

ABSTRACT
Intra‐host diversity is an intricate phenomenon related to immune evasion, antiviral resistance, and evolutionary leaps along transmission chains. SARS‐CoV‐2 intra‐host variation has been well‐evidenced from respiratory samples. However, data on systemic dissemination and diversification are relatively scarce and come from immunologically impaired patients. Here, the presence and variability of SARS‐CoV‐2 were assessed among 71 tissue samples obtained from multiple organs including lung, intestine, heart, kidney, and liver from 15 autopsies with positive swabs and no records of immunocompromise. The virus was detected in most organs in the majority of autopsies. All organs presented intra‐host single nucleotide variants (iSNVs) with low, moderate, and high abundances. The iSNV abundances observed within different organs indicate that the virus can mutate at one host site and subsequently spread to other parts of the body. In agreement with previous data from respiratory samples, our lung samples presented no more than 10 iSNVs each. But interestingly, when analyzing different organs we were able to detect between 11 and 45 iSNVs per case. Our results indicate that SARS‐CoV‐2 can replicate, and evolve in a compartmentalized manner, in different body sites, which agrees with the “viral reservoir” theory. We elaborate on how compartmentalized evolution in multiple organs may contribute to SARS‐CoV‐2 evolving so rapidly despite the virus having a proofreading mechanism.
Keywords: COVID‐19, intra‐host evolution, mutations, quasispecies, SARS‐CoV‐2, single‐nucleotide variants
1. Introduction
Coronaviruses causing mild to moderate illnesses have been known since about mid‐20th century. However, three concerning betacoronaviruses have been discovered in recent times, Middle East respiratory syndrome‐related coronavirus (MERS‐CoV), severe acute respiratory syndrome coronavirus (SARS‐CoV), and severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) [1]. They were responsible for an outbreak in 2012 (MERS‐CoV), an epidemic in 2002–2003 (SARS‐CoV), and a pandemic that started in about December 2019 (SARS‐CoV‐2). All three viruses can cause severe syndromes with high fatality rates (about 10%, 36%, and 4%, respectively) [2, 3]. The SARS‐CoV‐2 pandemic was rampant, with millions of people from hundreds of countries having been infected in a few months early in 2020. The discovery that a significant proportion of infections may become persistent has recently come into focus. Persistence can favor virus adaptation and the emergence of lineage‐defining mutations [4, 5]. Furthermore, it is common in patients with post‐acute sequelae of COVID‐19 (PASC), a concerning, yet poorly understood, multisystem syndrome that affects up to half of the diagnosed individuals [6, 7]. It is important to define whether the virus can replicate and mutate in a compartmentalized manner in different organs, as this could be related to the establishment of viral reservoirs in persistent infections, and systemic damage. Furthermore, compartmentalized mutation may increase intra‐host variability, impacting the virus's ability to make large evolutionary leaps, as will be shown and discussed throughout this work.
Intra‐host variability refers to the genetic diversity of a virus within a host. It plays a crucial role in a virus's ability to adapt to changing conditions [8], potentially leading to more severe disease outcomes [9, 10], contributing to viral persistence [11], and complicating treatment efforts [12, 13, 14]. Furthermore, monitoring intra‐host diversity is important for public health, as it can influence the emergence of new strains and impact epidemiology [15, 16]. Understanding intra‐host viral diversity is essential for developing effective diagnostics, treatments, and preventive measures.
Intra‐host variability has been well described in coronaviruses. For example, viruses from respiratory and enteric specimens from bovine coronavirus (BCoV) cases can differ to each other by about 30–80 non‐synonymous mutations [17, 18]. Furthermore, the viruses from both respiratory and enteric samples swiftly develop new mutations when cultured in vitro. De novo mutation has also been observed in vitro in avian infectious bronchitis virus (IBV), a chicken coronavirus. Remarkably, these IBV mutations were correlated with changes in pathogenicity, immunogenicity, and tropism [19, 20]. This prior knowledge justified carrying out analogous studies on SARS‐CoV‐2, which showed that this virus also develops genetic swarms [21, 22, 23, 24, 25, 26]. However, most of that information comes from respiratory samples. Data on other body sites are relatively few, and come from patients hospitalized with COVID‐19, and from immunocompromised individuals only. But importantly, viral cycle intermediaries have been detected in various tissues of people who died with COVID, suggesting that secondary replication may occur at multiple body sites [27, 28]. Similarly, organ‐exclusive iSNVs, much likely generated during secondary replication, have been detected in tissues from an immunocompromised patient who also died with COVID‐19 [29]. Here, we sought to detect the virus in various organs from individuals with no records of immunocompromise. Furthermore, we searched for evidence of multisystem secondary replication, which we did by identifying genetic differences between metagenomes from different organs.
2. Methods
2.1. Cases
Tissue samples were obtained from judicial autopsies performed between January and August 2022. SARS‐CoV‐2 infection was posthumously diagnosed in all the studied cases by routine real‐time PCR tests performed on nasopharyngeal swabs. Four individuals were hospitalized at the time of death, and no one had records of immune compromise. Forensic procedures and minimally invasive sample collection were performed as described previously [30]. For metagenomic analyses described below, tissue pieces of about 0.5 cm3 were equilibrated in RNAlater (Sigma‐Aldrich) as indicated by the manufacturer, and stored at −80°C until use. This study was approved by the Cuerpo Médico Forense (Corte Suprema de Justicia de la Nación Argentina).
2.2. Virus Detection
Tissue samples were homogenized in 600 μL of Dulbecco's modified Eagle medium, and clarified by centrifugation at ×12 000g for 10′ in a refrigerated centrifuge (4°C). RNA in the obtained supernatants was purified by a robot (chemagic 360, PerkinElmer) using chemagic Viral DNA/RNA kit H96 (PerkinElmer, Germany). For cDNA synthesis, 9 μL of purified RNA were used in a retro‐transcription reaction using 200 units of SuperScript IV Reverse Transcriptase (ThermoFisher) with random hexamers (60 ng), a final volume of 20 μL, following the manufacturer's recommendations. Real‐time PCR diagnosis was performed by DisCoVery SARS‐CoV‐2 RT‐PCR Detection Kit Rox (AP Biotech; Method 1) and SARS‐CoV‐2 RUO qPCR Primer & Probe Kit (Integrated DNA Technologies; Method 2). Furthermore, the presence of viral RNA was also tested by an end‐point PCR recommended by the World Health Organization (Method 3) [31, 32]. PCR products were analyzed by 1.8% agarose gel electrophoresis, GelRed staining, and UV visualization. Data were analyzed with Metacoder [33] and base graphical tools of the R statistical package [34]. Statistical concordance and agreement were analyzed with irr [35].
2.3. Highly Parallelized Sequencing
Amplification of cDNA was carried out using the NEBNext ARTIC SARS‐CoV‐2 RT‐PCR kit with the VarSkip Short v2 primer set. Illumina sequencing libraries were prepared using the tagmentation‐based and PCR‐based DNA Prep kit with a target insert size of 320 bp. Sequencing was performed on a NovaSeq 6000 sequencer producing 2 × 150 bp paired‐end reads. Demultiplexing and adapter trimming were done with bcl‐convert. Demultiplexed reads were quality controlled using fastp [36] with default settings and polyG/X trimming, low complexity filter, and base correction in overlapped regions enabled. Additionally, 18 and 3 bases were dismissed from the 5′ and 3′ ends, respectively, based on preliminary fastp analyses.
2.4. Bioinformatic Analyses
Reference alignment was performed with the mem routine of Burrows‐Wheeler Alignment tool [37]. Single nucleotide variants (SNVs) calling was performed with ivar [38] using either genome MN908947.3 from GenBank or each case consensus sequence as reference. To obtain consensus sequences from each case, we first generated a consensus sequence from each organ using samtools [39] and MN908947.3 as a reference. The consensus sequences of the organs from each case were then aligned with each other with MAFFT [40], and the obtained alignments were used to generate consensus sequences with the cons program of the EMBOSS package [41]. The obtained consensus genomes were classified with Nextclade [42]. A detailed analysis by Gu et al. showed that filtering out SNVs with frequencies below 0.025, and endorsed by less than 100 reads, brings data that is optimal for high sensitivity and minimal false discovery rates [43]. Therefore, we always accepted SNVs that (i) were present in all the organs of a case, (ii) had frequencies greater than 0.025, and (iii) were endorsed by at least 100 reads. In parallel, because one of our objectives was to compare the metagenomes of the different organs from each case, an additional filter was implemented aimed at equalizing detection power between organs. In this regard, the SNVs that did not pass criterion (i) described above, only were accepted as long as the corresponding genomic positions were covered by at least 500 reads in all organs. The filtered SNV data were analyzed with base R functions, and UpSet plots obtained by the R package UpSetR [44]. Rows of UpSet plots are equivalent to the sets in a Venn diagram, whereas columns are equivalent to their intersections. This type of charts can handle very large numbers of sets compared to Venn diagrams, for which depicting intersections for more than three to four sets is impractical [45, 46]. The distribution of the polymorphisms identified among the specimens studied, and their distribution throughout the SARS‐CoV‐2 genome, were analyzed using the Circos circular visualization tool [47]. The full workflow is schematized in Figure S1.
3. Results
Seventy‐one organ samples from 15 autopsies were tested for the presence of SARS‐CoV‐2 Since there are no validated diagnostic techniques for solid tissue samples, we used three different methods (M1‐3) to endorse and confirm these analyses. The virus was detected in different organs in all but one case, by at least one technique, and in cases from both hospitalized and nonhospitalized individuals (Figure 1; Table S1). The majority of intestine and lung samples ranked positive by the three methods (Figure 2). Likewise, most heart and kidney samples were positive, with the exception of some of them that ranked negative by Method 1. The liver samples presented the lower positivity rates independently of the implemented technique. Selected heart, intestine, kidney, liver, and lung specimens were submitted to metagenomic analyses, encompassing cases with diverse pathological findings (Figure 3). These analyses further confirmed the presence of the virus in all samples. In 18 of them, the obtained sequencing coverages and depths were adequate to perform SNV analyses across the whole virus genome (Table S2).
Figure 1.
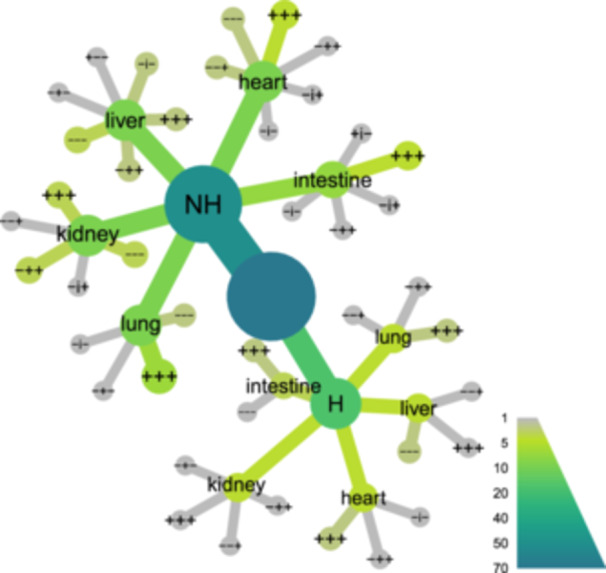
Characteristics of the organ samples studied here. Samples were hierarchically clustered and sorted by Metacoder. The node in the center includes all the samples. From there, in an outward direction, the samples are discriminated according to whether they come from hospitalized (H) or nonhospitalized (NH) individuals. Then it is indicated which organs the samples come from. The outermost part of the graph categorizes the samples according to methods 1, 2, and 3 results (+ positive; − negative; i indeterminate). For example, if a sample ranked positive by method 1, indeterminate by Method 2, and negative by Method 3, it was assigned to the category +i−. The nodes' size and color represent the number of samples in each category.
Figure 2.
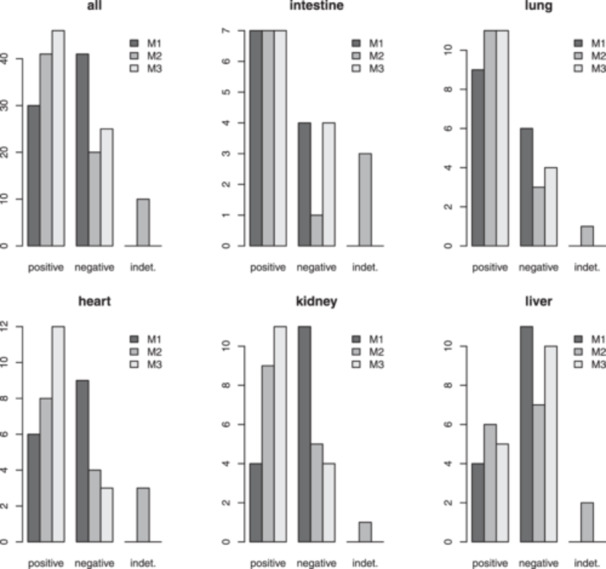
Evidence of the presence of SARS‐CoV‐2 RNA in solid organ samples. Results were confirmed with three different techniques (M1‐3). The y‐axis units are “number of samples.”
Figure 3.
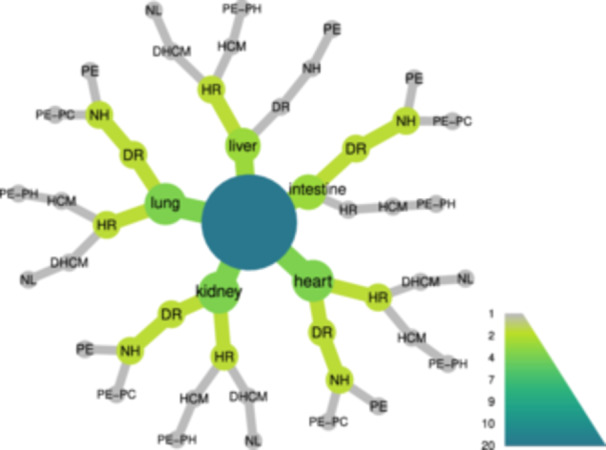
Characteristics of the organ samples submitted to metagenomic analyses. The first rank shows which organs the samples come from. Then anatomo‐pathological characteristics of the corresponding cases are provided, starting by radiological findings (DR, diffuse lung radiopacities; HR heterogeneous lung radiopacities), and followed by heart (DHCM, dilated hypertrophic cardiomyopathy; HCM, hypertrophic cardiomyopathy; NH, normal heart), and lungs (NL normal lungs; PC, pulmonary congestion; PE, pulmonary edema; PH, pulmonary hemorrhage) macroscopic findings. Node size and color codes are as in Figure 1.
Figure 4 displays the distributions and frequencies of the SNVs detected, relative to the MN908947.3 reference genome. Table S3 provides further details on their counterparts at the protein level, and their frequencies in each studied organ. SNVs detected in all reads of a case correspond to mutations specific to the corresponding viral subtype. For example, the Case 5 virus, which was classified by the Nextclade tool as BA.1.1, presented 64 SNVs relative to MN908947.3 (Intersections 1, 2, 5, 11, and 15 in Figure 4). Of them, 28 were absent in the rest of viruses (Intersection 15), which belonged to subtypes BA.4 (Case 18) and BA.5.2.1 (Cases 24 and 28). On the other hand, variants that have frequencies less than 1 reveal the presence of iSNVs, which are described in more detail below.
Figure 4.
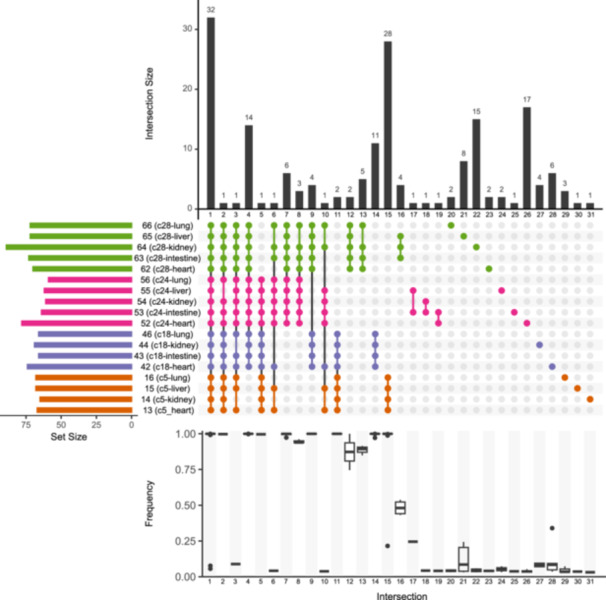
UpSet visualization of SARS‐CoV‐2 mutations in different organs, relative to MN908947.3 reference genome. The grid depicts mutation sets (columns) shared between organ specimens (rows). Colors indicate which case each specimen comes from (details in Table S1). The above barplot (Intersection size) shows the number of mutations in each intersection, that is, mutations shared by several specimens (Intersections 1–19) or present in a single specimen (Intersections 20–31). Alternative allele frequencies are summarized below the grid (e.g., the box plot below Intersection 16 summarizes the frequencies of the alternative alleles of four polymorphisms exclusive of Specimens 63–65). The barplot on the left (Set size) represent the number of iSNVs retrieved from each specimen.
Figures 5 and [Link], [Link], [Link], [Link] show how iSNV polymorphisms were distributed throughout the viral genome, their frequencies, and their effects at the protein level. Most iSNVs were present in only one organ or in just some organs of a case, suggesting the occurrence of tissue‐compartmentalized replication. The majority of alternative alleles presented low frequencies (~2.5%–5%). However, several abundant variants were as well observed. The abundant variants were sometimes detected in just one organ. Other times, besides the organ in which the variants presented the greater frequencies, they were detected at relatively low frequencies in all or several of the rest of organs from the corresponding cases (links in Figures 5 and [Link], [Link], [Link], [Link]). Importantly, a high‐frequency variant of the lung of Case 5 (Specimen 16), and most of the high‐frequency variants of the liver of Case 28 (Specimen 65), represented the dominant populations in these organs. This suggests that virus populations in these organs evolved independently of the populations from other anatomical compartments. Besides, these dominant variants were detected in other organs of these cases, although with lower frequencies (details in Figures S2 and S5), suggesting a possible dispersal from the organs in which they constituted the main population.
Figure 5.
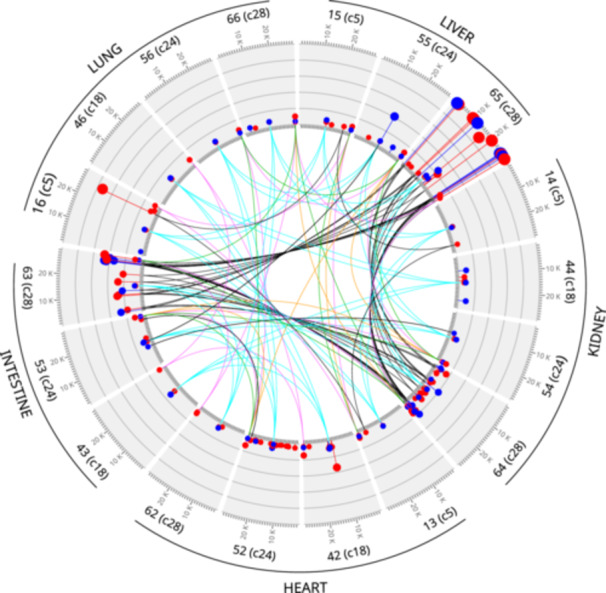
Distribution of iSNVs among lung, liver, kidney, heart, and intestine metagenomes. Gray segments represent the virus genome. Above each segment, it is indicated which specimen and case each metagenome corresponds to; for example, 46(c18) indicate the segment corresponds to the metagenome retrieved from Specimen 46 of case 18. Ticks on the inner and outer boundaries of segments indicate genome positions (1 tick = 1 kB). Intra‐host polymorphisms are represented by colored dots (red: non‐synonymous substitution; blue: synonymous substitution). Grids on the segments are used to display alternative allele frequencies. Each grid line correspond to 0.2 (20%) abundance units. The lines in the innermost part of the graph (namely “links”) connect polymorphic genome positions affected by same iSNVs. Colored links correspond to iSNVs present in all cases (cyan polymerase D533N; green nucleocapsid D415D; magenta nucleocapsid D415G; orange nucleocapsid D415E; MN908947.3 coordinates). Please see also Figures [Link], [Link], [Link], [Link].
The polymorphisms detected affected 22 genes corresponding to structural, nonstructural, and accessory proteins (Supplementary Figures [Link], [Link], [Link], [Link]). In agreement with previous studies, the effect of these mutations on their protein counterparts is compatible with the action of positive selection [5, 43, 48]. In this regard, we observed 2·36 times more non‐synonymous than synonymous iSNVs across all coding regions. Also, the rations of per‐gene non‐synonymous to synonymous polymorphisms observed in this work were statistically indistinguishable from those observed by Liu et al. [48] On the other side, while most respiratory specimens studied in previous works presented less than 10 iSNVs [22, 23, 24, 43, 48], we detected between 11 and 45 iSNVs per case. We attribute this difference to the fact that we sampled sites other than the respiratory system.
4. Discussion
In this work, we focused on investigating whether SARS‐CoV‐2 can colonize different organs and whether it can replicate in such organs. For this, we studied tissues from judicial autopsies of cases with postmortem detection of SARS‐CoV‐2. We could detect the virus in multiple organs from the majority of studied cases using three different methods and metagenomic analyses. These results agree with previous findings that the virus can colonize systems other than the respiratory one [49, 50, 51, 52]. So far there is evidence that, in addition to the gastrointestinal and respiratory systems, SARS‐CoV‐2 can reach the genitourinary, lymphoid, cardiovascular, central nervous, and endocrine systems [27, 53]. In general, these data correspond to severe COVID‐19 cases, in which viremia is exacerbated, or to immunocompromised individuals [54]. The autopsies analyzed in this work were carried out for issues related to judicial processes. Thus, the specific causes that led to death in each case are not available due to legal regulations in our country. However, although all the decedents studied had a diagnosis of COVID‐19, only four of them were hospitalized when they died. Furthermore, none of them had records of immunocompromise. Therefore, the data reported here support the concept that multi‐organ spread may occur in all SARS‐CoV‐2 infections. As mentioned in the Introduction, colonization of multiple organs could be related to the establishment of extrapulmonary reservoirs that may play a role in viral persistence.
The ability of SARS‐CoV‐2 to vary within patients was discovered early after the virus emerged [22], and was confirmed many times since then. However, the vast majority of previous works studied viral populations from the respiratory system, because this is the system most damaged by COVID‐19, and because infectious particles are transmitted by aerosols from the airways. Data on intra‐host variability in other body sites are relatively scarce, and come from autopsies of immunocompromised patients only [29, 55]. In this study, we observed a large proportion of organ‐specific iSNVs, likely reflecting the presence of variants generated during secondary replication. Van Cleemput et al., who identified exclusive variants in several tissues from an immunocompromised patient, came to similar conclusions [29]. These are not the only evidence that SARS‐CoV‐2 can replicate throughout the body. Negative‐sense viral RNA, an intermediary of viral replication, has been detected in non‐respiratory tissues from donors who died of COVID‐19 [27]. In addition, subgenomic RNA, another marker of viral metabolism, has been detected in the cardiovascular, lymphoid, genitourinary, endocrine, ocular, nervous, and muscular systems of patients who died with severe COVID‐19 [28]. Considering these findings, together with the vast evidence of the presence of viral antigens and RNA in multiple tissues, and the association of many COVID‐19 cases with systemic conditions, it is reasonable to assume that in‐situ replication could be directly involved in multi‐organ pathogenesis. Multisystem replication could facilitate the virus to reach the lungs by mechanisms other than microaspiration, which may explain why peripheral signs are common in lung images from COVID‐19 patients [56, 57]. Broad‐spectrum tropism may respond to quasispecies‐mediated cyclical adaptation [58], which should be further investigated. Besides, suboptimal variants in one environment may constitute a low‐frequency reservoir for adaptation to the other environment, as observed in measles virus [59].
Interestingly, punctuated evolutionary shifts—where viruses are very distinct from each other despite being monophyletic—have been observed in samples from the upper respiratory tract (URT) collected over time from a same patient [4, 60]. The interpretation given by the authors of these studies was that the marked URT turnovers were driven by viral populations originated in other organs, in agreement with the “viral reservoir” theory. Our results are compatible with this interpretation, since we observed very different metagenomes in different organs. Moreover, most of the iSNVs that displayed high frequencies in an organ were also present in other organs of the same individual, albeit at relatively low frequencies. This suggests a source–sink relationship between the organs where such iSNVs were abundant and those where they were rare. Thus, mutations originating in a specific location in the body apparently can then spread to other body sites. As mentioned, SARS‐CoV‐2 is transmitted by droplets expelled from the airways. These droplets harbor very small fractions of the whole virus population. In consequence, transmission events can lead to evolutionary bottlenecks that produce evolutionary leaps [61, 62]. If variants originated throughout the body could reach the airways, as suggested by our data and the above‐cited studies, the number of droplet‐transmissible variants would be greater than if such variants came only from the respiratory system. Therefore, compartmentalized evolution has the potential to increase evolutionary leaps associated with transmission events. This may explain, at least in part, why SARS‐CoV‐2 evolves so rapidly despite its replication system having a proofreading mechanism. As mentioned in the Introduction, other coronaviruses can also evolve divergent variants in different host sites. This highlights the need to further investigate the extent to which compartmentalized mutation is important for understanding the epidemiology, evolution, and pathogenesis of these successful mammalian pathogens.
Author Contributions
Autopsy procedures and sample collection: Santiago Maffia‐Bizzozero. Sample processing and viral detection: Julieta M. Manrique, Leandro R. Jones, M. Victoria Delpino, and Jorge Quarleri. Genomic data generation: Julieta M. Manrique. Computational workflow design and implementation: Leandro R. Jones. Conceptualization and design of the study, funding acquisition, and project administration: Jorge Quarleri, Leandro R. Jones, M. Victoria Delpino, and Julieta M. Manrique. Writing of manuscript: Leandro R. Jones. All authors contributed to the article and approved the submitted version.
Conflicts of Interest
The authors declare no conflict of interest.
Supporting information
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Acknowledgments
This study was supported by Agencia Nacional de Promoción de la Investigación, el Desarrollo Tecnológico y la Innovación (Fondo para la Investigación Científica y Tecnológica, PICTO‐COVID‐SECUELAS 00005 and PICT‐2020 A03643), and Consejo Nacional de Investigaciones Científicas y Técnicas (PIP GI11220200102657CO).
Data Availability Statement
Illumina data have been deposited with links to BioProject PRJNA1144939 in the NCBI BioProject database. Any other data described in this study are available upon reasonable request from the authors.
References
- 1. Gorbalenya A. E., Baker S. C., Baric R. S., et al., “The Species Severe Acute Respiratory Syndrome‐Related Coronavirus: Classifying 2019‐nCoV and Naming It Sars‐Cov‐2,” Nature Microbiology 5 (2020): 536–544, 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cui J., Li F., and Shi Z.‐L., “Origin and Evolution of Pathogenic Coronaviruses,” Nature Reviews Microbiology 17 (2018): 181–192, 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. World Health Organization , WHO COVID‐19 Dashboard (Geneva, Switzerland: World Health Organization, 2024). [Google Scholar]
- 4. Chaguza C., Hahn A. M., Petrone M. E., et al., “Accelerated SARS‐CoV‐2 Intrahost Evolution Leading to Distinct Genotypes During Chronic Infection,” Cell Reports Medicine 4 (2023): 100943, 10.1016/j.xcrm.2023.100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ghafari M., Hall M., Golubchik T., et al., “Prevalence of Persistent SARS‐CoV‐2 in a Large Community Surveillance Study,” Nature 626 (2024): 1094–1101, 10.1038/s41586-024-07029-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Swank Z., Senussi Y., Manickas‐Hill Z., et al., “Persistent Circulating Severe Acute Respiratory Syndrome Coronavirus 2 Spike Is Associated With Post‐Acute Coronavirus Disease 2019 Sequelae,” Clinical Infectious Diseases 76 (2022): e487–e490, 10.1093/cid/ciac722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lupi L., Vitiello A., Parolin C., Calistri A., and Garzino‐Demo A., “The Potential Role of Viral Persistence in the Post‐Acute Sequelae of SARS‐CoV‐2 Infection (PASC),” Pathogens 13 (2024): 388, 10.3390/pathogens13050388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vignuzzi M., Stone J. K., Arnold J. J., Cameron C. E., and Andino R., “Quasispecies Diversity Determines Pathogenesis Through Cooperative Interactions in a Viral Population,” Nature 439 (2005): 344–348, 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Al Khatib H. A., Al Maslamani M. A., Coyle P. V., et al., “Inter‐Versus Intra‐Host Sequence Diversity of pH1N1 and Associated Clinical Outcomes,” Microorganisms 8 (2020): 133, 10.3390/microorganisms8010133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Y., Jiang N., Qi W., et al., “SARS‐CoV‐2 Intra‐Host Single‐Nucleotide Variants Associated With Disease Severity,” Virus Evolution 8, no. 2 (2022): veac106, 10.1093/ve/veac106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nowak M. A., Anderson R. M., McLean A. R., Wolfs T. F. W., Goudsmit J., and May R. M., “Antigenic Diversity Thresholds and the Development of AIDS,” Science 254 (1991): 963–969, 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- 12. Johnson J. A., Li J.‐F., Wei X., et al., “Minority HIV‐1 Drug Resistance Mutations Are Present in Antiretroviral Treatment–Naïve Populations and Associate With Reduced Treatment Efficacy,” PLoS Medicine 5 (2008): e158, 10.1371/journal.pmed.0050158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jones L. R., Sede M., Manrique J. M., and Quarleri J., “Hepatitis B Virus Resistance Substitutions: Long‐Term Analysis by Next‐Generation Sequencing,” Archives of Virology 161 (2016): 2885–2891, 10.1007/s00705-016-2959-8. [DOI] [PubMed] [Google Scholar]
- 14. Colson P., Delerce J., Pontarotti P., et al., “Resistance‐Associated Mutations to the Anti‐SARS‐CoV‐2 Agent Nirmatrelvir: Selection Not Induction,” Journal of Medical Virology 96 (2024): e29462, 10.1002/jmv.29462. [DOI] [PubMed] [Google Scholar]
- 15. Zhang Y., Jiang N., Qi W., et al., “Intra‐Host SARS‐CoV‐2 Single‐Nucleotide Variants Emerged During the Early Stage of COVID‐19 Pandemic Forecast Population Fixing Mutations,” Journal of Infection 84 (2022): 722–746, 10.1016/j.jinf.2022.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Landis J. T., Moorad R., Pluta L. J., et al., “Intra‐Host Evolution Provides for the Continuous Emergence of SARS‐CoV‐2 Variants,” mBio 14 (2023): e0344822, 10.1128/mbio.03448-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang X., Hasoksuz M., Spiro D., et al., “Quasispecies of Bovine Enteric and Respiratory Coronaviruses Based on Complete Genome Sequences and Genetic Changes After Tissue Culture Adaptation,” Virology 363 (2007): 1–10, 10.1016/j.virol.2007.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saif L. J. and Jung K., “Comparative Pathogenesis of Bovine and Porcine Respiratory Coronaviruses in the Animal Host Species and SARS‐CoV‐2 in Humans,” Journal of Clinical Microbiology 58 (2020): e01355‐20, 10.1128/jcm.01355-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu S., Zhang X., Gong L., et al., “Altered Pathogenicity, Immunogenicity, Tissue Tropism and 3′‐7kb Region Sequence of an Avian Infectious Bronchitis Coronavirus Strain after Serial Passage in Embryos,” Vaccine 27 (2009): 4630–4640, 10.1016/j.vaccine.2009.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hewson K. A., Scott P. C., Devlin J. M., Ignjatovic J., and Noormohammadi A. H., “The Presence of Viral Subpopulations in an Infectious Bronchitis Virus Vaccine With Differing Pathogenicity—A Preliminary Study,” Vaccine 30 (2012): 4190–4199, 10.1016/j.vaccine.2012.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karamitros T., Papadopoulou G., Bousali M., Mexias A., Tsiodras S., and Mentis A., “SARS‐CoV‐2 Exhibits Intra‐Host Genomic Plasticity and Low‐Frequency Polymorphic Quasispecies,” Journal of Clinical Virology 131 (2020): 104585, 10.1016/j.jcv.2020.104585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen Z., Xiao Y., Kang L., et al., “Genomic Diversity of Severe Acute Respiratory Syndrome–Coronavirus 2 in Patients With Coronavirus Disease 2019,” Clinical Infectious Diseases 71 (2020): 713–720, 10.1093/cid/ciaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lythgoe K. A., Hall M., Ferretti L., et al., “SARS‐CoV‐2 Within‐Host Diversity and Transmission,” Science (New York, N.Y.) 372, no. 6539 (2021): eabg0821, 10.1126/science.abg0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tonkin‐Hill G., Martincorena I., Amato R., et al., “Patterns of Within‐Host Genetic Diversity in SARS‐CoV‐2,” eLife 10 (2021): e66857, 10.7554/elife.66857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li B., Deng A., Li K., et al., “Viral Infection and Transmission in a Large, Well‐Traced Outbreak Caused by the SARS‐CoV‐2 Delta Variant,” Nature Communications 13 (2022): 460, 10.1038/s41467-022-28089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hou M., Shi J., Gong Z., et al., “Intra‐ vs. Interhost Evolution of SARS‐CoV‐2 Driven by Uncorrelated Selection—The Evolution Thwarted,” Molecular Biology and Evolution 40 (2023): msad204, 10.1093/molbev/msad204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delorey T. M., Ziegler C. G. K., Heimberg G., et al., “COVID‐19 Tissue Atlases Reveal SARS‐CoV‐2 Pathology and Cellular Targets,” Nature 595 (2021): 107–113, 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stein S. R., Ramelli S. C., Grazioli A., et al., “SARS‐CoV‐2 Infection and Persistence in the Human Body and Brain at Autopsy,” Nature 612 (2022): 758–763, 10.1038/s41586-022-05542-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Cleemput J., van Snippenberg W., Lambrechts L., et al., “Organ‐Specific Genome Diversity of Replication‐Competent SARS‐CoV‐2,” Nature Communications 12 (2021): 6612, 10.1038/s41467-021-26884-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maffia‐Bizzozero S., Cevallos C., Lenicov F. R., et al., “Viable SARS‐CoV‐2 Omicron Sub‐Variants Isolated From Autopsy Tissues,” Frontiers in Microbiology 14 (2023): 1192832, 10.3389/fmicb.2023.1192832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. World Health Organization . Laboratory Testing for Coronavirus Disease (COVID‐19) in Suspected Human Cases. Interim Guidance 19 March 2020. WHO Reference Number: WHO/COVID‐19/laboratory/2020.5.
- 32. Shirato K., Nao N., Katano H., et al., “Development of Genetic Diagnostic Methods for Detection for Novel Coronavirus 2019 (nCoV‐2019) in Japan,” Japanese Journal of Infectious Diseases 73 (2020): 304–307, 10.7883/yoken.jjid.2020.061. [DOI] [PubMed] [Google Scholar]
- 33. Foster Z. S. L., Sharpton T. J., and Grünwald N. J., “Metacoder: An R Package for Visualization and Manipulation of Community Taxonomic Diversity Data,” PLoS Computational Biology 13 (2017): e1005404, 10.1371/journal.pcbi.1005404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.“R: A Language and Environment for Statistical Computing,” R Core Team, 2024, https://www.R-project.org/.
- 35. Gamer M., Lemon J., Fellows I., and Singh P. (2019). “irr: Various Coefficients of Interrater Reliability and Agreement. R Package Version 0.84.1,” https://CRAN.R-project.org/package=irr.
- 36. Chen S., “Ultrafast One‐Pass FASTQ Data Preprocessing, Quality Control, and Deduplication Using Fastp,” iMeta 2 (2023): e107, 10.1002/imt2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li H. and Durbin R., “Fast and Accurate Short Read Alignment With Burrows–Wheeler Transform,” Bioinformatics 25 (2009): 1754–1760, 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grubaugh N. D., Gangavarapu K., Quick J., et al., “An Amplicon‐Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and iVar,” Genome Biology 20 (2019): 8, 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H., Handsaker B., Wysoker A., et al., “The Sequence Alignment/Map Format and Samtools,” Bioinformatics 25 (2009): 2078–2079, 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Katoh K. and Standley D. M., “MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability,” Molecular Biology and Evolution 30 (2013): 772–780, 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rice P., Longden I., and Bleasby A., “EMBOSS: The European Molecular Biology Open Software Suite,” Trends in Genetics 16 (2000): 276–277, 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 42. Aksamentov I., Roemer C., Hodcroft E., and Neher R., “Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes,” Journal of Open Source Software 6 (2021): 3773, 10.21105/joss.03773. [DOI] [Google Scholar]
- 43. Gu H., Quadeer A. A., Krishnan P., et al., “Within‐Host Genetic Diversity of SARS‐CoV‐2 Lineages in Unvaccinated and Vaccinated Individuals,” Nature Communications 14 (2023): 1793, 10.1038/s41467-023-37468-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Conway J. R., Lex A., and Gehlenborg N., “UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties,” Bioinformatics 33 (2017): 2938–2940, 10.1093/bioinformatics/btx364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lex A. and Gehlenborg N., “Sets and Intersections,” Nature Methods 11 (2014): 779, 10.1038/nmeth.3033. [DOI] [Google Scholar]
- 46. Lex A., Gehlenborg N., Strobelt H., Vuillemot R., and Pfister H., “UpSet: Visualization of Intersecting Sets,” IEEE Transactions on Visualization and Computer Graphics 20 (2014): 1983–1992, 10.1109/tvcg.2014.2346248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krzywinski M., Schein J., Birol İ., et al., “Circos: An Information Aesthetic for Comparative Genomics,” Genome Research 19 (2009): 1639–1645, 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu P., Cai J., Tian H., et al., “Intra‐Host Genetic Diversity of SARS‐CoV‐2 Omicron Variants in Children,” Journal of Infection 88 (2024): 106132, 10.1016/j.jinf.2024.106132. [DOI] [PubMed] [Google Scholar]
- 49. Perisetti A., Goyal H., Gajendran M., Boregowda U., Mann R., and Sharma N., “Prevalence, Mechanisms, and Implications of Gastrointestinal Symptoms in COVID‐19,” Frontiers in Medicine 7 (2020): 588711, 10.3389/fmed.2020.588711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trypsteen W., Van Cleemput J., Snippenberg W., Gerlo S., and Vandekerckhove L., “On the Whereabouts of SARS‐CoV‐2 in the Human Body: A Systematic Review,” PLoS Pathogens 16 (2020): e1009037, 10.1371/journal.ppat.1009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xia J., Tong J., Liu M., Shen Y., and Guo D., “Evaluation of Coronavirus in Tears and Conjunctival Secretions of Patients With SARS‐CoV‐2 Infection,” Journal of Medical Virology 92 (2020): 589–594, 10.1002/jmv.25725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang W., Du R.‐H., Li B., et al., “Molecular and Serological Investigation of 2019‐nCoV Infected Patients: Implication of Multiple Shedding Routes,” Emerging Microbes & Infections 9 (2020): 386–389, 10.1080/22221751.2020.1729071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Proal A. D., VanElzakker M. B., Aleman S., et al., “SARS‐CoV‐2 Reservoir in Post‐Acute Sequelae of COVID‐19 (PASC),” Nature Immunology 24 (2023): 1616–1627, 10.1038/s41590-023-01601-2. [DOI] [PubMed] [Google Scholar]
- 54. Hogan C. A., Stevens B. A., Sahoo M. K., et al., “High Frequency of SARS‐CoV‐2 RNAemia and Association With Severe Disease,” Clinical Infectious Diseases 72 (2020): e291–e295, 10.1093/cid/ciaa1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lee J.‐S., Yun K. W., Jeong H., et al., “SARS‐CoV‐2 Shedding Dynamics and Transmission in Immunosuppressed Patients,” Virulence 13 (2022): 1242–1251, 10.1080/21505594.2022.2101198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Donohue R. C., Pfaller C. K., and Cattaneo R., “Cyclical Adaptation of Measles Virus Quasispecies to Epithelial and Lymphocytic Cells: To V, or Not to V,” PLoS Pathogens 15 (2019): e1007605, 10.1371/journal.ppat.1007605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Darwish H. S., Habash M. Y., and Habash W. Y., “Chest Computed Tomography Imaging Features in Patients With Coronavirus Disease 2019 (COVID‐19),” Journal of International Medical Research 49 (2021): 030006052110106, 10.1177/03000605211010631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salvatore M. M., Capaccione K. M., Saqi A., Overdevest J. B., Patrizio R., and Gudis D. A., “Characteristic Patterns of SARS‐CoV‐2 on Chest CT Suggests a Hematologic Pathway for Viral Entry Into the Lung,” Clinical Imaging 89 (2022): 92–94, 10.1016/j.clinimag.2022.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fournelle D., Mostefai F., Brunet‐Ratnasingham E., et al., “Intra‐Host Evolution Analyses in an Immunosuppressed Patient Supports SARS‐CoV‐2 Viral Reservoir Hypothesis,” Viruses 16 (2024): 342, 10.3390/v16030342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jones L. R. and Manrique J. M., “Quantitative Phylogenomic Evidence Reveals a Spatially Structured SARS‐CoV‐2 Diversity,” Virology 550 (2020): 70–77, 10.1016/j.virol.2020.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang D., Wang Y., Sun W., et al., “Population Bottlenecks and Intra‐Host Evolution During Human‐To‐Human Transmission of SARS‐CoV‐2,” Frontiers in Medicine 8 (2021): 585358, 10.3389/fmed.2021.585358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang D, Wang Y, Sun W et al. “Population Bottlenecks and Intra‐host Evolution During Human‐to‐Human Transmission of SARS‐CoV‐2.” Frontiers in Medicine. (2021): 8, 10.3389/fmed.2021.585358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Supporting information.
Data Availability Statement
Illumina data have been deposited with links to BioProject PRJNA1144939 in the NCBI BioProject database. Any other data described in this study are available upon reasonable request from the authors.