

Abstract
Inflammatory myopathies, collectively known as myositis, are heterogeneous disorders characterised by muscle inflammation, and frequently accompanied by extramuscular manifestations that affect the skin, lung, and joints. Patients with inflammatory myopathies were previously classified as having dermatomyositis if characteristic rashes accompanied the muscle involvement, and as having polymyositis if no rashes were present. Five main types of inflammatory myopathies are now widely recognised: dermatomyositis, immune-mediated necrotising myopathy, sporadic inclusion-body myositis, overlap myositis (including antisynthetase syndrome), and polymyositis. The discovery of autoantibodies that are specifically associated with characteristic clinical phenotypes has been instrumental to the understanding of inflammatory myopathies. Treatment is still largely based on expert opinion, but several studies have shown effectiveness of different therapies in various subsets of inflammatory myopathies. These advances will undoubtedly improve the outcomes of patients with inflammatory myopathies.
Introduction
Inflammatory myopathies, collectively known as myositis, are a heterogeneous group of rare diseases that affect multiple organs and systems, including the muscles, skin, lungs, and joints. Although universally accepted classification criteria for inflammatory myopathies have not yet been established, the five most recognised types of inflammatory myopathies are dermatomyositis, immune-mediated necrotising myopathy, overlap myositis (including antisynthetase syndrome), sporadic inclusion-body myositis, and polymyositis.1–3 Inflammatory myopathy-specific autoantibodies can be used to classify patients with inflammatory myopathies in homogeneous phenotypic subsets. However, the importance of these autoantibodies has not yet been translated to inflammatory-myopathy classification criteria, probably because of their recent development.1,4
Treatment options for inflammatory myopathies are broad, but their choice and combination are still largely based on expert opinion.3 Randomised controlled trials are providing evidence for treatment in specific subsets of patients, but the nature of the diseases, which are rare and heterogeneous, complicates enrolling adequate numbers of patients with homogeneous phenotypes and designing appropriate studies. This Review describes the classification and treatment of the most common types of inflammatory myopathies in adults.
Classification
Dermatomyositis
Patients with dermatomyositis typically present with proximal muscle weakness and cutaneous manifestations that develop over weeks to months. However, some patients with a dermatomyositis rash have little or no muscle involvement, as shown by the absence of weakness and muscle enzyme elevation, and of electromyography (EMG), MRI, and muscle biopsy findings. Some clinicians consider these hypomyopathic or amyopathic forms of dermatomyositis to be different subtypes of myositis,2 but, for simplicity, we will include them in this section.
The pathognomonic skin features of dermatomyositis include a violaceous periorbital, often oedematous, rash (ie, a heliotrope rash; figure 1, appendix) and erythematous lesions on the extensor surfaces of the joints (ie, Gottron’s papules; figure 1). Usually, muscle enzymes are elevated and EMG reveals a myopathic pattern (myopathic motor units with fibrillations and spontaneous sharp waves).5 As in other types of inflammatory myopathies, MRI scans in patients with dermatomyositis can reveal intramuscular T2 hyperintensities caused by muscle inflammation or necrosis.6 Additionally, patients with dermatomyositis often have T2 hyperintensities around individual muscles as a result of fascial involvement, a feature seen less frequently in other types of inflammatory myopathies.6 Myalgia and pruritus can also be important symptoms of the disease in some patients.7
Figure 1: Clinical features and pathological findings of dermatomyositis.
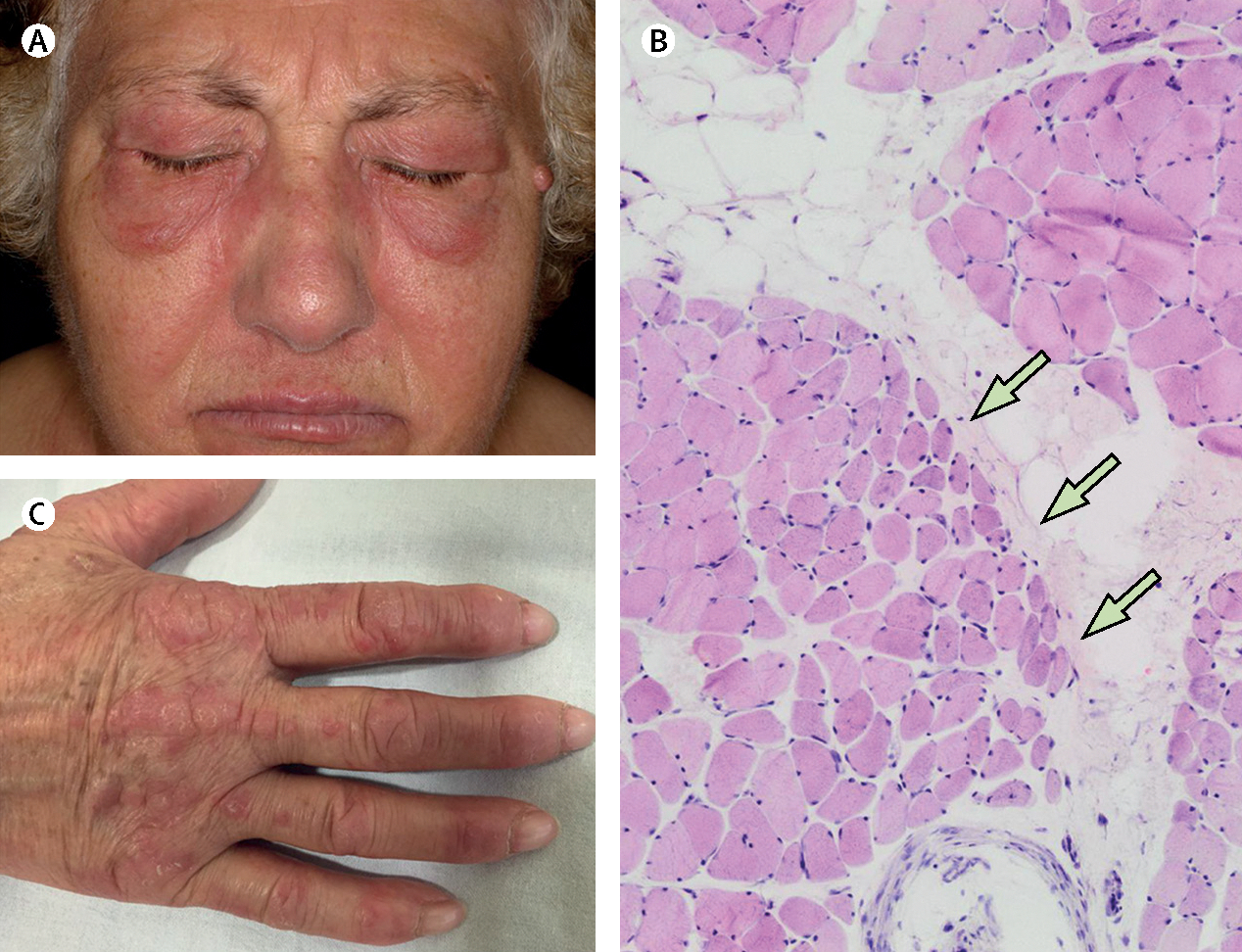
A women aged 67 years presented with muscle weakness. She had (A) a heliotrope rash and (C) Gottron’s papules, and a muscle biopsy revealed (B) perifascicular atrophy. The patient was positive for anti-Mi2 autoantibodies and was diagnosed with dermatomyositis. Arrows indicate perifascicular atrophy.
Perifascicular atrophy is a highly specific feature of muscle biopsies in patients with dermatomyositis (specificity >90%);8 however, this feature lacks sensitivity (25–50%; figure 1).8,9 Data support that the expression of perifascicular human myxovirus resistance protein 1 and retinoic acid-inducible gene 1 have higher diagnostic sensitivity (71% and 50%) than perifascicular atrophy with equivalent specificity.8,10 Additionally, tissue biopsies from patients with dermatomyositis often have cellular infiltrates consisting predominantly of plasmacytoid dendritic cells, B cells, CD4 T cells, and macrophages.11 These cells often surround medium sized blood vessels and invade the perimysium.11 However, up to 16% of dermatomyositis biopsies do not have infiltrates but have prominent necrosis that is pathologically indistinguishable from immune-mediated necrotising myopathy.9 The deposition of membrane attack complex and presence of microtubular inclusions on intramuscular capillaries is an early manifestation of dermatomyositis; as the disease progresses, capillary dropout can also occur.9 Furthermore, as with other inflammatory myopathies, class-1 major histocompatibility complex is usually upregulated on the sarcolemma of muscle fibres. In patients with dermatomyositis, class-1 major histo compatibility complex upregulation and other pathological findings (eg, myofibre degeneration and regeneration, and necrosis) can be especially prominent in perifascicular regions.11
Based on a prevalence study, approximately 70% of patients with dermatomyositis have a dermatomyositis-specific autoantibody,4 which are associated with a unique clinical phenotype (table 1). Autoantibodies recognising the Mi2 nuclear antigen have been associated with classic dermatomyositis features, including proximal muscle weakness and severe skin manifestations.12 Patients with dermatomyositis and autoantibodies recognising nuclear matrix protein (NXP)2 are more likely than patients with other types of autoantibodies to present with both proximal and distal muscle weakness, subcutaneous oedema, and dysphagia.13 Furthermore, patients who are positive for anti-NXP2 autoantibodies are more prone to developing calcinosis, which are painful deposits of calcium in the soft tissues, than patients with other types of autoantibodies.13 Patients with dermatomyositis who are positive for anti-NXP2 or anti-transcription intermediary factor (TIF)-1 autoantibodies are at increased risk of malignancy within 3 years of their diagnosis; as such, doing comprehensive cancer screening13–15 or PET-CT scans is particularly important in these patients.33
Table 1:
Clinical characteristics of the main clinical and phenotype-specific autoantibody groups in inflammatory myopathies
| Clinical features | Type of organ involvement and severity |
|||
|---|---|---|---|---|
| Muscle | Lung | Skin | ||
|
| ||||
| Dermatomyositis | ||||
| Anti-Mi2 autoantibodies12 | Mild-to-moderate muscle involvement with classical skin rash | Moderate | None | Moderate |
| Anti-NXP2 autoantibodies13,14 | Mild-to-moderate muscle involvement with myalgia, classical skin rash, calcinosis, distal extensor weakness and oedema, and dysphagia; increased risk of cancer | Moderate | None | Moderate |
| Anti-TIFl autoantibodies14,15 | Strong association with cancer; mild muscle involvement with marked skin involvement, occasionally this type of myositis can present as clinically amyopathic dermatomyositis | Mild | None | Moderate |
| Anti-SAE autoantibodies16 | Mild-to-moderate muscle involvement with classical skin rash | Mild | None | Moderate |
| Anti-MDA5 autoantibodies17–19 | Severe skin rash with no muscle involvement (hypomyopathic or amyopathic dermatomyositis) and occasionally highly lethal forms of rapidly progressive interstitial lung disease | None or mild | Severe | Severe |
| Antibody-negative dermatomyositis20 | Mild-to-moderate muscle involvement with classical skin rash | Mild | Unknown | Moderate |
| Immune-mediated necrotising myopathy | ||||
| Anti-SRP autoantibodies21–23 | Severe muscle involvement, dysphagia, and 20% of patients with lung involvement with no skin lesions | Severe | Mild | None |
| Anti-HMGCR autoantibodies24–26 | Exclusive severe muscle involvement; statin-exposed patients | Severe | None | None |
| Antibody-negative immune-mediated necrotising myopathy | Strong association with cancer | Unknown | Unknown | None |
| Sporadic inclusion-body myositis3,27 | Older (>50 years) patients with prominant distal and quadriceps involvement; slow progression and refractory to treatment | Severe | None | None |
| Overlap myositis | ||||
| Antisynthetase syndrome | ||||
| Anti-Jo1 autoantibodies28,29 | Mild-to-moderate muscle involvement with progressive lung involvement and possible mild dermatomyositis skin rash (~50% of patients); other characteristic cutaneous features (eg, mechanic's hands and Raynaud syndrome) | Moderate | Moderate | Mild |
| Anti-PL7 autoantibodies28 | Symptoms are similar to those of anti-Jo1 autoantibody-positive myositis with more severe lung involvement | Moderate | Severe | Mild |
| Anti-PL12 autoantibodies28 | Severe lung involvement with mild muscle weakness | Mild | Severe | Mild |
| Anti-Pm/Scl autoantibodies30 | Mild myositis and scleroderma features with muscle weakness, interstitial lung disease, and skin involvement | Mild | Mild | Mild |
| Anti-Ku autoantibodies31 | Mild muscle involvement and interstitial lung disease | Mild | Mild | Mild |
| Anti-U1RNP autoantibodies32 | Myositis, scleroderma, and systemic lupus erythematosus features; glomerulonephritis and pulmonary hypertension are possible | Mild | Mild | Mild |
| Polymyositis | Diagnosis of exclusion; heterogeneous clinical features | Unknown | Unknown | Unknown |
NXP2=nuclear matrix protein 2. TIF1=transcription intermediary factor 1. SAE=small ubiquitin-like modifier activating enzyme. MDA5=melanoma differentiation-associated gene 5. SRP=signal recognition particle. HMGCR=3-hydroxy 3-methylglutaryl coenzyme A reductase. Jo1=histidyl tRNA synthetase. PL7=threonyl tRNA synthetase. PL12=alanyl tRNA synthetase. Pm/Scl=anti-polymyositis-scleromyositis, EXOSC9 and EXOSC10 antigens. U1RNP=U1 ribonucleoprotein. The severity of the organ involvement is based on comparative studies of muscle, lung, and skin involvement in different myositis autoantibody groups.
Patients with dermatomyositis who have autoantibodies recognising small ubiquitin-like modifier activating enzyme or melanoma differentiation-associated gene 5 (MDA5) tend to have more substantial skin than muscle involvement.16–19 Along with the typical skin manifestations of dermatomyositis, patients who are positive for anti-MDA5 autoantibodies are prone to developing ulcers, often on the flexor surface of the digits and palm (figure 2).19 Most patients with anti-MDA5 autoantibodies are hypomyopathic or amyopathic.17–19 Furthermore, unlike patients with other dermatomyositis autoantibodies, those who are anti-MDA5 positive frequently develop a rapidly progressive and sometimes lethal form of interstitial lung disease.17,18 All patients with myositis who are suspected to have interstitial lung disease should initially be evaluated using pulmonary function tests (including carbon monoxide diffusion and inspiratory and expiratory pressures) and high-resolution CT scans. Monitoring of interstitial lung disease should rely on periodical pulmonary function tests, and subsequent high-resolution CT scans should be restricted to evaluating patients with evolving pulmonary issues.
Figure 2: Clinical features and radiological findings of anti-melanoma differentiation-associated gene 5 syndrome.
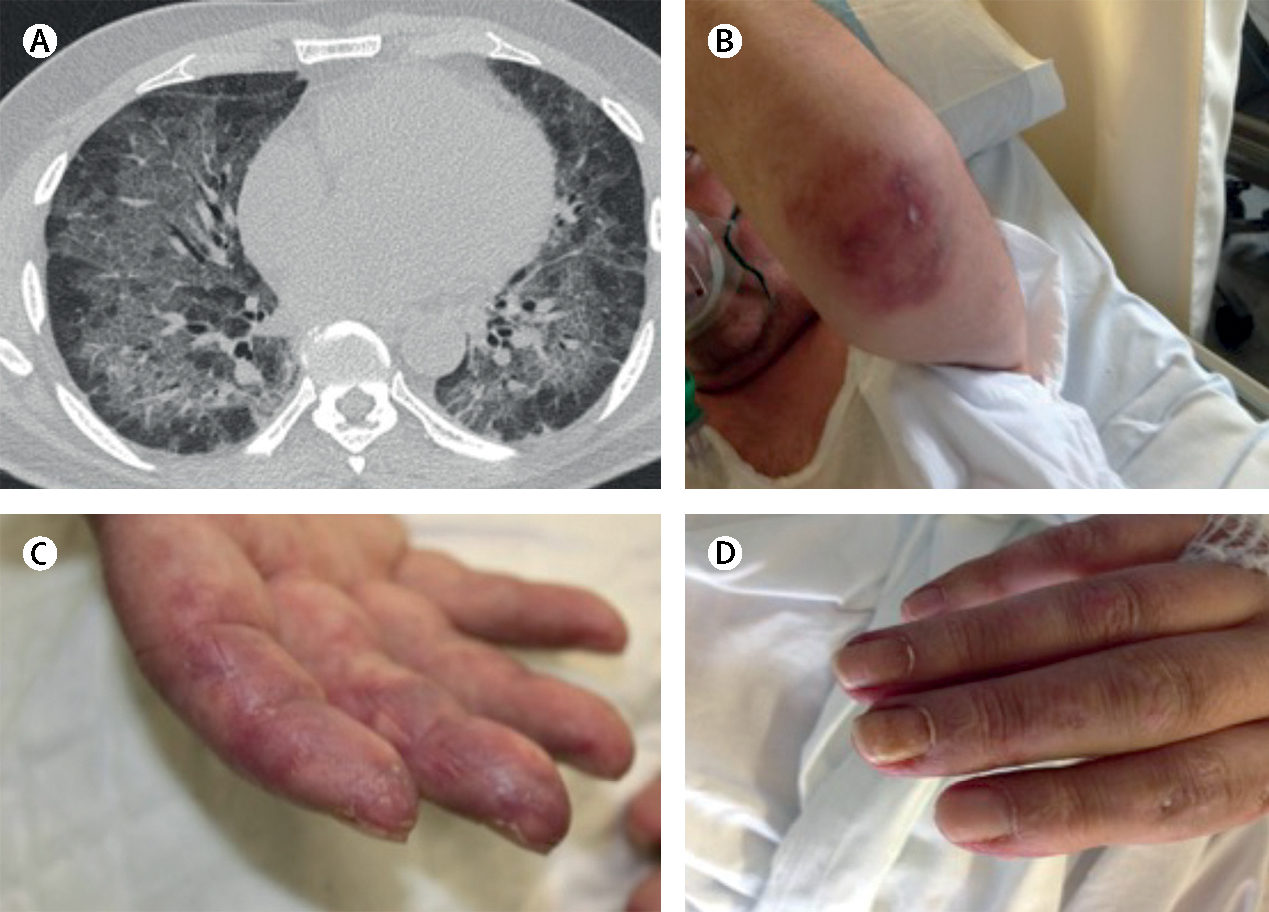
A man aged 52 years presented with progressive dyspnoea that had been present for 2 months. At the emergency room, he was hypoxemic with a ratio of arterial oxygen partial pressure to fractional inspired oxygen of less than 300 (normal >500). The patient was positive for high concentrations of anti-melanoma differentiation-associated gene-5 autoantibodies . (A) A high-resolution chest CT scan showed alveolar infiltrate in both lungs. Infectious and neoplastic causes were ruled out. (B) Characteristic skin lesion ulcer on elbow, (C) fingers suggesting vasculopathy , and (D) erythematous skin changes due to dermatomyositis. The patient was intubated and mechanic ventilation was started at the intensive care unit. After treatment with polymyxin-B haemoperfusion, glucocorticoids, tacrolimus, plasmapheresis, and intravenous immunoglobulin, the patient’s general condition improved to the extent that the orotracheal tube was withdrawn and he was discharged to a conventional ward.
A combination of genetic risk factors and exposure to environmental factors is presumably required to trigger dermatomyositis. Indeed, several immunogenetic risk factors, including certain class-2 HLA alleles, have been implicated in dermatomyositis pathogenesis.34 Exposure to ultraviolet light is also a known risk factor for developing dermatomyositis.35 However, the majority of people with known genetic risk factors, even those with high ultraviolet light exposure, never develop dermatomyositis. An increased number of mutations and loss of heterozygosity in TIF1 genes in tumours from patients with dermatomyositis who are positive for anti-TIF1 autoantibodies15 have been reported.36 This observation suggests that mutations in TIF1 genes might generate neoantigens that could trigger autoimmunity.
Whatever the cause, once a patient has developed dermatomyositis, it is unclear what mechanisms maintain muscle damage and weakness. Some evidence, including membrane attack complex deposition on endothelial cells, suggests that part of the muscle damage could be caused by hypoperfusion due to endothelial destruction.11 Additionally, the presence of plasmacytoid dendritic cells, potent sources of interferon, along with the increased expression of type-1 interferon-inducible proteins in the perifascicular area, suggest that interferon could somehow mediate perifascicular atrophy.8,37 However, further studies are required to clarify the role of these proteins in dermatomyositis muscle dysfunction.
Immune-mediated necrotising myositis
Immune-mediated necrotising myopathy is a distinct type of inflammatory myopathy characterised by proximal muscle weakness, exceptionally high muscle enzyme concentrations, myopathic EMG findings, and muscle biopsies showing necrosis or regeneration with minimal lymphocytic infiltrates and no perifascicular atrophy (appendix). Typical immune-mediated necrotising-myopathy muscle biopsies also include class-1 major histocompatibility complex upregulation, M2-macrophage infiltration, and membrane attack complex deposition on non-necrotic fibres (appendix).23,38 Extramuscular manifestations are rare and generally mild when they occur.21,22,25
Around two thirds of patients with immune-mediated necrotising myopathy have autoantibodies recognising either signal recognition particle (SRP) or 3-hydroxy 3-methylglutaryl coenzyme-A reductase (HMGCR). However, about 20% of patients who are positive for anti-SRP and anti-HMGCR have lymphocytic infiltrates in their muscle biopsies, but are otherwise indistinguishable from their counterparts with biopsies that reveal necrotising tissue.22,26,39 Patients with proximal muscle weakness, elevated creatine kinase concentrations, and anti-SRP autoantibodies are defined as having anti-SRP myopathy, but patients with proximal muscle weakness, elevated muscle creatine kinase concentrations, and anti-HMGCR autoantibodies are defined as having anti-HMGCR myopathy.40 According to this classification, specific biopsy features are not required to classify patients with autoantibody-positive immune-mediated necrotising myopathy.40 By contrast, a necrotising biopsy is still required to classify a patient as having autoantibody-negative immune-mediated necrotising myopathy.40
Anti-SRP and anti-HMGCR myopathy share many features, including similar muscle biopsy findings and high creatine kinase concentrations, and minimal extramuscular manifestations.23 Furthermore, in both types of myopathy, young patients seem to have more aggressive and refractory muscle disease.21,25 However, differences between these two immune-mediated necrotising-myopathy subtypes have been reported. First, anti-HMGCR myopathy is associated with statin exposure,24 whereas anti-SRP myopathy is not associated with use of statins.21,22 Second, patients who are positive for anti-SRP autoantibodies tend to have more severe muscle weakness than those with anti-HMGCR autoantibodies.21,23 Third, although virtually all patients with anti-HMGCR and anti-SRP autoantibodies show some degree of necrosis,22,26 those with anti-SRP autoantibodies have a higher number of necrotic muscle fibres than those with anti-HMGCR autoantibodies.39 Fourth, the presence of interstitial lung disease, although uncommon in both groups, is more common in patients with anti-SRP autoantibodies (13–22) than in those with anti-HMGCR autoantibodies (<5).21–23,25 Fifth, patients with anti-HMGCR myopathy and autoantibody-negative immune-mediated necrotising myopathy might have an increased risk of malignancy, unlike those with anti-SRP myopathy.41 Finally, anti-HMGCR myopathy has rarely been associated with cardiac involvement.23 By contrast, early cross-sectional studies in patients with anti-SRP myopathy suggested a high prevalence of cardiac manifestations in these patients,42,43 although these findings have not been supported by cohort studies.21–23 In patients with suspected cardiac involvement, an electrocardiogram and an echocardiogram should be performed. A gadolinium-enhanced MRI scan can assess active myocardial inflammation and, in selected patients, an endomyocardial biopsy can confirm the diagnosis.44
Regarding the immunogenetic risk factors in immune-mediated necrotising myopathy, one study45 suggested that class-2 HLA-allele DRB1*08:03 is associated with anti-SRP myopathy. Additionally, several case-control studies45–47 have supported that DRB1*11:01 is an immunogenetic risk factor for anti-HMGCR myopathy. This allele is present in about 70% of people with anti-HMGCR autoantibodies, but only in about 15% of the general population. Moreover, statins, which inhibit the enzymatic activity of HMGCR and increase HMGCR production, are a risk factor for anti-HMGCR myopathy.23–25 Given these observations, increased HMGCR production has been proposed to contribute to breaking tolerance for developing anti-HMGCR myopathy.48 Alternatively, in some patients, statins binding to HMGCR could generate a neoepitope that would have a role in triggering the immune response against HMGCR. In either scenario, DRB1*11:01 could have a role by presenting the relevant HMGCR peptides. Further work is required to show the relevance of these mechanisms in anti-HMGCR myopathy.
The mechanisms underlying myofibre necrosis in immune-mediated necrotising myopathy remain to be elucidated; however, some clues to these mechanisms have emerged. For example, given that the membrane attack complex deposits on the surface of non-necrotic fibres, anti-SRP and anti-HMGCR autoantibodies have been proposed to be directly pathogenic.39 A study49 showed that these autoantibodies can induce muscle atrophy, increase concentrations of reactive oxygen species and cytokines (eg, tumour necrosis factor and interleukin 6), and impair myoblast fusion (by decreasing the production of interleukin 4 and interleukin 13) of cultured muscle cells. However, anti-SRP and anti-HMGCR autoantibodies did not induce necrosis, and further studies are needed to show that these two autoantibodies are pathogenic in vivo.49
Sporadic inclusion-body myositis
As with other types of inflammatory myopathies, patients with sporadic inclusion-body myositis present with muscle weakness, and usually have elevated creatine kinase concentrations and myopathic EMG features, which could indicate chronicity. However, sporadic inclusion-body myositis is different from other inflammatory myopathies in numerous ways. First, twice as many men as women are affected by sporadic inclusion-body myositis; other inflammatory myopathies are more frequent in women.3 Second, sporadic inclusion-body myositis usually affects patients older than 50 years,3 but other types of inflammatory myopathies can also develop in patients younger than 50 years, including in children.2 Third, in most patients with sporadic inclusion-body myositis, the disease is slowly progressive, with weakness occurring over the course of years.3 In other inflammatory myopathies, progressive weakness can occur over weeks or months.3 Fourth, many patients with sporadic inclusion-body myositis have an asymmetric pattern of muscle weakness; symmetric weakness is the rule in patients with other inflammatory myopathies.3 Fifth, patients with sporadic inclusion-body myositis usually have prominent knee extensor weakness along with distal weakness, including of the deep finger flexors, wrist flexors, and ankle dorsiflexors; arm abductors and hip flexors can also be affected, but are frequently stronger than more distal muscles.27 By contrast, unless the disease is in an advanced stage, few other patients with inflammatory myopathies have substantial distal weakness.3,13 Sixth, these patients might also have progressive dysphagia,3 which can lead to bronchoaspiration and can be studied using video-fluoroscopy. Seventh, although autoantibodies recognizing cytosolic 5′-nucleotidase 1A (NT5C1a) are present in 30–60% of patients with sporadic inclusion-body myositis, these types of autoantibodies can also be found in 5–10% of patients with polymyositis, 15–20% of patients with dermatomyositis, and 10% of patients with systemic lupus erythematosus and 12% with Sjögren’s syndrome.50–53 Unlike other types of inflammatory myopathies, sporadic inclusion-body myositis is not associated with any myositis-specific autoantibody.54 Eighth, no clear evidence shows that immunosuppression benefits patients with sporadic inclusion-body myositis, whereas patients with other inflammatory myopathies usually do respond to immunosuppression.3 Finally, compared with patients with other inflammatory myopathies, those with sporadic inclusion-body myositis have the most characteristic MRI scan pattern, with severe involvement of the anterior compartment of the thigh.6,55
Muscle biopsies from patients with sporadic inclusion-body myositis are histologically unique in that they often include coexisting inflammation, mitochondrial dysfunction, and abnormal protein aggregation (figure 3).9 The inflammatory infiltrate is comprised of CD8 T cells that surround and invade non-necrotic fibres. A study56 has shown that these cells express CD57, a surface marker of T-cell aggressiveness, and that most (22 [58%] of 38) patients with sporadic inclusion-body myositis meet criteria for T-cell large granular lymphocytic leukaemia. Although this association has yet to be validated, it would explain the refractory nature and advanced age of these patients.56 Doing a muscle MRI scan to select the site of muscle biopsy is important in patients with sporadic inclusion-body myositis (as well as all types of inflammatory myopathies) because this method increases the diagnostic accuracy of the pathology.57
Figure 3: Clinical features and pathological findings of sporadic inclusion-body myositis.
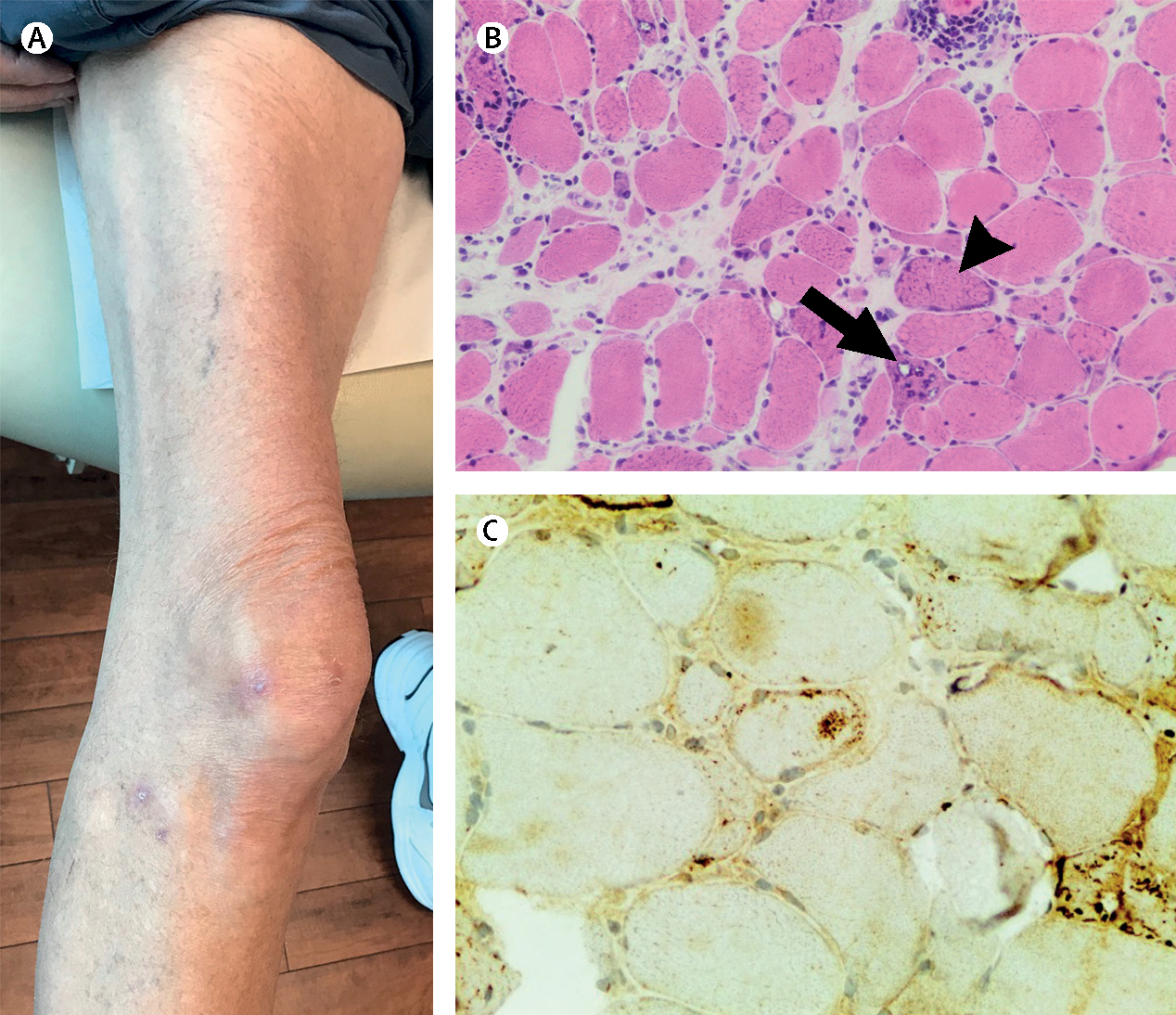
A man aged 65 years presented with slowly progressing muscle weakness that included weakness of distal muscle groups. (A) Quadriceps were markedly atrophied and the muscle biopsy showed (B) fibres with rimmed vacuoles (black arrow), ragged-red fibres (black arrowhead), and (C) p62 positivity. Quadriceps image courtesy of Dr Tom Lloyd (Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, MD, USA).
An increased number of cytochrome oxidase-negative muscle fibres and the presence of so-called ragged-red fibres suggest that mitochondrial damage has an important role in sporadic inclusion-body myositis (figure 3).11 Additionally, a study58 showed that mitochondrial DNA is depleted and that mitochondrial fusion proteins are dysregulated in the muscle of patients with sporadic inclusion-body myositis. Furthermore, an increased frequency of mitochondrial DNA deletions was also reported in the muscles of these patients.59
Rimmed vacuoles, best visualised by Gomori-trichrome staining, are a histological hallmark of biopsies from patients with sporadic inclusion-body myositis (figure 3).3 Although some patients with hereditary myopathies also have rimmed vacuoles, their presence can help in distinguishing sporadic inclusion-body myositis from other inflammatory myopathies, because these vacuoles are not present in other types of inflammatory myopathies.11 How the rimmed vacuoles of sporadic inclusion-body myositis are formed remains unknown. However, nuclear membrane proteins are found within rimmed vacuoles, suggesting that these vacuoles could be the remnants of degenerated myonuclei.60 Another study61 showed that proteins accumulating in rimmed vacuoles are related to protein folding and autophagy, suggesting that impaired autophagic function could be implicated in their formation.
Several other cytoplasmic inclusions, which are pathophysiologically relevant and important for diagnosis, are also found in muscle biopsies of patients with sporadic inclusion-body myositis. The tubofilamentous inclusions seen by electron microscopy gave rise to the name inclusion-body myositis.11 Cytoplasmic accumulations of amyloid can be visualised using Congo red and polarised light.11 These structures include β-amyloid-related and amyloidogenic-related molecules β-secretase 1 and γ-secretase, which are increased in the plasma of patients with sporadic inclusion-body myositis.62 Cytoplasmic aggregations of other proteins, including phosphorylated neurofilaments, p62, and TAR DNA-binding protein 43 can also be found in muscle biopsies of patients with sporadic inclusion-body myositis (figure 3).11
Although not specific to sporadic inclusion-body myositis, anti-NT5C1a autoantibodies have been associated with increased severity and mortality in these patients.53,63 Additionally, anti-NT5C1a autoantibodies might directly cause muscle damage.64 Therefore, the presence of such autoantibodies along with the presence of aggressive T cells in muscle biopsies suggest that autoimmunity might be relevant in the pathology of sporadic inclusion-body myositis. Alternatively, the presence of cytoplasmic inclusions suggests that an underlying degenerative process drives the progression of the disease. This hypothesis is reinforced by the observation that immunosuppression is not effective in patients with sporadic inclusion-body myositis.3 Future investigations will elucidate whether successful treatment strategies will need to target the immune system, protein aggregation pathways, or both.
Overlap myositis
Overlap myositis is a type of autoimmune myopathy associated with other connective tissue diseases. Autoimmune myopathy can also occur in patients presenting with features of other autoimmune diseases, such as systemic lupus erythematosus, rheumatoid arthritis, Sjögren’s syndrome, or systemic sclerosis.30–32 Many of these patients also have autoantibodies that are associated with characteristic phenotypes.30–32
The most representative form of overlap myositis is antisynthetase syndrome, with autoantibodies targeting aminoacyl tRNA synthetases, which are enzymes that conjugate an aminoacid to its cognate tRNA.28,29 Autoantibodies that recognise histidyl tRNA synthetase (anti-Jo1), threonyl tRNA synthetase (anti-PL7), and alanyl tRNA synthetase (anti-PL12) are most common.28,29 Patients with any one of these autoantibodies can be defined as having antisynthetase syndrome, and typically present with one or more of the following features: an inflammatory myopathy, interstitial lung disease, arthritis, Raynaud syndrome, fever, or hyperkeratotic radial-finger lesions known as mechanic’s hands (figure 4).3 Patients with antisynthetase syndrome can also have skin rashes similar to dermatomyositis.3 Notably, not all patients with antisynthetase syndrome have muscle weakness. Indeed, whereas about 90% of patients who have anti-Jo1 autoantibodies have an inflammatory myopathy, up to 50% of patients with anti-PL12 autoantibodies present with interstitial lung disease but no muscle involvement.28 Furthermore, patients who are positive for anti-Jo1 autoantibodies have more severe weakness than patients with anti-PL7 and anti-PL12 autoantibodies who have more severe lung involvement (table 1).28,29
Figure 4: Clinical features and radiological and pathological findings of antisynthetase syndrome.
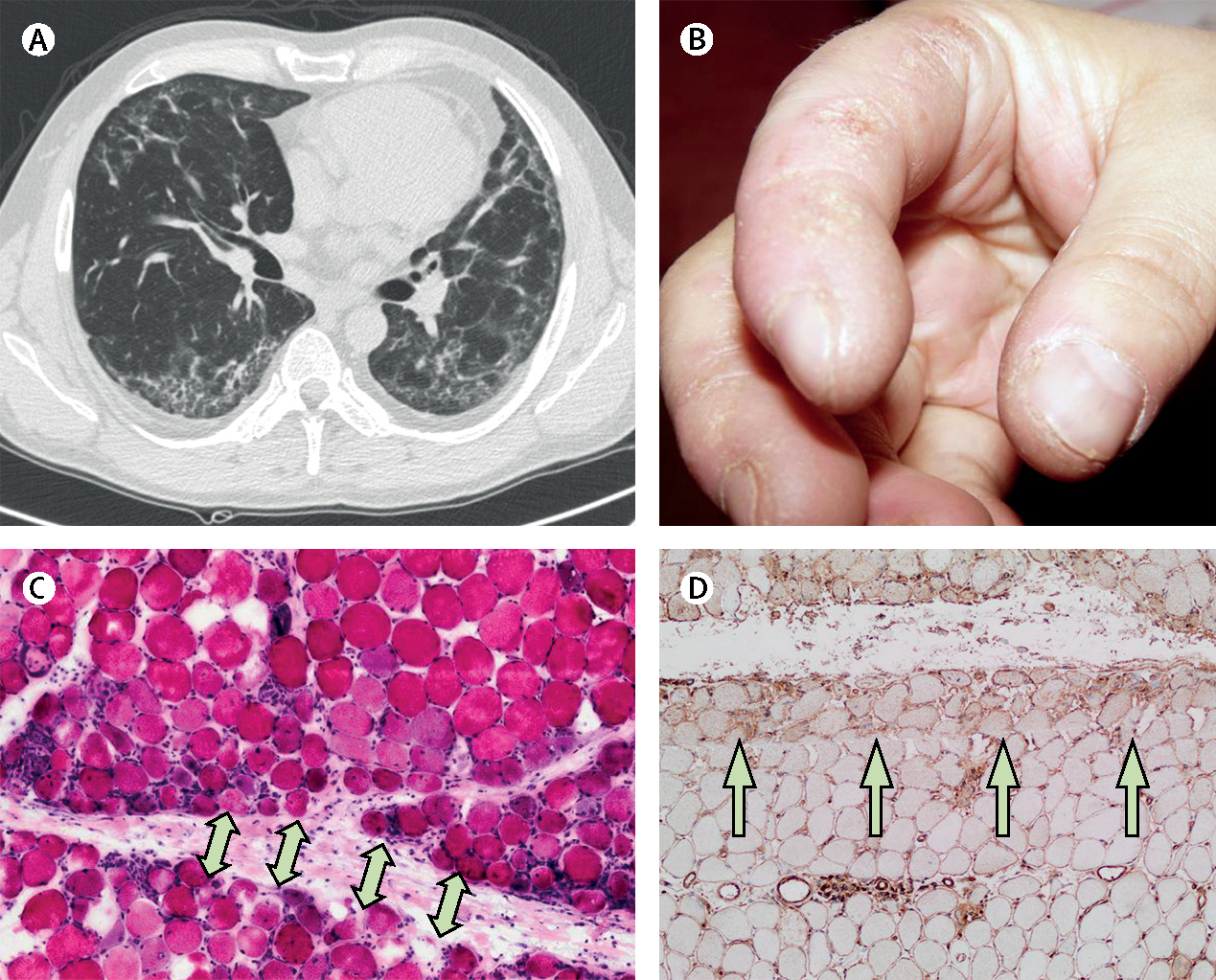
A woman aged 45 years presented with muscle weakness and dyspnoea. (A) A high-resolution chest CT scan showed interstitial lung disease. She had crackles in both lung bases and (B) mechanic’s hands. Muscle biopsy showed (C) necrotic and regenerating muscle fibres in the perifascicular area (arrows) and (D) prominent class-1 major histocompatibility complex positivity predominantly in the perifascicular area (arrows). Serum was positive for anti-Jo1 antibodies. Jo1=histidyl tRNA synthetase.
The features of myopathic antisynthetase syndrome are similar to those of dermatomyositis, including proximal muscle weakness, elevated muscle enzymes, and myopathic EMG.3 Patients with antisynthetase syndrome often have hyperintensities on intramuscular T2-weighted MRI scans, but a specific MRI scan pattern has not been described.65 Muscle biopsies can reveal perifascicular atrophy similar to dermatomyositis. However, compared with dermatomyositis, the muscle biopsies from patients with antisynthetase syndrome can show an increased number of perifascicular necrotic fibers.66,67 Furthermore, most of these biopsies show nuclear actin aggregation, an electron microscopy feature that is not seen in other inflammatory myopathies.68 However, little is known about what triggers and maintains autoimmunity in antisynthetase syndrome, and further research is warranted.
Anti-polymyositis-Scl or anti-Ku autoantibodies are associated with inflammatory myopathies in patients with systemic sclerosis.30,31 Similarly, patients with connective tissue disease who are positive for anti-U1 ribonucleoprotein can have myositis along with additional systemic sclerosis features, such as sclerodactyly.32 These patients can also have systemic lupus erythematosus-like features, such as glomerulonephritis, usually accompanied by anti-DNA autoantibodies (table 1).32 Sporadic inclusion-body myositis can be associated with Sjögren’s syndrome, especially in women with a genetic predisposition.69
Polymyositis
Polymyositis is defined by the presence of muscle weakness, elevated creatine phosphokinase concentrations, myopathic EMG features, and inflammatory CD8 T-cell infiltrates on muscle biopsy, with none of the characteristic accompanying features of the other above-mentioned groups (ie, dermatomyositis, immune-med iated necrotising myopathy, overlap myositis, or sporadic inclusion-body myositis). Many patients previously classified as having polymyositis could now be considered to have antisynthetase syndrome without a rash, immune-mediated necrotising myopathy, or sporadic inclusion-body myositis on the basis of characteristic clinical manifestations, serological features, and muscle biopsy findings.70–72 Even for patients who truly have polymyositis, the condition remains a diagnosis of exclusion, and these patients should be closely monitored for new clinical features suggesting alternative diagnoses.
Management
Treatment for inflammatory myopathies remains a challenge. The low prevalence, wide phenotypic heterogeneity, and variable course of these diseases complicate the assessment of different treatment approaches, which explains the absence of standardised therapeutic guidelines. For the same reasons, treatment should be multidisciplinary and managed by experienced clinicians. Tailored physical exercise programmes and rehabilitation under the supervision of a physical therapist are safe in all types of inflammatory myopathies, and are generally recommended to increase strength and reduce disability.73,74 In this regard, two randomised controlled trials73,74 have shown the utility of physical exercise intervention as a complementary treatment even in patients with acute inflammatory myopathies.
Non-inclusion-body myositis
Dermatomyositis, immune-mediated necrotising myopathy, overlap myositis, and polymyositis are considered to be non-inclusion-body myositis. A systematic Cochrane review75 highlighted the scarcity of high-quality randomised controlled trials for the treatment of inflammatory myopathies; although immunosuppressive agents are the primary form of treatment for inflammatory myopathies, few randomised controlled trials have been done in the field. Thus, treatment is largely based on historical clinical practice and case series, and fundamentally guided by the opinion of experts. Glucocorticoids are first-line drugs in the treatment of inflammatory myopathies, but are rarely used as a monotherapy because of their side-effects, such as osteoporosis, hypertension, or weight gain.76 The most commonly prescribed corticosteroid is prednisone, which is typically started at a dose of 0·5–1 mg/kg per day to a maximum dose of 80–100 mg per day. In severe cases, daily intravenous methylprednisolone pulses of 500 mg to 1 g per day for 3–5 days can be used initially (table 2). Prednisone is maintained for 4–6 weeks and is then decreased. Neither the initial dose nor the speed at which corticosteroid treatment should be decreased have been adequately investigated, and so treatment is based on clinical judement.
Table 2:
Treatment options for inflammatory myopathies other than sporadic inclusion-body myositis
| Doses and treatment suggestions | Treatment for | Side-effects | |
|---|---|---|---|
|
| |||
| Immunosuppressive or immunomodulatory drugs | |||
| Corticosteroids3 | Prednisone 0·5–1 mg/kg per day. Consider adding 500 mg to 1 g intravenous methylprednisolone pulses once per day for 3 days for severe cases | All patients and all manifestations | Hypertension, hyperglycaemia, hyperlipidaemia, osteoporosis, infections, and cataracts |
| Azathioprine3 | 2–3 mg/kg per day | Predominantly myositis | Gastrointestinal symptoms, myelosuppression, leukaemia pancreatitis, infections, and liver toxicity |
| Methotrexate3,77 | Up to 25 mg per week | Arthritis; use with caution in interstitial lung disease | Stomatitis, gastrointestinal symptoms, leucopoenia, liver toxicity, infections, and lung toxicity |
| Ciclosporin78 | Up to 5 mg/kg per day | Skin involvement (panniculitis and dermatomyositis skin rashes) and interstitial lung disease | Renal insufficiency, anaemia, infections, and hypertension |
| Tacrolimus79 | 0·06 mg/kg per day | Interstitial lung disease | Hypertension, renal insufficiency, gastrointestinal symptoms, infections, and tremor |
| Mycophenolate mofetil80 | 2–3 g per day | Interstitial lung disease | Gastrointestinal symptoms, myelosuppression, infections, and hypertension |
| Cyclophosphamide81 | Intravenous 0·5–1 g/m2 per month or 10–15 mg/kg per month for 6–12 months | Interstitial lung disease | Myelosuppression, myeloproliferative disorders, haemorrhagic cystitis, bladder cancer, infections, and infertility |
| Intravenous immunoglobulins82,83 | 2 g per kg every 4–6 weeks | Dysphagia and severe disease refractory to other treatments | Hypotension, anaphylaxis, headache, aseptic meningitis, blood clots, infections, and renal toxicity |
| Biological agents | |||
| Rituximab21,40,84,85 | 1 g given twice within a 2-week interval; maintenance with either one or two doses of 0·5–1 g rituximab on the basis of the patient’s clinical situation and their CD19 and CD20 counts (usually given every 6–9 months) | Rapidly progressive interstitial lung disease and severe cases of inflammatory myopathies | Infusion-related reaction, infections, and progressive multifocal leukoencephalopathy |
| Abatacept86 | 750 mg intravenously every 4 weeks (if patient’s weight <60 kg, 500 mg; if patient’s weight >100 kg, 1000 mg) | Consider for refractory disease | Infusion reactions and infections |
| Tocilizumab87 | 8 mg per kg intravenously every 4 weeks or 162 mg per week subcutaneously | Consider for refractory disease | Liver toxicity, neutropaenia, thrombocytopaenia, infections, hyperlipidaemia, and intestinal perforation |
| Physical exercise73,74 | Aerobic and resistance-tailored programmes (about 4 weeks after starting medical treatment or as soon as the patient can cope with exercise) | All patients, as a coadjuvant therapy | Not described |
Other immunosuppressive drugs that are used for treatment of inflammatory myopathies include methotrexate (10–25 mg per week orally or subcutaneously) and azathioprine (2–3 mg/kg per day) in patients with normal thiopurine methyltransferase activity, and mycophenolate mofetil (2–3 g per day divided into two doses), ciclosporin (3–4 mg/kg per day), tacrolimus (0·06 mg/kg per day), and intravenous immunoglobulins.3 These drugs are normally used as corticosteroid-sparing agents from the time of diagnosis. Some clinical settings can warrant the selection of different drugs. For example, methotrexate, which is useful for muscle and joint manifestations,77 should be carefully used in patients with myositis-associated interstitial lung disease because of the potential lung toxicity of this drug.77 Calcineurin inhibitors (eg, ciclosporin and tacrolimus) improve dermatomyositis skin manifestations, and along with mycophenolate are recommended by experts in myositis-associated interstitial lung disease.78–80 However, these drugs need to be administered with caution in elderly patients with hypertension because of the potential renal toxicity and management difficulties in hypertensive patients and their dose should be guided by serum concentrations of the drug. Cyclophosphamide can be used in patients with severe or rapidly progressive interstitial lung disease as in other autoimmune diseases, such as systemic sclerosis,81 but can impair fertility.
Intravenous immunoglobulins (2 g/kg per month administered during 2–5 days) have shown efficacy in an randomised controlled trial82 and in a retrospective study83 for the management of dermatomyositis. Intravenous immunoglobulins also seem to be effective in immune-mediated necrotising myopathy, especially in patients with anti-HMGCR autoantibodies.40,88 A preliminary case series89 of 19 patients suggests that subcutaneous immunoglobulins could be an alternative to intravenous administration, although no comparative studies are yet available (table 2).
Because of the existence of refractory forms of non-inclusion-body myositis, there has been growing interest in assessing the potential of several biological agents in inflammatory myopathies. The efficacy of one of these agents, rituximab (a monoclonal antibody targeting the CD20 antigen on B lymphocytes), was assessed in 124 patients with refractory dermatomyositis and 76 patients with polymyositis in a randomised, double-blind, placebo-controlled trial,84 the only randomised controlled trial done to date with this agent. Although the drug did not induce faster improvement when introduced early versus late in the course of the clinical trial, 161 (83%) of 195 patients with refractory disease with longitudinal data (regardless of the study branch) met the International Myositis Assessment & Clinical Studies Group definition of improvement. However, because the trial design allowed for treatment with other immunosuppressive drugs, the study did not establish whether rituximab was causing that improvement. Nevertheless, a post-hoc analysis of the trial and further case series and cohort studies suggested beneficial effects of this drug in patients with antisynthetase syndrome, and in those with anti-Mi2 or anti-SRP autoantibodies.21,40,85 Although the efficacy of rituximab for patients with myositis-associated interstitial lung disease is only based on two small uncontrolled studies90,91 and the aforementioned trial,84 rituximab is usually preferred to cyclophosphamide on account of the better tolerance and side-effect profile of this drug. Rituximab is usually administered as two 1-g doses 2 weeks apart (table 2).
Evidence supporting the use of anti-tumour necrosis factor in inflammatory myopathies is inconclusive since studies on etanercept and infliximab have shown conflicting results.92–94 Although some patients might respond to these therapies, more studies are necessary to define the role of these drugs in inflammatory myopathies. In a pilot study86 of 20 patients with inflammatory myopathies, abatacept, a fusion protein that inhibits T-cell costimulation, showed beneficial effects in reducing disease activity and increasing the number of regulatory T cells in muscle biopsies of these patients; however, these results need to be supported by further studies with larger sample sizes. Some case reports have reported the efficacy of tocilizumab (an antagonist of interleukin 6),87 anakinra (antiinterleukin 1),95 alemtuzumab (anti-CD52),96 and tofacitinib97 and ruxolitinib98 (janus-kinase inhibitors) in inflammatory myopathies, but confirmatory studies are required for these treatments to be widely used in clinical practice.
Sporadic inclusion-body myositis
No pharmacological therapy has been shown to be effective for sporadic inclusion-body myositis, and consequently treatment of this form of myositis remains largely supportive. Immunosuppressive drugs such as corticosteroids, azathioprine, methotrexate, etanercept, anakinra, or interferon β are not effective in sporadic inclusion-body myositis.99,100 Alemtuzumab is an anti-T-cell agent that has shown a trend towards a reduction of key biomarkers, such as interleukin 1β or class-1 major histocompatibility complex, in a pilot study that has not been confirmed so far,101 although larger trials are needed given the trends towards reduction. Two small clinical trials, one that investigated bimagrumab102 (a human monoclonal antibody that blocks the myostatin pathway) and the other that investigated follistatin103 (another inhibitor of myostatin locally delivered using an adenovirus), showed improved thigh muscle volume and performance on the 6-min walking test in patients with sporadic inclusion-body myositis; however, these drugs did not significantly improve muscle strength. An randomised controlled trial104 on the efficacy of sirolimus did not meet the primary outcome (improved quadriceps strength), but patients who received the drug did show improvement on the 6-min walking test.
Other drugs with alternative mechanisms of action have been investigated. Oxandrolone, an anabolic steroid, and simvastatin, a cholesterol-lowering agent, were not shown to be effective in small clinical trials.105,106 A large randomised controlled trial (NCT02753530) is underway with arimoclomol, a drug that prevents aberrant protein–protein interaction and promotes adequate protein folding. Another randomised controlled trial (NCT02483845) investigating natalizumab is ongoing.
Severe manifestations of the disease
Some patient conditions require different therapeutic management, such as when the patient presents with severe weakness, dysphagia, or rapidly progressive interstitial lung disease. Severe weakness is characteristic of patients with immune-mediated necrotising myopathy, but can also occur in all types of inflammatory myopathies.3 Additionally, severe muscle weakness can cause acute complications, such as development of restrictive lung disease due to respiratory muscle weakness, or severe dysphagia if oropharyngeal muscles are involved.3 Evidence suggests that atrophy and fatty replacement of muscle tissue is established early after the onset of disease, and thus, delayed treatment can lead to long-term disability.6 Thus, in these situations a three-drug initial regimen should be considered, and should include high-dose corticosteroids with an initial intravenous bolus, a secondary agent (usually azathioprine, methotrexate, tacrolimus, or mycophenolate mofetil), and intravenous immunoglobulins. Rituximab should be considered in patients with refractory disease (panel).40
Given the risk of bronchoaspiration, patients with inflammatory myopathies with dysphagia other than inclusion body myositis should receive a three-drug regimen that is similar to that given to patients with severe weakness. Intravenous immunoglobulins have been shown to improve dysphagia107 and should therefore be considered in patients with substantial dysphagia. In selected patients, mainly those with sporadic inclusion-body myositis but also those with extremely severe cases of dysphagia in other types of inflammatory myopathies, local therapies such as cricopharyngeal myotomy, pharyngoesophageal balloon dilatation, and injection of botulinum toxin into the upper oesophageal sphincter have shown a reasonable benefit in improving life-threatening dysphagia.108,109 Swallowing physical therapy could also help to avoid complications in patients with myositis and dysphagia.
Rapidly progressive interstitial lung disease is a hallmark of hypomyopathic or amyopathic dermatomyositis associated with anti-MDA5 autoantibodies, but can also occur with other types of inflammatory myopathies, such as the antisynthetase syndrome.28 In most cohorts studied, about 50–60% of patients with anti-MDA5-positive inflammatory myopathies develop interstitial lung disease soon after the onset of disease, and most of these patients (>85%) will have rapidly progressive forms of the illness.17,18 Because 30–50% of patients who develop rapidly progressive interstitial lung disease will die during the first year after the onset of lung disease,17,18 patients who are positive for anti-MDA5 autoantibodies, even those with mild interstitial lung disease, should be intensively treated from disease onset with glucocorticoids and a second-line immunosuppressant agent (eg, tacrolimus or mycophenolate mofetil), and should be followed closely. When interstitial lung disease progression is detected, immediate and intensive treatment should be initiated. Different therapeutic strategies include methylprednisolone pulses, immunosuppressants (tacrolimus, cyclophosphamide, or rituximab), courses of polymyxin-B-immobilised fibre-column haemo-perfusion, plasmapheresis, and intravenous immunoglobulins.110 Lung transplantation should be considered as a last-resort treatment in patients with myositis-associated interstitial lung disease, whether rapidly progressive or not.111
Conclusions and future directions
Five main types of inflammatory myopathies are now widely recognised: dermatomyositis, immune-mediated necrotising myopathy, sporadic inclusion-body myositis, overlap myositis, and polymyositis (table 1). Furthermore, within each type, specific autoantibodies further divide patients into even more homogeneous subtypes. For example, although patients in the immune-mediated necrotising-myopathy subgroup share some clinical characteristics (eg, necrotising muscle biopsies), those with anti-HMGCR autoantibodies have different environmental and immunogenetic risk factors, and might respond optimally to different treatments than patients with anti-SRP autoantibodies. This broad classification scheme is likely to require revision as more is learnt about inflammatory myopathies. For example, if new autoantibodies associated with dermatomyositis and immune-mediated necrotising myopathy are discovered, the number of patients with autoantibody-negative dermatomyositis and immune-mediated necrotising myopathy will decrease.
Therapies for inflammatory myopathies are broadly immunosuppressive rather than targeting specific pathogenic pathways. Moreover, past clinical trials have been hampered by the inclusion of heterogeneous patient populations. Even if imperfect, this widely accepted classification scheme should define more homogeneous patient populations, which will improve the power of clinical trials to identify effective treatments for the different forms of inflammatory myopathies. However, transformative changes in the way we treat inflammatory myopathies will probably require a deep understanding of disease mechanisms to reveal novel therapeutic targets. In this regard, recent investigations are promising, such as those establishing the pathogenic role of specific autoantibodies in immune-mediated necrotising myopathy,40 the association between sporadic inclusion-body myositis and large granular lymphocytic leukaemia,56 or the link between tumour TIF1 mutations and paraneoplastic anti-TIF1 myositis.36 As our understanding of inflammatory myopathy disease mechanisms expands, we expect that additional therapeutic targets will be identified.
Supplementary Material
Panel: Sequential treatment approach and treatment of severe manifestations of the disease.
Inflammatory myopathies
Initial treatment
Treatment for severe or refractory cases of disease
Dysphagia
Rapidly progressive interstitial lung disease
Pulses of methylprednisolone followed by systemic corticosteroids along with a second-line treatment agent (eg, tacrolimus, mycophenolate mofetil, cyclophosphamide, or rituximab). Additionally, the following treatments should be considered: two sessions of polymyxin in 24 h, daily plasmapheresis over the course of 3 days and on alternate days thereafter until the completion of seven sessions, and 400 mg intravenous immunoglobulin per kg after each plasmapheresis session
Search strategy and selection criteria.
Articles for this Review were identified by searches of PubMed between March 1, 2003, and March 31, 2018, and references from relevant articles. The search terms “dermatomyositis”, “polymyositis”, “myositis”, “autoantibodies”, “inclusion-body myositis”, “antisynthetase”, and “immune-mediated necrotising myositis” were used. There were no language restrictions. The final reference list was generated on the basis of relevance to the topics covered in this Review.
Footnotes
Declaration of interests
ALM has a patent on an anti-HMGCR assay. AS-O’C, IP-F, ET-A, JCM, and JMG-J declare no competing interests.
Contributor Information
Albert Selva-O’Callaghan, Systemic Autoimmune Diseases Unit, Vall d’Hebron General Hospital, Universitat Autònoma de Barcelona, Barcelona, Spain.
Iago Pinal-Fernandez, Muscle Disease Unit, Laboratory of Muscle Stem Cells and Gene Regulation, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD, USA; Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Ernesto Trallero-Araguás, Rheumatology Unit, Vall d’Hebron General Hospital, Universitat Autònoma de Barcelona, Barcelona, Spain.
José César Milisenda, Internal Medicine Department, Hospital Clinic, Universitat de Barcelona, Barcelona, Spain; Centro de Investigación Médica en Red Enfermedades Raras.
Josep Maria Grau-Junyent, Internal Medicine Department, Hospital Clinic, Universitat de Barcelona, Barcelona, Spain; Centro de Investigación Médica en Red Enfermedades Raras.
Andrew L Mammen, Muscle Disease Unit, Laboratory of Muscle Stem Cells and Gene Regulation, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD, USA; Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
References
- 1.Senecal JL, Raynauld JP, Troyanov Y. Editorial: a new classification of adult autoimmune myositis. Arthritis Rheumatol 2017; 69: 878–84. [DOI] [PubMed] [Google Scholar]
- 2.Lundberg IE, Tjarnlund A, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis 2017; 76: 1955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalakas MC. Inflammatory muscle diseases. N Engl J Med 2015; 373: 393–94. [DOI] [PubMed] [Google Scholar]
- 4.Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med 2016; 280: 8–23. [DOI] [PubMed] [Google Scholar]
- 5.Paganoni S, Amato A. Electrodiagnostic evaluation of myopathies. Phys Med Rehabil Clin N Am 2013; 24: 193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinal-Fernandez I, Casal-Dominguez M, Carrino JA, et al. Thigh muscle MRI in immune-mediated necrotising myopathy: extensive oedema, early muscle damage and role of anti-SRP autoantibodies as a marker of severity. Ann Rheum Dis 2017; 76: 681–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirani Z, Kucenic MJ, Carroll CL et al. Pruritus in adult dermato-myositis. Clin Exp Dermatol 2004; 29: 273–76 [DOI] [PubMed] [Google Scholar]
- 8.Suarez-Calvet X, Gallardo E, Pinal-Fernandez I, et al. RIG-I expression in perifascicular myofibers is a reliable biomarker of dermatomyositis. Arthritis Res Ther 2017; 19: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinal-Fernandez I, Casciola-Rosen LA, Christopher-Stine L, Corse AM, Mammen AL. The prevalence of individual histopathologic features varies according to autoantibody status in muscle biopsies from patients with dermatomyositis. J Rheumatol 2015; 42: 1448–54. [PMC free article] [PubMed] [Google Scholar]
- 10.Uruha A, Nishikawa A, Tsuburaya RS, et al. Sarcoplasmic MxA expression: a valuable marker of dermatomyositis. Neurology 2017; 88: 493–500. [DOI] [PubMed] [Google Scholar]
- 11.Dalakas MC. Muscle biopsy findings in inflammatory myopathies. Rheum Dis Clin North Am 2002; 28: 779–98. [DOI] [PubMed] [Google Scholar]
- 12.Ghirardello A, Zampieri S, Iaccarino L, et al. Anti-Mi-2 antibodies. Autoimmunity 2005; 38: 79–83. [DOI] [PubMed] [Google Scholar]
- 13.Albayda J, Pinal-Fernandez I, Huang W, et al. Antinuclear matrix protein 2 autoantibodies and edema, muscle disease, and malignancy risk in dermatomyositis patients. Arthritis Care Res (Hoboken) 2017; 69: 1771–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiorentino DF, Chung LS, Christopher-Stine L, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis Rheum 2013; 65: 2954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trallero-Araguas E, Rodrigo-Pendas JA, Selva-O’Callaghan A, et al. Usefulness of anti-p155 autoantibody for diagnosing cancer-associated dermatomyositis: a systematic review and meta-analysis. Arthritis Rheum 2012; 64: 523–32. [DOI] [PubMed] [Google Scholar]
- 16.Ge Y, Lu X, Shu X, Peng Q, Wang G. Clinical characteristics of anti-SAE antibodies in Chinese patients with dermatomyositis in comparison with different patient cohorts. Sci Rep 2017; 7: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labrador-Horrillo M, Martinez MA, Selva-O’Callaghan A, et al. Anti-MDA5 antibodies in a large Mediterranean population of adults with dermatomyositis. J Immunol Res 2014; 2014: 290797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009; 60: 2193–200. [DOI] [PubMed] [Google Scholar]
- 19.Narang NS, Casciola-Rosen L, Li S, Chung L, Fiorentino DF. Cutaneous ulceration in dermatomyositis: association with anti-melanoma differentiation-associated gene 5 antibodies and interstitial lung disease. Arthritis Care Res (Hoboken) 2015; 67: 667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodoki L, Budai D, Nagy-Vincze M, Griger Z, Betteridge Z, Dankó K. Comparison of clinical characteristics and laboratory parameters of patients with dermatomyositis-specific autoantibodies and autoantibody negative patients. Orv Hetil 2015; 156: 1451–59. [DOI] [PubMed] [Google Scholar]
- 21.Pinal-Fernandez I, Parks C, Werner JL, et al. Longitudinal course of disease in a large cohort of myositis patients with autoantibodies recognizing the signal recognition particle. Arthritis Care Res (Hoboken) 2017; 69: 263–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki S, Nishikawa A, Kuwana M, et al. Inflammatory myopathy with anti-signal recognition particle antibodies: case series of 100 patients. Orphanet J Rare Dis 2015; 10: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe Y, Uruha A, Suzuki S, et al. Clinical features and prognosis in anti-SRP and anti-HMGCR necrotising myopathy. J Neurol Neurosurg Psychiatry 2016; 87: 1038–44. [DOI] [PubMed] [Google Scholar]
- 24.Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010; 62: 2757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tiniakou E, Pinal-Fernandez I, Lloyd TE, et al. More severe disease and slower recovery in younger patients with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Rheumatology (Oxford) 2017; 56: 787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63: 713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology 2014; 83: 426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinal-Fernandez I, Casal-Dominguez M, Huapaya JA, et al. A longitudinal cohort study of the anti-synthetase syndrome: increased severity of interstitial lung disease in black patients and patients with anti-PL7 and anti-PL12 autoantibodies. Rheumatology (Oxford) 2017; 56: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trallero-Araguas E, Grau-Junyent JM, Labirua-Iturburu A, et al. Clinical manifestations and long-term outcome of anti-Jo1 antisynthetase patients in a large cohort of Spanish patients from the GEAS-IIM group. Semin Arthritis Rheum 2016; 46: 225–31. [DOI] [PubMed] [Google Scholar]
- 30.Guillen-Del Castillo A, Pilar Simeon-Aznar C, Fonollosa-Pla V, et al. Good outcome of interstitial lung disease in patients with scleroderma associated to anti-PM/Scl antibody. Semin Arthritis Rheum 2014; 44: 331–37. [DOI] [PubMed] [Google Scholar]
- 31.Rigolet A, Musset L, Dubourg O, et al. Inflammatory myopathies with anti-Ku antibodies: a prognosis dependent on associated lung disease. Medicine (Baltimore) 2012; 91: 95–102. [DOI] [PubMed] [Google Scholar]
- 32.Escola-Verge L, Pinal-Fernandez I, Fernandez-Codina A, et al. Mixed connective tissue disease and epitope spreading: a historical cohort study. J Clin Rheumatol 2017; 23: 155–59. [DOI] [PubMed] [Google Scholar]
- 33.Selva-O’Callaghan A, Grau JM, Gamez-Cenzano C, et al. Conventional cancer screening versus PET/CT in dermatomyositis/polymyositis. Am J Med 2010; 123: 558–62. [DOI] [PubMed] [Google Scholar]
- 34.Miller FW, Chen W, O’Hanlon TP, et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun 2015; 16: 470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mamyrova G, Rider LG, Ehrlich A, et al. Environmental factors associated with disease flare in juvenile and adult dermatomyositis. Rheumatology (Oxford) 2017; 56: 1342–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinal-Fernandez I, Ferrer-Fabregas B, Trallero-Araguas E, et al. Tumour TIF1 mutations and loss of heterozygosity related to cancer-associated myositis. Rheumatology (Oxford) 2018; 57: 388–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenberg SA, Pinkus JL, Pinkus GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005; 57: 664–78. [DOI] [PubMed] [Google Scholar]
- 38.Chung T, Christopher-Stine L, Paik JJ, Corse A, Mammen AL. The composition of cellular infiltrates in anti-HMG-CoA reductase-associated myopathy. Muscle Nerve 2015; 52: 189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allenbach Y, Arouche-Delaperche L, Preusse C, et al. Necrosis in anti-SRP(+) and anti-HMGCR(+) myopathies: role of autoantibodies and complement. Neurology 2018; 90: e507–17. [DOI] [PubMed] [Google Scholar]
- 40.Allenbach Y, Mammen AL, Stenzel W, Benveniste O, Immune-Mediated Necrotizing Myopathies Working Group. 224th ENMC International Workshop: clinico-sero-pathological classification of immune-mediated necrotizing myopathies Zandvoort, The Netherlands, Oct 14–16, 2016. Neuromuscul Disord 2018; 28: 87–99. [DOI] [PubMed] [Google Scholar]
- 41.Allenbach Y, Keraen J, Bouvier AM, et al. High risk of cancer in autoimmune necrotizing myopathies: usefulness of myositis specific antibody. Brain 2016; 139: 2131–35. [DOI] [PubMed] [Google Scholar]
- 42.Targoff IN, Johnson AE, Miller FW. Antibody to signal recognition particle in polymyositis. Arthritis Rheum 1990; 33: 1361–70. [DOI] [PubMed] [Google Scholar]
- 43.Kao AH, Lacomis D, Lucas M, Fertig N, Oddis CV. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum 2004; 50: 209–15. [DOI] [PubMed] [Google Scholar]
- 44.Chen F, Peng Y, Chen M. Diagnostic Approach to Cardiac Involvement in Idiopathic Inflammatory Myopathies. Int Heart J 2018; 59: 256–62. [DOI] [PubMed] [Google Scholar]
- 45.Ohnuki Y, Suzuki S, Shiina T, et al. HLA-DRB1 alleles in immune-mediated necrotizing myopathy. Neurology 2016; 87: 1954–55. [DOI] [PubMed] [Google Scholar]
- 46.Mammen AL, Gaudet D, Brisson D, et al. Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res (Hoboken) 2012; 64: 1233–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Limaye V, Bundell C, Hollingsworth P, et al. Clinical and genetic associations of autoantibodies to 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase in patients with immune-mediated myositis and necrotizing myopathy. Muscle Nerve 2015; 52: 196–203. [DOI] [PubMed] [Google Scholar]
- 48.Selva-O’Callaghan A, Alvarado-Cardenas M, Pinal-Fernández I, et al. Statin-induced myalgia and myositis: an update on pathogenesis and clinical recommendations. Expert Rev Clin Immunol 2018; 14: 215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arouche-Delaperche L, Allenbach Y, Amelin D, et al. Pathogenic role of anti-signal recognition protein and anti-3-Hydroxy-3-methylglutaryl-CoA reductase antibodies in necrotizing myopathies: myofiber atrophy and impairment of muscle regeneration in necrotizing autoimmune myopathies. Ann Neurol 2017; 81: 538–48. [DOI] [PubMed] [Google Scholar]
- 50.Lloyd TE, Christopher-Stine L, Pinal-Fernandez I, et al. Cytosolic 5’-nucleotidase 1A as a target of circulating autoantibodies in autoimmune diseases. Arthritis Care Res (Hoboken) 2016; 68: 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herbert MK, Stammen-Vogelzangs J, Verbeek MM, et al. Disease specificity of autoantibodies to cytosolic 5’-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann Rheum Dis 2016; 75: 696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muro Y, Nakanishi H, Katsuno M, Kono M, Akiyama M. Prevalence of anti-NT5C1A antibodies in Japanese patients with autoimmune rheumatic diseases in comparison with other patient cohorts. Clin Chim Acta 2017; 472: 1–4. [DOI] [PubMed] [Google Scholar]
- 53.Lilleker JB, Rietveld A, Pye SR, et al. Cytosolic 5’-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann Rheum Dis 2017; 76: 862–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rojana-Udomsart A, Bundell C, James I, et al. Frequency of autoantibodies and correlation with HLA-DRB1 genotype in sporadic inclusion-body myositis (s-IBM): a population control study. J Neuroimmunol 2012; 249: 66–70. [DOI] [PubMed] [Google Scholar]
- 55.Tasca G, Monforte M, De Fino C, Kley RA, Ricci E, Mirabella M. Magnetic resonance imaging pattern recognition in sporadic inclusion-body myositis. Muscle Nerve 2015; 52: 956–62. [DOI] [PubMed] [Google Scholar]
- 56.Greenberg SA, Pinkus JL, Amato AA, Kristensen T, Dorfman DM. Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain 2016; 139: 1348–60. [DOI] [PubMed] [Google Scholar]
- 57.Van De Vlekkert J, Maas M, Hoogendijk JE, De Visser M, Van Schaik IN. Combining MRI and muscle biopsy improves diagnostic accuracy in subacute-onset idiopathic inflammatory myopathy. Muscle Nerve 2015; 51: 253–58. [DOI] [PubMed] [Google Scholar]
- 58.Catalan-Garcia M, Garrabou G, Moren C, et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin Sci (Lond) 2016; 130: 1741–51. [DOI] [PubMed] [Google Scholar]
- 59.Rygiel KA, Tuppen HA, Grady JP, et al. Complex mitochondrial DNA rearrangements in individual cells from patients with sporadic inclusion body myositis. Nucleic Acids Res 2016; 44: 5313–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Greenberg SA, Pinkus JL, Amato AA. Nuclear membrane proteins are present within rimmed vacuoles in inclusion-body myositis. Muscle Nerve 2006; 34: 406–16. [DOI] [PubMed] [Google Scholar]
- 61.Guttsches AK, Brady S, Krause K, et al. Proteomics of rimmed vacuoles define new risk allele in inclusion body myositis. Ann Neurol 2017; 81: 227–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Catalan-Garcia M, Garrabou G, Moren C, et al. BACE-1, PS-1 and sAPPbeta levels are increased in plasma from sporadic inclusion body myositis patients: surrogate biomarkers among inflammatory myopathies. Mol Med 2015; 21: 817–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goyal NA, Cash TM, Alam U, et al. Seropositivity for NT5c1A antibody in sporadic inclusion body myositis predicts more severe motor, bulbar and respiratory involvement. J Neurol Neurosurg Psychiatry 2016; 87: 373–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tawara N, Yamashita S, Zhang X, et al. Pathomechanisms of anti-cytosolic 5’-nucleotidase 1A autoantibodies in sporadic inclusion body myositis. Ann Neurol 2017; 81: 512–25. [DOI] [PubMed] [Google Scholar]
- 65.Andersson H, Kirkhus E, Garen T, Walle-Hansen R, Merckoll E, Molberg O. Comparative analyses of muscle MRI and muscular function in anti-synthetase syndrome patients and matched controls: a cross-sectional study. Arthritis Res Ther 2017; 19: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mescam-Mancini L, Allenbach Y, Hervier B, et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain 2015; 138: 2485–92. [DOI] [PubMed] [Google Scholar]
- 67.Noguchi E, Uruha A, Suzuki S, et al. Skeletal muscle involvement in antisynthetase syndrome. JAMA Neurol 2017; 74: 992–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stenzel W, Preusse C, Allenbach Y, et al. Nuclear actin aggregation is a hallmark of anti-synthetase syndrome-induced dysimmune myopathy. Neurology 2015; 84: 1346–54. [DOI] [PubMed] [Google Scholar]
- 69.Rojana-udomsart A, Needham M, Luo YB, et al. The association of sporadic inclusion body myositis and Sjogren’s syndrome in carriers of HLA-DR3 and the 8.1 MHC ancestral haplotype. Clin Neurol Neurosurg 2011; 113: 559–63. [DOI] [PubMed] [Google Scholar]
- 70.van der Meulen MF, Bronner IM, Hoogendijk JE, et al. Polymyositis: an overdiagnosed entity. Neurology 2003; 61: 316–21. [DOI] [PubMed] [Google Scholar]
- 71.Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 2008; 70: 418–24. [DOI] [PubMed] [Google Scholar]
- 72.Vilela VS, Prieto-Gonzalez S, Milisenda JC, Selva-O’Callaghan A, Grau JM. Polymyositis, a very uncommon isolated disease: clinical and histological re-evaluation after long-term follow-up. Rheumatol Int 2015; 35: 915–20. [DOI] [PubMed] [Google Scholar]
- 73.Alemo Munters L, Dastmalchi M, Andgren V, et al. Improvement in health and possible reduction in disease activity using endurance exercise in patients with established polymyositis and dermatomyositis: a multicenter randomized controlled trial with a 1-year open extension followup. Arthritis Care Res (Hoboken) 2013; 65: 1959–68. [DOI] [PubMed] [Google Scholar]
- 74.Alexanderson H, Munters LA, Dastmalchi M, et al. Resistive home exercise in patients with recent-onset polymyositis and dermatomyositis—a randomized controlled single-blinded study with a 2-year followup. J Rheumatol 2014; 41: 1124–32. [DOI] [PubMed] [Google Scholar]
- 75.Gordon PA, Winer JB, Hoogendijk JE, Choy EH. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev 2012; 8: CD003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.van de Vlekkert J, Hoogendijk JE, de Haan RJ, et al. Dexa myositis trial. Oral dexamethasone pulse therapy versus daily prednisolone in sub-acute onset myositis, a randomised clinical trial. Neuromuscul Disord 2010; 20: 382–89. [DOI] [PubMed] [Google Scholar]
- 77.Vencovsky J, Jarosova K, Machacek S, et al. Cyclosporine A versus methotrexate in the treatment of polymyositis and dermatomyositis. Scand J Rheumatol 2000; 29: 95–102. [DOI] [PubMed] [Google Scholar]
- 78.Go DJ, Park JK, Kang EH, et al. Survival benefit associated with early cyclosporine treatment for dermatomyositis-associated interstitial lung disease. Rheumatol Int 2016; 36: 125–31. [DOI] [PubMed] [Google Scholar]
- 79.Labirua-Iturburu A, Selva-O’Callaghan A, Martinez-Gomez X, Trallero-Araguas E, Labrador-Horrillo M, Vilardell-Tarres M. Calcineurin inhibitors in a cohort of patients with antisynthetase-associated interstitial lung disease. Clin Exp Rheumatol 2013; 31: 436–39. [PubMed] [Google Scholar]
- 80.Morganroth PA, Kreider ME, Werth VP. Mycophenolate mofetil for interstitial lung disease in dermatomyositis. Arthritis Care Res (Hoboken) 2010; 62: 1496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barnes H, Holland AE, Westall GP, Goh NS, Glaspole IN. Cyclophosphamide for connective tissue disease-associated interstitial lung disease. Cochrane Database Syst Rev 2018; 1: CD010908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 1993; 329: 1993–2000. [DOI] [PubMed] [Google Scholar]
- 83.Kampylafka EI, Kosmidis ML, Panagiotakos DB, Dalakas M, Moutsopoulos HM, Tzioufas AG. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol 2012; 30: 397–401. [PubMed] [Google Scholar]
- 84.Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum 2013; 65: 314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aggarwal R, Bandos A, Reed AM, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol 2014; 66: 740–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tjarnlund A, Tang Q, Wick C, et al. Abatacept in the treatment of adult dermatomyositis and polymyositis: a randomised, phase IIb treatment delayed-start trial. Ann Rheum Dis 2018; 77: 55–62. [DOI] [PubMed] [Google Scholar]
- 87.Narazaki M, Hagihara K, Shima Y, Ogata A, Kishimoto T, Tanaka T. Therapeutic effect of tocilizumab on two patients with polymyositis. Rheumatology (Oxford) 2011; 50: 1344–46. [DOI] [PubMed] [Google Scholar]
- 88.Mammen AL, Tiniakou E. Intravenous Immune Globulin for Statin-Triggered Autoimmune Myopathy. N Engl J Med 2015; 373: 1680–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cherin P, Belizna C, Cartry O, et al. Long-term subcutaneous immunoglobulin use in inflammatory myopathies: a retrospective review of 19 cases. Autoimmun Rev 2016; 15: 281–86. [DOI] [PubMed] [Google Scholar]
- 90.Allenbach Y, Guiguet M, Rigolet A, et al. Efficacy of rituximab in refractory inflammatory myopathies associated with anti-synthetase auto-antibodies: an open-label, phase II trial. PLoS One 2015; 10: e0133702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Andersson H, Sem M, Lund MB, et al. Long-term experience with rituximab in anti-synthetase syndrome-related interstitial lung disease. Rheumatology (Oxford) 2015; 54: 1420–28. [DOI] [PubMed] [Google Scholar]
- 92.Chen D, Wang XB, Zhou Y, Zhu XC. Efficacy of infliximab in the treatment for dermatomyositis with acute interstitial pneumonia: a study of fourteen cases and literature review. Rheumatol Int 2013; 33: 2455–58. [DOI] [PubMed] [Google Scholar]
- 93.Muscle Study Group. A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol 2011; 70: 427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schiffenbauer A, Garg M, Castro C, et al. A randomized, double-blind, placebo-controlled trial of infliximab in refractory polymyositis and dermatomyositis. Semin Arthritis Rheum 2018; 47: 858–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zong M, Dorph C, Dastmalchi M, et al. Anakinra treatment in patients with refractory inflammatory myopathies and possible predictive response biomarkers: a mechanistic study with 12 months follow-up. Ann Rheum Dis 2014; 73: 913–20. [DOI] [PubMed] [Google Scholar]
- 96.Ruck T, Bittner S, Kuhlmann T, Wiendl H, Meuth SG. Long-term efficacy of alemtuzumab in polymyositis. Rheumatology (Oxford) 2015; 54: 560–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kurtzman DJ, Wright NA, Lin J, et al. Tofacitinib citrate for refractory cutaneous dermatomyositis: an alternative treatment. JAMA Dermatol 2016; 152: 944–45. [DOI] [PubMed] [Google Scholar]
- 98.Hornung T, Janzen V, Heidgen FJ, Wolf D, Bieber T, Wenzel J. Remission of recalcitrant dermatomyositis treated with ruxolitinib. N Engl J Med 2014; 371: 2537–38. [DOI] [PubMed] [Google Scholar]
- 99.Benveniste O, Guiguet M, Freebody J, et al. Long-term observational study of sporadic inclusion body myositis. Brain 2011; 134: 3176–84. [DOI] [PubMed] [Google Scholar]
- 100.Kosmidis ML, Alexopoulos H, Tzioufas AG, Dalakas MC. The effect of anakinra, an IL1 receptor antagonist, in patients with sporadic inclusion body myositis (sIBM): a small pilot study. J Neurol Sci 2013; 334: 123–25. [DOI] [PubMed] [Google Scholar]
- 101.Schmidt K, Kleinschnitz K, Rakocevic G, Dalakas MC, Schmidt J. Molecular treatment effects of alemtuzumab in skeletal muscles of patients with IBM. BMC Neurol 2016; 16: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Amato AA, Sivakumar K, Goyal N, et al. Treatment of sporadic inclusion body myositis with bimagrumab. Neurology 2014; 83: 2239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mendell JR, Sahenk Z, Al-Zaidy S, et al. Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol Ther 2017; 25: 870–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Benveniste O, Hogrel JY, Annoussamy M, et al. Rapamycin vs. placebo for the treatment of inclusion body myositis: improvement of the 6 min walking distance, a functional scale, the fvc and muscle quantitative MRI. Arthritis Rheumatol 2017; 69 (suppl 10): (abstr). [Google Scholar]
- 105.Rutkove SB, Parker RA, Nardin RA, Connolly CE, Felice KJ, Raynor EM. A pilot randomized trial of oxandrolone in inclusion body myositis. Neurology 2002; 58: 1081–87. [DOI] [PubMed] [Google Scholar]
- 106.Sancricca C, Mora M, Ricci E, Tonali PA, Mantegazza R, Mirabella M. Pilot trial of simvastatin in the treatment of sporadic inclusion-body myositis. Neurol Sci 2011; 32: 841–47. [DOI] [PubMed] [Google Scholar]
- 107.Cherin P, Pelletier S, Teixeira A, et al. Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology 2002; 58: 326. [DOI] [PubMed] [Google Scholar]
- 108.Murata KY, Kouda K, Tajima F, Kondo T. Balloon dilation in sporadic inclusion body myositis patients with dysphagia. Clin Med Insights Case Rep 2013; 6: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schrey A, Airas L, Jokela M, Pulkkinen J. Botulinum toxin alleviates dysphagia of patients with inclusion body myositis. J Neurol Sci 2017; 380: 142–47. [DOI] [PubMed] [Google Scholar]
- 110.Teruya A, Kawamura K, Ichikado K, Sato S, Yasuda Y, Yoshioka M. Successful polymyxin B hemoperfusion treatment associated with serial reduction of serum anti-CADM-140/MDA5 antibody levels in rapidly progressive interstitial lung disease with amyopathic dermatomyositis. Chest 2013; 144: 1934–36. [DOI] [PubMed] [Google Scholar]
- 111.Morisset J, Johnson C, Rich E, Collard HR, Lee JS. Management of myositis-related interstitial lung disease. Chest 2016; 150: 1118–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.