

Abstract
Patients with familial adenomatous polyposis (FAP) harbor mutations in the APC gene and will develop adenoma and early colorectal cancer. There is no validated treatment, and animal models are not sufficient to study FAP. Our aim was to investigate the early events associated with FAP using the intestinal organoid model in a single‐center study using biopsies from nonadenomatous and adenomatous colonic mucosa of FAP patients and from healthy controls (HCs). We analyzed intestinal stem cell (ISC) activity and regulation through organoid development and expression of mRNA and proteins, as well as within colonic crypts. We used several compounds to regulate the signaling pathways controlling ISCs, such as WNT, EGFR, PI3K‐AKT, TGF‐β, yes‐associated protein (YAP), and protease‐activated receptors. In addition to their high proliferative capacity, nonadenomatous and adenomatous organoids were characterized by cysts and cysts with buds, respectively, suggesting abnormal maturation. Adenomatous organoids were enriched in the stem cell marker LGR5 and dependent on EGF and TGF‐β for their growth. Downstream of EGFR, AKT, β‐catenin, and YAP were found to be activated in the adenomatous organoids. While the p110β isoform of PI3K was predominant in adenomatous organoids and essential for their growth, p110α was associated with the immature state of nonadenomatous organoids. We conclude that organoids offer a relevant model for studying FAP, and this work highlights abnormal behaviors of immature cells in both nonadenomatous and adenomatous mucosa of FAP patients, which could be targeted therapeutically. © 2024 The Author(s). The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: intestinal polyposis, intestinal stem cells, intestinal organoids, EGF, PI3K
Introduction
Patients with familial adenomatous polyposis (FAP) harbor mutations in the adenomatous polyposis coli (APC) gene, with an autosomal‐dominant inheritance. They will develop adenomatous polyps in the colon during their second decade and colorectal cancer before the age of 40. Since there is no treatment, they undergo a colonoscopy follow‐up to perform a prophylactic colectomy when the risk of cancer will be high, that is, in the presence of high‐grade dysplasia, in situ carcinoma, and large and/or hundreds of polyps [1]. No model adequately reproduces human FAP. Apc‐deficient mice develop polyps in the small bowel instead of the colon, as well as with microadenoma, delayed adenocarcinoma, and few metastases [2, 3]. Organoids have been used to study combinatorial oncogenic transformation in the colon (i.e. APC, P53, KRAS mutations) and to perform genetic/epigenetic or pharmacological comparisons between healthy controls (HCs) and FAP patients [4, 5, 6, 7, 8]. However, no published organoid model recapitulates the functional and morphological characteristics of FAP pathology, in particular differences between adenomatous and nonadenomatous mucosa. Thus, colonic organoids from FAP patients could be useful to improve FAP knowledge and to identify effective drug treatments.
The organoid culture is based on intestinal stem cell (ISC) properties, mainly controlled by the Wnt/APC/β‐catenin pathway and requires several growth factors, such as WNT3a, EGF, and R‐spondin [9, 10]. There are two kinds of ISCs, located at the bottom of the crypt, the crypt base columnar (CBC) LGR5+ (leucine‐rich repeat‐containing G‐protein coupled receptor 5) cells and the cells located at or near the +4 position (+4 ISC) having high phenotypic plasticity [11]. CBCs are highly proliferative, while +4 ISCs exhibit slow proliferation or quiescence and are more resistant to injury. ISC proliferation and differentiation are controlled by Wnt and EGF signaling [12]. Local gradients result in the control of CBC proliferation by Wnt/APC/β‐catenin pathway and progenitors located near the +4 position by EGFR signaling [13].
The transition between CBCs and progenitors of absorptive or secretory differentiated intestinal epithelial cells requires a controlled balance between Wnt and EGF signaling [12]. Such cross‐regulation may involve the transcriptional co‐activator yes‐associated protein (YAP), involved in the coordination of cell adhesion and proliferation [14]. Several signaling pathways, including phosphoinositide 3‐kinase (PI3K)‐AKT‐mTOR and Ras‐Raf‐MEK‐ERK, can be involved downstream of EGFR and require tight control by phosphatases PTEN and DUSP6, respectively [15, 16, 17]. Interestingly, these pathways have been implicated in epithelial regeneration following injury, involving the production of growth factors, morphogens, and their regulators (EGF, WNT, TGF‐β, R‐spondin) within the crypt microenvironment [18]. The pathophysiology of FAP involves a supercompetitor phenotype of APC‐mutant ISCs, which inhibits the growth of normal ISCs by secreting Wnt antagonists while stimulating mutant cells through Wnt and EGF signaling [19]. Indeed, one study demonstrated the necessity of EGFR signaling in the development of adenomas in Apc Min mice [20].
Studies have reported that APC inactivation and EGFR signaling promote the expression of the cyclooxygenase COX‐2, which contributes to the development of adenoma and colorectal cancer [21]. COX‐2 inhibitors (sulindac or celecoxib) were effective, but they are not recommended due to their side effects. Thus, new drugs for FAP chemoprevention should have a plausible mechanism of action, be well tolerated, and maintain their effectiveness [22]. Several drugs have been tested or are currently under investigation in the context of FAP (https://clinicaltrials.gov), inhibiting Wnt/APC/β‐catenin (pyrvinium, azithromycin), EGFR (erlotinib), mTOR (rapamycin), or the metabolism of cancer stem cells (eflornithine, niclosamide, metformin). However, clinical trials indicate that the efficacy of these treatments may be limited to particular segments of the gut, and their potential long‐term adverse effects remain unknown.
Studies with organoids hold the potential to reveal new targets for preventing the carcinogenic transformation of ISCs and to perform drug testing [23]. One group analyzed the gene expression and epigenetic profile of colonic organoids from several FAP patients [6, 7]. FAP organoids were obtained from nonadenomatous tissue and these studies have unveiled significant genetic and epigenetic disparities when contrasted with organoids from healthy individuals. Here, our objective was to evaluate the utility of human colonic organoids to study the early events that occur in FAP pathophysiology and to identify therapeutic targets. We designed a trial using colonoscopic biopsies from both adenomatous (A) and nonadenomatous (NA) areas of FAP patients and from HCs. We characterized a new structure, a cyst with buds, representative of adenoma in vitro. Notably, EGF is important, while its deprivation led to A‐organoid mortality and NA‐organoid maturation. Moreover, we found differences downstream of EGFR involving MAPK and PI3K‐AKT pathways, as well as TGF‐β.
Materials and methods
Patients
Colonic biopsies were obtained at Toulouse University Hospital, France, from patients included in the protocol NCT02874365, approved by a French ethics committee (the South‐West and Oversea IV ethics committee). The patients and/or their parents provided an inform consent. During colonoscopy, four biopsies from all nonadenomatous and adenomatous tissues of the FAP patients or HCs. The main characteristics of the patients are shown in supplementary material, Table S1.
Organoid culture experiments
We adapted the protocol of Jung and Sato [9, 10]. Biopsies were incubated for 4 × 5 min in antibiotics and antifungal solutions, then incubated in 3 × 5 min in baths of dithiothreitol (10 mm, sterile solution in PBS) at room temperature. Then the mucosal pieces were incubated in EDTA, 8 mm, for 1 h at 4 °C with gentle agitation. The supernatant was replaced by 10 ml of PBS. Crypt isolation required strong manual agitation; 2 min at 40× g centrifugation at 4 °C within 5% FCS; three washes in the base medium (advanced DMEM/F12 supplemented with HEPES 10 mm, Glutamax 2 mm, and 5% FCS). Finally, the pellet was resuspended in 5 ml in order to count the number of crypts.
The crypts were resuspended in a volume of Matrigel hESC‐qualified Matrix (Corning, Amsterdam, the Netherlands) at 4 °C in order to obtain a concentration of 50 crypts in 25 μl, which was deposited in wells of 48‐well plates, previously heated to 37 °C. For immunostaining, 15‐well Ibidi plates (CliniSciences, Nanterre, France) were used in which 10 μl of Matrigel containing 20 crypts was seeded. The Matrigel was polymerized for 20 min at 37 °C, then the day (D) 0 complete medium was added (supplementary material, Table S2), which is a mixture of DMEM/F12 supplemented with 10 mm HEPES, 2 mm Glutamax, 1× B‐27 without vitamin A, 1× N2, 10 mm nicotinamide, 1 mm N‐acetylcysteine, 10 nm PGE2, 50 ng/ml h‐EGF, 100 ng/ml h‐Noggin, 1 μg/ml h‐R‐spondin 1, 10 nm gastrin, 10 μm SB202190 (p38MAPK inhibitor), and 0.5 μm LY2157299 (TGF‐βR inhibitor). Recombinant WNT3a was added to HC organoid culture only (supplementary material, Table S2), and in some experiments, WNT3a‐conditioned medium from subcutaneous connective tissue transfected with a WNT3a expression vector (ATCC®, L WNT‐3a, n°CRL‐2648) was added for both FAP and HC organoid cultures. The medium was changed every 2–3 days for maintenance medium, i.e. the D0 complete medium without N‐acetylcysteine and LY2157299 (supplementary material, Table S2, 250 μl/well in 48‐well plates, 50 μl/well in Ibidi plates).
The growth and morphology of organoids were followed every 2 days from D2 to D9 under a wide‐field transmission microscope (Apotome Zeiss, 10× lens; Carl Zeiss AG, Jena, Germany). The size (largest diameter) of the organoids and the width of their epithelial monolayer were assessed using ImageJ (https://imagej.net/ij/, U.S. National Institutes of Health, Bethesda, MD) Cystic/budding/dead structures were counted manually using brightfield microscopy.
Immunostaining
Organoids were fixed at the end of culture with 3.7% paraformaldehyde for 40 min. Then the permeabilization of structures was performed with 0.5% Triton x100 (in HBSS) for 40 min followed by blocking of nonspecific antigenic sites with 3% BSA (in HBSS) for 1.5 h. Wells were washed and incubated with primary antibodies at 4 °C overnight. Secondary antibodies were incubated for 1 h at 37 °C. Nuclei and actin were labeled respectively using DAPI (Thermo Fisher Scientific, Illkirch, France, ref. D21490, 1/100) and phalloidin Alexa Fluor‐647 (Thermo Fisher Scientific ref. A22287, diluted to 1/500 in the DAPI solution) was performed (incubation for 20 min at room temperature). After extensive washing, a drop of liquid Vectashield mounting medium was applied. The antibodies used are listed in supplementary material, Table S3.
The slides were examined using a Zeiss 710 confocal microscope with 20× and 63× lenses. For each experiment, control wells were incubated under the same conditions, without any antibody (autofluorescence control) and with secondary antibodies alone. Acquired images (single plane at the larger diameter of each organoid) were analyzed and fluorescence quantified with ImageJ software.
Proteomic screening, western blotting, and transcriptomic analysis
At the end of the organoid culture, the medium was replaced by 250 μl Cell Recovery Solution (Corning Catalogue No. 354253) and 48‐well plates were put on ice for 45 min. After dissolution of the Matrigel, organoids from 10 wells of the same assay were collected by pipetting and mixed in 10 ml of the base medium (DMEM/F12 supplemented with HEPES 10 mm, Glutamax 2 mm) and centrifuged for 5 min at 40× g at 4 °C. The organoid pellet was resuspended in 350 μl of lysis buffer RP1 (Macherey‐Nagel, Hoerdt, France, ref. 740934.50) and stored at −80 °C until protein and RNA were extracted using a Macherey‐Nagel kit (Macherey‐Nagel, ref. 740933.250).
Protein expression of different EGF pathway actors was quantified using the PathScan® EGFR Signaling Antibody Array Kit (Cell Signaling Technology, Catalogue No. 12622, Ozyme, Saint‐Quentin‐Yvelines, France). The chemiluminescent reaction was measured using a ChemiDoc™ XRS (Bio‐Rad, Marnes‐la‐Coquette, France) and presented as heatmap (GraphPad Prism, Boston, MA, USA). Western blots for Pthr202/tyr204‐ERK1/2, total ERK, and actin were performed as follows: extracted proteins were quantified and conserved in Laemmli sample buffer before boiling and migration on a precast gel stain‐free Any Kd™ (Bio‐Rad Catalogue No. 4568125). After transfer to nitrocellulose membrane (membrane Hybond C‐super; Merck Millipore, Molsheim, France) and blocking of nonspecific sites with PBS containing 5% fat‐free milk powder and 1% BSA, membranes were probed overnight at 4 °C with appropriate antibodies (supplementary material, Table S3) in Tris‐buffered saline supplemented with 5% BSA and 0.1% Tween 20. After incubation for 1 h at room temperature with either anti‐mouse or anti‐rabbit IgG antibody coupled to horseradish peroxidase (supplementary material, Table S3), detection was achieved using a chemiluminescent substrate (ECL Cytiva; Thermo Fisher Scientific). Images were captured and analyzed using Image Lab software from Bio‐Rad.
The quantity and quality of RNAs were assessed using a Nanodrop (NanoPhotometer® P‐330 Implen, Munich, Germany). The desired amount of RNA (between 0.5 and 5 μg in 14 μl water) was removed and added to 4 μl reaction buffer and 2 μl enzyme (RT Life Technology Fermentas, Catalogue No. K1642, Thermo Fisher Scientific). Reverse transcription was performed using GeneAmp® PCR System 9700. Quantitative PCR was performed using Fluidigm technology (GENOTOUL facility, Toulouse, France). The primers were selected using PrimerBlast software, ordered from Eurogentec (Angers, France), and had PCR efficiency >85% (supplementary material, Table S4). The minus delta Ct was calculated from reference genes (mean, HPRT, GAPDH). Fold‐changes of mRNA levels were calculated using the comparative 2−ΔΔCt method using the mean of HC data as a control.
Statistical analysis
Statistical analyses and graphs were performed using GraphPad Prism software (version 10.0, GraphPad Software, Boston, MA, USA). The quantitative variables were compared by the one‐way ANOVA test or t‐test. The graphical representation includes the SEM or median. The significance threshold was p < 0.05.
Results
Using a WNT3a‐conditioned medium, we found a growth of FAP (A, NA) and HC organoids from D2 to D9 (Figure 1A). However, the Wnt/APC/β‐catenin pathway remained active in FAP because of the APC mutation. As expected, the growth of A and NA organoids was not altered by WNT3a starvation, while it was blocked in HC organoids (Figure 1A). Later, we used only recombinant WNT3a for HC organoids (Figure 1B). Regardless of the culture conditions, A were larger than NA organoids.
Figure 1.
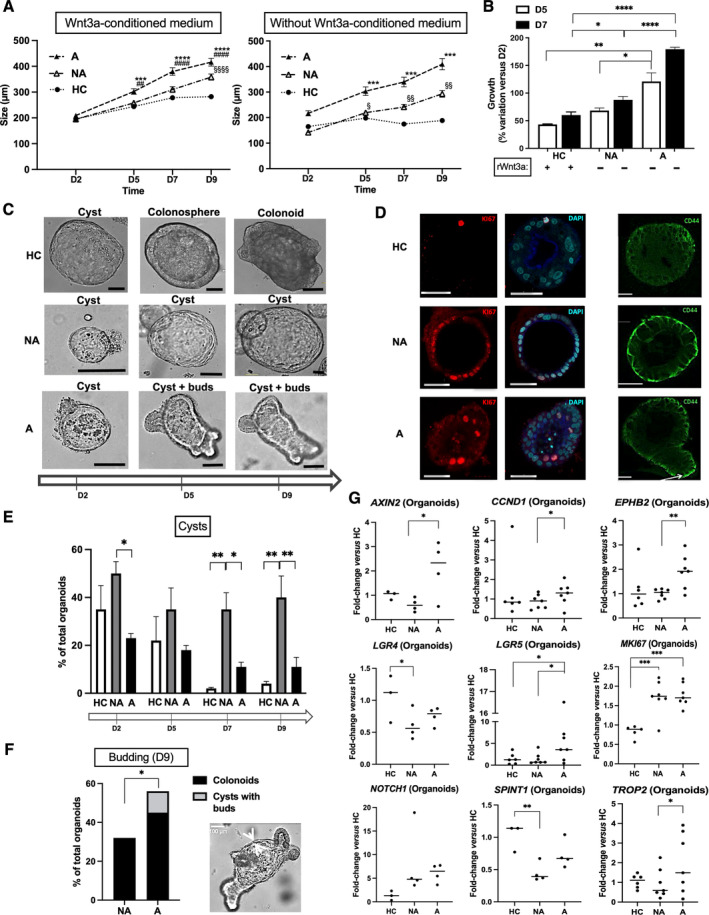
Characterization of FAP organoids. (A and B) In contrast to organoids from healthy controls (HCs), FAP‐organoid growth (NA, nonadenomatous; A, adenomatous) was independent of WNT3a‐conditioned medium or recombinant WNT3a. (A) n patients: HC = 6, NA/A = 7 (with WNT3a‐conditioned medium), NA/A = 4 (without WNT3a‐conditioned medium), organoid size mean ± SEM (diameter, around 200–300 organoids per condition), ANOVA comparison of A to HC: ***p < 0.001 ****p < 0.0001, A to NA ## p < 0.01 #### p < 0.0001, NA to HC § p < 0.05 §§ p < 0.01 §§§§ p < 0.0001. (B) n patients: HC = 3, NA/A = 3, percentage variation of organoid size from D2 mean ± SEM (diameter at D2, mean ± SEM: HC 144 ± 18 μm, NA 117 ± 16 μm, A 76 ± 17 μm; around 10 organoids per condition), ANOVA comparison: *p < 0.05 **p < 0.01 ****p < 0.0001. (C) From D2 to D9 of culture, HC organoids develop as immature forms with a thin monolayer boarding a central lumen (cysts) to mature forms as colonospheres with a larger and polarized monolayer and then as colonoids with buds of neo‐crypts in formation. NA organoids show a delayed maturation with very large cysts. A organoid culture contains unique structures as cysts with buds. Scale bar, 100 μm. (D) Expression of proliferation marker Ki67 (immunolabeling) is increased in FAP organoids compared to HC. Labeling of nuclei by DAPI is shown. CD44 labeling is high in NA organoids and present in buds of A organoids. Representative of two independent experiments. Scale bar, 50 μm. (E) n patients: HC = 3, NA/A = 3, percentage cysts of total organoids mean ± SEM, ANOVA comparison: *p < 0.05 **p < 0.01. (F) n patients: NA/A = 3, percentage budding structures of total organoids mean ± SEM, paired (NA to A) t‐test comparison: *p < 0.05. A typical cyst with buds, structure unique to A organoid culture, is shown. (G) At D7 of culture, mRNA from A organoids compared to HC and/or NA organoids is enriched in markers of CBC stem cells (EPHB2, LGR5), β‐catenin transcriptional targets (AXIN2, CCND1), marker of cell proliferation (MKI67), and β‐catenin interacting partners (NOTCH1, TROP2). Compared to HC, NA organoids are characterized by both an increase of MKI67 and a decrease of LGR4 and SPINT1 mRNA, two guardians of the crypt integrity. n patients: HC = 3–6, NA/A = 4–7, ANOVA comparison between HC and NA/A: *p < 0.05 **p < 0.01 ***p < 0.001, paired t‐test comparison between NA and A: *p < 0.05 **p < 0.01.
Different kinds of colonic organoids have been reported in the literature for HC patients: cysts, colonospheres, and colonoids respectively characterized by a thin monolayer and a thick monolayer of epithelial cells without or with the presence of buds; maturity progresses from cyst to colonosphere and to colonoid (Figure 1C) [24]. Indeed, the thick monolayer corresponds to colonic cells with an apical–basal orientation and the buds being new crypts in the process of formation. In the culture of NA organoids, cysts were frequent (Figure 1C) and associated with high Ki67 (proliferative marker) and CD44 (immaturity marker) labeling compared to HC (Figure 1D). The NA cystic phenotype was maintained at 40% of total organoids until D9, whereas HC and A cystic organoids decreased from D2 (40 and 20%, respectively) to D9 (5 and 10%, respectively) (Figure 1E). Surprisingly, in A organoid culture, early on we found a new structure characterized by the presence of buds in cysts (Figure 1C). A organoids were highly labeled for Ki67 compared to HC, and buds in cysts were rich in CD44 (Figure 1D). Of note, budding cysts were absent in the organoid culture from NA biopsies (Figure 1F). These data suggest that the epithelial maturation may be delayed in NA organoids, whereas in A organoids, budding is abnormally independent of maturation.
A large transcriptomic study allowed to highlight genes whose mRNA expression varied in a similar fashion in both FAP organoids and crypts compared to HC (Figure 1G and supplementary material, Figure S1A). As expected, markers of proliferation (MKI67), of CBC stem cells (EPHB2, LGR5), β‐catenin transcriptional targets (AXIN2, CCND1) and β‐catenin interacting partners (NOTCH1, TROP2) were increased in A organoids. Interestingly, NA organoids were characterized by both an increase of MKI67 and a decrease of LGR4 and SPINT1 mRNA, two guardians of the crypt integrity (Figure 1G). [25, 26] Further comparison with HC has shown that the mRNA expression of several genes was changed only in FAP organoids (supplementary material, Figure S1B) or in FAP crypts (supplementary material, Figure S1C). A organoids displayed increased expression of genes associated with tumor survival (NOTCH2, TNFA) and transformation (ITGB2, PROM1, VIM). Whereas both NA‐ and A‐crypts had decreased expression of CDX2 and increased expression of MUC2 and MYC, they were respectively characterized by decreased expression of ALD1HA1, ATOH1, BMI1, ETS2, SOX9 and increased expression of CD166, CLDN1, DUSP6, FZD4, IL33, ITGA6, ITGB4, MUC5A, PI3, TIMP1, TIMP2. Overall, these observations are in accordance with the critical role of the microenvironment in the crypt regulation [13]. The organoid model pointing out the capacities of tumor transformation of adenomatous ISC, and crypts highlighting defects in the integrity of NA‐mucosa and invasive capacities of A‐mucosa. Based on the individualized data of these changes in gene expression in FAP organoids and crypts compared to HC (supplementary material, Table S1), we established a graphic relationship between genes (supplementary material, Figure S1D). Interestingly, from an immature (LGR5) and proliferative (CCND1) phenotype characteristic of the evolution from NA to A and common to the analyses in crypts and organoids, this analysis points the attention to potential key molecular actors of this evolution, interacting with the microenvironment or the signaling pathways. Of note, in our study the expression of 38 genes did not vary in FAP crypts and organoids compared to HC (supplementary material, Figure S1E).
Then we aimed to investigate FAP ISC regulation. Given the important role of EGFR signaling in the establishment of adenomas in Apc Min‐mice [20], we investigated the EGF pathway in our organoid model. First, among EGFR partners (EGF, EREG, AREG, ERBB1, ERBB3, LRIG1), only EGF was found decreased in FAP organoids, but not crypts, compared to HC (Figure 2A), suggesting a strong dependence of FAP ISCs and progenitors to the EGF supplied by the microenvironment. Under EGF starvation in the culture medium from D2, the expression of the aforementioned genes was not changed, except for an increase in the EGFR modulator LRIG1 in NA organoids (Figure 2A). Furthermore, whereas starvation of EGF did not change the expression of most genes studied (supplementary material, Figure S2), it induced a loss of changes in the mRNA expression associated with the FAP phenotype: AXIN2, NOTCH2, VIM mRNA in A compared to NA and/or HC organoids; ITGB2, LGR4, SPINT1, TIMP1 mRNA in NA compared to A and/or HC organoids (Figure 2B). In FAP organoids, EGF deprivation increased the expression of genes involved in crypt differentiation: SOX9, A organoids; ATOH1, CDH1, EFNB2, F10, GSK3B, KRT20, MUC2, NANOG, OCLN, PI3, NA organoids (Figure 2B). Moreover, EGF deprivation significantly blocked the growth of A organoids compared to HC and NA organoids (Figure 3A). Finally, an important finding was that the absence of EGF increased the mortality of A organoids compared to NA organoids (Figure 3B) and HC organoids (supplementary material, Figure S3A). Overall, these data strongly suggest that EGF plays a critical role in the FAP pathogenesis in both NA and A mucosa.
Figure 2.
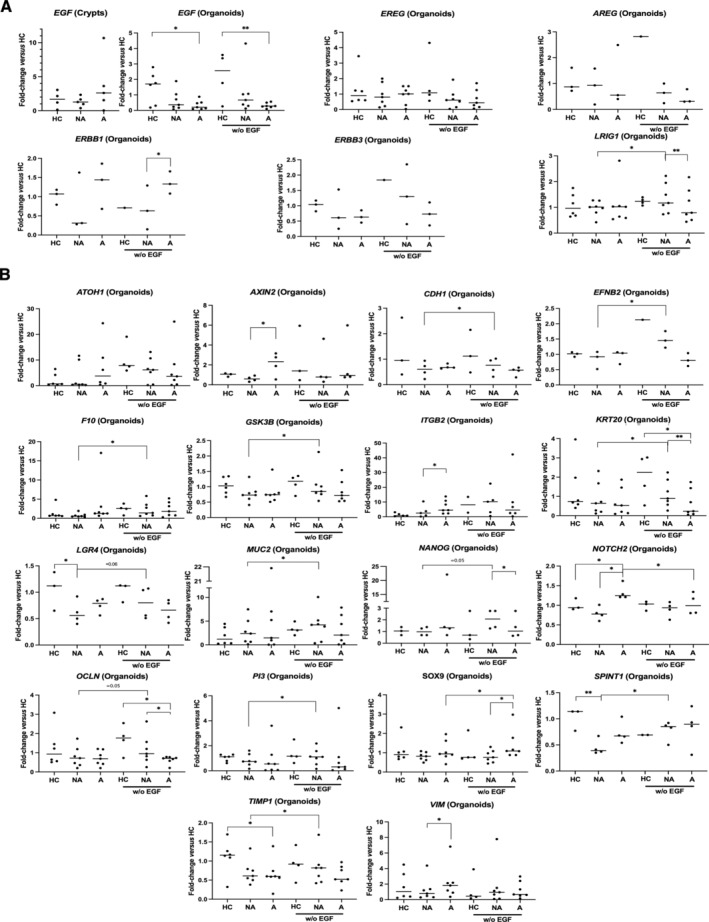
Transcriptional regulation by EGF in FAP organoids. (A) At D7 of culture, data on the EGFR pathway show a decrease of EGF mRNA in FAP organoids (NA, nonadenomatous; A, adenomatous) but not crypts compared to healthy controls (HCs). Under EGF starvation in the culture medium from D2, no change in the expression of several genes (EGF, EREG, AREG, ERBB1, ERBB3) was measured, except an increase for the EGFR regulator LRIG1 (NA organoids). (B) Starvation of EGF induced a loss of changes in the mRNA expression associated with the FAP phenotype: AXIN2, NOTCH2, VIM mRNA in A compared to NA and/or HC organoids; ITGB2, LGR4, SPINT1, TIMP1 mRNA in NA compared to A and/or HC organoids. In FAP organoids, EGF‐deprivation increased the expression of genes involved in crypt differentiation (SOX9, A organoids; ATOH1, CDH1, EFNB2, F10, GSK3B, KRT20, MUC2, NANOG, OCLN, PI3, NA organoids). n patients: crypts, HC = 4, NA/A = 5; organoids, HC = 3–6, NA/A = 3–7. ANOVA comparison between HC and NA/A: *p < 0.05 **p < 0.01, paired t‐test comparison between NA and A, NA and NA without EGF, A and A without EGF: *p < 0.05 **p < 0.01.
Figure 3.
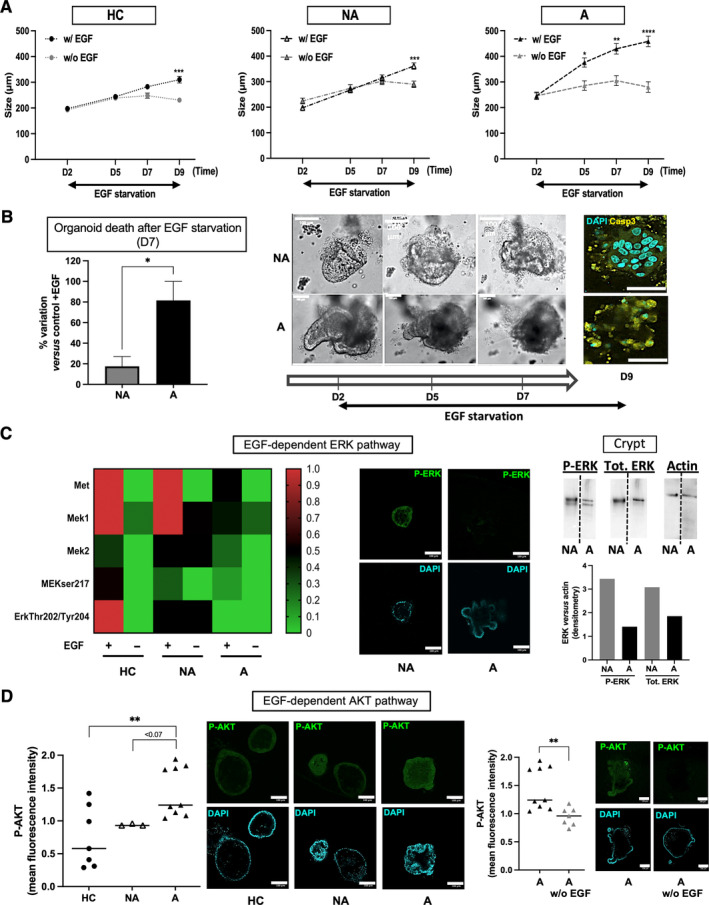
EGF dependence of FAP organoids. (A) Compared to healthy controls (HCs) and NA organoids, A organoid growth is greatly more dependent on the presence of EGF in the culture medium. EGF was starved from D2 of the organoid culture since it was required for organoid closure between D0 and D2. n patients: HC = 6 NA/A = 3, organoid size mean ± SEM (diameter, around 100–300 organoids per condition), ANOVA comparison of +EGF to −EGF: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (B) EGF starvation induces death in A organoids. The number of dead organoids (example is shown with brightfield image of black and dense structure in A culture) versus total number of organoids was calculated at D7 of culture, and variation of this percentage in EGF‐starved conditions is shown. n patients: NA/A = 2, percentage variation mean ± SEM (around 50 organoids per condition), NA to A t‐test comparison: *p < 0.05. Immunofluorescence (IF): labeling for active Caspase‐3 (yellow) and nuclei by DAPI at D9 of culture is shown. (C) EGF‐dependent ERK pathway is decreased in FAP organoids. Heatmap of protein expression in EGF signaling pathway (D7 of culture) is shown. n patients: HC = 3, NA/A = 2, normalization of expression to HC MEK1. IF: labeling for Pthr202/tyr204‐ERK1/2 (green) and nuclei by DAPI at D7 of culture is shown. WB: western blotting for Pthr202/tyr204‐ERK1/2 (P‐ERK), total ERK (Tot. ERK) and actin, and their quantification, in NA and A crypts are shown. (D) EGF‐dependent AKT pathway is increased in FAP organoids. IF: labeling for Pser473‐AKT (green) and nuclei with DAPI at D7 of culture is shown and quantified. n patients: HC = 2, NA/A = 2, NA/A to HC ANOVA comparison, and A‐EGF to A t‐test comparison: **p < 0.01. Scale bar, 100 μm.
It was thus important to investigate signaling pathways downstream of EGFR. A proteomic screening (supplementary material, Figure S3B) showed that the MEK/ERK pathway was strictly dependent on EGF in HC organoids (Figure 3C). The MEK/ERK activity declined in NA organoids and was abolished in A organoids, and the impact of EGF starvation in the culture medium was limited (Figure 3C). The expression of the hepatocyte growth factor receptor (MET), capable of promoting adenoma formation and interfering with the EGFR signaling pathway [27], decreased in A organoids but remained at an intermediate level of expression compared to HC and NA organoids (Figure 3C). These findings were confirmed by immunostaining and western blotting, showing a decreased activation of ERK in A mucosa compared to NA mucosa (Figure 3C and supplementary material, Figure S3C). Further, activated AKT labeling was increased in A compared to NA and HC organoids and dependent on the presence of EGF in the culture medium (Figure 3D), in contrast to HC organoids (supplementary material, Figure S3D).
We then looked for a possible synergy of EGFR with signaling partners. It has been shown that cells with APC mutations could still depend on Wnt/Frizzled signaling pathway [28]. Accordingly, our data show that the Wnt signaling potentiator R‐spondin 1 is required in the culture medium to promote FAP‐organoid growth (Figure 4A). Moreover, an inhibitor of Wnt secretion (IWP12, Sigma‐Aldrich Catalogue No. SML0677, Saint‐Quentin‐Fallavier, France) induced death and growth inhibition in NA and A organoids, respectively (Figure 4B). As shown in supplementary material, Figure S1E, we did not observe changes in WNT5A mRNA expression in FAP crypts and organoids compared to HC ones. However, the Wnt receptor Frizzled 4, which can mediate the canonical activity of WNT5a, was found to have increased in A crypts compared to HC (supplementary material, Figure S1C). Altogether these data suggest that WNT5a, known to play a critical role in crypt regeneration, may regulate cell survival and growth in APC‐deficient ISCs of NA and A areas, respectively [29].
Figure 4.
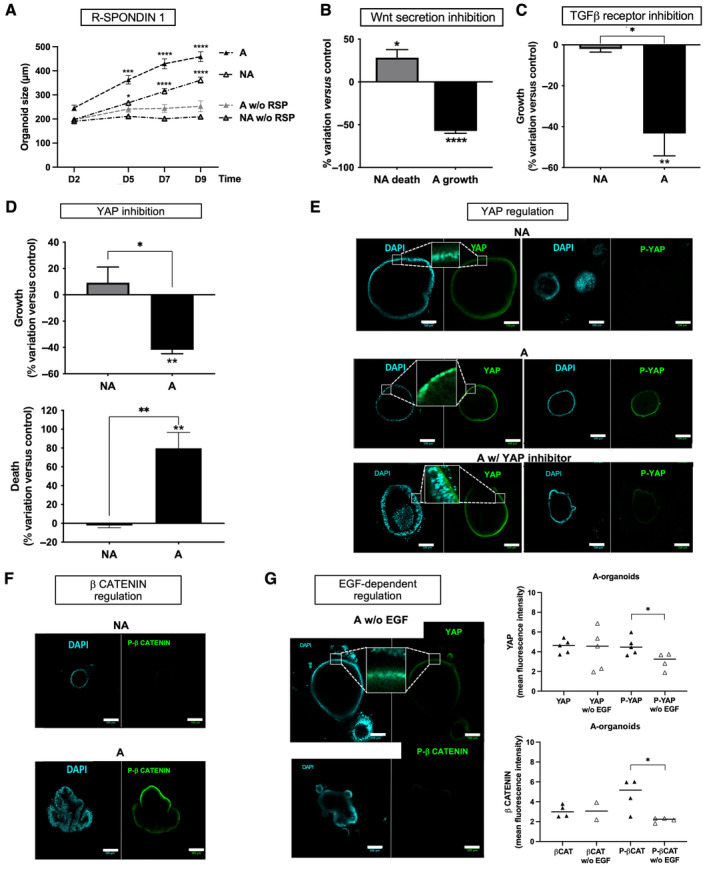
Signaling partners of EGF in FAP organoids. (A) R‐spondin 1 (RSP) starvation from D0 of the culture had a negative impact on FAP (NA, nonadenomatous; A, adenomatous) organoid growth. n patients: NA/A = 5, organoid size mean ± SEM (diameter, around 100–300 organoids per condition), ANOVA comparison of A with RSP to A without RSP and NA with RSP to NA without RSP: *p < 0.05 ***p < 0.001 ****p < 0.0001. (B) Wnt secretion inhibition using IWP12 (5 μm) impaired survival of NA organoids and growth of A organoids. n patients: NA/A = 3, percentage variation of organoid death and size between D2 and D7 of culture compared to control (0.02% DMSO), mean ± SEM calculated through analysis of around 10 organoids per condition, t‐test comparison with IWP12 to without IWP12: *p < 0.05, ****p < 0.0001. (C) TGF‐β receptor is implicated in growth of FAP adenoma. Addition of the TGF‐β receptor I inhibitor LY2157299 (LY, 0.5 μm) along the culture of FAP organoids induced a decrease of A organoid growth. n patients: NA/A = 3, percentage variation of organoid size between D2 and D7 of culture compared to control (without treatment), mean ± SEM calculated through analysis of around 10 organoids per condition, t‐test comparison with LY to without LY: **p < 0.01 A to NA *p < 0.05. (D) YAP controls growth and survival of adenoma. Addition of YAP inhibitor Verteporfin (VP, 0.5 μm) to FAP organoid culture inhibited growth/survival of A organoids. n patients: NA/A = 2 (growth) NA/A = 3 (survival), percentage variation of organoid size/death between D2 and D7 of culture compared to control (0.02% DMSO), mean ± SEM calculated through analysis of around 10 organoids per condition, t‐test comparison with VP to without VP: **p < 0.01 A to NA *p < 0.05 **p < 0.01. (E) YAP was activated (nuclear localization and Ptyr357 phosphorylation) specifically in FAP A organoids. Immunofluorescence (IF): labeling for YAP or P‐YAP (green) and nuclei using DAPI at D7 of culture is shown as well as Verteporfin's (YAP inhibitor) negative impact on it. n patients: NA/A = 2, images representative of five structures per condition and zoomed to show nuclear localization of YAP. (F) An activated form of β‐catenin (Pser552‐β‐catenin, P‐β CATENIN) is specifically found in A organoids. IF: labeling of Pser552‐β‐catenin (green) and nuclei by DAPI at D7 of culture is shown. Representative images from NA/A = 2 patients. (G) Activated forms of YAP (nuclear localization and Tyr357‐phosphorylation) and β‐catenin (Pser552‐β CATENIN) are dependent on EGF. IF quantification of total (YAP, βCAT) and phosphorylated forms (P‐YAP, P‐βCAT) of YAP and β‐catenin in FAP A organoids are shown, as well as representative images of the impact of EGF starvation on that labeling. IF: labeling of YAP/Pser552‐β‐catenin (green) and nuclei by DAPI at D7 of culture is shown. n patients: A = 2, t‐test comparison: *p < 0.05. Scale bar, 100 μm.
In the intestinal crypt, TGF‐β is an important signaling partner of EGF and WNT5A. Its role in tumorigenesis is ambiguous, acting either as tumor suppressor or prometastatic factor [30]. We did not observe a significant difference in its mRNA expression between HC and FAP crypts/organoids (supplementary material, Figure S1E). As shown in Figure 4C, the pharmacological inhibition of the TGF‐β receptor did not affect the size of NA organoids, whereas it significantly decreased by around 40% the size of A organoids. These data show that growth of adenoma is dependent on both EGF and TGF‐β.
YAP is an important signaling target of morphogens (WNTs, TGF‐β) and EGF in the intestinal crypt, and its inhibition by Verteporfin (Sigma‐Aldrich ref. SML0534) induced specifically growth inhibition and death of A organoids (Figure 4D). Moreover, YAP was found at nuclear locations and phosphorylated on the residue tyrosine 357 specifically in A organoids (Figure 4E). This regulation of the active form of YAP was abolished by Verteporfin (Figure 4E).
AKT can phosphorylate serine552‐β‐catenin to promote its nuclear localization in a context of increased ISC proliferation, crypt fission, and adenoma formation [31, 32]. Our results show a specific labeling of Pser552‐β‐catenin in buds of A organoids, not found in NA organoids (Figure 4F). Importantly, both active forms of YAP and β‐catenin were under the control of EGF in A organoids (Figure 4G). TGF‐β receptor inhibition also blocked Serine‐552‐β‐catenin phosphorylation (supplementary material, Figure S4). Thus, the modulation of β‐catenin activity in adenoma is dependent on both EGF and TGF‐β. Taken together, these data support a clear role of EGF and its partners in adenoma formation through the regulation of β‐catenin in APC‐deficient ISCs.
AKT is activated downstream of PI3K, and inhibitors targeting PI3K isoforms are now being considered for several pathologies [33, 34]. A quantitative PCR study of the expression of p110α and p110β isoforms of PI3K showed a specific increase of p110β mRNA in FAP crypts, but not organoids, compared to HC (Figure 5A). However, immunolabeling revealed an enrichment of p110α and p110β in NA and A organoids, respectively (Figure 5B). Furthermore, p110β was specifically enriched in buds of A organoids (Figure 5B). Pharmacological inhibitors of p110α (A66, Selleckchem, Catalogue No. S2636, Euromedex, Souffelweyersheim, France) and p110β(AZD6482, Selleckchem, Catalogue No. S1462) reduced A‐organoid growth (Figures 5C and supplementary material, S5B). Moreover, A organoid death was observed by microscopy following AZD treatment (Figure 5C). In contrast, p110α inhibition increased NA organoid growth, associated with epithelial maturation, as evidenced by an enlarged width of epithelial monolayer (Figure 5C). Of note, the expression of the regulatory p85 subunit of PI3K, associated with p110 isoforms, was not different between NA and A organoids and not significantly changed upon treatment with pharmacological inhibitors of p110 isoforms (supplementary material, Figure S5A). Taken together, these results suggest p110 isoforms of PI3K are interesting therapeutic targets in FAP.
Figure 5.
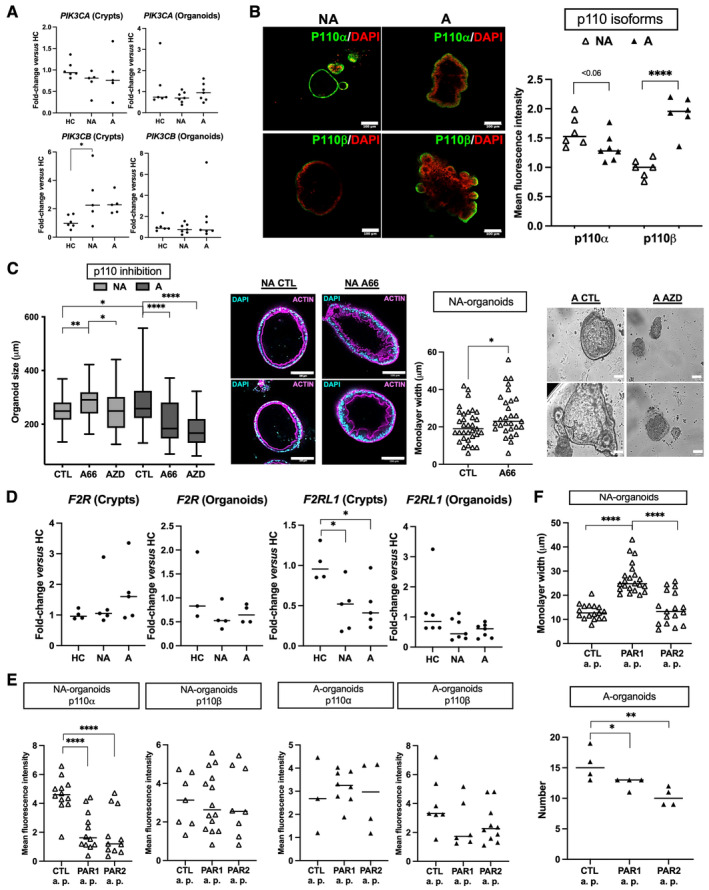
p110 isoforms of PI3K as pharmacological targets in FAP. (A) p110 isoforms (α, PIK3CA; β, PIK3CB) of PI3K were studied through their mRNA expression in crypts and organoids (D7 of culture). Only PIK3CB was increased in FAP (NA, nonadenomatous; A, adenomatous) crypts compared to healthy controls (HCs). n patients: HC = 6, NA/A = 5 (crypts) or 7 (organoids). (B) Immunolabeling for p110 isoforms in FAP organoids. Immunofluorescence (IF): quantification and representative images of labeling are presented. IF: labeling of p110α and p110β (green) and nuclei using DAPI at D7 of culture is shown. n patients: NA/A = 2, ANOVA comparison: ****p < 0.0001. (C) Pharmacological inhibition of p110α (A66, 0.5 μm) and p110β (AZD6482, 0.5 μm) induces NA organoid maturation and A organoid death, respectively. Growth (n patients: NA/A = 3, diameter size of around 40 organoids analyzed per condition) and epithelial monolayer width (n patients: NA = 4) quantifications are shown, as well as representative images at D7 of culture (n patients: NA/A = 3) of actin labeling and organoid death (brightfield images). IF: labeling of actin (magenta) and nuclei using DAPI is shown. Organoid size: ANOVA comparison versus control (CTL, 0.005% DMSO) or between NA with A66 and NA with AZD: *p < 0.05, **p < 0.01, ****p < 0.0001. Epithelial monolayer width: t‐test comparison, *p < 0.05. (D) Protease‐activated receptors (PAR1, F2R; PAR2, F2RL1) were studied through their mRNA expression in crypts and organoids (D7 of culture). Only F2RL1 was decreased in FAP (NA and A) crypts compared to HCs. n patients: HC = 4 (crypts) and 3–6 (organoids), NA/A = 5 (crypts) and 4–7 (organoids). (E) PAR1 and PAR2 agonist peptides (PAR1 a.p. and PAR2 a.p., respectively TFLLR‐NH2 and SLIGRL‐NH2, 100 μm) or control peptides (Ctl a.p., mixture of reversed sequences, 100 μm) were added daily from D2 to D5 of organoid culture and IF of p110 isoforms (α, β) and its quantification were performed. n patients: NA = 3, A = 2. ANOVA comparison: ****p < 0.0001. (F) PAR1 and PAR2 activation was performed as in E. The measurements of the epithelial monolayer width in NA organoids and of the number of A organoids are shown. n patients: NA = 3, A = 4, ANOVA comparison: *p < 0.05 **p < 0.01 ****p < 0.0001. Scale bar, 100 μm.
Finally, considering the role of the protease‐activated receptor PAR1 in impeding adenoma formation in APC Min/+ mice [35], we measured mRNA expression of genes encoding PAR1 and PAR2 (respectively F2R and F2RL1). Compared to HC, only F2RL1 mRNA was decreased both in NA and A crypts (Figure 5D). Then we investigated the impact of PAR1 and PAR2 activation using agonist peptides (TFLLR‐NH2 and SLIGRL‐NH2, respectively; Genscript, Piscataway, NJ, USA) on p110‐isoform expression and FAP‐organoid behavior. Compared to control peptides (reversed sequences, Genscript or Ezbiolab Inc., Carmel, IN, USA), we measured a specific decrease of p110α level in NA organoids induced by both PAR1 and PAR2 agonist peptides (Figure 5E). However, only PAR1 activation proved capable of enhancing NA organoid maturation (Figure 5F), whereas PAR2 activation was the most efficient at decreasing the p110β level in A organoids, which was associated with a decrease in the number of organoids and budding (Figures 5F and supplementary material, S5C). Once again, these results highlight the distinct phenotypes between NA and A areas and strongly suggest that pharmacological tools aimed at modulating p110‐isoform expression/activation could be of therapeutic interest in FAP.
Discussion
Our results confirmed that the organoid culture was a relevant model for studying FAP pathophysiology, in particular in initial events involved in the pathogenesis and in the nonadenomatous to adenomatous transition. Our demonstration included (1) delayed maturation of NA mucosa regulated by EGF, (2) EGF‐dependent survival of adenoma, (3) progressive switch of ERK to AKT activity from HC to NA and A, (4) EGF control of β‐catenin through AKT in adenomas, (5) specific implication of p110β subtype of PI3K in adenoma development, and (6) downregulation of PAR2 (F2RL1) mRNA in both NA and A, which is among 16 genes molecular prognostic score is being established for gastrointestinal tumors [36].
We identified a specific organoid structure exclusive to A organoids: a budding cyst. To our knowledge, this structure has rarely been described in the literature but could correspond to the patient‐derived organoid type 4 from colorectal cancer enriched in LGR5 mRNA [37]. Unlike normal budding linked to epithelial polarization and differentiation, this early budding could share mechanisms with the developmental crypt fission under the control of ISCs and Wnt signaling [38]. Indeed, in A organoids, we found the active β‐catenin form phosphorylated on serine552, implicated in crypt fission/regeneration and polyposis [31, 32]. Although mutated stem cells could promote tumorigenesis by crypt fission, it seems insufficient to explain the carcinogenic risk [39]. However, we can postulate that a budding cyst might represent an endpoint, analogous to adenoma in human histopathology, within FAP organoid culture, which is useful for assessing the effectiveness of a potential drug.
Our study revealed differences in terms of signaling pathways involved and ISC fate between NA and A organoids. According to the aims of treatment, it should be of importance, either halting the progression to adenoma or preventing the transition to adenocarcinoma. We confirmed previous data showing that the Wnt/Frizzled signaling pathway was still operative in APC‐mutated cells [28] and controls survival of NA organoids and growth of A organoids. Interestingly, the two genes TIMP2 and VIM whose expressions were respectively increased in A‐crypt and A‐organoid analyses have been linked to the consensus molecular signature 4 (CMS4) of bad tumor prognosis, related to Wnt‐receptor signaling, not APC mutation [40]. However, in the absence of EGF or after the blockade of TGF‐βR, we reported a specific and significant effect on A organoid growth that correlated with decreased P‐Ser552‐β‐catenin. Moreover, from HC to NA and A organoids, EGF signaling was progressively switched from ERK to AKT activity in correlation with delayed maturation in NA‐ and PI3K‐dependent survival in A organoids. The specific impact of EGF starvation on A‐organoid survival may result from compensatory mechanisms in HC and NA organoids such as PGE2‐dependent EGFR transactivation and downstream MAPK activation [41, 42]. Interestingly, we showed that the expression of the ERK inhibitor DUSP6 increased in A crypts compared to HC and NA and is an important connective node between genes differentially expressed between NA and A mucosa (supplementary material, Figure S1D).
Thus, the strategy could be to target the PI3K/AKT pathway, in particular p110β (as in other cancers) [43], when dealing with FAP adenomas and the differentiation process to prevent adenoma development. Here, it should be noted that we observed no bias in our data interpretation relative to differences in the ages of FAP and HC patients. Interestingly, we measured a specific and EGF‐dependent decrease of LGR4 mRNA in NA organoids, an important regulator of crypt metabolism [25], and others have measured an increase in IGFL1 expression in NA organoids, within the same age range as HC [6]. Also, it has been shown in an intestinal organoid model expressing oncogenic β‐catenin and p110α that the latter supports the adaptative metabolism of tumor cells [44]. Altogether, these data suggest that specific metabolic changes could occur early in APC‐deficient ISCs, since it has been shown that APC controls pyruvate metabolism in mitochondria and, therefore, intestinal differentiation [45]. Metabolic changes occurring in FAP NA areas could be therapeutically targeted, and our results show that the inhibition of p110α, as well as PAR1 activation, can induce the differentiation of NA‐ISCs.
Finally, despite limitations of the organoid model, our work allows us to propose a design for the phenotypes of FAP organoids (Figure 6). First, survival pathways differ between NA and A ISCs, with the former relying on a Wnt receptor pathway and the latter depending on the EGF receptor pathway. Second, the main defect in NA ISCs could be a deficiency in their capacities of differentiation associated with a metabolic switch, whereas A ISCs display mainly high proliferative and migratory capacities. Thus, early chemoprevention proposed for FAP patients could be based on strategies enhancing the differentiation of NA ISCs by targeting p110α or metabolic pathways (which require further investigation). For advanced FAP cases, targeting p110β and/or TGF‐β and/or FZD4 may be considered.
Figure 6.
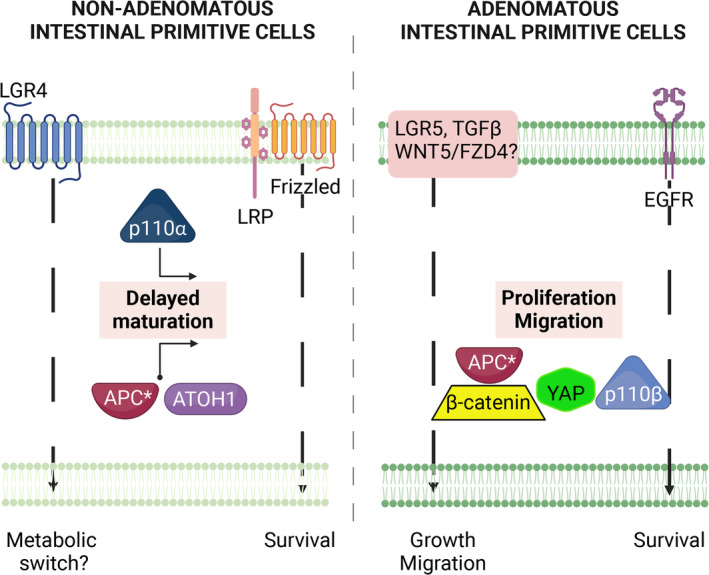
Schematic synthesis of data and hypothesis. Organoids from nonadenomatous (NA) and adenomatous (A) biopsies displayed different phenotypes during investigations, summarized as delayed maturation in intestinal primitive cells from NA and overactive proliferation and migration in A organoids. Delayed maturation in intestinal primitive cells from NA seems to impact secretory differentiation (ATOH1 pathway) and may be linked to APC and LGR4 dysfunctions as well as to p110α activation. In the proliferation and migration of intestinal primitive cells from A, in addition to APC dysfunction, EGFR, and TGF‐β (TGF‐β), but probably also LGR5‐ and Wnt‐dependent pathways, play a critical role by regulating p110β, β‐catenin, and YAP. Notably, both NA and A areas differ in their survival pathways. APC*, mutant APC. Figure created with BioRender.com
Author contributions statement
EM and CR‐S designed and directed the project. MQ, NV, CR, CR‐S, FB, M‐PG, JG‐G and AF designed the methodology. NL, AB, MQ, CR, DB, SK, NS‐T, DH, KB, J‐PD, AnB, LA, CR‐S and EM performed the experiments. EM received financial support for the project. MQ and AA contributed to obtaining and registering the samples. EM and CR‐S wrote the manuscript. EM, CR‐S, FB, M‐PG and JG‐G reviewed and edited the manuscript. All authors read and agreed to the published version of the manuscript.
Supporting information
Figure S1. Transcriptomic study of FAP crypts and organoids
Figure S2. Impact of EGF starvation on gene transcription in FAP organoids
Figure S3. EGF‐dependent ERK and AKT activation in FAP organoids
Figure S4. Implication of the TGF‐β receptor in activation of β‐catenin in adenomatous organoids
Figure S5. Impact of p110 and PAR modulation on FAP organoid development
Table S1. Description of human samples used in analyses
Table S2. Organoid culture medium components
Table S3. Primary and secondary antibodies
Table S4. Quantitative PCR primers
Acknowledgements
The authors thank Arthur Amalvy (Laboratoire Informatique d'Avignon, France) for the graphical representation of genes' relationships. Emmanuel Mas received a grant from the Rotary Club of Portet sur Garonne, INSERM, Toulouse University Hospital, Reference Center for Rare Digestive Disorders.
No conflicts of interest were declared.
Data availability statement
The data that support the findings of this study are available upon request from the corresponding author (EM). Raw data are not publicly available because they contain information that could compromise the privacy of research participants.
References
- 1. Hyer W, Cohen S, Attard T, et al. Management of familial adenomatous polyposis in children and adolescents: position paper from the ESPGHAN polyposis working group. J Pediatr Gastroenterol Nutr 2019; 68: 428–441. [DOI] [PubMed] [Google Scholar]
- 2. Moser AR, Dove WF, Roth KA, et al. The MIN (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol 1992; 116: 1517–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jackstadt R, Sansom OJ. Mouse models of intestinal cancer. J Pathol 2016; 238: 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drost J, van Jaarsveld RH, Ponsioen B, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015; 521: 43–47. [DOI] [PubMed] [Google Scholar]
- 5. Li X, Nadauld L, Ootani A, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med 2014; 20: 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Devall MA, Eaton S, Ali MW, et al. Insights into early onset colorectal cancer through analysis of normal colon organoids of familial adenomatous polyposis patients. Cancers (Basel) 2022; 14: 4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Devall MA, Eaton S, Ali MW, et al. DNA methylation analysis of normal colon organoids from familial adenomatous polyposis patients reveals novel insight into colon cancer development. Clin Epigenetics 2022; 14: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kanth P, Hazel MW, Schell JC, et al. Evaluation of EGFR and COX pathway inhibition in human colon organoids of serrated polyposis and other hereditary cancer syndromes. Fam Cancer 2024; 23: 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 2009; 459: 262–265. [DOI] [PubMed] [Google Scholar]
- 10. Jung P, Sato T, Merlos‐Suárez A, et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med 2011; 17: 1225–1227. [DOI] [PubMed] [Google Scholar]
- 11. Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010; 327: 542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Basak O, Beumer J, Wiebrands K, et al. Induced quiescence of Lgr5+ stem cells in intestinal organoids enables differentiation of hormone‐producing enteroendocrine cells. Cell Stem Cell 2017; 20: e174. [DOI] [PubMed] [Google Scholar]
- 13. Medema JP, Vermeulen L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 2011; 474: 318–326. [DOI] [PubMed] [Google Scholar]
- 14. Zhang Z, Zhang F, Davis AK, et al. CDC42 controlled apical‐basal polarity regulates intestinal stem cell to transit amplifying cell fate transition via YAP‐EGF‐mTOR signaling. Cell Rep 2022; 38: 110009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abud HE, Chan WH, Jardé T. Source and impact of the EGF family of ligands on intestinal stem cells. Front Cell Dev Biol 2021; 9: 685665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richmond CA, Shah MS, Deary LT, et al. Dormant intestinal stem cells are regulated by PTEN and nutritional status. Cell Rep 2015; 13: 2403–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beaudry K, Langlois MJ, Montagne A, et al. Dual‐specificity phosphatase 6 deletion protects the colonic epithelium against inflammation and promotes both proliferation and tumorigenesis. J Cell Physiol 2019; 234: 6731–6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hageman JH, Heinz MC, Kretzschmar K, et al. Intestinal regeneration: regulation by the microenvironment. Dev Cell 2020; 54: 435–446. [DOI] [PubMed] [Google Scholar]
- 19. van Neerven SM, de Groot NE, Nijman LE, et al. Apc‐mutant cells act as supercompetitors in intestinal tumour initiation. Nature 2021; 594: 436–441. [DOI] [PubMed] [Google Scholar]
- 20. Roberts RB, Min L, Washington MK, et al. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci U S A 2002; 99: 1521–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eisinger AL, Prescott SM, Jones DA, et al. The role of cyclooxygenase‐2 and prostaglandins in colon cancer. Prostaglandins Other Lipid Mediat 2007; 82: 147–154. [DOI] [PubMed] [Google Scholar]
- 22. Kemp Bohan PM, Mankaney G, Vreeland TJ, et al. Chemoprevention in familial adenomatous polyposis: past, present and future. Fam Cancer 2021; 20: 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crespo M, Vilar E, Tsai SY, et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat Med 2017; 23: 878–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stelzner M, Helmrath M, Dunn JC, et al. A nomenclature for intestinal in vitro cultures. Am J Physiol Gastrointest Liver Physiol 2012; 302: G1359–G1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li Z, Zhang W, Mulholland MW. LGR4 and its role in intestinal protection and energy metabolism. Front Endocrinol (Lausanne) 2015; 6: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Danielsen ET, Olsen AK, Coskun M, et al. Intestinal regulation of suppression of tumorigenicity 14 (ST14) and serine peptidase inhibitor, Kunitz type ‐1 (SPINT1) by transcription factor CDX2. Sci Rep 2018; 8: 11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Joosten SPJ, Zeilstra J, van Andel H, et al. MET signaling mediates intestinal crypt‐villus development, regeneration, and adenoma formation and is promoted by stem cell CD44 isoforms. Gastroenterology 2017; 153: 1040–1053.e4. [DOI] [PubMed] [Google Scholar]
- 28. Voloshanenko O, Erdmann G, Dubash TD, et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun 2013; 4: 2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miyoshi H, Ajima R, Luo CT, et al. Wnt5a potentiates TGF‐β signaling to promote colonic crypt regeneration after tissue injury. Science 2012; 338: 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Batlle E, Massagué J. Transforming growth factor‐β signaling in immunity and cancer. Immunity 2019; 50: 924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dekaney CM, Gulati AS, Garrison AP, et al. Regeneration of intestinal stem/progenitor cells following doxorubicin treatment of mice. Am J Physiol Gastrointest Liver Physiol 2009; 297: G461–G470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He XC, Yin T, Grindley JC, et al. PTEN‐deficient intestinal stem cells initiate intestinal polyposis. Nat Genet 2007; 39: 189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pons‐Tostivint E, Thibault B, Guillermet‐Guibert J. Targeting PI3K signaling in combination cancer therapy. Trends Cancer 2017; 3: 454–469. [DOI] [PubMed] [Google Scholar]
- 34. Vanhaesebroeck B, Perry MWD, Brown JR, et al. PI3K inhibitors are finally coming of age. Nat Rev Drug Discov 2021; 20: 741–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adams GN, Sharma BK, Rosenfeldt L, et al. Protease‐activated receptor‐1 impedes prostate and intestinal tumor progression in mice. J Thromb Haemost 2018; 16: 2258–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shimizu H, Nakayama KI. A universal molecular prognostic score for gastrointestinal tumors. NPJ Genom Med 2021; 6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Okamoto T, Natsume Y, Doi M, et al. Integration of human inspection and artificial intelligence‐based morphological typing of patient‐derived organoids reveals interpatient heterogeneity of colorectal cancer. Cancer Sci 2022; 113: 2693–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dudhwala ZM, Hammond PD, Howarth GS, et al. Intestinal stem cells promote crypt fission during postnatal growth of the small intestine. BMJ Open Gastroenterol 2020; 7: e000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Will OCC, Deheragoda M, Phillips RKS, et al. The role of cell proliferation and crypt fission in adenoma aggressiveness: a comparison of ileoanal pouch and rectal adenomas in familial adenomatous polyposis. Colorectal Dis 2011; 13: 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Michels BE, Mosa MH, Grebbin BM, et al. Human colon organoids reveal distinct physiologic and oncogenic Wnt responses. J Exp Med 2019; 216: 704–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buchanan FG, DuBois RN. Connecting COX‐2 and Wnt in cancer. Cancer Cell 2006; 9: 6–8. [DOI] [PubMed] [Google Scholar]
- 42. Wang D, Buchanan FG, Wang H, et al. Prostaglandin E2 enhances intestinal adenoma growth via activation of the Ras‐mitogen‐activated protein kinase cascade. Cancer Res 2005; 65: 1822–1829. [DOI] [PubMed] [Google Scholar]
- 43. Whale AD, Colman L, Lensun L, et al. Functional characterization of a novel somatic oncogenic mutation of PIK3CB. Signal Transduct Target Ther 2017; 2: 17063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Riemer P, Rydenfelt M, Marks M, et al. Oncogenic β‐catenin and PIK3CA instruct network states and cancer phenotypes in intestinal organoids. J Cell Biol 2017; 216: 1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sandoval IT, Delacruz RG, Miller BN, et al. A metabolic switch controls intestinal differentiation downstream of adenomatous polyposis coli (APC). Elife 2017; 6: e22706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Transcriptomic study of FAP crypts and organoids
Figure S2. Impact of EGF starvation on gene transcription in FAP organoids
Figure S3. EGF‐dependent ERK and AKT activation in FAP organoids
Figure S4. Implication of the TGF‐β receptor in activation of β‐catenin in adenomatous organoids
Figure S5. Impact of p110 and PAR modulation on FAP organoid development
Table S1. Description of human samples used in analyses
Table S2. Organoid culture medium components
Table S3. Primary and secondary antibodies
Table S4. Quantitative PCR primers
Data Availability Statement
The data that support the findings of this study are available upon request from the corresponding author (EM). Raw data are not publicly available because they contain information that could compromise the privacy of research participants.