

Abstract
Macrophages are critical mediators of tissue homeostasis, cell proliferation, and tumor metastasis. Tumor-associated macrophages (TAMs) are generally associated with tumor-promoting immunosuppressive functions in solid tumors. Here we examined the transcriptional landscape of adaptor molecules downstream of Toll-like receptors in human cancers and found that higher expression of MYD88 correlated with tumor progression. In murine melanoma, MyD88, but not Trif, was essential for tumor progression, angiogenesis, and maintaining the immunosuppressive phenotype of TAMs. Additionally, MyD88 expression in myeloid cells drove melanoma progression. The MyD88/interleukin-1 receptor (IL-1R) axis regulated programmed cell death (PD)-1 expression on TAMs by promoting recruitment of NF-κBp65 to the Pdcd1 promoter. Furthermore, a combinatorial immunotherapy approach combining the MyD88 inhibitor with anti–PD-1 blockade elicited strong anti-tumor effects. Thus, the MyD88/IL-1R axis maintains the immunosuppressive function of TAMs and promotes tumor growth by regulating PD-1 expression.
Significance
Findings indicate that MyD88 regulates TAM immunosuppressive activity, suggesting that macrophage-mediated immunotherapy by combining MYD88 inhibitors with PD-1 blockade could result in better treatment outcomes in a wide variety of cancers.
Keywords: Carcinoma, Immunosuppression, Immunotherapy, IL-1β, IL-1R, Melanoma, MYD88, PD-1, PD-L1, Tumor-associated macrophages
Graphical Abstract
The melanoma tumor microenvironment (TME) contains many infiltrated macrophages and are considered non-immunogenic with minimal cytotoxic T cells. These tumor-associated macrophages (TAMs) are thought to be associated with promoting tumor growth by suppressing effector T-cell responses to tumor cells and secretion of angiogenic factors. This is mediated by MyD88/IL-1R signaling resulting in recruitment of NF-κB at the PD-1 promoter. In the absence of MyD88, NF-κB recruitment at the PD-1 promoter is substantially reduced resulting in enhanced infiltration of activated CD8+ T cells in the TME. This cumulative effect possibly results in the decreased tumor growth and angiogenesis.
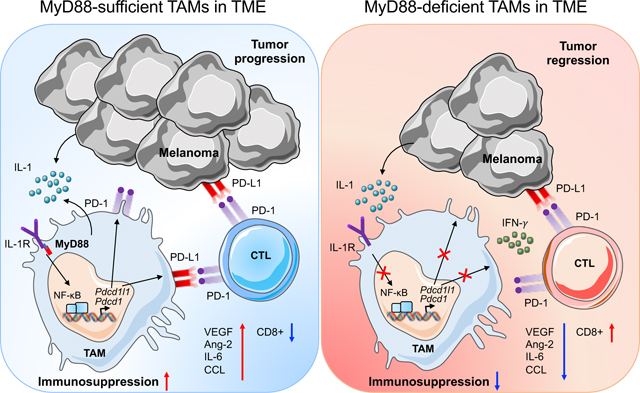
Introduction
Tumor-tissue myeloid cells play important roles in both anti-tumor immunity and tumor progression. These myeloid cells are actively recruited into the tumors to accelerate tumor progression by interacting with the tumor microenvironment (TME). Tumor-associated macrophages (TAMs) can promote growth by suppressing effector T-cell responses to tumor cells (1,2). These TAMs are considered phenotypically and functionally distinct from tissue-resident macrophages (3). Recent studies have demonstrated that the TME can influence the gene expression program of TAMs to maintain their immunosuppressive functions (2,4,5). It has long been known that in human solid tumors high macrophage infiltration is associated with poor prognosis (6,7). These observations led to the largely accepted view that the presence of TAMs correlates with poor prognosis in human cancers. Depending upon the stimulus, these TAMs can polarize towards an inflammatory ‘M1’ or pro-tumor ‘M2’ state (8).
The receptor programmed cell death 1 (PD-1) is known to be highly expressed by tumor-specific cytotoxic T lymphocytes and has been well studied in the context of malignancies associated with impaired T-cell function (9,10). Growing evidence suggests that PD-1 is also expressed on TAMs and that these PD-1+ TAMs express an M2-like tumor-promoting phenotype, whereas PD-1− TAMs show more of an M1-like inflammatory phenotype (11). Given that M2-like TAMs create an immunosuppressive microenvironment at the tumor site, they may serve as a potential therapeutic target in cancer (12).
Toll-like receptors (TLRs) and their downstream intracellular adaptor molecules have been explored widely in the context of inflammation and tumorigenesis (13,14). One of the adaptor molecules MyD88 associates with all TLRs, with the exception of TLR3, as well as with all receptors for the interleukin (IL)-1 family of cytokines (13,14). In addition to its role in promoting sterile inflammation in inflammatory skin disease models (15,16), MyD88-dependent signaling regulates the expression of several genes responsible for intestinal tumorigenesis and plays a critical role in both spontaneous and carcinogen-induced tumor development (17,18). MyD88 was also found to prevent lesion formation in a mouse model of colitis-associated cancer (CAC) as a result of altered colonic homeostasis (19). MyD88 has been shown to play a cell-autonomous role in Ras-mediated transformation via its interaction with the canonical mitogen-activated protein (MAP) kinase extracellular signal-regulated kinase (ERK) (20). Similar to MyD88, the involvement of IL-1 receptor (IL-1R) signaling in inflammation and tumorigenesis has also been studied extensively (21). Dysregulation of the IL-1R signaling pathway is often associated with human malignancies, such as breast cancer, glioma, and hepatic cancer (22–24). The CANTOS clinical trial has also suggested the involvement of IL-1β in the incidence of lung cancer and lung cancer mortality in addition to its role in inflammation (25). Despite their well-studied roles in innate immunity and inflammation, how IL-1R and MyD88 signaling regulate the function of TAMs in modulating tumorigenesis is not yet known.
In the present study, we examined the transcriptional landscape of intracellular adaptors of TLRs in human cancers and found that human MYD88 expression correlated with tumor progression. In addition, using well-established mouse models of melanoma and colon carcinoma, we found that mouse MyD88 was essential for tumor progression and angiogenesis. Interestingly, MyD88 deficiency led to impaired recruitment of F4/80+CD11b+ macrophages into the tumors. These observations motivated us to examine how MyD88 regulates the recruitment of F4/80+CD11b+ macrophages in the TME and the underlying mechanism for tumor progression. By deleting MyD88 specifically in the myeloid compartment, we showed that MyD88 expression in TAMs is important to drive PD-1 expression and reduce infiltration of CD8+ T cells in the TME, possibly regulating melanoma growth. A macrophage suppression assay further confirmed that these tumor-polarized wild-type (WT) macrophages exerted immunosuppressive effects on CD8+ T cells which can be rescued in tumor-polarized MyD88−/− or Il1r−/− macrophages. Mechanistically, we found that in macrophages the IL-1R/MyD88 axis promoted the recruitment of NF-κBp65 to the CR-C region of the Pdcd1 promoter. Combining the MyD88 inhibitor (MyD88i) with anti–PD-1 blockade resulted in improved outcomes compared with MyD88i treatment alone. Taken together, MyD88, an intracellular adaptor protein downstream of IL-1R, maintains the immunosuppressive functions of TAMs and promote tumor growth by regulating PD-1 expression.
Materials and Methods
Mice
MyD88−/− (ref. (26)), Il1r−/− (ref. (27)), Il1b−/− (ref. (28)), and Trif−/− (ref. (29)) mutant mice have been described previously. MyD88fl/fl mice were purchased from Jackson (30). MyD88fl/flLysMCre+ mice were generated by crossing MyD88fl/fl mice with LysMCre+ (ref. (31)) mice. Male and female 6–12-week-old mice (littermates) were used in this study unless otherwise mentioned. All mice were kept in specific pathogen-free conditions within the Animal Resource Center at St. Jude Children’s Research Hospital. All the animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of St. Jude Children’s Research Hospital, Memphis, TN. All the methods were performed in strict accordance with the relevant guidelines and regulations.
Cell lines
The mouse melanoma cell line B16-F10 (ATCC® CRL-6475™) and MC38 colon adenocarcinoma cell line (Kerafast #ENH204) were a kind gift from Prof. Osamu Takeuchi, Kyoto University, Japan. Mycoplasma detection was performed in Dr. Takeuchi’s lab by fixing the cells in an ice-cold mixture of acetic acid and methanol (freshly prepared 1:3 ratio) followed by DNA staining (Hoechst 33258). Cells were cultured right away upon receipt, and stocks were kept in liquid nitrogen. Cells were thawed and passaged 3–4 times before experiments. All cells were cultured in a humidified, 5% CO2 incubator at 37°C, and grown in DMEM with 10% fetal bovine serum (FBS) and 100 U mL−1 penicillin/streptomycin (Life Technologies).
Tumor model
Male and female 6–12-week-old mice were shaved on their lower back and engrafted with B16-F10 melanoma cells or MC38 colon adenocarcinoma cells by subcutaneously injecting 1 × 106 cells in 200 μL PBS. Tumors were used for flow cytometry and IHC at the end point of the experiment (day 15). Tumors were measured with digital calipers, and tumor volume was calculated using the formula: volume = (length × width2) × ½.
Histopathology
Formalin-fixed tumors were processed and embedded in paraffin by standard techniques, sectioned at 4 μm, mounted on positively charged glass slides (Superfrost Plus; Thermo Fisher Scientific) and dried at 60°C for 20 min. The two different antibodies used to detect macrophages were anti-Iba1 (1:300 dilution, CP290A; Biocare Medical, RRID:AB_10578940) and a rat monoclonal anti-F4/80 (1:500, clone BM8; Thermo Fisher Scientific, RRID:AB_1548747). All sections were examined by a pathologist blinded to the experimental group assignments. All images were acquired using light microscopy (Nikon Eclipse Ni Widefield Microscope).
Flow cytometry
Antibodies against F4/80 (BM8; RRID:AB_893481); CD45 (30-F11; RRID:AB_312973); Gr-1 (RB6–8C5; RRID:AB_313370); Ly-6C (HK1.4; RRID:AB_1186133); Ly-6G (1A8; RRID:AB_1877163); CD3ε (145–2C11; RRID:AB_312671); B220 (RA3–6B2; RRID:AB_312996); CD4 (RM4–5/GK1.5; RRID:AB_312715); CD8 (53–6.7; RRID:AB_312751); CD62L (MEL–14; RRID:AB_313094); CD44 (IM7; RRID:AB_312963); CD69 (H1.2F3; RRID:AB_313111); CD19 (1D3; RRID:AB_2629816); NK1.1 (PK136; RRID:AB_313394); CD279/PD-1 (29F.1A12; RRID:AB_10680238); CD274/PD-L1 (10F.9G2; RRID:AB_2073556); IFN-γ (XMG1.2; RRID:AB_315399) were purchased from BioLegend. The antibody against CD11b (M1/70; RRID:AB_1582236) was purchased from Invitrogen. Single-cell suspensions were prepared from the spleen by passing through a cell strainer to remove cell debris. To deplete the red blood cells, an equal volume of ACK red blood cell lysis buffer was added to the cells and incubated at room temperature for 5 min. For staining, cells were washed in ice cold flow cytometry buffer (0.5% (vol/vol) FCS and 2 mM EDTA in PBS, pH 7.5), then incubated with each antibody for 30 min, washed twice and resuspended in an appropriate volume of flow cytometry buffer. Tumors were harvested, dissociated with scissors and then digested with 10 mL HBSS containing 10 μg/mL DNase I (Sigma; #D4527) and 25 μg/mL Liberase™ (LIBTM-RO Roche; #5401119001) for 40 min at 37°C with continuous shaking. After dissociation, tumor suspensions were filtered through a 40-μm filter, put on ice, washed with cold PBS, and resuspended in flow cytometer buffer. Cells were blocked with anti-mouse CD16/32 antibody (2.4G2; BD Biosciences; RRID:AB_394656) for 15 minutes on ice, before being stained with antibody panel below. For cell sorting of F4/80+CD11b+ TAMs, a single cell suspension of whole tumor was stained with antibodies specific for mouse CD45, CD3 and B220, NK1.1, F4/80 and CD11b. The cells were sorted as live, CD45+CD3−B220−NK1.1−F4/80+CD11b+ (TAMs) on Reflection (i-Cyt), at the Core Instrumentation Facility of the Department of Immunology at St. Jude Children’s Research Hospital. For staining of BMDMs, 2 × 106 cells were stained for anti-CD279/PD-1 and anti-CD274/PD-L1. Flow cytometry data were acquired on the LSR Fortessa or LSR II (BD Biosciences) and analyzed using Flowjo software (Tree Star); RRID: SCR_008520.
ELISA
ELISA was performed on the serum samples collected from tumor bearing mice according to the manufacturer’s instructions using the MAGPMAG-24K kit (Millipore).
Chromatin immunoprecipitation (ChIP)
ChIP was performed by using the EZ-Magna ChIP™ assay kit (Millipore; 17–10086) as described elsewhere with little modification (32). Briefly, BMDMs (5 × 106 cells) were co-cultured with B16-F10 melanoma cells for 48 hours using transwell plates (Costar, Corning) and were fixed with 1% formaldehyde (Sigma; F8775) for 10 min at 37°C. Cells were then washed twice with ice cold PBS and resuspended in the lysis buffer supplied with the kit. Lysates were sonicated using the ultrasonicator (Covaris S2) to obtain DNA fragments with a peak in size between 150 and 300 bp. Lysates pre-cleared with the protein A/G beads (provided with the kit) were incubated with anti–NF-κBp65 (D14E12) XP® (Cell Signaling Technologies, #8242; RRID:AB_10859369) or normal mouse IgG (negative control; provided with the kit) and immune-precipitated at 4°C overnight. Beads were washed once with low salt buffer, high salt buffer, LiCl wash buffer, and twice with TE buffer. Immune complexes were extracted with elution buffer containing proteinase K for 2 hours at 62°C followed by a 10-minute incubation at 95°C. DNA was then purified using spin columns provided with the kit. The purified DNA was quantified and used for qPCR analysis to assess the presence of target sequences. Quantitative RT-PCR was performed with SYBR green qPCR mix (4368706; Applied Biosystems) in an Applied Biosystems 7500. Primers used for amplifying the CR-C region of the Pdcd1 promoter, were published previously (33). ChIP values were normalized against the input and expressed as relative enrichment of the material precipitated by the indicated antibody on the Pdcd1 promoter [relative quantification using the comparative Ct method (2−ΔΔCt)]. Error bars indicate mean ± standard deviation. The results are representative of at least two independent experiments.
Quantitative PCR Analysis
Total RNA was extracted using TRIzol (15596026; Thermo Fisher Scientific) and converted into cDNA by using the High-Capacity cDNA Reverse Transcription Kit (4368814, Applied Biosystems). Real-time quantitative PCR was performed on an Applied Biosystems 7500 real-time PCR instrument with 2× SYBR Green (4368706; Applied Biosystems). To determine the relative induction of cytokine mRNA in response to various stimuli, the mRNA expression level of each gene was normalized to the expression level of Gapdh mRNA. The following primer pairs were used for quantitative PCR analysis: MmPdcd1 (Cd279) forward, 5′-CGGTTTCAAGGCATGGTCATTGG-3’, and reverse, 5′-TCAGAGTGTCGTCCTTGCTTCC-3’; MmPdcd1lg1 (Cd274) forward, 5′-TGCGGACTACAAGCGAATCACG-3’, and reverse, 5′-CTCAGCTTCTGGATAACCCTCG-3’; MmGapdh forward, 5′-CGTCCCGTAGACAAAATGGT-3′, and reverse, 5′-TTGATGGCAACAATCTCCAC-3′. Error bars indicate mean ± standard deviation. The results are representative of at least two independent experiments.
Macrophage B16 co-culture assay
Macrophages (1 × 106) from WT, MyD88−/−, Il1r−/− or Trif−/− mice were treated with LPS (Invivogen; #tlrl-smlps) or recombinant IFN-γ (Peprotech; #315–05) for 4 hours or co-cultured with 1 × 106 B16 tumor cells in a 12-well transwell plate (Corning® #3401) for 48 hours. PD-1 and PD-L1 expression was measured by flow cytometry.
Macrophage suppression assay
Macrophages (1 × 106) from WT, MyD88−/− or Il1r−/− mice were co-cultured with 1 × 106 B16 tumor cells in a 12-well transwell plate for 48 hours. Sort-purified splenic CD8+ T cells from WT mice were plated at a density of 1 × 106 cells together with tumor cell polarized macrophages and stimulated with anti-CD3 (5 μg/mL) and anti-CD28 (1 μg/mL). Intracellular expression of IFN-γ on these CD8+ T cells was assessed by flow cytometry after 2 days.
Analysis of TCGA expression data
Gene expression data (FPKM) for MYD88, TIRAP, TICAM1 and TICAM2 were downloaded from TCGA and analyzed by two-way ANOVA in GraphPad Prism v7.0. Differences were considered statistically significant when P < 0.05. ns not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Microarray data analysis
Transcripts were profiled for TAMs obtained from WT and MyD88−/− mice. Total RNA (100 ng) was converted into biotin-labeled cDNA by using an Affymetrix Whole Transcript Plus Expression kit (Thermo Fisher Scientific, 902281) and was hybridized to an Affymetrix Clariom S Mouse Genechip Array (Thermo Fisher Scientific, 902930). After chips were stained and washed, array signals were normalized and transformed into log2 transcript expression values by using the robust multi-array average algorithm (Affymetrix Expression Console v1.1) (34). Differential expression was defined by application of a threshold of FDR < 0.1 using the Cyber-T t-test (35). Lists of differentially expressed transcripts were analyzed for ‘functional enrichment’ by using the DAVID bioinformatics database (36) and Ingenuity Pathways Analysis software (QIAGEN). Pathways with altered activity levels were identified by using the Gene Set Enrichment Analysis (GSEA) (37) with curated pathways obtained from The Broad Institute (http://software.broadinstitute.org/gsea/msigdb/index). Gene expression data generated in this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) and is accessible through GEO Series accession number GSE137046. Microarray datasets generated from human melanoma and normal skin samples [GSE15605 (ref. (38)); GSE7553 (ref. (39)); GSE46517 (ref. (40))] were downloaded from the GEO repository (www.ncbi.nlm.nih.gov/geo/). Individual expression profiles (log2 signal) for MYD88 and TICAM1 were extracted from each dataset and investigated for differential expression between normal skin and primary melanoma by using Welch’s t-test (GraphPad Prism software, v7.0).
MyD88-inhibitor and anti–PD-1 treatment
For in vivo treatment, mice were treated with intra-tumor delivery of 500 μM of MyD88i (Novus Biologicals; NBP2–29328) or antenna peptide or 15 mg/kg InVivoPlus anti-mouse PD-1 (clone RMP 1–14; Bio X Cell; RRID:AB_10949053) resuspended in PBS on day 3, 7 and 10 post-tumor injection. For combination immunotherapy, anti-mouse PD-1 was used at a concentration of 15 mg/kg along with MyD88i resuspended in PBS on day 3, 7 and 10 post-tumor injection.
Statistical analysis
All results are presented as mean ± standard deviation. Statistical analysis between multiple samples was performed using the two-way ANOVA with Sidak’s or Tukey’s or Dunnett’s multiple comparison test and one-way ANOVA with Dunnett’s multiple comparison test. qPCR data were analyzed by the unpaired t-test with Welch’s correction or Kruskal-Wallis test. All the analysis was done using GraphPad Prism software (version 7.0); RRID:SCR_002798. No statistical methods were used to predetermine sample size. Differences were considered statistically significant when p < 0.05. ns not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Results
Transcriptional landscape of MYD88 expression in human cancers
The Cancer Genome Atlas (TCGA) project covers the genome-wide effect of individual genes on tumor growth (41). Understanding the genomic mutations in tumors is helping to improve patient diagnostics and treatment outcomes. To this end, the human pathology atlas provides a comprehensive database to explore the prognostic role of each protein-coding gene in different cancers (42) (https://www.proteinatlas.org/humanproteome/pathology). Given that the role of TLRs and IL-1Rs has been extensively studied in various cancers, we investigated the human pathology atlas to understand the expression patterns of all four adaptors MYD88, TIRAP, TICAM1, and TICAM2, that act downstream of TLRs in the most frequently occurring cancers. Among all the cancers we analyzed (melanoma, breast, colorectal, ovarian, lung and stomach cancer), we found MYD88 to be the most highly expressed compared with the other adaptors for TLRs (Fig. 1A and Supplementary Table S1).
Fig. 1: Transcriptional landscape of MYD88 expression in human cancers.
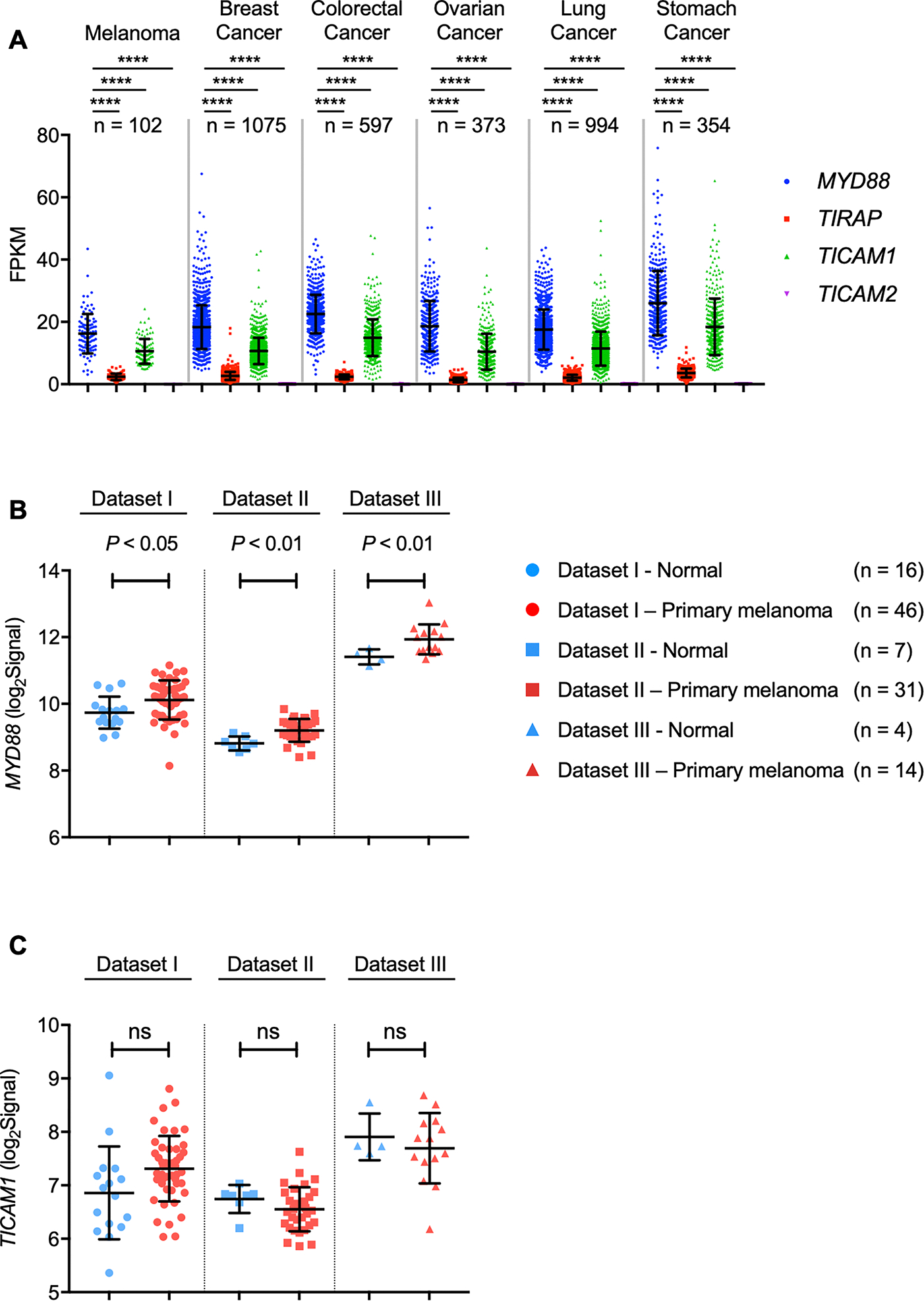
A, Expression profiles of MYD88, TIRAP, TICAM1 and TICAM2 in six human cancers. Expression data (FPKM) were downloaded from TCGA and analyzed by two-way ANOVA with Tukey’s multiple comparison test (****P < 0.0001).
B–C, Comparison of expression of MYD88 (B) and TICAM1 (C) between normal skin and primary melanoma tumors. Microarray data from three separate studies [Dataset I (ref. (38)), Dataset II (ref. (40)), Dataset III (ref. (39))] were downloaded from GEO, and individual gene profiles were analyzed within each study using a two-tailed Welch’s t-test.
Melanoma is regarded as a serious form of skin cancer originating in melanocytes, and it is considered dangerous because of its ability to rapidly spread to other organs if not treated at an early stage. Since we observed that in addition to MYD88, TICAM1 was also expressed highly among all the cancers analyzed, we investigated the profiles of these adaptors in published microarray studies of human normal and primary melanoma tissues (38–40). Within each study, we found MYD88 was consistently expressed at a higher level in primary melanoma than in normal tissue (Fig. 1B). However, TICAM1 (Fig. 1C) was not observed to be differentially expressed between tumor and normal samples. These results indicate that MYD88 is highly expressed across a broad range of different cancer types, and that MYD88 expression levels tend to be higher than those of other adaptor molecules downstream of TLRs. Further, in several human melanoma studies, MYD88 showed increased expression in primary tumors compared with normal tissue, suggesting MYD88 expression might be associated with melanoma progression.
MyD88 promotes melanoma tumor progression by regulating TAM infiltration and angiogenesis
Based on the expression differences observed between MYD88 in primary melanoma and normal skin, we further investigated whether deletion of MyD88 affected tumorigenicity in vivo by using a mouse tumor model. In an acute solid tumor model, 1 × 106 B16-F10 melanoma cells were subcutaneously injected into WT and MyD88−/− mice; mice were monitored for 2 weeks for tumor growth. We found that MyD88−/− mice exhibited significantly impaired tumor growth and tumor weight compared with WT mice (Fig. 2A–C). Given that the presence of TAMs correlates with poor prognosis in human cancers, we performed immunohistochemistry (IHC) on tumors harvested from MyD88−/− and WT mice. IHC staining showed massive infiltration of F4/80+ myeloid cells in the tumor tissues in WT mice (Fig. 2D). Surprisingly, this infiltration of F4/80+ cells was substantially ameliorated in the tumors harvested from MyD88−/− mice (Fig. 2D). Next, we performed staining for the pan-macrophage marker Iba-1, and, consistent with what we observed with the F4/80+ staining, tumors from WT mice showed increased infiltration of Iba-1+ cells, which was drastically reduced in the tumors from MyD88−/− mice (Fig. 2E).
Fig. 2: MyD88 promotes tumor progression by regulating TAM infiltration and angiogenesis.
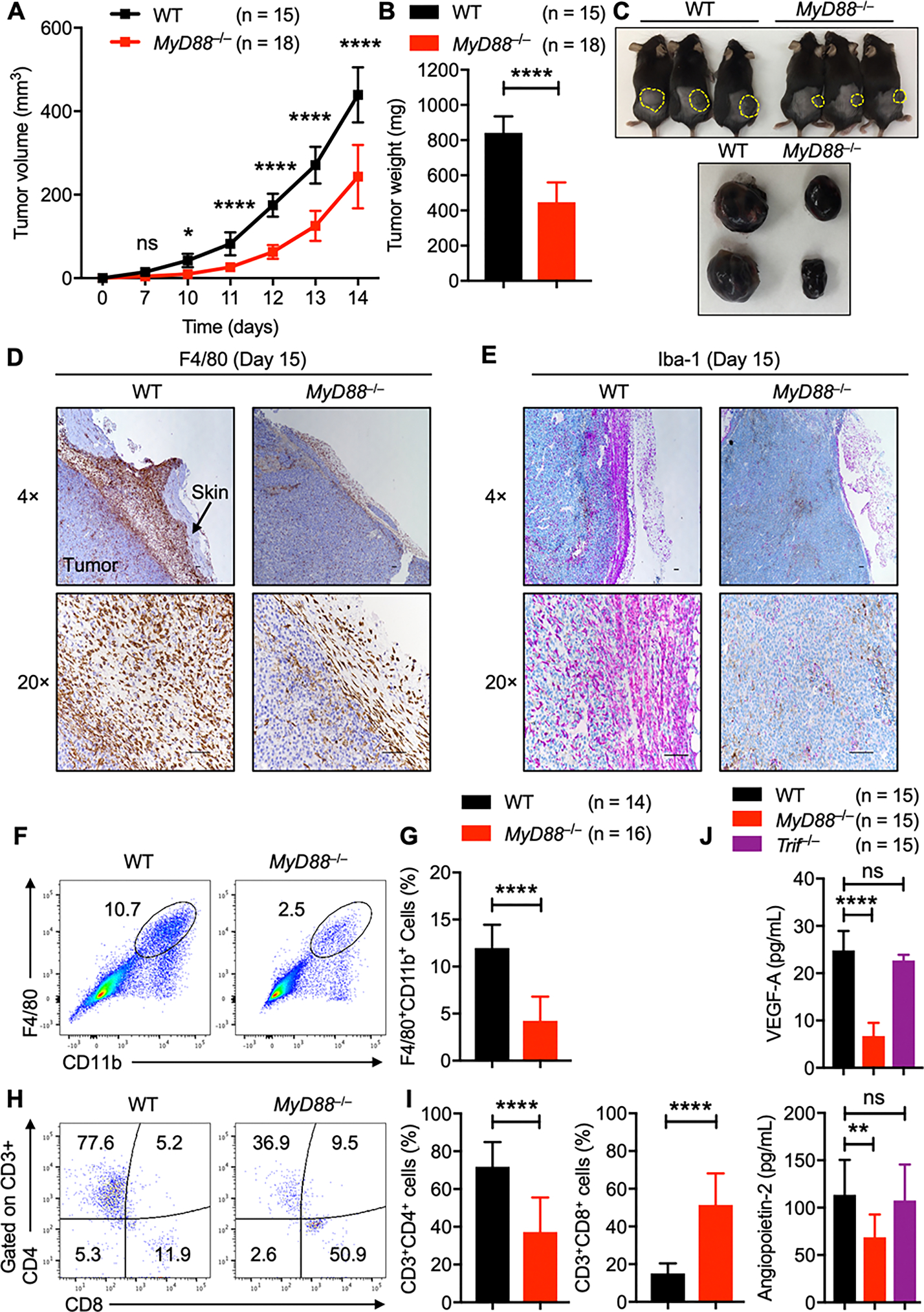
A–C, B16-F10 melanoma cells were injected into WT and MyD88−/− mice. (A) Mean tumor volume in WT (n = 15) and MyD88−/− (n = 18) mice. (B) Tumor weights of WT (n = 15) and MyD88−/− (n = 18) mice, 2 weeks after tumor cell injection. (C) Representative pictures of tumors from WT and MyD88−/− mice.
D–E, Representative images from immunohistochemistry staining of tumors harvested from WT (n = 6) and MyD88−/− (n = 6) mice stained with (D) anti-F4/80 and (E) anti-Iba-1. (Scale bar, 100 μm).
F–I, Flow cytometry analysis of tumors harvested from WT (n = 14) and MyD88−/− (n = 16) mice. (F) Pseudocolor plots of F4/80+CD11b+ macrophage infiltration in tumors. (G) Quantification of F4/80+CD11b+ macrophage infiltration in tumors. (H) Pseudocolor plots of CD3+CD4+ and CD3+CD8+ T cell infiltration in tumors. (I) Quantification of CD3+CD4+ and CD3+CD8+ T-cell infiltration in tumors.
(J) Quantification of VEGF-A and angiopoietin-2 by ELISA in the serum harvested from tumor-bearing WT (n = 15), MyD88−/− (n = 15), and Trif−/− (n = 15) mice.
Data are presented as mean ± SD. (A) Two-way ANOVA with Sidak’s multiple comparison test, (B, G and I) unpaired t-test with Welch’s correction and (J) one-way ANOVA with Dunnett’s multiple comparison test were used to determine the significance between the two groups analyzed. ns not significant, * P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Since Trif is a mouse homologue of human TICAM1, we next subcutaneously injected 1 × 106 B16-F10 melanoma cells into WT and Trif−/− mice. Trif−/− mice did not show any difference in the tumor development and tumor weight compared with WT mice (Supplementary Fig. S1A–C). Consistent with having no defect in the tumor growth, IHC staining showed comparable levels of infiltration by F4/80+ myeloid cells in the tumors harvested from WT and Trif−/− mice (Supplementary Fig. S1D), suggesting that the reduced tumor growth phenotype we had observed in MyD88−/− mice was specific to MyD88.
To check the extent of immune cell infiltration, we subjected the tumor samples to flow cytometry analysis. Cellular analysis showed higher levels of F4/80+CD11b+ macrophage cell infiltration in tumors from WT mice than in those from MyD88−/− mice (Fig. 2F and G). Melanomas are considered less-immunogenic than other tumors, with minimal CD8+ T-cell infiltration. Consistent with this, we found that tumors from WT mice showed low levels of CD3+CD8+ T-cell infiltration; however, MyD88−/− mice showed an increase in the CD3+CD8+ T-cell population (Fig. 2H and I). A subtle defect in the recruitment of the Gr1+CD11b+ granulocyte population was also observed in the tumors from MyD88−/− mice, but no difference was noticed in the B220+ (B cells) and NK1.1+ (NK) cell populations compared with the tumors from WT mice (Supplementary Fig. S2A and B). Since the spleen has been identified as a reservoir of immune cells which can play a significant role in the inflammatory response that follows acute injury(43), we analyzed the immune cell population in spleens from tumor-bearing WT and MyD88−/− mice. As shown in Supplementary Fig. S2C–H, no significant differences were observed between myeloid, B-, or overall T-cell populations in WT and MyD88−/− mice. In contrast, the CD3+CD8+ T-cell population was slightly increased in the spleens harvested from tumor-bearing MyD88−/− mice compared with WT mice (Supplementary Fig. S2I and J). These T cells analyzed from tumor-bearing MyD88−/− mice tended to be more activated compared with those from WT mice (Supplementary Fig. S2K and L).
We next investigated the effect of MyD88 deletion on angiogenesis, a process necessary to ensure a sufficient supply of oxygen and nutrients for expanding solid tumors. We measured the levels of vascular endothelial growth factor (VEGF)-A and angiopoietin-2 in the serum from the tumor-bearing WT, MyD88−/−, and Trif−/− mice. While the production of VEGF-A and angiopoietin-2 were comparable between WT and Trif−/− mice, MyD88−/− mice produced significantly less VEGF-A and angiopoietin-2 (Fig. 2J), implying that the process of angiogenesis is altered in the absence of MyD88. Collectively, these results suggest that MyD88 signaling promotes tumor progression by driving the infiltration of TAMs in the TME, possibly suppressing CD8+ T-cell recruitment and activation in addition to angiogenesis in melanoma-bearing mice.
MyD88 regulates PD-1/PD-L1 expression on TAMs
The receptor PD-1 is one of the best-studied and clinically most successful immune checkpoint drug targets, and its primary function is widely understood in the context of T cells. However, recent studies suggest that in addition to T cells, the TAMs also express PD-1 (11). We hypothesized that the tumor phenotype observed in the MyD88−/− mice could be due to a defect in the PD-1 expression on these TAMs. To assess the expression of PD-1, we performed PD-1 staining on the TAMs harvested from WT, MyD88−/−, and Trif−/− mice. In accordance with the previous reports, TAMs harvested from WT mice expressed both PD-1 and PD-L1 (Fig. 3A). MyD88−/− TAMs showed reduced expression of PD-1 and PD-L1 (Fig. 3A). On the contrary, TAMs harvested from Trif−/− mice did not show any reduction in PD-1 or PD-L1 expression when compared with TAMs from WT mice (Fig. 3B).
Fig. 3: MyD88 regulates PD-1/PD-L1 expression on TAMs.
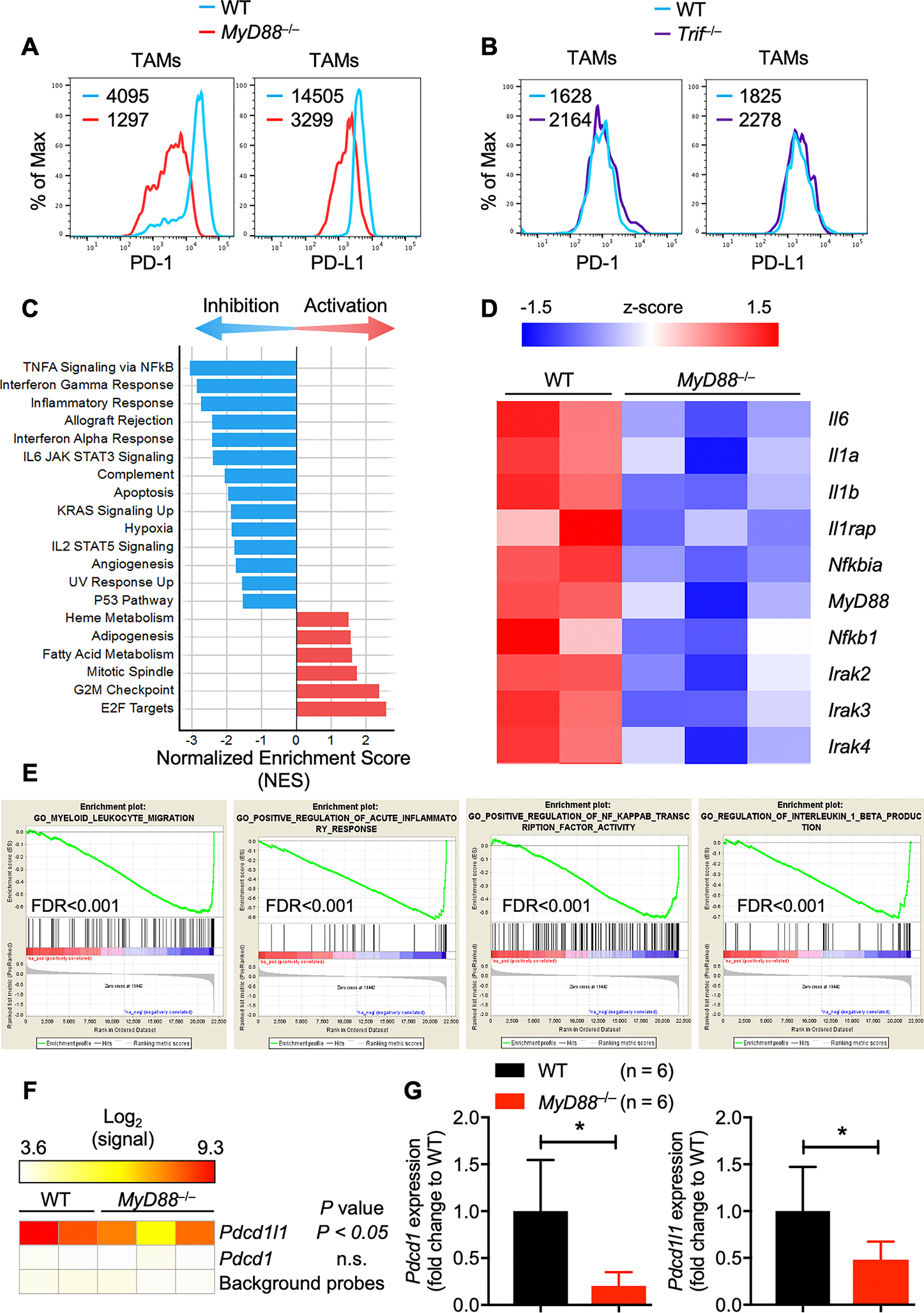
A, Flow cytometry analysis of PD-1 and PD-L1 expression on F4/80+CD11b+ TAMs in tumors harvested from WT and MyD88−/− mice.
B, Flow cytometry analysis of PD-1 and PD-L1 expression on F4/80+CD11b+ TAMs in tumors harvested from WT and Trif−/− mice.
C, Altered pathway activity in MyD88−/− TAMs. TAM profiles from WT and MyD88−/− mice were analyzed by Gene Set Enrichment Analysis (GSEA). Hallmark pathways with FDR < 0.05 showing inhibition (blue) or activation (red) are shown by the normalized enrichment score (NES).
D, Inhibition of key inflammatory genes in MyD88−/− TAMs. Microarray profiles (z-score) of NF-κB and IL-1 responsive genes are shown for WT and MyD88−/− TAMs.
E, Inhibition of Gene Ontology (GO) biological processes in MyD88−/− TAMs. Myeloid-leukocyte migration, acute inflammatory responses, NF-κB transcription factor activity and IL-1β production enrichment plots (normalized enrichment score < −2 and FDR q-value < 0.001 in MyD88−/− TAMs compared with WT TAMs).
F, Expression profile of Pdcd1 and Pdcd1l1 in MyD88−/− TAMs. Profiles were compared between WT and MyD88−/− samples by ANOVA, and P values are reported in the figure.
G, Quantitatve PCR analysis of Pdcd1 and Pdcd1l1 mRNA expression in F4/80+CD11b+ TAMs sorted from WT (n = 6) and MyD88−/− (n = 6) mice. All samples were normalized to the expression of the endogenous control Gapdh. Data are presented as mean ± SD. Unpaired t-test, with Welch’s correction was used to determine the statistical significance. *P < 0.05.
To comprehensively examine the effect of MyD88 deficiency in F4/80+CD11b+ TAMs, we sort purified F4/80+CD11b+ macrophages from the tumors of WT and MyD88−/− mice (gating strategy; Supplementary Fig. S3) and examined the genome-wide changes in gene expression by microarray analysis. As anticipated, all the major NF-κB and IL-1R target pathways (Fig. 3C, GSEA panels) and genes (Fig. 3D, heatmap of NF-κB and IL-1 genes) were downregulated in TAMs harvested from B16 tumor-transplanted MyD88−/− mice compared with those from WT mice. Among the gene ontology biological processes, myeloid-leukocyte migration, acute inflammatory responses, NF-κB transcription factor activity and IL-1β production were highly downregulated (normalized enrichment score < −2 and FDR q-value < 0.001) (Fig. 3E). Since we observed a decrease in PD-1 and PD-L1 expression on the F4/80+CD11b+ TAMs from MyD88−/− mice, we looked for a corresponding decrease in transcript levels based on the microarray data. Consistent with the flow cytometry analysis, F4/80+CD11b+ TAMs harvested from WT mice showed robust expression of Pdcd1l1, and this expression was substantially reduced in TAMs from MyD88−/− mice (Fig. 3F). However, the microarray data did not provide any insight into Pdcd1 expression, as the probe signals for all TAM samples were the same as background measurements (Fig. 3F). Therefore, we performed qPCR to directly assess Pdcd1 and Pdcd1l1 transcription in WT and MyD88−/− TAMs. The qPCR data confirmed the microarray results, showing a significant reduction in Pdcd1l1 expression in MyD88−/− TAMs compared with WT (Fig. 3G). Further, MyD88−/− TAMs also showed substantially reduced Pdcd1 expression compared with TAMs from WT mice (Fig. 3G). Taken together, the microarray and qPCR data corroborate the FACS findings and show substantially reduced transcription of Pdcd1 and Pdcd1l1 mRNA in the TAMs from MyD88−/− mice. These results suggest that MyD88 is a critical regulator of PD-1/PD-L1 expression in F4/80+CD11b+ TAMs.
IL-1R signaling in TAMs drives PD-1/PD-L1 expression in melanoma
Given that IL-1R signaling is the most downregulated in F4/80+CD11b+ TAMs harvested from MyD88−/− mice compared with WT mice (Fig. 3D), we hypothesized that TAMs from Il1r−/− mice should behave similarly to the TAMs from MyD88−/− mice. We transferred 1 × 106 B16-F10 melanoma cells subcutaneously into WT and Il1r−/− mice; mice were monitored for 2 weeks for tumor growth. We found that Il1r−/− mice exhibited significantly impaired tumor growth compared to WT control mice (Fig. 4A and B). Since IL-1β is a well-known inflammation-modulating ligand that is processed through IL-1R, we further speculated that Il1b−/− mice would phenocopy Il1r−/− mice. Consistent with the findings in Il1r−/− mice, Il1b−/− mice had a reduction in the tumor size and weight compared to WT mice (Fig. 4C and D). Since the major defects we found with MyD88−/− mice were the impaired infiltration of F4/80+CD11b+ TAMs and the reduced PD-1 and PD-L1 expression on TAMs, we performed flow cytometry analysis of tumors from Il1r−/− and Il1b−/− mice to check for these defects. Consistent with the results seen in MyD88−/− mice, Il1r−/− and Il1b−/− mice showed impaired infiltration of F4/80+CD11b+ TAMs in the TME (Fig. 4E and F). Compared with the infiltration in WT mice, CD3+CD8+ T-cell infiltration was increased in the tumors from Il1r−/− and Il1b−/− mice (Fig. 4G and H), suggesting that IL-1β could possibly be driving signaling through the IL-1R-MyD88 axis, thereby regulating myeloid cell infiltration and tumor progression. The Gr1+CD11b+ granulocyte population was found to be slightly reduced in the tumors from both Il1r−/− and Il1b−/− mice, whereas the NK cell population was slightly increased only in Il1b−/− mice compared with the tumors from WT mice (Supplementary Fig. S4A and B). Next, we analyzed the expression of PD-1 and PD-L1 on the F4/80+CD11b+ TAMs from Il1r−/− and Il1b−/− mice by flow cytometry. F4/80+CD11b+ TAMs from Il1r−/− and Il1b−/− mice showed decreased PD-1 and PD-L1 expression compared to TAMs from WT mice (Fig. 4I–4L). To understand whether there was a decrease at the transcriptional level, we sort purified F4/80+CD11b+ TAMs from Il1r−/− and Il1b−/− mice (gating strategy; Supplementary Fig. S3) and performed qPCR to determine the changes in the expression of Pdcd1 and Pdcd1l1 mRNA. Consistent with the flow cytometry data, we observed reductions in the Pdcd1 and Pdcd1l1 mRNA levels from Il1r−/− and Il1b−/− TAMs compared with TAMs from WT mice (Fig. 4M and N). These results suggest that IL-1β–mediated IL-1R signaling can drive melanoma progression by inducing PD-1 and PD-L1 expression on TAMs.
Fig. 4: IL-1R signaling in TAMs drives PD-1/PD-L1 expression in melanoma.
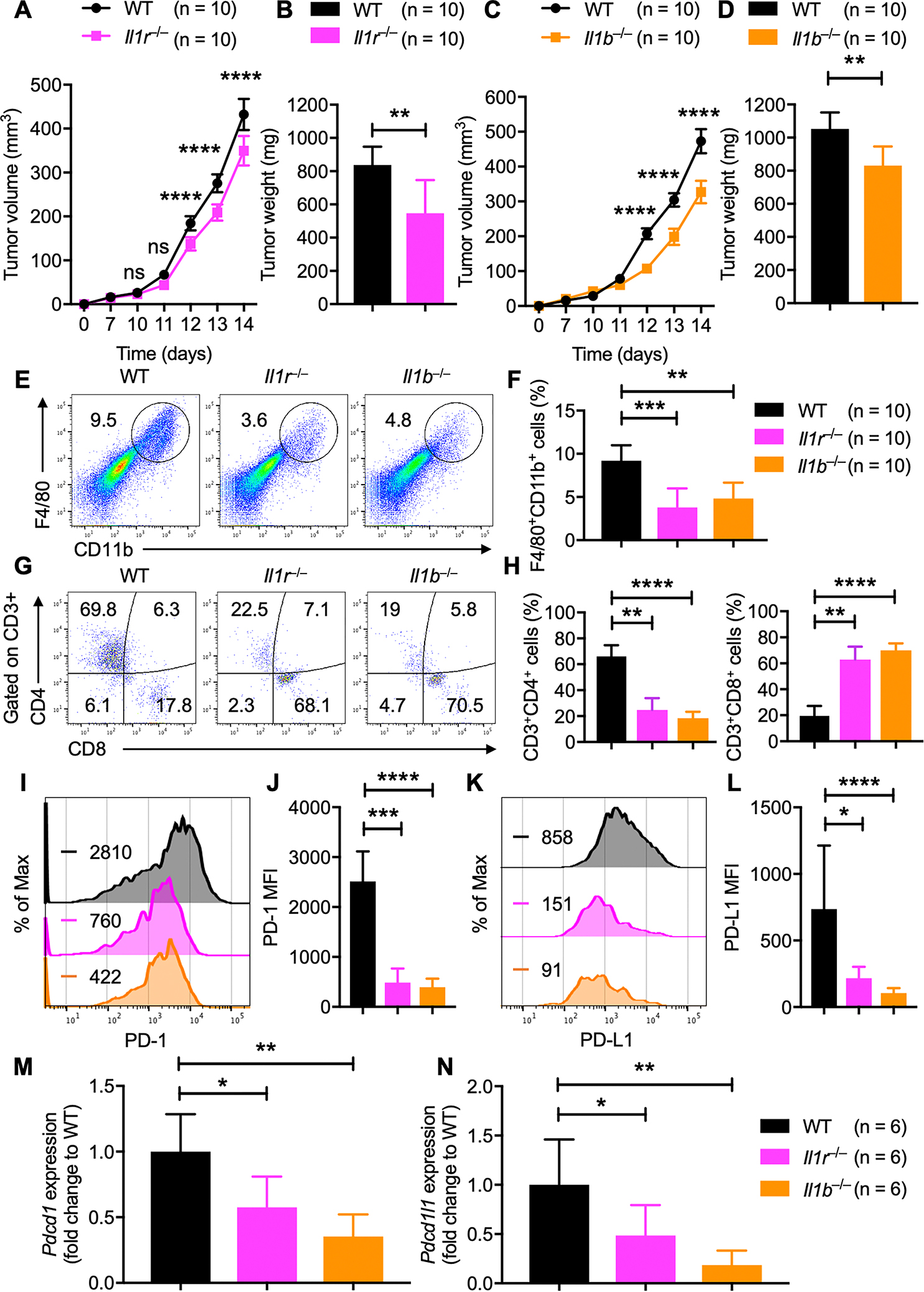
A–B, B16-F10 melanoma cells were injected into WT and Il1r−/− mice. (A) Mean tumor volume in WT (n = 10) and Il1r−/− (n = 10) mice. (B) Tumor weights of WT (n = 10) and Il1r−/− (n = 10) mice, 2 weeks after tumor cell injection.
C–D, B16-F10 melanoma cells were injected into WT and Il1b−/− mice. (C) Mean tumor volume in WT (n = 10) and Il1b−/− (n = 10) mice. (D) Tumor weights of WT (n = 10) and Il1b−/− (n = 10) mice, 2 weeks after tumor cell injection.
E–H, Flow cytometry analysis of tumors harvested from WT (n = 10), Il1r−/− (n = 10) and Il1b−/− (n = 10) mice. (E) Pseudocolor plots of F4/80+CD11b+ macrophage infiltration in tumors. (F) Quantification of F4/80+CD11b+ macrophage infiltration in tumors. (G) Pseudocolor plots of CD3+CD4+ and CD3+CD8+ T-cell infiltration in tumors. (H) Quantification of CD3+CD4+ and CD3+CD8+ T-cell infiltration in tumors.
I–L, Flow cytometry analysis of PD-1 and PD-L1 expression on F4/80+CD11b+ TAMs in tumors harvested from WT (n = 10), Il1r−/− (n = 10) and Il1b−/− (n = 10) mice. (I) Histogram of PD-1 expression. (J) MFI of PD-1 expression from TAMs. (K) Histogram of PD-L1 expression. (L) MFI of PD-L1 expression from TAMs.
M–N, Quantitatve PCR analysis of (M) Pdcd1 and (N) Pdcd1l1 mRNA expression in F4/80+CD11b+ TAMs sorted from WT (n = 6), Il1r−/− (n = 6) and Il1b−/− (n = 6) mice. All samples were normalized to the expression of the endogenous control Gapdh.
Data are presented as mean ± SD. (A, C) Two-way ANOVA with Sidak’s multiple comparison test, (B, D, M, N) unpaired t-test with Welch’s correction and (F, H, J, L) Kruskal-Wallis test were used to determine the statistical significance. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We next analyzed the immune cell populations in spleens from tumor-bearing WT, Il1r−/− and Il1b−/− mice by flow cytometry. As shown in Supplementary Fig. S4C–J, no significant differences were observed in myeloid, B-cell or CD4+ or CD8+ T-cell populations from WT, Il1r−/−, and Il1b−/− mice. Although no significant difference was found between activated T-cell populations, naïve T-cell populations were significantly reduced in tumor-bearing Il1r−/− and Il1b−/− mice compared with WT mice (Supplementary Fig. S4K and L). These results suggest that IL-1R signaling upstream of MyD88 drives the infiltration of inflammatory macrophages in the TME possibly suppressing CD8+ T cell recruitment and activation.
Myeloid cell-intrinsic MyD88 is sufficient to promote melanoma progression
Since the major defect in MyD88−/− mice was observed with the macrophage population in the tumors, we hypothesized that MyD88 in macrophages is playing an essential role in promoting tumorigenesis. To dissect the functional significance of MyD88 in myeloid cells, we crossed MyD88fl/fl mice with LysMCre mice to deplete MyD88 specifically in the myeloid cell compartment. Next, we subcutaneously injected 1 × 106 B16-F10 melanoma cells into WT, MyD88fl/+LysMCre+ (hereafter denoted as MyD88Ctrl), and MyD88fl/flLysMCre+ (hereafter denoted as MyD88ΔMye) littermate mice, and tumor growth was monitored. Consistent with the results in MyD88−/− mice, myeloid cell-specific deletion of MyD88 caused significantly impaired tumor growth compared with that observed in WT and MyD88Ctrl mice (Fig. 5A–C). These data prompted us to hypothesize that myeloid cell-specific expression of MyD88 is sufficient to drive progression of melanoma.
Fig. 5: MyD88 in myeloid cells is sufficient to promote melanoma progression.
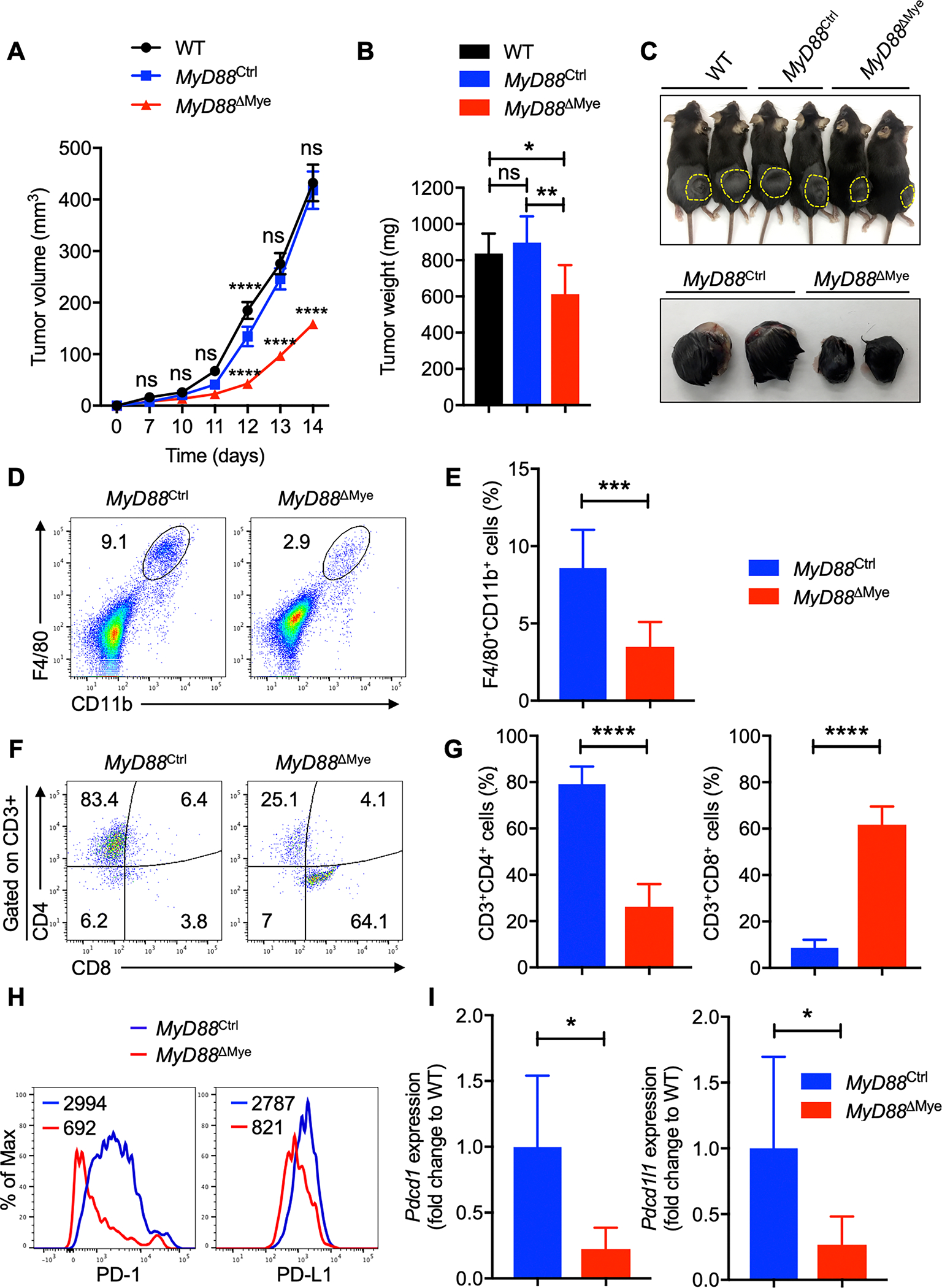
A–C, B16-F10 melanoma cells were injected into WT, MyD88Ctrl and MyD88ΔMye mice. (A) Mean tumor volume in WT (n = 7), MyD88Ctrl (n = 7) and MyD88ΔMye (n = 7) mice. (B) Tumor weights of WT (n = 7), MyD88Ctrl (n = 7), and MyD88ΔMye (n = 7) mice, 2 weeks after tumor cell injection. (C) Representative pictures of tumors from WT, MyD88Ctrl and MyD88ΔMye mice.
D–G, Flow cytometry analysis of tumors harvested from MyD88Ctrl (n = 9) and MyD88ΔMye (n = 13) mice. (D) Pseudocolor plots of F4/80+CD11b+ macrophage infiltration in tumors. (E) Quantification of F4/80+CD11b+ macrophage infiltration in tumors. (F) Pseudocolor plots of CD3+CD4+ and CD3+CD8+ T-cell infiltration in tumors. (G) Quantification of CD3+CD4+ and CD3+CD8+ T-cell infiltration in tumors.
H, Flow cytometry analysis of PD-1 and PD-L1 expression on F4/80+CD11b+ TAMs in tumors harvested from MyD88Ctrl and MyD88ΔMye mice.
I, Quantitatve PCR analysis of Pdcd1 and Pdcd1l1 mRNA expression in F4/80+CD11b+ TAMs sorted from MyD88Ctrl (n = 6) and MyD88ΔMye (n = 6) mice. All samples were normalized to expression of the endogenous control Gapdh.
Data are presented as mean ± SD. (A) Two-way ANOVA with Sidak’s multiple comparison test and (B, E, G, I) unpaired t-test with Welch’s correction were used to determine the statistical significance. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To investigate the immune cell population infiltrating the tumors, we performed flow cytometry analysis on the tumors harvested from MyD88Ctrl and MyD88ΔMye mice. Consistent with the results observed in the MyD88−/− mice, MyD88ΔMye mice showed impaired recruitment of F4/80+CD11b+ TAMs in the TME (Fig. 5D and E). Interestingly, we observed an increase in the CD3+CD8+ T-cell infiltration in the tumors, probably contributing to the reduced tumor growth in these mice (Fig. 5F and G). Next, we analyzed PD-1 and PD-L1 expression on F4/80+CD11b+ TAMs from MyD88Ctrl and MyD88ΔMye mice. This analysis showed reduced expression of PD-1 and PD-L1 on F4/80+CD11b+ TAMs from MyD88ΔMye mice compared with the TAMs from MyD88Ctrl mice (Fig. 5H). To understand if the defect is at the mRNA level, we sort purified F4/80+CD11b+ TAMs from MyD88Ctrl and MyD88ΔMye mice (gating strategy; Supplementary Fig. S3) and performed qPCR to determine the changes in the expression of Pdcd1 and Pdcd1l1 mRNA. Consistent with the results of the flow cytometry data, we observed reductions in the Pdcd1 and Pdcd1l1 mRNA levels from MyD88ΔMye TAMs compared with the levels from TAMs sort purified from MyD88Ctrl mice (Fig. 5I). These data suggest that myeloid cell-specific expression of MyD88 drives PD-1/PD-L1 expression on F4/80+CD11b+ TAMs thereby regulating melanoma growth.
The Gr1+CD11b+ granulocyte population was also found to be reduced in the tumors from MyD88ΔMye mice, whereas B- and NK-cell populations showed no defects compared with the tumors from MyD88Ctrl mice (Supplementary Fig. S5A and B). Further analysis of immune cell populations in spleens from tumor-bearing MyD88Ctrl and MyD88ΔMye mice by flow cytometry revealed no significant differences in myeloid, B-cell or CD3+CD4+ or CD3+CD8+ T-cell populations from MyD88Ctrl and MyD88ΔMye mice (Supplementary Fig. S5C–J). In accordance with the trend observed in the spleens of tumor-bearing MyD88−/− mice, MyD88ΔMye mice also showed a significant increase in the activated T-cell population compared with MyD88Ctrl mice (Supplementary Fig. S5K and L). These results suggest that MyD88 in myeloid cells is driving the infiltration of TAMs in the TME, creating an immunosuppressive environment to favor melanoma progression.
MyD88 promotes tumor progression in a syngeneic MC38 colon carcinoma tumor model by regulating TAM infiltration
Since MYD88 is highly expressed in a wide variety of human cancers (Fig. 1A), we hypothesized that MyD88 and IL-1R would have roles in other tumor types in addition to melanoma. To test this, we expanded our studies to use another well-accepted murine tumor model, the MC38 colon carcinoma system. In this solid tumor model, 1 × 106 MC38 colon carcinoma cells were subcutaneously injected into WT, MyD88−/− and Il1r−/− mice; mice were monitored for 18 days for tumor growth. We found that MyD88−/− and Il1r−/− mice exhibited significantly impaired tumor growth compared with WT mice (Fig. 6A). Consistent with the findings in MyD88−/− and Il1r−/− mice injected with B16-F10 melanoma cells, MyD88−/− and Il1r−/− mice injected with MC38 tumor cells also displayed reduced infiltration of F4/80+CD11b+ TAMs in the TME (Fig. 6B).
Fig. 6: MyD88 promotes tumor progression in a syngeneic MC38 colon carcinoma tumor model by regulating TAM infiltration.
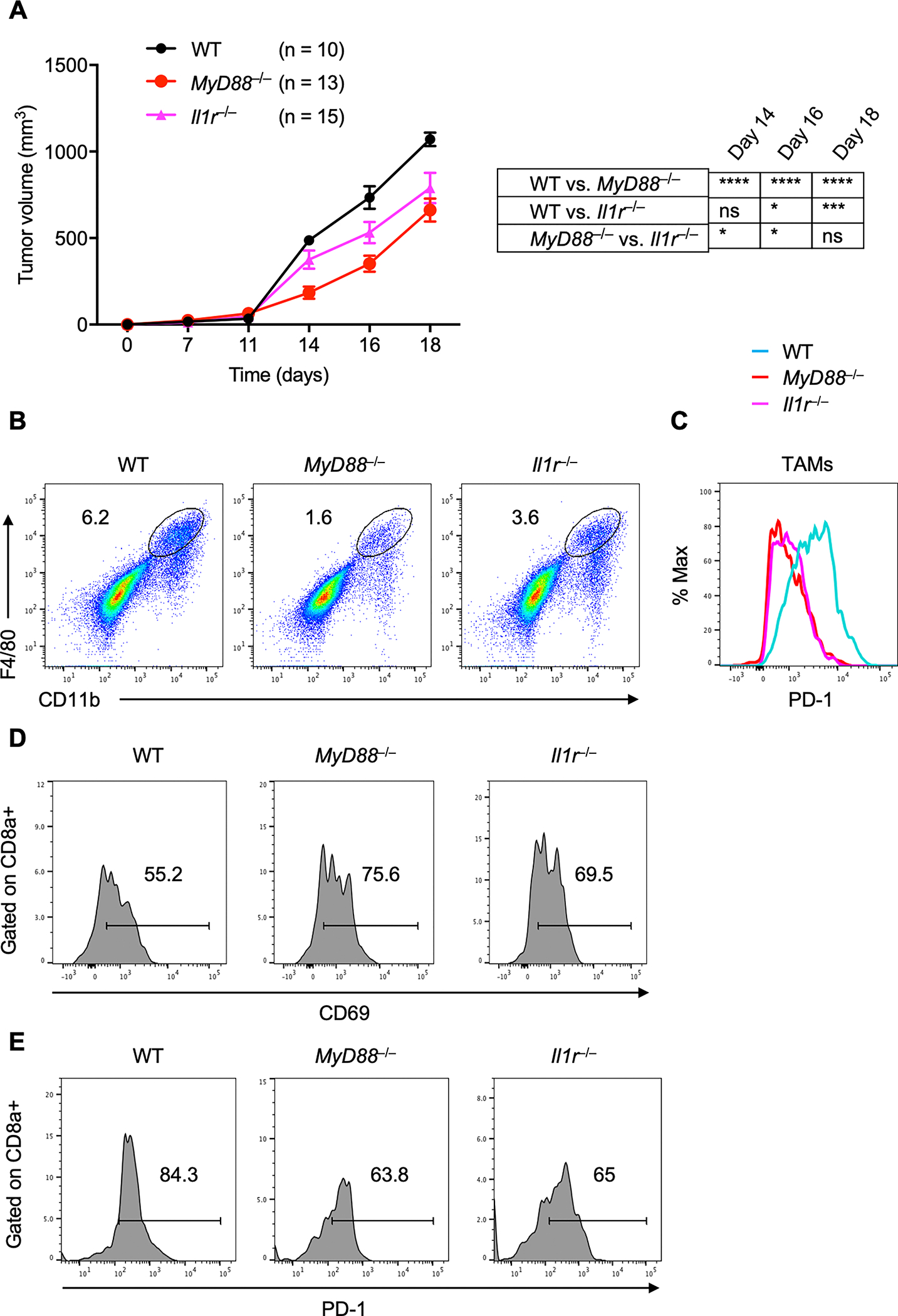
A, MC38 colon adenocarcinoma cells were injected into WT (n = 10), MyD88−/− (n = 13) and Il1r−/− (n = 15) mice, and mean tumor volume was measured.
B, Flow cytometry analysis (pseudocolor plots) of F4/80+CD11b+ macrophage infiltration in tumors harvested from WT, MyD88−/− and Il1r−/− mice.
C, Flow cytometry analysis (histogram) of PD-1 expression on F4/80+CD11b+ TAMs in tumors harvested from WT, MyD88−/− and Il1r−/− mice.
D–E, Flow cytometry analysis (histogram) of CD69 (D) and PD-1 (E) expression on CD3+CD8+ T-cells in tumors harvested from WT, MyD88−/− and Il1r−/− mice.
Data are presented as mean ± SD. (A) Two-way ANOVA with Sidak’s multiple comparison test was used to determine the significance between the two groups analyzed. ns not significant, *P < 0.05, ***P < 0.001, ****P < 0.0001.
Next, we analyzed PD-1 expression on F4/80+CD11b+ TAMs from MyD88−/− and Il1r−/− mice. This analysis showed reduced expression of PD-1 on F4/80+CD11b+ TAMs from MyD88−/− and Il1r−/− mice compared with the TAMs from WT mice (Fig. 6C). Next, we sought to analyze the activation status of CD8+ T cells infiltrating the TME. Compared with the WT mice, CD8+ T cells from the TME of MyD88−/− and Il1r−/− mice tended to show a more activated phenotype (Fig. 6D). The CD8+ T cells from MyD88−/− and Il1r−/− mice also showed less PD-1 expression (Fig. 6E), suggesting possible crosstalk between macrophages and T cells in the TME. Taken together, these data suggest that the absence of MyD88 and IL-1R leads to reduction in the MC38 tumor burden similar to that seen in melanoma by limiting the infiltration of F4/80+CD11b+ TAMs and promoting the recruitment of activated cytotoxic T lymphocytes, thereby creating an anti-tumor microenvironment, and shows that the IL-1R/MyD88 axis has implications in multiple tumor types.
IL-1R/MyD88 axis is critical for NF-κB recruitment on the PD-1 promoter in macrophages
One mechanism whereby PD-1 expression might be regulated is by tumor or immune cell secreted cytokines. Previous reports suggest that LPS or IFNγ can stimulate PD-1 or PD-L1 expression on bone marrow-derived macrophages (BMDMs) (33). We first examined the activation of PD-1 or PD-L1 on BMDMs upon LPS or IFN-γ stimulation (Fig. 7A). In addition to LPS and IFNγ, we also performed co-culture of BMDMs with B16-F10 melanoma cells. Compared with LPS or IFNγ stimulation, co-culture of BMDMs with B16-F10 melanoma cells resulted in a strong induction of PD-1 expression (Fig. 7A). Next, we examined the changes in the PD-L1 expression upon LPS or IFN-γ stimulation or co-culture with B16-F10 melanoma cells. Both LPS and IFN-γ stimulation and co-culture with B16-F10 melanoma cells showed increased PD-L1 expression (Fig. 7B), compared with basal expression in WT BMDMs. To investigate whether BMDMs from MyD88−/− or Il1r−/− mice showed a defect in PD-1 expression upon co-culture with B16-F10 melanoma cells, we subjected BMDMs harvested from MyD88−/−, Il1r−/− or Trif−/− mice to co-culture experiments. Consistent with the defect observed in MyD88-deficient F4/80+CD11b+ TAMs, BMDMs from MyD88−/− or Il1r−/− mice showed a reduction in PD-1 expression upon co-culture with tumor cells compared with WT or Trif−/− BMDMs (Fig. 7C and D).
Fig. 7: IL-1R/MyD88 axis controls recruitment of NF-κB to the PD-1 promoter.
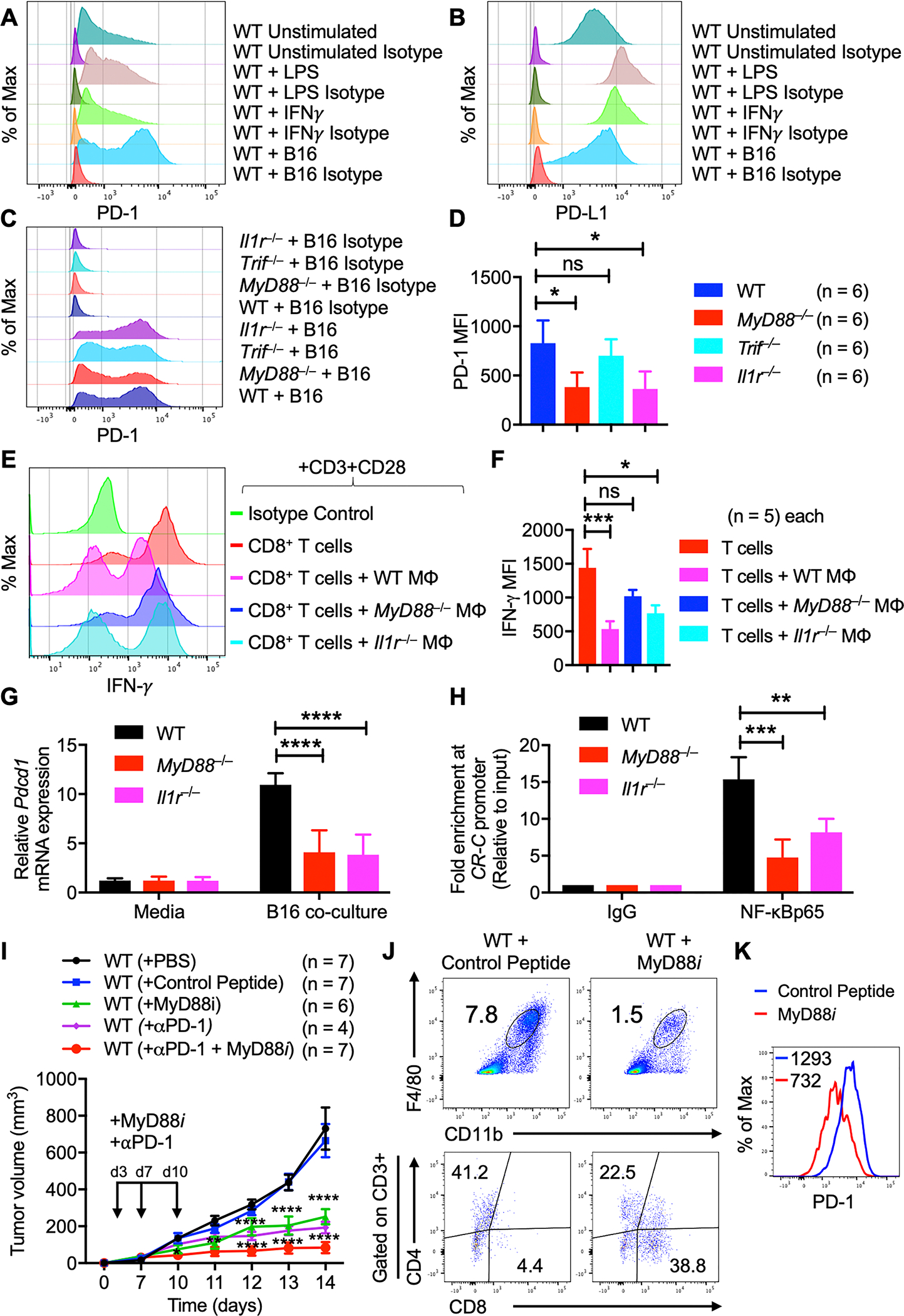
A–B, Flow cytometry analysis of WT BMDMs stimulated with LPS or IFN-γ or co-cultured with B16-F10 melanoma cells. (A) Histogram of PD-1 expression on BMDMs. (B) Histogram of PD-L1 expression on BMDMs.
C–D, Flow cytometry analysis of BMDMs from WT, MyD88−/−, Il1r−/− and Trif−/− mice co-cultured with B16-F10 melanoma cells for 48 h. (C) Histogram of PD-1 expression on BMDMs. (D) Quantification of PD-1 expression on BMDMs.
E–F, Macrophage suppression assay. (E) Flow cytometry analysis of IFN-γ expression on activated CD3+CD8+ T cells from WT mice co-cultured with B16-F10 polarized macrophages from WT (n = 5), MyD88−/− (n = 5) and Il1r−/− (n = 5) mice. (F) Quantification of IFN-γ expression on CD3+CD8+ T cells.
G, Quantitatve PCR analysis of Pdcd1 mRNA expression in BMDMs from WT, MyD88−/− and Il1r−/− mice co-cultured with B16-F10 melanoma cells for 48 h. All samples were normalized to expression of the endogenous control Gapdh.
H, ChIP experiments were performed with chromatin prepared from WT, MyD88−/− and Il1r−/− BMDMs co-cultured with B16-F10 melanoma cells for 48 h. Antibodies against NF-κBp65 and IgG were used. Precipitated DNA was quantified by real-time PCR using primers specific for the CR-C region of the Pdcd1 promoter. ChIP values were normalized against the input and expressed as relative enrichment of the material precipitated by the indicated antibody on specific promoter [relative quantification using the comparative Ct method (2−ΔΔCt)].
I, B16-F10 melanoma cells were injected into WT mice with or without MyD88 inhibitor (MyD88i) treatment and in combination with anti–PD-1 blockade. Mean tumor volume 2 weeks after tumor cell injection in WT + PBS (n = 7), WT + control peptide (n = 7), WT + MyD88i (n = 6), WT + anti–PD-1 (n = 4) and WT + MyD88i + anti–PD-1 (n = 7) mice.
J, Flow cytometry analysis of tumors harvested from WT mice (with or without MyD88i). Pseudocolor plots of F4/80+CD11b+ macrophage infiltration and CD3+CD4+ and CD3+CD8+ T-cell infiltration in tumors.
K, Flow cytometry analysis (histogram) of PD-1 expression on F4/80+CD11b+ TAMs in tumors harvested from WT mice (with or without MyD88i).
Data are presented as mean ± SD. (D, F) Kruskal-Wallis test with Dunn’s multiple comparison test, (G) two-way ANOVA with Dunnett’s multiple comparison test and (H, I) two-way ANOVA with Sidak’s multiple comparison test were used to determine the statistical significance. ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To address the direct effect of these macrophages on T-cell suppression, we designed a macrophage suppression assay. We co-cultured macrophages from WT, MyD88−/− or Il1r−/− mice with B16 tumor cells. After polarizing the macrophages in this way, we cultured them with activated (CD3+CD28+) splenic CD8+ T cells and assessed the IFN-γ expression on these T cells by flow cytometry. In accordance with our previous results, activated T cells from WT mice showed elevated IFN-γ expression (Fig. 7E and F). When these T cells were co-cultured with polarized macrophages from WT mice, the IFN-γ expression was significantly reduced, and this reduction was rescued when the T cells were co-cultured with macrophages harvested from MyD88−/− or Il1r−/− mice (Fig. 7E and F). These data suggest that MyD88 and IL-1R from myeloid cells have direct effects on T-cell function in the context of the tumor.
Next, we isolated RNA from WT, MyD88−/− and Il1r−/− BMDMs upon co-culture with B16-F10 melanoma cells and performed qPCR to determine the changes in the expression of Pdcd1 mRNA. Consistent with the flow cytometry data, Pdcd1 mRNA levels from MyD88−/− or Il1r−/− BMDMs were significantly reduced compared with those from WT BMDMs (Fig. 7G), suggesting that the TME may act on the gene expression program of macrophages. To examine whether MyD88 controls tumor cell-induced Pdcd1 gene expression at the transcriptional level, we performed chromatin-immunoprecipitation (ChIP) using an anti–NF-κBp65 antibody in BMDMs co-cultured with B16-F10 melanoma cells. As shown in Fig. 7H, WT BMDMs showed recruitment of NF-κBp65 to the CR-C region of the Pdcd1 promoter in response to the tumor cell co-culture. However, this recruitment was severely impaired in the absence of MyD88 or IL-1R (Fig. 7H). These results indicate that the MyD88/IL-1R axis is required for the recruitment of NF-κBp65 to the CR-C region of the Pdcd1 promoter in macrophages co-cultured with tumor cells.
Given that anti–PD-1 combinatorial immunotherapy is being used in the clinic to improve the therapeutic outcome in various cancers, we sought to determine whether MyD88 inhibition using a MyD88 inhibitor (MyD88i) could result in a better treatment outcome in mice with melanoma. WT mice treated with the control antenna peptide did not show any reduction in tumor growth, whereas WT mice treated with either MyD88i or anti–PD-1 had significantly reduced tumor size (Fig. 7I). Next, we undertook a combinatorial immunotherapy approach combining the MyD88i with anti–PD-1 blockade. The mice treated with both MyD88i with anti–PD-1 had significantly reduced tumor growth compared with those treated with MyD88i alone (Fig. 7I). Flow cytometry analysis of the tumors from WT mice treated with control and MyD88i peptides showed reduced F4/80+CD11b+ macrophage infiltration, increased CD8+ T-cell recruitment and less PD-1 expression on these TAMs (Fig. 7J and K), consistent with the data obtained from MyD88−/− and MyD88ΔMye mice. Collectively, these results suggest that MyD88 inhibition may potentially provide an anti-tumor effect, and this inhibition coupled with immune checkpoint blockade may further improve therapeutic efficacy in melanoma.
Discussion
Cancer is a leading cause of death worldwide and new cancer cases are on the rise globally (44). Understanding the molecular mechanisms behind the development and progression of individual cancers is a major unmet need. The onset and progression of melanoma is often characterized by the presence of an inflammatory microenvironment with prevalent immunosuppressive myeloid cell types and, to a certain extent, lymphoid cells (45). Understanding the interaction between tumor cells and immune cells is extremely important considering the fact that there are growing instances of remarkable resistance to current immunotherapeutic strategies (46). Although, checkpoint blockade immunotherapies have focused on the role of receptor-ligand interactions to regulate T-cell suppression, less is known about the role of these molecules in the context of macrophages. In the current study, we identified the unknown cell-intrinsic function of an adaptor molecule MyD88 downstream of IL-1R signaling in regulating PD-1/PD-L1 expression on a subset of myeloid cells called TAMs to control melanoma progression and growth.
Macrophages have been shown to promote metastasis in advanced tumors (47). Blocking their recruitment with an anti-CSF1 receptor (CSF1R) antibody leads to a reduction in the number of TAMs and in their immunosuppressive activity (48). The blockade of CSF1R signaling also results in the upregulation of CTLA4 and PD-L1 on T cells and tumor cells, respectively. These immune checkpoint molecules get upregulated as a result of enhanced inflammatory signaling in the TME (49), implying that macrophages in the TME can be polarized towards an anti-tumor function.
Several studies have suggested an important role for MYD88 in tumor development. High expression of MYD88 has been correlated with poor prognosis in several tumors, such as hepatocellular carcinoma and colorectal and ovarian cancers (50). Using a DSS model and mammary tumor transplantations, cell-autonomous functions of MyD88 have also been shown to be involved in tumor progression (51). Ablation of Tet2 (Ten-Eleven-Translocation-2), a tumor suppressor gene in myeloid cells has been shown to suppress melanoma growth in vivo and the expression of Tet2 is dependent on the IL-1R/MyD88 pathway (52). Recent studies have also linked a gain-of-function driver mutation (L265P) in MYD88 that triggers IRAK-mediated NF-κB signaling to Waldenström’s macroglobulinemia and the activated B-cell-like (ABC) subtype of diffuse large B-cell lymphoma (DLBCL) (53,54). Despite the fact that MyD88 is ubiquitously expressed, the precise mechanisms whereby MyD88 acts intrinsically in macrophages to sustain cancer progression remain poorly understood. Analysis of TCGA mRNA expression data revealed an important role for MYD88 as a tumor-promoting adaptor molecule. Human melanoma datasets further emphasized the contribution of MYD88 in driving tumorigenesis. However, these data need careful interpretation as the tissue samples analyzed consist of heterogeneous populations, including immune cells, and it is challenging to ascertain the cell type-specific role of MYD88 in this context. Our data provide compelling evidence that MyD88 acts in a cell type-specific manner to promote an immunosuppressive pro-tumorigenic tissue microenvironment. Using a MyD88-specific inhibitor, we showed a marked decrease in melanoma progression in WT mice transplanted with B16 melanoma. These results strongly emphasize that inhibition of the MyD88 signaling pathway could be a new potential therapeutic target in melanoma.
Targeting PD-1 in the context of tumor/T cell crosstalk has shown impressive anti-tumor effects and clinical benefits in numerous cancers. Despite these encouraging clinical results, the majority of patients did not benefit from anti-PD-1/PD-L1 therapy, with some responders relapsing after a period of response (46). This could be due to inadequate information on the regulation of these immune checkpoint molecules in the innate immune cells. Thus, a complete understanding of the role played by TAMs and their crosstalk with the TME is necessary to develop new combinatorial immunotherapy approaches. Our data provide a previously unknown link between the transcriptional regulation of PD-1 by the adaptor molecule MyD88 in TAMs which will open new avenues for combination therapies. Combining MYD88 inhibitors with PD-1 or PD-L1 blockade in the context of macrophage-mediated immunotherapy could result in a better treatment outcome in a wide variety of cancers.
Supplementary Material
Acknowledgments
We would like to thank all the members of Kanneganti lab for their critical comments and suggestions and Rebecca Tweedell, PhD, for scientific editing and writing support. We also would like to thank Dr. Yongqiang Feng, PhD (St. Jude Immunology faculty) for help with ultrasonicator (Covaris S2) and St. Jude Immunology FACS core facility for cell sorting. This work was supported by the National Institutes of Health grants CA253095, AR056296, AI124346, and AI101935 and by the American Lebanese Syrian Associated Charities to T.-D. Kanneganti. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest disclosure
The authors declare that they have no conflict of interest.
References
- 1.Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol 2015;36:229–39 [DOI] [PubMed] [Google Scholar]
- 2.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity 2014;41:49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science 2014;344:921–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014;513:559–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Church SE, Galon J. Tumor Microenvironment and Immunotherapy: The Whole Picture Is Better Than a Glimpse. Immunity 2015;43:631–3 [DOI] [PubMed] [Google Scholar]
- 6.Poh AR, Ernst M. Targeting Macrophages in Cancer: From Bench to Bedside. Front Oncol 2018;8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One 2012;7:e50946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sica A, Schioppa T, Mantovani A, Allavena P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer 2006;42:717–27 [DOI] [PubMed] [Google Scholar]
- 9.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol 2006;27:195–201 [DOI] [PubMed] [Google Scholar]
- 11.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017;545:495–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tartey S, Takeuchi O. Pathogen recognition and Toll-like receptor targeted therapeutics in innate immune cells. Int Rev Immunol 2017;36:57–73 [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010;140:805–20 [DOI] [PubMed] [Google Scholar]
- 15.Gurung P, Fan G, Lukens JR, Vogel P, Tonks NK, Kanneganti TD. Tyrosine Kinase SYK Licenses MyD88 Adaptor Protein to Instigate IL-1alpha-Mediated Inflammatory Disease. Immunity 2017;46:635–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma BR, Karki R, Lee E, Zhu Q, Gurung P, Kanneganti TD. Innate immune adaptor MyD88 deficiency prevents skin inflammation in SHARPIN-deficient mice. Cell Death Differ 2019;26:741–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salcedo R, Cataisson C, Hasan U, Yuspa SH, Trinchieri G. MyD88 and its divergent toll in carcinogenesis. Trends Immunol 2013;34:379–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 2007;317:124–7 [DOI] [PubMed] [Google Scholar]
- 19.Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med 2010;207:1625–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coste I, Le Corf K, Kfoury A, Hmitou I, Druillennec S, Hainaut P, et al. Dual function of MyD88 in RAS signaling and inflammation, leading to mouse and human cell transformation. J Clin Invest 2010;120:3663–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mantovani A, Barajon I, Garlanda C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol Rev 2018;281:57–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dinarello CA. An Interleukin-1 Signature in Breast Cancer Treated with Interleukin-1 Receptor Blockade: Implications for Treating Cytokine Release Syndrome of Checkpoint Inhibitors. Cancer Res 2018;78:5200–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarassishin L, Casper D, Lee SC. Aberrant expression of interleukin-1beta and inflammasome activation in human malignant gliomas. PLoS One 2014;9:e103432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su B, Luo T, Zhu J, Fu J, Zhao X, Chen L, et al. Interleukin-1beta/Iinterleukin-1 receptor-associated kinase 1 inflammatory signaling contributes to persistent Gankyrin activation during hepatocarcinogenesis. Hepatology 2015;61:585–97 [DOI] [PubMed] [Google Scholar]
- 25.Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017;390:1833–42 [DOI] [PubMed] [Google Scholar]
- 26.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998;9:143–50 [DOI] [PubMed] [Google Scholar]
- 27.Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, et al. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol 1997;159:3364–71 [PubMed] [Google Scholar]
- 28.Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, et al. Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med 1998;187:1463–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003;301:640–3 [DOI] [PubMed] [Google Scholar]
- 30.Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity 2008;29:272–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 1999;8:265–77 [DOI] [PubMed] [Google Scholar]
- 32.Tartey S, Matsushita K, Vandenbon A, Ori D, Imamura T, Mino T, et al. Akirin2 is critical for inducing inflammatory genes by bridging IkappaB-zeta and the SWI/SNF complex. EMBO J 2014;33:2332–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bally AP, Lu P, Tang Y, Austin JW, Scharer CD, Ahmed R, et al. NF-kappaB regulates PD-1 expression in macrophages. J Immunol 2015;194:4545–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003;4:249–64 [DOI] [PubMed] [Google Scholar]
- 35.Kayala MA, Baldi P. Cyber-T web server: differential analysis of high-throughput data. Nucleic Acids Res 2012;40:W553–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 2007;35:W169–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raskin L, Fullen DR, Giordano TJ, Thomas DG, Frohm ML, Cha KB, et al. Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J Invest Dermatol 2013;133:2585–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riker AI, Enkemann SA, Fodstad O, Liu S, Ren S, Morris C, et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics 2008;1:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabbarah O, Nogueira C, Feng B, Nazarian RM, Bosenberg M, Wu M, et al. Integrative genome comparison of primary and metastatic melanomas. PLoS One 2010;5:e10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 2013;45:1113–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 43.Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity 2013;39:806–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108 [DOI] [PubMed] [Google Scholar]
- 45.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140:883–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nowicki TS, Hu-Lieskovan S, Ribas A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J 2018;24:47–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012;12:253–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 2010;116:3955–63 [DOI] [PubMed] [Google Scholar]
- 49.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 2014;74:5057–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Je EM, Kim SS, Yoo NJ, Lee SH. Mutational and expressional analyses of MYD88 gene in common solid cancers. Tumori 2012;98:663–9 [DOI] [PubMed] [Google Scholar]
- 51.Scheeren FA, Kuo AH, van Weele LJ, Cai S, Glykofridis I, Sikandar SS, et al. A cell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia and oncogenesis. Nat Cell Biol 2014;16:1238–48 [DOI] [PubMed] [Google Scholar]
- 52.Pan W, Zhu S, Qu K, Meeth K, Cheng J, He K, et al. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017;47:284–97 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011;470:115–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med 2012;367:826–33 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.