

Abstract
The hyper-unstable Chr9p21 locus, harbouring the interferon gene cluster, oncogenes and C9orf72, is linked to multiple diseases. C9orf72 (GGGGCC)n expansions (C9orf72Exp) are associated with incompletely penetrant amyotrophic lateral sclerosis, frontotemporal dementia and autoimmune disorders. C9orf72Exp patients display hyperactive cGAS-STING-linked interferon immune and DNA damage responses, but the source of immunostimulatory or damaged DNA is unknown. Here, we show C9orf72Exp in pre-symptomatic and amyotrophic lateral sclerosis-frontotemporal dementia patient cells and brains cause the folate-sensitive chromosomal fragile site, FRA9A. FRA9A centers on >33 kb of C9orf72 as highly compacted chromatin embedded in an 8.2 Mb fragility zone spanning 9p21, encompassing 46 genes, making FRA9A one of the largest fragile sites. C9orf72Exp cells show chromosomal instability, heightened global- and Chr9p-enriched sister-chromatid exchanges, truncated-Chr9s, acentric-Chr9s and Chr9-containing micronuclei, providing endogenous sources of damaged and immunostimulatory DNA. Cells from one C9orf72Exp patient contained a highly rearranged FRA9A-expressing Chr9 with Chr9-wide dysregulated gene expression. Somatic C9orf72Exp repeat instability and chromosomal fragility are sensitive to folate deficiency. Age-dependent repeat instability, chromosomal fragility and chromosomal instability can be transferred to CNS and peripheral tissues of transgenic C9orf72Exp mice, implicating C9orf72Exp as the source. Our results highlight unappreciated effects of C9orf72 expansions that trigger vitamin-sensitive chromosome fragility, adding structural variations to the disease-enriched 9p21 locus, and likely elsewhere.
Graphical Abstract
Graphical Abstract.

Introduction
Expansions of 30–7950 GGGGCC repeats in the C9orf72 gene, which resides in 9p21 (C9orf72Exp), are the most common cause of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD) and numerous seemingly unrelated diseases including inflammation dysregulation, autoimmune diseases, over-expression of type I IFN genes (1–6) and melanoma (7). Most C9orf72Exp carriers are asymptomatic at 40 years of age, some will develop ALS, FTD, another co-occurring disease, or a mixture of these, while many can be disease-free into their 80s and 90s (8,9). This incomplete penetrance and clinical diversity suggest the involvement of genetic and environmental modifiers and/or cumulative events.
Many studies on C9orf72Exp disease have focused on the expanded repeat RNA and Repeat-Associated Non-AUG RAN-peptides, where many model systems are devoid of the endogenous chromosomal expanded repeat. Repeat expansions downregulate C9orf72Exp expression leading to reduced C9ORF72 protein levels (10,11). Stimulator of Interferon Genes (STING), which with cyclic GMP-AMP synthase (cGAS) activates the innate immune response following sensing of cytosolic double-stranded DNA that could arise from viruses, damaged mitochondria or damaged chromosomes leaked from micronuclei (MN). In C9orf72Exp individuals, decreased C9ORF72 leads to STING-hyperactivated IFN expression and inflammation in vulnerable neurons (12), but the source of the immunostimulatory DNA is unknown (4,13,14). Accumulating evidence supports the role of an activated DNA damage response (DDR) in C9orf72Exp disease (15–25), yet the source of the damaged DNA is unknown. Massive post-natal inter- and intra-tissue repeat length instability is evident in post-mortem tissues of C9orf72Exp individuals with the largest somatic expansions in the brain (26–30). Surprisingly, there have been no prior cytogenetic or chromosomal instability studies of C9orf72Exp. The recent discontinued clinical programs of antisense oligonucleotides targeting the sense-strand C9orf72 mRNA, which successfully reduced the RAN-peptides, but resulted in no clinical benefit or trended toward greater clinical decline for the cohort, highlight our poor understanding of C9orf72Exp (31–36). A recent meeting focused on the post-trials path forward for C9orf72Exp highlighted key areas needing attention: ‘Critical information in our understanding of the biology around the C9orf72 expansion is still lacking, including what molecular and cellular changes occur at presymptomatic stages…studies of genetics and lifestyles might be useful’ (31). The unknown biology of the C9orf72Exp has been highlighted as a focus area (31–36). Factors that may determine disease penetrance, clinical variation, disease onset, progression and severity, are indispensable for family life decisions as well as clinical trial design (6,9,31,37–41).
Genomic context is important, as C9orf72Exp occurs on a risk haplotype, which could influence repeat length instability, leading to reduced transcription and its retention of the intron containing the expanded GGGGCC repeat (42–45) (Figure 1). Many of the co-occurring C9orf72Exp symptoms can arise independent of a C9orf72Exp and are associated with other 9p21 genes. For example, the type I interferon (IFN) cluster of 17 genes, including IFNk adjacent to C9orf72, are linked to autoimmune diseases (e.g. dysregulated inflammation, lupus, rheumatoid arthritis, diabetes and multiple sclerosis) (1,3–5,7). The enhancer-rich 9p21 gene desert is associated with IFN signaling (46,47). CDKN2A/B, EMICERI and IFNs are linked to melanoma (48), a cancer recently associated with C9orf72Exp (7). LINGO2, TOPORS, APTX and DNAJA1 have been associated with neurodevelopmental, neurodegenerative and motoneuron disease (49).
Figure 1.

The 9p21 neighborhood, associated genes, diseases and instability hotspots. See the text.
The 9p21 locus is hyper-prone to various recurrent structural rearrangements (50–54) (Figure 1). 9p deletion/duplication syndrome includes 9p21.2 as a breakpoint hotspot (55,56). Instability in cancers is suspected to involve chromosomal fragile sites although the hyper-unstable 9p21 has no molecularly mapped fragile site (57). ‘Rare’ fragile sites can be present in as few as a single individual, as initially reported for FRA9A (58), to ≤5% of individuals and are linked to partially penetrant neurological disorders, as reviewed (59). Most rare fragile sites are folate-sensitive fragile sites (FSFS), induced in the absence of folate or by the antimetabolite 5-fluorodeoxyuridine (FUdR). Ten of the ∼30 FSFS have been mapped to gene-specific expanded (CGG)n repeats, including FRAXA, FRAXE, FRAXF, FRA2A, FRA7A, FRA10A, FRA11A, FRA11B, FRA12A and FRA16A. FSFS are prone to DNA breaks making them mutation hotspots—mutations that can alter clinical presentation, which is well-characterized for the CGG-expanded FRAXA/FMR1, reviewed in (59). Through >75 years of study, many isolated case reports, diverse mutation forms (varying CGG expansion sizes, contiguous gene deletions, gene duplications, intra- and inter-chromosomal recombination and point mutations), and FMR1 mosaic epimutations have been identified as the cause of numerous seemingly unrelated diseases, many incompletely penetrant (59). Individual case studies with deep clinical, neuropathological and molecular phenotyping can be informative of disease etiology. Thus, the instability of FSFS can impact disease manifestation and clinical presentation. Our objective was to better understand the biology of the endogenous expanded C9orf72 repeat in the context of 9p21 in presymptomatic and disease states. We assessed whether the expanded C9orf72 repeat, GGGGCCGGGGCCGGGGCCGGGGCC…, which contains the CGG-motif common to all mapped FSFS, is a FSFS.
Materials and methods
In silico datasets and analyses
Sequence feature and epigenetic datasets used in Figure 2 and Supplementary Figure S1 are described as follows. Repeat annotations and %GC tracks were obtained from the UCSC genome browser (hg19). Wavelet-summarized repliseq data (ENCODE) was obtained in bigWig format from GEO:GSE34399. Erythroblast SNS-seq data and replication origins were obtained in bedGraph and bed format, respectively, from GEO:GSE6197. Imputed %DNA methylation for GM12878 was obtained from the roadmap epigenomics data portal (https://egg2.wustl.edu/roadmap/data/byFileType/signal/consolidatedImputed/DNAMethylSBS/E116-DNAMethylSBS.imputed.FractionalMethylation.signal.bigwig). Pre-calculated second-order DNA shape features were obtained from the Rohs lab GBShape database.
Figure 2.

DNA sequence, structure, topological-associated domain (TAD), CpG island and Repli-Chip analysis for all 10 CGG-FSFS. (A) Boxplots comparing average %GC, helix twist, propeller twist, major groove width (MGW), SNS-seq (short nascent strand sequencing) signal and number of origins of replication initiation (ORIs) at CGG/CCG-mapped FSFS and C9orf72 versus 944 control genome-wide CGG/GGC repeats, calculated for windows ±50 kb and ±500 kb from the repeat center. Control repeat regions were further separated as genic or non-genic based on overlap with an annotated transcript. Median repeat length for FSFS is 8.7; all FSFS have GC content of 100% (AT = 0 except FRAXF, GC = 0.826). Repeat length req of >4.5 for 944 CGG/CCG control regions yielded a median length of 8.7, same as mapped FSFS. Asterisk denotes significance based on a two-sided Wilcoxon test (*P < 0.05; **P < 0.01). Each individual FSFS is represented as a black dot. The C9orf72 (GGGGCC)n is represented by a red dot. (B and C) Topological domain, DNA replication timing tracks, CpG islands and CTCF sites for the contiguous FRAXA, FRAXE and FRAXF compared to the same for C9orf72. Shown are the contour density plots depicting the number of CTCF sites and CpG-islands in 100 kb windows centered on the repeat, representing boundaries with normal-length, matched repeats. Repli-seq data were obtained in bigwig format (GSE61972). H1 ESC = H1 Embryonic Stem Cells; accession:ENCFF000KUF; NP = Neuronal progenitor cells (BG01 Fibroblast-derived); accession:ENCFF907YXU; LCL = GM06990 lymphoblastoid cells; accession:ENCFF000KUA. Early (positive) and late (negative) replication was determined by subtracting the genome-wide median score from all values. To conserve space, only the FSFS gene is indicated, while other UCSC genes are condensed into one line. (D) Summary of enrichment of each fragile site (indicated in blue fonts) for CpG islands and CTCF sites. Points are colored according to density. For analyses of FRA2A, FRA7A, FRA10A, FRA11A, FRA11B, FRA12A and FRA16A, see Supplementary Figure S1.
Cell culture and fragile site induction
All cytogenetic studies were performed using Epstein-Barr virus-transformed lymphoblastoid cell lines from C9orf72-ALS patients, expansion carriers and control individuals provided by Dr Guy Rouleau (McGill University). One of the patients, P4, displayed signs of FTD. All patients provided written informed consent. Cells were cultured in RPMI 1640 media supplemented with 15% fetal bovine serum (FBS) (ThermoFisher Scientific), 2 mM L-glutamine, Pen/Strep (100 IU/ml, 100 μg/ml) and 1 mM sodium pyruvate. Two modes of FSFS induction were used to demonstrate fragility at 9p21.2: (i) 24 h with 0.1 μM FUdR (an anti-folate) or (ii) 14 days with low-folate Media 199 (Gibco, Cat #: 11150–059) supplemented with 2% fetal calf serum (FCS), 2 mM L-glutamine and Pen/Strep (100 IU/ml, 100 μg/ml). Cells in 199 media were treated with 100 nM methotrexate for 16 h, washed and then treated with 0.01 μM bromodeoxyuridine (BrdU) for a total of 3 h prior to harvest. All cells were treated with 6 μg/ml ethidium bromide (EtBr) and 0.05 μg/ml KaryoMAX (Gibco; Cat #: 15 212 012) prior to harvest. Cells were then centrifuged and resuspended in 0.075 M KCl solution for 30 min at 37°C followed by several washes with fixative solution (3:1 methanol:acetic acid). Long-term FUdR culture experiments were performed in either RPMI 1640 media (as described above) or Roswell Park Memorial Institute Medium (RPMI) 1640 media + 0.1 μM FUdR for a total of 8 weeks. Media and FUdR were replaced every 2–3 days. Cells were harvested at 1-, 2-, 4-, 6- and 8-week time points for DNA purification, and Southern blot was run as described below.
Metaphase chromosome and FISH analysis
Slide preparations for both cytogenetic and fluorescent in situ hybridization (FISH) analysis of metaphase spreads were made from fixed cell suspensions using a Model CDS 5 Cytogenetic Drying chamber according to standard methods. For 199 media conditions, breaks and gaps were quantified on total Giemsa stained metaphases and subsequently G-banded to karyotype each metaphase and confirm fragile site on 9p-arm. SpectrumGreen and SpectrumOrange-conjugated dUTP probes (RP11-15P13, RP11-274I21, RP11-1149M23, RP11-491J7, RP11-945G22, RP11-1079P2, RP11-360B21 and RP11-696J10) from RPCI-11 human BAC library were generated by The Centre for Applied Genomics (TCAG, PGCRL, Toronto, Canada) using the Vysis nick translation kit (Cat #07J00-001; Abbott Molecular, Illinois, USA) in accordance with manufacturer’s instructions. Chromosomes were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). One hundred metaphases per sample were scored for each condition using either brightfield microscopy, Olympus IX81 quorum spinning disk confocal microscope or Zeiss Axioplan 2 fluorescence microscope. Images were collected using a CCD camera (Hamamatsu C9100-13 EM-CCD, Hamamatsu, Japan) (Princeton Instruments Pentamax, Roper Scientific, New Jersey, USA) and analyzed using Volocity (Perkin Elmer, Massachusetts, USA) or CytoVision (Leica Biosystems, Germany) software.
Southern blot analysis of C9orf72 repeat expansion size
Southern blot analysis was performed to size C9orf72 repeat expansions as this method is the gold standard for sizing large (GGGGCC)n repeat expansions (28). Southern blotting provides an estimate of non-bias expansion sizes (60). We used the Southern blotting method (28,61) with 241 bp probe, which anneals 153 bp upstream of the (GGGGCC)n repeat tract, and used selected restriction-endonuclease digestions of genomic DNA (as noted in the figures), which permit sensitive detection of repeat size and length heterogeneity at molar levels. The 241 bp probe was produced by polymerase chain reaction (PCR) using primers (Fwd: 5′-AGAACAGGACAAGTTGCCCC-3′ and Rev: 5′-AACACACACCTCCTAAACCC-3′) as published (28,61). For mapping boundaries of MNase accessibility an additional probe was used (see below). Molecular weight markers were used to estimate the size of repeats. Distances between molecular weight markers were measured using ImageLab software (BioRad) and plotted against the known number of base pairs for each marker. A best-fit linear regression was calculated in Microsoft Excel and the equation of this line was used to calculate the size of each band on the blot. Different restriction enzyme combinations were used to cut the genomic DNA, as noted in each figure, hence resulting in different flank repeat sizes.
Methylation analysis of the C9orf72 repeat
Methylation analyses were done in blinded experiments using the same DNA prep as for Southern blot. Genomic DNA was bisulfite converted using the EZ DNA Methylation-Lightning™ Kit (Zymo). We estimated the number of methylated CpG-sites at the CpG-island 5′ of the (GGGGCC)n repeat using bisulfite sequencing as reported previously (62). To estimate the methylation of the (GGGGCC)n repeat itself, we used a qualitative GGGGCC-methylation assay, sensitive enough to detect repeat methylation in a mixture containing ∼5% high-methylated DNA standard (63). We always observed methylation mosaicism for expanded repeat samples, revealed by the presence of the amplification product of the expansion in both methylated (blue) and unmethylated (green) channels (64).
Micronucleus and nuclear bud detection
MN and nuclear bud (NBud) analyses were done as previously described with some modifications (65–67). Briefly, cells were treated with 0.5 μM FUdR for a total of 24 h, with cytochalasin B (4 μg/ml) being added 3 h after addition of FUdR. Cells were briefly incubated in 0.075 M KCl, fixed with Carnoy’s fixative and slides with sparsely spread cells were prepared using a Model CDS 5 Cytogenetic Drying chamber (Thermotron, Michigan, USA). SpectrumGreen (RP11-696J10) and SpectrumOrange (RP11-672M4)-conjugated dUTP probes from RPCI-11 human BAC library were generated by The Centre for Applied Genomics (TCAG, PGCRL, Toronto, Canada) using the Vysis nick translation kit (Cat #07J00-001; Abbott Molecular, Illinois, USA) in accordance with manufacturer’s instructions. Nuclei were counterstained with DAPI. At least 950 binucleated cells were scored per condition for the presence of MN and NBuds and Chr9 signals, as previously described (66). Two blinded and independent experiments were carried out for each data asset. Statistical analyses were done using Fisher’s exact test for the combined values of MN and NBud events as they are believed to arise from the same phenomenon (66,68). Images were captured using an Olympus BX61 fluorescence microscope. Images were collected using a CCD camera (Hamamatsu C9100-13 EM-CCD, Hamamatsu, Japan) (Princeton Instruments Pentamax, Roper Scientific, New Jersey, USA) and analyzed using ASI SpotScan software (Applied Spectral Imaging, California, USA).
Sister-chromatid exchange (SCE) assay
Cells were incubated in the presence of 10 μM BrdU for two cell cycle periods and pulsed with 0.1 μg/ml of KaryoMAX (Gibco; Cat #: 15 212 012) for the last 2 h before being harvested. FUdR (0.1 μM) was added for the last cell cycle. Harvested cells were treated with 0.075 M KCl for 20 min and subsequently fixed with methanol:acetic acid (3:1) for at least 30 min. Cells were fixed onto glass slides. Dried slides were incubated with 10 μg of Hoechst 33 258 per ml in phosphate buffer (pH 6.8) for 20 min, followed by rinsing with MacIlvaine solution (164 mM Na2HPO4, 16 mM citric acid [pH 7.0]). Slides were irradiated with UV light for 2 h and incubated in 2X SSC solution at 62°C for 1 h before staining with 5% Giemsa solution and subsequent microscopy. At least 100 metaphase cells were evaluated for sister-chromatid exchange (SCE) events genome-wide per genotype. For Chr9-specific SCEs, slides were destained with xylene solution followed by methanol:acetic acid (3:1) and then standard FISH procedures, as described above were followed using SpectrumOrange-dUTP conjugated RP11-696J10 probe. At least 30 of the same Giemsa-stained metaphases were relocalized using fluorescence microscopy and imaged to allow for quantification of SCEs specifically on the 9p- and 9q-arms.
Construction of plasmids and yeast strains
An XbaI-HindIII fragment containing a region of the human C9orf72 gene with an expanded (GGGGCC)n repeat and 547 bp of flanking sequence (from pcDNA3.1 (+)c9-200 + obtained from R.H. Brown; CF stock #435) was cloned into the MCS of plasmid pEP1 to create plasmids pEP1- (GGGGCC)n. pEP1 was made by cloning the ADE2 gene from pRS402 into the BsaAI and NsiI sites of pHZ1; pHZ1 was made by cloning the G4T4 sequence into the SmaI site of pYIP5. The CAG repeat was cloned into pEP1 XhoI and XbaI sites. Repeat lengths in plasmid clones were confirmed by sequencing. Repeat lengths of the (GGGGCC)n plasmids were ∼16, 29 and 36 for plasmids 504, 484 and 485, respectively; the exact number of repeats varied with different sequencing runs. Plasmids were linearized with XcmI and recombined into the URA3 marked yeast artificial chromosome (YAC) CF1 in strain VPS105 (CFY #289) to create the new GGGGCC-ADE2-URA3 and CAG-ADE2-URA3 YACs (see Supplementary Figure S7A and D), which were confirmed for YAC structure by PCR. CAG tract lengths were confirmed by sequencing of PCR amplicons but (GGGGCC)n tract lengths could only be determined approximately by Southern blot since amplification from genomic DNA was unsuccessful, and are therefore reported to the nearest 5 bp.
YAC fragility assay
End loss of the repeat-ADE2-URA3 YACs was determined by an established protocol (69) except that 1 ml of an overnight culture inoculated with a specific colony was plated on media containing 5-Fluoro-Orotic Acid (FOA) and a low amount of adenine but lacking leucine (FOA-Leu low Ade) to determine the number of mutants, and a dilution was plated on yeast complete media lacking leucine (YC-Leu) for a total cell count. Rates were determined by the method of the median. Another 1 ml of the same culture was used to isolate genomic DNA for Southern blot to assess repeat length. For reasons not completely understood, but that could include reduction of transcription through the repeat tract, the insertion of the ADE2 marker resulted in a lower rate of FOAR compared to previously published values for the CAG-85 YAC without the ADE2 gene. End loss was confirmed in a subset of colonies (usually one from each FOA-Leu plate) by PCR (retention of the sequence to the left of the G4T4 telomere seed and loss of sequence to the right of the repeat tract; Supplementary Figure S7).
Southern blot—yeast
Yeast genomic DNA was digested with NheI and XbaI restriction enzymes, separated by horizontal gel electrophoresis and blotted to a nylon membrane using a standard Southern protocol. Either the Roche Molecular Weight Marker VI or VII was run as a size marker. For the preparation of the probe, a 301 bp fragment adjacent to the (GGGGCC)n repeat was PCR amplified from DNA of plasmid #485 as a template, using Primers #1402 (ALSsouthernFOR) and #1403 (ALSsouthernREV). Probes were labeled using the DIG High-Prime DNA Labeling and Detection Starter Kit I (Cat #: 11 745 832 910; Roche). Repeat tract estimation was performed as previously described (Southern blot analysis of repeat expansion size).
Chromatin accessibility in cellulo with MNase
Human lymphoblast cell lines from C9orf72-expansion carriers were assessed for MNase accessibility, as outlined in Figure 5B. Briefly, cell pellets were washed in ice-cold 1X PBS, re-pelleted and permeabilized by ice-cold NP-40 lysis buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40, 0.15 mM spermine, 0.5 mM spermidine) for 5 min on ice. Nuclei were collected at 200 × g (10 min, 4°C), washed in 1X MNase digestion buffer (10X stock: 100 mM Tris pH 7.4, 150 mM NaCl, 600 mM KCl, 5 mM spermidine, 1.5 mM spermine), re-pelleted and subsequently resuspended in 1X MNase digestion buffer supplemented with 1 mM CaCl2. Treatment followed with MNase for 5 min at 22°C, at differing concentrations (refer to Figure 5 and Supplementary Figure S4A—all serial dilutions). All digestion reactions were stopped with an equal volume of Stop solution (0.625 mg/ml proteinase K, 20 mM EDTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid), also known as egtazic acid), 20 mM EGTA, 1% SDS diluted in 1X MNase buffer) and incubated at 37°C for a minimum of 1 h. Reaction conditions were optimized to ensure over-digestion by MNase, which diminishes MNase cleavage preference. DNA was purified by phenol/chloroform extraction, RNase A treatment and ethanol precipitation. A small amount of purified DNA was assessed for completeness of MNase digestion, by its conversion to mono-, di- and tri-nucleosome sized fragments as resolved by electrophoresis stained with EtBr and visualized by UV imaging (Figure 5B, left panel; Supplementary Figure S4A). DNA samples were analyzed by Southern blot as described.
Figure 5.

C9orf72Exp locus is MNase-inaccessible. (A) Schematic of the region of unusual chromatin organized as determined by MNase accessibility analysis in C9orf72-ALS patient cells and brain regions. C9orf72 (GGGGCC)n-N repeat, CpG-islands (hatched boxes), long-2 kb and intermediate 435 bp promoters (green and red arrows, respectively), exons 1a (yellow dot) and 1b (orange dot), sense and antisense transcription start sites, restriction sites with distances from the beginning and end of the (GGGGCC)N tract, and probes for upstream (green) and downstream (blue) mapping of MNase accessibility. Cryptic splice site transcripts C1-C4 derived from the C9orf72Exp allele are indicated with their distance from the end of the repeat tract (42,45). (B) Workflow for MNase assay. (C) MNase treatment of chromatinized DNA from a cell line (P6) with a large methylated C9orf72Exp that had undergone single-cell cloning (scc) to isolate a single expanded allele, ensuring a single expansion size and facilitating clarity. EtBr staining of the MNase-treated DNA reveals over-digestion of the DNA to mono-, di- and tri-nucleosomal, sized fragments. The same DNA was digested with EcoRI/BamHI, to release the repeat fragment having 697 bp upstream and 1714 bp downstream of the repeat, then assessed by Southern blotting using the blue probe. Resistance of only the expanded allele occurs, even at high concentration of MNase (200U). Red ‘R’ indicates the region with reduced MNase accessibility that is about to lose the probed region indicated in the schematic at right. (D) Sample P3 with a mixture of heterogeneous repeat allele lengths shows that with increasing repeat size, MNase accessibility is decreased, as quantified by densitometric analysis for each allele size (colours coordinate between graph and Southern blot). In panels D–F the repeat was released with AflII/PciI, leaving 404 bp upstream and 689 bp downstream allowing increased resolution. NB, van Blitterswijk uses XbaI/XbaI to release the repeat, leaving 1751 bp upstream and 608 bp downstream of the repeat, the greater amount of flanking sequence reduces electrophoretic resolution. (E) Methylation is associated with increased MNase inaccessibility as assessed in cell lines (P6 [ssc] and P7) containing similar sized repeats but differing methylation status. The non-expanded allele is digested at the same rate in both lines. (F) MNase treatment of post-mortem (orbito)frontal cortex and cerebellum from an asymptomatic 90-year-old father and his ALS-FTD-affected daughter, previously deep-phenotyped for lifestyle choices, clinical, neuropathological and molecular biomarkers (29,70) (summarized in Supplementary Table S2). MNase concentrations are doubled between each lane (2.5U in lane 2 up to 40U). Expanded allele in both tissues is MNase resistant relative to the equimolar non-expanded allele, which serves as an internal control. See Supplementary Figure S4 for MNase controls and mapping experiments.
Chromatin accessibility in post-mortem tissues with MNase
We conducted MNase accessibility analysis using post-mortem brain tissues (orbito-frontal cortex and cerebellum) of an asymptomatic 90-year-old male and his 59-year-old ALS-FTD-affected daughter, previously deep-phenotyped (summarized in Supplementary Table S2) (29,70). The asymptomatic male carried 70-repeat C9orf72 expansion in the blood and was deceased at 90 years of age. Tissue was Dounce homogenized using 1X MNase digestion buffer, 10% NP-40 was added to final concentration 0.5%, incubated 5 min on ice, filtered through a 40 μm strainer and centrifuged. Nuclei pellets were washed in MNase digestion buffer and incubated with different concentrations of MNase. Remaining steps were performed as described above.
Digestibility of C9orf72 BAC DNA in vitro with MNase
A bacterial artificial chromosome (BAC) containing ∼160 (GGGGCC)•(GGCCCC) repeats, prepared in the lab of Piet de Jong, was restricted by NheI and XhoI giving rise to a shorter fragment of ∼1750 base pairs, which was gel-purified. Half (by volume) of the purified product underwent CpG-methylation by M.SssI methyltransferase supplemented with 640 μM S-adenosylmethionine (New England Biolabs) and purified using the QIAEX II kit (Qiagen). Completeness of methylation was checked by the inability to be digested by the methyl-sensitive HpaII but successful digestion by the methyl-insensitive MspI. Subsequently, in parallel, equal concentrations (∼100 ng/reaction) of the non-methylated and methylated fragments were treated with increasing levels of MNase (0U–0.1U–1U–5U–25 U) for 5 min at 22°C in 1X MNase digestion buffer. The MNase reaction was halted by addition of 6X purple loading dye (New England Biolabs) and electrophoresed immediately on a 1% agarose gel at 50 V for 20 min, followed by 100 V for 100 min. Gene Ruler Mix (Thermo Scientific) was loaded in the first lane. Electrophoresed products were stained with EtBr, visualized by UV imaging (Supplementary Figure S4B, left hand side), and DNA fragments were transferred to a positively charged nylon membrane, and probed with a 177 bp probe, annealing just upstream of the GGGGCC repeat tract (Supplementary Table S1). To maximize our ability to assess that loss of signal corresponded to MNase digestion of the GGGGCC repeat fragment, we developed the 177 bp probe which anneals immediately adjacent to the repeat. The 177 probe anneals 7 bp upstream of the GGGGCC repeat tract, while the 241 bp probe anneals 153 bp upstream. The 177 bp probe was produced by PCR using primers (Fwd: 5′-GAGCAGGTGTGGGTTTAGGA-3′ and Rev: 5′-CGACTCCTGAGTTCCAGAGC-3′) (Supplementary Table S1). A phosphor image of the Southern blot was obtained using the Typhoon system (Supplementary Figure S4B, right hand side), and the rate of digestion of probed sequence was calculated using densitometric analysis in ImageJ. The percentage of DNA resistant to MNase (therefore, the fraction of undigested ‘starting fragment’ BAC DNA) was plotted as a function of MNase units of activity (concentration) in GraphPad Prism 8 (Supplementary Figure S4B).
MNase controls
The MNase resistance we observe is not a reflection of the sequence preference of MNase or the sequence composition of the C9orf72 region. The MNase resistance spanning the mutant C9orf72 locus supports altered chromatin packaging of the non-repetitive flanks, as the 241 bp probe anneals 153 bp upstream of the (GGGCC)n repeat, and resistance is still evident (Figure 5). The relative resistance of the expanded allele compared to the internal control of the non-mutant C9orf72 allele, provides strong support for altered chromatin accessibility. MNase is known to display a cleavage sequence preference, preferring 5′-NpT and 5′-NpA sites (71,72), yet is capable of digesting G-C only sequences and can digest most calf thymus DNA with over-digestion, which diminishes MNase cleavage preference (71). All of our nuclear digests were completed to over-digestion. The C9orf72 sequences flanking the repeat, that are probed herein, contain an extensive number of the preferred 5′-NpT and 5′-NpA sites (Supplementary Figure S2B, green font). That protein-free genomic DNA from C9orf72 expansion carriers is digested at an equal rate relative to the internal equimolar control of the non-expanded C9orf72 allele, supports that the flanks can be digested (Supplementary Figure S4A). The MNase digest of the purified (GGGGCC)160 repeat DNA further confirms that the pure GC-rich repeat can be digested by MNase as evidenced by the EtBr-stained gel (Supplementary Figure S4B). As such, the reduced digestion of the expanded allele in its native chromatinized state represents a chromatin-mediated inaccessibility.
To rule out possible artefacts such as differences in the sensitivity between EtBr staining and Southern probe hybridization, or loss of DNA during Southern transfer, or an uneven transfer or membrane retention of DNA, an aliquot of the same samples was run on a high-resolution gel, ethidium-stained, hybridized with the C9orf72-specific probe, imaged, and then the blot was stripped and re-probed using radiolabeled total genomic DNA as the probe (Supplementary Figure S4A). The hybridization pattern from total genomic DNA probe is very similar to the EtBr-stained gel: most of the genome has been digested to completion to mono-, di-, tri- and tetra-nucleosome sized fragments (Supplementary Figure S7A). In contrast, hybridization with the C9orf72 probe shows dramatic MNase resistance of the expanded allele, while the non-expanded allele appears to be digested at the same rate as the rest of the genome.
Control MNase digestions of protein-free genomic DNA of unmethylated and methylated C9orf72-ALS samples, compared to the chromatinized samples of the same cell lines showed that MNase digestion occurs similarly for both alleles at extremely low MNase concentrations (0.01 Units) in protein-free conditions, yet the expanded allele is strongly resistant in chromatinized samples, supporting that this resistance is due to an unusual chromatin state (Supplementary Figure S4B and C).
There is a striking difference in MNase digestion between the ethidium-stained bulk genomic DNA, and the Southern blot detected C9orf72 expanded allele, which is internally controlled for by its non-expanded homolog (Figure 5). See above notes and Supplementary Figure S4 for controls. The C9orf72 region showed reduced presence in the mono-, di- and tri-nucleosomal digested regions, relative to the rest of the genome (compare Southern blot with ethidium stain, Figure 4A).
Figure 4.

C9orf72Exp causes FRA9A folate-sensitive fragile site that maps across 9p21. (A) Fragile site forms (indicated), with examples of non-fragile chromosomes from the same metaphase spreads. Showing DAPI-stained and paired FISH-probed chromosomes from FUdR-treated cells. 9q-arm FISH probe (9q32∼34.11; RP11-696J10) in green. FISH probe #2 at C9orf72 (panel D–F) in red. Magnification is the same for all chromosome images within each panel, facilitating comparison within the panel. Control Chr9 in upper left of panel (A) is from the same metaphase as the leftmost isochromatid Chr9. Full metaphase spreads can be provided upon request. Notably, the control ‘No fragile site’ Chr9 in lower left of panel A is from the same metaphase as the leftmost ‘Despiralized/stretched’ Chr9, revealing the lengthening of the stretched Chr9. (B) Representative example of the same chromosome (same magnification) with or without indicated FISH probes. (C) Two representative examples (same magnification) of isochromatids with FISH signal of C9orf72-containing probe 4 split over the fragile site. (D) 9p21 location of FISH probes with regional summary of fragile site location. (E) Quantification of fragility for 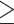 100 metaphases under two folate-stressed conditions. (D) Cells were described in Figure 3. (F) Fragile site breakage regions calculated from number of breaks relative to FISH signal found centromeric, telomeric or spanning across probes used. Percentage of telomerically located FISH signal relative to the break is on the bottom row. Frequencies of probes found spanning or on each side of the break were used to calculate the percent breakage within each region in panel (D).
100 metaphases under two folate-stressed conditions. (D) Cells were described in Figure 3. (F) Fragile site breakage regions calculated from number of breaks relative to FISH signal found centromeric, telomeric or spanning across probes used. Percentage of telomerically located FISH signal relative to the break is on the bottom row. Frequencies of probes found spanning or on each side of the break were used to calculate the percent breakage within each region in panel (D).
Control MNase digests using cloned C9orf72 expanded (GGGGCC)160 repeat and flanks (±CpG-methylation) were performed to assess the impact of MNase sequence selectivity and the influence of CpG-methylation on digestibility. As shown in Supplementary Figure S4B, the expanded repeat itself can be completely digested by MNase, and under the conditions used, the effect of sequence and CpG-methylation had limited effects on MNase digestibility, as previously reported (71,72). This is consistent with the extensive number of the MNase-preferred 5′-NpT and 5′-NpA sites (71,72) in the C9orf72 sequences flanking the repeat that are probed in the Southern blot (Supplementary Figure S2B, green font). These controls, coupled with the equal rate of MNase digestion of the protein-free C9orf72 expanded allele in genomic DNA relative to the internal control of the non-expanded C9orf72 allele (Supplementary Figure S4A,ii), demonstrate that the expanded repeat and flanks can be digested by MNase. Thus, the inaccessibility of MNase to the chromatinized C9orf72 expanded allele and flanks (where the Southern probe binds) is due to its unusual chromatin compaction, and is not due to an inherent inability of MNase to cleave the expanded (GGGGCC)n DNA sequence.
Further mapping of the MNase accessibility in the chromatinized C9orf72 expanded cells, with and without aberrant methylation, was performed. Essentially, for large expansions the mutant C9orf72 allele is less accessible to MNase ∼3300 bp upstream and ∼2500 bp downstream of the expanded repeat. Decreased accessibility is further exacerbated by aberrant methylation (detailed in Supplementary Figure SD4A,iii). The absolute boundaries of decreased MNase accessibility varied with repeat expansions.
Mapping MNase accessibility boundaries
We mapped the boundaries of MNase resistance in patient cell lines with no expansions (WT), expansions (P1 and P7) and expansions with methylation (P3 and P6), using a series of post-MNase restriction digests upstream and downstream of the repeat; fragments were detected by Southern blotting using either the 241 or 177 bp probe, as indicated. Upstream mapping was carried out with a post-MNase genomic digestion using BanI, which cleaves just downstream of the repeat, in combination with either one of a series of restriction digests progressing along the upstream flank, as far as 3335 bp upstream (AflII, EcoRI, PvuII and SphI) (Supplementary Figure S4C). These samples were assessed by Southern blot, detected with the 177 bp probe (Supplementary Figure S4C upper panel, schematic). For each restriction fragment, the non-expanded allele signal was completely lost while the expanded allele was either resistant or partially digested to a shorter, distinct, nearly repeat-only containing fragment. This might suggest that the repeat tract itself is the most resistant to MNase accessibility. The amount of MNase-resistant starting material decreases progressively with increasing distance upstream from the repeat (Supplementary Figure S4C). Thus, the MNase resistance of the mutant allele extends upstream of the repeat at least ∼1252 bp to the PvuII site, where some of the full-length fragment is still inaccessible to MNase, but diminishes further upstream ∼3335 bp to the SphI site (Supplementary Figure S4C). Downstream mapping was conducted using a post-MNase genomic digestion with AflII, which cleaves upstream of the repeat and beside the region detected by the 241 bp probe. AflII digestion was paired with one of a series of restriction digests progressing along the downstream flank, as far as 3750 bp downstream (BanI, BstYI, BamHI, PvuII or PsiI) (Supplementary Figure S4C, lower panel, schematic). For each restriction fragment, the expanded allele was resistant and progressively digested to a shorter, distinct nearly repeat-only containing fragment, again suggesting that the repeat tract is the most MNase resistant region. Resistance in the downstream region was more evident for the methylated allele and for larger expansions. For the larger methylated sample, P3 (∼3000 repeats), the MNase resistance of the mutant allele extends downstream of the repeat at least ∼2440 bp to the PvuII site, where some of the full-length fragment is still inaccessible to MNase, but diminishes further downstream ∼3750 bp to the PsiI site (Supplementary Figure S4C). For the shorter methylated expansion P6 (∼770 repeats), the MNase resistance of the mutant allele extends downstream of the repeat at least ∼1714 bp to the BamHI site, where some of the full-length fragment is still inaccessible to MNase, but diminishes further downstream ∼3750 bp to the PsiI site (Supplementary Figure S4C). Thus, the MNase resistance of the mutant allele extends at least ∼2440 bp downstream from the repeat to the PvuII site, where some of the full-length starting fragment is still resistant downstream ∼3750 bp to the distal PsiI site (Supplementary Figure S4C). The methylated and expanded allele was more resistant than the non-methylated expanded allele. In summary, for large expansions some of the mutant C9orf72 alleles can be resistant to MNase ∼1250 bp, diminishing through to ∼3300 bp upstream and downstream resistance is ∼2440 bp, diminishing through to ∼3700 bp away from the expanded repeat, and resistance is further exacerbated by aberrant methylation.
Southern blot—HTT
Southern blotting for the HTT allele was followed as described (73), modifying the probe to be 226 bp, which anneals 143 bp upstream of the (CAG)n repeat tract, and used selected restriction-endonuclease digestions of genomic DNA—permitting sensitive detection of repeat size and length heterogeneity at molar levels. The 226 bp probe was produced by PCR using primers (Fwd: 5′-CTTGCTGTGTGAGGCAGAAC-3′ and Rev: 5′-CGCAGGTAAAAGCAGAACCT-3′). Molecular weight markers were run alongside the DNA (Gene Ruler Mix, Thermo Scientific and Roche Molecular Weight Marker II).
Chromatin accessibility in cellulo with MNase
An HD patient fibroblast cell line, with an HTT expansion of (CAG)21/180 (GM09197) was assessed for MNase accessibility at HTT. Cell pellets were washed in ice-cold 1X PBS, re-pelleted and permeabilized by ice-cold NP-40 lysis buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40, 0.15 mM spermine, 0.5 mM spermidine) for 5 min. on ice. Nuclei were collected at 200 × g (10 min, 4°C), washed in 1X MNase digestion buffer (10 X stock: 100 mM Tris pH 7.4, 150 mM NaCl, 600 mM KCl, 5 mM spermidine, 1.5 mM spermine), re-pelleted and subsequently resuspended in 1X MNase digestion buffer supplemented with 1 mM CaCl2. Treatment followed with MNase for 5 min at 22°C, at differing concentrations (0.5–20U). All digestion reactions were stopped with an equal volume of Stop solution (0.625 mg/ml proteinase K, 20 mM EDTA, 20 mM EGTA and 1% sodium dodecyl sulfate [SDS] diluted in 1X MNase buffer) and incubated at 37°C for a minimum of 1 h. Reaction conditions were optimized to ensure over-digestion by MNase, which diminishes MNase cleavage preference. DNA was purified by phenol/chloroform extraction, RNase A treatment and ethanol precipitation. A small amount of purified DNA was assessed for completeness of MNase digestion, by its conversion to mono-, di- and tri-nucleosome sized fragments as resolved by electrophoresis stained with EtBr and visualized by UV imaging. Approximately 12 μg of each DNA sample was then restriction digested to completion to release the repeat-containing fragment and flanking regions—this increases resolution by electrophoresis. Following neutral transfer, the HTT containing regions were detected by Southern blot as described above.
RNA-seq library preparation, sequencing and analysis
Total RNA from the lymphoblastoid cell lines was isolated using the hot TRIzol extraction protocol (74). Briefly, samples dissolved in TRIzol were incubated in a thermomixer set to 55°C and 1000 rpm for 10 min. After cooling to ambient temperature, RNA was isolated by using TRIzol Reagent and the Direct-zol RNA MiniPrep Kit with DNase treatment according to manufacturer’s provided protocol (Zymo Research). RNA was quantitated using Qubit RNA BR Assay Kit (Thermo Fisher Scientific). Strand-specific, rRNA-depleted RNA-seq libraries were prepared using the KAPA Stranded RNA-seq Kit with RiboErase HMR (Kapa Biosystems) per manufacturer’s instructions, except for the use of custom Illumina-compatible index primers to allow multiplexing. Library size distribution was assessed using the High Sensitivity NGS Fragment Analysis Kit (DNF-747) on a Fragment Analyzer (Agilent). 2 × 150 paired-end sequencing was performed using an Illumina NextSeq 500. Reads were aligned to the genome assembly GRCh38 (hg38). Salmon was used for transcript expression quantification and differential gene expression analysis was performed using DESeq2 (75,76). A heat map was generated in R using the ggplot2 package. Graph was generated and statistical analysis was performed by using GraphPad Prism 8.
Developing murine tissue-derived cell lines
Mouse tissues for cytogenetic analysis of chromosomes followed published protocols (77–86). Mouse care, sacrifice and tissue harvesting was done on the same day, including three wild-type and three transgenic mice, all 5 months of age. Mice aged 5 months, when cell proliferation in the central nervous system (CNS) and peripheral organs had ceased at 2 months of age (77–79). Ear, lung, tail, cerebrum, cerebellum, brain stem, kidney, liver and spleen were harvested. A section of each tissue was manually macerated then treated with DMEM (Dulbecco's Modified Eagle Medium) + Trypsin-EDTA for 1 h in a 37°C incubator. Cells were collected, spun down and then seeded in a T25 flask with DMEM media supplemented with 20% FBS and 2 mM L-glutamate. Two flasks were prepared: one with 1% penicillin/streptomycin (Pen/Strep) [100 IU/ml, 100 μg/ml]) added and one without. Once confluent, cells were seeded in a new T25 in DMEM (supplemented with 15% fetal bovine serum, Pen/Strep, and 2 mM L-glutamate to begin fragile site induction. To avoid cell type selection and cell type loss due to an inability to adapt to cell culture, harvested tissues were purposely cultured for very short terms following tissue harvesting. When possible, cells were grown to 80% confluency. Avoiding high cell densities, or long periods at confluency, has been shown to diminish FRAXA expression (83). Tissues were harvested and immediately made into cultures. These cultures were grown and induced to express the fragile sites and cytogenetically assessed. There was neither transit delays nor storage of cell lines until fragile sites were induced in metaphase. All procedures, including colony housing and tissue harvesting, were done in-house. Precautions were taken to avoid delays following tissue harvesting, tissue freezing, extended culturing and storage of cells, as these have been noted to diminish fragile site expression (83–86).
Results
9p21 shares sequence and epigenetic features with (CGG)n FSFS
The C9orf72 locus and each of the CGG-expanded FSFS share many sequence, structural, functional and epigenetic features (Figure 2A–D and Supplementary Figure S1A–J). Nine of the 10 CGG-FSFS plus C9orf72 colocalize to boundaries between topologically associating domains and were enriched with CTCF sites and CpG-islands (Figure 2D). Thus, C9orf72 and CGG-FSFS share with 22 other disease-associated repeat expansion loci these epigenetic features (63). Unlike other disease-associated repeats, both the C9orf72 GGGGCC and all CGG-FSFS share G4-quadruplex forming repeats (87–89).
Variations in C9orf72 repeat expansion size, CpG-methylation and disease state permit assessment of contribution to fragility
To study the connection between C9orf72Exp and fragility, we characterized lymphoblastoid cells from C9orf72Exp families, where some carriers were presymptomatic and some presented with ALS or ALS/FTD (Figure 3A). Sizing C9orf72 expansions by Southern blots shows repeat length heterogeneity within and between individuals, with expansions ranging from ∼300 to ∼4400 repeats (Figure 3B and Supplementary Table S1). All individuals carried (GGGGCC)2 on the non-expanded allele.
Figure 3.

C9orf72Exp cells, repeat lengths, CpG-methylation and disease state. (A) ALS/FTD families with Southern blot vertically aligned for each individual. Females are represented by circles, males by squares, and disease state is as per the insert legend. Colors of text and boxes around symbols indicate the expansion, methylation and affected status for each sample. Percentages of cells showing FRA9A expression are indicated for each individual in the pedigree. ‘P#’ refers to patient number, ‘E’, ‘M’ and ‘A’ refer to Expanded, Methylated and Affected, is summarized at right, and this coding system is used throughout the text. Southern blot of C9orf72 (GGGGCC)n repeat expansion (‘Materials and methods’ section) used EcoRI/BamHI to release the repeat-containing fragment leaving 697 and 1714 bp upstream and downstream of the (GGGGCC)n/N repeat. (B) Schematic of Southern blot probe location, CpG-methylation status at the GGGGCC repeat and CpG-islands (filled boxes denote methylation). For raw CpG methylation data, see Supplementary Figure S2.
Most of our cell lines were CpG methylated in the expanded repeat, with varying levels in flanking CpG-islands, and no methylation in the non-expanded allele (Figure 3B and Supplementary Figure S2), consistent with previous findings of aberrant methylation and levels of methylation mosaicism in C9orf72Exp carriers (62,90). We found an absence of methylation, likely arising from methylation mosaicism, in cells of a presymptomatic C9orf72Exp (P1) and ALS-affected individuals (P7 and P8). No obvious differences were observed between cells from presymptomatic and affected C9orf72Exp individuals. Methylation status was stable, consistent with longitudinally collected samples from C9orf72Exp carriers (91). Control cells from individuals without C9orf72Exp had no methylation at C9orf72. Thus, our C9orf72Exp cell line collection permitted assessing fragility in the presence/absence of methylation and with/without disease of donors (Figure 3A, see right-hand chart).
C9orf72 expansion is the molecular cause of a FSFS at 9p21 (FRA9A)
Fragility was assessed cytogenetically (Figure 4A–D). FUdR-induced fragile sites were observed and localized to 9p21 by FISH, in all eight C9orf72Exp cell lines. Fragility was only observed in C9orf72Exp cells, with FUdR-treatment and only on one Chr9, consistent with FRA9A (58). Fragility presented in various forms including gaps (isochromatid and chromatid), constrictions and despiralized regions (Figure 4A). Full metaphase spreads and zoomed-in versions, with greater resolution are available upon request. With the exception of the despiralized form, these are similar to forms of FRAXA (59). The despiralized Chr9 was similar to the despiralized heterochromatin in immunodeficiency, centromeric instability and facial anomalies (ICF) syndrome (92). Lengthening of the despiralized FRA9A is evident when compared to the non-fragile Chr9 from the same metaphase spread (Figure 4A, control Chr9 in lower left is from the same metaphase as the leftmost stretched FRA9A). These cytogenetic observations reveal a fragile site, FRA9A, at 9p21.
We next quantified fragility. Established FXS/FRAXA diagnostic guidelines (93,94) dictate that fragile site frequencies of 3% or 5% within 100 metaphases yields confidence levels of 95% or 99%. Typically, FXS individuals show fragility at 3–15% but never >50% of CGG expanded ChrXs (94,95). We observed 9p21 fragility at 7–14% across C9orf72Exp cell lines (Figure 4E). Fragility was evident in C9orf72Exp cells of presymptomatic and ALS/FTD-affected individuals. Chromosomal assignment and band location of fragility was confirmed by Giemsa-banding, trypsinization and karyotyping (Supplementary Figure S3). Fragility at 9p21 was observed on only one of the Chr9 homologs in a given metaphase of any one C9orf72Exp cell line, consistent with each being heterozygously expanded. Fragility at 9p21 was only detected with FUdR treatment. Similar levels of fragility arose using folate-free media (M199) as for FUdR, revealing induction by different methods of folate stress (Supplementary Figure S3A). Fragility could not be induced without C9orf72 expansions in two control cell lines (100 metaphases each). Thus, fragility depends on folate perturbation and the presence of the expanded repeat. As few as 300 repeats (P3, 9%) could express FRA9A at similar levels as 4400 repeats (P4, 8%). The localized fragility, limited to one Chr9, in C9orf72Exp cells is consistent with previous FS mapping to autosomes that concluded fragility arises on the expanded allele (96,97). We validated that the C9orf72 repeat expansion is the molecular cause of FRA9A fragility through gene-transfer experiments (below).
We further mapped fragility using seven FISH probes spanning the outer edges of 9p21, in eight C9orf72Exp cell lines, >100 metaphases each, with probe signals being telomeric, spanning or centromeric to the break (Figure 4B, D and F). Breaks were detected across 9p21.1–9p21.3 chromosomal bands, ∼8.2 Mb, with ≥83% of breaks at a ∼1 Mb stretch of 9p21.2 encompassing C9orf72 (Figure 4D). Splitting of the FISH signal to both sides of the break was evident for the GGGGCC-containing probe 4, supporting that the expanded repeat causes fragility (Figure 4C). Probes telomeric to C9orf72 preferentially fluoresced telomeric of the break, with the same trend for centromeric probes (Figure 4F). The telomeric boundary could be considered at probe 2, which straddles 9p21.2–9p21.3, with fluorescence both telomeric and spanning fragility. A centromeric boundary could not be defined, as probe 7 was detected telomeric, spanning and centromeric to breaks, suggesting fragility extended centromeric of probe 7. Additional centromeric probes, being pericentromeric, hampered break detection. Thus, within the limits of FISH resolution, FRA9A maps most intensely to C9orf72, but has a wide zone of breakage (summarized in Figure 4D). FRA9A’s fragility core of enriched breakage includes C9orf72 covering ∼500 kb (plus the repeat), spanning the C9orf72Exp risk haplotype, where breaks can extend ∼2 Mb and ∼6.2 Mb telomerically and centromerically (Table 1). This makes FRA9A one of the largest fragile sites, spanning ∼8.2 Mb over 9p21.1–21.3, encompassing 46 genes (Table 1), similar in size to FRA1H, FRA3B, FRA4F, FRA7B, FRA9E, FRA11G and FRAXF, spanning up to 12.2 Mb over multiple chromosome bands, encompassing 10–110 genes (Table 1) (98–107). Moreover, CpG-methylation of C9orf72Exp was not required, as fragility was observed in cells ± methylation (Figure 4E). Thus, neither symptomatic state of blood donors nor CpG-methylation were required for FRA9A expression. That we detected FRA9A fragility at 9p21 in C9orf72Exp, lymphoblastoid cells, confirms previous observations that FRAXA expression was similarly detected lymphoblastoid cells as in freshly-collected peripheral blood lymphocyte cultures (108). Thus, FRA9A and FRAXA are distinct from the fragility at 11q23, which depends upon the EBV encoded protein EBNA1 (109–112).
Table 1.
Large fragile sites with variably located fragility cores (increased breakage) and gene numbers
|
C9orf72Exp allele assumes a compact chromatin conformation, which is enhanced by expansion size and CpG-methylation
Since constricted fragile sites on metaphase chromosomes may be the result of localized unusual chromatin condensation, we next determined whether the mutant C9orf72Exp locus assumes an unusual chromatinized state relative to the non-mutant allele using micrococcal nuclease (MNase) accessibility, which is widely used as an indication of regional chromatin compaction. We were able to electrophoretically resolve MNase accessibility on the mutant from the non-mutant alleles by Southern blotting. MNase can directly reveal both inaccessible and accessible regions for each allele, while sequencing-based methods of chromatin accessibility, like ATAC-seq, must impute inaccessibility and cannot resolve alleles. Permeabilized cells were exposed to MNase, which preferentially cuts DNA between nucleosomes; DNA was de-proteinized, isolated, restriction digested releasing the repeat-containing fragment, electrophoretically resolved and detected by the Southern blotting (Figure 5A and B, blue probe). Over-digestion by MNase to mono-, di- and tri-nucleosome sized DNA fragments was evident by ethidium staining (Figure 5C, left panel). A striking resistance to MNase accessibility is seen exclusively for the C9orf72Exp allele, contrasting the non-expanded allele, which serves as an internal equimolar control, completely degraded at low MNase concentrations (Figure 5C, compare lanes 4 and 5). The expanded allele resisted digestion at much higher MNase concentrations (Figure 5, compare lanes 5–10). MNase resistance extended beyond the expansion into the flanking regions of the mutant allele. Increasing concentrations of MNase (>50 Units) led to partial and progressive degradation of the expanded allele to a distinct shorter size, losing ∼1 kb of flanks from the full-length EcoRI-BamHI restriction fragment, which subsequently digested the probed region (Figure 5C, schematic). Conversely, on the non-mutant allele, the same flanking regions and non-expanded repeat were completely digested at low MNase concentrations, along with the rest of the genome (<10 Units, Figure 5C, lanes 5–10). The poor digestion of the C9orf72Exp mutant allele relative to the internal sequence control of the equimolar non-mutant allele strongly suggests the formation of an unusual chromatinized state of the mutant C9orf72 allele. Control experiments confirmed that the inaccessibility of MNase to the C9orf72Exp allele was due to its chromatinized state, and not due to an inherent inability of MNase to cleave the expanded non-chromatinized (GGGGCC)n sequence (Supplementary Figure S4A and B, ‘Materials and methods’ section). Six cell lines showed MNase resistance of the C9orf72Exp allele, regardless of presymptomatic or symptomatic state of the blood donors (Figure 5C–E).
Mapping the boundaries of MNase-resistance using multi-probe and multi-restriction digests revealed the extent and distribution of MNase inaccessibility over the C9orf72Exp allele. Maximal MNase resistance spanned a total of >33 kb for an allele with 4400 repeats (P4), with MNase inaccessibility extending ∼3300 bp upstream and ∼3700 bp downstream of the repeat (summarized in Figure 5A to scale, Supplementary Figure S4C, see ‘Materials and methods’ section). Mapping in six C9orf72Exp cell lines showed little variation of the absolute boundaries of MNase inaccessibility with expansion size or presence of disease. Together, these data confirm that the C9orf72Exp allele assumes a large region of compact chromatin. Maximal MNase inaccessibility covers the upstream CpG-island, the C9orf72 promoter, transcription start sites (sense and anti-sense), the expanded repeat, cryptic splice sites C1-4, the downstream CpG-island, beyond exon 2, and falls within FRA9A’s fragility core.
Repeat length and CpG-methylation enhanced MNase-resistance. Analysis of a cell line that harbored a mosaic heterogenous mixture of expansion sizes ranging between ∼300 and 4400 repeat units demonstrated that repeat expansion size directly correlates with MNase resistance (Figure 5D, P4). Longer expansions were more resistant to MNase than shorter expansions, which were more resistant than the non-expanded alleles (Figure 5D, lanes 3 and 4, quantified in Figure 5D). CpG-methylation exacerbated inaccessibility. Two cell lines with similar expansion sizes (∼770 repeats) with or without aberrant methylation at the mutant C9orf72 (P6 [isolated single cell clone] and P7, respectively) showed that methylation further enhanced MNase resistance of the expanded allele (Figure 5E and Supplementary Figure S4A, iii). Larger expansions and methylation enhance chromatin compaction of the C9orf72 locus.
We assessed chromatin compaction of post-mortem brains of two C9orf72Exp individuals, previously deep-phenotyped for lifestyle choices, clinical, neuropathological, molecular and genetic biomarkers (29,70). One was a 90-year old male asymptomatic for ALS or FTD, with a distinct ∼70 repeats in blood, no signs of neurodegeneration, overexpression of C9orf72/C9ORF72 and devoid of TDP-43 neuropathology (summarized in Supplementary Table S2). The other was his daughter with 300–3000 repeats in blood who had ALS at age 57 and died at 59 and autopsy showing neurodegeneration and TDP-43 inclusions (29) (detailed in Supplementary Table S2). The asymptomatic father had a striking heterogenous mosaic range of 13–3500 and 13–1500 repeats in the frontal cortex and cerebellum, respectively, with CpG-methylation likely triggered by large (>70 repeats) expansions (Figure 5F, lanes 0). In contrast, the affected daughter showed very large distinct expansions of mostly ∼4400 and ∼1600 CpG-methylated repeats in the frontal cortex and cerebellum, respectively, where the intensity of the expansions matched that of the non-expanded allele (Figure 5F, lanes 0). Chromatin was MNase inaccessible on only the expanded C9orf72 alleles in both brain regions of both individuals, while the non-expanded (GGGGCC)2 allele was rapidly digested by MNase (Figure 5F). MNase inaccessibility for both individuals appeared more severe in the cerebellum relative to the frontal cortex, which contrasts the expectation that the longer expansions in the frontal cortex would make it more MNase resistant, but may reflect the longer expansions and/or the increased levels of deacetylated and trimethylated histone modifications in the frontal cortex over the cerebellum, relative to the acetylated histones on the non-expanded allele of C9orf72Exp carriers (113). The shorter expansions in the father’s brain were more MNase sensitive than the larger expansions. Quantifying MNase inaccessibility between asymptomatic and symptomatic brains is hampered by the repeat length variations they presented, which we have shown to affect chromatin accessibility (Figure 5D). Expression of the C9orf72 transcript in the asymptomatic C9orf72Exp father was dramatically increased 2.5- to 7-fold in the frontal cortex, cerebellum, spinal cord, blood and four other tissues compared to the affected daughter’s tissues, whose C9orf72 levels were twice less than an individual devoid of expansions (29,70). We suggest that the increased transcription in the father’s brain arose from the shorter range of expansion sizes (13–1200 repeats) which were less MNase resistant than the longer expansions, which is consistent with both the over expression in the father's blood, which contained predominantly 70 repeats, and with the low transcription in the daughter’s brain that contained only very large expansions. This interpretation is consistent with the 2.5- to 5-fold increased expression of intermediate C9orf72 repeats in human brains, blood and knockin induced pluripotent stem cell- (iPSC-) derived neural progenitor cells (91,114). This parallels the 5- to 10-fold increased expression from the premutation CGG expansions at the FMR1 repeat in hemizygous males, which also show compact chromatin (115,116). We conclude that C9orf72 expansions and flanks assume a highly compact chromatin in cells and brain regions of C9orf72Exp carriers. MNase resistance is evident on all C9orf72Exp alleles in a given sample, which differs from the 7–14% of C9orf72Exp cells cytogenetically showing FRA9A-Chr9s, like FXS, only a portion of the CGG-expanded ChrXs show FRAXA (94,95). In this manner, MNase resistance might be considered a proxy for fragility. Towards assessing whether MNase resistance was linked to the unusual chromatin compaction of cytogenetic fragility or merely linked to expanded repeats, we assessed the MNase accessibility of the HTT locus in Huntington’s disease patient cells with (CAG)21/180, which is known to not be a cytogenetically detectable fragile site (117). MNase treatment and Southern blotting resolved the mutant and non-mutant HTT alleles and both showed the same MNase accessibility when most of the genome was sensitive (Supplementary Figure S5, lanes 3–5), suggesting that MNase inaccessibility does not extend to this non-fragile mutant locus. As part of another study, we extended the MNase inaccessibility to the CGG-expanded FMR1 gene the molecular cause for the fragile site FRAXA. We find MNase resistance of only the CGG expanded allele (in cells from male and female carriers of the CGG expansion) (not shown). That the CGG expanded FMR1, which forms FRAXA, but not the non-fragile CAG expanded HTT, forms an MNase resitant region supports our suggestion that MNase inaccessibility of the C9orf72Exp can be considered a proxy for chromosomal fragility. We suggest that the MNase resistance of the C9orf72Exp locus in human brains is a reflection of chromosomal fragility (Figure 5F).
Increased Chr9-containing micronuclei in C9orf72Exp cells
MN are indicators of chromosomal double-strand breaks (DSBs) including fragile sites (118,65–66,68), where MN often harbor broken chromosomes to be eliminated or reintegrated. NBuds are chromatin-containing protrusions linked to the nucleus by a nucleoplasmic stalk (65,66). Both MN and NBuds can arise from chromosome breaks. NBuds can become MN (65). These subnuclear structures can be enriched with tandem gene amplifications and satellite repeats (92,119–120). Cytoplasmic release of MN DNA triggers an immune response via DNA sensing by cGAS-STING that activates IFN expression (118), a pathway hyperactivated in vulnerable neurons of C9orf72Exp carriers (4,12–14). Given these connections, we assessed MN and NBuds and whether they contained Chr9 in C9orf72Exp cells. C9orf72Exp cells showed elevated MN/NBud levels compared to control cells, which were further increased with folate stress (Figure 6A). More NBuds over MNs contained Chr9 in C9orf72Exp cells compared to control cells. The modest increase in MN with Chr9 could be due to loss of fragile chromosome-containing MN during folate-perturbation, as suggested for FRAXA cells (Figure 6B) (121,122). Depending upon whether the breakpoint is centromeric or telomeric of C9orf72, the presence of the C9orf72-containing FISH signal alone or the 9q-arm FISH signal alone, supports subnuclear inclusion of an acentric or truncated Chr9, respectively, while the presence of both C9orf72- and q-arm signals supports the inclusion of truncated Chr9 or the full-Chr9 (Figure 6B). An absence of either FISH signal could reflect an absence of any Chr9, or an acentric Chr9 broken telomeric of C9orf72. When Chr9 signals were present in MN/NBuds, the proportion of the C9orf72 signal alone, 9q-arm alone, or the two together occurred at 1:1:5, respectively. Increased Chr9-containing MN/NBuds were evident in C9orf72Exp cells of presymptomatic and symptomatic individuals. Cumulatively, these data support increased chromosomal damage in C9orf72Exp cells.
Figure 6.

C9orf72Exp cells display chromosomal instability. (A) Quantification of MN and NBuds in binucleated cells without FISH, left, and with C9orf72 and 9q-FISH probes, middle, in WT and C9orf72Exp cells (P6 (ssc) and P7) containing similar sized repeats (∼770) but differing methylation status. Data are means of two independent experiments. Dots show data of each experiment. Statistical analyses using Fisher’s exact test (*P < 0.05, **P < 0.01, ***P < 0.001). Representative images of MN and NBuds in cytokinesis-blocked cells treated with FUdR for 24 h that contain no FISH signal (hollow arrows), a C9orf72-signal (red arrows) or 9q-signal (green arrows). (B) Depending upon site of breakage, C9orf72-FISH probes can have varying interpretations, left. Example of bona fide chromosome breaks in C9orf72Exp line P4 with acentric terminal C9orf72-containing fragment in same metaphase spread as a truncated-Chr9 that is free of C9orf72-signal, middle. Example of truncated-Chr9 with C9orf72-signal, and control Chr9 from same spread, right. (C) Levels of global SCEs in WT and C9orf72Exp cells ± FUdR. Methylation/clinical status is as per Figure 3. Each datapoint represents a single metaphase. At least 100 mitotic cells were analyzed per condition in each experiment. Statistical analyses were performed by Student’s t-test (P < 0.01). NS: not significant. See also Supplementary Figure S6B and D. (D) C9orf7Exp line P8 with complex highly rearranged derivative-Chr9, schematic, see text and Supplementary Figure S7A. (E) Example of FRA9A-expressing derivative-Chr9 and control Chr9 in same spread with C9orf72- (red) and 9q-FISH probes (green) (100% = 100/100 metaphases). (F) Only the derivative-Chr9 expresses FRA9A coincident with the C9orf72-FISH signal, revealing derivative-Chr9 to be the C9orf72Exp-Chr9, (+FUdR, 7% = 7/100, 99% confidence). (G) Breakpoint analysis (top) and gene expression (binned/gene) along Chr9 in each of the C9orf72 Exp cells with trisomic (red dots) and monosomic (blue dots) in duplicated and deleted regions of the derivative-Chr9 (P8). Breakpoint and copy number change analysis in P8 was by whole genome sequencing, see Supplementary Table S3. An interactive html file detailing mis-expressed genes can be accessed here https://data.cyverse.org/dav-anon/iplant/home/ljsznajder/FRA9A/Chromosome9.html), also presented in Supplementary Table S4. See Supplementary Figure S7B–D for statistical significance of differential gene expression.
We also observed increased nuclear blebs (NBlebs) in C9orf72Exp compared to control cells (Figure 6A). NBlebs are chromatin-containing nuclear herniations (65–66,123–124), without an obvious constriction between the nucleus and the protruding nuclear material. NBlebs may in some instances lead to NBuds, which may lead to MN (65). NBlebs are hallmarks of the DNA damage accumulating progeroid syndromes and can be enriched with genomic regions with perturbed chromatin and DNA damage (γH2AX) (123–127). In C9orf72Exp cells, NBlebs contained Chr9 at higher levels than in cells without expansions (Supplementary Figure S6A), which is consistent with reports of NBlebs being enriched with regions having altered chromatin compaction and damaged DNA (123–125).
C9orf72Exp and double-strand breakage
Direct support that fragile sites are prone to DNA breakage is the cytogenetic manifestation of a truncated chromosome or acentric chromosome fragment broken at the fragile site (128,129). We observed truncated Chr9s broken at 9p21, where the truncated p21→pter acentric arm, containing the C9orf72-FISH signal, remained within the same metaphase spread (Figure 6B). We observed numerous instances of truncated Chr9s with the C9orf72 signal at the broken end, in the absence of their acentric p21→pter acentric arms, which had likely been lost. 9p21-truncated Chr9s without C9orf72-signal were also observed, consistent with FRA9A’s broad ∼8.2 Mb breakage zone (Figure 6B, see truncated Chr9 broken centromeric and telomeric of the C9orf72-FISH signal). Our cytogenetic findings, coupled with the increased Chr9-containing MN/NBuds, is convincing evidence that double-strand DNA breaks occur at FRA9A, consistent with the γ-H2AX foci in cells, spinal cord and brains of C9orf72Exp carriers (23,24).
We further explored the susceptibility of GGGGCC repeats to double-strand DNA breaks using a yeast system that quantifies rates of chromosome end-loss (69). A (GGGGCC)60 inserted into a yeast chromosome was extremely unstable, yielding mixed populations with 15–60 repeat units (Supplementary Figure S7A–F). Rates of breakage showed a significant length-dependent increase: Tracts of 35–60 repeats broke 6–16-fold more frequently compared to (GGGGCC)15, which incurred breaks at 10-fold higher rates than a (CAG)85 tract analyzed in the same assay (Supplementary Figure S7B and C). Thus, even short GGGGCC repeats can be highly prone to DNA breakage. Instability of a (GGGGCC)70 tract in humans was reported (29).
Sister-chromatid exchange and recombinant events in C9orf72 expansion cells
An increased incidence of SCEs occurs at rare and common fragile sites (130). We analyzed SCE formation in C9orf72Exp. Unexpectedly, C9orf72Exp cells exhibit an increase, up to 2-fold, in spontaneous genome-wide SCEs/cell (∼7 up to 10–15/metaphase), compared to control cells without expansions (Figure 6C). Increased SCEs were evident in C9orf72Exp cells from presymptomatic and symptomatic individuals (Figure 6C, compare P1, P5 with P6, all greater than WT). The increased SCEs did not require FUdR. This increase is significant, yet modest relative to 10-fold increased SCE/metaphase in individuals with the chromosome instability Bloom syndrome. Bloom individuals present high SCEs (131–134), increased MN (132,135), a self-DNA cGAS-STING-mediated IFN over-expression (135), and often succumb to cancer before age 30 due to a coincident 10-fold increased mutation rate (134,136,137). Chemical and genetic factors can drive SCEs (134,138,139). In all instances, SCEs predominantly occur at common (140–142) and rare fragile sites (130,143–146), regardless of whether the fragile site is being induced or not (142,147). Consistent with this, the SCEs levels in C9orf72Exp cells were mildly but not significantly increased by FUdR (Figure 6C). Through FISH mapping SCEs were significantly enriched on Chr9p, where C9orf72 resides (Supplementary Figure S6B–D). Thus, the GGGGCC-expanded FRA9A, like the CGG-expanded FRAXA, the AT-repeat-expanded FRA16B, and the EBNA1-motif repeat 11q23 fragile site, incurs increased SCEs (110,112,130,146,148). These findings extend the forms of genetic instability and the sources of DNA damage in C9orf72Exp cells to include global and focal SCEs.
Unexpectedly, one of the C9orf72Exp cell lines (P8) showed complex rearrangements restricted to one Chr9, clearly evident in every metaphase (100/100 observed) (Figure 6D–F). Specifically, one Chr9 showed a duplication of nearly the entire 9q-arm (9qter-q21.11 fused to 9p23, Figure 6C), a terminal deletion of 9pter-p23, and duplications of 9p23, 9p12, 9p11.2, 9q13, 9q21.11, and deletions in 9q13, making a derivative-Chr9 (see CNV and whole-genome sequencing analysis, Figure 6D–E; Supplementary Figure S8A and Supplementary Table S3). Breakpoint pattern was reminiscent to 9p deletion/duplication syndrome (Figure 6G, top) (55,149). Importantly, only the derivative-Chr9 expressed FRA9A with FUdR in 7% (7/100) metaphases (Figure 6F), indicating it arose from the C9orf72Exp Chr9. Strikingly every metaphase showed the same rearrangements, which were evident prior to FUdR treatment. In our previous examinations of thousands of metaphases from control cells, we have not observed such homogeneity of rearrangements, making it highly unlikely that this derivative-Chr9 arose by cell line establishment or culturing. It is also highly unlikely that this individual, who was neurotypical until presenting with bulbar onset ALS at age 56, had inherited this complex set of rearrangements constitutionally in all tissues, as congenitally such rearrangements would be incompatible with neurotypical life (55,149). Rather than inherited, it is likely the rearrangements arose somatically in the blood of the C9orf72Exp individual, which is supported by our observation of similar rearrangements of the C9orf72 transgene-containing Chr6 in mouse tissues (below). Whole genome sequencing revealed the derivative-C9orf72Exp-Chr9 presented a unique state with  13 breakpoints clustered on 9p, with direct and inverted junctions, confined to a single chromosome, as such, fulfilling the criteria of chromothripsis (150–152). It is possible the FRA9A-expressing C9orf72Exp-Chr9 isolated in a micronucleus, was shattered and reassembled as the derivative-Chr9 (153). A single experimentally targeted DSB can induce chromothripsis to the targeted chromosome (154,155). The derivative-Chr9 may have arisen by stepwise alterations. The origin of the derivative-C9orf72Exp-Chr9 could not be discerned as the patient was deceased. The complex chromosomal rearrangements of the mutant C9orf72Exp FRA9A-expressing Chr9 could have downstream impact.
13 breakpoints clustered on 9p, with direct and inverted junctions, confined to a single chromosome, as such, fulfilling the criteria of chromothripsis (150–152). It is possible the FRA9A-expressing C9orf72Exp-Chr9 isolated in a micronucleus, was shattered and reassembled as the derivative-Chr9 (153). A single experimentally targeted DSB can induce chromothripsis to the targeted chromosome (154,155). The derivative-Chr9 may have arisen by stepwise alterations. The origin of the derivative-C9orf72Exp-Chr9 could not be discerned as the patient was deceased. The complex chromosomal rearrangements of the mutant C9orf72Exp FRA9A-expressing Chr9 could have downstream impact.
While we cannot exclude the possibility that the highly rearranged derrivarive-Chr9 in the C9orf72Exp cell line (P8) arose during cell line establishment or passage, it is difficult to argue away the coincidences that the rearrangements we observe were limited to only one Chr9—the Chr9 with the C9orf72Exp expressing FRA9A (Figure 6D–F), and that the breakpoints were non-random but aligned with the breakpoints of 9p-duplication/deletion syndrome (Figure 6G) (55). We also observed numerous forms of chromosome rearrangements of murine chromosomes harboring the human C9orf72 expansion, including one reminiscent of the der-Chr9 we observed in the human cell (see murine cytogenetics described below). Moreover, the cytogenetic observation of thousands of human EBV-transformed cells have not presented such chromosomal instability (59), asides from the EBV-induced fragility at 11q23, which depends upon the EBV protein EBNA1 (109–112).
Gene expression across most of the derivative-Chr9 is dysregulated. Expression analysis by RNA-seq comparing P8 to other C9orf72Exp cells, revealed trisomic and monosomic expression levels of many genes corresponding to the large duplicated and deleted regions (Figure 6G, see red and blue dots, for significance see Supplementary Figure S8B–D, an interactive html file detailing mis-expressed genes can be accessed here: https://data.cyverse.org/dav-anon/iplant/home/ljsznajder/FRA9A/Chromosome9.html), also presented in Supplementary Table S4. Amongst the dysregulated genes along the der-Chr9 in P8, many are also dysregulated in vulnerable neurons of C9orf72Exp ALS patients, including ALS- and inflammation-linked genes (https://www.biorxiv.org/content/10.1101/2023.12.22.573083v1, Supplementary Table S4). Breakpoint and copy number changes analyzed in P8 are presented in Supplementary Table S4.
Cell culture in FUdR increases repeat instability
We tested whether repeat length instability could be generated in C9orf72Exp patient cells under fragile site inducing conditions. We observed that expansion tracts shifted towards shorter repeat lengths over culture time in FUdR-treated cells (Supplementary Figure S9). In contrast, under uninduced conditions, the expanded repeat showed a general shift towards longer repeat sizes or the more predominant repeat length (Supplementary Figure S9). FUdR affected the length of only the mutant C9orf72Exp allele. FUdR-induced changes were consistent in three different C9orf72Exp cell lines and did not appear to depend upon methylation or clinical status (Supplementary Figure S9). These results support repeat size variations being generated under folate-perturbing fragile site inducing conditions.
Tissue-specific somatic repeat expansions, chromosomal fragility and chromosomal instability in C9orf72Exp mice
Tissue-specific somatic repeat instability, FRA9A chromosome fragility and chromosome instability were recapitulated in C9orf72Exp-Transgenic (Tg) mice (C9orf72Exp-Tg) harboring a single copy of the human C9orf72 with a GGGGCC expansion integrated into murine Chr6 (156). Using this mouse as a tool for instability, we isolated cells from various organs of C9orf72Exp-Tg and control non-Tg mice, aged 5 months, when cell proliferation in the CNS and peripheral organs had ceased at 2 months of age (77–79) (see ‘Materials and methods’ section). To retain as close a representation of cell types, harvested tissues were cultured for short periods sufficient for chromosome preparation (1–4 weeks, with no storage). Tissues from three mice inheriting ∼620–690 repeats (tail at 2 weeks, aged 5 months) showed a heterogenous smear of 586–1488 repeat sizes, with some of the greatest expansions in the CNS (Figure 7A). This supports massive post-natal inter- and intra-tissue repeat expansions in non-proliferating murine tissues, as observed in C9orf72Exp humans (26–30). Expansions were evident as early as 2-weeks (Shum et al., in preparation).
Figure 7.

C9orf72 repeat instability, chromosomal fragility and chromosomal instability in C9orf72Exp-Tg-mice. Southern blots for C9orf72 transgene in C9-500 mouse tissues. C9-500 mouse tissues were sized for repeat size via Southern blot in three C9orf72Exp Tg-mice. The tail was the most stable tissue. (A) Somatic repeat instability by Southern blots in tissues of three 5-month-old C9orf72Exp-Transgenic mice harboring a single copy of the human C9orf72 gene with a GGGGCC expansion integrated into murine Chr6 (156). (B) Cells isolated from tissues of the mice shown in panel (A) were grown for 1–4 weeks sufficient for metaphase spreads for fragility analysis with 1 μM FUdR with FISH analysis. A murine-specific green probe identified mouse Chr6, and human-specific red probe identified the C9orf72Exp transgene. Pictured are FSFS in the CNS (i), the periphery (ii) and quantified in panel (iii). (C) Chromosomal instability at the C9orf72Exp transgene was assessed in the same tissues used in panel (C), using the same FISH probes.
Chromosomal fragility of the C9orf72Exp transgene occurred in all tissues at frequencies of 5% to > 35% (99% confidence, Figure 7Bi–iii). Fragility presented as despiralized regions, constrictions, chromatid and isochromatid, breaks, with split transgene-specific C9orf72 FISH signals, as we observed in human C9orf72Exp cells (Figure 5B,i). Only chromosomes from the transgenic mice showed FUdR-induced fragile sites, where fragility localized to the expanded repeat of the integrated transgene, but never to the non-transgenic Chr6 (Supplementary Figure S10). Thus, FRA9A fragility can be transferred to this Tg-mouse thereby validating the C9orf72 repeat expansion as the molecular cause of FRA9A.
Chromosomal instability was evident on the C9or72Exp-Tg-containing Chr6, including acentrics, truncations, duplications, translocations, deletions and multi-branched chromosomes (Figure 7C). In one instance the C9orf72Exp-Tg translocated itself from murine Chr6 to another chromosome (Figure 7C), and in another the C9orf72Exp-Tg was deleted (Figure 7C). We observed a rearranged chromosome with a large duplication (Figure 7C), reminiscent of the derivative-C9orf72Exp-Chr9 in ALS patient, P8 (Figure 6D–G). Chromosomal fragility and instability were observed in all tissues of all three C9or72Exp-Tg mice.
While rearranged chromosomes were rare relative to fragility, neither were ever observed in the non-transgenic murine Chr6 at 6qE3, the site of transgene integration, or in any control non-transgenic tissues (three replicate mice). The absence of fragility at 6qE3 in control non-transgenic tissues is consistent with 6qE3 not being an endogenous fragile site in mice (80). Together our data support the ability of the C9orf72Exp-Transgene to incur post-natal somatic tissue-specific repeat expansions, display chromosomal fragility and chromosomal instability, as in humans. Our findings provide insight into fragile site expression and rearrangement in the CNS and periphery.
Discussion
Here, we demonstrate that C9orf72Exp is the molecular cause of the rare folate-sensitive FRA9A, cytogenetically observed more than four decades ago (58,157). This reveals previously unconsidered avenues to study genotype-phenotype relationships for co-occurring disease states associated with C9orf72Exp and attributes linked to the 9p21 region. First, folate-sensitivity suggests a possible gene-environment interaction. Second, FRA9A may serve as a DNA source to activate DDR and IFN pathways. Third, FRA9A chromosomal instability and global SCEs may increase somatic mutations at 9p21 and beyond.
Disease penetrance and gene–environment
Gene–environment interactions have long been suspected for ALS, FTD and the incompletely penetrant co-occurring phenotypes of C9orf72Exp (158). Folate-sensitive induction of C9orf72Exp repeat instability, brain region-specific repeat expansions, FRA9A fragility, chromosomal instability and methylation-sensitive chromatin compaction, which we have shown can arise in cells and tissues of both presymptomatic and symptomatic individuals, may have clinical implications. Lifestyle, including nutritional choices, of C9orf72Exp carriers can affect disease onset and progression (40). Proper levels of the linked vitamins B9 (folate) and B12 are protectors from ALS (159). Individuals with folate deficiencies, nutritionally, pharmaceutically or genetically induced, are associated with chromosomal fragility, localized chromosomal rearrangements (160–169), megaloblastic/pernicious anemia, ALS- and FTD-like symptoms (170,171), reviewed (59). In such cases these presentations were reversed upon B12 supplementation—indicating that chromosomal instability was induced by folate-stress. Somatic GGGGCC repeat instability, FRA9A fragility and global increase of SCEs depend upon C9orf72 expansion and are folate-sensitive and can occur in cells from presymptomatic C9orf72Exp individuals. C9orf72Exp carriers, in folate-deficient states, may be predisposed to repeat instability, FRA9A fragility and chromosomal instability, which if exacerbated over time may impact gene dysregulation, altering the susceptibility of phenotypic variations amongst C9orf72Exp carriers. Fragile sites and SCEs can be induced in the blood and myeloid cells by lifestyle choices, including cigarette smoking, alcohol consumption and diet (59,172–176). Tobacco and alcohol consumption by presymptomatic C9orf72Exp carriers affected disease age-of-onset (40). Various clinically used agents (177), including some considered for use in ALS/FTD, C9orf72Exp (178) and fragile X syndrome (179), like phenytoin or decitabine can induce fragile sites (59,180), induce SCEs (181,182) and modify chromosome condensation (183,184). Age-dependent ongoing repeat expansions in the brain have been suggested as a contributor to disease (26,70,185,186) and could be folate sensitive. Additionally, CpG-methylation of C9orf72Exp (185,187) and acceleration of genome-specific DNA methylation age (epigenetic clock) are modifiers of disease onset and progression in C9orf72Exp carriers (188,189). Chromatin compaction of C9orf72Exp is sensitive to the degree of expansion and CpG-methylation (Figure 5). Efforts to prophylactically slow the epigenetic clock are a central focus (190–192). The C9orf72 expansion was recently reported to be bound by the chromatin remodeler DAXX (193). Coincidentally, DAXX, is known to associate with satellite and telomere repeats and its downregulation led to a global increase in MNase sensitivity (194–196). Environmental contributions to C9orf72Exp diseases must be considered.
Various genetic, molecular and environmental aspects have been demonstrated to modify disease presentation in C9orf72Exp carriers (3,4,7,8,40,42,45,197–204). The 90-year-old male C9orf72Exp carrier presented herein (Figure 5) is likely to be truly asymptomatic, as a long presymptomatic phase with evident degeneration often detected decades before ALS or FTD symptom manifestation (205,206), pathology that was not present in his brain (70). Clinical differences between the truly asymptomatic father and symptomatic daughter studied herein could not be explained by either genetic modifiers (TMEM106B, ATXN2 and Finnish haplotype), both expressed similar burdens of p62 inclusions, similar burdens of sense and antisense RNA foci, similar burdens of all five types of RAN-peptides (GA > GP > GR > PA/PR, except for the cerebellum, which showed more RAN-peptide inclusions (∼7-fold) and sense RNA foci (∼2-fold) in the asymptomatic father compared to the ALS/FTD-affected daughter) (29,70) (summarized in Supplementary Table S2). Also, neither lifestyle choices, life events, nor autoimmunity, could explain their phenotypic differences (29,70) (Supplementary Table S2). However, compared to the daughter, in the brain the father showed extreme repeat length mosaicism (13–3500 repeats) (Figure 5D), with increased MNase accessibility of shorter expansions, increased C9orf72 expression (mRNA and C9ORF72-long, but not C9ORF72-short protein) (29,70), increased TDP-43 inclusions (29,70) and an epigenetic age ∼9 years younger than his chronologic age (the daughter was 3 years older) (70,188) (summarized in Supplementary Table S2). Thus, these somatic variations deserve further attention in pre-, asymptomatic and symptomatic C9orf72Exp carriers.
FRA9A as a DNA source to activate the DDR and IFN pathways
Both a spontaneous activated DDR (15–25) and a hyperactivated IFN cGAS-STING-associated immune response occur in C9orf72Exp patient cells and brains (4,12–14). The source of the damaged DNA or DNA-immunostimulant to activate either DDR or IFN response is unknown. DDR in cells and brains of C9orf72Exp, evident as increased γ-H2AX, phosphorylated-ATM, cleaved Poly(ADP-ribose) polymerase-1 (PARP-1) and 53BP1, is coincident with DSBs (22–24). We reveal that DSBs arise at C9orf72Exp via heightened FRA9A fragility, broken/rearranged Chr9s, increased genome-wide SCEs, increased MN and Chr9-containing MN. The increased C9ORF72 protein levels in multiple tissues of the asymptomatic father shown to have massive mosaic somatic expansions (Figure 5F and Supplementary Table S2; (29,70)) may suppress an autoimmune response by increasing STING levels. Models of the C9orf72Exp-linked DDR activation involve RAN-peptide induced p53-target gene activation, inhibition or mislocalization of the DSB DNA repair proteins ATM, 53BP1, TDP-43, FUS, Ku80 and PARP-1 (15–25). Other non-RAN-peptide DDR paths have received little attention.
The unusual >33kb of compact chromatin at FRA9A’s core fragility zone (Figure 5A) could be susceptible to DSBs, which would contribute to DDR activation. Supporting this possibility is that experimentally-induced locus-specific compacted chromatin of an artificial ∼10 kb stretch of ∼250 tandem arrays of LacO, which becomes a fragile site, has been demonstrated to activate a DDR in the absence of damage (207–210). A contribution of FRA9A fragility to DDR can act in parallel to RAN-peptide induced DDR (23,24). Disruption of higher-order chromosomal organization may also contribute to DDR and IFN responses. Long-range chromosomal interactions involving 9p21 are activated in the inflammatory response. All IFN genes at 9p21 are transcriptionally inactive until immunostimulated by viral infection or cytosolic self-DNAs, whereupon dramatic chromatin (211) and multiple intra-9p21, inter-arm (9p21-9q34.2), and inter-chromosomal (9p21-4p14 and 9p21-18q21.1) interactions arise to activate IFN cluster expression and inflammation (47,212). Reduced levels of the C9ORF72 protein in C9orf72Exp patients, being unable to supress STING, leads to a sustained hyperactivation of IFN in brains (213). Upon FRA9A breakage the C9orf72Exp-Chr9 activates DDR and is then sequestered to MN whose contents can be released to the cytosol to activate IFN through cGAS-STING, and this sequestration may disrupt other inter- and intra-Chr9 interactions (214). That disruption of intra-/inter-chromosomal associations can drive DDR and IFN cascades is supported by the experimental disruption of such interactions leading to chromosome sequestration to MN (215,216). CGG expansions at FMR1, the cause of FRAXA, disrupt intra- and inter-chromosome interactions (217). The 2-fold increase in global SCEs in C9orf72Exp cells we observe may contribute to DDR and MN. MN enriched with the DSB repair protein, TDP-43, have been detected in brains of ALS patients (218). Our suggestion that DNA damage induced by FRA9A fragility can serve as a DNA-immunostimulant in the CNS is consistent with reports of in vivo DNA damage by ATM-deficiency or brain trauma that activates an IFN response in motor neurons (219–221). Our working model that the C9orf72Exp acts as a chromatin compacted FSFS, is an inducer of DNA damage at many locations including on Chr9 and is suited to non-cycling neurons.
We suggest that the cytoplasmic release of DNA from FRA9A-enhanced MN, may further tip the balance by exacerbating the stimulation of the aberrantly hyperactive cGAS-STING initiated IFN expression in C9orf72Exp carriers (4,13,14), which may explain the co-morbid partially-penetrant autoimmunity (1–6). FRA9A broken-Chr9s and increased global SCEs might also further exacerbate the DDR cascade, as DNA-immunostimulation enhances sense and antisense transcription from C9orf72 (42) whose RAN-peptide products would feedback upon the DDR by suppressing DSB DNA repair proteins ATM, p53, TDP-43, FUS, Ku80 and PARP-1 (15–25). The neuroinflammatory and autoimmune phenotypes of C9orf72Exp carriers is complex (14), and the effects of either FRA9A altered chromatin or MNs containing broken Chr9s upon the IFN response is unknown.
FRA9A chromosomal instability, mosaic somatic mutations and 9p21-associated diseases
A reasonable prediction from our findings is that the C9orf72/FRA9A fragile site may, like other fragile sites, predispose to mosaic deletions, duplications or rearrangements resulting in altered expression of genes on 9p21 or throughout Chr9. Such mosaic chromosomal variations may vary between tissues, accumulate with age, and lead to phenotypic variability. In the case of FMR1/FRAXA, phenotypic variability can arise through mosaic genetic or chromosomal instability (deletions, duplications and rearrangements) extending beyond the FMR1 CGG-repeat, as revealed through decades of isolated case reports (59). Similar phenomena may arise at the mutant C9orf72, as suggested by our observation of truncated-Chr9s broken at C9orf72, acentric-Chr9s, chromosomal instability of the C9orf72Exp allele and heightened levels of genome-wide SCEs. Detection of somatic mosaic levels of chromosomal instability will likely require more focused studies. For example, the dramatic Chr9 rearrangements, evident by FISH in every metaphase of the ALS/C9orf72Exp line P8, escaped detection by repeat-primed PCR and by Southern blot assessment of the repeat size (compare Figure 3B with Figure 6F). Indeed, mosaic mutations at various loci (reviewed in (222–224)) including FMR1 (225–230) required focused attention. Mosaic mutations may be confined to specific cell types, as recently revealed for Purkinje cells with large somatic CAG-expansions, amongst stable tracts in the cerebellum of Huntington’s disease patients (231,232). One might expect an increase in somatic mutation rates throughout the genome in C9orf72Exp patients, as the increased 2-fold global SCEs we observed may have a coincident increase in mutation rate, similar to the 10-fold increased mutation rates in BLM individuals who show a 10-fold increase of SCEs (131,132,136,137). C9orf72Exp cells from presymptomatic individuals show folate-sensitive repeat instability, FRA9A fragility, chromosomal instability, MNase-resistance, increased levels of MN/NBuds and global SCEs. Long-term display of these features may exacerbate disease-onset and affect penetrance. Our findings support that the C9orf72Exp patients may incur mosaic mutations at under-appreciated levels, and recent findings support increased somatic mutations in brains and spinal cords of C9orf72Exp individuals (233, https://www.biorxiv.org/content/10.1101/2023.11.30.569436v1).
Knowledge gaps remain for FRA9A/C9orf72Exp. In particular, the co-occurrence of many diverse phenotypes in C9orf72 expansion carriers is unclear. That FRA9A encompasses the IFN cluster, CDKN2A, IFNk, LINGO2, TOPORS, APTX, DNAJA1 and SMU1 genes, each independently associated with symptoms that can co-occur in C9orf72Exp carriers, provide new insights into the possible involvement of chromosomal instability (Figure 1). An increase in ALS occurring in cancer survivors, particularly melanoma, has been repeatedly reported (7,234–238). While a correlation of C9orf72Exp in cancer predisposition has not been studied in large populations, C9orf72Exp carriers have a significantly higher risk of melanoma (7). C9orf72Exp carriers may succumb to cancer prior to onset or awareness of ALS. That both CDKN2A and the IFN gene cluster, frequently deleted in melanoma, are encompassed in FRA9A’s fragility zone introduces the possible involvement of C9orf72Exp. Mosaic somatic mutations that extend beyond the C9orf72 repeat may affect expression of genes encompassed by FRA9A or beyond. Thus, revisiting the C9orf72/9p21 locus with the awareness that it is, and can share the attributes of, a chromosomal fragile site should provide key insights into the genotype–phenotype relationships for the many and varied presentations associated with the 9p21 region.
Supplementary Material
Acknowledgements
This study is dedicated to the memory of Dr Stephen T. Warren (1953–2021), a pioneer and advocate of Fragile X syndrome, a mentor and friend. This study is also dedicated to Robert K. Ranum, a joyful, persevering individual, and friend. We thank Robert H. Brown for providing a YAC-C9orf72 clone, John Volpe, Shamima Keka Islam for counting micronuclei, Lazar Joksimovic and Raymond Wong for his expertise in the preparation of metaphase spreads. We thank Olivia Beck and Lieve Wiericx for sharing their expertise in cytogenetic fragile site recognition and Edwin Reyniers and Stephen W. Scherer, Jeff MacDonald, Mehdi Zarrei, Mehdi Layeghifard and Bhooma Thiruvahindrapuram for help with the der-Chr9 data, and Michael Fenech for support regarding micronuclei, NBuds and NBlebs.
Author contributions: Conceptualization: C.E.P. and R.F.K.; Figure 1: C.E.P.; Figure 2: M.L., M.D.W. and C.E.P.; Figure 3: M.M., M.H.M.S., C.E.P., P.A.D., G.A.R., E.K. and M.Z.; Figure 4: M.M., I.v.d.W., S.S., R.F.K. and C.E.P.; Figure 5: M.H.M.S., T.K.P. and C.E.P. (sample collection by L.Z., J.K., A.A., P.M.c.G., P.M.c.K. and J.R.); Figure 6: M.M.*, M.H.M.S., N.S., S.L., K.Y., M.M.†, M.K., R.K.C.Y., P.M.c.K., J.R., L.S., M.S.S. and C.E.P.; Figure 7: N.S. and B.M. (training by M.M.*, M.H.M.S. and T.K.P.); Table 1: M.M.*, M.K. and C.E.P.; Figure S1: M.L., M.D.W. and C.E.P.; Supplementary Figure S2: M.M.*, M.Z., E.R. and M.H.M.S.; Supplementary Figure S3: M.M.*, I.v.d.W., S.S. and R.F.K.; Supplementary Figure S4: M.H.M.S.; Supplementary Figure S5: M.M.*; Supplementary Figure S6: M.M.*, M.M.†, S.L., N.S., K.Y., M.H.M.S. and C.E.P.; Supplementary Figure S7: T.A., E.A.P. and C.H.F.; Supplementary Figure S8: R.K.C.Y., R.F.K., M.M.* and L.S.; Supplementary Figure S9: N.S.; Supplementary Figure S10: B.M. (training by M.M.*); Supplementary Table S1: M.H.M.S.; Supplementary Table S2: L.Z., J.K., A.A., P.M.c.G., P.M.c.K., J.R. and C.E.P.; Supplementary Table S3: L.S., M.K., P.Mc.K., R.K.C.Y., J.R. and C.E.P.; Supplementary Table S4: M.K. and R.K.C.Y.; BAC DNA for patient C9orf72 provided by C.J., P.J.de.J.; ALS patient cells provided by P.A.D. and G.A.R.; Autopsy tissues prepared by L.Z., P.M.K. and J.R.; Intellectual input and experimental design: C.E.P., R.F.K., E.C., R.W., M.D.W., M.S.S. and E.R.; Supervision: C.E.P. and R.F.K.; Writing—original draft: MM*, CEP; Writing—review and editing: C.E.P., M.H.M.S., N.S., M.S.S. and M.M.†. All authors provided feedback on the manuscript; M.M.* = Mila Mirceta; M.M.† = Mohiuddin Mohiuddin.
Contributor Information
Mila Mirceta, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Monika H M Schmidt, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Natalie Shum, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Tanya K Prasolava, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada.
Bryanna Meikle, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Stella Lanni, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada.
Mohiuddin Mohiuddin, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada.
Paul M McKeever, Tanz Centre for Research of Neurodegenerative Diseases, University of Toronto, 60 Leonard Avenue, Toronto, M5T 2S8, Canada.
Ming Zhang, Tanz Centre for Research of Neurodegenerative Diseases, University of Toronto, 60 Leonard Avenue, Toronto, M5T 2S8, Canada; The First Rehabilitation Hospital of Shanghai, Department of Medical Genetics, School of Medicine, Tongji University, Shanghai, 200090, China; State Key Laboratory of Cardiology and Medical Innovation Center, Shanghai East Hospital, School of Medicine, Tongji University, Shanghai, China; Advanced Study, Tongji University, Shanghai, 200092, China.
Minggao Liang, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Ilse van der Werf, Department of Medical Genetics, University of Antwerp, Belgium.
Stefaan Scheers, Department of Medical Genetics, University of Antwerp, Belgium.
Patrick A Dion, Montreal Neurological Institute-Hospital, McGill University, 3801 University Avenue, Montreal, Quebec, H3A 2B4, Canada; Department of Neurology and Neurosurgery, McGill University, 3801 University Avenue, Montreal, Quebec, H3A 2B4, Canada.
Peixiang Wang, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada.
Michael D Wilson, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Theresa Abell, Department of Biology, Tufts University, 200 Boston Avenue, Medford, MA 02155, USA.
Elliot A Philips, Department of Biology, Tufts University, 200 Boston Avenue, Medford, MA 02155, USA.
Łukasz J Sznajder, Department of Molecular Genetics and Microbiology, Center for NeuroGenetics and the Genetics Institute, College of Medicine, University of Florida, 2033 Mowry Road, Gainesville, FL 32610-3610, USA; Department of Chemistry and Biochemistry, University of Nevada, 4003-4505 South Maryland Parkway, Las Vegas, NV 89154, USA.
Maurice S Swanson, Department of Molecular Genetics and Microbiology, Center for NeuroGenetics and the Genetics Institute, College of Medicine, University of Florida, 2033 Mowry Road, Gainesville, FL 32610-3610, USA.
Mustafa Mehkary, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Mahreen Khan, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Katsuyuki Yokoi, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada.
Christine Jung, BACPAC Resource Center, Children’s Hospital Oakland Research Institute, 25129 NE 42nd Pl, Redmond, WA 98053, USA.
Pieter J de Jong, BACPAC Resource Center, Children’s Hospital Oakland Research Institute, 25129 NE 42nd Pl, Redmond, WA 98053, USA.
Catherine H Freudenreich, Department of Biology, Tufts University, 200 Boston Avenue, Medford, MA 02155, USA.
Philip McGoldrick, Tanz Centre for Research of Neurodegenerative Diseases, University of Toronto, 60 Leonard Avenue, Toronto, M5T 2S8, Canada.
Ryan K C Yuen, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Agessandro Abrahão, Sunnybrook Health Sciences Centre, 2075 Bayview Avenue, North York, Toronto, ON, M4N 3M5, Canada.
Julia Keith, Sunnybrook Health Sciences Centre, 2075 Bayview Avenue, North York, Toronto, ON, M4N 3M5, Canada.
Lorne Zinman, Sunnybrook Health Sciences Centre, 2075 Bayview Avenue, North York, Toronto, ON, M4N 3M5, Canada.
Janice Robertson, Tanz Centre for Research of Neurodegenerative Diseases, University of Toronto, 60 Leonard Avenue, Toronto, M5T 2S8, Canada.
Ekaterina Rogaeva, Tanz Centre for Research of Neurodegenerative Diseases, University of Toronto, 60 Leonard Avenue, Toronto, M5T 2S8, Canada.
Guy A Rouleau, Montreal Neurological Institute-Hospital, McGill University, 3801 University Avenue, Montreal, Quebec, H3A 2B4, Canada; Department of Neurology and Neurosurgery, McGill University, 3801 University Avenue, Montreal, Quebec, H3A 2B4, Canada; Department of Human Genetics, McGill University, 3801 University Avenue, Montreal, Quebec, H3A 2B4, Canada.
R Frank Kooy, Department of Medical Genetics, University of Antwerp, Belgium.
Christopher E Pearson, Program of Genetics and Genome Biology, Peter Gilgan Centre for Research and Learning, The Hospital for Sick Children, 686 Bay Street, Toronto, M5G 0A4, Canada; Department of Molecular Genetics, University of Toronto, 1 King's College Circle, Toronto, ON, M3S 1A8, Canada.
Inclusion and ethics statement
All collaborators of this study who have fulfilled the criteria for authorship required by NAR Molecular Medicine and Oxford University Press journals are included as authors. Each author’s contribution is detailed in the Author Contributions list. Expectations for collaborators were agreed upon ahead of the study’s initiation, and authorship placement was agreed upon by each collaborator. This study was conducted in collaboration with researchers and physicians from distinct cultural, ethnic and racial backgrounds. The findings of this study apply to members of the population globally, as well as locally in the regions where patient recruitment occurred. The research does not result in stigmatization, incrimination, discrimination or personal risk to participants who provided samples. Local and regional research publications relevant to our study was considered and included in our bibliography.
The Hospital for Sick Children Research Ethics Board oversight and approval for the herein study was obtained. REB for human tissue, cell and animal studies: #1000056560.
Data availability
Datasets generated during the current study supporting its findings have been deposited or made available as indicated: RNASeq gene expression data—available on Gene Expression Ombinus (GEO), accession GSE262674. Whole genome sequencing data for the der-Chr9 (P8) sample is available in the European Genome-Phenome Archive (EGA) upon approval by the Data Access Committee. Correspondence and requests for materials should be addressed to Dr Christopher E. Pearson (mail to: cepearson.sickkids@gmail.com). Mouse models presented in this study are commercially available from Jackson Laboratories (FVB/NJ-Tg(C9orf72)500Lpwr/J).
Supplementary data
Supplementary Data are available at NARMME Online.
Funding
ALS Canada-Brain Canada 2020 Discovery Grant [HSC0005599 to C.E.P., E.R., G.A.R.]; ALS Canada Doctoral Research Award 2016 [to M.H.M.S.]; Weston Family Foundation [TR202268 to C.E.P.]; W. Garfield Weston Foundation [CNV0001757 to C.E.P.]; Natural Sciences and Engineering Research Councilof Canada (NSERC) [RGPIN-2019–07014 to M.D.W.], National Institutes of Health [GM122880 and GM144215 to C.H.F. and NS048843 to M.S.S.]; Muscular Dystrophy Association Development Grant [MDA546770 to L.S.], National Natural Science Foundation of China [82071430 to M.Z., E.R.]; James Hunter and Family ALS Initiative (to J.R., E.R.); Canadian Consortium on Neurodegeneration in Aging (to G.A.R., E.R.); ALS Association Milton Safenowitz Fellowhipand ALS Double Play Christopher Chiu Fellowship [P.M.M.]; ALS Society of Canada Project Grant [to J.R.], ERA-LEARN (E-Rare-3/Canadian Institute for Health Research) grant (063-REPETOMICS) [J.R.].
Conflict of interest statement. M.S.S. is a Scientific Advisory Board member of Skyhawk Therapeutics and Tacit Therapeutics. For all other authors, no conflict declared.
References
- 1. Atanasio A., Decman V., White D., Ramos M., Ikiz B., Lee H.-C., Siao C.-J., Brydges S., LaRosa E., Bai Y. et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep. 2016; 6:23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Breevoort S., Gibson S., Figueroa K., Bromberg M., Pulst S. Expanding clinical spectrum of C9ORF72-related disorders and promising therapeutic strategies: a review. Neurol. Genet. 2022; 8:e670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fredi M., Cavazzana I., Biasiotto G., Filosto M., Padovani A., Monti E., Tincani A., Franceschini F., Zanella I. C9orf72 intermediate alleles in patients with amyotrophic lateral sclerosis, systemic lupus erythematosus, and rheumatoid arthritis. Neuromol. Med. 2019; 21:150–159. [Google Scholar]
- 4. McCauley M.E., O’Rourke J.G., Yáñez A., Markman J.L., Ho R., Wang X., Chen S., Lall D., Jin M., Muhammad A.K.M.G. et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 2020; 585:96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller Z.A., Sturm V.E., Camsari G.B., Karydas A., Yokoyama J.S., Grinberg L.T., Boxer A.L., Rosen H.J., Rankin K.P., Gorno-Tempini M.L. et al. Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts: completing the picture. Neurol. Neuroimmunol. Neuroinflamm. 2016; 3:e301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van der Ende E.L., Jackson J.L., White A., Seelaar H., van Blitterswijk M., Van Swieten J.C. Unravelling the clinical spectrum and the role of repeat length in C9ORF72 repeat expansions. J. Neurol. Neurosurg. Psychiatry. 2021; 92:502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tábuas-Pereira M., Almendra L., Almeida M.R., Durães J., Pinho A., Matos A., Negrão L., Geraldo A., Santana I. Increased risk of melanoma in C9ORF72 repeat expansion carriers: a case-control study. Muscle Nerve. 2019; 59:362–365. [DOI] [PubMed] [Google Scholar]
- 8. Chiò A., Mazzini L., D’Alfonso S., Corrado L., Canosa A., Moglia C., Manera U., Bersano E., Brunetti M., Barberis M. et al. The multistep hypothesis of ALS revisited: the role of genetic mutations. Neurology. 2018; 91:e635–e642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Wijk I.F., Van Eijk R.P.A., Van Boxmeer L., Westeneng H.-J., Van Es M.A., Van Rheenen W., Van Den Berg L.H., Eijkemans M.J.C., Veldink J.H Assessment of risk of ALS conferred by the GGGGCC hexanucleotide repeat expansion in C9orf72 among first-degree relatives of patients with ALS carrying the repeat expansion. Amyotroph. Lateral Scler. Frontotemporal Degener. 2024; 25:188–196. [DOI] [PubMed] [Google Scholar]
- 10. Waite A.J., Bäumer D., East S., Neal J., Morris H.R., Ansorge O., Blake D.J. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging. 2014; 35:1779.e5–e1779. [Google Scholar]
- 11. Xiao S., MacNair L., McLean J., McGoldrick P., McKeever P., Soleimani S., Keith J., Zinman L., Rogaeva E., Robertson J. C9orf72 isoforms in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Brain Res. 2016; 1647:43–49. [DOI] [PubMed] [Google Scholar]
- 12. Marques C., Held A., Dorfman K., Sung J., Song C., Kavuturu A.S., Aguilar C., Russo T., Oakley D.H., Albers M.W. et al. Neuronal STING activation in amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. 2024; 147:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pang W., Hu F. C9ORF72 suppresses JAK-STAT mediated inflammation. Iscience. 2023; 26:106579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Masrori P., Beckers J., Gossye H., Van Damme P. The role of inflammation in neurodegeneration: novel insights into the role of the immune system in C9orf72 HRE-mediated ALS/FTD. Mol. Neurodegener. 2022; 17:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Y., Liu L., Chen H., Yang Y., Mu C., Ren H., Liu Y., Yu L., Fang Q., Wang G. et al. Disrupted phase behavior of FUS underlies poly-PR-induced DNA damage in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2023; 33:64–77. [DOI] [PubMed] [Google Scholar]
- 16. Braems E., Bercier V., Van Schoor E., Heeren K., Beckers J., Fumagalli L., Dedeene L., Moisse M., Geudens I., Hersmus N. et al. HNRNPK alleviates RNA toxicity by counteracting DNA damage in C9orf72 ALS. Acta Neuropathol. 2022; 144:465–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Konopka A., Atkin J.D. DNA damage, defective DNA repair, and neurodegeneration in amyotrophic lateral sclerosis. Front. Aging Neurosci. 2022; 14:786420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maor-Nof M., Shipony Z., Lopez-Gonzalez R., Nakayama L., Zhang Y.-J., Couthouis J., Blum J.A., Castruita P.A., Linares G.R., Ruan K. et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell. 2021; 184:689–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andrade N.S., Ramic M., Esanov R., Liu W., Rybin M.J., Gaidosh G., Abdallah A., Del’Olio S., Huff T.C., Chee N.T. et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020; 15:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nihei Y., Mori K., Werner G., Arzberger T., Zhou Q., Khosravi B., Japtok J., Hermann A., Sommacal A., Weber M. et al. Poly-glycine-alanine exacerbates C9orf72 repeat expansion-mediated DNA damage via sequestration of phosphorylated ATM and loss of nuclear hnRNPA3. Acta Neuropathol. 2020; 139:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lopez-Gonzalez R., Yang D., Pribadi M., Kim T.S., Krishnan G., Choi S.Y., Lee S., Coppola G., Gao F.-B. Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD. Proc. Natl Acad. Sci. U.S.A. 2019; 116:9628–9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Konopka A., Atkin J.D. The emerging role of DNA damage in the pathogenesis of the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2018; 19:E3137. [Google Scholar]
- 23. Walker C., Herranz-Martin S., Karyka E., Liao C., Lewis K., Elsayed W., Lukashchuk V., Chiang S.-C., Ray S., Mulcahy P.J. et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat. Neurosci. 2017; 20:1225–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Farg M.A., Konopka A., Soo K.Y., Ito D., Atkin J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017; 26:2882–2896. [DOI] [PubMed] [Google Scholar]
- 25. Lopez-Gonzalez R., Lu Y., Gendron T.F., Karydas A., Tran H., Yang D., Petrucelli L., Miller B.L., Almeida S., Gao F.-B. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron. 2016; 92:383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Blitterswijk M., DeJesus-Hernandez M., Niemantsverdriet E., Murray M.E., Heckman M.G., Diehl N.N., Brown P.H., Baker M.C., Finch N.A., Bauer P.O. et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): A cross-sectional cohort study. Lancet Neurol. 2013; 12:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nordin A., Akimoto C., Wuolikainen A., Alstermark H., Jonsson P., Birve A., Marklund S.L., Graffmo K.S., Forsberg K., Brännström T. et al. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum. Mol. Genet. 2015; 24:3133–3142. [DOI] [PubMed] [Google Scholar]
- 28. Akimoto C., Volk A.E., van Blitterswijk M., Van den Broeck M., Leblond C.S., Lumbroso S., Camu W., Neitzel B., Onodera O., van Rheenen W. et al. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J. Med. Genet. 2014; 51:419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xi Z., van Blitterswijk M., Zhang M., McGoldrick P., McLean J.R., Yunusova Y., Knock E., Moreno D., Sato C., McKeever P.M. et al. Jump from pre-mutation to pathologic expansion in C9orf72. Am. J. Hum. Genet. 2015; 96:962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xi Z., Yunusova Y., van Blitterswijk M., Dib S., Ghani M., Moreno D., Sato C., Liang Y., Singleton A., Robertson J. et al. Identical twins with the C9orf72 repeat expansion are discordant for ALS. Neurology. 2014; 83:1476–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sattler R., Traynor B.J., Robertson J., Van Den Bosch L., Barmada S.J., Svendsen C.N., Disney M.D., Gendron T.F., Wong P.C., Turner M.R. et al. Roadmap for C9ORF72 in frontotemporal dementia and amyotrophic lateral sclerosis: report on the C9ORF72 FTD/ALS summit. Neurol. Ther. 2023; 12:1821–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cammack A.J., Balendra R., Isaacs A.M. Failure of C9orf72 sense repeat-targeting antisense oligonucleotides: lessons learned and the path forward. Brain. 2024; 147:2607–2609. [DOI] [PubMed] [Google Scholar]
- 33. Petri S. Targeting C9orf72 in people with ALS. Lancet Neurol. 2024; 23:850–852. [DOI] [PubMed] [Google Scholar]
- 34. van den Berg L.H., Rothstein J.D., Shaw P.J., Babu S., Benatar M., Bucelli R.C., Genge A., Glass J.D., Hardiman O., Libri V. et al. Safety, tolerability, and pharmacokinetics of antisense oligonucleotide BIIB078 in adults with C9orf72-associated amyotrophic lateral sclerosis: a phase 1, randomised, double blinded, placebo-controlled, multiple ascending dose study. Lancet Neurol. 2024; 23:901–912. [DOI] [PubMed] [Google Scholar]
- 35. Wave Life Sciences Announces Topline Results from Phase 1b/2a FOCUS-C9 Study of WVE-004 for C9orf72-associated Amyotrophic Lateral Sclerosis and Frontotemporal Dementia - Wave Life Sciences. (25 November 2024, date last accessed)https://www.neurologylive.com/view/wave-life-sciences-discontinues-c9orf72-als-frontotemporal-dementia-agent-wve-004-after-disappointing-phase-1b-2a-findings.
- 36. Tran H., Moazami M.P., Yang H., McKenna-Yasek D., Douthwright C.L., Pinto C., Metterville J., Shin M., Sanil N., Dooley C. et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 2022; 28:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saracino D., Le Ber I. How can we define the presymptomatic C9orf72 disease in 2022? An overview on the current definitions of preclinical and prodromal phases. Rev. Neurol. (Paris). 2022; 178:426–436. [DOI] [PubMed] [Google Scholar]
- 38. Saracino D., Le Ber I. Clinical update on C9orf72: frontotemporal dementia, amyotrophic lateral sclerosis, and beyond. Adv. Exp. Med. Biol. 2021; 1281:67–76. [DOI] [PubMed] [Google Scholar]
- 39. Mandrioli J., Zucchi E., Martinelli I., Van der Most L., Gianferrari G., Moglia C., Manera U., Solero L., Vasta R., Canosa A. et al. Factors predicting disease progression in C9ORF72 ALS patients. J. Neurol. 2023; 270:877–890. [DOI] [PubMed] [Google Scholar]
- 40. Westeneng H.-J., van Veenhuijzen K., van der Spek R.A., Peters S., Visser A.E., van Rheenen W., Veldink J.H., van den Berg L.H. Associations between lifestyle and amyotrophic lateral sclerosis stratified by C9orf72 genotype: a longitudinal, population-based, case-control study. Lancet Neurol. 2021; 20:373–384. [DOI] [PubMed] [Google Scholar]
- 41. Querin G., Biferi M.G., Pradat P.-F. Biomarkers for C9orf7-ALS in symptomatic and pre-symptomatic patients: state-of-the-art in the new era of clinical trials. J. Neuromuscul. Dis. 2022; 9:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rizzu P., Blauwendraat C., Heetveld S., Lynes E.M., Castillo-Lizardo M., Dhingra A., Pyz E., Hobert M., Synofzik M., Simón-Sánchez J. et al. C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol. Commun. 2016; 4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Niblock M., Smith B.N., Lee Y.-B., Sardone V., Topp S., Troakes C., Al-Sarraj S., Leblond C.S., Dion P.A., Rouleau G.A. et al. Retention of hexanucleotide repeat-containing intron in C9orf72 mRNA: implications for the pathogenesis of ALS/FTD. Acta Neuropathol. Commun. 2016; 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sznajder Ł.J., Thomas J.D., Carrell E.M., Reid T., McFarland K.N., Cleary J.D., Oliveira R., Nutter C.A., Bhatt K., Sobczak K. et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl Acad. Sci. U.S.A. 2018; 115:4234–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ben-Dor I., Pacut C., Nevo Y., Feldman E.L., Reubinoff B.E. Characterization of C9orf72 haplotypes to evaluate the effects of normal and pathological variations on its expression and splicing. PLoS Genet. 2021; 17:e1009445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sarkar M.K., Hile G.A., Tsoi L.C., Xing X., Liu J., Liang Y., Berthier C.C., Swindell W.R., Patrick M.T., Shao S. et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann. Rheum. Dis. 2018; 77:1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Harismendy O., Notani D., Song X., Rahim N.G., Tanasa B., Heintzman N., Ren B., Fu X.-D., Topol E.J., Rosenfeld M.G. et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature. 2011; 470:264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Joung J., Engreitz J.M., Konermann S., Abudayyeh O.O., Verdine V.K., Aguet F., Gootenberg J.S., Sanjana N.E., Wright J.B., Fulco C.P. et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. 2017; 548:343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ahel I., Rass U., El-Khamisy S.F., Katyal S., Clements P.M., McKinnon P.J., Caldecott K.W., West S.C. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006; 443:713–716. [DOI] [PubMed] [Google Scholar]
- 50. Lim G., Karaskova J., Beheshti B., Vukovic B., Bayani J., Selvarajah S., Watson S.K., Lam W.L., Zielenska M., Squire J.A. An integrated mBAND and submegabase resolution tiling set (SMRT) CGH array analysis of focal amplification, microdeletions, and ladder structures consistent with breakage-fusion-bridge cycle events in osteosarcoma. Genes Chromosomes Cancer. 2005; 42:392–403. [DOI] [PubMed] [Google Scholar]
- 51. Fletcher J.A., Gebhardt M.C., Kozakewich H.P. Cytogenetic aberrations in osteosarcomas. Nonrandom deletions, rings, and double-minute chromosomes. Cancer Genet. Cytogenet. 1994; 77:81–88. [DOI] [PubMed] [Google Scholar]
- 52. Giollant M., Bertrand S., Verrelle P., Tchirkov A., du Manoir S., Ried T., Mornex F., Doré J.F., Cremer T., Malet P. Characterization of double minute chromosomes’ DNA content in a human high grade astrocytoma cell line by using comparative genomic hybridization and fluorescence in situ hybridization. Hum. Genet. 1996; 98:265–270. [DOI] [PubMed] [Google Scholar]
- 53. Satinover D.L., Vance G.H., Van Dyke D.L., Schwartz S. Cytogenetic analysis and construction of a BAC contig across a common neocentromeric region from 9p. Chromosoma. 2001; 110:275–283. [DOI] [PubMed] [Google Scholar]
- 54. Jee K.J., Kim Y.T., Kim K.R., Aalto Y., Knuutila S. Amplification at 9p in cervical carcinoma by comparative genomic hybridization. Anal. Cell. Pathol. 2001; 22:159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sams E.I., Ng J.K., Tate V., Claire Hou Y.-C., Cao Y., Antonacci-Fulton L., Belhassan K., Neidich J., Mitra R.D., Cole F.S. et al. From karyotypes to precision genomics in 9p deletion and duplication syndromes. HGG Adv. 2022; 3:100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shapiro J.A. How chaotic is genome chaos?. Cancers (Basel). 2021; 13:1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bignell G.R., Greenman C.D., Davies H., Butler A.P., Edkins S., Andrews J.M., Buck G., Chen L., Beare D., Latimer C. et al. Signatures of mutation and selection in the cancer genome. Nature. 2010; 463:893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sutherland G.R., Jacky P.B., Baker E., Manuel A. Heritable fragile sites on human chromosomes. X. New folate-sensitive fragile sites: 6p23, 9p21, 9q32, and 11q23. Am. J. Hum. Genet. 1983; 35:432–437. [PMC free article] [PubMed] [Google Scholar]
- 59. Mirceta M., Shum N., Schmidt M.H.M., Pearson C.E. Fragile sites, chromosomal lesions, tandem repeats, and disease. Front. Genet. 2022; 13:985975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Van Mossevelde S., van der Zee J., Cruts M., Van Broeckhoven C. Relationship between C9orf72 repeat size and clinical phenotype. Curr. Opin. Genet. Dev. 2017; 44:117–124. [DOI] [PubMed] [Google Scholar]
- 61. DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011; 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xi Z., Zhang M., Bruni A.C., Maletta R.G., Colao R., Fratta P., Polke J.M., Sweeney M.G., Mudanohwo E., Nacmias B. et al. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015; 129:715–727. [DOI] [PubMed] [Google Scholar]
- 63. Sun J.H., Zhou L., Emerson D.J., Phyo S.A., Titus K.R., Gong W., Gilgenast T.G., Beagan J.A., Davidson B.L., Tassone F. et al. Disease-associated short tandem repeats co-localize with chromatin domain boundaries. Cell. 2018; 175:224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xi Z., Rainero I., Rubino E., Pinessi L., Bruni A.C., Maletta R.G., Nacmias B., Sorbi S., Galimberti D., Surace E.I. et al. Hypermethylation of the CpG-island near the C9orf72 G₄C₂-repeat expansion in FTLD patients. Hum. Mol. Genet. 2014; 23:5630–5637. [DOI] [PubMed] [Google Scholar]
- 65. Utani K., Okamoto A., Shimizu N. Generation of micronuclei during interphase by coupling between cytoplasmic membrane blebbing and nuclear budding. PLoS One. 2011; 6:e27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fenech M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007; 2:1084–1104. [DOI] [PubMed] [Google Scholar]
- 67. Migliore L., Coppedè F., Fenech M., Thomas P. Association of micronucleus frequency with neurodegenerative diseases. Mutagenesis. 2011; 26:85–92. [DOI] [PubMed] [Google Scholar]
- 68. Fenech M., Kirsch-Volders M., Natarajan A.T., Surralles J., Crott J.W., Parry J., Norppa H., Eastmond D.A., Tucker J.D., Thomas P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011; 26:125–132. [DOI] [PubMed] [Google Scholar]
- 69. Polleys E.J., Freudenreich C.H. Methods to study repeat fragility and instability in Saccharomyces cerevisiae. Methods Mol. Biol. 2018; 1672:403–419. [DOI] [PubMed] [Google Scholar]
- 70. McGoldrick P., Zhang M., van Blitterswijk M., Sato C., Moreno D., Xiao S., Zhang A.B., McKeever P.M., Weichert A., Schneider R. et al. Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology. 2018; 90:e323–e331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dingwall C., Lomonossoff G.P., Laskey R.A. High sequence specificity of micrococcal nuclease. Nucleic Acids Res. 1981; 9:2659–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hörz W., Altenburger W. Sequence specific cleavage of DNA by micrococcal nuclease. Nucleic Acids Res. 1981; 9:2643–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bastepe M., Xin W. Huntington disease: molecular diagnostics approach. Curr. Protoc. Hum. Genet. 2015; 87:9.26.1–9.26.23. [Google Scholar]
- 74. Nutter C.A., Bubenik J.L., Oliveira R., Ivankovic F., Sznajder Ł.J., Kidd B.M., Pinto B.S., Otero B.A., Carter H.A., Vitriol E.A. et al. Cell-type-specific dysregulation of RNA alternative splicing in short tandem repeat mouse knockin models of myotonic dystrophy. Genes Dev. 2019; 33:1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017; 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Semenov M. Proliferative capacity of adult mouse brain. Int. J. Mol. Sci. 2021; 22:3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bordiuk O.L., Smith K., Morin P.J., Semënov M.V. Cell proliferation and neurogenesis in adult mouse brain. PLoS One. 2014; 9:e111453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lui J.C., Baron J. Mechanisms limiting body growth in mammals. Endocr. Rev. 2011; 32:422–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sanz M.M., Jenkins E.C., Brown W.T., Davisson M.T., Kevin M.J., Roderick T.H., Silverman W.P., Wisniewski H.M. Mouse chromosome fragility. Am. J. Med. Genet. 1986; 23:491–509. [DOI] [PubMed] [Google Scholar]
- 81. Angelosanto F.A. Tissues other than bone marrow that can be used for cytogenetic analyses. Environ. Mol. Mutagen. 1995; 25:338–343. [DOI] [PubMed] [Google Scholar]
- 82. Bayani J., Squire J.A. Preparation of cytogenetic specimens from tissue samples. Curr. Protoc. Cell Biol. 2004; Chapter 22(1):Unit 22.2. [Google Scholar]
- 83. Krawczun M.S., Lele K.P., Jenkins E.C., Brown W.T. Fragile X expression increased by low cell-culture density. Am. J. Med. Genet. 1986; 23:467–473. [DOI] [PubMed] [Google Scholar]
- 84. Fonatsch C., Schwinger E. Frequency of fragile X chromosomes, fra(X), in lymphocytes in relation to blood storage time and culture techniques. Hum. Genet. 1983; 64:39–41. [DOI] [PubMed] [Google Scholar]
- 85. Brookwell R., Daniel A., Turner G., Fishburn J. The fragile X(q27) form of X-linked mental retardation: FUdR as an inducing agent for fra(X)(q27) expression in lymphocytes, fibroblasts, and amniocytes. Am. J. Med. Genet. 1982; 13:139–148. [DOI] [PubMed] [Google Scholar]
- 86. Daniel A., Ekblom L., Phillips S. Constitutive fragile sites 1p31, 3p14, 6q26, and 16q23 and their use as controls for false-negative results with the fragile(X). Am. J. Med. Genet. 1984; 18:483–491. [DOI] [PubMed] [Google Scholar]
- 87. Reddy K., Zamiri B., Stanley S.Y., Macgregor R.B., Pearson C.E. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem. 2013; 288:9860–9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zamiri B., Mirceta M., Bomsztyk K., Macgregor R.B., Pearson C.E. Quadruplex formation by both G-rich and C-rich DNA strands of the C9orf72 (GGGGCC)8•(GGCCCC)8 repeat: effect of CpG methylation. Nucleic Acids Res. 2015; 43:10055–10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fry M., Loeb L.A. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl Acad. Sci. U.S.A. 1994; 91:4950–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xi Z., Zinman L., Moreno D., Schymick J., Liang Y., Sato C., Zheng Y., Ghani M., Dib S., Keith J. et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 2013; 92:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jackson J.L., Finch N.A., Baker M.C., Kachergus J.M., DeJesus-Hernandez M., Pereira K., Christopher E., Prudencio M., Heckman M.G., Thompson E.A. et al. Elevated methylation levels, reduced expression levels, and frequent contractions in a clinical cohort of C9orf72 expansion carriers. Mol. Neurodegen. 2020; 15:7. [Google Scholar]
- 92. Xu G.L., Bestor T.H., Bourc’his D., Hsieh C.L., Tommerup N., Bugge M., Hulten M., Qu X., Russo J.J., Viegas-Péquignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999; 402:187–191. [DOI] [PubMed] [Google Scholar]
- 93. Hecht F., Sutherland G.R. Detection of the fragile X chromosome and other fragile sites. Clin. Genet. 1984; 26:301–303. [DOI] [PubMed] [Google Scholar]
- 94. Veenema H., Beverstock G.C., de Koning T., Pearson P.L., van de Kamp J.J The fragile X-chromosome: an evaluation of the results in a routine cytogenetic laboratory in the period 1981-1986. Clin. Genet. 1988; 33:410–417. [DOI] [PubMed] [Google Scholar]
- 95. Cantú E.S., Jacobs P.A. Fragile (X) expression: relationship to the cell cycle. Hum. Genet. 1984; 67:99–102. [DOI] [PubMed] [Google Scholar]
- 96. Yu S., Mangelsdorf M., Hewett D., Hobson L., Baker E., Eyre H.J., Lapsys N., Le Paslier D., Doggett N.A., Sutherland G.R. et al. Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell. 1997; 88:367–374. [DOI] [PubMed] [Google Scholar]
- 97. Nancarrow J.K., Kremer E., Holman K., Eyre H., Doggett N.A., Le Paslier D., Callen D.F., Sutherland G.R., Richards R.I. Implications of FRA16A structure for the mechanism of chromosomal fragile site genesis. Science. 1994; 264:1938–1941. [DOI] [PubMed] [Google Scholar]
- 98. Bosco N., Pelliccia F., Rocchi A. Characterization of FRA7B, a human common fragile site mapped at the 7p chromosome terminal region. Cancer Genet. Cytogenet. 2010; 202:47–52. [DOI] [PubMed] [Google Scholar]
- 99. Callahan G., Denison S.R., Phillips L.A., Shridhar V., Smith D.I. Characterization of the common fragile site FRA9E and its potential role in ovarian cancer. Oncogene. 2003; 22:590–601. [DOI] [PubMed] [Google Scholar]
- 100. Curatolo A., Limongi Z.M., Pelliccia F., Rocchi A. Molecular characterization of the human common fragile site FRA1H. Genes Chromosomes Cancer. 2007; 46:487–493. [DOI] [PubMed] [Google Scholar]
- 101. Rozier L., El-Achkar E., Apiou F., Debatisse M. Characterization of a conserved aphidicolin-sensitive common fragile site at human 4q22 and mouse 6C1: possible association with an inherited disease and cancer. Oncogene. 2004; 23:6872–6880. [DOI] [PubMed] [Google Scholar]
- 102. Becker N.A., Thorland E.C., Denison S.R., Phillips L.A., Smith D.I. Evidence that instability within the FRA3B region extends four megabases. Oncogene. 2002; 21:8713–8722. [DOI] [PubMed] [Google Scholar]
- 103. Fechter A., Buettel I., Kuehnel E., Savelyeva L., Schwab M. Common fragile site FRA11G and rare fragile site FRA11B at 11q23.3 encompass distinct genomic regions. Genes Chromosomes Cancer. 2007; 46:98–106. [DOI] [PubMed] [Google Scholar]
- 104. Hirst M.C., Rack K., Nakahori Y., Roche A., Bell M.V., Flynn G., Christadoulou Z., MacKinnon R.N., Francis M., Littler A.J. A YAC contig across the fragile X site defines the region of fragility. Nucleic Acids Res. 1991; 19:3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Verkerk A.J., Eussen B.H., Van Hemel J.O., Oostra B.A. Limited size of the fragile X site shown by fluorescence in situ hybridization. Am. J. Med. Genet. 1992; 43:187–191. [DOI] [PubMed] [Google Scholar]
- 106. Hirst M.C., Barnicoat A., Flynn G., Wang Q., Daker M., Buckle V.J., Davies K.E., Bobrow M. The identification of a third fragile site, FRAXF, in Xq27–q28 distal to both FRAXA and FRAXE. Hum. Mol. Genet. 1993; 2:197–200. [DOI] [PubMed] [Google Scholar]
- 107. Ritchie R.J., Knight S.J., Hirst M.C., Grewal P.K., Bobrow M., Cross G.S., Davies K.E. The cloning of FRAXF: trinucleotide repeat expansion and methylation at a third fragile site in distal Xqter. Hum. Mol. Genet. 1994; 3:2115–2121. [DOI] [PubMed] [Google Scholar]
- 108. Abruzzo M.A., Hunt P.A., Mayer M., Jacobs P.A., Wang J.C., Erbe R.W. A comparison of fragile X expression in lymphocyte and lymphoblastoid cultures. Am. J. Hum. Genet. 1986; 38:533–539. [PMC free article] [PubMed] [Google Scholar]
- 109. Sutherland G.R., Baker E., Callen D.F. A BrdU-enhanceable fragile site or viral modification site at 11q23.1 in lymphoblastoid cultures. Cytogenet. Cell Genet. 1988; 47:201–203. [DOI] [PubMed] [Google Scholar]
- 110. Seki N., Tsuji H., Takahashi E., Yamauchi M., Saito T., Hashimoto T., Yamamoto K., Hori T. Induction of a BrdU-enhanceable fragile site-like lesion and sister chromatid exchanges at 11q23.1 in EBV-transformed lymphoblastoid cell lines. Cytogenet. Cell Genet. 1992; 61:95–98. [DOI] [PubMed] [Google Scholar]
- 111. Sutherland G.R., Baker E. Forgotten fragile sites and related phenomena. Cytogenet. Genome Res. 2003; 100:89–91. [DOI] [PubMed] [Google Scholar]
- 112. Li J.S.Z., Abbasi A., Kim D.H., Lippman S.M., Alexandrov L.B., Cleveland D.W. Chromosomal fragile site breakage by EBV-encoded EBNA1 at clustered repeats. Nature. 2023; 616:504–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Belzil V.V., Bauer P.O., Prudencio M., Gendron T.F., Stetler C.T., Yan I.K., Pregent L., Daughrity L., Baker M.C., Rademakers R. et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013; 126:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cali C.P., Patino M., Tai Y.K., Ho W.Y., McLean C.A., Morris C.M., Seeley W.W., Miller B.L., Gaig C., Vonsattel J.P.G. et al. C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol. 2019; 138:795–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tassone F., Hagerman R.J., Taylor A.K., Gane L.W., Godfrey T.E., Hagerman P.J. Elevated Levels of FMR1 mRNA in Carrier Males: A New Mechanism of Involvement in the Fragile-X Syndrome. Am. Hum. Genet. 2000; 66:6–15. [Google Scholar]
- 116. Eberhart D.E., Warren S.T. Nuclease sensitivity of permeabilized cells confirms altered chromatin formation at the fragile X locus. Somat. Cell Mol. Genet. 1996; 22:435–441. [DOI] [PubMed] [Google Scholar]
- 117. Beverstock G.C., Mol A., Wienhofer E. Absence of significant autosomal lesions in Huntington’s disease. Ann. Hum. Genet. 1985; 49:283–290. [DOI] [PubMed] [Google Scholar]
- 118. Krupina K., Goginashvili A., Cleveland D.W. Causes and consequences of micronuclei. Curr. Opin. Cell Biol. 2021; 70:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Shimizu N., Itoh N., Utiyama H., Wahl G.M. Selective entrapment of extrachromosomally amplified DNA by nuclear budding and micronucleation during S phase. J. Cell Biol. 1998; 140:1307–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Guttenbach M., Schmid M. Exclusion of specific human chromosomes into micronuclei by 5-azacytidine treatment of lymphocyte cultures. Exp. Cell Res. 1994; 211:127–132. [DOI] [PubMed] [Google Scholar]
- 121. Bjerregaard V.A., Garribba L., McMurray C.T., Hickson I.D., Liu Y. Folate deficiency drives mitotic missegregation of the human FRAXA locus. Proc. Natl Acad. Sci. U.S.A. 2018; 115:13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Duncan A.M. Enhanced sensitivity of lymphoblastoid cells from individuals carrying the mutation for the fragile X syndrome to the clastogenic effects of FUdR. Mutat. Res. 1986; 173:201–205. [DOI] [PubMed] [Google Scholar]
- 123. Shah P., Wolf K., Lammerding J. Bursting the bubble - nuclear envelope rupture as a path to genomic instability?. Trends Cell Biol. 2017; 27:546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Srivastava N., Nader G.P., de F., Williart A., Rollin R., Cuvelier D., Lomakin A., Piel M. Nuclear fragility, blaming the blebs. Curr. Opin. Cell Biol. 2021; 70:100–108. [DOI] [PubMed] [Google Scholar]
- 125. Stephens A.D., Liu P.Z., Banigan E.J., Almassalha L.M., Backman V., Adam S.A., Goldman R.D., Marko J.F. Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell. 2018; 29:220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bercht Pfleghaar K., Taimen P., Butin-Israeli V., Shimi T., Langer-Freitag S., Markaki Y., Goldman A.E., Wehnert M., Goldman R.D. Gene-rich chromosomal regions are preferentially localized in the lamin B deficient nuclear blebs of atypical progeria cells. Nucleus. 2015; 6:66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Niedernhofer L.J., Gurkar A.U., Wang Y., Vijg J., Hoeijmakers J.H.J., Robbins P.D. Nuclear genomic instability and aging. Annu. Rev. Biochem. 2018; 87:295–322. [DOI] [PubMed] [Google Scholar]
- 128. Fitchett M., Seabright M. Deleted X chromosomes in patients with the fragile X syndrome. J. Med. Genet. 1984; 21:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Verdyck P., Berckmoes V., De Vos A., Verpoest W., Liebaers I., Bonduelle M., De Rycke M. Chromosome fragility at FRAXA in human cleavage stage embryos at risk for fragile X syndrome. Am. J. Med. Genet. A. 2015; 167A:2306–2313. [DOI] [PubMed] [Google Scholar]
- 130. Wenger S.L., Hennessey J.C., Steele M.W. Increased sister chromatid exchange frequency at Xq27 site in affected fragile X males. Am. J. Med. Genet. 1987; 26:909–914. [DOI] [PubMed] [Google Scholar]
- 131. van Wietmarschen N., Merzouk S., Halsema N., Spierings D.C.J., Guryev V., Lansdorp P.M. BLM helicase suppresses recombination at G-quadruplex motifs in transcribed genes. Nat. Commun. 2018; 9:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Chan K.L., Palmai-Pallag T., Ying S., Hickson I.D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 2009; 11:753–760. [DOI] [PubMed] [Google Scholar]
- 133. Chaganti R.S., Schonberg S., German J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc. Natl Acad. Sci. U.S.A. 1974; 71:4508–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Cunniff C., Bassetti J.A., Ellis N.A. Bloom’s syndrome: clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol. Syndromol. 2017; 8:4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Gratia M., Rodero M.P., Conrad C., Bou Samra E., Maurin M., Rice G.I., Duffy D., Revy P., Petit F., Dale R.C. et al. Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J. Exp. Med. 2019; 216:1199–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Suzuki T., Yasui M., Honma M. Mutator phenotype and DNA double-strand break repair in BLM helicase-deficient human cells. Mol. Cell. Biol. 2016; 36:2877–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Warren S.T., Schultz R.A., Chang C.C., Wade M.H., Trosko J.E. Elevated spontaneous mutation rate in Bloom syndrome fibroblasts. Proc. Natl Acad. Sci. U.S.A. 1981; 78:3133–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wenger S.L. Chemical induction of sister chromatid exchange at fragile sites. Cancer Genet. Cytogenet. 1995; 85:72–74. [DOI] [PubMed] [Google Scholar]
- 139. Kumar J.V., Saraswathi T., Ranganathan K., Umadevi K., Joshua E., Rooban T. Sister chromatid exchanges in smokers and smokers with alcohol habit. J. Oral Maxillofac. Pathol. 2012; 16:338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Glover T.W., Stein C.K. Induction of sister chromatid exchanges at common fragile sites. Am. J. Hum. Genet. 1987; 41:882–890. [PMC free article] [PubMed] [Google Scholar]
- 141. Fuster C., Mirò R., Templado C., Barrios L., Egozcue J. Can sister chromatid intercrossings be considered as prelesions?. Hum. Genet. 1988; 79:179–180. [DOI] [PubMed] [Google Scholar]
- 142. Gaddini L., Pelliccia F., Limongi M.Z., Rocchi A. Study of the relationships between common fragile sites, chromosome breakages and sister chromatid exchanges. Mutagenesis. 1995; 10:257–260. [DOI] [PubMed] [Google Scholar]
- 143. Gregory P., Wang N., Howard-Peebles P.N. Analysis of sister chromatid exchanges in fra (X) individuals. Am. J. Med. Genet. 1986; 23:563–566. [DOI] [PubMed] [Google Scholar]
- 144. Branda R.F., Arthur D.C., Woods W.G., Danzl T.J., King R.A. Folate metabolism and chromosomal stability in the fragile X syndrome. Am. J. Med. 1984; 77:602–611. [DOI] [PubMed] [Google Scholar]
- 145. Suleimanova D.G., Kuleshov N.P. [Spontaneous and induced chromosome instability in patients with fragile X syndrome]. Genetika. 1987; 23:504–509. [PubMed] [Google Scholar]
- 146. Schmid M., Feichtinger W., Haaf T. The fragile site (16)(q22). II. Sister chromatid exchanges. Hum. Genet. 1987; 76:365–368. [DOI] [PubMed] [Google Scholar]
- 147. Hirsch B. Sister chromatid exchanges are preferentially induced at expressed and nonexpressed common fragile sites. Hum. Genet. 1991; 87:302–306. [DOI] [PubMed] [Google Scholar]
- 148. Lukusa T., Meulepas E., Fryns J.P., Van den Berghe H., Cassiman J.J. Spontaneous’ FRA16B is a hot spot for sister chromatid exchanges. Hum. Genet. 1991; 87:583–586. [DOI] [PubMed] [Google Scholar]
- 149. Recalcati M.P., Bellini M., Norsa L., Ballarati L., Caselli R., Russo S., Larizza L., Giardino D. Complex rearrangement involving 9p deletion and duplication in a syndromic patient: genotype/phenotype correlation and review of the literature. Gene. 2012; 502:40–45. [DOI] [PubMed] [Google Scholar]
- 150. Korbel J.O., Campbell P.J. Criteria for inference of chromothripsis in cancer genomes. Cell. 2013; 152:1226–1236. [DOI] [PubMed] [Google Scholar]
- 151. Kinsella M., Patel A., Bafna V. The elusive evidence for chromothripsis. Nucleic Acids Res. 2014; 42:8231–8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Krupina K., Goginashvili A., Cleveland D.W. Scrambling the genome in cancer: causes and consequences of complex chromosome rearrangements. Nat. Rev. Genet. 2024; 25:196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Zhang C.-Z., Spektor A., Cornils H., Francis J.M., Jackson E.K., Liu S., Meyerson M., Pellman D. Chromothripsis from DNA damage in micronuclei. Nature. 2015; 522:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.-Z., Weiss M.J., Pellman D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021; 53:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Urnov F.D. CRISPR-Cas9 can cause chromothripsis. Nat. Genet. 2021; 53:768–769. [DOI] [PubMed] [Google Scholar]
- 156. Pattamatta A., Nguyen L., Olafson H.R., Scotti M.M., Laboissonniere L.A., Richardson J., Berglund J.A., Zu T., Wang E.T., Ranum L.P.W. Repeat length increases disease penetrance and severity in C9orf72 ALS/FTD BAC transgenic mice. Hum. Mol. Genet. 2021; 29:3900–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kähkönen M., Tengström C., Alitalo T., Matilainen R., Kaski M., Airaksinen E. Population cytogenetics of folate-sensitive fragile sites. II. Autosomal rare fragile sites. Hum. Genet. 1989; 82:3–8. [DOI] [PubMed] [Google Scholar]
- 158. Vasta R., Chia R., Traynor B.J., Chiò A. Unraveling the complex interplay between genes, environment, and climate in ALS. EBioMedicine. 2022; 75:103795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Goncharova P.S., Davydova T.K., Popova T.E., Novitsky M.A., Petrova M.M., Gavrilyuk O.A., Al-Zamil M., Zhukova N.G., Nasyrova R.F., Shnayder N.A. Nutrient effects on motor neurons and the risk of amyotrophic lateral sclerosis. Nutrients. 2021; 13:3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Cingam S.R., Koshy N., Veillon D., Peddi P. Reversal of isolated 20q deletion with vitamin B12 replacement in a patient with pernicious anaemia. BMJ Case Rep. 2017; 2017:bcr2016218689. [Google Scholar]
- 161. Wollman M.R., Penchansky L., Shekhter-Levin S. Transient 7q- in association with megaloblastic anemia due to dietary folate and vitamin B12 deficiency. J. Pediatr. Hematol. Oncol. 1996; 18:162–165. [DOI] [PubMed] [Google Scholar]
- 162. Goh K. Vitamin B12 deficiency in an 18p–patient. Arch. Pathol. Lab. Med. 1981; 105:164. [PubMed] [Google Scholar]
- 163. Parmentier S., Meinel J., Oelschlaegel U., Mohr B., Ehninger G., Schaich M., Platzbecker U. Severe pernicious anemia with distinct cytogenetic and flow cytometric aberrations mimicking myelodysplastic syndrome. Ann. Hematol. 2012; 91:1979–1981. [DOI] [PubMed] [Google Scholar]
- 164. Chintagumpala M.M., Dreyer Z.A., Steuber C.P., Cooley L.D. Pancytopenia with chromosomal fragility: vitamin B12 deficiency. J. Pediatr. Hematol. Oncol. 1996; 18:166–170. [DOI] [PubMed] [Google Scholar]
- 165. Maltby E.L., Higgins S. Folate sensitive site at 10q23 and its expression as a deletion. J. Med. Genet. 1987; 24:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Morel C.F., Duncan A.M.V., Désilets V. A fragile site at 10q23 (FRA10A) in a phenytoin-exposed fetus: A case report and review of the literature. Prenat. Diagn. 2005; 25:318–321. [DOI] [PubMed] [Google Scholar]
- 167. De Leon-Luis J., Santolaya-Forgas J., May G., Tonk V., Shelton D., Galan I. Prenatal diagnosis of FRA10A: a case report and literature review. Am. J. Med. Genet. A. 2005; 136:63–65. [DOI] [PubMed] [Google Scholar]
- 168. Ozisik Y.Y., Meloni A.M., Stone J.F., Sandberg A.A., Surti U. Spontaneous expression of the chromosome fragile site at 10q23 in leiomyoma. Cancer Genet. Cytogenet. 1994; 74:73–75. [DOI] [PubMed] [Google Scholar]
- 169. Sarafidou T., Kahl C., Martinez-Garay I., Mangelsdorf M., Gesk S., Baker E., Kokkinaki M., Talley P., Maltby E.L., French L. et al. Folate-sensitive fragile site FRA10A is due to an expansion of a CGG repeat in a novel gene, FRA10AC1, encoding a nuclear protein. Genomics. 2004; 84:69–81. [DOI] [PubMed] [Google Scholar]
- 170. Sashindran V.K., Aggarwal V., Khera A. Prevalence of Vitamin B12 deficiency in elderly population (>60 years) presenting with dementia to outpatient department. Med. J. Armed Forces India. 2022; 78:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Pinto W.B.V.d.R., Souza P.V.S.d., Rogério R.M., Pedroso J.L., Barsottini O.G.P Vitamin B12 deficiency mimicking neuroimaging features of motor neuron disease. Arq. Neuropsiquiatr. 2014; 72:85. [DOI] [PubMed] [Google Scholar]
- 172. Chen A.T., Reidy J.A., Annest J.L., Welty T.K., Zhou H.G. Increased chromosome fragility as a consequence of blood folate levels, smoking status, and coffee consumption. Environ. Mol. Mutagen. 1989; 13:319–324. [DOI] [PubMed] [Google Scholar]
- 173. Barale R., Chelotti L., Davini T., Del Ry S., Andreassi M.G., Ballardin M., Bulleri M., He J., Baldacci S., Di Pede F. et al. Sister chromatid exchange and micronucleus frequency in human lymphocytes of 1,650 subjects in an Italian population: II. Contribution of sex, age, and lifestyle. Environ. Mol. Mutagen. 1998; 31:228–242. [DOI] [PubMed] [Google Scholar]
- 174. Maffei F., Fimognari C., Castelli E., Stefanini G.F., Forti G.C., Hrelia P. Increased cytogenetic damage detected by FISH analysis on micronuclei in peripheral lymphocytes from alcoholics. Mutagenesis. 2000; 15:517–523. [DOI] [PubMed] [Google Scholar]
- 175. Demirhan O., Tastemir D. Cytogenetic effects of ethanol on chronic alcohol users. Alcohol Alcohol. 2008; 43:127–136. [DOI] [PubMed] [Google Scholar]
- 176. Ueda K., Sakai C., Ishida T., Morita K., Kobayashi Y., Horikoshi Y., Baba A., Okazaki Y., Yoshizumi M., Tashiro S. et al. Cigarette smoke induces mitochondrial DNA damage and activates cGAS-STING pathway: application to a biomarker for atherosclerosis. Clin. Sci. (Lond.). 2023; 137:163–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Hesdorffer C.S., Longo D.L. Drug-induced megaloblastic anemia. N. Engl. J. Med. 2015; 373:1649–1658. [DOI] [PubMed] [Google Scholar]
- 178. Czuppa M., Dhingra A., Zhou Q., Schludi C., König L., Scharf E., Farny D., Dalmia A., Täger J., Castillo-Lizardo M. et al. Drug screen in iPSC-Neurons identifies nucleoside analogs as inhibitors of (G4C2)n expression in C9orf72 ALS/FTD. Cell Rep. 2022; 39:110913. [DOI] [PubMed] [Google Scholar]
- 179. Chiurazzi P., Pomponi M.G., Pietrobono R., Bakker C.E., Neri G., Oostra B.A. Synergistic effect of histone hyperacetylation and DNA demethylation in the reactivation of the FMR1 gene. Hum. Mol. Genet. 1999; 8:2317–2323. [DOI] [PubMed] [Google Scholar]
- 180. Lamparska K., Clark J., Babilonia G., Bedell V., Yip W., Smith S.S. 2’-Deoxyriboguanylurea, the primary breakdown product of 5-aza-2’-deoxyribocytidine, is a mutagen, an epimutagen, an inhibitor of DNA methyltransferases and an inducer of 5-azacytidine-type fragile sites. Nucleic Acids Res. 2012; 40:9788–9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Feichtinger W., Schmid M. Increased frequencies of sister chromatid exchanges at common fragile sites (1)(q42) and (19)(q13). Hum. Genet. 1989; 83:145–147. [DOI] [PubMed] [Google Scholar]
- 182. Rodríguez-Reyes R., Morales-Ramírez P. The in vivo induction of sister chromatid exchange by the demethylating agent 5-aza-2’-deoxycytidine. Mutagenesis. 2011; 26:551–554. [DOI] [PubMed] [Google Scholar]
- 183. Haaf T., Ott G., Schmid M. Differential inhibition of sister chromatid condensation induced by 5-azadeoxycytidine in human chromosomes. Chromosoma. 1986; 94:389–394. [DOI] [PubMed] [Google Scholar]
- 184. Davidson S., Crowther P., Radley J., Woodcock D. Cytotoxicity of 5-aza-2’-deoxycytidine in a mammalian cell system. Eur. J. Cancer. 1992; 28:362–368. [DOI] [PubMed] [Google Scholar]
- 185. Gijselinck I., Van Mossevelde S., van der Zee J., Sieben A., Engelborghs S., De Bleecker J., Ivanoiu A., Deryck O., Edbauer D., Zhang M. et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry. 2016; 21:1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Kojak N., Kuno J., Fittipaldi K.E., Khan A., Wenger D., Glasser M., Donnianni R.A., Tang Y., Zhang J., Huling K. et al. Somatic and intergenerational G4C2 hexanucleotide repeat instability in a human C9orf72 knock-in mouse model. Nucleic Acids Res. 2024; 52:5732–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Russ J., Liu E.Y., Wu K., Neal D., Suh E., Irwin D.J., McMillan C.T., Harms M.B., Cairns N.J., Wood E.M. et al. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 2015; 129:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Zhang M., Tartaglia M.C., Moreno D., Sato C., McKeever P., Weichert A., Keith J., Robertson J., Zinman L., Rogaeva E. DNA methylation age-acceleration is associated with disease duration and age at onset in C9orf72 patients. Acta Neuropathol. 2017; 134:271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Zhang M., McKeever P.M., Xi Z., Moreno D., Sato C., Bergsma T., McGoldrick P., Keith J., Robertson J., Zinman L. et al. DNA methylation age acceleration is associated with ALS age of onset and survival. Acta Neuropathol. 2020; 139:943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Galkin F., Kovalchuk O., Koldasbayeva D., Zhavoronkov A., Bischof E. Stress, diet, exercise: common environmental factors and their impact on epigenetic age. Ageing Res. Rev. 2023; 88:101956. [DOI] [PubMed] [Google Scholar]
- 191. Fitzgerald K.N., Hodges R., Hanes D., Stack E., Cheishvili D., Szyf M., Henkel J., Twedt M.W., Giannopoulou D., Herdell J. et al. Potential reversal of epigenetic age using a diet and lifestyle intervention: a pilot randomized clinical trial. Aging (Albany NY). 2021; 13:9419–9432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Sae-Lee C., Corsi S., Barrow T.M., Kuhnle G.G.C., Bollati V., Mathers J.C., Byun H.-M. Dietary intervention modifies DNA methylation age assessed by the epigenetic clock. Mol. Nutr. Food Res. 2018; 62:e1800092. [DOI] [PubMed] [Google Scholar]
- 193. Liu Y., Huang Z., Liu H., Ji Z., Arora A., Cai D., Wang H., Liu M., Simko E.A.J., Zhang Y. et al. DNA-initiated epigenetic cascades driven by C9orf72 hexanucleotide repeat. Neuron. 2023; 111:1205–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Rapkin L.M., Ahmed K., Dulev S., Li R., Kimura H., Ishov A.M., Bazett-Jones D.P. The histone chaperone DAXX maintains the structural organization of heterochromatin domains. Epigenetics Chromatin. 2015; 8:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. He Q., Kim H., Huang R., Lu W., Tang M., Shi F., Yang D., Zhang X., Huang J., Liu D. et al. The Daxx/Atrx complex protects tandem repetitive elements during DNA hypomethylation by promoting H3K9 trimethylation. Cell Stem Cell. 2015; 17:273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Scott W.A., Dhanji E.Z., Dyakov B.J.A., Dreseris E.S., Asa J.S., Grange L.J., Mirceta M., Pearson C.E., Stewart G.S., Gingras A.-C. et al. ATRX proximal protein associations boast roles beyond histone deposition. PLoS Genet. 2021; 17:e1009909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. van Blitterswijk M., Mullen B., Nicholson A.M., Bieniek K.F., Heckman M.G., Baker M.C., DeJesus-Hernandez M., Finch N.A., Brown P.H., Murray M.E. et al. TMEM106B protects C9ORF72 expansion carriers against frontotemporal dementia. Acta Neuropathol. 2014; 127:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. van Blitterswijk M., Mullen B., Heckman M.G., Baker M.C., DeJesus-Hernandez M., Brown P.H., Murray M.E., Hsiung G.-Y.R., Stewart H., Karydas A.M. et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol. Aging. 2014; 35:2421. [Google Scholar]
- 199. van Blitterswijk M., Mullen B., Wojtas A., Heckman M.G., Diehl N.N., Baker M.C., DeJesus-Hernandez M., Brown P.H., Murray M.E., Hsiung G.-Y.R. et al. Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol. Neurodegener. 2014; 9:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Pliner H.A., Mann D.M., Traynor B.J. Searching for Grendel: origin and global spread of the C9ORF72 repeat expansion. Acta Neuropathol. 2014; 127:391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Biasiotto G., Zanella I. The effect of C9orf72 intermediate repeat expansions in neurodegenerative and autoimmune diseases. Mult. Scler. Relat. Disord. 2019; 27:42–43. [DOI] [PubMed] [Google Scholar]
- 202. Burberry A., Suzuki N., Wang J.-Y., Moccia R., Mordes D.A., Stewart M.H., Suzuki-Uematsu S., Ghosh S., Singh A., Merkle F.T. et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 2016; 8:347ra93. [Google Scholar]
- 203. Béland L.-C., Markovinovic A., Jakovac H., De Marchi F., Bilic E., Mazzini L., Kriz J., Munitic I. Immunity in amyotrophic lateral sclerosis: blurred lines between excessive inflammation and inefficient immune responses. Brain Commun. 2020; 2:fcaa124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Burberry A., Wells M.F., Limone F., Couto A., Smith K.S., Keaney J., Gillet G., van Gastel N., Wang J.-Y., Pietilainen O. et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature. 2020; 582:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Bertrand A., Wen J., Rinaldi D., Houot M., Sayah S., Camuzat A., Fournier C., Fontanella S., Routier A., Couratier P. et al. Early cognitive, structural, and microstructural changes in presymptomatic C9orf72 carriers younger than 40 years. JAMA Neurol. 2018; 75:236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Querin G., Bede P., El Mendil M.M., Li M., Pélégrini-Issac M., Rinaldi D., Catala M., Saracino D., Salachas F., Camuzat A. et al. Presymptomatic spinal cord pathology in c9orf72 mutation carriers: a longitudinal neuroimaging study. Ann. Neurol. 2019; 86:158–167. [DOI] [PubMed] [Google Scholar]
- 207. Burgess R.C., Burman B., Kruhlak M.J., Misteli T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014; 9:1703–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Jacome A., Fernandez-Capetillo O. Lac operator repeats generate a traceable fragile site in mammalian cells. EMBO Rep. 2011; 12:1032–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Soutoglou E., Dorn J.F., Sengupta K., Jasin M., Nussenzweig A., Ried T., Danuser G., Misteli T. Positional stability of single double-strand breaks in mammalian cells. Nat. Cell Biol. 2007; 9:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Soutoglou E., Misteli T. Activation of the cellular DNA damage response in the absence of DNA lesions. Science. 2008; 320:1507–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Freaney J.E., Zhang Q., Yigit E., Kim R., Widom J., Wang J.-P., Horvath C.M. High-density nucleosome occupancy map of human chromosome 9p21-22 reveals chromatin organization of the type I interferon gene cluster. J. Interferon Cytokine Res. 2014; 34:676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Apostolou E., Thanos D. Virus infection induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell. 2008; 134:85–96. [DOI] [PubMed] [Google Scholar]
- 213. Pang W., Hu F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2021; 157:334–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Quinodoz S.A., Ollikainen N., Tabak B., Palla A., Schmidt J.M., Detmar E., Lai M.M., Shishkin A.A., Bhat P., Takei Y. et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell. 2018; 174:744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Jagannathan M., Yamashita Y.M. Function of junk: pericentromeric satellite DNA in chromosome maintenance. Cold Spring Harb. Symp. Quant. Biol. 2017; 82:319–327. [DOI] [PubMed] [Google Scholar]
- 216. Jagannathan M., Cummings R., Yamashita Y.M. A conserved function for pericentromeric satellite DNA. Elife. 2018; 7:e34122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Malachowski T., Chandradoss K.R., Boya R., Zhou L., Cook A.L., Su C., Pham K., Haws S.A., Kim J.H., Ryu H.-S. et al. Spatially coordinated heterochromatinization of long synaptic genes in fragile X syndrome. Cell. 2023; 186:5840–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Droppelmann C.A., Campos-Melo D., Moszczynski A.J., Amzil H., Strong M.J. TDP-43 aggregation inside micronuclei reveals a potential mechanism for protein inclusion formation in ALS. Sci. Rep. 2019; 9:19928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Fritsch L.E., Ju J., Gudenschwager Basso E.K., Soliman E., Paul S., Chen J., Kaloss A.M., Kowalski E.A., Tuhy T.C., Somaiya R.D. et al. Type I interferon response is mediated by NLRX1-cGAS-STING signaling in brain injury. Front. Mol. Neurosci. 2022; 15:852243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Song X., Ma F., Herrup K. Accumulation of cytoplasmic DNA due to ATM deficiency activates the microglial viral response system with neurotoxic consequences. J. Neurosci. 2019; 39:6378–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Quek H., Luff J., Cheung K., Kozlov S., Gatei M., Lee C.S., Bellingham M.C., Noakes P.G., Lim Y.C., Barnett N.L. et al. Rats with a missense mutation in Atm display neuroinflammation and neurodegeneration subsequent to accumulation of cytosolic DNA following unrepaired DNA damage. J. Leukoc. Biol. 2017; 101:927–947. [DOI] [PubMed] [Google Scholar]
- 222. Lupski J.R. Somatic cell structural variant mutagenesis and neurologic disease. Cell Genom. 2023; 3:100376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Schuy J., Grochowski C.M., Carvalho C.M.B., Lindstrand A. Complex genomic rearrangements: an underestimated cause of rare diseases. Trends Genet. 2022; 38:1134–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Campbell I.M., Shaw C.A., Stankiewicz P., Lupski J.R. Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 2015; 31:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Coffee B., Ikeda M., Budimirovic D.B., Hjelm L.N., Kaufmann W.E., Warren S.T. Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: a case report and review of the literature. Am. J. Med. Genet. A. 2008; 146A:1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Gómez-Rodríguez M.J., Morales-Conejo M., Arteche-López A., Sánchez-Calvín M.T., Quesada-Espinosa J.F., Gómez-Manjón I., Palma-Milla C., Lezana-Rosales J.M., Pérez de la Fuente R., Martin-Ramos M.-L. et al. Fragile X syndrome caused by maternal somatic mosaicism of FMR1 gene: case report and literature review. Genes (Basel). 2022; 13:1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Gonçalves T.F., dos Santos J.M., Gonçalves A.P., Tassone F., Mendoza-Morales G., Ribeiro M.G., Kahn E., Boy R., Pimentel M.M.G., Santos-Rebouças C.B. Finding FMR1 mosaicism in Fragile X syndrome. Expert Rev. Mol. Diagn. 2016; 16:501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Hensel C.H., Vanzo R.J., Martin M.M., Ling L., Aliaga S.M., Bui M., Francis D.I., Twede H., Field M.H., Morison J.W. et al. Abnormally methylated FMR1 in absence of a detectable full mutation in a U.S.A patient cohort referred for fragile X testing. Sci. Rep. 2019; 9:15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Prawer Y., Hunter M., Cronin S., Ling L., Aliaga Vera S., Fahey M., Gelfand N., Oertel R., Bartlett E., Francis D. et al. Prenatal diagnosis of fragile X syndrome in a twin pregnancy complicated by a complete retraction. Genes (Basel). 2018; 9:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Pandelache A., Francis D., Oertel R., Dickson R., Sachdev R., Ling L., Gamage D., Godler D.E. Detection of cryptic fragile X full mutation alleles by southern blot in a female and her foetal DNA via chorionic villus sampling, complicated by mosaicism for 45,X0/46,XX/47,XXX. Genes (Basel). 2021; 12:798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Mätlik K., Baffuto M., Kus L., Deshmukh A.L., Davis D.A., Paul M.R., Carroll T.S., Caron M.-C., Masson J.-Y., Pearson C.E. et al. Cell type specific CAG repeat expansions and toxicity of mutant Huntingtin in human striatum and cerebellum. Nat. Genet. 2024; 56:383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Pressl C., Mätlik K., Kus L., Darnell P., Luo J.-D., Paul M.R., Weiss A.R., Liguore W., Carroll T.S., Davis D.A. et al. Selective vulnerability of layer 5a corticostriatal neurons in Huntington’s disease. Neuron. 2023; 112:924–941. [Google Scholar]
- 233. Ziff O.J., Neeves J., Mitchell J., Tyzack G., Martinez-Ruiz C., Luisier R., Chakrabarti A.M., McGranahan N., Litchfield K., Boulton S.J. et al. Integrated transcriptome landscape of ALS identifies genome instability linked to TDP-43 pathology. Nature Commun. 2023; 14:2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Albert S.M. Neurodegenerative disease and cancer: a critical role for melanoma?. Neuroepidemiology. 2010; 35:305–306. [DOI] [PubMed] [Google Scholar]
- 235. Baade P.D., Herrero Hernández E., Freedman D.M., Smithers B.M., Fritschi L. No role for melanoma treatment in the association between melanoma and amyotrophic lateral sclerosis or Parkinson’s disease. Neuroepidemiology. 2010; 35:303–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Baade P.D., Fritschi L., Freedman D.M. Mortality due to amyotrophic lateral sclerosis and Parkinson's disease among melanoma patients. Neuroepidemiology. 2007; 28:16–20. [DOI] [PubMed] [Google Scholar]
- 237. Freedman D.M., Curtis R.E., Daugherty S.E., Goedert J.J., Kuncl R.W., Tucker M.A. The association between cancer and amyotrophic lateral sclerosis. Cancer Causes Control. 2013; 24:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Freedman D.M., Travis L.B., Gridley G., Kuncl R.W. Amyotrophic lateral sclerosis mortality in 1.9 million US cancer survivors. Neuroepidemiology. 2005; 25:176–180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets generated during the current study supporting its findings have been deposited or made available as indicated: RNASeq gene expression data—available on Gene Expression Ombinus (GEO), accession GSE262674. Whole genome sequencing data for the der-Chr9 (P8) sample is available in the European Genome-Phenome Archive (EGA) upon approval by the Data Access Committee. Correspondence and requests for materials should be addressed to Dr Christopher E. Pearson (mail to: cepearson.sickkids@gmail.com). Mouse models presented in this study are commercially available from Jackson Laboratories (FVB/NJ-Tg(C9orf72)500Lpwr/J).