

Abstract
The allylation of isochromans at the α-position via aerobic DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) catalysis is described. This process involves the DDQ oxidation of various isochromans under mild conditions to generate oxocarbenium intermediates, which are effectively stabilized in equilibration in the presence of acid before undergoing allylation. Molecular oxygen and tert-butyl nitrite are employed as an environmentally benign oxidant and mediator, respectively, in the catalytic cycle. The transformation of an allylated product to biologically active molecules has also been successfully demonstrated.
A direct C–H activation of isochromans using a DDQ/TBN/O2 catalytic system, followed by addition of allylstannane in the presence of methanesulfonic acid, provides various α-allyl substituted isochromans efficiently.
Introduction
Isochromans, also known as 3,4-dihydro-1H-2-benzopyrans, are commonly found in biologically active compounds. In particular, α-substituted isochromans serve as key scaffolds in agents targeting neurotransmitter regulation (Fig. 1). For instance, sonepiprazole is an antipsychotic isochroman analogue that acts as a selective dopamine receptor D4 antagonist.1 U-54537 is another D4 antagonist,2 while Penidicitrinin B is a natural product known for its potent antioxidant activity.3 Additionally, compound 4 functions as a calcium channel receptor antagonist,4 and PNU-109291 is a highly selective 5-HT1D agonist.5 These structural motifs are thus pharmaceutically important, and various synthetic methods to α-substituted isochromans have been developed.
Fig. 1. Representative biologically active α-substituted isochromans.

Due to the significance of α-substituted isochromans, extensive efforts have been made toward their synthesis. One of the general approaches involves the formation of carbon–oxygen bond through intramolecular electrophilic aromatic substitution reactions of oxonium intermediate (Scheme 1a).6 While this strategy provides easy access to α-substituted isochromans, the installation of functionalities typically requires multiple, labor-intensive steps. Alternatively, the coupling reaction of isochromans with nucleophiles at the α-position via an oxocarbenium intermediate, generated either by acid-catalyzed elimination from α-activating substrates or direct oxidative C–H activation, offers a direct synthetic route to α-substituted isochromans (Scheme 1b).7
Scheme 1. General methods for the synthesis of α-substituted isochromans.

Since Li's group first reported oxidative α-functionalization of isochromans as an atom- and step-economical strategy,8 various oxidative C–C bond forming reactions using carbon nucleophiles such as ketones, allyl species, acetylenes, and aryl rings have been developed.9 Both metal and organic oxidants have been employed for α-functionalization, with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) being particularly effective in promoting C–H activation of isochromans.10 Initially, α-substituted isochromans were synthesized using stoichiometric amounts of DDQ, but this method posed challenges in purification for large-scale synthesis and raised toxicity concerns. Only a few examples of DDQ catalyzed α-functionalization of isochromans have been reported. For example, Floreancig reported DDQ-catalyzed synthesis of α-substituted isochromans using MnO2 as a co-oxidant,11 while Muramatsu and Li demonstrated organocatalytic arylation, alkylation, and amidation with a DDQ/PIFA co-oxidant system.12 Although DDQ-catalyzed systems are well-established for alcohol and benzylic oxidations,13 their application to direct C–H activation of isochromans remains underexplored (Scheme 2).
Scheme 2. DDQ catalyzed α-substitution of isochromans.

Recently, we reported aerobic DDQ-catalyzed α-allylation of tetrahydroisoquinolines using NaNO2 and molecular oxygen.14 In line with our ongoing interest in DDQ catalyzed C–H activation,15 we sought to extend this strategy to synthesize α-substituted isochromans. In this approach, we envisioned that DDQ-catalyzed oxidation of isochroman under aerobic conditions would generate an oxocarbenium intermediate, which could then react with neutral allyl nucleophile to afford α-substituted isochromans. Herein, we report the aerobic oxidative C–H allylation of isochromans using DDQ catalysis.
Results and discussion
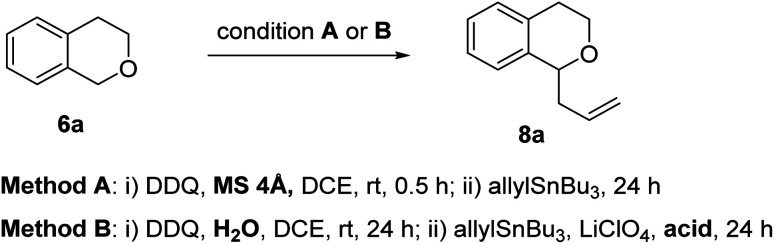
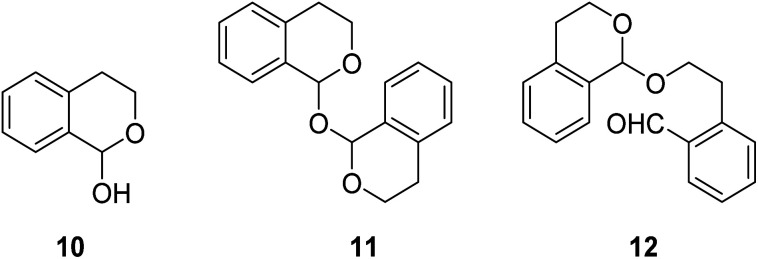
Initially, we begin with the oxidative allylation of isochromans using a stoichiometric amount of DDQ while screening reaction conditions. As demonstrated in Table 1, the oxidative allylations were investigated under both anhydrous and hydrous conditions. DDQ-promoted allylations of isochroman 6a with allyl stannane at room temperature after treatment with molecular sieves, yielded little or no product 8a (entries 1–3). Increasing the reaction temperature slightly improved the yield, reaching only 18%. We speculated that such low yields could be attributed to the instability of the oxocarbenium intermediate. The Liu group discovered that DDQ oxidation of isochroman in the presence of H2O afforded a mixture of hemiacetal 10 and dimerized acetals 11 and 12, which reversibly formed oxocarbenium ion upon treatment with acid.16 Based on this study, we further explored the allylation reactions using an acid as a catalyst under hydrous condition. When H2O (1.1 equiv.) was added to the mixture of isochroman and DDQ, followed by addition of allyl stannane in the presence of TFA, the desired α-allyl isochroman was obtained in 28% yield. Notably, LiClO4 was used to activate oxocarbenium ion.17 Finally, the reaction was performed at 80 °C in the presence of methanesulfonic acid (MSA) or p-chlorobenzenesulfonic acid (CBSA) to afford 8a in approximately 60% yield. Thus, Brønsted acid played an important role in the formation and activation of the oxocarbenium ion in this DDQ-stoichiometric allylation process.
Initial screening of reactions conditions in the presence of a stoichiometric amount of DDQa.
| |||||
|---|---|---|---|---|---|
| Entry | Method | Acid | Solvent | Temp (°C) | Yieldb (%) |
| 1 | A | CH3CN | 25 | NRe | |
| 2 | A | DCE | 25 | 7 | |
| 3c | A | DCE | 25 | 11 | |
| 4 | A | DCE | 80 | 18 | |
| 5 | B | TFA | DCE | 25 | 10 |
| 6 | B | TFA | DCE | 80 | 28 |
| 7 | B | MSA | DCE | 80 | 59 |
| 8d | B | CBSA | DCE | 80 | 58 |
| |||||
Reaction conditions: (Method A) (i) 6a (0.15 mmol), DDQ (0.22 mmol), MS 4 Å (20 mg), solvent (0.1 M), rt, 0.5 h; (ii) allyltributyl stannane (0.22 mmol), 24 h. (Method B) (i) 6a (0.37 mmol), DDQ (0.37 mmol), H2O (0.41 mmol), DCE (0.1 M), rt, 24 h; (ii) allyltributyl stannane (0.75 mmol), LiClO4 (0.37 mmol), acid (0.075 mmol), 24 h.
Isolated yield.
LiClO4 (0.5 equiv.) was added.
The reaction mixture was stirred for 6 h before allylstannane was added.
No reaction.
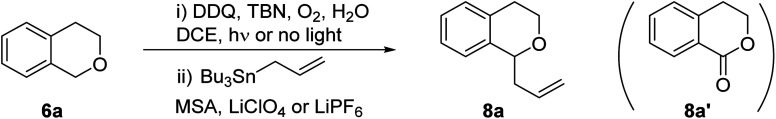
Encouraged by these preliminary results, we turned our attention to optimize the reaction conditions for catalytic DDQ allylation of isochroman. Considering eco-friendly process for catalytic allylation, we have selected molecular oxygen as an oxidant. However, TBN (tert-butyl nitrite) was also required as a mediator to regenerate DDQ from 2,3-dichloro-5,6-dicyanohydroquinone (DDQH2) due to high oxidation potential of DDQH2.18 Thus, DDQ and TBN co-catalytic system was explored under both photochemical and thermal conditions as shown in Table 2. First, the allylation of isochroman with allylstannane was performed in the presence of DDQ and TBN under molecular oxygen along upon blue led irradiation to give 8a in low yield. The yield of the reactions did not increase when the amount of reagents was changed or LiClO4 was used as an activating additive, whereas the use of LiPF6 improved the reaction yield up to 30% (entry 5). Interestingly, the yield of allylated product further increased when the reactions proceeded without water (entries 6 and 7). In most cases, however, we observed unidentified side products under photochemical reaction conditions, which is presumably due to strong oxidizing capability of DDQ, resulted from increased reduction potential of 3DDQ*  .19 On the contrary, the reactions without light irradiation generally proceeded smoothly to afford the desired allylated isochroman in higher yields compared to the results from the photochemical reactions (entries 8–11). In particular, we observed the highest yield of 8a when the allylation was performed without water (entry 10). It is noted that a small amount of lactone 8a′ was also formed in some cases by simple oxidation of the corresponding lactol 10, generated from addition of water to oxocarbenium intermediate (entry 11). We observed the yield decreased when the reaction was performed in the presence of molecular sieves, which indicated that water, generated during oxidation of DDQH2 to DDQ in the catalytic cycle (vide infra), is crucial to promote reversible formation of oxocarbenium under the optimized reaction condition (entry 12). The yield was dropped without LiPF6 additive (entry 13), and no reaction occurred without both MSA and LiPF6 (entry 14). The allylated product 8a was produced in low yield without oxygen bubbling (entry 15).
.19 On the contrary, the reactions without light irradiation generally proceeded smoothly to afford the desired allylated isochroman in higher yields compared to the results from the photochemical reactions (entries 8–11). In particular, we observed the highest yield of 8a when the allylation was performed without water (entry 10). It is noted that a small amount of lactone 8a′ was also formed in some cases by simple oxidation of the corresponding lactol 10, generated from addition of water to oxocarbenium intermediate (entry 11). We observed the yield decreased when the reaction was performed in the presence of molecular sieves, which indicated that water, generated during oxidation of DDQH2 to DDQ in the catalytic cycle (vide infra), is crucial to promote reversible formation of oxocarbenium under the optimized reaction condition (entry 12). The yield was dropped without LiPF6 additive (entry 13), and no reaction occurred without both MSA and LiPF6 (entry 14). The allylated product 8a was produced in low yield without oxygen bubbling (entry 15).
Optimization of DDQ/TBN-catalyzed allylationa.
| ||||||
|---|---|---|---|---|---|---|
| Entry | DDQ (mol%) | TBN (equiv.) | H2O (equiv.) | Additive (equiv.) | Blue LED | Yieldb (%) |
| 1 | 10 | 0.5 | 2.0 | LiClO4 (1.0) | On | 8 |
| 2 | 10 | 1.0 | 2.0 | LiClO4 (1.0) | On | 12 |
| 3 | 10 | 2.0 | 2.0 | LiClO4 (1.0) | On | 12 |
| 4 | 10 | 1.0 | 2.0 | LiClO4 (0.5) | On | 25 |
| 5 | 10 | 0.2 | 2.0 | LiPF6 (0.5) | On | 30 |
| 6 | 10 | 1.0 | — | LiPF6 (0.5) | On | 37 |
| 7 | 20 | 0.2 | — | LiPF6 (0.5) | On | 36 |
| 8 | 20 | 1.0 | 2.0 | LiPF6 (0.5) | Off | 39 |
| 9 | 20 | 0.2 | 2.0 | LiPF6 (0.5) | Off | 63 |
| 10 | 20 | 0.2 | — | LiPF6 (0.5) | Off | 76 |
| 11 | 30 | 0.2 | 2.0 | LiPF6 (0.5) | Off | 51cd |
| 12 | 20 | 0.2 | — | LiPF6 (0.5) | Off | 50e |
| 13 | 20 | 0.2 | — | —f | Off | 16 |
| 14 | 20 | 0.2 | — | —g | Off | NRh |
| 15 | 20 | 0.2 | — | LiPF6 (0.5) | Off | 41i |
Reaction conditions: (i) 1a (0.37 mmol), DDQ, TBN, H2O, O2 bubbling (1 h), blue LED (18W) irradiation or no light, DCE (3.7 mL), rt, 24 or 36 h; (ii) allyltributyl stannane (0.75 mmol), additives, MSA (0.075 mmol), 80 °C, 24 h.
Isolated yield.
65% conversion.
8a′ was produced in 3% yield.
The reaction was performed in the presence of molecular sieves 4 Å.
The reaction was performed without LiPF6 only.
The reaction was performed without both LiPF6 and MSA.
No reaction.
The reaction was performed without oxygen bubbling.
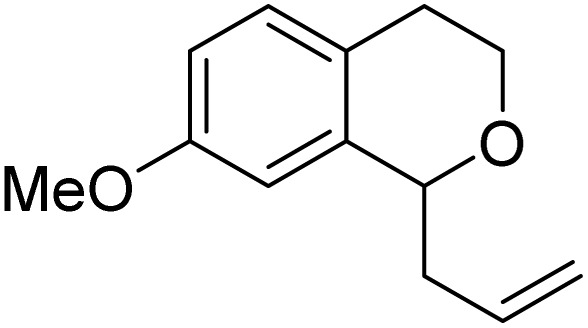
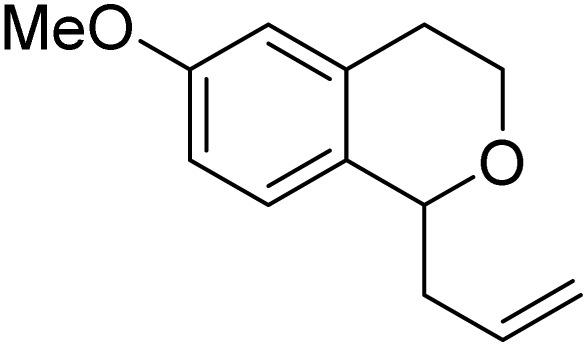
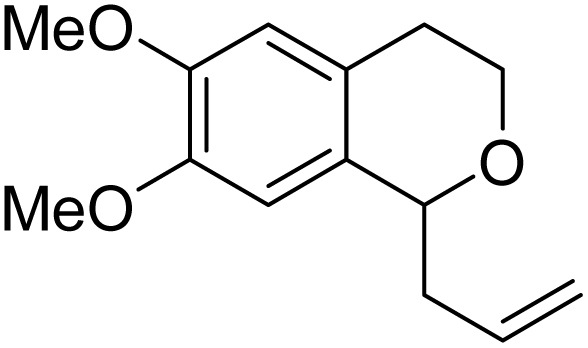
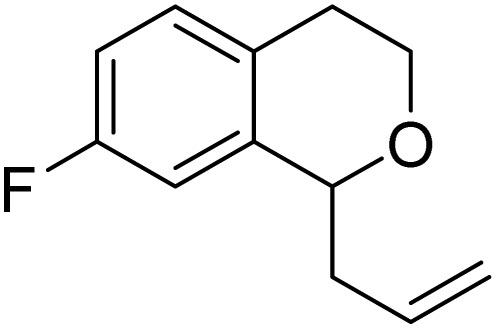
With the optimized reaction conditions established, we investigated the substrate scope of DDQ-catalyzed aerobic allylation of various isochromans, as shown in Table 3. In general, electron-rich isochromans turned out to be the best substrates for this oxidative α-allylation process. Allylation of isochromans 6b–6d, which contain methyl substituents either on the phenyl ring or the tetrahydropyran ring, afforded the corresponding products 8b–8d in good yields (entries 1–3). When substrates with high electron donating groups, such as 6- or 7-methoxy isochromans 6e/6f and 6,7-dimethoxy isochroman 6g, were subjected to the standard reaction conditions, the desired products 8e–8g were obtained in moderate yields (entries 4–6). It should be noticed that most of electron-rich substrates also formed lactones 8′ as side products via oxidation of lactol intermediates (e.g., 10). In particular, the highly activated substrate 6g produced lactone 8g′ in significant yield, reflecting its enhanced susceptibility to oxidation. In the case of halogen-substituted isochromans 6h–6l, the reactions proceeded slowly, yielding α-allylisochromans 8h–8l in relatively low yields due to incomplete consumption of the starting materials (entries 7–11). Moreover, the reactions of isochromans 6m, 6n and 6o, containing acetoxy, cyano and aldehyde groups, yielded little or no desired products, indicating that electron-deficient substrates are not well-suited to this DDQ-catalyzed allylation process (entries 12–14). Importantly, no lactones 8′ were observed in the reactions of electron-deficient substrates, which could confirm that oxocarbenium intermediates were not well generated likely to their low reactivity toward oxidation. These results demonstrate that the presence of electron-donating groups is critical for achieving efficient oxidative allylation in this DDQ/TBN/O2 catalytic system.
Scope of DDQ/TBN-catalyzed aerobic allylation of isochromans 6a.
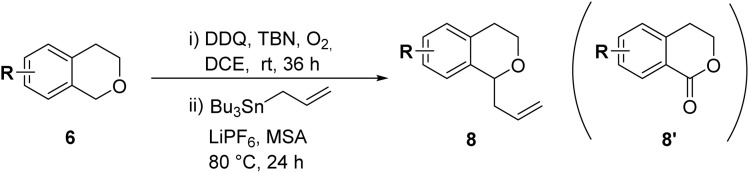
| ||||
|---|---|---|---|---|
| Entry | Product | Yield (%) | ||
| 8b | 8′ | |||
| 1 |
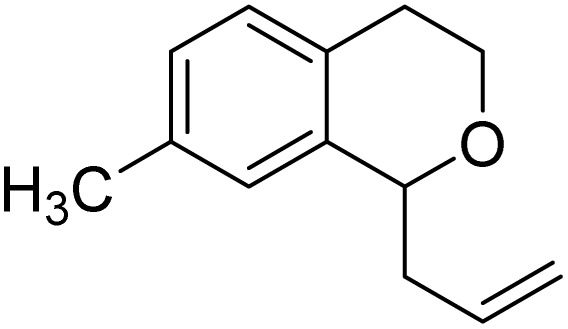
|
8b | 76 | 8 |
| 2 |
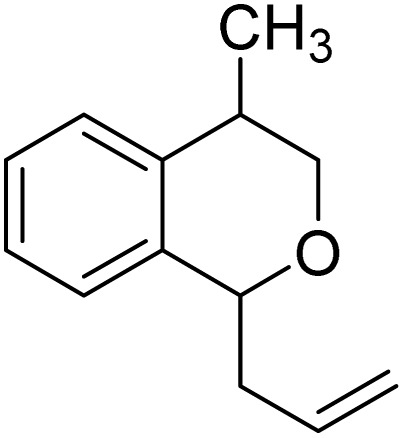
|
8c | 80 (2.2 : 1)c | 7 |
| 3 |
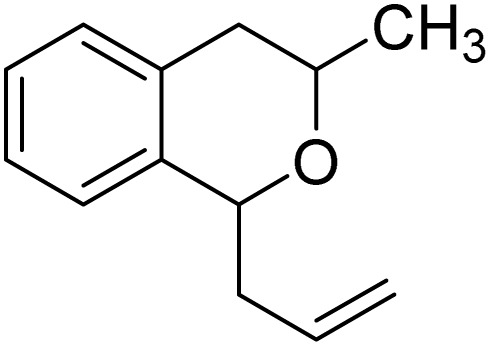
|
8d | 72 (2.9 : 1)c | 10 |
| 4 |
|
8e | 50 (75) | 14 |
| 5 |
|
8f | 66 (90) | — |
| 6 |
|
8g | 48 | 42 |
| 7 |
|
8h | 18 (30) | — |
| 8 |
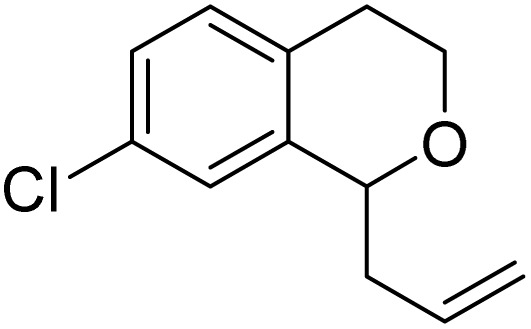
|
8i | 33 (54) | — |
| 9 |
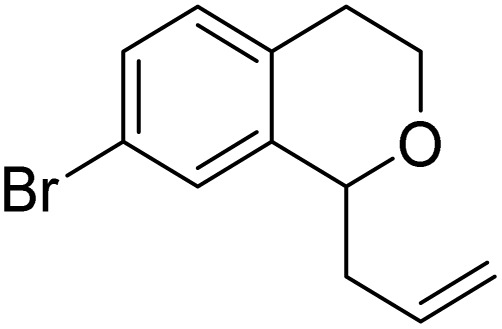
|
8j | 34 (49) | — |
| 10 |
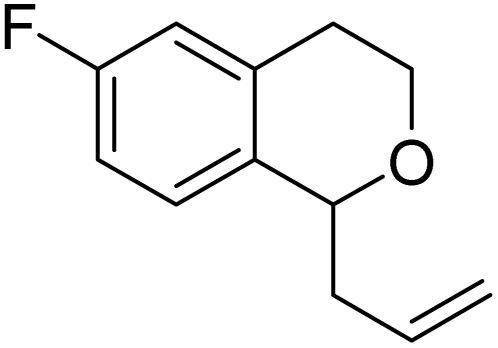
|
8k | 41 (59) | — |
| 11 |
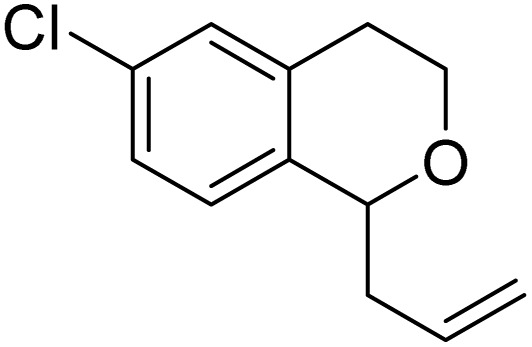
|
8l | 37 (52) | — |
| 12 |
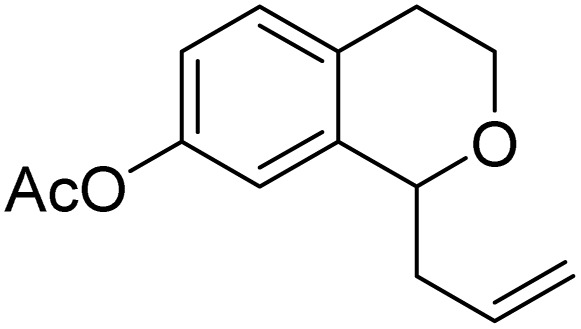
|
8m | 12 (22) | — |
| 13 |
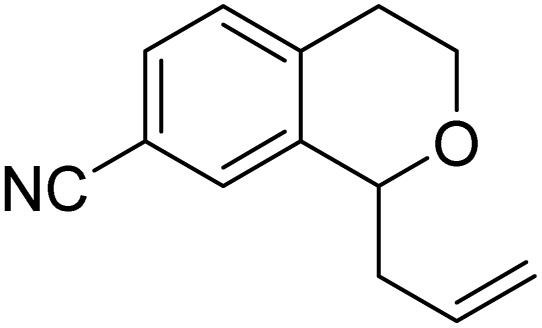
|
8n | NRd | — |
| 14 |
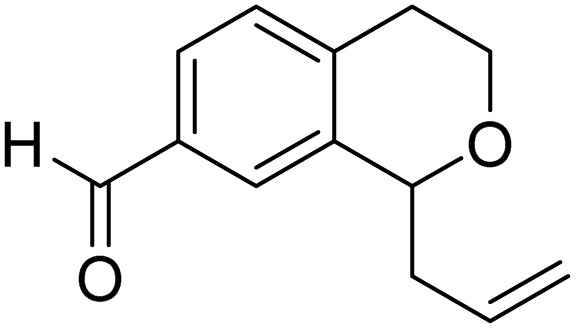
|
8o | NRd | — |
Reaction conditions: (i) 6 (0.37 mmol), DDQ (0.074 mmol), TBN (0.074 mmol), O2, DCE (3.7 mL), 0 °C or rt, 36 h; (ii) allyltributyl stannane (0.75 mmol), LiPF6 (0.19 mmol), MSA (0.075 mmol), 80 °C, 24 h.
Isolated yield. Yields in the parentheses are based upon recovered starting material.
Only the diastereomeric ratio was obtained by the analysis of 1H NMR spectrum.
No reaction.
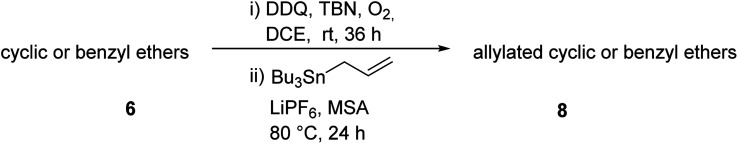
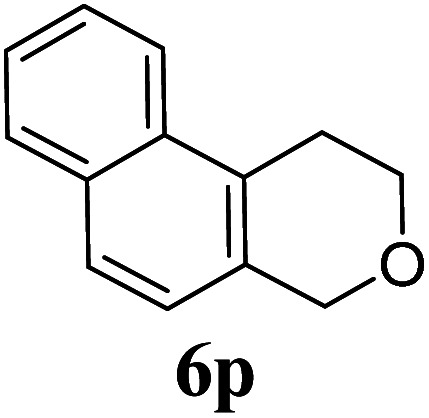
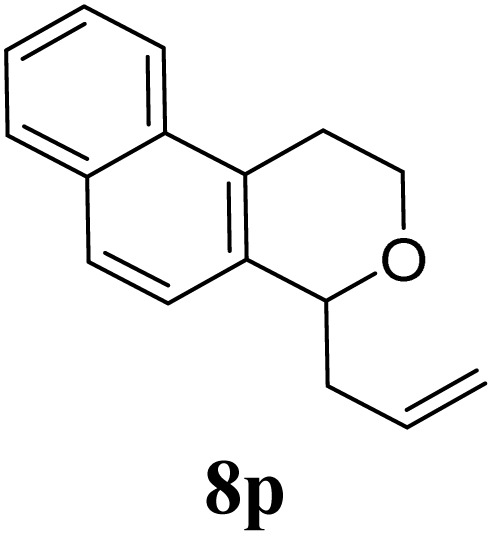
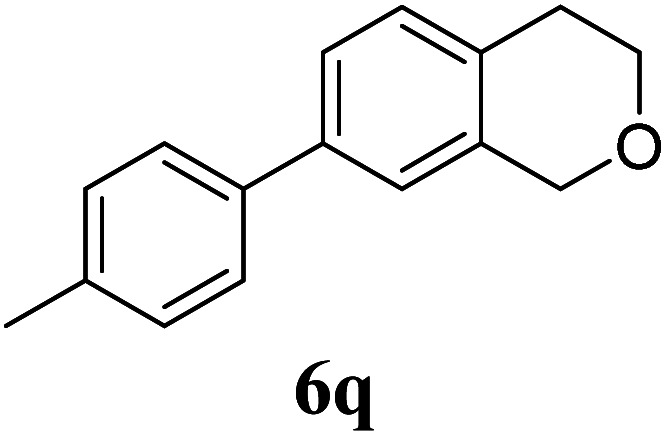
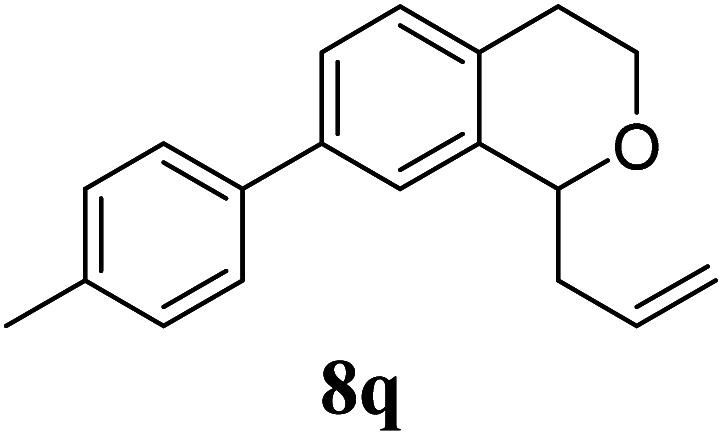
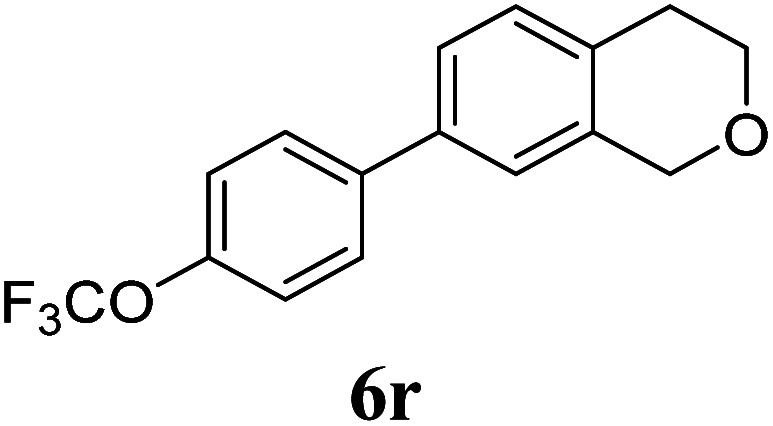
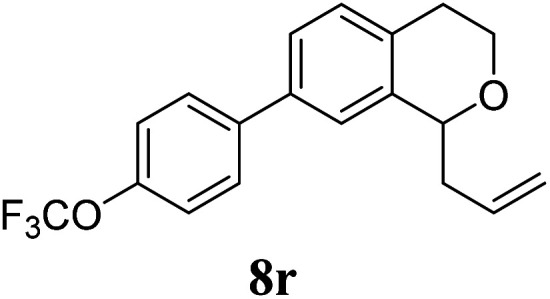
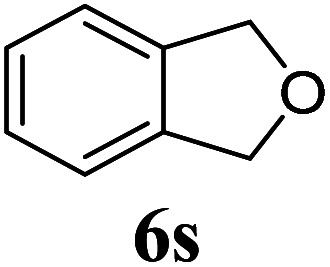
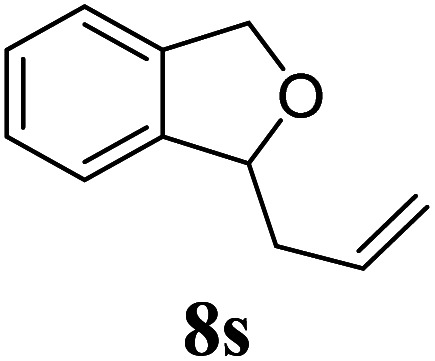
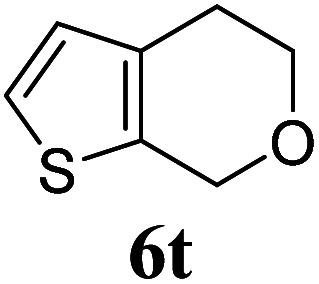
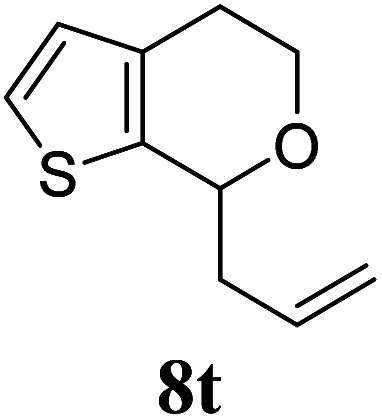
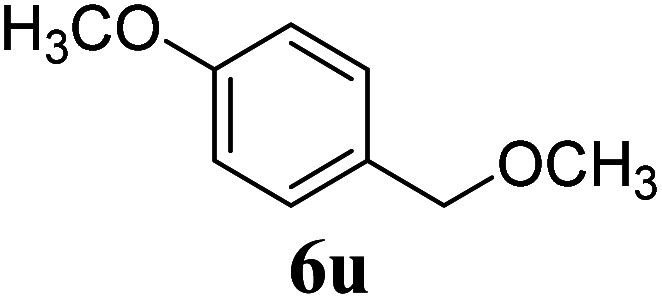
Under the same reaction conditions, the oxidative allylation of other cyclic and acyclic ethers was investigated as summarized in Table 4. Benzoisochroman 6p and 7-aryl isochromans 6q/6r underwent allylation efficiently, affording the corresponding products 8p–8r. However, 8r, having trifluoromethoxyphenyl at the 7-position, was obtained in low yield, with a large amount of starting material remaining. In contrast, isochroman analogues such as 1,3-dihydroisobenzofuran 6s and thienopyran 6t were not well-tolerated, giving 8s and 8t in only 29% and 30% yield, respectively. Additionally, no desired product was observed when simple benzyl ether 6u was subjected to the reaction.
DDQ/TBN-catalyzed aerobic allylation of other cyclic or acyclic ethers 6a.
| ||||
|---|---|---|---|---|
| Entry | Substrate | Product | Yield (%) | |
| 8b | 8′ (lactone) | |||
| 1 |
|
|
86 | 8 |
| 2 |
|
|
49 (62) | 21 |
| 3 |
|
|
31 (77) | 17 |
| 4 |
|
|
29 | — |
| 5 |
|
|
30 (33) | |
| 6 |
|
|
NRc | |
Reaction conditions: (i) 6 (0.37 mmol), DDQ (0.074 mmol), TBN (0.074 mmol), O2, DCE (3.7 mL), 0 °C or rt, 36 h; (ii) allyltributyl stannane (0.75 mmol), LiPF6 (0.19 mmol), MSA (0.075 mmol), 80 °C, 24 h.
Isolated yield.
No reaction.
A plausible mechanism for the allylation of isochroman in the DDQ/TBN/O2 catalytic system is outlined in Scheme 3. According to Floreancig and colleagues,20 the reaction begins with hydride abstraction from isochroman 6a by either DDQ (thermal) or excited 3DDQ* (photochemical), generating an oxocarbenium ion 9, which then forms intermediate 10, 11, or 12 in the presence of H2O. Methanesulfonic acid (MSA) promotes the degradation of these intermediates, regenerating the oxocarbenium ion.16a LiPF6 may further assist in their decomposition, forming an oxocarbenium+-PF6− ion pair that enhances nucleophilic addition of allylstannane.16b In the catalytic cycle, nitrogen monoxide (NO), derived from the thermal or photochemical decomposition of TBN, is converted to NO2 in the presence of O2. NO2, in turn, oxidizes DDQH2 back to DDQ, producing H2O to facilitate the formation of intermediates 10–12. As the water is generated within the catalytic cycle, the reaction proceeds without addition of water. Notably, in electron-rich substrates, lactones 8′ are frequently formed through the oxidation of intermediate 10. In contrast, electron-poor isochromans do not undergo this oxidation, owing to their limited conversion to oxocarbenium intermediate 9. These findings indicate that activated isochromans bearing electron-donating substituents are suited for the current oxidative allylation process.
Scheme 3. Proposed mechanism of DDQ-catalyzed aerobic allylation.

Next, we investigated the further transformation of allylated product to biologically useful isochroman derivatives as depicted in Scheme 4. Thus, U-54537 (2), known as D4 antagonist, was selected as a target molecule. Pd-catalyzed amination of N-Boc-piperazine 13, followed by removal of Boc protective group, afforded 1-(4-fluorophenyl)piperazine 15 in 68% yield. Oxidative cleavage of 8g using modified Lemieux–Johnson oxidation (OsO4, NaIO4 and 2,6-lutidine)21 proceeded to give the corresponding aldehyde 16, which was subjected to reductive amination with piperazine 15 in the presence of NaBH(OAc)3 to produce the target molecule 2 successfully.
Scheme 4. Synthesis of U-54537 (2) starting from α-allyl isochroman 8g.

Conclusions
In this study, we have developed the oxidative allylation of isochromans using DDQ/TBN/O2 catalytic system. This process showed a direct oxidative functionalization of various isochroman derivatives at the benzylic α-position. Several key features of this method highlighted: (1) DDQ is employed as a thermo or photochemical organocatalyst to activate C–H bond at the α-position of isochromans; (2) the formation of reactive oxocarbenium species is controlled by DDQ-oxidation capacity, along with the reversible generation of stable intermediates promoted by water, acid and additive; (3) molecular oxygen serves as an environmentally benign oxidant in the catalytic cycle. Additionally, the synthetic utility of the allylated product was demonstrated by its transformation into biologically active molecule.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was financially supported by the NRF grant funded by Korean Government (MSIT) (NRF-2022R1A2C1005110). This work was partially supported by the GRRC program of Gyeonggi province [GRRC-Hanyang2020 (B02)].
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07586b
Notes and references
- TenBrink R. E. Bergh C. L. Duncan J. N. Harris D. W. Huff R. M. Lahti R. A. Lawson C. F. Lutzke B. S. Martin I. J. Rees S. A. Schlachter S. K. Sih J. C. Smith M. W. J. Med. Chem. 1996;39:2435–2437. doi: 10.1021/jm960084f. [DOI] [PubMed] [Google Scholar]
- McCall J. M. McCall R. B. TenBrink R. E. Kamdar B. V. Humphrey S. J. Sethy V. H. Harris D. W. Daenzer C. J. Med. Chem. 1982;25:75–81. doi: 10.1021/jm00343a015. [DOI] [PubMed] [Google Scholar]
- Liu H.-C. Du L. Zhu T.-J. Li D.-H. Geng M.-Y. Gu Q.-Q. Helv. Chim. Acta. 2010;93:2224–2230. [Google Scholar]
- Combourieu M. and Laigle J.-C., European Pat., Appl. EP0450689A1, 1991
- Ennis M. D. Ghazal N. B. Hoffman R. L. Smith M. W. Schlachter S. K. Lawson C. F. Im W. B. Pregenzer J. F. Svensson K. A. Lewis R. A. Hall E. D. Sutter D. M. Harris L. T. McCall R. B. J. Med. Chem. 1998;41:2180–2183. doi: 10.1021/jm980137o. [DOI] [PubMed] [Google Scholar]
- (a) Wünsch B. Zott M. Liebigs Ann. Chem. 1992;1992:39–45. [Google Scholar]; (b) Larghi E. L. Kaufman T. S. Synthesis. 2006:187–220. [Google Scholar]; (c) Guiso M. Marra C. Cavarischia C. Tetrahedron Lett. 2001;42:6531–6534. [Google Scholar]; (d) Yang G. Li K. Zeng K. Li Y. Yu T. Liu Y. RSC Adv. 2021;11:10610–10614. doi: 10.1039/d1ra01004b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tanaka N. Mitton-Fry S. R. Hwang M. U. Zhu J. L. Scheidt K. A. ACS Catal. 2024;14:7949–7955. [Google Scholar]
- (a) Reisman S. E. Doyle A. G. Jacobsen E. N. J. Am. Chem. Soc. 2008;130:7198–7199. doi: 10.1021/ja801514m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maity P. Srinivas H. D. Watson M. P. J. Am. Chem. Soc. 2011;133:17142–17145. doi: 10.1021/ja207585p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schneider U. Dao H. T. Kobayashi S. Org. Lett. 2010;12:2488–2491. doi: 10.1021/ol100450s. [DOI] [PubMed] [Google Scholar]; (d) Ghobrial M. Schnürch M. Mihovilovic M. D. J. Org. Chem. 2011;76:8781–8793. doi: 10.1021/jo201511d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xiang M. Meng Q.-Y. Li J.-X. Zheng Y.-W. Ye C. Li Z.-J. Chen B. Tung C.-H. Wu L.-Z. Chem.–Eur. J. 2015;21:18080–18084. doi: 10.1002/chem.201503361. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Li C.-J. J. Am. Chem. Soc. 2006;128:4242–4243. doi: 10.1021/ja060050p. [DOI] [PubMed] [Google Scholar]
- (a) Ghobrial M. Harhammer K. Mihovilovic M. D. Schnürch M. Chem. Commun. 2010;46:8836–8838. doi: 10.1039/c0cc02491k. [DOI] [PubMed] [Google Scholar]; (b) Richter H. García Mancheño O. Eur. J. Org Chem. 2010;4460:4460–4467. [Google Scholar]; (c) Richter H. Rohlmann R. García Mancheño O. Chem.–Eur. J. 2011;17:11622–11627. doi: 10.1002/chem.201101786. [DOI] [PubMed] [Google Scholar]; (d) Park S. J. Price J. R. Todd M. H. J. Org. Chem. 2012;77:949–955. doi: 10.1021/jo2021373. [DOI] [PubMed] [Google Scholar]; (e) Liu X. Sun B. Xie Z. Qin X. Liu L. Lou H. J. Org. Chem. 2013;78:3104–3112. doi: 10.1021/jo4000674. [DOI] [PubMed] [Google Scholar]; (f) Tambe S. D. Cho E. J. Bull. Korean Chem. Soc. 2022;43:1226–1230. [Google Scholar]; (g) Pan X. Hu Q. Chen W. Liu X. Sun B. Huang Z. Zeng Z. Wang L. Zhao D. Ji M. Liu L. Lou H. Tetrahedron. 2014;70:3447–3451. [Google Scholar]; (h) Chen W. Xie Z. Zheng H. Lou H. Liu L. Org. Lett. 2014;16:5988–5991. doi: 10.1021/ol503004a. [DOI] [PubMed] [Google Scholar]; (i) Peng Z. Wang Y. Yu Z. Wu H. Fu S. Song L. Jiang C. Adv. Synth. Catal. 2019;361:2048–2053. [Google Scholar]; (j) Cao H. Long C.-J. Yang D. Guan Z. He Y.-H. J. Org. Chem. 2023;88:4145–4154. doi: 10.1021/acs.joc.2c02616. [DOI] [PubMed] [Google Scholar]; (k) Atriardi S. R. Kim J.-Y. Anita Y. Woo S. K. Bull. Korean Chem. Soc. 2023;44:50–53. [Google Scholar]
- (a) Mao Y. Cao M. Pan X. Huang J. Li J. Xu L. Liu L. Org. Chem. Front. 2019;6:2028–2031. [Google Scholar]; (b) Chandrasekhar S. Sumithra G. Yadav J. S. Tetrahedron Lett. 1996;37:1645–1646. [Google Scholar]; (c) Zhang Y. Li C.-J. Angew. Chem., Int. Ed. 2006;45:1949–1952. doi: 10.1002/anie.200503255. [DOI] [PubMed] [Google Scholar]; (d) Xu Y.-C. Kohlman D. T. Liang S. X. Erikkson C. Org. Lett. 1999;1:1599–1602. [Google Scholar]; (e) Correia C. A. Li C.-J. Heterocycles. 2010;82:555–562. [Google Scholar]; (f) Peng Z. Wang Y. Yu Z. Zhao D. Song L. Jiang C. J. Org. Chem. 2018;83:7900–7906. doi: 10.1021/acs.joc.8b00776. [DOI] [PubMed] [Google Scholar]; (g) Cao M. Mao Y. Huang J. Ma Y. Liu L. Tetrahedron Lett. 2019;60:1075–1078. [Google Scholar]
- Liu L. Floreancig P. E. Org. Lett. 2010;12:4686–4689. doi: 10.1021/ol102078v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Muramatsu W. Nakano K. Li C.-J. Org. Lett. 2013;15:3650–3653. doi: 10.1021/ol401534g. [DOI] [PubMed] [Google Scholar]; (b) Muramatsu W. Nakano K. Org. Lett. 2014;16:2042–2045. doi: 10.1021/ol5006399. [DOI] [PubMed] [Google Scholar]
- (a) Alsharif M. A. Raja Q. A. Majeed N. A. Jassas R. S. Alsimaree A. A. Sadiq A. Naeem N. Mughal E. U. Alsantali R. I. Moussa Z. RSC Adv. 2021;11:29826–29858. doi: 10.1039/d1ra04575j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wendlandt A. E. Stahl S. S. Angew. Chem., Int. Ed. 2015;54:14638–14658. doi: 10.1002/anie.201505017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Na S. Lee A. Bull. Korean Chem. Soc. 2023;44:921–925. [Google Scholar]; (d) Bosque I. Chinchilla R. Gonzalez-Gomez J. C. Guijarro D. Alonso F. Org. Chem. Front. 2020;7:1717–1742. [Google Scholar]; (e) Natarajan P. König B. Eur. J. Org Chem. 2021:2145–2161. [Google Scholar]
- Jung S. Yoon S. Lee J. K. Min S.-J. ACS Omega. 2022;7:32562–32568. doi: 10.1021/acsomega.2c04154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Jo H. Hassan A. H. E. Jung S. Y. Lee J. K. Cho Y. S. Min S.-J. Org. Lett. 2018;20:1175–1178. doi: 10.1021/acs.orglett.8b00098. [DOI] [PubMed] [Google Scholar]; (b) Park S. Yoon S. Min S.-J. Bull. Korean Chem. Soc. 2021;42:525–528. [Google Scholar]; (c) Cho H. Jang S. Lee K. Cha D. Min S.-J. Org. Lett. 2023;25:8558–8563. doi: 10.1021/acs.orglett.3c03335. [DOI] [PubMed] [Google Scholar]
- (a) Meng Z. Sun S. Yuan H. Lou H. Liu L. Angew. Chem., Int. Ed. 2014;53:543–547. doi: 10.1002/anie.201308701. [DOI] [PubMed] [Google Scholar]; (b) Xia Q. Wang Q. Yan C. Dong J. Song H. Li L. Liu Y. Wang Q. M. Liu X. Song H. Chem.–Eur. J. 2017;23:10871–10877. doi: 10.1002/chem.201701755. [DOI] [PubMed] [Google Scholar]
- (a) Hayashi Y. Mukaiyama T. Chem. Lett. 1987;16:1811–1814. [Google Scholar]; (b) Hayashi Y. Itoh T. Ishikawa H. Angew. Chem., Int. Ed. 2011;50:3920–3924. doi: 10.1002/anie.201006885. [DOI] [PubMed] [Google Scholar]
- (a) Shen Z. Dai J. Xiong J. He X. Mo W. Hu B. Sun N. Hu X. Adv. Synth. Catal. 2011;353:3031–3038. [Google Scholar]; (b) Ma J. Hu Z. Li M. Zhao W. Hu X. Mo W. Hu B. Sun N. Shen Z. Tetrahedron. 2015;71:6733–6739. [Google Scholar]; (c) Rusch F. Schober J.-C. Brasholz M. ChemCatChem. 2016;8:2881–2884. [Google Scholar]; (d) Dahiya A. Sahoo A. K. Alam T. Patel B. K. Chem.–Asian J. 2019;14:4454–4492. doi: 10.1002/asia.201901072. [DOI] [PubMed] [Google Scholar]
- (a) Ohkubo K. Fujimoto A. Fukuzumi S. J. Am. Chem. Soc. 2013;135:5368–5371. doi: 10.1021/ja402303k. [DOI] [PubMed] [Google Scholar]; (b) Hubig S. M. Bockman T. M. Kochi J. K. J. Am. Chem. Soc. 1997;119:2926–2935. [Google Scholar]
- Miller J. L. Lawrence J.-M. I. A. Rodriguez del Rey F. O. Floreancig P. E. Chem. Soc. Rev. 2022;51:5660–5690. doi: 10.1039/d1cs01169c. [DOI] [PubMed] [Google Scholar]
- Yu W. Mei Y. Kang Y. Hua Z. Jin Z. Org. Lett. 2004;6:3217–3219. doi: 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this article have been included as part of the ESI.†