

Abstract
Duchenne muscular dystrophy (DMD) is caused by the absence of the full form of the dystrophin protein, which is essential for maintaining the structural integrity of muscle cells, including those in the heart and respiratory system. Despite progress in understanding the molecular mechanisms associated with DMD, myocardial insufficiency persists as the primary cause of mortality, and existing therapeutic strategies remain limited. This study investigates the hypothesis that a dysregulation of the biological communication between infiltrating macrophages (MPs) and neurocardiac junctions exists in dystrophic cardiac tissue. In a mouse model of DMD (mdx), this phenomenon is influenced by the over‐release of chondroitin sulfate proteoglycan‐4 (CSPG4), a key inhibitor of nerve sprouting and a modulator of the neural function, by MPs infiltrating the cardiac tissue and associated with dilated cardiomyopathy, a hallmark of DMD. Givinostat, the histone deacetylase inhibitor under current development as a clinical treatment for DMD, is effective at both restoring a physiological microenvironment at the neuro‐cardiac junction and cardiac function in mdx mice in addition to a reduction in cardiac fibrosis, MP‐mediated inflammation, and tissue CSPG4 content. This study provides novel insight into the pathophysiology of DMD in the heart, identifying potential new biological targets. © 2024 The Author(s). The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: cardiac innervation, dilated cardiomyopathy, DMD heart failure, macrophages, proteoglycans, extracellular matrix, inflammation, cardiac fibrosis
Introduction
Duchenne muscular dystrophy (DMD) is an X‐linked disorder caused by mutations in the dystrophin gene, a key protein connecting the internal cytoskeleton to the extracellular matrix (ECM) [1]. Although skeletal muscle impairment is most prominent, DMD also adversely affects myocardial and nervous system function [2].
Research on DMD has advanced in managing heart dysfunction by addressing molecular imbalances in cardiomyocytes (CMs), such as increased intracellular calcium (Ca2+), dysfunctional SarcoEndoplasmic Reticulum Calcium ATPase (SERCA) activity, and elevated ROS production [3]. Despite several therapeutic approaches targeting dystrophin or utrophin transcription [4], preventing disease progression remains elusive due to poorly understood maladaptive cardiac function often attributed to excessive fibrosis.
Chronic immune system activity, particularly involving T lymphocytes and macrophages (MPs), plays a critical role in DMD pathogenesis, with an inflammatory phenotype predominant [5]. However, the interplay between pro‐ (M1) and anti‐inflammatory (M2) MPs generates a complex inflammatory feedback loop. Chronic activation of M1 MPs also triggers an excessive response from M2 MPs, leading to the overproduction of ECM [6, 7]. This imbalance significantly contributes to cardiac fibrosis, exacerbating the loss of myocardium function [8]. Additionally, membrane instability due to Ca2+ dysregulation (caused by dystrophin absence) damages the cardiac muscle and induces chronic permeabilization to inflammatory infiltrate, exacerbating the massive production of extracellular fluid containing ECM from the fibrotic tissue [9]. Dysfunctional mitophagy, oxidative stress, CM death, and mechanical impairment are also described in dystrophic cardiac tissue [10].
Beyond inflammation, dystrophin serves as a crucial link between motor neurons and skeletal muscles [11]. Its absence in DMD disrupts synaptic structure and function, affecting the cyclic AMP (cAMP) compartmentalization and beta‐adrenoceptors stimulation [12]. Dystrophic mice (mdx mice) exhibit decreased sympathetic innervation density in the heart [2].
This study proposes a novel perspective on DMD's cardiac pathology, focusing on the perturbed communication between the immune system and the neuro‐cardiac junction (NCJ).
Inflammatory MP infiltrates and alterations in proteins controlling synaptic function have been observed in mdx mouse hearts. An excessive release of chondroitin sulfate proteoglycan‐4 (CSPG4) within the infiltrate was identified. This proteoglycan, known to inhibit ocular muscle innervation and to be overexpressed during skeletal muscle regeneration in DMD patients [13], also regulates neuronal networks [14]. Co‐culture experiments with chimeric antigen receptor T lymphocytes (CARTs) targeting CSPG4 demonstrate that MPs produce this proteoglycan, inhibiting axon growth in a 3D model of NCJ via a paracrine mechanism.
Considering that dilated cardiomyopathy in DMD results from tissue denervation and muscle tone loss [15], it is conceivable that cardiac function impairment might stem from the immune system altering stromal proteins controlling the communication with the NCJ. Most DMD studies focus on neuromuscular junctions, overlooking the potentially interesting link between inflammation and NCJ via ECM proteoglycans.
Understanding this interaction could reveal new therapeutic targets and intervention areas, to provide novel insights into DMD pathology and management.
Materials and methods
Ethical approval
All procedures involving living animals were performed in conformity with the experimental protocols approved by the Ministry of Health (Protocol 717/2018 and Protocol 1188/2020). All manipulations were carried out by qualified personnel and under the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. The methods used for euthanasia were isoflurane inhalation (1.2–2.0% in 100% oxygen) or tiletamine/zolazepam (60 mg/kg) and xylazine (5 mg/kg).
Mice and treatments
Three‐ and 10‐month‐old dystrophic (C57BL/10‐mdx) and healthy wild type (WT) mice (C57BL/10) were used. The mdx mice were divided into two groups. The first group (Givinostat) received a daily i.p. injection of givinostat (Giv) dissolved in saline (5 mg/kg/day) for 60 days. The second group (control) received saline.
Cardiac characterization
Echocardiogram
Mice were anesthetized with isoflurane (1.2–2.0% in 100% oxygen) and maintained at 37 °C. Imaging was performed on days 0, 1, 3, 7, 15, 30, and 60 using VisualSonics Vevo 3100 (Fujifilm, Amsterdam, The Netherlands), as previously described [16, 17].
Hemodynamic
Anesthetized mice with isoflurane inhalation. A polyethylene catheter was inserted into the right carotid artery and connected to a transducer to measure left ventricular (LV) systolic and diastolic pressure.
Histological analysis
The animals were anesthetized with tiletamine/zolazepam and xylazine. Hearts were perfused with 50 mm KCl via abdominal aortic cannulation, then excised and either embedded in Tissue‐Tek O.C.T. (Catalogue No. 361603E, VWR International S.r.l, Milan, Italy) or paraffin. Hearts in O.C.T. were dipped in cold isobutane and sectioned at 8 μm using a cryostat. For paraffin sections, hearts were fixed in 10% neutral‐buffered formalin, dehydrated with alcohol, embedded in paraffin, and sectioned into 2‐μm slices using a microtome. Frozen heart tissues were warmed to room temperature (RT) for 20 min, fixed in ice‐cold acetone for 5 min, then washed in PBS. Paraffin sections were deparaffinized in xylene and rehydrated with descending alcohols. Antigen retrieval was done using citrate buffer (0.1 m, pH 6.0). Frozen and paraffin sections were blocked for 30 min at RT with 5% bovine serum albumin (BSA, Catalogue No. A1470, Sigma‐Aldrich, Saint Louis, MO, USA), incubated overnight at 4 °C with primary antibodies: anti‐cardiac troponin T (cTNNT, 1:100, Abcam, ab33589, Cambridge, UK), anti‐Connexin 43 (Cx43, 1:100, Cell Signaling Technology (CST), #3512, Danvers, MA, USA), anti‐tyrosine hydroxylase (TH, 1:100, Sigma‐Aldrich, ab152), anti‐Synapsin 1 (SYN1, 1:100, CST, #6710, Danvers, MA, USA), anti‐MPs F4/80 (F4/80, 1:100, Biorad, MCA497G, Segrate, Milan, Italy), anti‐CSPG4 (1:200, Novus Biologicals, NBP266979 or 1:50, Abingdon, Oxfordshire, UK, Santa Cruz, SC‐80003, Dallas, TX, USA), and anti‐fibronectin 1 (FN1; 1:50, Santa Cruz, SC‐271098) and incubated with fluorescent‐conjugated secondary antibodies (1:1000, Invitrogen, Waltham, MA, USA) for 1 h. Nuclei were counterstained with DAPI (Catalogue No. D1306, Invitrogen). Images were acquired using a confocal microscope TCS SP5 (Leica Microsystems, Wetzlar, Germany). The fibrotic index was determined using Masson's Trichrome assay (Catalogue No. AUS240, Bioptica, Milan, Italy) staining, following the manufacturer's protocol. Scar size was expressed as the ratio of the fibrotic area to the total area. CX43 quantification was performed as described previously [16]. Innervation density was evaluated using TH and SYN1 antibody staining. All images were analyzed using ImageJ [Rasband, W.S. (1997–2015) ImageJ, National Institutes of Health, Bethesda, MD, USA, http://imagej.nih.gov/ij].
Immunofluorescence analysis
MPs were fixed in 4% paraformaldehyde for 10 min, permeabilized with 0.1% TRITON X‐100 (Sigma‐Aldrich) in PBS for 10 min, and blocked in 5% BSA‐PBS for 45 min [18]. Samples were incubated with primary antibody (F4/80, 1:100, or CSPG4, 1:200) at 4 °C overnight, followed by secondary antibodies (1:1000, Invitrogen) for 2 h at RT. Nuclei were counterstained with DAPI. Images were acquired using a high‐resolution spinning disk confocal microscope (CREST XlightV2/VCS) involving processing 100× images with background suppression using ROI‐MaxPI visualization in Nis Elements GA software.
Bone marrow‐derived MP isolation
MPs isolated from bone marrow (femur and tibia) were cultivated (1 × 106) in DMEM (Catalogue No. 11965092, Gibco, Waltham, MA, USA) with 20% FBS (Catalogue No. 10270106, Gibco) 1% L‐glutamine (200 mm, Catalogue No. 25030081, Gibco), 1% P/S (penicillin and streptomycin stock solution 10,000 U/ml, Catalogue No. 15140122, Gibco), 0.5% sodium pyruvate (100 mm, Catalogue No. 11360070, Gibco), 0.1% β‐mercaptoethanol (Catalogue No. 21985023, Gibco), and 20 ng/ml MP‐stimulating growth factor (mouse M‐CSF premium grade, 130‐101‐700, Miltenyi Biotec, Bergisch Gladbach, Germany). After 7 days cells were stimulated with 100 ng/ml LPS (Catalogue No. L2012, Sigma‐Aldrich) or 10 ng/ml IL‐4 (Catalogue No. 130‐097‐757, Miltenyi Biotec) for 24 h to induce M1 and M2 phenotypes, respectively. MPs were analyzed by fluorescence‐activated cell sorting (FACS) or co‐culture experiments with chimeric antigen receptor T cells (CARTs).
3D neuro‐cardiac junction generation
After 7 days differentiated WT and mdx MPs were treated with 40 μg/ml of CSPG4‐blocking antibody (BAb) (Santa Cruz, SC‐80003) for 24 h [19]. Supernatants were collected, centrifuged at 3800 rpm (Eppendorf Centrifuge 5804R, A‐4‐81, Catalogue No. 5805000010), concentrated (VIVASPIN tubes cutoff 30 kDa, Catalogue No. VS0621, Sartorius, Göttingen, Germany), and equilibrated in HEPES 20 mm (1 m, Catalogue No. 15630080, Gibco) and NaCl 150 mm buffer at 3000 rpm (Eppendorf Centrifuge 5804R, A‐4‐81, Catalogue No. 5805000010) at 15 °C. The desalted mixture was loaded into gel filtration column chromatography for isolating chondroitin sulfate proteoglycans (CSPGs) from other protein pools. Eluted fractions with similar protein profiles were analyzed using an ELISA against CSPG4 (Catalogue No. MOEB1952, Assay Genie, Dublin, Ireland). The fractions containing CSPG4 were used for hydrogel functionalization.
Neonatal dorsal root ganglia isolation
The dorsal root ganglia (DRG) were isolated from the vertebral column of WT pups and digested in 1.25 mg/ml collagenase A (Catalogue No. 10103578001, Roche, Basel, Switzerland) and 2.5 mg/ml dispase II (Catalogue No. D4693‐1G, Sigma‐Aldrich) for 15 min at 37 °C. Then the pellet was resuspended in Neurobasal‐A Medium (Catalogue No. 10888022, Thermo Fisher Scientific, Waltham, MA, USA), with B27 (Catalogue No. 108889038, Thermo Fisher Scientific) and 100 ng/ml nerve growth factor (NGF) 2.5S (115AFC60, Sigma‐Aldrich).
Neonatal CM and cardiac fibroblast isolation
Hearts were predigested in 0.1 mg/ml trypsin (Sigma‐Aldrich) overnight at 4 °C, then incubated with 0.5 mg/ml collagenase II (Catalogue No. CLS‐2, Worthington Biochemical, Lakewood, NJ, USA). Cell suspensions were filtered with a 70‐μm strainer, centrifuged at 800 rpm (Eppendorf Centrifuge 5804R, A‐4‐81, Catalogue No. 5805000010) for 5 min at 4 °C, and resuspended in DMEM/M119 (31153‐026, Gibco) medium with 5% FBS, 10% HS, 2 mm L‐glutamine, and 1% P/S. Cells were plated for 3 h to allow nonmyocytes to attach, then the CM‐enriched suspension was plated on 0.1% gelatin‐coated dishes at 1.5 × 104 cells/cm2.
3D NCJs were bio‐assembled using PEG‐FB as a scaffold [20, 21]. Four PEG‐FB bioinks were produced using MPs supernatants: WT, WT + CSPG4 BAb, mdx, and mdx + CSPG4 BAb. 18 × 106 WT CMs/ml and 6 × 106 WT CFs/ml were resuspended in bio‐inks to create 100 μl 3D bulks. Irgacure 2959 (0.01% w/v) was used as a photoinitiator (VLIG00000101, Twin Helix, London, UK), and the bulks were UV cross‐linked (365 nm, 4–5 mW/cm2) for 5 min. After 24 h, 5 × 106 WT DRG were seeded on the top surface of each bulk construct and cultured in a co‐culture medium with 100 ng/ml NGF for 7 days. The 3D NCJ models were fixed with 4% paraformaldehyde and stained with TUBB3 (1:100, Biolegend, San Diego, CA, USA), cTNNT (1:100, Ab8295, Abcam), and ER‐TR7 (1:100, sc‐73355, Santa Cruz).
Co‐culture experiments with CART
Engineered CSPG4 CARTs and control (cCARTs) were provided by Dotti [22]. CARTs were maintained in Iscove's Modified Dulbecco's Medium (Catalogue No. 31980030, Gibco) with 1% P/S, 10% FBS, IL‐15 (5 ng/ml, 130‐095‐762, Miltenyi Biotec), and IL‐7 (10 ng/ml, 130‐095‐361, Miltenyi Biotec) for 5 days. For co‐culture experiments, 1 × 104 MPs and 5 × 103 CF per cm2 were plated in six‐well plates with CSPG4.CARTs or cCART at 2:1 (effector‐to‐target ratio) in serum‐ and interleukin‐free CAR‐T medium. After 3 days, residual MPs and CFs were collected and evaluated by FACS for F4/80 and CD140b expression.
Flow cytometry (FACS)
MPs were incubated at 4 °C for 10 min with the following fluorochrome‐conjugated antibodies: CSPG4/AN2‐PE (1:100, Miltenyi Biotec, 130‐116‐376), F4/80‐APC (1:100, Miltenyi Biotec, 130‐116‐547), CD11b APC/Cyanine7 (1:100, Invitrogen, 130‐113‐803, Carlsbad, CA, USA), CD80 eFluor450 (1:100, Invitrogen, 48‐0801‐82), CD206 Alexa‐647 (1:100, Invitrogen, 53‐2061‐82), CD140b‐APC‐Vio770 (1:100, Miltenyi Biotec, 130‐105‐119). Cell viability was determined using the LIVE/DEAD fixable aqua dead cell stain kit (1:1000, Invitrogen, L34957). Data were acquired using a BD FACSCanto II flow cytometer using BD FACSDiva software (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed using Flowjo version 10.8 (BD Biosciences).
FACSymphony
Hearts were predigested with 2.5 U/ml collagenase type II (Catalogue No. CLS‐2, Worthington Biochemical) for 30 min at 37 °C and then centrifuged at RT for 5 min. A second incubation with 1.5 U/ml collagenase D (Catalogue No. 11088882001, Sigma‐Aldrich) and dispase II (Catalogue No. 22615200, Sigma‐Aldrich) was performed for 60 min. The pellet was filtered through 40‐μm cell strainers and centrifuged at 1200 rpm at RT (Eppendorf Centrifuge 5804R, A‐4‐81, Catalogue No. 5805000010) for 5 min. Cell suspensions were incubated with Fixable stain 780 (Catalogue No. 565388, Thermo Fisher Scientific), washed, and stained with primary antibodies for 10 min on ice: CD45/BUV737 (1:100, BD Biosciences, Catalogue No. 748371, San Jose, CA, USA), CD11b /BUV563 (1:100, BD, Catalogue No. 741242), F4/80/PE‐CF594 (1:100, BD Biosciences, Catalogue No. 565613), CD80/BV615 (1:100, BD Biosciences, Catalogue No. 751328), CD206/Alexa Fluor 488 (1:100, BD Biosciences, Catalogue No. 565250), Ly6C/BV605 (1:100, BD Biosciences, Catalogue No. 563011), Ly6G/BB790 (1:100, custom BD Biosciences), and CSPG4/PE (1:100, Miltenyi Biotec, Catalogue No. 130‐116‐440). Cells counts were acquired using a FACSymphony A5 (BD Biosciences) and analyzed by FlowJo (version 10.9). The FlowJo plugin flowAI was used for quality control and filtering out debris and nonviable cells. A random downsampling to 5000 events per sample focused on live cells. Samples were concatenated into a dataset of 60,000 cells. Dimensionality reduction was performed using the t‐SNE algorithm in FlowJo with default parameters: interaction value of 1000, perplexity value of 30, and the Barnes–Hut gradient algorithm. The gating strategy is detailed in supplementary material, Figure S1F.
Quantitative real‐time PCR
Total RNA from hearts was isolated using TRIZOL reagent (Invitrogen, Life Technologies, 15596026, Carlsbad, CA, USA) following the manufacturer's instructions. Samples were treated with DNase I (Invitrogen, 2358609) following the protocol and the RNA reverse transcribed into cDNA using a TaqMan Reverse Transcription Reagents kit (Applied Biosystems, N8080234, Foster City, CA, USA). Gene expression was evaluated by quantitative real‐time PCR (RT‐qPCR) with SYBR green (SensiFAST SYBR Lo‐ROX kit, Meridian Bioscience, BIO‐94005, Cincinnati, OH, USA) on a Real‐Time PCR Instrument (A28135 A28140, Applied Biosystems Quantstudio 5 real‐time PCR system). The thermal cycling conditions for all primers (supplementary material, Table S2) were as follows: 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Data are presented as relative expression normalized to the reference transcript EIF2A and the WT group as control.
Western blot
Proteins were extracted from tissues using lysis buffer (50 mm TRIS HCl pH 7.5), 0.6 m sucrose (Catalogue No. S0389, Sigma‐Aldrich), 50% glycerol (Catalogue No. G5516, Sigma‐Aldrich), 1% TRITON, 50 mm NaCl, 10 mm NaF, 2 mm sodium orthovanadate (Na3VO4, Catalogue No. 450243 Sigma‐Aldrich), 1 mm PMSF (Catalogue No. 36978, Thermo Scientific, Waltham, MA, USA), 5 mm β‐glycerophosphate (Catalogue No. G9422, Sigma‐Aldrich), 1000X protease inhibitors (Catalogue No. 04693116001, Roche) for 30 min on ice, sonicated, and centrifuged at 12,000 × g for 15 min at 4 °C. Proteins were resolved using SDS‐PAGE, transferred to nitrocellulose membranes (Catalogue No. GE10600079, Amersham Protran, Johns Creek, GA, USA), blocked with 5% nonfat dry milk powder (Catalogue No. 1.15363, Merck Millipore, Billerica, MA, USA) in PBS 1× for 45 min, and incubated at 4 °C overnight with primary antibodies: tyrosine hydroxylase (1:1000, ab152), Synapsin 1 (1:1000, Cell Signaling Technology, #5297), CSPG4 (1:1000, Cell Signaling Technology, #52635), and Vinculin (1:500, V9131, Sigma‐Aldrich), followed by incubation with secondary antibodies (anti‐Rabbit NA934AV, 1:5000; anti‐Mouse LNXa931/AE, 1:10000, GE Healthcare Life Sciences, Havelock, NE, USA) for 1 h at RT. Immunocomplexes were detected by chemiluminescence (ECL kit; Merck Millipore) and analyzed by densitometry using ImageJ.
Statistics
GraphPad Prism Software (version 8.4.2; GraphPad, Boston, MA, USA) was used for graphing and statistical analysis. Data were expressed as mean ± SEM. Significance for two experimental groups was determined using unpaired single‐tail Student's t‐tests, whereas differences among more groups were calculated as one‐way and two‐way ANOVA with multiple comparisons testing (Tukey). p values: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Increased fibrosis and NCJ alterations in dystrophic hearts
Histological analysis of cardiac tissues from young and aged (3 and 10 months, respectively) mdx mice (a DMD model) confirmed severe and progressive ventricular fibrosis, with a higher fibrotic index compared to WTs (Figure 1A). Genes encoding for collagen 1 and 3 (Col1a1, Col3a1) and fibronectin (Fn1), associated with fibrosis cardiac tissue stiffness [23], were significantly upregulated in both young and aged dystrophic mice with respect to controls (Figure 1B). However, Tgfb1 and transcription factors Twist1 and Twist2, involved in collagen production and ECM deposition [24], were significantly increased only in aged dystrophic mice (Figure 1B).
Figure 1.
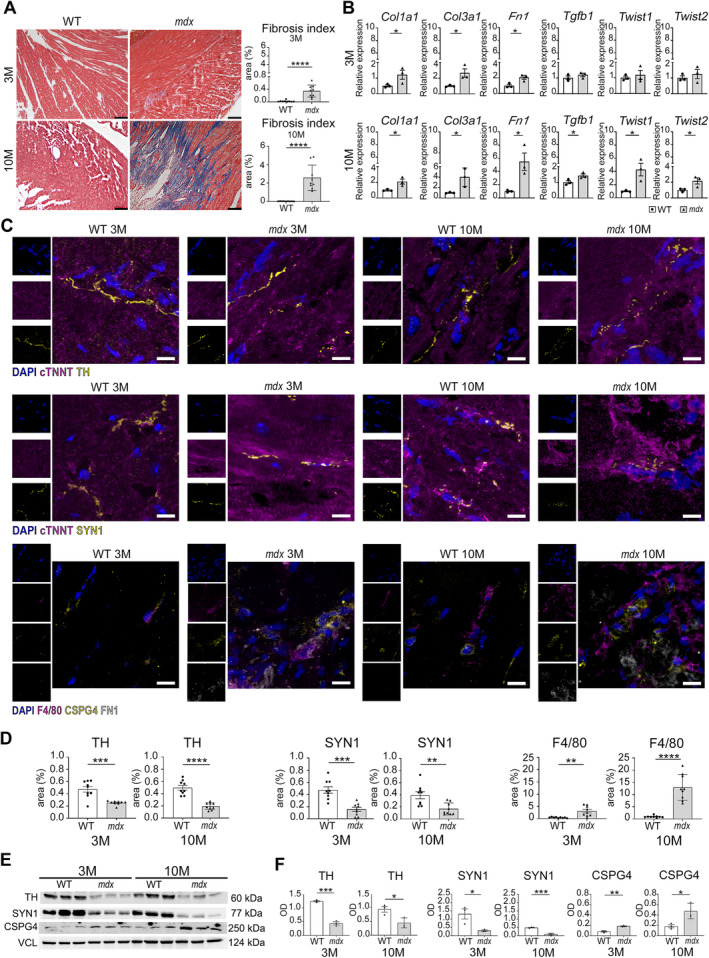
Cardiac fibrosis and innervation. (A) Masson's trichrome staining of young (3‐month‐old) and old (10‐month‐old) WT and mdx mouse hearts. Scale bars, 100 μm. The graph shows the area as a percentage of the fibrosis index (fibrotic area/whole area). N = 3 biological replicates. (B) qPCR for fibrosis‐related transcripts in 3‐ and 10‐month‐old mdx and WT hearts (Col1a1, Col3a1, Fn1, Tgfb1, Twist 1, Twist 2). N = 3 biological replicates. (C) Confocal images for tyrosine hydroxylase (TH), Synapsin 1 (SYN1), MPs (F4/80), proteoglycan CSPG4, and fibronectin 1 (FN1) on myocardium sections of 3‐ and 10‐month‐old mice. The markers TH, SYN1, and CSPG4 are labeled in yellow, cardiac troponin (cTNNT) and F4/80 are in magenta, while FN1 is in white. Scale bars, 10 μm. Nuclei are counterstained with DAPI in blue. (D) Charts indicate positive area expressed as percentage of TH, SYN1, and F4/80 on whole area. N = 3 sections per N = 3 biological replicates. (E and F) Western blot analysis of TH, SYN1, and CSPG4 in mdx and WT hearts of young and old mice. Graphs show optical density (OD) of protein bands normalized to vinculin (VCL). N = 3 biological replicates. Error bars show SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 were calculated using Student's t‐test.
Confocal microscopy revealed a disorganized distribution of synaptic terminals and a decreased percentage of the positive area to TH and SYN1 (markers of dopaminergic neurons and synaptic vesicles, respectively) in mdx mice at 3 and 10 months (Figure 1C,D). Interestingly, the percentage of the positive area for F4/80 (a unique marker of mouse microglia and MPs) significantly and progressively increased in both young and mainly in adult mdx mice (Figure 1C,D). Notably, CSPG4 staining was observed both around and overlapped on F4/80 positive cells within the fibrotic area labeled with Fn1 antibody (Figure 1C). Western blotting analysis confirmed increased CSPG4 and decreased TH and SYN1 in both young and aged mdx mice (Figure 1E,F).
Bone marrow‐derived MPs can produce CSPG4
Given the concurrent presence of MPs and CSPG4 in the cardiac tissue of mdx mice, we investigated whether MPs produced CSPG4. Bone marrow‐derived monocytes (essential for muscle regeneration in DMD and known contributors of CSPG4 [25]) from both WTs and mdx mice were differentiated in vitro into three MP phenotypes (M0, M1, M2) by means of LPS and IL‐4 (supplementary material, Figure S1A,B). CSPG4 expression in these phenotypes was confirmed by FACS analysis and high‐resolution immunofluorescence (supplementary material, Figure S1C–E). Next, we co‐cultured the M0, M1, and M2 populations with the engineered CARTs targeting CSPG4 (CSPG4‐CARTs), designed to exert cytolytic activity against cells expressing CSPG4. The CSPG4‐CARTs showed significantly increased cytolytic activity toward all MP populations (Figure 2A). Cardiac fibroblasts (CFs), known producers of proteoglycans in the cardiac tissue [26], did not reproduce this effect (Figure 2A). Supernatants from WT and aged dystrophic MP‐derived cultures were tested for CSPG4 levels by ELISA, which revealed that old mdx mice MPs excreted higher CSPG4 levels than WTs (Figure 2B).
Figure 2.
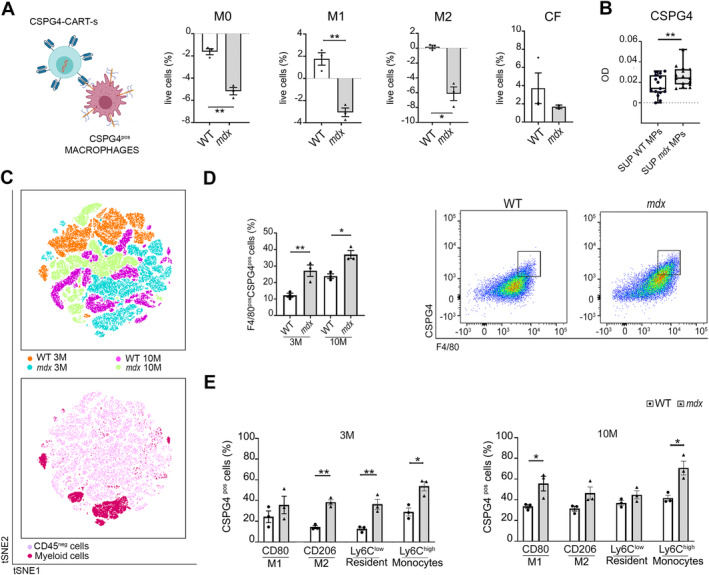
MPs as main producers of CSPG4. (A) Graphic illustration of CSPG4‐CAR‐Ts cells against MPs expressing CSPG4. Created with Biorender.com. Diagrams quantifying the percentage of live cells of bone marrow‐derived MPs of both WT and mdx M0 (nonactivated), M1 (pro‐inflammatory) and M2 (anti‐inflammatory), or CFs after exposure to CSPG4.CARTs compared to cells co‐cultured with control CART (cCART). N = 3 biological replicates. (B) ELISA assay for CSPG4 in supernatants (SUP) isolated from WT and mdx MPs. The chart illustrates the optical density (OD) of CSPG4 in the two groups. Each dot represents the eluted fractions, N = 16. (C) FACSymphony of cardiac tissues. The first chart represents a scatter plot of t‐SNE relative to concatenated total cells from 3‐ or 10‐month‐old WT and mdx hearts. The second chart illustrates the distribution of CD45neg cells (light pink cluster) and myeloid cells (dark pink cluster) in all groups. (D) Graph of FACS analysis relative to number of total MPs (F4/80) positive for CSPG4 in 3‐ or 10‐month‐old WT and mdx hearts. (E) Expression of CSPG4 in subpopulations of MPs: CD80pos (M1), CD206 pos (M2), Ly6Clow (resident), and Ly6Chigh (monocytes) in 3‐ or 10‐month‐old WT and mdx hearts. Error bars show SEM. *p < 0.05, **p < 0.01 calculated using Student's t‐test and one‐way ANOVA.
Additionally, cardiac tissues from young and old WT and mdx mice were analyzed using FACSymphony. The unsupervised analysis was performed on the same number of events per sample (15,000 events for experimental groups) (tSNE scatter plot, Figure 2C, top), representing the distribution of concatenated total cells of all experimental groups (blue and greenish for 3‐ and 10‐month‐old mdx; orange and magenta for 3‐ and 10‐month‐old WT), and showed that the cluster of myeloid cells (dark pink, Figure 2C, bottom) was more enriched between young and old mdx mice compared to WTs.
The deep analysis revealed a higher percentage of total MPs positive for CSPG4 (F4/80pos/CSPG4pos) in mdx mice at both 3 and 10 months of age compared to controls (Figure 2D). A focused analysis of MP subpopulations showed a significant increase in M2 (CD206pos/CSPG4pos), resident MPs (Ly6Clow/CSPG4pos), and recruited monocytes (Ly6Chigh/CSPG4pos) [27] in young dystrophic mice compared to controls (Figure 2E). A nonsignificant trend was observed in the pro‐inflammatory M1 subset (Figure 2E). In aged mice, higher percentages of M1 (CD80high/CSPG4pos) and recruited monocytes (Ly6Chigh/CSPG4pos) were found (Figure 2E).
CSPG4 released by MP‐inhibited axon growth in a 3D model of NCJ
To verify the effect of CSPG4 released by MPs on the biological cross‐talk between the heart and nervous system, we recapitulated in vitro NCJ using 3D multicellular constructs.
Supernatants of WT and mdx MPs, untreated or treated with CSPG4‐blocking antibody (BAb), were employed to reconstitute polyethylene glycol‐fibrinogen (PEG‐FB) biomaterial. Four 3D NCJ models were generated: WT, WT + CSPG4 BAb, mdx, and mdx + CSPG4 BAb. The NCJ consisted of neonatal CMs and CFs isolated from pups, embedded in conditioned PEG‐FB. DRG were plated on the top of the 3D structures (Figure 3A). We quantified the area occupied by TUBB3‐labeled neuron endings (24) within the 3D models. Results showed a significant restoration in the mdx 3D NCJ treated with CSPG4 BAb compared to untreated mdx 3D NCJ (Figure 3C,D). The growth of DRG in the mdx 3D NCJ was inhibited with respect to WT, as expected (Figure 3C,D). The result confirmed that CSPG4 released from dystrophic MPs altered the communication between cardiac tissue and synaptic terminals by paracrine activity.
Figure 3.
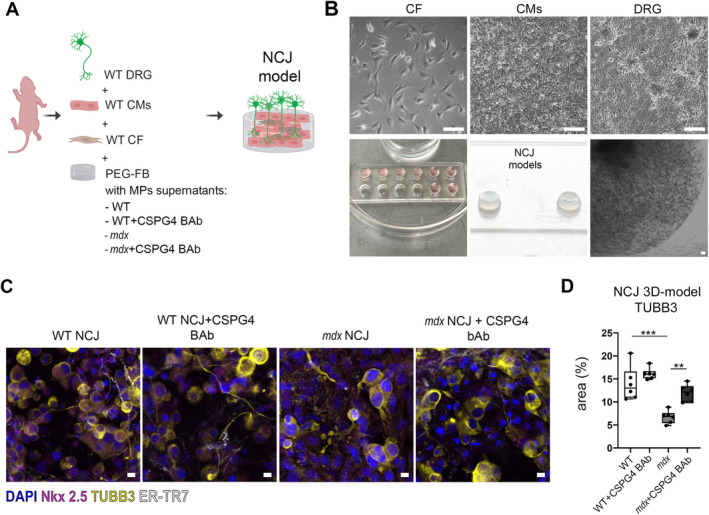
MPs release CSPG4 in media and inhibit axon growth of dorsal root ganglia. (A) Rendering of 3D NCJ generation. The picture was created with Biorender.com and illustrates the composition of the 3D NCJ model: WT dorsal root ganglia (DRG), WT cardiomyocytes (CMs), and WT CFs were encapsulated in PEG‐FB hydrogel conditioned with the supernatants derived from MP cultures of WT, WT + CSPG4 blocking antibody (BAb), mdx, and mdx + CSPG4 (BAb). (B) Brightfield images of CFs, CMs, and DRG after 1 day of culture (upper panel) and 3D constructs of NCJ models after 7 days (lower panel). Scale bars, 100 μm. (C) Confocal images of 3D NCJ models: WT, WT + CSPG4 BAb, mdx, mdx + CSPG4 BAb. DRG are labeled with tubulin beta 3 (TUBB3, in yellow), CMs with cardiac troponin (cTNNT, in magenta), and CFs with fibroblast marker (ER‐TR7, in white). (D) The graph indicates the area (%) occupied by neuron endings labeled by TUBB3 inside the 3D NCJ models. N = 3 biological replicates, N = 2 sections/constructs. Scale bars, 10 μm. **p < 0.01, ***p < 0.001 were determined using one‐way ANOVA.
Givinostat modulates CSPG4 and MP phenotype and enhances cardiac function
As we previously demonstrated, givinostat, a pan‐histone deacetylase inhibitor currently in phase III clinical trial for DMD [28], effectively reduces inflammation and cardiac fibrosis [16]. We investigated whether givinostat also affected CSPG4 in the heart and modulated the MP phenotype. Results showed that givinostat significantly enhanced the percentage of the positive area to both TH and SYN1 in the cardiac tissue of aged mdx mice compared to the untreated dystrophic group (Figure 4A,B). Additionally, there was a significant decrease in the MP‐covered area (F4/80pos) around and overlapping CSPG4 in the fibrotic regions of aged mdx treated with givinostat with respect to the mdx saline group (Figure 4A,B). Staining for TH revealed intact synaptic terminals in WT, unlike disorganized distribution within the fibrotic area in mdx mice overlapping with MPs and CSPG4 (Figure 4A, lower panel). Similar results were observed in young mdx mice with a decrease in F4/80 positive area (supplementary material, Figure S2A,B). FACSymphony showed a significant reduction of the percentage of CSPG4pos MPs positive in cardiac tissues of young and aged mdx mice treated with givinostat compared to controls (supplementary material, Figure S2C).
Figure 4.
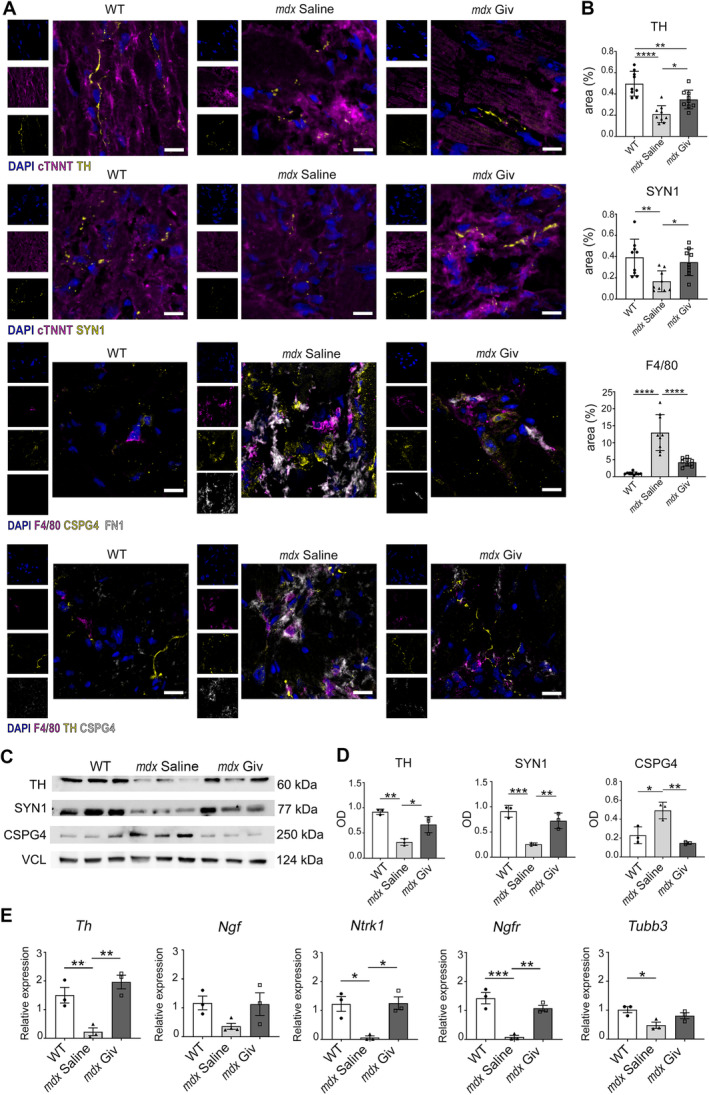
Cardiac innervation in old mdx and WT mice. (A) Immunofluorescence staining for tyrosine hydroxylase (TH), Synapsin 1 (SYN1), total MPs (F4/80) and proteoglycan (CSPG4), fibronectin 1 (FN1) on cardiac sections from 10‐month‐old mice. The markers TH and SYN1 are labeled in yellow, while cardiac troponin (cTNNT) and F4/80 are in magenta, FN1 in white, and CSPG4 in yellow or white. DAPI nuclear counterstaining is in blue. Scale bars, 10 μm. (B) Charts indicate area expressed as percentage of TH, SYN1, and F4/80 (positive area/whole area). N = 3 sections per N = 3 biological replicates. (C and D) Western blot analysis for TH, SYN1, and CSPG4 quantified as optical density (OD) of protein bands normalized to vinculin (VCL). N = 3 biological replicates. (E) qPCR relative to markers for neurotransmitter production (Th), survival (Ngf, Ntrk1, and Ngfr) and axon guidance (Tubb3) in DRG. N = 3 biological replicates. Error bars show SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 were calculated using one‐way ANOVA.
Western blot analysis confirmed these results (Figure 4C,D). Importantly, Th, high‐ and low‐affinity Ngf receptors, neurotrophic receptor tyrosine kinase 1 (Ntrk1), and nerve growth factor receptor (Ngfr) were upregulated in DRG isolated from aged dystrophic mice treated with givinostat (Figure 4E), while Tubb3 levels remained unaltered (Figure 4E).
Connexin‐43 expression, crucial for myocardial gap junction formation and overexpressed in dystrophic pathology [29], was restored in aged mdx animals after givinostat treatment (Figure 5A,B).
Figure 5.
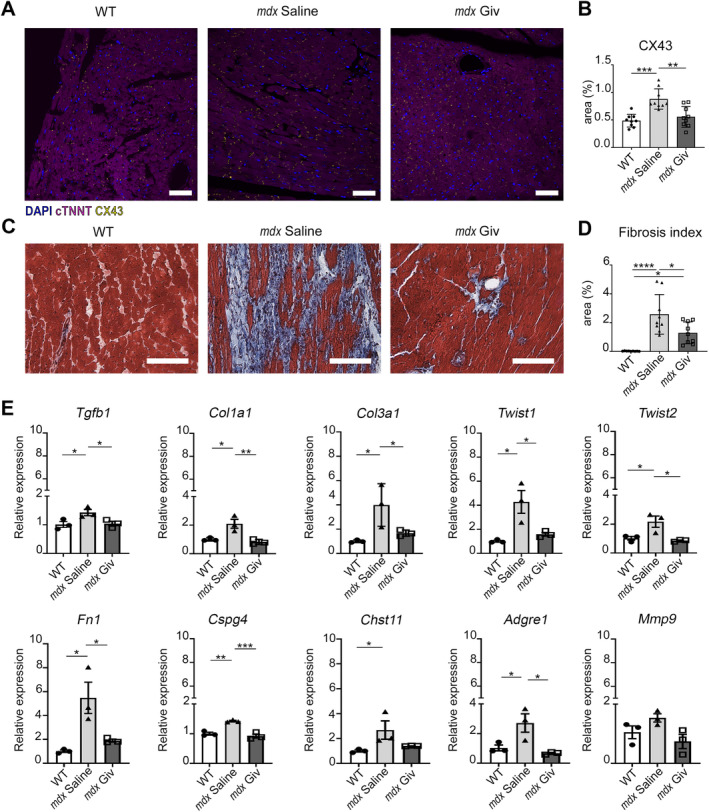
Histological analysis and gene profile of cardiac tissue after treatment with givinostat in old mdx and WT mice. (A) Confocal images of immunostaining for gap junctions (CX43) and cardiomyocytes positive for cardiac troponin (cTNNT), N = 3 sections per N = 3 biological replicates. Scale bars, 50 μm. (B) Charts indicate area as percentage of CX43 (positive area/whole area). (C and D) Representative Masson's trichrome images of WT, mdx saline, and mdx Giv hearts and fibrosis index quantification (fibrotic area/whole area) after 60 days of treatment. Scale bars, 100 μm. N = 3 biological replicates. (E) qPCR of fibrosis‐related genes (Tgfb1, Col1a1, Col3a1, Twist1, Twist2, Fn1), cell surface proteoglycan (Cspg4); the enzyme catalyzes the transfer of sulfate groups on CSPG4 chains (Chst11) and inflammatory markers (Adgre1 and Mmp9). N = 3 biological replicates. Error bars show SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 were calculated using one‐way ANOVA and Student's t‐test.
Cardiac fibrotic area and phenotype significantly reduced (Figure 5C,D), with downregulated transcriptional levels of Tgfb1, Col1a1, Col3a1, Twist1, Twist2 Fn1, Cspg4, Chst11 (the enzyme catalyzing the transfer of sulfate groups on CSPG4 chains), Adgre1, and Mmp9 (Figure 5E). Givinostat had no such effect in young mice (supplementary material, Figure S3A,B).
Echocardiographic analysis showed functional heart recovery with significant enhancement of the fractional shortening (FS) and ejection fraction (EF) in aged mdx mice upon givinostat treatment (Figure 6A,B). Furthermore, left ventricular end‐diastolic (LVEDV) and stroke volume (LVESV) indicated cardiac remodeling changes (supplementary material, Table S1). Hemodynamic analysis confirmed improved systolic function in the mdx givinostat group, with recovery of end‐systolic pressure (P es), maximum pressure (P max), and maximum and minimum rate of pressure change (Max Dp/Dt and Min Dp/Dt) (Figure 6C). No significant changes in body weight, heart weight, or ratio of heart weight to tibia length were found between givinostat‐treated and mdx saline groups (supplementary material, Figure S4A).
Figure 6.
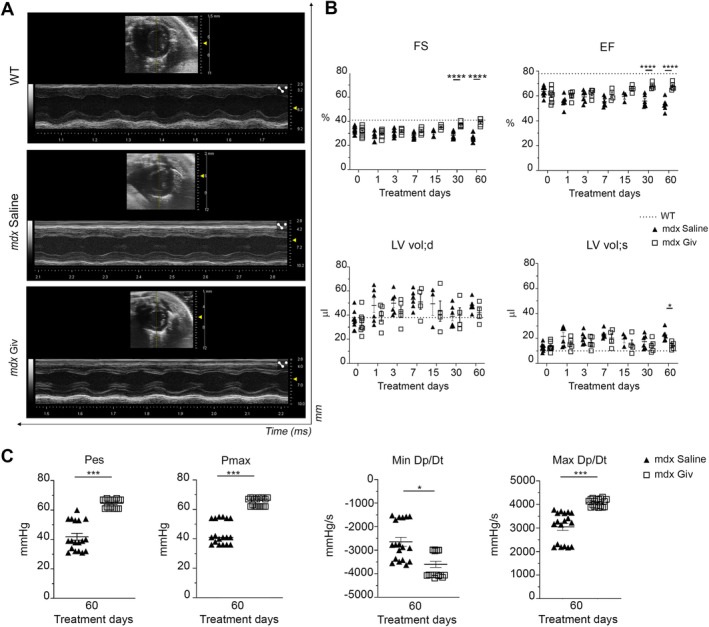
Impact of givinostat on dystrophic cardiomyopathy. (A) Echocardiographic M‐mode images of parasternal long axis view of anterior and posterior walls of left ventricle of WT, mdx saline, and mdx Giv (Vevo 3100, VisualSonics). (B) Echocardiographic parameters: fractional shortening (FS), ejection fraction (EF), and left ventricular end‐diastolic/systolic volume (LV vol;d LV vol;s) in 10‐month‐old mice. The x‐axis shows givinostat or saline treatment on different days (0, 1, 3, 7, 30, and 60). A range of a minimum of three to a maximum of six biological replicates. (C) Hemodynamic analysis including end‐systolic pressure (P es), maximum pressure (P max), minimum rate of pressure change (Min Dp/Dt), and maximum (Max Dp/Dt). N = 3 biological replicates, N = 6 measurements/mouse. Error bars show SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 were calculated using two‐way ANOVA.
Discussion
CSPG4 is expressed at peripheral nerves and triggers cytoskeletal rearrangements [30]. The role of CSPG4 in the cardiac tissue of DMD has not been previously explored. Only a few studies have reported the upregulated expression of CSPG4 in the sarcolemma and the neuromuscular junction of human postnatal skeletal muscle in DMD and other severe dystrophies [31, 32, 33].
CSPG4 is also a marker for mesenchymal progenitor cells involved in fibroblast/adipogenic differentiation in skeletal muscle [34]. The accumulation of CSPG4 is recognized to inhibit the progression of nerve endings within the heart. However, our study is the first to show CSPG4 overproduction at NCJ in mdx mice cardiac tissue, with CSPG4 protein levels increasing with age. Older mdx mice show higher CSPG4 production than their young counterparts, whereas in human dystrophic skeletal muscles, CSPG4 expression inversely correlates with age [33], suggesting differences between skeletal and cardiac tissue and between murine and human models.
Our data confirm that MP‐driven inflammatory processes are dysregulated in DMD: both pro‐ and anti‐inflammatory MP responses are imbalanced, dynamic, and disease‐stage‐specific [35]. MPs are the most important cell population found altered in human dystrophic skeletal muscles of human patients and mdx mice. The role of MP subsets is still debated and tissue‐dependent due to different signals within the inflammatory microenvironment, polarizing the inflammatory response. Our study focused on cardiac MPs modulating CSPG4 and their contribution to fibrosis. While CSPG4 is expressed in all MP populations, our data highlight that the percentages of different subsets positive for CSPG4 in dystrophic mice hearts vary with disease progression. In young dystrophic mice, all subsets contribute to CSPG4 expression, coherent with the necrotic phase of the disease [36] as the cardiac tissue attempts to accelerate repair. In aged mice (late stage of the disease for the mdx model) [37], we observed a chronic inflammatory phase characterized by a higher percentage of the pro‐inflammatory M1 MPs and a higher percentage of Ly6Chigh marker, indicating persistent recruitment of bone marrow‐derived monocytes. This suggests a chronic progression of the inflammatory process, driven by MPs in early disease stages, fostering progressive accumulation of CSPG4, unfavorable ECM remodeling, and disrupted NCJ communication. As DMD progresses, the pro‐inflammatory and recruiting MP phenotype dominates, negatively impacting cardiac function and generating dilated cardiomyopathy, a delayed phenomenon in mdx mice [38, 39]. Notably, our data indicate that the dystrophic MPs' ability to excrete CSPG4 has a paracrine effect on the cardiac microenvironment, reducing synaptic terminals in the 3D model of NCJ. However, it remains unclear whether denervation precedes dilated cardiomyopathy and fibrosis or whether these events occur concurrently. Denervation likely follows inflammatory damage, with CSPG4 as the biological mediator. Accordingly, after ischemia‐reperfusion injury, the infarcted area shows sympathetic denervation, increased CSPG4, and inhibited axon growth [40]. In other contexts, pathological tissue inflammation upregulates MPs expressing CSPG4 [41].
Previously, we demonstrated that givinostat reduced inflammation and fibrosis in a mouse model of acute myocardial infarction, improving cardiac performance [16]. The impact of givinostat on dystrophic dilated cardiomyopathy is still unknown. We have already shown that givinostat has a broad‐spectrum and anti‐inflammatory effect with anti‐fibrotic consequences [16]. The present study demonstrates that by decreasing MP‐driven inflammation, givinostat also reduces the content of CSPG4 in dystrophic cardiac tissue. The effect of givinostat on the NCJ is likely indirect, fostering a favorable microenvironment for restoring cardiac sympathetic innervation, crucial for cardiac repair, regeneration, and function [42].
We observed cardiac function improvement within 30–60 days, consistent with the mdx preclinical model pathophysiology [43]. We speculate that givinostat might exert a long‐term stabilizing action on cardiac function, parallel to a sustained reduction of cardiac inflammation and fibrosis over time.
Conclusions
We cannot exclude that additional partners such as microvasculature‐associated vascular mural cells [44, 45] and adult cardiac pericytes [46] may contribute to CSPG4 release, beyond MPs in dystrophic cardiac tissue, contributing to ECM deposition.
Our data offer insights into the interplay among inflammation, fibrosis, and the nervous system in cardiomyopathy, identifying new pharmacological targets.
Author contributions statement
RR conceptualized the study and designed the experiments. RR and CB acquired funding for the project and supervised all generation, collection, and analyses of research data. MM performed all the experiments, analyzed the data and prepared the figures and tables. RR, CB, EDF and MM wrote and edited the manuscript. FM, MC, RZ, VDP, NF, CP, MT and MB conducted the in vitro experiments and supported in data analysis. AS, SM, FM and MC performed the in vivo experiments. EL and GD provided CAR‐T cells. MGC and MC conducted experiments using flow cytometry and analyzed the data. CC carried out high‐resolution immunofluorescence on cells. DS supplied biomaterial for the 3D culture. DDS, FB, PM, AC, YT, CL and EDF provided experimental support. All authors approved the final version of the manuscript.
Supporting information
Figure S1. FACS analysis of bone marrow‐derived MPs from and FACSymphony gating strategy
Figure S2. Cardiac innervation in young mice
Figure S3. Cardiac fibrosis in young mice
Figure S4. Body weight, heart weight, and heart weight to tibia length ratio and evaluation of dystrophin expression
Table S1. Echocardiographic parameters
Table S2. List of primers used for mouse genes
Acknowledgements
This work was supported by Fondazione Regionale per la Ricerca Biomedica (Regione Lombardia), Project ID 1731651; Italian Ministry of Health, Project starting grant code SG‐2019‐12368961; Duchenne Parent Project ONLUS (Italy); ITALFARMACO; Fondazione ROCHE (nonconditional contribution). The authors thank Fondazione Roma for the VisualSonics Vevo 3100. EDF is supported by the grant Progetto ECS 0000024 Rome Technopole, CUP B83C2200282000820006, PNRR Missione 4 Componente 2 Investimento 1.5, funded by the European Union—Next Generation EU.
No conflicts of interest were declared.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Nowak KJ, Davies KE. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep 2004; 5: 872–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lombardi L, Stefano MED, Paggi P. Components of the NGF signaling complex are altered in mdx mouse superior cervical ganglion and its target organs. Neurobiol Dis 2008; 32: 402–411. [DOI] [PubMed] [Google Scholar]
- 3. Shannon TR. Ryanodine receptor Ca2+ sensitivity and excitation‐contraction coupling in muscular dystrophy and heart failure: similar and yet different. Am J Physiol Heart Circ Physiol 2009; 297: H1965–H1966. [DOI] [PubMed] [Google Scholar]
- 4. Canonico F, Chirivi M, Maiullari F, et al. Focus on the road to modelling cardiomyopathy in muscular dystrophy. Cardiovasc Res 2022; 118: 1872–1884. [DOI] [PubMed] [Google Scholar]
- 5. Tripodi L, Villa C, Molinaro D, et al. The immune system in Duchenne muscular dystrophy pathogenesis. Biomedicines 2021; 9: 1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kharraz Y, Guerra J, Pessina P, et al. Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed Res Int 2014; 2014: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tulangekar A, Sztal TE. Inflammation in Duchenne muscular dystrophy‐exploring the role of neutrophils in muscle damage and regeneration. Biomedicines 2021; 9: 1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gallardo FS, Córdova‐Casanova A, Brandan E. The linkage between inflammation and fibrosis in muscular dystrophies: the axis autotaxin‐lysophosphatidic acid as a new therapeutic target? J Cell Commun Signal 2021; 15: 317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klingler W, Jurkat‐Rott K, Lehmann‐Horn F, et al. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol 2012; 31: 184–195. [PMC free article] [PubMed] [Google Scholar]
- 10. Bez Batti Angulski A, Hosny N, Cohen H, et al. Duchenne muscular dystrophy: disease mechanism and therapeutic strategies. Front Physiol 2023; 14: 1183101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rüdolf R, Khan MM, Labeit S, et al. Degeneration of neuromuscular junction in age and dystrophy. Front Aging Neurosci 2014; 6: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brescia M, Chao Y‐C, Koschinski A, et al. Multi‐compartment, early disruption of cGMP and cAMP signalling in cardiac myocytes from the mdx model of Duchenne muscular dystrophy. Int J Mol Sci 2020; 21: 7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ilieva KM, Cheung A, Mele S, et al. Chondroitin sulfate proteoglycan 4 and its potential As an antibody immunotherapy target across different tumor types. Front Immunol 2017; 8: 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sakry D, Neitz A, Singh J, et al. Oligodendrocyte precursor cells modulate the neuronal network by activity‐dependent ectodomain cleavage of glial NG2. PLoS Biol 2014; 12: e1001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ng SY, Ljubicic V. Recent insights into neuromuscular junction biology in Duchenne muscular dystrophy: impacts, challenges, and opportunities. EBioMedicine 2020; 61: 103032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Milan M, Pace V, Maiullari F, et al. Givinostat reduces adverse cardiac remodeling through regulating fibroblasts activation. Cell Death Dis 2018; 9: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pegoli G, Milan M, Manti PG, et al. Role of Cdkn2a in the Emery‐Dreifuss muscular dystrophy cardiac phenotype. Biomolecules 2021; 11: 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Paolis V, Maiullari F, Chirivì M, et al. Unusual association of NF‐κB components in tumor‐associated macrophages (TAMs) promotes HSPG2‐mediated immune‐escaping mechanism in breast cancer. Int J Mol Sci 2022; 23: 7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sardone F, Santi S, Tagliavini F, et al. Collagen VI‐NG2 axis in human tendon fibroblasts under conditions mimicking injury response. Matrix Biol 2016; 55: 90–105. [DOI] [PubMed] [Google Scholar]
- 20. Chirivì M, Maiullari F, Milan M, et al. Tumor extracellular matrix stiffness promptly modulates the phenotype and gene expression of infiltrating T lymphocytes. Int J Mol Sci 2021; 22: 5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maiullari F, Costantini M, Milan M, et al. A multi‐cellular 3D bioprinting approach for vascularized heart tissue engineering based on HUVECs and iPSC‐derived cardiomyocytes. Sci Rep 2018; 8: 13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pellegatta S, Savoldo B, Ianni ND, et al. Constitutive and TNFα‐inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: implications for CAR‐T cell therapy. Sci Transl Med 2018; 10: eaao2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cowling RT, Kupsky D, Kahn AM, et al. Mechanisms of cardiac collagen deposition in experimental models and human disease. Transl Res 2019; 209: 138–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu J, Lamouille S, Derynck R. TGF‐beta‐induced epithelial to mesenchymal transition. Cell Res 2009; 19: 156–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Y, Hammel G, Shi M, et al. Myelin debris stimulates NG2/CSPG4 expression in bone marrow‐derived macrophages in the injured spinal cord. Front Cell Neurosci 2021; 15: 651827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hall C, Gehmlich K, Denning C, et al. Complex relationship between cardiac fibroblasts and cardiomyocytes in health and disease. J Am Heart Assoc 2021; 10: e019338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang P, Liu L, Sun L, et al. Immunological feature and transcriptional signaling of Ly6C monocyte subsets from transcriptome analysis in control and Hyperhomocysteinemic mice. Front Immunol 2021; 12: 632333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mercuri E, Vilchez JJ, Boespflug‐Tanguy O, et al. Safety and efficacy of givinostat in boys with Duchenne muscular dystrophy (EPIDYS): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Neurol 2024; 23: 393–403. [DOI] [PubMed] [Google Scholar]
- 29. Gonzalez JP, Ramachandran J, Himelman E, et al. Normalization of connexin 43 protein levels prevents cellular and functional signs of dystrophic cardiomyopathy in mice. Neuromuscul Disord 2018; 28: 361–372. [DOI] [PubMed] [Google Scholar]
- 30. Nicolosi PA, Dallatomasina A, Perris R. Theranostic impact of NG2/CSPG4 proteoglycan in cancer. Theranostics 2015; 5: 530–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maciej‐Hulme ML, Melrose J, Farrugia BL. Arthritis and Duchenne muscular dystrophy: the role of chondroitin sulfate and its associated proteoglycans in disease pathology and as a diagnostic marker. Am J Physiol Cell Physiol 2023; 324: C142–C152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blake MR, Parrish DC, Staffenson MA, et al. Chondroitin sulfate proteoglycan 4,6 sulfation regulates sympathetic nerve regeneration after myocardial infarction. Elife 2022; 11: e78387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Petrini S, Tessa A, Carrozzo R, et al. Human melanoma/NG2 chondroitin sulfate proteoglycan is expressed in the sarcolemma of postnatal human skeletal myofibers. Abnormal expression in merosin‐negative and Duchenne muscular dystrophies. Mol Cell Neurosci 2003; 23: 219–231. [DOI] [PubMed] [Google Scholar]
- 34. Takeuchi S, Nakano S‐I, Nakamura K, et al. Roles of chondroitin sulfate proteoglycan 4 in fibrogenic/adipogenic differentiation in skeletal muscle tissues. Exp Cell Res 2016; 347: 367–377. [DOI] [PubMed] [Google Scholar]
- 35. Petrof BJ. Macrophage plasticity in Duchenne muscular dystrophy: a nexus of pathological remodelling with therapeutic implications. J Physiol 2022; 600: 3455–3464. [DOI] [PubMed] [Google Scholar]
- 36. Morotti M, Garofalo S, Cocozza G, et al. Muscle damage in dystrophic mdx mice is influenced by the activity of Ca2+‐activated KCa3.1 channels. Life (Basel) 2022; 12: 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li W, Liu W, Zhong J, et al. Early manifestation of alteration in cardiac function in dystrophin deficient mdx mouse using 3D CMR tagging. J Cardiovasc Magn Reson 2009; 11: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen G, Jiang H, Yao Y, et al. Macrophage, a potential targeted therapeutic immune cell for cardiomyopathy. Front Cell Dev Biol 2022; 10: 908790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Quinlan JG, Hahn HS, Wong BL, et al. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord 2004; 14: 491–496. [DOI] [PubMed] [Google Scholar]
- 40. Gardner RT, Habecker BA. Infarct‐derived chondroitin sulfate proteoglycans prevent sympathetic reinnervation after cardiac ischemia‐reperfusion injury. J Neurosci 2013; 33: 7175–7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stallcup WB, You W, Kucharova K, et al. NG2 proteoglycan‐dependent contributions of pericytes and macrophages to brain tumor vascularization and progression. Microcirculation 2016; 23: 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. White IA, Gordon J, Balkan W, et al. Sympathetic reinnervation is required for mammalian cardiac regeneration. Circ Res 2015; 117: 990–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Consalvi S, Mozzetta C, Bettica P, et al. Preclinical studies in the mdx mouse model of Duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med 2013; 19: 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stallcup WB. The NG2 proteoglycan in pericyte biology. In Pericyte Biology ‐ Novel Concepts Advances in Experimental Medicine and Biology (Vol. 1109), Birbrair A (ed). Springer International Publishing: Cham, 2018; 5–19. [DOI] [PubMed] [Google Scholar]
- 45. Trembley MA, Velasquez LS, De Mesy Bentley KL, et al. Myocardin‐related transcription factors control the motility of epicardium‐derived cells and the maturation of coronary vessels. Development 2015; 142: 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Quijada P, Park S, Zhao P, et al. Cardiac pericytes mediate the remodeling response to myocardial infarction. J Clin Invest 2023; 133: e162188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. FACS analysis of bone marrow‐derived MPs from and FACSymphony gating strategy
Figure S2. Cardiac innervation in young mice
Figure S3. Cardiac fibrosis in young mice
Figure S4. Body weight, heart weight, and heart weight to tibia length ratio and evaluation of dystrophin expression
Table S1. Echocardiographic parameters
Table S2. List of primers used for mouse genes
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.