

ABSTRACT
The Gram-positive pathogen Listeria monocytogenes is able to promote its entry into a diverse range of mammalian host cells by triggering plasma membrane remodeling, leading to bacterial engulfment. Upon cell invasion, L. monocytogenes disrupts its internalization vacuole and translocates to the cytoplasm, where bacterial replication takes place. Subsequently, L. monocytogenes uses an actin-based motility system that allows bacterial cytoplasmic movement and cell-to-cell spread. L. monocytogenes therefore subverts host cell receptors, organelles and the cytoskeleton at different infection steps, manipulating diverse cellular functions that include ion transport, membrane trafficking, post-translational modifications, phosphoinositide production, innate immune responses as well as gene expression and DNA stability.
Listeria monocytogenes is a facultative intracellular pathogen that has the capacity to actively invade and multiply within mammalian cells. Intracellular replication of L. monocytogenes within mononuclear cells was noted in the 1926 publication by Murray and colleagues reporting on this bacterial pathogen for the first time (1). In the 1960s, the seminal work of Mackaness that identified the main actors of cellular immunity against bacterial intracellular pathogens took advantage of the L. monocytogenes intracellular lifestyle as a model (2). In the late 1980s and early 1990s, major L. monocytogenes virulence factors involved in bacterial adaptation to intracellular life were molecularly characterized (3–7) and the precise stages of the L. monocytogenes intracellular life-cycle were morphologically identified (8, 9). Since then, cellular effectors involved in the infection process have been also identified and characterized (10–12). In this article, we review the molecular mechanisms driving L. monocytogenes adaptation to the mammalian host cell intracellular environment.
ADAPTATION TO INTRACELLULAR LIFE: GENERALITIES
L. monocytogenes is able to invade and proliferate within macrophages and epithelial nonphagocytic cells. For entry into the latter, bacterial surface molecules (InlA, InlB) interact with cellular ligands, activating signaling cascades that lead to internalization of the pathogen within a membrane-bound compartment (Fig. 1). Residency in the internalization vacuole is prevented by secretion of a pore-forming toxin (listeriolysin O [LLO]) and two phospholipases (PlcA, PlcB) that disrupt the vacuolar membrane, promoting L. monocytogenes translocation to the host cell cytoplasm. In this intracellular location, L. monocytogenes activates several bacterial metabolic pathways that favor the uptake of cellular resources sustaining bacterial proliferation. The pathogen also displays several strategies to escape cytoplasmic innate immune responses, which include the polymerization of actin by a bacterial surface protein (ActA), allowing L. monocytogenes to spread to neighboring cells. In secondary infected cells, L. monocytogenes is located in a double-membrane compartment that is disrupted by the same set of secreted enzymes that favor lysis of the primary internalization vacuole. L. monocytogenes translocation to the cytoplasm of secondary infected cells leads to a new bacterial replication cycle and further spread to other cells in infected tissues. The intracellular lifestyle is therefore critical for L. monocytogenes virulence: it allows escape from extracellular host defense mechanisms, including the complement or antibodies, and hinders detection by patrolling cell populations, e.g., neutrophils; in macrophages, cytoplasmic translocation allows escape from degradative components of the phagocytic cascade, while it provides access to a “Trojan horse” host cell population that can safely transport bacteria to distant locations within the infected organism.
FIGURE 1.
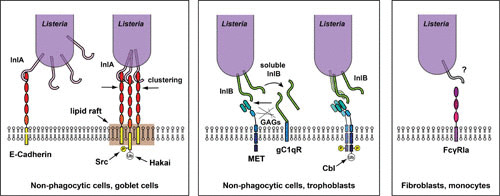
Cellular receptors for L. monocytogenes in host cells. The receptor for InlA in nonphagocytic polarized cells (including goblet cells) is the transmembrane molecule E-cadherin. Interaction takes place between the InlA leucine-rich repeats (LRRs) and the first extracellular domain of E-cadherin, leading to phosphorylation and ubiquitylation of the cytoplasmic domain of E-cadherin by the kinase Src and the ubiquitin ligase Hakai, respectively. Clustering of E-cadherin requires the presence of lipid rafts (left panel). Via its C-terminal glycine-tryptophan (GW) repeats, InlB interacts with the receptor for the globular part of the C1q complement component (gC1qR) and glycosaminoglycans, which enable interaction of the N-terminal LRRs of InlB with the tyrosine receptor kinase Met in nonphagocytic cells (including trophoblasts). Met dimerization upon interaction with InlB leads to autophosphorylation and recruitment of the ubiquitin ligase Cbl, which ubiquitylates the cytoplasmic tail of Met (center panel). In fibroblasts and monocytes, a function for the FcγRIA receptor has been described for L. monocytogenes internalization, via interaction with a still unidentified L. monocytogenes surface molecule (right panel). Modified from reference 12.
THE CELLULAR INVASION PROCESS
Upon L. monocytogenes contact with host target tissues, cellular invasion is morphologically characterized by a localized extension of the plasma membrane around invading bacteria, triggering bacterial internalization within a tight vacuolar compartment. Mechanistically, bacterial surface proteins interact with host cell receptors that are posttranslationally modified (phosphorylation, ubiquitylation), favoring the recruitment of protein adaptors and enzymes that contribute to actin polymerization, the key molecular event required for plasma membrane reorganization. Depending on the invaded cell type, bacterial modulation of the phosphoinositide (PI) metabolism is also critical to trigger cortical actin polymerization.
InlA is the archetypal member of a family of L. monocytogenes surface proteins named internalins, which are characterized by the presence of N-terminal leucine-rich repeats (LRR) which mediate interaction with host cell ligands (6, 13). The internalins InlA and InlB, encoded within a single locus in the L. monocytogenes genome, are the two major surface molecules driving bacterial entry into host cells. More than 20 other internalins have been identified, but they do not necessarily participate in the cell invasion process, contributing instead to diverse functions including cell-to-cell spread and escape from innate immune responses (i.e., InlC [14, 15]) and escape from autophagy (i.e., InlK [16]). The pore-forming toxin LLO, the actin polymerizing factor ActA, and other bacterial surface proteins have been described as supporting bacterial entry independently of the InlA and InlB invasion pathways (see below).
InlA/E-Cadherin-Mediated Entry
InlA displays a C-terminal LPXTG domain that favors covalent binding to the L. monocytogenes cell wall (6). The LRR domain of InlA interacts with the cellular receptor E-cadherin (17, 18), a transmembrane glycoprotein present in the adherens junctions of polarized tissues (e.g., the intestine and the placenta). E-cadherin normally plays a key role in the maintenance of tissue stability, and while the ectodomain participates in most cases in homotypic interactions (E-cadherin/E-cadherin intercellular binding), the cytoplasmic domain interacts with the actin cytoskeleton machinery. By subverting the E-cadherin physiological function, L. monocytogenes promotes cortical actin polymerization and plasma membrane rearrangements, favoring cellular invasion and traversal of the intestinal and the feto-maternal barriers (19, 20). InlA access to intestinal E-cadherin mostly occurs at the level of goblet cells, which expose this cellular receptor to bacteria during mucus secretion (21). Exposure of E-cadherin to L. monocytogenes during apoptotic cell extrusion at the tip of intestinal villi has also been documented (22). The interaction between InlA and E-cadherin is species-specific (23). A proline at position 16 allows interaction between InlA and human E-cadherin, while a glutamic acid at the same position, as observed in the mouse E-cadherin, does not allow InlA binding. A transgenic mouse model specifically expressing the human E-cadherin in the murine intestine allows a more efficient animal infection through the oral route, demonstrating the pivotal role of InlA in the crossing of the intestinal barrier (19).
In in vitro polarized cellular systems, lipid rafts are critical for InlA-dependent E-cadherin clustering (24). InlA binding promotes two successive posttranslational modifications in the cytoplasmic tail of E-cadherin: phosphorylation by the host kinase Src, followed by ubiquitylation by the ubiquitin ligase Hakai (25). These events are critical for the recruitment of a clathrin coat via the adaptor Dab2; the coat is stabilized by tyrosine phosphorylation of the clathrin heavy chain, followed by sequential recruitment of the protein adaptor Hip1R, which in turn coordinates recruitment of actin; myosin VI and unconventional myosin VIIa provide the pulling force that finally leads to bacterial internalization (26, 27). Interestingly, the nonmuscle myosin heavy chain IIA is specifically phosphorylated by Src upon L. monocytogenes infection and restricts bacterial entry (28). Several other molecules modulate actin association to the E-cadherin cytoplasmic site during L. monocytogenes InlA-dependent invasion: β- and α-catenins provide a physical link between E-cadherin and actin filaments during bacterial entry (29, 30), while cortactin and Src participate in the activation of the Arp2/3 complex, a major actin nucleator (31), highlighting the exploitation of adheren junctions and classical E-cadherin endocytosis components by L. monocytogenes during invasion of polarized tissues (32, 33). In the intestinal barrier, the constitutive PI 3-kinase activity is required for promoting actin polymerization during L. monocytogenes cell entry; in the placenta, PI 3-kinase activity is not constitutive, and InlB is required for PI 3-kinase activation and InlA-mediated cell invasion (34, 35).
InlB/Met-Mediated Entry
InlB was identified as a second L. monocytogenes invasion protein (6, 36). InlB allows L. monocytogenes entry into nonpolarized epithelial cells in vitro (37), and it cooperates with InlA during placental invasion in vivo (34, 35). In nonpregnant animals, InlB expression is associated with an increase of necrotic foci in the liver and spleen (38). The C-terminal region of InlB is characterized by the presence of glycine-tryptophan (GW) repeats that favor loose binding to bacterial membrane-tethered lipoteichoic acids (39, 40) and pedptidoglycan-bound wall teichoic acids (41). At the surface of host cells, the GW repeats mediate binding to the receptor of the globular part of the complement component C1q (42) and to glycosaminoglycans (43). The N-terminal region displays LRRs that are critical for cell invasion (44, 45) and bind the hepatocyte growth factor receptor Met (46) in a species-specific manner (47). Met expression is modulated by epithelial keratins, which promote InlB-mediated L. monocytogenes infection of epithelial cells (48). Met is a tyrosine kinase receptor, and its interaction with InlB leads to Met autophosphorylation and recruitment of the protein adaptors Gab1, Shc, Cbl, and CrkII (49–51), which play key roles in the activation of PI 3-kinase (52–54). Production of PI(3,4,5)P3 and its accumulation in lipid rafts leads to Rac1 activation (24, 55) and recruitment of Ena/VASP, WAVE, and N-WASP, which activate the Arp2/3 complex promoting actin polymerization in a tightly regulated manner (56, 57). The serine/threonine kinases mTOR and protein kinase C-α also control actin polymerization downstream of the InlB/Met interaction (58). The host 5′-phosphatase OCRL restricts L. monocytogenes entry by reducing PI(3,4,5)P3 levels and actin polymerization at bacterial entry foci (59). Production of PI 4P by type II PI 4-kinases, downstream of the tetraspanin CD81, is also critical for L. monocytogenes entry into host cells (60, 61). The PI 3-kinase adaptor Cbl also displays ubiquitin ligase activity and promotes ubiquitylation of Met upon InlB binding, leading to modulation of actin polymerization via clathrin recruitment (62–64). InlB also modulates exocytosis and favors the delivery of the endocytic GTPase dynamin 2 to bacterial entry sites (65). Finally, the septin cytoskeleton is recruited during cell invasion by L. monocytogenes in an InlB-dependent manner (66), and it controls the anchorage of Met to the cortical actin cytoskeleton (67–69).
Additional Adhesion/Entry Effectors
L. monocytogenes displays other surface and secreted molecules that can modulate adhesion and entry into host cells by indirectly affecting the surface exposure of InlA or InlB, by behaving as adhesins, by directly binding putative cellular receptors, and/or by activating cellular pathways that lead to actin rearrangements and bacterial engulfment. For example, the internalins InlE, InlG, and InlH support the InlA-dependent-invasion pathway in Caco-2 cells and might modulate the bacterial cell wall organization, consequently affecting InlA exposure (70). On the other hand, InlJ favors bacterial adhesion but not invasion in a specific subset of polarized epithelial cells (71, 72). A role for InlF in cell adhesion and invasion has been detected only upon inhibition of the RhoA/Rho kinase pathway (73, 74).
Several L. monocytogenes autolysins modulate cell adhesion and/or entry processes: Ami is involved in cell adhesion (75, 76), and Auto has been implicated in entry (77), while IspC is required for adhesion and/or invasion in a cell line-dependent manner (78). The lipoteichoic acid modifiers GtcA and DltA (79, 80), the lipoprotein LpeA (81), the prolipoprotein transferase Lgt (82), and the lysylphosphatidylglycerol modifier MprF (83) play roles in host cell adhesion or invasion probably by modulating the bacterial surface charge and/or by altering the organization of bacterial surface proteins. The surface protein ActA, involved in cytoplasmic actin-based motility (see below), has been proposed to favor host cell invasion through interaction with heparan sulfate (84, 85). Additional L. monocytogenes surface adhesins or invasins include Vip (86, 87), Lap (88), LapB (89), and FbpA (90).
LLO, a secreted cholesterol-dependent pore-forming toxin that is required for L. monocytogenes vacuolar escape (see below), is also secreted by extracellular bacteria and induces a transient influx of extracellular calcium within host cells that correlates with increased cell invasion (91). Mitochondrial fragmentation also correlates with the LLO-dependent calcium influx, and it has been proposed that L. monocytogenes modulates the bioenergetic state of resting cells to trigger cell invasion (92). LLO has been recently proposed to mediate L. monocytogenes entry into epithelial cells in a Ca2+/K+-, cholesterol-, dynamin-, tyrosine kinase- and actin-dependent but InlA/InlB- and clathrin-independent manner (93, 94). A broad-range phospholipase C of L. monocytogenes, PlcB (see below), has been reported to induce a calcium influx required for efficient bacterial internalization in macrophages (95).
THE VACUOLAR STAGE
The modulation of the actin cytoskeleton and the rearrangements of the plasma membrane upon L. monocytogenes interaction with its host cell receptors lead to bacterial engulfment and internalization in a membrane-bound vacuole (Fig. 2). In the intestinal epithelium, and particularly in the goblet cells, L. monocytogenes does not escape from this compartment and is directly transcytosed to the lamina propria, where the bacteria disseminate systemically (21). In other cell types, L. monocytogenes is able to disrupt its containing compartment and translocates to the host cell cytoplasm. The cholesterol-dependent pore-forming toxin LLO, together with two bacterial phospholipases, are the major bacterial effectors controlling L. monocytogenes vacuolar escape. L. monocytogenes residency and persistence in vacuolar compartments have also been described (96, 97) (see below).
FIGURE 2.
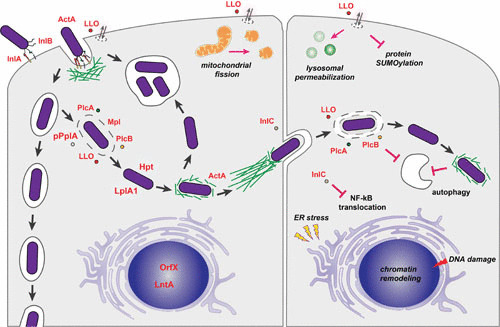
L. monocytogenes intracellular stages. L. monocytogenes is able to induce its entry into nonphagocytic cells mainly via the interaction of InlA and InlB with host cells receptors that promote actin recruitment, remodeling of the plasma membrane, and bacterial engulfment. The surface molecule ActA and the secreted pore-forming toxin LLO have also been implicated in the early L. monocytogenes entry steps (left cell, upper left). In goblet cells, upon internalization, L. monocytogenes is localized in a vacuole, and through transcytosis the bacterium is translocated to the lamina propria (left cell, left). In other cells, the combined activity of diverse virulence factors, including the pore-forming LLO, the metalloprotease Mpl, the phospholipases PlcA and PlcB, and the pheromone pPplA, favor disruption of the vacuole and L. monocytogenes release in the cytosol, where the bacteria takes advantage of host metabolites via the phosphate transporter Hpt and the lipoate protein ligase LplA. The surface protein ActA promotes actin-based motility, and the secreted protein InlC favors reduction of plasma membrane cortical tension, allowing L. monocytogenes to form protrusions and to invade neighboring cells. LLO and the phospholipases PlcA and PlcB contribute to the disruption of the double-membrane vacuole (right cell). L. monocytogenes has been observed in large spacious compartments that may arise rapidly after internalization of bacteria or upon decrease of ActA expression in already cytoplasmic bacteria (left cell, upper center). Extracellular LLO is able to modulate different cellular functions, including mitochondrial fission, lysosomal permeabilization, protein SUMOylation, ER stress, DNA damage, and chromatin remodeling. The phospholipases PlcA and PlcB, together with actin polymerization by ActA, have been implicated in the resistance to autophagy (195). The secreted molecule InlC prevents NF-κB translocation to the nucleus. Modified from reference 12.
LLO and Vacuolar Disruption
LLO is able to induce hemolysis in vitro, and early on, this activity was correlated with L. monocytogenes virulence (98). This toxin is encoded by the hly gene, located within a pathogenicity island that also encodes other important virulence factors, including two phospholipases, a metalloprotease, the actin polymerizing factor ActA, and the transcriptional activator PrfA (99). Inactivation of the hly gene coupled with electron microscopic observations subsequently demonstrated that LLO is required for bacterial escape from internalization vacuoles (100). LLO belongs to the family of cholesterol-dependent cytolysins which also includes perfringolysin O from Clostridium perfringens and streptolysin O from Streptococcus pyogenes (101). Perfringolysin O studies indicate that a conserved undecapeptide provides a structural conformation for a threonine-leucine pair at the C-terminal region of cholesterol-dependent cytolysins responsible for cholesterol binding (102, 103). Theoretically predicted to form large pores (20 to 80 monomers), based on initial perfringolysin O studies (104), electron microscopy and atomic force microscopy analyses indicate that LLO oligomers actually form arc-like structures that assemble into lineactants, and these heterogenous structures are responsible for membrane disruption and vacuolar rupture (105, 106). Membrane perforation by LLO not only facilitates L. monocytogenes translocation to the cytoplasm, but it also controls the vacuolar pH and calcium concentration, delaying the maturation of the bacteria-containing compartment and inhibiting lysosomal fusion (107, 108).
Several physical parameters and host molecules modulate the activity of LLO. Removal of LLO pores from the host cell plasma membrane is mediated by an LLO PEST-like sequence recognized by the clathrin adaptor Ap2a2, favoring pore endocytosis and protection of plasma membrane integrity (109). At 37°C and neutral pH, LLO undergoes denaturation, but it is in a stable conformation at acidic conditions (105). Consequently, within mammalian hosts, the LLO pore-forming activity is compartmentalized to slightly acidified bacteria-containing compartments (110, 111). Cytoplasmic LLO activity, which is cytotoxic to host cells and detrimental to intracellular L. monocytogenes (112), is limited by translational regulation of LLO synthesis (113) and by cytoplasmic LLO degradation by the ubiquitin system (114). LLO is activated by reducing agents (115), and within the vacuole of macrophages, the γ-interferon-inducible lysosomal thiol reductase is responsible for reducing and activating LLO (116). The increase in vacuolar chloride concentration mediated by the cystic fibrosis transmembrane conductance regulator has been proposed to enhance LLO oligomerization and L. monocytogenes cytoplasmic escape (117). LLO-disrupted vacuoles trigger the recruitment of the protein kinase C (PKC) ε, suggesting that this enzyme is involved in the recognition of damaged membrane organelles (118).
Phospholipases PlcA/PlcB and Vacuolar Disruption
L. monocytogenes secretes two phospholipases, a phosphatidylinositol-specific phospholipase C named PlcA (119) and a broad-range phospholipase C/sphingomyelinase named PlcB (120, 121), which is activated by the metalloprotease Mpl (122–124). Both enzymes have been shown to contribute to L. monocytogenes escape from primary vacuoles and from secondary vacuoles during bacterial cell-to-cell spread (4, 125–128).
PlcA from L. monocytogenes is the only bacterialphosphatidylinositol-specific phospholipase C that lacks a Vb β-strand that increases activity toward glycosylphosphatidylinositol-anchored proteins (129); interestingly, expression of this β-strand in PlcA impairs bacterial escape from vacuoles and cell-to-cell spread (130), suggesting that an L. monocytogenes adaptation to the intracellular environment requires reduced activity against glycosylphosphatidylinositol-anchored proteins. It has been proposed that PlcA translocating via LLO pores reaches the host cell cytoplasm, and in this compartment PlcA cleaves intracellular phosphatidylinositol into inositol phosphate and diacylglycerol (131); production of diacylglycerol, which might also take place through activation of host phospholipases C and D in an LLO-dependent signaling pathway (132), leads to activation of PKC βI and βII, which are required for vacuolar disruption (133). Because both PKC βI and βII are recruited to the L. monocytogenes internalization vacuole (134), it is speculated that the phosphorylation of PKC βI and βII targets at the surface of the bacteria-containing compartment is critical for a still unidentified signaling cascade leading to vacuolar rupture (133).
PlcB maturation and activation by the metalloprotease Mpl requires acidification of the vacuole (135, 136), and as has been observed for LLO, compartmentalization of this phospholipase C activity is critical for intracellular survival of L. monocytogenes (137). Both PlcA and PlcB have been found to activate NADPH oxidase during L. monocytogenes infection, which might be harmful to internalized bacteria via the production of reactive oxygen species; however, modulation of vacuolar maturation by LLO restricts NADPH oxidase localization to the L. monocytogenes-containing compartments (138).
Additional Mechanisms Regulating Vacuolar Disruption
Several other bacteria- and host-related mechanisms have been proposed to modulate the L. monocytogenes vacuolar stage. A recent study indicates that L. monocytogenes secretes a pheromone, pPplA, that triggers the production of an unknown factor that cooperates with LLO in facilitating vacuolar disruption (139): pPplA is processed from the N-terminal secretion signal sequence of the lipoprotein PplA; pPplA is secreted, accumulates in the space of the L. monocytogenes vacuole, and is then exported by the CtaP peptide transporter; cytoplasmic pPplA induces the production of a factor that accelerates vacuolar disruption mediated by LLO in a still unidentified manner (139).
Modulation of bacterial gene expression by “reversible lysogeny” has also been proposed to modulate L. monocytogenes vacuolar escape (140). The prophage A118 is inserted within the coding region of the gene comK, a master regulator of competence genes that are normally not expressed by L. monocytogenes; interestingly, during the bacterial vacuolar stage, A118 is excised, and this event allows reactivation of comK and expression of the competence machinery by L. monocytogenes; by a still unknown mechanism, the competence system promotes efficient bacterial translocation to the host cell cytoplasm. In this environment, the phage reinserts into comK (140).
Additional host factors have been reported to control L. monocytogenes vacuolar residency. Rab5a was shown to control the accelerated maturation of L. monocytogenes-containing vacuoles (141, 142); the product of the gene Lmo2459 was subsequently shown to induce the specific ADP ribosylation of Rab5a, inhibiting its activation and reverting its bactericidal functions (143). The activity of the cytosolic cysteine protease calpain has been shown to be required for efficient L. monocytogenes vacuolar escape, but the targets of this protease remain to be identified (144).
THE CYTOPLASMIC STAGE
By translocating from the vacuolar stage toward the cytoplasm, L. monocytogenes escapes cellular degradative mechanisms associated with phagosomal pathways. On the other hand, L. monocytogenes must adapt its metabolism to nutrients and metabolites found in this novel intracellular compartment and must also escape from additional innate immunity defenses including autophagy. The hexose phosphate transporter Hpt and the actin-polymerizing surface protein ActA play key roles in the survival of L. monocytogenes in the host cell cytoplasm.
Utilization of Host Metabolites
Glucose-1-phosphate is the primary degradation product of glycogen and is broadly available within mammalian cells. The observation that L. monocytogenes uses glucose-1-phosphate as a carbon source in a PrfA-dependent manner suggested that related hexose phosphates could be important growth substrates for intracellular bacteria (145). In silico analysis of the L. monocytogenes genome identified the gene hpt as encoding a hexose phosphate transporter responsible for the uptake of glucose-6-phosphate in the cytoplasm of host cells, playing a key role in L. monocytogenes in vivo virulence (146). A subsequent screen for identification of additional genes required for bacterial intracellular replication recognized lplA1 as a lipoate protein ligase that could potentially use host-derived lipoic acid to modify bacterial substrates (147). LplA1 was later confirmed to be essential for intracellular growth of L. monocytogenes under limiting concentrations of available small mammalian lipoylated peptides (148). A genetic screen led to the discovery that the menaquinone synthesis intermediate 1,4-dihydroxy-2-naphtoate is required for L. monocytogenes cytosolic survival, but full-length menaquinone is not (149).
Cytoplasmic Innate Immune Responses
Autophagy is a cellular mechanism responsible for protein turnover and removal of abnormal or superfluous subcellular components. The pioneering work of Rich et al. (150) demonstrated that cytoplasmic and metabolically arrested L. monocytogenes can be targeted for destruction by the autophagic machinery. Different mechanisms have been proposed to participate in the active escape of cytoplasmic L. monocytogenes from autophagy: polymerization of actin by the surface protein ActA favors cytoplasmic motility and avoidance of autophagosomes (151); polymerized actin or Arp2/3 sequestering by ActA may also act as a protective physical barrier preventing the accumulation of signaling molecules (i.e., ubiquitin) that are required for autophagy activation (152, 153). PlcA and PlcB have also been implicated in autophagosomal avoidance (151, 154, 155), and recent studies suggest that these PLCs decrease cytoplasmic levels of PI 3P, causing stalling of preautophagosomal structures and preventing efficient targeting of cytosolic bacteria (156). The surface internalin InlK has also been proposed to recruit the major vault protein and to protect cytoplasmic L. monocytogenes from autophagic recognition (16), but these results have been recently challenged using a different L. monocytogenes strain (157).
Cytoplasmic L. monocytogenes secretes small molecules leading to activation of an IRF3-dependent cytosolic pathway, resulting in type I interferon activation (158). One of these small molecules, cyclic-di-AMP, is sufficient to activate production of interferon β in macrophages (159). Sensing of tri-phosphorylated RNA via RIG-I and a MAVS-dependent pathway triggers type I interferon production in epithelial cells (160, 161). L. monocytogenes cytoplasmic DNA is recognized through STING, TBK1, IRF3, and IRF7, leading to the upregulation of the di-ubiquitin-like protein ISG15 and ISGylation of endoplasmic reticulum (ER) and Golgi proteins, which correlate with increased secretion of cytokines that counteract infection (162). L. monocytogenes also activates the type III interferon pathway (163).
Persistence
It is increasingly recognized that bacterial pathogens may persist within host tissues in a dormant state that allows resistance to antibiotics and subsequent reinfection. In macrophages of severe combined immunodeficient (SCID) mice, L. monocytogenes can persist in large compartments termed spacious Listeria-containing phagosomes (SLAPS), which are formed in an LLO-dependent manner (96). LC3-associated phagocytosis has been proposed to precede the formation of SLAPS (164). A recent study indicates that in epithelial cells, cytoplasmic L. monocytogenes bacteria in which ActA production is halted are trapped in acidic vacuoles that are not associated with classical autophagosomal markers and in which bacteria enter a viable but nonculturable state (97). These studies indicate that L. monocytogenes may persist in different host cellular populations, favoring the asymptomatic carriage of this pathogen.
CELL-TO-CELL SPREAD
Actin-based motility allows L. monocytogenes not only to escape autophagy but also to reach neighboring cells within infected tissues, favoring cell-to-cell spread and bacterial dissemination in organs, avoiding exposure to humoral immunity. Motile L. monocytogenes first induces the formation of a membrane protrusion in the primary infected cell that is accompanied by membrane internalization in the neighboring bystander cell, leading to bacterial entrapment in a double-membrane vacuole that is then disrupted (9). Several bacterial virulence factors, including ActA, the internalin InlC, the pore-forming toxin LLO, and the phospholipases PlcA and PlcB, participate at different stages of L. monocytogenes cell-to-cell spread.
Cytoplasmic Actin-Based Motility
The surface protein ActA is sufficient to trigger actin polymerization at the surface of L. monocytogenes (7). The central region of ActA contains four short proline-rich repeats that bind members of the enabled/vasodilator-stimulated phosphoprotein (Ena/VASP) family; these molecules contribute to the persistence of speed/directionality of bacterial movement by recruiting profilin, which provides polymerization-competent actin monomers (165). The N-terminal region of ActA recruits the Arp2/3 complex which drives actin nucleation (166, 167). The Arp2/3 complex is formed of seven subunits, and it has been traditionally considered a single molecular entity (168). A genome-wide small interfering RNA screen demonstrated that different Arp2/3 complexes are required to control L. monocytogenes cell invasion and actin-based motility: the Arp2, Arp3, ARPC2, and ARPC3 subunits are conserved, but the ARPC1B subunit is only involved in cell invasion, while the ARPC1A subunit is required for actin-based motility, and the ARPC4 subunit is dispensable for cell invasion, while the ARPC5 subunit is dispensable both for cell invasion and actin-based motility (169). Multiple actin cross-linking proteins, actin filament-capping or -severing proteins, and protein scaffolds are recruited to the L. monocytogenes actin tail (170). Cryo-electron tomography of actin tails has demonstrated that actin bundling is critical for ensuing actin-based motility (171).
Cortical Actin Rearrangements and Protrusion Formation
In mammalian tissues, cortical membrane tension represents a physical barrier for motile L. monocytogenes, inhibiting the deformation of the plasma membrane into protrusions. InlC, a secreted member of the internalin family devoid of a cell wall anchoring motif (13), perturbs apical cell junctions by interacting with the protein adaptor Tuba, inhibiting the recruitment of the actin regulatory protein N-WASP and COPII proteins and therefore relieving cortical membrane tension favoring L. monocytogenes protrusion formation (14, 172, 173). The downregulation by L. monocytogenes of the small GTPase Cdc42, another Tuba interactor, is also required for efficient protrusion formation (174). Within the protrusion, the membrane-cystoskeletal linker ezrin has been proposed to support the formation and stabilization of protrusions (175). Arp2/3 drives actin polymerization at the proximal L. monocytogenes rear-end within protrusions, but at distal locations the recruitment of Rho GTPases activate diaphanous-related formins which promote the formation of unbranched actin filaments (176). Inhibition of actin polymerization by components of the AIP1-dependent actin disassembly machinery (177) and ActA processing by the metalloprotease Mpl (178) are proposed mechanisms for the resolution of membrane protrusions into double membrane vacuoles. Efficient cell-to-cell spread can be facilitated by the exofacial exposure of phosphatidylserine at the tip of protrusions promoted by the pore-forming activity of LLO, which leads to phosphatidylserine binding by the TIM-4 receptor in macrophages and protrusion internalization (179).
Lysis of Secondary Vacuoles
Internalization of L. monocytogenes-induced protrusions into neighboring bystander cells leads to bacterial localization within a double-membrane compartment (9). Initial studies suggested that phospholipases PlcA and PlcB, together with LLO, contributed to cell-to-cell spread (4, 125, 127), and a more specific contribution of PlcB to double-membrane vacuolar rupture was suggested (128). A current model proposes that PlcA and PlcB contribute to the disruption of the inner membrane of the spreading vacuole, while LLO participates more precisely in the disruption of the outer membrane of this vacuole (180). Bacterial translocation to the cytoplasmic space of neighboring cells allows L. monocytogenes to start a new infection process.
MODULATION OF CELLULAR, ORGANELLAR, AND NUCLEAR FUNCTIONS
L. monocytogenes is able to modulate a broad range of cellular functions, even before being internalized within host cells. The pore-forming toxin LLO, which plays a major role in vacuolar escape (see above) is able to modulate from the extracellular space the function of mitochondria, the ER, lysosomes, protein posttranslational modifications, and DNA stability. Several bacterial nucleomodulins have been identified which directly affect the transcription of host genes involved in the control of immune responses.
LLO Influence on Mitochondria
Mitochondria are critical organelles involved in the generation of chemical energy in eukaryotic cells. As mentioned above, extracellular LLO triggers the influx of calcium, which leads to transient fission of mitochondria, triggering a bioenergetic change of host cells that is beneficial for L. monocytogenes host cell invasion (92). Interestingly, atypical mitochondrial fission through a Drp1-independent fragmentation process has been associated with L. monocytogenes cellular infection (181).
LLO Influence on the ER
The unfolded protein response is a signaling cascade that maintains the function of the ER under stress conditions. L. monocytogenes activates the unfolded protein response in an LLO-dependent manner prior to bacterial entry into host cells (182). The induction of ER stress by drugs such as thapsigargin or tunicamycin leads to a decrease in bacterial intracellular numbers, suggesting that the unfolded protein response represents an innate immune response to bacterial infection (182).
LLO Influence on Lysosomes
The integrity of lysosomes has been shown to be compromised by extracellular LLO, which induces permeabilization and release of lysosome content, including cathepsins, which remain transiently active in the cytoplasm (183). The functional significance of this finding for L. monocytogenes infection and survival remains to be identified.
LLO Influence on Protein Posttranslational Modifications
Posttranslational modifications allow the rapid modification of the activity of cellular proteins. Sumoylation is an essential posttranslational modification that is impaired by L. monocytogenes through the proteasome-independent degradation of the E2 enzyme Ubc9 following calcium influx mediated by extracellular LLO (184). The downregulation of cellular protein sumoylation, together with the proteasome-dependent degradation of some sumoylated proteins, favors bacterial infection in vitro and in vivo (184). Histone modifications are also associated with the L. monocytogenes infection process (185) (see below).
LLO Influence on DNA Stability
L. monocytogenes modulates general DNA stability in host cells in different manners. L. monocytogenes induces DNA strand breaks and simultaneously blocks the DNA damage response through degradation of the sensor Mre11 in an extracellular LLO-dependent manner (186), promoting a cell cycle delay that favors bacterial intracellular replication (187). Interestingly, it has been also reported that LLO-induced calcium influx leads to the proteasomal degradation of the human telomerase reverse transcriptase, an event that is detrimental to bacterial replication (188).
Bacterial Influence on Gene Expression
LntA is the first nucleomodulin discovered in L. monocytogenes (189): it targets the chromatin repressor BAHD1 and fine-tunes transcription of interferon-stimulated genes, which is required for efficient in vivo infection (190, 191). More recently, the nucleomodulin OrfX has been shown to interact and decrease the levels of the regulatory protein RybP, dampening the oxidative response in macrophages probably through modulation of host transcriptional responses (192). The secreted internalin InlC interferes with innate immune responses by targeting the IκB kinase subunit IKKα, inhibiting NF-κB translocation to the nucleus (15). LLO modulates gene transcription with opposite effects for infection: LLO induces a dramatic dephosphorylation of histone H3 and deacetylation of histone H4 that leads to reduced transcriptional activity of key immunity host genes (185); LLO has also been shown to modulate the functionality of the promyelocytic leukemia protein nuclear bodies, activating a signaling response that decreases L. monocytogenes infection (193). Finally, an InlB/PI 3-kinase pathway is required for the SIRT2-dependent deacetylation of histone H3 on lysine 18, which is involved in efficient bacterial infection in vitro and in vivo (194).
CONCLUSIONS
The study of the interactions of L. monocytogenes with eukaryotic host cells during bacterial invasion, intracellular growth, and cell-to-cell spread has proven to be fundamental to better understanding the exquisite adaptation of this bacterial pathogen to mammalian hosts. Indeed, L. monocytogenes is able to hijack multiple cellular functions including receptor signaling, membrane trafficking, cytoskeletal rearrangements, organellar dynamics, DNA stability, and gene transcription. The work reviewed in this article also highlights that L. monocytogenes is an extraordinary tool to manipulate and to unravel host cell signaling cascades, in particular, innate immune responses that allow us to expand our understanding of the control of bacterial intracellular infections.
ACKNOWLEDGMENTS
We thank members of the Cossart lab for helpful discussions. We apologize to those people who have contributed to advancements in the field of listeriosis but whose work was not cited in our article due to space limitations.
J.P.C. received support from the Institut Pasteur Transversal Research Program PTR521 and from National Research Agency (ANR) grant ANR-15-CE15-0017, StopBugEntry. Work in P.C.’s laboratory received financial support from Institut Pasteur, INSERM, INRA, ANR-13-IFEC-0004-02 ERANET Infect-ERA PROANTILIS, the French government’s Investissement d’Avenir program, the Laboratoire d’Excellence Integrative Biology of Emerging Infectious Diseases (ANR-10-LABX-62-IBEID), the European Research Council (ERC) (advanced grant H2020-ERC-2014-ADG 670823-BacCellEpi), the Human Frontier Science Program Organization (RGP011/2013), the Fondation le Roch les Mousquetaires, and the Fondation Balzan. P.C. is a senior international research scholar at the Howard Hughes Medical Institute.
Contributor Information
Javier Pizarro-Cerdá, Unité Interactions Bactéries-Cellules, Institut Pasteur, Paris F-75015, FRANCE; INSERM U604, Paris F-75015, FRANCE; INRA USC2020, Paris F-75015, FRANCE.
Pascale Cossart, Unité Interactions Bactéries-Cellules, Institut Pasteur, Paris F-75015, FRANCE; INSERM U604, Paris F-75015, FRANCE; INRA USC2020, Paris F-75015, FRANCE.
Vincent A. Fischetti, The Rockefeller University, New York, NY
Richard P. Novick, Skirball Institute for Molecular Medicine, NYU Medical Center, New York, NY
Joseph J. Ferretti, Department of Microbiology & Immunology, University of Oklahoma Health Science Center, Oklahoma City, OK
Daniel A. Portnoy, Department of Molecular and Cellular Microbiology, University of California, Berkeley, Berkeley, CA
Miriam Braunstein, Department of Microbiology and Immunology, University of North Carolina-Chapel Hill, Chapel Hill, NC.
Julian I. Rood, Infection and Immunity Program, Monash Biomedicine Discovery Institute, Monash University, Melbourne, Australia
REFERENCES
- 1.Murray E, Webb RA, Swan M. 1926. A disease of rabbits characterized by a large mononuclear leucocytosis, caused by a hitherto undescribed bacillus Bacterium monocytogenes (n.sp.). J Pathological Biol 29:407–439 10.1002/path.1700290409. [PubMed] [DOI] [Google Scholar]
- 2.Mackaness GB. 1962. Cellular resistance to infection. J Exp Med 116:381–406 10.1084/jem.116.3.381. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mengaud J, Chenevert J, Geoffroy C, Gaillard JL, Cossart P. 1987. Identification of the structural gene encoding the SH-activated hemolysin of Listeria monocytogenes: listeriolysin O is homologous to streptolysin O and pneumolysin. Infect Immun 55:3225–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun AN, Camilli A, Portnoy DA. 1990. Isolation of Listeria monocytogenes small-plaque mutants defective for intracellular growth and cell-to-cell spread. Infect Immun 58:3770–3778. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domann E, Leimeister-Wächter M, Goebel W, Chakraborty T. 1991. Molecular cloning, sequencing, and identification of a metalloprotease gene from Listeria monocytogenes that is species specific and physically linked to the listeriolysin gene. Infect Immun 59:65–72. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaillard JL, Berche P, Frehel C, Gouin E, Cossart P. 1991. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from Gram-positive cocci. Cell 65:1127–1141 10.1016/0092-8674(91)90009-N. [DOI] [PubMed] [Google Scholar]
- 7.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. 1992. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68:521–531 10.1016/0092-8674(92)90188-I. [DOI] [PubMed] [Google Scholar]
- 8.Mounier J, Ryter A, Coquis-Rondon M, Sansonetti PJ. 1990. Intracellular and cell-to-cell spread of Listeria monocytogenes involves interaction with F-actin in the enterocytelike cell line Caco-2. Infect Immun 58:1048–1058. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tilney LG, Portnoy DA. 1989. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol 109:1597–1608 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cossart P. 2011. Illuminating the landscape of host-pathogen interactions with the bacterium Listeria monocytogenes. Proc Natl Acad Sci U S A 108:19484–19491 10.1073/pnas.1112371108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rolhion N, Cossart P. 2017. How the study of Listeria monocytogenes has led to new concepts in biology. Future Microbiol 12:621–638 10.2217/fmb-2016-0221. [PubMed] [DOI] [PubMed] [Google Scholar]
- 12.Radoshevich L, Cossart P. 2017. Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat Rev Micro 16:32–46. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Bierne H, Sabet C, Personnic N, Cossart P. 2007. Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect 9:1156–1166 10.1016/j.micinf.2007.05.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 14.Rajabian T, Gavicherla B, Heisig M, Müller-Altrock S, Goebel W, Gray-Owen SD, Ireton K. 2009. The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat Cell Biol 11:1212–1218 10.1038/ncb1964. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gouin E, Adib-Conquy M, Balestrino D, Nahori M-A, Villiers V, Colland F, Dramsi S, Dussurget O, Cossart P. 2010. The Listeria monocytogenes InlC protein interferes with innate immune responses by targeting the IkappaB kinase subunit IKKalpha. Proc Natl Acad Sci U S A 107:17333–17338 10.1073/pnas.1007765107. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dortet L, Mostowy S, Samba-Louaka A, Gouin E, Nahori M-A, Wiemer EAC, Dussurget O, Cossart P. 2011. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog 7:e1002168 10.1371/journal.ppat.1002168. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mengaud J, Ohayon H, Gounon P, Mege R-M, Cossart P. 1996. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 84:923–932 10.1016/S0092-8674(00)81070-3. [DOI] [PubMed] [Google Scholar]
- 18.Lecuit M, Ohayon H, Braun L, Mengaud J, Cossart P. 1997. Internalin of Listeria monocytogenes with an intact leucine-rich repeat region is sufficient to promote internalization. Infect Immun 65:5309–5319. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lecuit M, Vandormael-Pournin S, Lefort J, Huerre M, Gounon P, Dupuy C, Babinet C, Cossart P. 2001. A transgenic model for listeriosis: role of internalin in crossing the intestinal barrier. Science 292:1722–1725 10.1126/science.1059852. [PubMed] [DOI] [PubMed] [Google Scholar]
- 20.Lecuit M, Nelson DM, Smith SD, Khun H, Huerre M, Vacher-Lavenu M-C, Gordon JI, Cossart P. 2004. Targeting and crossing of the human maternofetal barrier by Listeria monocytogenes: role of internalin interaction with trophoblast E-cadherin. Proc Natl Acad Sci U S A 101:6152–6157 10.1073/pnas.0401434101. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikitas G, Deschamps C, Disson O, Niault T, Cossart P, Lecuit M. 2011. Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of goblet cell accessible E-cadherin. J Exp Med 208:2263–2277 10.1084/jem.20110560. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pentecost M, Otto G, Theriot JA, Amieva MR. 2006. Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog 2:e3 10.1371/journal.ppat.0020003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lecuit M, Dramsi S, Gottardi C, Fedor-Chaiken M, Gumbiner B, Cossart P. 1999. A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J 18:3956–3963 10.1093/emboj/18.14.3956. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seveau S, Bierne H, Giroux S, Prévost MC, Cossart P. 2004. Role of lipid rafts in E-cadherin- and HGF-R/Met-mediated entry of Listeria monocytogenes into host cells. J Cell Biol 166:743–753 10.1083/jcb.200406078. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonazzi M, Veiga E, Pizarro-Cerdá J, Cossart P. 2008. Successive post-translational modifications of E-cadherin are required for InlA-mediated internalization of Listeria monocytogenes. Cell Microbiol 10:2208–2222 10.1111/j.1462-5822.2008.01200.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Bonazzi M, Vasudevan L, Mallet A, Sachse M, Sartori A, Prevost MC, Roberts A, Taner SB, Wilbur JD, Brodsky FM, Cossart P. 2011. Clathrin phosphorylation is required for actin recruitment at sites of bacterial adhesion and internalization. J Cell Biol 195:525–536 10.1083/jcb.201105152. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sousa S, Cabanes D, El-Amraoui A, Petit C, Lecuit M, Cossart P. 2004. Unconventional myosin VIIa and vezatin, two proteins crucial for Listeria entry into epithelial cells. J Cell Sci 117:2121–2130 10.1242/jcs.01066. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Almeida MT, Mesquita FS, Cruz R, Osório H, Custódio R, Brito C, Vingadassalom D, Martins M, Leong JM, Holden DW, Cabanes D, Sousa S. 2015. Src-dependent tyrosine phosphorylation of non-muscle myosin heavy chain-IIA restricts Listeria monocytogenes cellular infection. J Biol Chem 290:8383–8395 10.1074/jbc.M114.591313. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lecuit M, Hurme R, Pizarro-Cerdá J, Ohayon H, Geiger B, Cossart P. 2000. A role for alpha-and beta-catenins in bacterial uptake. Proc Natl Acad Sci U S A 97:10008–10013 10.1073/pnas.97.18.10008. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sousa S, Cabanes D, Archambaud C, Colland F, Lemichez E, Popoff M, Boisson-Dupuis S, Gouin E, Lecuit M, Legrain P, Cossart P. 2005. ARHGAP10 is necessary for alpha-catenin recruitment at adherens junctions and for Listeria invasion. Nat Cell Biol 7:954–960 10.1038/ncb1308. [PubMed] [DOI] [PubMed] [Google Scholar]
- 31.Sousa S, Cabanes D, Bougnères L, Lecuit M, Sansonetti P, Tran-Van-Nhieu G, Cossart P. 2007. Src, cortactin and Arp2/3 complex are required for E-cadherin-mediated internalization of Listeria into cells. Cell Microbiol 9:2629–2643 10.1111/j.1462-5822.2007.00984.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 32.Bonazzi M, Kühbacher A, Toledo-Arana A, Mallet A, Vasudevan L, Pizarro-Cerdá J, Brodsky FM, Cossart P. 2012. A common clathrin-mediated machinery co-ordinates cell-cell adhesion and bacterial internalization. Traffic 13:1653–1666 10.1111/tra.12009. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pizarro-Cerdá J, Bonazzi M, Cossart P. 2010. Clathrin-mediated endocytosis: what works for small, also works for big. BioEssays 32:496–504 10.1002/bies.200900172. [PubMed] [DOI] [PubMed] [Google Scholar]
- 34.Gessain G, Tsai YH, Travier L, Bonazzi M, Grayo S, Cossart P, Charlier C, Disson O, Lecuit M. 2015. PI3-kinase activation is critical for host barrier permissiveness to Listeria monocytogenes. J Exp Med 212:165–183 10.1084/jem.20141406. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Disson O, Grayo S, Huillet E, Nikitas G, Langa-Vives F, Dussurget O, Ragon M, Le Monnier A, Babinet C, Cossart P, Lecuit M. 2008. Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455:1114–1118 10.1038/nature07303. [PubMed] [DOI] [PubMed] [Google Scholar]
- 36.Dramsi S, Biswas I, Maguin E, Braun L, Mastroeni P, Cossart P. 1995. Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol Microbiol 16:251–261 10.1111/j.1365-2958.1995.tb02297.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 37.Lingnau A, Domann E, Hudel M, Bock M, Nichterlein T, Wehland J, Chakraborty T. 1995. Expression of the Listeria monocytogenes EGD inlA and inlB genes, whose products mediate bacterial entry into tissue culture cell lines, by PrfA-dependent and -independent mechanisms. Infect Immun 63:3896–3903. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quereda JJ, Rodríguez-Gómez IM, Meza-Torres J, Gomez-Laguna J, Nahori MA, Dussurget O, Carrasco L, Cossart P, Pizarro-Cerdá J. 2018. Reassessing the role of Internalin B in Listeria monocytogenes virulence using the epidemic strain F2365. Clin Microbiol Infect. Epub ahead of print. 10.1016/j.cmi.2018.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braun L, Dramsi S, Dehoux P, Bierne H, Lindahl G, Cossart P. 1997. InlB: an invasion protein of Listeria monocytogenes with a novel type of surface association. Mol Microbiol 25:285–294 10.1046/j.1365-2958.1997.4621825.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 40.Jonquières R, Bierne H, Fiedler F, Gounon P, Cossart P. 1999. Interaction between the protein InlB of Listeria monocytogenes and lipoteichoic acid: a novel mechanism of protein association at the surface of Gram-positive bacteria. Mol Microbiol 34:902–914 10.1046/j.1365-2958.1999.01652.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 41.Carvalho F, Sousa S, Cabanes D. 2018. l-Rhamnosylation of wall teichoic acids promotes efficient surface association of Listeria monocytogenes virulence factors InlB and Ami through interaction with GW domains. Environ Microbiol 43:1. [DOI] [PubMed] [Google Scholar]
- 42.Braun L, Ghebrehiwet B, Cossart P. 2000. gC1q-R/p32, a C1q-binding protein, is a receptor for the InlB invasion protein of Listeria monocytogenes. EMBO J 19:1458–1466 10.1093/emboj/19.7.1458. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonquières R, Pizarro-Cerdá J, Cossart P. 2001. Synergy between the N- and C-terminal domains of InlB for efficient invasion of non-phagocytic cells by Listeria monocytogenes. Mol Microbiol 42:955–965 10.1046/j.1365-2958.2001.02704.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 44.Braun L, Ohayon H, Cossart P. 1998. The InIB protein of Listeria monocytogenes is sufficient to promote entry into mammalian cells. Mol Microbiol 27:1077–1087 10.1046/j.1365-2958.1998.00750.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 45.Braun L, Nato F, Payrastre B, Mazié JC, Cossart P. 1999. The 213-amino-acid leucine-rich repeat region of the Listeria monocytogenes InlB protein is sufficient for entry into mammalian cells, stimulation of PI 3-kinase and membrane ruffling. Mol Microbiol 34:10–23 10.1046/j.1365-2958.1999.01560.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 46.Shen Y, Naujokas M, Park M, Ireton K. 2000. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 103:501–510 10.1016/S0092-8674(00)00141-0. [DOI] [PubMed] [Google Scholar]
- 47.Khelef N, Lecuit M, Bierne H, Cossart P. 2006. Species specificity of the Listeria monocytogenes InlB protein. Cell Microbiol 8:457–470 10.1111/j.1462-5822.2005.00634.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 48.Cruz R, Pereira-Castro I, Almeida MT, Moreira A, Cabanes D, Sousa S. 2018. Epithelial keratins modulate cMet expression and signaling and promote InlB-mediated Listeria monocytogenes infection of HeLa cells. Front Cell Infect Microbiol 8:146 10.3389/fcimb.2018.00146. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ireton K, Payrastre B, Cossart P. 1999. The Listeria monocytogenes protein InlB is an agonist of mammalian phosphoinositide 3-kinase. J Biol Chem 274:17025–17032 10.1074/jbc.274.24.17025. [PubMed] [DOI] [PubMed] [Google Scholar]
- 50.Sun H, Shen Y, Dokainish H, Holgado-Madruga M, Wong A, Ireton K. 2005. Host adaptor proteins Gab1 and CrkII promote InlB-dependent entry of Listeria monocytogenes. Cell Microbiol 7:443–457 10.1111/j.1462-5822.2004.00475.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 51.Basar T, Shen Y, Ireton K. 2005. Redundant roles for Met docking site tyrosines and the Gab1 pleckstrin homology domain in InlB-mediated entry of Listeria monocytogenes. Infect Immun 73:2061–2074 10.1128/IAI.73.4.2061-2074.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ireton K, Payrastre B, Chap H, Ogawa W, Sakaue H, Kasuga M, Cossart P. 1996. A role for phosphoinositide 3-kinase in bacterial invasion. Science 274:780–782 10.1126/science.274.5288.780. [PubMed] [DOI] [PubMed] [Google Scholar]
- 53.Dokainish H, Gavicherla B, Shen Y, Ireton K. 2007. The carboxyl-terminal SH3 domain of the mammalian adaptor CrkII promotes internalization of Listeria monocytogenes through activation of host phosphoinositide 3-kinase. Cell Microbiol 9:2497–2516 10.1111/j.1462-5822.2007.00976.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Jiwani S, Wang Y, Dowd GC, Gianfelice A, Pichestapong P, Gavicherla B, Vanbennekom N, Ireton K. 2012. Identification of components of the host type IA phosphoinositide 3-kinase pathway that promote internalization of Listeria monocytogenes. Infect Immun 80:1252–1266 10.1128/IAI.06082-11. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seveau S, Tham TN, Payrastre B, Hoppe AD, Swanson JA, Cossart P. 2007. A FRET analysis to unravel the role of cholesterol in Rac1 and PI 3-kinase activation in the InlB/Met signalling pathway. Cell Microbiol 9:790–803 10.1111/j.1462-5822.2006.00832.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 56.Bierne H, Gouin E, Roux P, Caroni P, Yin HL, Cossart P. 2001. A role for cofilin and LIM kinase in Listeria-induced phagocytosis. J Cell Biol 155:101–112 10.1083/jcb.200104037. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bierne H, Miki H, Innocenti M, Scita G, Gertler FB, Takenawa T, Cossart P. 2005. WASP-related proteins, Abi1 and Ena/VASP are required for Listeria invasion induced by the Met receptor. J Cell Sci 118:1537–1547 10.1242/jcs.02285. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.Bhalla M, Law D, Dowd GC, Ireton K. 2017. Host serine/threonine kinases mTOR and protein kinase C-α promote InlB-mediated entry of Listeria monocytogenes. Infect Immun 85:85 10.1128/IAI.00087-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kühbacher A, Dambournet D, Echard A, Cossart P, Pizarro-Cerdá J. 2012. Phosphatidylinositol 5-phosphatase oculocerebrorenal syndrome of Lowe protein (OCRL) controls actin dynamics during early steps of Listeria monocytogenes infection. J Biol Chem 287:13128–13136 10.1074/jbc.M111.315788. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pizarro-Cerdá J, Payrastre B, Wang Y-J, Veiga E, Yin HL, Cossart P. 2007. Type II phosphatidylinositol 4-kinases promote Listeria monocytogenes entry into target cells. Cell Microbiol 9:2381–2390 10.1111/j.1462-5822.2007.00967.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 61.Tham TN, Gouin E, Rubinstein E, Boucheix C, Cossart P, Pizarro-Cerdá J. 2010. Tetraspanin CD81 is required for Listeria monocytogenes invasion. Infect Immun 78:204–209 10.1128/IAI.00661-09. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Veiga E, Cossart P. 2005. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat Cell Biol 7:894–900 10.1038/ncb1292. [PubMed] [DOI] [PubMed] [Google Scholar]
- 63.Veiga E, Guttman JA, Bonazzi M, Boucrot E, Toledo-Arana A, Lin AE, Enninga J, Pizarro-Cerdá J, Finlay BB, Kirchhausen T, Cossart P. 2007. Invasive and adherent bacterial pathogens co-Opt host clathrin for infection. Cell Host Microbe 2:340–351 10.1016/j.chom.2007.10.001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pizarro-Cerdá J, Cossart P. 2009. Listeria monocytogenes membrane trafficking and lifestyle: the exception or the rule? Annu Rev Cell Dev Biol 25:649–670 10.1146/annurev.cellbio.042308.113331. [PubMed] [DOI] [PubMed] [Google Scholar]
- 65.Van Ngo H, Bhalla M, Chen D-Y, Ireton K. 2017. A role for host cell exocytosis in InlB-mediated internalisation of Listeria monocytogenes. Cell Microbiol 19:e12768 10.1111/cmi.12768. [PubMed] [DOI] [PubMed] [Google Scholar]
- 66.Pizarro-Cerdá J, Jonquières R, Gouin E, Vandekerckhove J, Garin J, Cossart P. 2002. Distinct protein patterns associated with Listeria monocytogenes InlA- or InlB-phagosomes. Cell Microbiol 4:101–115 10.1046/j.1462-5822.2002.00169.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 67.Mostowy S, Nam Tham T, Danckaert A, Guadagnini S, Boisson-Dupuis S, Pizarro-Cerdá J, Cossart P. 2009. Septins regulate bacterial entry into host cells. PLoS One 4:e4196 10.1371/journal.pone.0004196. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mostowy S, Danckaert A, Tham TN, Machu C, Guadagnini S, Pizarro-Cerdá J, Cossart P. 2009. Septin 11 restricts InlB-mediated invasion by Listeria. J Biol Chem 284:11613–11621 10.1074/jbc.M900231200. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mostowy S, Janel S, Forestier C, Roduit C, Kasas S, Pizarro-Cerdá J, Cossart P, Lafont F. 2011. A role for septins in the interaction between the Listeria monocytogenes INVASION PROTEIN InlB and the Met receptor. Biophys J 100:1949–1959 10.1016/j.bpj.2011.02.040. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bergmann B, Raffelsbauer D, Kuhn M, Goetz M, Hom S, Goebel W. 2002. InlA- but not InlB-mediated internalization of Listeria monocytogenes by non-phagocytic mammalian cells needs the support of other internalins. Mol Microbiol 43:557–570 10.1046/j.1365-2958.2002.02767.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Sabet C, Lecuit M, Cabanes D, Cossart P, Bierne H. 2005. LPXTG protein InlJ, a newly identified internalin involved in Listeria monocytogenes virulence. Infect Immun 73:6912–6922 10.1128/IAI.73.10.6912-6922.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sabet C, Toledo-Arana A, Personnic N, Lecuit M, Dubrac S, Poupel O, Gouin E, Nahori M-A, Cossart P, Bierne H. 2008. The Listeria monocytogenes virulence factor InlJ is specifically expressed in vivo and behaves as an adhesin. Infect Immun 76:1368–1378 10.1128/IAI.01519-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dramsi S, Dehoux P, Lebrun M, Goossens PL, Cossart P. 1997. Identification of four new members of the internalin multigene family of Listeria monocytogenes EGD. Infect Immun 65:1615–1625. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kirchner M, Higgins DE. 2008. Inhibition of ROCK activity allows InlF-mediated invasion and increased virulence of Listeria monocytogenes. Mol Microbiol 68:749–767 10.1111/j.1365-2958.2008.06188.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Milohanic E, Pron B, Berche P, Gaillard JL, European Listeria Genome Consortium. 2000. Identification of new loci involved in adhesion of Listeria monocytogenes to eukaryotic cells. Microbiology 146:731–739 10.1099/00221287-146-3-731. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Milohanic E, Jonquières R, Cossart P, Berche P, Gaillard JL. 2001. The autolysin Ami contributes to the adhesion of Listeria monocytogenes to eukaryotic cells via its cell wall anchor. Mol Microbiol 39:1212–1224 10.1111/j.1365-2958.2001.02208.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 77.Cabanes D, Dussurget O, Dehoux P, Cossart P. 2004. Auto, a surface associated autolysin of Listeria monocytogenes required for entry into eukaryotic cells and virulence. Mol Microbiol 51:1601–1614 10.1111/j.1365-2958.2003.03945.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 78.Wang L, Lin M. 2008. A novel cell wall-anchored peptidoglycan hydrolase(autolysin), IspC, essential for Listeria monocytogenes virulence: genetic and proteomic analysis. Microbiology 154:1900–1913 10.1099/mic.0.2007/015172-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Promadej N, Fiedler F, Cossart P, Dramsi S, Kathariou S. 1999. Cell wall teichoic acid glycosylation in Listeria monocytogenes serotype 4b requires gtcA, a novel, serogroup-specific gene. J Bacteriol 181:418–425. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abachin E, Poyart C, Pellegrini E, Milohanic E, Fiedler F, Berche P, Trieu-Cuot P. 2002. Formation of d-alanyl-lipoteichoic acid is required for adhesion and virulence of Listeria monocytogenes. Mol Microbiol 43:1–14 10.1046/j.1365-2958.2002.02723.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 81.Réglier-Poupet H, Pellegrini E, Charbit A, Berche P. 2003. Identification of LpeA, a PsaA-like membrane protein that promotes cell entry by Listeria monocytogenes. Infect Immun 71:474–482 10.1128/IAI.71.1.474-482.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Machata S, Tchatalbachev S, Mohamed W, Jänsch L, Hain T, Chakraborty T. 2008. Lipoproteins of Listeria monocytogenes are critical for virulence and TLR2-mediated immune activation. J Immunol 181:2028–2035 10.4049/jimmunol.181.3.2028. [PubMed] [DOI] [PubMed] [Google Scholar]
- 83.Thedieck K, Hain T, Mohamed W, Tindall BJ, Nimtz M, Chakraborty T, Wehland J, Jänsch L. 2006. The MprF protein is required for lysinylation of phospholipids in listerial membranes and confers resistance to cationic antimicrobial peptides (CAMPs) on Listeria monocytogenes. Mol Microbiol 62:1325–1339 10.1111/j.1365-2958.2006.05452.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 84.Suárez M, González-Zorn B, Vega Y, Chico-Calero I, Vázquez-Boland JA. 2001. A role for ActA in epithelial cell invasion by Listeria monocytogenes. Cell Microbiol 3:853–864 10.1046/j.1462-5822.2001.00160.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 85.Alvarez-Domínguez C, Vázquez-Boland JA, Carrasco-Marín E, López-Mato P, Leyva-Cobián F. 1997. Host cell heparan sulfate proteoglycans mediate attachment and entry of Listeria monocytogenes, and the listerial surface protein ActA is involved in heparan sulfate receptor recognition. Infect Immun 65:78–88. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cabanes D, Sousa S, Cebriá A, Lecuit M, García-del Portillo F, Cossart P. 2005. Gp96 is a receptor for a novel Listeria monocytogenes virulence factor, Vip, a surface protein. EMBO J 24:2827–2838 10.1038/sj.emboj.7600750. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martins M, Custódio R, Camejo A, Almeida MT, Cabanes D, Sousa S. 2012. Listeria monocytogenes triggers the cell surface expression of Gp96 protein and interacts with its N terminus to support cellular infection. J Biol Chem 287:43083–43093 10.1074/jbc.M112.422568. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jagadeesan B, Fleishman Littlejohn AE, Amalaradjou MAR, Singh AK, Mishra KK, La D, Kihara D, Bhunia AK. 2011. N-terminal Gly(224)-Gly(411) domain in Listeria adhesion protein interacts with host receptor Hsp60. PLoS One 6:e20694 10.1371/journal.pone.0020694. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reis O, Sousa S, Camejo A, Villiers V, Gouin E, Cossart P, Cabanes D. 2010. LapB, a novel Listeria monocytogenes LPXTG surface adhesin, required for entry into eukaryotic cells and virulence. J Infect Dis 202:551–562 10.1086/654880. [PubMed] [DOI] [PubMed] [Google Scholar]
- 90.Dramsi S, Bourdichon F, Cabanes D, Lecuit M, Fsihi H, Cossart P. 2004. FbpA, a novel multifunctional Listeria monocytogenes virulence factor. Mol Microbiol 53:639–649 10.1111/j.1365-2958.2004.04138.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 91.Dramsi S, Cossart P. 2003. Listeriolysin O-mediated calcium influx potentiates entry of Listeria monocytogenes into the human Hep-2 epithelial cell line. Infect Immun 71:3614–3618 10.1128/IAI.71.6.3614-3618.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stavru F, Cossart P. 2011. Listeria infection modulates mitochondrial dynamics. Commun Integr Biol 4:364–366 10.4161/cib.4.3.15506. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vadia S, Arnett E, Haghighat A-C, Wilson-Kubalek EM, Tweten RK, Seveau S. 2011. The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathog 7:e1002356 10.1371/journal.ppat.1002356. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vadia S, Seveau S. 2014. Fluxes of Ca2+ and K+ are required for the listeriolysin O-dependent internalization pathway of Listeria monocytogenes. Infect Immun 82:1084–1091 10.1128/IAI.01067-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wadsworth SJ, Goldfine H. 1999. Listeria monocytogenes phospholipase C-dependent calcium signaling modulates bacterial entry into J774 macrophage-like cells. Infect Immun 67:1770–1778. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Birmingham CL, Canadien V, Kaniuk NA, Steinberg BE, Higgins DE, Brumell JH. 2008. Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles. Nature 451:350–354 10.1038/nature06479. [PubMed] [DOI] [PubMed] [Google Scholar]
- 97.Kortebi M, Milohanic E, Mitchell G, Péchoux C, Prévost M-C, Cossart P, Bierne H. 2017. Listeria monocytogenes switches from dissemination to persistence by adopting a vacuolar lifestyle in epithelial cells. PLoS Pathog 13:e1006734 10.1371/journal.ppat.1006734. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seastone CV. 1935. Pathogenic organisms of the genus Listerella. J Exp Med 62:203–212 10.1084/jem.62.2.203. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mengaud J, Dramsi S, Gouin E, Vázquez-Boland JA, Milon G, Cossart P. 1991. Pleiotropic control of Listeria monocytogenes virulence factors by a gene that is autoregulated. Mol Microbiol 5:2273–2283 10.1111/j.1365-2958.1991.tb02158.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 100.Gaillard JL, Berche P, Mounier J, Richard S, Sansonetti P. 1987. In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect Immun 55:2822–2829. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dal Peraro M, van der Goot FG. 2016. Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol 14:77–92 10.1038/nrmicro.2015.3. [PubMed] [DOI] [PubMed] [Google Scholar]
- 102.Soltani CE, Hotze EM, Johnson AE, Tweten RK. 2007. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc Natl Acad Sci U S A 104:20226–20231 10.1073/pnas.0708104105. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farrand AJ, LaChapelle S, Hotze EM, Johnson AE, Tweten RK. 2010. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc Natl Acad Sci U S A 107:4341–4346 10.1073/pnas.0911581107. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shepard LA, Shatursky O, Johnson AE, Tweten RK. 2000. The mechanism of pore assembly for a cholesterol-dependent cytolysin: formation of a large prepore complex precedes the insertion of the transmembrane β-hairpins. Biochemistry 39:10284–10293 10.1021/bi000436r. [PubMed] [DOI] [PubMed] [Google Scholar]
- 105.Schuerch DW, Wilson-Kubalek EM, Tweten RK. 2005. Molecular basis of listeriolysin O pH dependence. Proc Natl Acad Sci U S A 102:12537–12542 10.1073/pnas.0500558102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ruan Y, Rezelj S, Bedina Zavec A, Anderluh G, Scheuring S. 2016. Listeriolysin O membrane damaging activity involves arc formation and lineaction: implication for Listeria monocytogenes escape from phagocytic vacuole. PLoS Pathog 12:e1005597 10.1371/journal.ppat.1005597. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Henry R, Shaughnessy L, Loessner MJ, Alberti-Segui C, Higgins DE, Swanson JA. 2006. Cytolysin-dependent delay of vacuole maturation in macrophages infected with Listeria monocytogenes. Cell Microbiol 8:107–119 10.1111/j.1462-5822.2005.00604.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shaughnessy LM, Hoppe AD, Christensen KA, Swanson JA. 2006. Membrane perforations inhibit lysosome fusion by altering pH and calcium in Listeria monocytogenes vacuoles. Cell Microbiol 8:781–792 10.1111/j.1462-5822.2005.00665.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen C, Nguyen BN, Mitchell G, Margolis SR, Ma D, Portnoy DA. 2018. The Listeriolysin O PEST-like sequence co-opts AP-2-mediated endocytosis to prevent plasma membrane damage during Listeria infection. Cell Host Microbe 23:786–795.e5 10.1016/j.chom.2018.05.006. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Beauregard KE, Lee KD, Collier RJ, Swanson JA. 1997. pH-dependent perforation of macrophage phagosomes by listeriolysin O from Listeria monocytogenes. J Exp Med 186:1159–1163 10.1084/jem.186.7.1159. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. 2002. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J Cell Biol 156:1029–1038 10.1083/jcb.200201081. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Glomski IJ, Decatur AL, Portnoy DA. 2003. Listeria monocytogenes mutants that fail to compartmentalize listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect Immun 71:6754–6765 10.1128/IAI.71.12.6754-6765.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schnupf P, Hofmann J, Norseen J, Glomski IJ, Schwartzstein H, Decatur AL. 2006. Regulated translation of listeriolysin O controls virulence of Listeria monocytogenes. Mol Microbiol 61:999–1012 10.1111/j.1365-2958.2006.05286.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 114.Schnupf P, Zhou J, Varshavsky A, Portnoy DA. 2007. Listeriolysin O secreted by Listeria monocytogenes into the host cell cytosol is degraded by the N-end rule pathway. Infect Immun 75:5135–5147 10.1128/IAI.00164-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Geoffroy C, Gaillard JL, Alouf JE, Berche P. 1987. Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect Immun 55:1641–1646. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Singh R, Jamieson A, Cresswell P. 2008. GILT is a critical host factor for Listeria monocytogenes infection. Nature 455:1244–1247 10.1038/nature07344. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Radtke AL, Anderson KL, Davis MJ, DiMagno MJ, Swanson JA, O’Riordan MX. 2011. Listeria monocytogenes exploits cystic fibrosis transmembrane conductance regulator (CFTR) to escape the phagosome. Proc Natl Acad Sci U S A 108:1633–1638 10.1073/pnas.1013262108. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shaughnessy LM, Lipp P, Lee K-D, Swanson JA. 2007. Localization of protein kinase C epsilon to macrophage vacuoles perforated by Listeria monocytogenes cytolysin. Cell Microbiol 9:1695–1704 10.1111/j.1462-5822.2007.00903.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mengaud J, Braun-Breton C, Cossart P. 1991. Identification of phosphatidylinositol-specific phospholipase C activity in Listeria monocytogenes: a novel type of virulence factor? Mol Microbiol 5:367–372 10.1111/j.1365-2958.1991.tb02118.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 120.Geoffroy C, Raveneau J, Beretti JL, Lecroisey A, Vázquez-Boland JA, Alouf JE, Berche P. 1991. Purification and characterization of an extracellular 29-kilodalton phospholipase C from Listeria monocytogenes. Infect Immun 59:2382–2388. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vázquez-Boland JA, Kocks C, Dramsi S, Ohayon H, Geoffroy C, Mengaud J, Cossart P. 1992. Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread. Infect Immun 60:219–230. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mengaud J, Geoffroy C, Cossart P. 1991. Identification of a new operon involved in Listeria monocytogenes virulence: its first gene encodes a protein homologous to bacterial metalloproteases. Infect Immun 59:1043–1049. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Poyart C, Abachin E, Razafimanantsoa I, Berche P. 1993. The zinc metalloprotease of Listeria monocytogenes is required for maturation of phosphatidylcholine phospholipase C: direct evidence obtained by gene complementation. Infect Immun 61:1576–1580. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bitar AP, Cao M, Marquis H. 2008. The metalloprotease of Listeria monocytogenes is activated by intramolecular autocatalysis. J Bacteriol 190:107–111 10.1128/JB.00852-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Camilli A, Tilney LG, Portnoy DA. 1993. Dual roles of plcA in Listeria monocytogenes pathogenesis. Mol Microbiol 8:143–157 10.1111/j.1365-2958.1993.tb01211.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marquis H, Doshi V, Portnoy DA. 1995. The broad-range phospholipase C and a metalloprotease mediate listeriolysin O-independent escape of Listeria monocytogenes from a primary vacuole in human epithelial cells. Infect Immun 63:4531–4534. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Smith GA, Marquis H, Jones S, Johnston NC, Portnoy DA, Goldfine H. 1995. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect Immun 63:4231–4237. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gründling A, Gonzalez MD, Higgins DE. 2003. Requirement of the Listeria monocytogenes broad-range phospholipase PC-PLC during infection of human epithelial cells. J Bacteriol 185:6295–6307 10.1128/JB.185.21.6295-6307.2003. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Moser J, Gerstel B, Meyer JE, Chakraborty T, Wehland J, Heinz DW. 1997. Crystal structure of the phosphatidylinositol-specific phospholipase C from the human pathogen Listeria monocytogenes. J Mol Biol 273:269–282 10.1006/jmbi.1997.1290. [PubMed] [DOI] [PubMed] [Google Scholar]
- 130.Wei Z, Zenewicz LA, Goldfine H. 2005. Listeria monocytogenes phosphatidylinositol-specific phospholipase C has evolved for virulence by greatly reduced activity on GPI anchors. Proc Natl Acad Sci U S A 102:12927–12931 10.1073/pnas.0501725102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sibelius U, Chakraborty T, Krögel B, Wolf J, Rose F, Schmidt R, Wehland J, Seeger W, Grimminger F. 1996. The listerial exotoxins listeriolysin and phosphatidylinositol-specific phospholipase C synergize to elicit endothelial cell phosphoinositide metabolism. J Immunol 157:4055–4060. [PubMed] [PubMed] [Google Scholar]
- 132.Goldfine H, Wadsworth SJ, Johnston NC. 2000. Activation of host phospholipases C and D in macrophages after infection with Listeria monocytogenes. Infect Immun 68:5735–5741 10.1128/IAI.68.10.5735-5741.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Poussin MA, Goldfine H. 2005. Involvement of Listeria monocytogenes phosphatidylinositol-specific phospholipase C and host protein kinase C in permeabilization of the macrophage phagosome. Infect Immun 73:4410–4413 10.1128/IAI.73.7.4410-4413.2005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wadsworth SJ, Goldfine H. 2002. Mobilization of protein kinase C in macrophages induced by Listeria monocytogenes affects its internalization and escape from the phagosome. Infect Immun 70:4650–4660 10.1128/IAI.70.8.4650-4660.2002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Marquis H, Goldfine H, Portnoy DA. 1997. Proteolytic pathways of activation and degradation of a bacterial phospholipase C during intracellular infection by Listeria monocytogenes. J Cell Biol 137:1381–1392 10.1083/jcb.137.6.1381. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Marquis H, Hager EJ. 2000. pH-regulated activation and release of a bacteria-associated phospholipase C during intracellular infection by Listeria monocytogenes. Mol Microbiol 35:289–298 10.1046/j.1365-2958.2000.01708.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yeung PSM, Na Y, Kreuder AJ, Marquis H. 2007. Compartmentalization of the broad-range phospholipase C activity to the spreading vacuole is critical for Listeria monocytogenes virulence. Infect Immun 75:44–51 10.1128/IAI.01001-06. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lam GY, Fattouh R, Muise AM, Grinstein S, Higgins DE, Brumell JH. 2011. Listeriolysin O suppresses phospholipase C-mediated activation of the microbicidal NADPH oxidase to promote Listeria monocytogenes infection. Cell Host Microbe 10:627–634 10.1016/j.chom.2011.11.005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xayarath B, Alonzo F III, Freitag NE. 2015. Identification of a peptide-pheromone that enhances Listeria monocytogenes escape from host cell vacuoles. PLoS Pathog 11:e1004707 10.1371/journal.ppat.1004707. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rabinovich L, Sigal N, Borovok I, Nir-Paz R, Herskovits AA. 2012. Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 150:792–802 10.1016/j.cell.2012.06.036. [PubMed] [DOI] [PubMed] [Google Scholar]
- 141.Alvarez-Dominguez C, Stahl PD. 1999. Increased expression of Rab5a correlates directly with accelerated maturation of Listeria monocytogenes phagosomes. J Biol Chem 274:11459–11462 10.1074/jbc.274.17.11459. [PubMed] [DOI] [PubMed] [Google Scholar]
- 142.Prada-Delgado A, Carrasco-Marín E, Peña-Macarro C, Del Cerro-Vadillo E, Fresno-Escudero M, Leyva-Cobián F, Alvarez-Dominguez C. 2005. Inhibition of Rab5a exchange activity is a key step for Listeria monocytogenes survival. Traffic 6:252–265 10.1111/j.1600-0854.2005.00265.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 143.Alvarez-Dominguez C, Madrazo-Toca F, Fernandez-Prieto L, Vandekerckhove J, Pareja E, Tobes R, Gomez-Lopez MT, Del Cerro-Vadillo E, Fresno M, Leyva-Cobián F, Carrasco-Marín E. 2008. Characterization of a Listeria monocytogenes protein interfering with Rab5a. Traffic 9:325–337 10.1111/j.1600-0854.2007.00683.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 144.Lopez-Castejon G, Corbett D, Goldrick M, Roberts IS, Brough D. 2012. Inhibition of calpain blocks the phagosomal escape of Listeria monocytogenes. PLoS One 7:e35936 10.1371/journal.pone.0035936. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ripio MT, Brehm K, Lara M, Suárez M, Vázquez-Boland JA. 1997. Glucose-1-phosphate utilization by Listeria monocytogenes is PrfA dependent and coordinately expressed with virulence factors. J Bacteriol 179:7174–7180 10.1128/jb.179.22.7174-7180.1997. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chico-Calero I, Suárez M, González-Zorn B, Scortti M, Slaghuis J, Goebel W, Vázquez-Boland JA, European Listeria Genome Consortium. 2002. Hpt, a bacterial homolog of the microsomal glucose- 6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc Natl Acad Sci U S A 99:431–436 10.1073/pnas.012363899. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.O’Riordan M, Moors MA, Portnoy DA. 2003. Listeria intracellular growth and virulence require host-derived lipoic acid. Science 302:462–464 10.1126/science.1088170. [PubMed] [DOI] [PubMed] [Google Scholar]
- 148.Keeney KM, Stuckey JA, O’Riordan MXD. 2007. LplA1-dependent utilization of host lipoyl peptides enables Listeria cytosolic growth and virulence. Mol Microbiol 66:758–770 10.1111/j.1365-2958.2007.05956.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen GY, McDougal CE, D’Antonio MA, Portman JL, Sauer J-D. 2017. A genetic screen reveals that synthesis of 1,4-dihydroxy-2-naphthoate (DHNA), but not full-length menaquinone, is required for Listeria monocytogenes cytosolic survival. MBio 8:e00119-17 10.1128/mBio.00119-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rich KA, Burkett C, Webster P. 2003. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol 5:455–468 10.1046/j.1462-5822.2003.00292.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 151.Birmingham CL, Canadien V, Gouin E, Troy EB, Yoshimori T, Cossart P, Higgins DE, Brumell JH. 2007. Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3:442–451 10.4161/auto.4450. [PubMed] [DOI] [PubMed] [Google Scholar]
- 152.Perrin AJ, Jiang X, Birmingham CL, So NSY, Brumell JH. 2004. Recognition of bacteria in the cytosol of Mammalian cells by the ubiquitin system. Curr Biol 14:806–811 10.1016/j.cub.2004.04.033. [PubMed] [DOI] [PubMed] [Google Scholar]
- 153.Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, Kakizuka A, Sztul E, Chakraborty T, Sasakawa C. 2009. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol 11:1233–1240 10.1038/ncb1967. [PubMed] [DOI] [PubMed] [Google Scholar]
- 154.Py BF, Lipinski MM, Yuan J. 2007. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 3:117–125 10.4161/auto.3618. [PubMed] [DOI] [PubMed] [Google Scholar]
- 155.Mitchell G, Ge L, Huang Q, Chen C, Kianian S, Roberts MF, Schekman R, Portnoy DA. 2015. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect Immun 83:2175–2184 10.1128/IAI.00110-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tattoli I, Sorbara MT, Yang C, Tooze SA, Philpott DJ, Girardin SE. 2013. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J 32:3066–3078 10.1038/emboj.2013.234. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cemma M, Lam GY, Stöckli M, Higgins DE, Brumell JH. 2015. Strain-specific interactions of Listeria monocytogenes with the autophagy system in host cells. PLoS One 10:e0125856 10.1371/journal.pone.0125856. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Crimmins GT, Herskovits AA, Rehder K, Sivick KE, Lauer P, Dubensky TW Jr, Portnoy DA. 2008. Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc Natl Acad Sci U S A 105:10191–10196 10.1073/pnas.0804170105. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Woodward JJ, Iavarone AT, Portnoy DA. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705 10.1126/science.1189801. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Abdullah Z, Schlee M, Roth S, Mraheil MA, Barchet W, Böttcher J, Hain T, Geiger S, Hayakawa Y, Fritz JH, Civril F, Hopfner KP, Kurts C, Ruland J, Hartmann G, Chakraborty T, Knolle PA. 2012. RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J 31:4153–4164 10.1038/emboj.2012.274. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hagmann CA, Herzner AM, Abdullah Z, Zillinger T, Jakobs C, Schuberth C, Coch C, Higgins PG, Wisplinghoff H, Barchet W, Hornung V, Hartmann G, Schlee M. 2013. RIG-I detects triphosphorylated RNA of Listeria monocytogenes during infection in non-immune cells. PLoS One 8:e62872 10.1371/journal.pone.0062872. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Radoshevich L, Impens F, Ribet D, Quereda JJ, Nam Tham T, Nahori M-A, Bierne H, Dussurget O, Pizarro-Cerdá J, Knobeloch K-P, Cossart P. 2015. ISG15 counteracts Listeria monocytogenes infection. eLife 4:4 10.7554/eLife.06848. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bierne H, Travier L, Mahlakõiv T, Tailleux L, Subtil A, Lebreton A, Paliwal A, Gicquel B, Staeheli P, Lecuit M, Cossart P. 2012. Activation of type III interferon genes by pathogenic bacteria in infected epithelial cells and mouse placenta. PLoS One 7:e39080 10.1371/journal.pone.0039080. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lam GY, Cemma M, Muise AM, Higgins DE, Brumell JH. 2013. Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy 9:985–995 10.4161/auto.24406. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Auerbuch V, Loureiro JJ, Gertler FB, Theriot JA, Portnoy DA. 2003. Ena/VASP proteins contribute to Listeria monocytogenes pathogenesis by controlling temporal and spatial persistence of bacterial actin-based motility. Mol Microbiol 49:1361–1375 10.1046/j.1365-2958.2003.03639.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 166.Welch MD, Iwamatsu A, Mitchison TJ. 1997. Actin polymerization is induced by Arp2/3 protein complex at the surface of Listeria monocytogenes. Nature 385:265–269 10.1038/385265a0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 167.Welch MD, Rosenblatt J, Skoble J, Portnoy DA, Mitchison TJ. 1998. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science 281:105–108 10.1126/science.281.5373.105. [PubMed] [DOI] [PubMed] [Google Scholar]
- 168.Pizarro-Cerdá J, Chorev DS, Geiger B, Cossart P. 2017. The diverse family of Arp2/3 complexes. Trends Cell Biol 27:93–100 10.1016/j.tcb.2016.08.001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kühbacher A, Emmenlauer M, Rämo P, Kafai N, Dehio C, Cossart P, Pizarro-Cerdá J. 2015. Genome-wide siRNA screen identifies complementary signaling pathways involved in Listeria infection and reveals different actin nucleation mechanisms during Listeria cell invasion and actin comet tail formation. MBio 6:e00598–e15 10.1128/mBio.00598-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Van Troys M, Lambrechts A, David V, Demol H, Puype M, Pizarro-Cerdá J, Gevaert K, Cossart P, Vandekerckhove J. 2008. The actin propulsive machinery: the proteome of Listeria monocytogenes tails. Biochem Biophys Res Commun 375:194–199 10.1016/j.bbrc.2008.07.152. [PubMed] [DOI] [PubMed] [Google Scholar]
- 171.Jasnin M, Asano S, Gouin E, Hegerl R, Plitzko JM, Villa E, Cossart P, Baumeister W. 2013. Three-dimensional architecture of actin filaments in Listeria monocytogenes comet tails. Proc Natl Acad Sci U S A 110:20521–20526 10.1073/pnas.1320155110. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Polle L, Rigano LA, Julian R, Ireton K, Schubert W-D. 2014. Structural details of human tuba recruitment by InlC of Listeria monocytogenes elucidate bacterial cell-cell spreading. Structure 22:304–314 10.1016/j.str.2013.10.017. [PubMed] [DOI] [PubMed] [Google Scholar]
- 173.Gianfelice A, Le PHB, Rigano LA, Saila S, Dowd GC, McDivitt T, Bhattacharya N, Hong W, Stagg SM, Ireton K. 2015. Host endoplasmic reticulum COPII proteins control cell-to-cell spread of the bacterial pathogen Listeria monocytogenes. Cell Microbiol 17:876–892 10.1111/cmi.12409. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rigano LA, Dowd GC, Wang Y, Ireton K. 2014. Listeria monocytogenes antagonizes the human GTPase Cdc42 to promote bacterial spread. Cell Microbiol 16:1068–1079 10.1111/cmi.12260. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pust S, Morrison H, Wehland J, Sechi AS, Herrlich P. 2005. Listeria monocytogenes exploits ERM protein functions to efficiently spread from cell to cell. EMBO J 24:1287–1300 10.1038/sj.emboj.7600595. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fattouh R, Kwon H, Czuczman MA, Copeland JW, Pelletier L, Quinlan ME, Muise AM, Higgins DE, Brumell JH. 2015. The diaphanous-related formins promote protrusion formation and cell-to-cell spread of Listeria monocytogenes. J Infect Dis 211:1185–1195 10.1093/infdis/jiu546. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Talman AM, Chong R, Chia J, Svitkina T, Agaisse H. 2014. Actin network disassembly powers dissemination of Listeria monocytogenes. J Cell Sci 127:240–249 10.1242/jcs.140038. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Alvarez DE, Agaisse H. 2016. The metalloprotease Mpl supports Listeria monocytogenes dissemination through resolution of membrane protrusions into vacuoles. Infect Immun 84:1806–1814 10.1128/IAI.00130-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Czuczman MA, Fattouh R, van Rijn JM, Canadien V, Osborne S, Muise AM, Kuchroo VK, Higgins DE, Brumell JH. 2014. Listeria monocytogenes exploits efferocytosis to promote cell-to-cell spread. Nature 509:230–234 10.1038/nature13168. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Alberti-Segui C, Goeden KR, Higgins DE. 2007. Differential function of Listeria monocytogenes listeriolysin O and phospholipases C in vacuolar dissolution following cell-to-cell spread. Cell Microbiol 9:179–195 10.1111/j.1462-5822.2006.00780.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 181.Stavru F, Palmer AE, Wang C, Youle RJ, Cossart P. 2013. Atypical mitochondrial fission upon bacterial infection. Proc Natl Acad Sci U S A 110:16003–16008. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pillich H, Loose M, Zimmer K-P, Chakraborty T. 2012. Activation of the unfolded protein response by Listeria monocytogenes. Cell Microbiol 14:949–964 10.1111/j.1462-5822.2012.01769.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 183.Malet JK, Cossart P, Ribet D. 2017. Alteration of epithelial cell lysosomal integrity induced by bacterial cholesterol-dependent cytolysins. Cell Microbiol 19:e12682 10.1111/cmi.12682. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ribet D, Hamon M, Gouin E, Nahori M-A, Impens F, Neyret-Kahn H, Gevaert K, Vandekerckhove J, Dejean A, Cossart P. 2010. Listeria monocytogenes impairs SUMOylation for efficient infection. Nature 464:1192–1195 10.1038/nature08963. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hamon MA, Batsché E, Régnault B, Tham TN, Seveau S, Muchardt C, Cossart P. 2007. Histone modifications induced by a family of bacterial toxins. Proc Natl Acad Sci U S A 104:13467–13472 10.1073/pnas.0702729104. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Samba-Louaka A, Pereira JM, Nahori M-A, Villiers V, Deriano L, Hamon MA, Cossart P. 2014. Listeria monocytogenes dampens the DNA damage response. PLoS Pathog 10:e1004470 10.1371/journal.ppat.1004470. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Leitão E, Costa AC, Brito C, Costa L, Pombinho R, Cabanes D, Sousa S. 2014. Listeria monocytogenes induces host DNA damage and delays the host cell cycle to promote infection. Cell Cycle 13:928–940 10.4161/cc.27780. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Samba-Louaka A, Stavru F, Cossart P. 2012. Role for telomerase in Listeria monocytogenes infection. Infect Immun 80:4257–4263 10.1128/IAI.00614-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lebreton A, Lakisic G, Job V, Fritsch L, Tham TN, Camejo A, Matteï P-J, Régnault B, Nahori M-A, Cabanes D, Gautreau A, Ait-Si-Ali S, Dessen A, Cossart P, Bierne H. 2011. A bacterial protein targets the BAHD1 chromatin complex to stimulate type III interferon response. Science 331:1319–1321 10.1126/science.1200120. [PubMed] [DOI] [PubMed] [Google Scholar]
- 190.Bierne H, Tham TN, Batsché E, Dumay A, Leguillou M, Kernéis-Golsteyn S, Régnault B, Seeler JS, Muchardt C, Feunteun J, Cossart P. 2009. Human BAHD1 promotes heterochromatic gene silencing. Proc Natl Acad Sci U S A 106:13826–13831 10.1073/pnas.0901259106. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lebreton A, Job V, Ragon M, Le Monnier A, Dessen A, Cossart P, Bierne H. 2013. Structural basis for the inhibition of the chromatin repressor BAHD1 by the bacterial nucleomodulin LntA. mBio 5:e00775-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Prokop A, Gouin E, Villiers V, Nahori M-A, Vincentelli R, Duval M, Cossart P, Dussurget O. 2017. OrfX, a nucleomodulin required for Listeria monocytogenes virulence. MBio 8:e01550-17 10.1128/mBio.01550-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ribet D, Lallemand-Breitenbach V, Ferhi O, Nahori M-A, Varet H, de Thé H, Cossart P. 2017. Promyelocytic leukemia protein (PML) controls Listeria monocytogenes infection. MBio 8:e02179-16 10.1128/mBio.02179-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Eskandarian HA, Impens F, Nahori MA, Soubigou G, Coppée JY, Cossart P, Hamon MA. 2013. A role for SIRT2-dependent histone H3K18 deacetylation in bacterial infection. Science 341:1238858 10.1126/science.1238858. [PubMed] [DOI] [PubMed] [Google Scholar]
- 195.Mitchell G, Cheng MI, Chen C, Nguyen BN, Whiteley AT, Kianian S, Cox JS, Green DR, McDonald KL, Portnoy DA. 2018. Listeria monocytogenes triggers noncanonical autophagy upon phagocytosis, but avoids subsequent growth-restricting xenophagy. Proc Natl Acad Sci U S A 115:E210–E217 10.1073/pnas.1716055115. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]