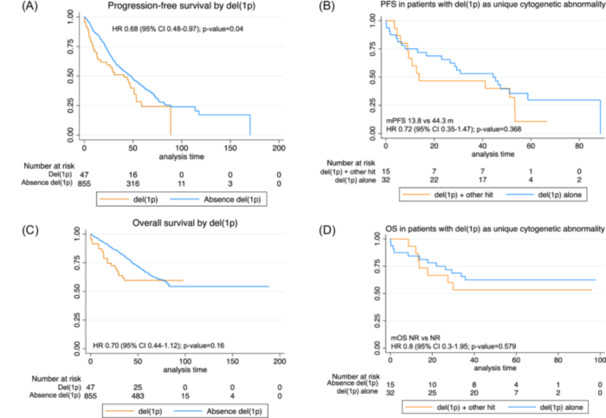
Survival curves by status of del(1p). (A) Progression‐free survival by overall presence or absence of del(1p). (B) Progression‐free survival in patients with del(1p) as a single cytogenetic abnormality. (C) Overall survival by del(1p). (D) Overall survival in patients with del(1p) as a single cytogenetic abnormality.
Impact of high‐risk genetic abnormalities on R‐ISS and R2‐ISS
As expected, patients with R‐ISS III had a significantly shorter median PFS as compared to patients with R‐ISS II or I (26.2 vs. 40.1 vs. 63.4 months, respectively, p < 0.01), and the HR for PFS was 2.6 and 1.6, using R‐ISS I as the reference group. Furthermore, the R‐ISS model discriminated three groups with significantly different OS, with a median of 39.2 versus 82.8 versus NR for R‐ISS III, II, and I, respectively, and HR of 2.1 and 4.5 for R‐ISS II and III, respectively (Figure 5A,C).
Figure 5.
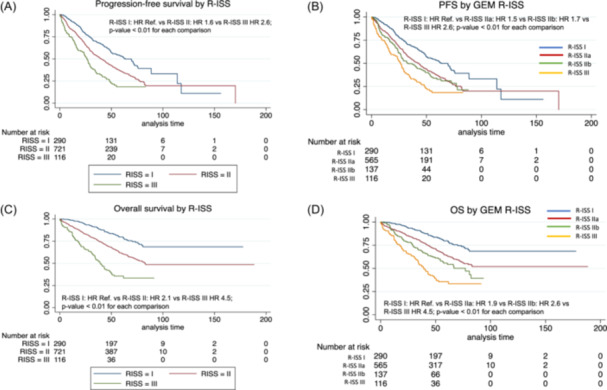
Survival curves by R‐ISS. (A) Pogression‐free survival by R‐ISS. (B) Progression‐free survival by R‐ISS in four categories considering group II split into two subgroups according to the absence or presence of high‐risk cytogenetic abnormalities (IIa and IIb, respectively). (C) Overall survival R‐ISS. (D) Overall survival by R‐ISS in four categories considering group II split into two subgroups according to the absence or presence of high‐risk cytogenetic abnormalities (IIa and IIb, respectively).
In order to investigate the impact of HR cytogenetic abnormalities on the survival of the R‐ISS II group, we re‐classified R‐ISS II into two subgroups according to the absence or presence of HR cytogenetic abnormalities (IIa and IIb, respectively), comparing them with either R‐ISS I or III, and subsequently, defining four groups. Interestingly, statistically significant differences in terms of PFS and OS were found among these groups. After applying this new model, 51% of patients were classified as R‐ISS IIa with a median PFS of 42.7 months, (HR: 1.5), and a median OS: NR, (HR: 1.9); whereas 12% were classified as R‐ISS IIb with a median PFS of 33.1 months, (HR: 1.7) and a median OS of 77 months (HR: 2.6); the rest of patients remained in their corresponding R‐ISS I or III groups, with the previously mentioned outcomes (Figure 5B,D).
Besides, R2‐ISS that incorporates gain(1q) could be applied in 889 patients. R2‐ISS I patients were 166 (18.7%), R2‐ISS II 271 (30.5%), R2‐ISS III 384 (43.2%), and R2‐ISS IV 68 (7.6%). Median PFS was 69.5 (95% CI, 58.5 to 80.4) versus 54.1 (95% CI: 47.5–60.6) versus 41.3 (95% CI: 35.2–47.4) versus 15.7 (95% CI: 9.0–22.3) months, respectively.
Median OS was NR in R2‐ISS I and II (95% CI: NR–NR) versus 88.2 (95% CI: NR–NR) versus 29.8 (95% CI: 19.1–40.6) in the R2‐ISS III and IV groups, respectively. The differences among the R2‐ISS groups were statistically significant (Supporting information S2: Figures S1A and S1B).
DISCUSSION
In this study, we analyzed the impact of different cytogenetic abnormalities in a large series of 1304 newly diagnosed MM patients who were enrolled in upfront trials conducted by the Spanish myeloma group. As previously described, the presence of high‐risk cytogenetic abnormalities (del(17p), t(4;14), and t(14;16)) is associated with inferior outcomes in terms of survival despite not having a negative impact in the response to frontline therapy. 4 , 5
Regarding t(4;14), in our series, this cytogenetic abnormality was detected in 12.2% of the patients and was associated with a negative impact in both PFS and OS. Interestingly, although patients harboring t(4;14) are usually considered as high risk, in our experience, this translocation was frequently associated with other genetic abnormalities (71% of the cases), and therefore, it is very difficult to evaluate the independent contribution of t(4;14). In this regard, the impact of t(4;14) in prognosis has been largely discuss by several groups highlighting the heterogeneity in the prognostic impact of this genetic abnormality. 7 , 16 , 17 , 18 Thus, the French group has analyzed the prognosis of t(4;14) and has shown that only patients with t(4;14 plus del(1p32) or t(4;14) in the absence of trisomy 3 or 5 or presence of trisomy 21 had an inferior PFS. 17 , 19 , 20 , 21 In addition, the breakpoint location on chromosome 4 may also play a role in modifying the prognosis impact of t(4;14). 22 Unfortunately, this interesting recent finding cannot be identified by a straightforward method, such as the FISH technique.
As in other studies, t(14;16) was a very uncommon abnormality, and the associated adverse prognosis could not be proven, likely due to the small number of cases that carried this abnormality in this study, resulting in a rather small sample size. 23 , 24 However, as stated in the R‐ISS model by IMWG, t(14;16) was confirmed to be a high‐risk cytogenetic abnormality in a retrospective study with the largest number of patients harboring this abnormality. 25 As in this last study, we found that t(14;16) is more frequent in women than in men.
Consistent with previous reports, we demonstrate that the presence of del(17p) is associated with a shorter PFS and OS. The impact of clonal fractions on survival has been evaluated in different studies. 17 , 26 , 27 , 28 , 29 In our study, the optimal cut‐off for del(17p) was ≥20%. We found no differences between 20% and 70% cut‐off points, in contrast with previous results reported by other groups that suggest 50%–60% as the most discriminative cut‐off values for prognostication, 26 , 27 , 28 , 30 , 31 , 32 although, in any of those, a difference in OS was shown. 32 , 33 Interestingly, when whole‐exome sequencing data were combined, it was shown that TP53 mutation was a necessary requirement to identify poor prognosis in patients with CCF > 55%, and no significant differences were displayed between groups with >55 and <55% if TP53 mutation is not present. 27 Unfortunately, TP53 mutations were not tested in our studies.
As previously described, del(17p) was associated with other adverse prognostic factors such as high bone marrow infiltration, high LDH, and ISS III. 28 This could reinforce the notion that MM with del(17p) has a unique pathogenic program. Indeed, MM with del(17p) responds initially well to treatment, but early relapses are the rule. According to our results, unlike other cytogenetic abnormalities, in patients with del(17p), the cosegregation with other hits does not have a significant impact on the outcome. This may mean that del(17p) by itself is a clear determinant of MM outcome. In fact, this group of patients should be considered as a different MM subtype with a dismal prognosis, for which more targeted clinical trials should be designed.
As it has been previously reported, gain(1q) is the most frequent cytogenetic abnormality found in MM. 32 In our study, patients with gain(1q) had an inferior PFS and OS as compared to patients without this genetic abnormality. We did not find any difference between gain(1q) and amp(1q), but these findings may be limited by the sample size. Nevertheless, the results in this regard are controversial. 34 , 35 Interestingly and consistent with other reports, 36 , 37 the negative impact of gain(1q) on PFS and OS was not observed upon analyzing only those patients with gain(1q) as a unique cytogenetic abnormality, suggesting that this is not an independent prognostic factor.
Similarly, the negative impact of del(1p) was also abrogated upon analyzing patients with del(1p) as a unique abnormality. This finding is in contrast with other reports, 37 and our analysis may be limited by the small number of patients with del(1p) in our series. In addition, we have not been able to evaluate the role of biallelic deletion that has been shown to identify patients with ultra‐high‐risk disease with a median OS of only 24 months. 38
Finally, we validated a simple modification on the R‐ISS. As everybody agrees, stage II is a dark box that includes many patients but with a heterogenous outcome. 37 , 39 The simple recategorization of group R‐ISS II into two subcategories according to the presence or absence of cytogenetic abnormalities could help to better predict the risk of these patients at presentation, and, accordingly, to individualize treatment with the goal of achieving the deepest response in these high‐risk patients.
In summary, we validate the prognostic impact of high‐risk cytogenetic abnormalities at diagnosis in a large series of MM patients. We have also demonstrated the importance of considering the co‐occurrence of high‐risk alterations to accurately assess their prognostic significance. Thus, while 17p deletion retains its adverse prognosis even when present as a solitary abnormality, with co‐segregation having minimal impact on outcomes, the negative prognosis of 1q gains were mitigated if this abnormality occurs as the sole aberration.
AUTHOR CONTRIBUTIONS
Veronica González‐Calle, Norma C. Gutiérrez, Maria V. Mateos, and Jesus F. San Miguel involved in the design of the study. Norma C. Gutiérrez, Maria J. Calasanz, Manuela Guijarro perfomed the cytogenetic analysis. Veronica González‐Calle and Paula Rodriguez‐Otero made the statistical analysis and wrote the manuscript. All authors contributed enrolling patients in the clinical trials and provided data. All authors reviewed and approved the manuscript.
CONFLICT OF INTEREST STATEMENT
Veronica González‐Calle: Consulting or Advisory Role: Janssen. Speakers' Bureau: Janssen, GlaxoSmithKline, Pfizer, Bristol Myers Squibb/Celgene. Travel, Accommodations, Expenses: Janssen, GlaxoSmithKline. Paula Rodriguez‐Otero declares honoraria derived from Consulting or advisory board role: Celgene‐BMS, Janssen, Roche, Abbvie, Pfizer, GSK, Sanofi, H3Biomedicine. Steering committee member: Celgene‐BMS, Regeneron, Janssen Speaker's bureau: Janssen, Celgene‐BMS, GSK, Sanofi, Abbvie. Travel grant: Pfizer, and is an Editor of HemaSphere. Miguel T. Hernández: Consulting or Advisory Role: Janssen, Sanofi/Aventis, Celgene/Bristol Myers Squibb, GlaxoSmithKline. Speakers' Bureau: Janssen, Celgene/Bristol Myers Squibb, GlaxoSmithKline. Research Funding: Celgene/Bristol Myers Squibb (Inst). Laura Rosiñol reports honoraria from Janssen, Celgene, Amgen, and Takeda. Joaquin Martínez‐López declares honoraria from consulting activities from Astellas Pharma, BMS, F. Hoffman‐La Roche, Janssen, Novartis, Sanofi. Research grant from BMS. Ana P. González‐Rodríguez reports honoraria from Janssen, Celgene, Takeda, and Amgen. JdlR is a consultant for Bristol‐Meyers Squibb, GlaxoSmithKline, Janssen, Menarini, Pfizer, Sanofi, and Takeda and is on the advisory board of GlaxoSmithKline, Janssen, Pfizer, and Sanofi. Maria V. Mateos: declares honoraria derived from lectures and advisory roles at Amgen, Celgene, BMS, GSK, Janssen, Pfizer, Regeneron, Sanofi, and Takeda. Anna Sureda reports consultancy for BMS, Celgene, Gilead, Janssen, MSD, Novartis, Sanofi, Takeda, and GSK; speakers bureau for Takeda; travel grants from BMS, Celgene, Janssen, Roche, Sanofi, and Takeda; research support from Takeda and BMS. Albert Oriol declares consultant activities from Amgen, Celgene, BMS, GSK, Janssen, and Sanofi. EMO reports consulting or an advisory role for Amgen, AbbVie, Bristol Myers Squibb, GlaxoSmithKline, Janssen, Karyopharm, Menarini‐Stemline, Mundipharma, Oncopeptides, Sanofi, Secura Bio, and Takeda; meeting and/or travel expenses from Bristol Myers Squibb, GlaxoSmithKline, Janssen, Lilly, and Sanofi; and honoraria from Amgen, Asofarma, Bristol Myers Squibb, GlaxoSmithKline, Janssen, MSD, Pfizer, Sanofi, and Takeda. Joan Bargay: honoraria for lectures and advisory boards from Janssen, BMS‐Celgene, Amgen, Takeda, Oncopeptides. Juan J. Lahuerta: Consulting or Advisory Role: Celgene, Amgen, Janssen‐Cilag, Sanofi. Travel, Accommodations, Expenses: Celgene, Pfizer. Jesus F. San Miguel declares consulting activities from Abbvie, Amgen, BMS, Celgene, F. Hoffman‐La Roche, GSK, HaemaLogiX, KaryoPharm, Merck, Novartis, Pfizer, Regeneron, Sanofi‐Aventis, Takeda, and SecuraBio. Maria V. Mateos: honoraria for lectures and advisory boards from Janssen, Celgene, Takeda, Amgen, GSK, Abbvie, Pfizer, Regeneron, Roche, Sanofi, Oncopeptides; The remaining authors declare no competing interests relative to this work.
FUNDING
This research received no funding.
Supporting information
Supporting information.
Supporting information.
DATA AVAILABILITY STATEMENT
Data that support the findings of this study are available on request from the corresponding author. Data are not publicly available due to privacy or ethical restrictions.
The Spanish Myeloma Group is open to the possibility of sharing the data used in this study for research projects as long as they do not interfere with the present or future objectives of the clinical trial. The interest and feasibility of any clinical or biological research proposal based on the data from this study must be approved by the board of directors of the Spanish Myeloma Group. In such a case, the data deposited in REDCap will be presented in anonymized CSV format. This availability is subject to the laws and provisions in force that regulate the development of clinical trials both in Spain and in the European Union.
Data are available on request from the corresponding author, Norma C. Gutierrez (normagu@usal.es).
REFERENCES
- 1. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335‐348. [DOI] [PubMed] [Google Scholar]
- 2. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28(5):1122‐1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shah UA, Mailankody S. Emerging immunotherapies in multiple myeloma. BMJ [Internet]. 2020;370:m3176. 10.1136/bmj.m3176 [DOI] [PubMed] [Google Scholar]
- 4. Cardona‐Benavides IJ, de Ramón C, Gutiérrez NC. Genetic abnormalities in multiple myeloma: prognostic and therapeutic implications. Cells. 2021;10(2):336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ross FM, Avet‐Loiseau H, Ameye G, et al. Report from the European Myeloma Network on interphase FISH in multiple myeloma and related disorders. Haematologica. 2012;97(8):1272‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538‐e548. [DOI] [PubMed] [Google Scholar]
- 7. Gutiérrez NC, Castellanos MV, Martín ML, et al. Prognostic and biological implications of genetic abnormalities in multiple myeloma undergoing autologous stem cell transplantation: t(4;14) is the most relevant adverse prognostic factor, whereas RB deletion as a unique abnormality is not associated with adverse prognosis. Leukemia. 2007;21(1):143‐150. [DOI] [PubMed] [Google Scholar]
- 8. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood. 2015;125(13):2095‐2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boyd KD, Ross FM, Chiecchio L, et al. A novel prognostic model in myeloma based on co‐segregating adverse FISH lesions and the ISS: analysis of patients treated in the MRC Myeloma IX trial. Leukemia. 2012;26(2):349‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosiñol L, Oriol A, Teruel AI, et al. Bortezomib and thalidomide maintenance after stem cell transplantation for multiple myeloma: a PETHEMA/GEM trial. Leukemia. 2017;31(9):1922‐1927. [DOI] [PubMed] [Google Scholar]
- 11. Rosiñol L, Oriol A, Teruel AI, et al. Superiority of bortezomib, thalidomide, and dexamethasone (VTD) as induction pretransplantation therapy in multiple myeloma: a randomized phase 3 PETHEMA/GEM study. Blood. 2012;120(8):1589‐1596. [DOI] [PubMed] [Google Scholar]
- 12. Rosiñol L, Oriol A, Rios R, et al. Bortezomib, lenalidomide, and dexamethasone as induction therapy prior to autologous transplant in multiple myeloma. Blood. 2019;134(16):1337‐1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mateos MV, Martínez‐López J, Hernández MT, et al. Sequential vs alternating administration of VMP and Rd in elderly patients with newly diagnosed MM. Blood. 2016;127(4):420‐425. [DOI] [PubMed] [Google Scholar]
- 14. Mateos MV, Oriol A, Martínez‐López J, et al. GEM2005 trial update comparing VMP/VTP as induction in elderly multiple myeloma patients: do we still need alkylators? Blood. 2014;124(12):1887‐1893. [DOI] [PubMed] [Google Scholar]
- 15. López‐Corral L, Gutiérrez NC, Vidriales MB, et al. The progression from MGUS to smoldering myeloma and eventually to multiple myeloma involves a clonal expansion of genetically abnormal plasma cells. Clin Cancer Res. 2011;17(7):1692‐1700. [DOI] [PubMed] [Google Scholar]
- 16. Paiva B, Calasanz MJ. Highs and lows of t(4;14) in multiple myeloma. Blood. 2023;141(13):1500‐1502. [DOI] [PubMed] [Google Scholar]
- 17. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood. 2015;125(13):2095‐2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stong N, Ortiz‐Estévez M, Towfic F, et al. The location of the t(4;14) translocation breakpoint within the NSD2 gene identifies a subset of patients with high‐risk NDMM. Blood [Internet]. 2023;141(13):1574‐1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood. 2015;125:2095‐2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Avet‐Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. 2007;109(8):3489‐3495. [DOI] [PubMed] [Google Scholar]
- 21. Chretien ML, Corre J, Lauwers‐Cances V, et al. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter? Blood. 2015;126(25):2713‐2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stong N, Ortiz‐Estévez M, Towfic F, et al. The location of the t(4;14) translocation breakpoint within the NSD2 gene identifies a subset of patients with high‐risk NDMM. Blood. 2033;141(13):1574‐1583. 10.1182/blood.2022016212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Avet‐Loiseau H, Malard F, Campion L, et al. Translocation t(14;16) and multiple myeloma: is it really an independent prognostic factor? Blood. 2011;117(6):2009‐2011. [DOI] [PubMed] [Google Scholar]
- 24. Mina R, Joseph NS, Gay F, et al. Clinical features and survival of multiple myeloma patients harboring t(14;16) in the era of novel agents. Blood Cancer J. 2020;10(4):40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldman‐Mazur S, Jurczyszyn A, Castillo JJ, et al. A multicenter retrospective study of 223 patients with t(14;16) in multiple myeloma. Am J Hematol. 2020;95(5):503‐509. [DOI] [PubMed] [Google Scholar]
- 26. Flynt E, Bisht K, Sridharan V, Ortiz M, Towfic F, Thakurta A. Prognosis, biology, and targeting of TP53 dysregulation in multiple myeloma. Cells. 2020;9(2):287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thakurta A, Ortiz M, Blecua P, et al. High subclonal fraction of 17p deletion is associated with poor prognosis in multiple myeloma. Blood. 2019;133(11):1217‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lakshman A, Painuly U, Rajkumar SV, et al. Natural history of multiple myeloma with de novo del(17p). Blood Cancer J. 2019;9(3):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shah V, Johnson DC, Sherborne AL, et al. Subclonal TP53 copy number is associated with prognosis in multiple myeloma. Blood. 2018;132(23):2465‐2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Merz M, Hielscher T, Wagner B, et al. Predictive value of longitudinal whole‐body magnetic resonance imaging in patients with smoldering multiple myeloma. Leukemia. 2014;28(9):1902‐1908. [DOI] [PubMed] [Google Scholar]
- 31. Neben K, Lokhorst HM, Jauch A, et al. Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood. 2012;119(4):940‐948. [DOI] [PubMed] [Google Scholar]
- 32. Avet‐Loiseau H, Attal M, Campion L, et al. Long‐term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long‐term survival. J Clin Oncol. 2012;30(16):1949‐1952. [DOI] [PubMed] [Google Scholar]
- 33. Merz M, Hielscher T, Seckinger A, et al. Baseline characteristics, chromosomal alterations, and treatment affecting prognosis of deletion 17p in newly diagnosed myeloma. Am J Hematol. 2016;91(11):E473‐E477. [DOI] [PubMed] [Google Scholar]
- 34. Walker BA, Mavrommatis K, Wardell CP, et al. A high‐risk, Double‐Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33(1):159‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sawyer JR, Tian E, Walker BA, et al. An acquired high‐risk chromosome instability phenotype in multiple myeloma: Jumping 1q Syndrome. Blood Cancer J [Internet]. 2019;9(8):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hao S, Lin P, Medeiros LJ, et al. Clinical implications of cytogenetic heterogeneity in multiple myeloma patients with TP53 deletion. Mod Pathol. 2017;30(10):1378‐1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schavgoulidze A, Lauwers‐Cances V, Perrot A, et al. Heterogeneity in long‐term outcomes for patients with Revised International Staging System stage II, newly diagnosed multiple myeloma. Haematologica. 2023;108(5):1374‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schavgoulidze A, Talbot A, Perrot A, et al. Biallelic deletion of 1p32 defines ultra‐high‐risk myeloma, but monoallelic del(1p32) remains a strong prognostic factor. Blood. 2023;141(11):1308‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. González‐Calle V, Slack A, Keane N, et al. Evaluation of Revised International Staging System (R‐ISS) for transplant‐eligible multiple myeloma patients. Ann Hematol. 2018;97(8):1453‐1462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Data Availability Statement
Data that support the findings of this study are available on request from the corresponding author. Data are not publicly available due to privacy or ethical restrictions.
The Spanish Myeloma Group is open to the possibility of sharing the data used in this study for research projects as long as they do not interfere with the present or future objectives of the clinical trial. The interest and feasibility of any clinical or biological research proposal based on the data from this study must be approved by the board of directors of the Spanish Myeloma Group. In such a case, the data deposited in REDCap will be presented in anonymized CSV format. This availability is subject to the laws and provisions in force that regulate the development of clinical trials both in Spain and in the European Union.
Data are available on request from the corresponding author, Norma C. Gutierrez (normagu@usal.es).