

Abstract
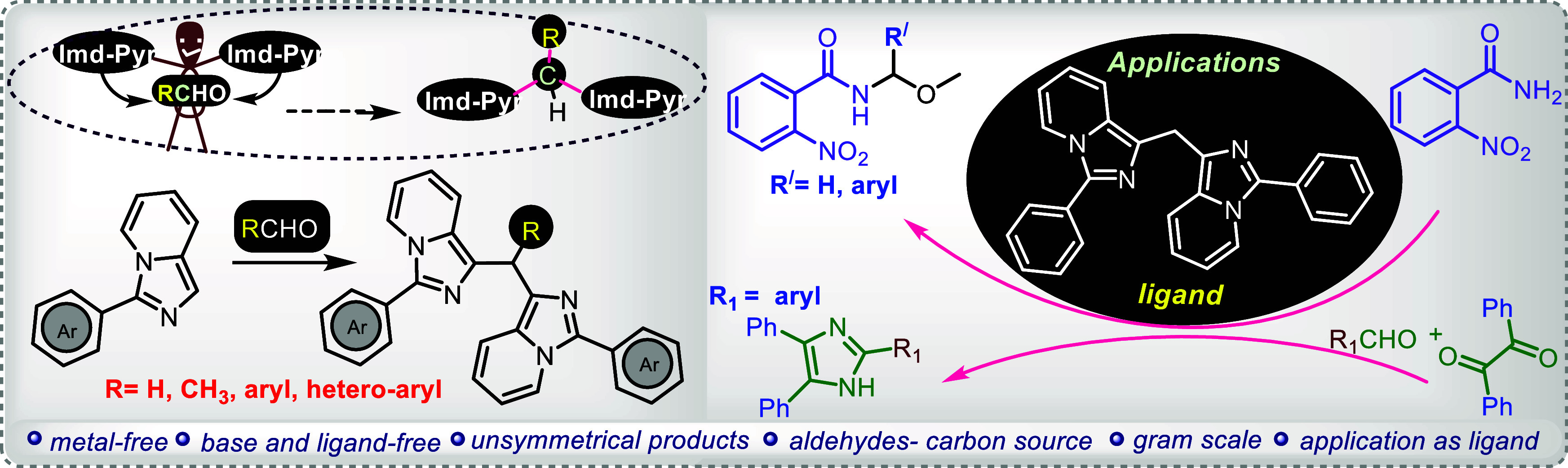
Formaldehyde has been used as a solvent and a source of carbon to insert a methylene group for bridging two imidazo[1,5-a]pyridine molecules without using any metal catalysis. This strategy has been extended on other alkyl-, aryl-, and heteroaryl aldehydes as well. This C(sp2)–C(sp3)–H–C(sp2) bond forming reaction proceeds via C(sp2)H functionalization of imidazo[1,5-a]pyridine and was applied on a wide range of substrates offering moderate to good yields of methylene-bridged/inserted bis-imidazo[1,5-a]pyridines. Most importantly, as an application, the bis-heteroarene product has been demonstrated as a ligand. The ligand-like behavior of bis-imidazo[1,5-a]pyridines has been demonstrated as an extension of current methodology. This reaction works well at the gram scale level.
Introduction
Over the past 2 decades, C–H activation has become a rapidly advancing field in homogeneous catalysis, significantly impacting synthetic organic chemistry. Due to the inert nature of C–H bonds, typically harsh reaction conditions have been required, which limits functional group tolerance.1a To address this, new comprehensive and innovative concepts for C–H functionalization have been introduced to achieve milder methodologies. “C–H functionalization” encompasses both the activation of C–H bonds and their subsequent transformation.1b Catalytic functionalization using transition metals has emerged as an atom-economical technique, creating new bonds without the need for activated precursors—a major drawback for large-scale synthesis.1c Replacing transition-metal catalysis with metal-free strategies offers an alternative that is both cost-effective and environmentally friendly. Aerobic oxidation and metal-free C–H functionalization have become complementary approaches to classical transition-metal-catalyzed transformations.1d
The unique chemical structure, flexibility, optical characteristics, and biological features of imidazo[1,5-a]pyridine nuclei and derivatives have garnered increasing interest in this scaffold. From materials science to the field of medicine, this class of aromatic heterocycles have a lot of potential in research.2a,2b The compound imidazo[1,5-a]pyridine is particularly unique in its nature. To improve the long-term stability or solubility, its optical and electrochemical behaviors, as well as biocompatibility of this core, positions 1 and 3 on the imidazo[1,5-a]pyridine skeleton have been frequently utilized by different strategies to manipulate these properties.2c
Various fused imidazo heterocycles are frequently found to be pharmacophores and display a variety of biological functions; they have attracted the attention of many chemists. A number of commercially available medications (Figure 1), including zolpidem(i), alpidem(i), miroprofen(ii), olprinone(iii), and minodronic acid(iv), include imidazo[1,2-a]pyridine, are being currently used.3a Similarly, the biological effects of imidazo[1,5-a]pyridine derivatives, which include antiviral, antibacterial, antifungal, and anti-inflammatory activities, make them a potentially helpful sector.3b Many organic compounds with an imidazo[1,5-a]pyridine moiety that have demonstrated a variety of biological effects, which are as follows a type-2 cannabinoid receptor (CB2R) agonists(v), inhibitors of phosphodiesterase 10A(vi), inhibitors of tubulin polymerization(vii), lead compound for anticancer chemotherapy(viii), and active against breast cancer cell line (MCF-7) (ix).
Figure 1.

Structures of some biologically active imidazopyridines.
Due to the wide range of biological activity displayed by bis(heteroaryl)methanes, chemists have recently developed an interest in the simplest and shortest synthetic strategies to construct these kinds of molecules. The useful bis(hetero)aryl compounds may have their utility in drug discovery; especially the toxicity could be tackled by offering the reduced dose of the candidate drug, as the required molecules will be available for its efficacy in double concentrations. Also, this strategy offers, patients’ compliance and dose tolerance.4a,4b−4c Our group is continuously engaged in developing synthetic strategies for construction of bis-heterocycles.5a,5b−5c
There are numerous studies on the synthesis of bis(indolyl)methanes6a employing aldehydes6b,6c−6d and their derivatives as precursors to methylene linkages but on the synthesis of various bis(heteroaryl) methanes, such as those of bisimidazo[1,5-a]pyridines7a,7b−7c a very few reports are available. In the literature for the synthesis of 1,1′-bisimidazopyridinylmethanes, DMSO,8a DMF,7b DMA,8c,8d PEG-400,8e etc. were used as the methylene source. Although these reagents have a few useful advantages, they are constrained by factors including the need for metal catalysts, inorganic acids, and bases, and inert atmospheres to catalyze the reaction, and also some others. Herein, we disclose a unique approach for the synthesis of bis(3-arylimidazo[1,5-a]pyridin-3-yl)methanes that involves functionalizing C(sp2)H bonds, using a conventional formaldehyde solution as both the carbon source for insertion of methylene and as a solvent as well. The proposed method allowed for a straightforward transition to produce 1,1′-bisimidazopyridinylmethanes at room temperature in a metal-free and aerobic environment.
The triarylmethane family of organic compounds is one of the most intriguing because of its special properties that can be used in organic materials, leuco dyes, and chemical biological probes.7c Therefore, using aryl aldehydes, we further extended this method to create bis(1-imidazo[1,5-a]pyridyl)arylmethanes. The reports on the synthesis (Scheme 1) of bis(1-imidazo[1,5-a]pyridyl)arylmethanes show the cyclization of N-thioacyl 1,2-aminoalcohols and their silyl ethers in the presence of pyridine and the interaction of imidazo[1,5-a]pyridines with aromatic aldehydes, both involving the use of iodine. It takes multiple steps to prepare N-thioacyl-1-(2-pyridyl)-1,2-aminoalcohols, which are complicated starting materials (Scheme 1A).7b,7c Another study uses iodine as a promoter to synthesize bis-imidazo[1,5-a]pyridines and its derivatives from pyridin-2-ylmethanamine and aldehydes (Scheme 1B).7a In contrast, the reaction in the current approach does not use any complicated starting material and does not require the inclusion of any additive or promoter. It is the simplest single step protocol developed under metal-free conditions (Scheme 1C).
Scheme 1. Synthesis of Bisimidazo[1,5-a]pyridines: Prior Art and Our Strategy.

Results and Discussion
A series of imidazo[1,5-a]pyridines was synthesized based on our earlier work.9 It was subjected to reaction with formaldehyde solution (37% in water) to serve as both reagent and solvent as well, at room temperature, producing the required bis product in good yields. Starting with the model substrate 3-phenylimidazo[1,5-a]pyridine and formaldehyde (HCHO) at room temperature, to our satisfaction, the desired product, bis(3-phenylimidazo[1,5-a]pyridin-1-yl)methane, was achieved with a good yield. The reaction was taken up for further optimization in (Table 1).
Table 1. Optimization of Reaction Conditionsa.

| entry | methylene source | solvent | reaction temp (°C) | reaction time (h) | product 3a (% yield)b | product 3aa (% yield)b |
|---|---|---|---|---|---|---|
| 1 | HCHO | rt | 2 | 11 | 14 | |
| 2 | HCHO | rt | 6 | 42 | 11 | |
| 3 | HCHO | rt | 12 | 86 | 5 | |
| 4 | HCHO | rt | 24 | 85 | 5 | |
| 5 | HCHO | rt | 48 | 85 | 4 | |
| 6 | HCHO | Me0H | rt | 12 | 83 | 7 |
| 7 | HCHO | EtOH | rt | 12 | 84 | 6 |
| 8 | HCHO | ACN | rt | 12 | 80 | 8 |
| 9 | HCHO | H2O | rt | 12 | 78 | 8 |
| 10 | HCHO | 0 | 12 | NR | NR | |
| 11 | HCHO | 60 | 12 | 74 | 10 | |
| 12 | DMSO | rt | 12 | NR | NR | |
| 13 | DMF | rt | 12 | NR | NR | |
| 14 | DMA | rt | 12 | NR | NR |
Reaction conditions: 1 (0.52 mmol, 1.0 equiv), 2a (12.36 mmol, 1.0 mL), solvent EtOH (0.5 mL, if required).
Isolated yields.
Initially, the evaluation of reaction time was performed; in this, the reaction was originally stirred for 2 h, and the yield of the desired product was very less. As a result, the reaction time was extended from 2 to 4 h and then to 6 h and so on, up to 48 h, in an observation, it was found that 12 h of reaction time produced the highest yield. Keeping the reaction going for longer than 12 h had no positive impact on the yield.
Then, we examined a variety of solvents, including MeOH, EtOH, ACN, and water, and found that all of them carried out the reaction without much change in the yields. Hence, EtOH was opted to use if at all the solvent was required for reaction; otherwise, HCHO was only picked as the primary solvent to conduct the reactions. The temperature conditions were also optimized; in this, the rise in temperature yielded decreased products. Moreover, the reaction was also agitated at 0 °C to understand the requirement of milder conditions; it has not given any product. Hence, choosing room temperature was the best option. Further, the reaction was performed with other methylene synthons like DMA, DMF, DMSO, and DESO and observed that none of them could insert methylene between two imidazo[1,5-a]pyridines at conditions as optimized above and mentioned in Table 1.
We studied a range of imidazo[1,5-a]pyridines using optimal conditions to further understand the applicability and depth of the current reaction. The effectiveness of the reactions was not much affected by the presence of electron-donating and electron-withdrawing substituents on the aromatic rings of imidazo[1,5-a]pyridines(Table 2). When the phenyl ring was replaced with pyridyl, a heteroaromatic ring also efficiently gave the product in good yields (3b). To further widen the substrate range, instead of HCHO, we employed acetaldehyde, various aromatic-aldehydes and heteroaryl carboxaldehydes, and we observed that apart from acetaldehyde(3l), no other alkyl aldehyde was found to engage in the reaction to produce the desired product, whereas aromatic and heteroaromatic aldehydes reacted without any difficulty(3m–3w). To further widen the substrate scope, unsubstituted imidazo[1,5-a]pyridine and alkyl substituted imidazo[1,5-a]pyridines like 3-propylimidazo[1,5-a]pyridine, 3-butylimidazo[1,5-a]pyridine, and 3-pentylimidazo[1,5-a]pyridine, were tried under the same reaction conditions. However, none of these substrates successfully yielded the desired results. Additionally, the applicability of the current method for the synthesis of unsymmetric bis-imidazo[1,5-a]pyridines(3x, 3y) was also explored by carrying out the reactions with equivalent amounts of two differently substituted imidazo[1,5-a]pyridines under optimal conditions. Unsymmetric bis-products were formed as the major product along with two symmetric bis-products as minor ones.
Table 2. Substrate Scope for Imidazo[1,5-a]pyridines and Aldehydesa.


Isolated yields, reaction conditions A (3a–3k): 1 (0.28–0.52 mmol, 1.0 equiv), 2a (12.36 mmol, 1.0 mL), and solvent EtOH (0.5 mL, if required), reaction conditions B (3l): 1 (0.52 mmol, 1.0 equiv) and 2b (9.27 mmol, 1.0 mL), reaction conditions C (3m–3w): 1 (0.47–0.52 mmol, 1.0 equiv), 2 (0.71–0.78 mmol, 1.5 equiv), and solvent EtOH (0.5 mL), reaction conditions D (3x): 1a (0.26 mmol, 1.0 equiv), 4-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one (0.26 mmol, 1 equiv), and 2a (12.36 mmol, 1.0 mL), and reaction conditions E (3y): 1b (0.26 mmol, 1.0 equiv), 1g (0.26 mmol, 1.0 equiv), 2g (0.39 mmol, 1.5 equiv), and solvent EtOH (0.5 mL).
The literature on imidazo[1,5-a]pyridine transition-metal complexes and imidazo[1,5-a]pyridine-derived carbenes (IPCs) is extensive. Numerous reports highlight the use of IPCs as auxiliary ligands in catalysis.10a The imidazo[1,5-a]pyridine framework can coordinate with various transition metal ions, including Ni, Co, Pd, and Cu, resulting in a huge number of metal complexes with a wide range of distinct coordination motifs.10b,10c Here, we have reported bis(3-phenylimidazo[1,5-a]pyridin-1-yl)methane acts as a ligand (L) in the presence of copper salt.10d,10e In the presence of copper salt and cesium carbonate as a base in the solvent MeOH,11c,11a−11b compound L was used for the first time as a ligand to synthesize N-(methoxy(aryl)methyl)-2-nitrobenzamide from 2-nitrobenzamide. We combined benzamide with HCHO, benzaldehyde, and 4-chlorobenzaldehyde to generate the corresponding product [Scheme 2a]. For the synthesis of 2-(aryl)-4,5-diphenyl-1H-imidazole, the role of L as a ligand was further expanded. ACN was used as the solvent, while benzil12a,12b was combined with benzaldehydes and ammonium carbamate in the presence of catalyst ferric oxide (Scheme 2b).
Scheme 2. Application of Method—Product Used as a Ligand.

Some control experiments were carried out to illustrate the plausible mechanism. Initially, the deuterium labeling experiment verified that methylene from the DCDO has been incorporated in between both the heterocycles. Free radical scavengers, such as TEMPO, BHT, and DPE, were used in the reaction to circumvent about possibility of the radical mediated mechanism, which produced a high yield of 3a, proving that the transition is predominantly an ionic one (Table 3).
Table 3. Control Experiments.

The reaction of 3-phenylimidazo[1,5-a]pyridine with HCHO was also carried out in the gram scale to obtain the required product with nearly identical efficacy as obtained at the lab scale in order to demonstrate the scalability of the present approach (Scheme 3).
Scheme 3. Gram Scale Synthesis of Bis(3-phenylimidazo[1,5-a]pyridin-1-yl)methane.

A plausible mechanism is presented in (Figure 2) based on prior findings and our own results from control studies. An unstable cation A is generated by adding 3-phenylimidazo[1,5-a]pyridine (1a) to HCHO, which is subsequently followed by the removal of a water molecule to generate intermediate B. Intermediate B is then attacked by a subsequent molecule of 1a to form cation C, which then triggers deprotonation to produce the necessary methylene-bridged imidazopyridine 3a.8a
Figure 2.

Plausible mechanism.
Conclusions
In conclusion, under metal-free conditions, we have demonstrated an effective process for the methylenation of imidazoheterocycles to produce symmetrical and nonsymmetrical methylene-bridged binary heteroaryls. The bridging procedure was tolerated by a wide variety of substituents, and they produced the corresponding products in good yields. By carrying out the reaction at the gram scale, we also demonstrated the scalability of the present protocol. The applicability of the method was demonstrated by using one of the bisimidazopyridines as the ligand for organic transformations.
Experimental Section
General Information
All reactions were performed in an oven-dried glass apparatus. Solvents were distilled in a standard way, and commercial reagents were used without any purification. Analytical TLC was performed on 60 F254 plates and visualized by exposure to ultraviolet light (UV-254 nm). Column chromatography was carried out with silica (100–200 mesh). NMR spectra for characterization of compounds were recorded on a Bruker Advance DPX FT-NMR 400 MHz instrument (1H) at 400 MHz and (13C) at 101 MHz, respectively. 19F NMR spectra were recorded at 377 MHz. Chemical shifts (δ) are reported in ppm, using the residual solvent peak in CDCl3 (δH = 7.26 and δC = 77.16 ppm), DMSO-d6 (δH = 2.50 and δC = 39.52 ppm), acetone-d6 (δH = 2.09 and δC = 30.60 and 205.87 ppm), and pyridine-d5 (δH = 7.22, 7.58, and 8.74 and δC = 123.87, 135.91, and 150.35 ppm) as an internal reference and coupling constants (J) are given in hertz (Hz). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. High-resolution mass spectroscopy (HRMS) were recorded using a Waters XEVO-G2-XS-Q-TOF mass spectrometer.
Experimental Details
Unless mentioned, reactions were performed in an open atmosphere at room temperature in a 25 or 50 mL round-bottomed flask.
General Procedure for the Synthesis of Bis(3-arylimidazo[1,5-a]pyridin-1-yl)methanes (3a–3k)
To 3-arylimidazo[1,5-a]pyridines (0.28–0.52 mmol, 1 equiv) was added 1 mL of aqueous solution of formaldehyde (12.36 mmol). The mixture was stirred at room temperature overnight. After complete conversion (product monitored by TLC), the reaction was quenched with water. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 × 20 mL, monitored by TLC). The combined organic layers were dried with anhydrous sodium sulfate. After removal of the solvent in vacuo, the residue was directly subjected to silica gel column chromatography by using the EtOAc/n-hexane mixture as an eluent.
General Procedure for the Synthesis of 1,1′-(Ethane-1,1-diyl)bis(3-phenylimidazo[1,5-a]pyridine) (3l)
To 3-phenylimidazo[1,5-a]pyridine (0.52 mmol, 1 equiv) was added 1 mL of an aqueous solution of acetaldehyde (9.27 mmol). The mixture was stir at room temperature overnight. After complete conversion (product monitored by TLC), the reaction was quenched with water. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 × 20 mL, monitored by TLC). The combined organic layers were dried with anhydrous sodium sulfate. After removal of the solvent in vacuo, the residue was subjected to silica gel column chromatography by using an EtOAc/n-hexane mixture as an eluent.
General Procedure for the Synthesis of 1,1′-(Arylmethylene)bis(3-arylimidazo[1,5-a]pyridine) (3m–3w)
To the solution of 3-arylimidazo[1,5-a]pyridines (0.47–0.52 mmol, 1 equiv) in EtOH (0.5 mL, when required), aryl aldehydes (0.71–0.78 mmol, 1.5 equiv) were added. The mixture was stirred at room temperature overnight. After complete conversion (product monitored by TLC), solvent EtOH was first evaporated and then the reaction was quenched with water. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 × 20 mL, monitored by TLC). The combined organic layers were dried with anhydrous sodium sulfate. After removal of the solvent in vacuo, the residue was subjected to silica gel column chromatography by using a EtOAc/n-hexane mixture as an eluent.
General Procedure for the Synthesis of 4-Methyl-2-phenyl-5-((3-phenylimidazo[1,5-a]pyridin-1-yl)methyl)-2,4-dihydro-3H-pyrazol-3-one (3x)
To 3-phenylimidazo[1,5-a]pyridine (0.26 mmol, 1 equiv), 1 mL solution of formaldehyde (12.36 mmol) was added with 4-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one (0.26 mmol, 1 equiv). The mixture was stirred at room temperature overnight. After complete conversion (product monitored by TLC), the reaction was quenched with water. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 × 20 mL, monitored by TLC). The combined organic layers were dried with anhydrous sodium sulfate. After removal of the solvent in vacuo, the residue was subjected to silica gel column chromatography by using the EtOAc/n-hexane mixture as an eluent.
General Procedure for the Synthesis of 3-(3-Fluorophenyl)-1-((3-fluorophenyl)(3-(pyridin-2-yl)imidazo[1,5-a]pyridin-1-yl)methyl)imidazo[1,5-a]pyridine (3y)
To solution of 3-(pyridin-2-yl)imidazo[1,5-a]pyridine (0.26 mmol, 1 equiv) in EtOH (0.5 mL), 3-(3-fluorophenyl)imidazo[1,5-a]pyridine (0.26 mmol, 1 equiv), 3-fluorobenzaldehyde (0.39 mmol, 1.5 equiv) was added. The mixture was stirred at room temperature overnight. After complete conversion (product monitored by TLC), solvent EtOH was first evaporated and then the reaction was quenched with water. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 × 20 mL, monitored by TLC). The combined organic layers were dried with anhydrous sodium sulfate. After removal of the solvent in vacuo, the residue was subjected to silica gel column chromatography by using a EtOAc/n-hexane mixture as an eluent.
General Procedure for the Synthesis of N-(Methoxymethyl)-2-nitrobenzamide (5a)
To the solution of 2-nitrobenzamide (0.30 mmol, 1 equiv) in MeOH (1.0 mL), 1 mL of aqueous solution of HCHO (37% in water) was added with CuCl (10 mol %, 0.1 equiv) as a catalyst, Cs2CO3 (0.45 mmol, 1.5 equiv) as the base and compound 3a (5 mol %, 0.05 equiv) as the ligand. The mixture was stirred at reflux for10 h. After complete conversion (product monitored by TLC), the solvent was removed by using a rotary evaporator and then the reaction was quenched with water. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 × 20 mL, monitored by TLC). The combined organic layers were dried with anhydrous sodium sulfate. After removal of the solvent in vacuo, the residue was subjected to silica gel column chromatography by using a EtOAc/n-hexane mixture as an eluent.
General Procedure for the Synthesis of N-(Methoxy(aryl)methyl)-2-nitrobenzamide (5b, 5c)
To the solution of 2-nitrobenzamide (0.30 mmol, 1 equiv) in MeOH (1.0 mL), aryl aldehyde (0.30 mmol) was added with CuCl (10 mol %, 0.1 equiv) as a catalyst, Cs2CO3 (0.45 mmol, 1.5 equiv) as a base, and compound 3a (5 mol %, 0.05 equiv) as a ligand. The mixture was stirred at reflux for 8 h. After complete conversion (product monitored by TLC), the solvent was removed by using a rotary evaporator and then the reaction was quenched with water. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 × 20 mL, monitored by TLC). The combined organic layers were dried with anhydrous sodium sulfate. After removal of the solvent in vacuo, the residue was subjected to silica gel column chromatography by using a EtOAc/n-hexane mixture as an eluent.
General Procedure for the Synthesis of 2-(Aryl)-4,5-diphenyl-1H-imidazole (7a, 7b)
To the solution of benzil (0.24 mmol, 1 equiv) in ACN (1.0 mL) were added aryl aldehyde (0.24 mmol) and ammonium carbamate (0.48 mmol, 2 equiv) with Fe3O4 (10 mol %, 0.1 equiv) as a catalyst and compound 3a (10 mol %, 0.1 equiv) as a ligand. The mixture was stirred at reflux for 4 h. After complete conversion (product monitored by TLC), the solvent was removed by using a rotary evaporator and then the reaction was quenched with water. The phases were separated and the aqueous phase was extracted with ethyl acetate (2 × 20 mL, monitored by TLC). The combined organic layers were dried with anhydrous sodium sulfate. After removal of the solvent in vacuo, the residue was subjected to silica gel column chromatography by using a EtOAc/n-hexane mixture as an eluent.
Bis(3-phenylimidazo[1,5-a]pyridin-1-yl)methane (3a)
Compound 3a was obtained as a dark colored solid (89 mg, 86%), mp = 210–212 °C. 1H NMR (400 MHz, CDCl3, δ): 8.15 (d, J = 7.20 Hz, 2H), 7.79–7.76 (m, 4H), 7.54–7.47 (m, 6H), 7.39 (t, J = 7.44 Hz, 2H), 6.59–6.56 (m, 2H), 6.47–6.44 (m, 2H), 4.68 (s, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 136.5, 131.1, 130.4, 129.0, 128.5, 128.3, 128.0, 121.1, 119.1, 117.6, 113.1, 27.1 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C27H21N4, 401.1766; found, 401.1758.
Bis(3-(pyridin-2-yl)imidazo[1,5-a]pyridin-1-yl)methane (3b)
Compound 3b was obtained as a dark blue solid (86 mg, 83%), mp = 198–200 °C·1H NMR (400 MHz, pyridine-d5, δ): 9.99 (td, J = 7.28, 1.05 Hz, 2H), 8.64 (qd, J = 4.88, 0.90 Hz, 2H), 7.58 (td, J = 8.10, 0.98 Hz, 2H), 7.91–7.88 (m, 2H), 7.69–7.65 (m, 2H), 7.11–7.07 (m, 2H), 6.78–6.74 (m, 2H), 6.66–6.63 (m, 2H), 4.93 (s, 2H) ppm; 13C{1H} NMR (101 MHz, pyridine-d5, δ): 151.9, 148.8, 136.9, 134.1, 132.8, 130.5, 126.4, 122.1, 121.8, 119.8, 118.7, 114.1, 27.7 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C25H19N6, 403.1671; found, 403.1678.
Bis(3-(perfluorophenyl)imidazo[1,5-a]pyridin-1-yl)methane (3c)
Compound 3c was obtained as a white solid (81 mg, 79%), mp = 266–268 °C. 1H NMR (400 MHz, CDCl3, δ): 7.62 (d, J = 9.12 Hz, 2H), 7.56 (d, J = 7.16 Hz, 2H), 6.77–6.73 (m, 2H), 6.65–6.61 (m, 2H), 4.72 (s, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 146.5 (m), 144.0 (m), 143.2 (m), 140.7 (m), 139.4 (m), 136.9 (m), 132.3, 129.4, 121.7, 121.3, 119.0, 114.1, 105.9 (m), 27.7 ppm; 19F NMR (377 MHz, CDCl3): δ −137.3 to −137.4, −151.7 to −151.9, −160.6 to −160.7 (m) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C27H11N4F10, 581.0824; found, 581.0826.
Bis(3-(3,5-dimethylphenyl)imidazo[1,5-a]pyridin-1-yl)methane (3d)
Compound 3d was obtained as a dark colored semisolid (79 mg, 77%). 1H NMR (400 MHz, CDCl3, δ): 8.16 (d, J = 7.34 Hz, 2H), 7.44 (d, J = 9.11 Hz, 2H), 7.40 (s, 4H), 7.04 (s, 2H), 6.55–6.51 (m, 2H), 6.44 (t, J = 6.74 Hz, 2H), 4.68 (s, 2H), 2.39 (s, 12H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 138.6, 136.7, 131.0, 130.1, 130.4, 130.2, 128.2, 125.7, 121.4, 119.1, 117.3, 112.9, 27.6, 21.5 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C31H29N4, 457.2392; found, 457.2390.
Bis(3-(3,5-dibromophenyl)imidazo[1,5-a]pyridin-1-yl)methane (3e)
Compound 3e was obtained as a dark colored solid (76 mg, 75%), mp = 240–242 °C. 1H NMR (400 MHz, CDCl3, δ): 8.15 (d, J = 7.20 Hz, 2H), 7.88 (d, J = 1.63 Hz, 4H), 7.67 (t, J = 1.53 Hz, 2H), 6.56 (d, J = 9.11 Hz, 2H), 6.71–6.67 (m, 2H), 6.60 (t, J = 6.75 Hz, 2H), 4.62 (s, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 133.8, 133.6, 133.4, 131.9, 129.2, 129.1, 123.5, 120.9, 119.2, 118.6, 114.2, 27.2 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C27H17N4Br4, 712.8187; found, 712.8170.
Bis(3-(3-chlorophenyl)imidazo[1,5-a]pyridin-1-yl)methane (3f)
Compound 3f was obtained as a dark colored semi solid (79 mg, 77%). 1H NMR (400 MHz, CDCl3, δ): 8.15 (d, J = 7.24 Hz, 2H), 7.79 (t, J = 1.76 Hz, 2H), 7.67 (td, J = 7.64, 1.30 Hz, 2H), 7.54 (td, J = 9.16, 1.13 Hz, 2H), 7.42 (t, J = 7.82 Hz, 2H), 7.38–7.35 (m, 2H), 6.65–6.61 (m, 2H), 6.54–6.51 (m, 2H), 4.65 (s, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 134.9 (d, J = 1.61 Hz), 132.2, 131.5, 130.1, 128.7, 128.3, 127.8, 125.7, 120.9, 119.0, 118.0, 113.5, 27.2 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C27H19N4Cl2, 469.0987; found, 469.0978.
Bis(3-(3-fluorophenyl)imidazo[1,5-a]pyridin-1-yl)methane (3g)
Compound 3g was obtained as a pale white solid (76 mg, 74%), mp = 202–204 °C. 1H NMR (400 MHz, CDCl3, δ): 7.74 (dt, J = 7.44, 1.70 Hz, 2H), 7.71–7.68 (m, 2H), 7.53 (d, J = 9.15 Hz, 2H) 7.46–7.41 (m, 2H), 7.30 (dt, J = 7.54, 1.06 Hz, 2H), 7.24–7.19 (m, 2H), 6.65–6.61 (m, 2H), 6.53–6.49 (m, 2H), 4.73 (s, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 161.2, 158.7, 132.3 (d, J = 3.15 Hz), 131.6, 131.4, 130.6 (d, J = 8.21 Hz), 128.6, 124.8 (d, J = 3.34 Hz), 122.1 (d, J = 6.83 Hz), 118.7, 118.3 (d, J = 14.52 Hz), 117.8, 116.1 (d, J = 21.62 Hz), 112.8, 27.4 ppm; 19F NMR (377 MHz, CDCl3): δ −110.8 to −110.9 (m) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C27H19N4F2, 437.1578; found, 437.1574.
Bis(3-(3-nitrophenyl)imidazo[1,5-a]pyridin-1-yl)methane (3h)
Compound 3h was obtained as a dark orange solid (74 mg, 71%), mp = 226–228 °C. 1H NMR (400 MHz, DMSO-d6 and Acetone-d6, δ): 8.55 (s, 2H), 8.51 (d, J = 7.19 Hz, 2H), 8.25 (d, J = 7.79 Hz, 2H), 8.20 (dd, J = 8.12, 1.81 Hz, 2H), 7.79–7.74 (m, 4H), 6.86–6.82 (m, 2H), 6.75 (t, J = 6.53 Hz, 2H), 4.56 (s, 2H) ppm; 13C{1H} NMR (101 MHz, DMSO-d6 and Acetone-d6, δ): 148.2, 133.4, 132.8, 131.7, 131.5, 130.5, 128.7, 122.3, 121.8, 121.7, 119.0, 118.4, 114.1, 25.7 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C21H19N6O4, 491.1468; found, 491.1470.
Bis(3-(4-methoxyphenyl)imidazo[1,5-a]pyridin-1-yl)methane (3i)
Compound 3i was obtained as a dark colored semi solid (78 mg, 76%). 1H NMR (400 MHz, CDCl3): δ 8.08 (d, J = 7.28 Hz, 2H), 7.72–7.69 (m, 4H), 7.48 (d, J = 9.23 Hz, 2H), 7.05–7.01 (m, 4H), 6.56–6.52 (m, 2H), 6.45–6.42 (m, 2H), 4.66 (s, 2H), 3.87 (s, 6H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 159.9, 136.4, 130.4, 129.6, 127.9, 122.6, 121.1, 119.1, 117.5, 114.5, 113.1, 55.4, 26.8 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C29H25N4O2, 461.1978; found, 461.1975.
Bis(3-(2-fluorophenyl)imidazo[1,5-a]pyridin-1-yl)methane (3j)
Compound 3j was obtained as a green semi solid (72 mg, 70%). 1H NMR (400 MHz, CDCl3, δ): 8.17 (d, J = 7.25 Hz, 2H), 7.58–7.56 (m, 4H), 7.51–7.43 (m, 4H), 7.11–7.06 (m, 2H), 6.65–6.61 (m, 2H), 6.54–6.50 (m, 2H), 4.65 (s, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 164.3, 161.9, 135.2 (d, J = 2.83 Hz), 132.5 (d, J = 8.39 Hz), 131.5, 130.6 (d, J = 8.48 Hz), 128.7, 123.4 (d, J = 2.88 Hz), 121.0, 119.2, 118.0, 115.3 (d, J = 21.23 Hz), 114.9 (d, J = 22.83 Hz), 113.6, 27.2 ppm; 19F NMR (377 MHz, CDCl3): δ −111.9 to −112.0 (m) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C27H19N4F2, 437.1578; found, 437.1576.
Bis(3-(benzo[d][1,3]dioxol-5-yl)imidazo[1,5-a]pyridin-1-yl)methane (3k)
Compound 3k was obtained as a green solid (85 mg, 83%), mp = 193–195 °C. 1H NMR (400 MHz, CDCl3, δ): 8.06 (d, J = 7.24 Hz, 2H), 7.49 (d, J = 9.20 Hz, 2H), 7.23–7.20 (m, 4H), 6.91 (d, J = 8.39 Hz, 2H), 6.56–6.52 (m, 2H), 6.45–6.41 (m, 2H), 6.00 (s, 4H), 4.62 (s, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 148.2, 147.9, 136.1, 130.7, 128.0, 124.2, 121.9, 121.1, 119.1, 117.5, 113.1, 108.8, 108.7, 101.4, 27.0 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C29H21N4O4, 489.1563; found, 489.1552.
1,1′-(Ethane-1,1-diyl)bis(3-phenylimidazo[1,5-a]pyridine) (3l)
Compound 3l was obtained as a dark colored semisolid (78 mg, 73%). 1H NMR (400 MHz, CDCl3, δ): 8.13 (td, J = 7.24, 1.00 Hz, 2H), 7.80–7.78 (m, 4H), 7.56 (td, J = 9.22, 1.14 Hz, 2H), 7.52–7.48 (m, 4H), 7.42–7.37 (m, 2H), 6.55–6.51 (m, 2H), 6.45–6.41 (m, 2H), 5.01 (q, J = 7.35 Hz, 1H), 2.03 (d, J = 7.35 Hz, 3H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 136.3, 136.1, 130.6, 129.0, 128.4, 128.1, 127.4, 121.1, 119.4, 117.3, 113.0, 33.3, 20.7 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C28H23N4, 415.1923; found, 415.1920.
1,1′-(Phenylmethylene)bis(3-phenylimidazo[1,5-a]pyridine) (3m)
Compound 3m was obtained as a green solid (101 mg, 82%), mp = 130–132 °C. 1H NMR (400 MHz, CDCl3, δ): 8.15 (d, J = 7.24 Hz, 2H), 7.77 (d, J = 7.83 Hz, 4H), 7.54–7.46 (m, 8H), 7.40–7.36 (m, 2H), 7.31 (t, J = 7.59 Hz, 2H), 7.23–7.20 (m, 1H), 6.57–6.53 (m, 2H), 6.45–6.41 (m, 3H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 143.1, 136.6, 133.9, 130.5, 128.8 (d, J = 5.36 Hz), 128.5, 128.3, 128.1 (d, J = 6.34 Hz), 126.2, 121.1, 119.7, 117.8, 113.0, 44.8 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H25N4, 477.2079; found, 477.2072.
1,1′-((4-(Trifluoromethyl)phenyl)methylene)bis(3-phenylimidazo[1,5-a]pyridine) (3n)
Compound 3n was obtained as a green solid (98 mg, 70%), mp = 124–126 °C. 1H NMR (400 MHz, CDCl3, δ): 8.19 (d, J = 7.27 Hz, 2H), 7.78–7.76 (m, 4H), 7.61–7.56 (m, 4H), 7.53–7.48 (m, 6H), 7.43–7.39 (m, 2H), 6.63–6.59 (m, 2H), 6.52–6.48 (m, 2H), 6.37 (s, 1H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 147.3, 137.0, 133.0, 130.5, 129.1, 129.0, 128.7, 128.6, 128.0, 125.2 (q, J = 3.85 Hz), 121.3, 119.5, 118.2, 113.2, 44.7 ppm; 19F NMR (377 MHz, CDCl3): δ −62.2 (s) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H24N4F3, 545.1953; found, 545.1953.
4-(Bis(3-phenylimidazo[1,5-a]pyridin-1-yl)methyl)benzaldehyde (3o)
Compound 3o was obtained as a green solid (104 mg, 80%), mp = 142–144 °C. 1H NMR (400 MHz, CDCl3, δ): 9.96 (s, 1H), 8.19 (d, J = 7.23 Hz, 2H), 7.81–7.76 (m, 6H), 7.65 (d, J = 8.25 Hz, 2H), 7.55 (td, J = 9.23, 1.16 Hz, 2H), 7.49 (t, J = 7.60 Hz, 4H), 7.39 (t, J = 7.39 Hz, 2H), 6.62–6.58 (m, 2H), 6.49 (t, J = 6.78 Hz, 2H), 6.40 (s, 1H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 192.3, 150.5, 137.0, 134.7, 132.8, 130.4, 129.8, 129.5, 129.0, 128.7, 128.6, 128.2, 121.3, 119.4, 118.2, 113.2, 45.1 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H25N4O, 505.2028; found, 505.2028.
1,1′-(m-Tolylmethylene)bis(3-phenylimidazo[1,5-a]pyridine) (3p)
Compound 3p was obtained as a green semisolid (89 mg, 71%). 1H NMR (400 MHz, CDCl3): δ 8.16 (dd, J = 7.24, 0.95 Hz, 2H), 7.78–7.76 (m, 4H), 7.52–7.46 (m, 6H), 7.40–7.36 (m, 2H), 7.34–7.32 (m, 2H), 7.19 (t, J = 7.61 Hz, 1H), 7.02 (d, J = 7.56 Hz, 1H) 6.57–6.53 (m, 2H), 6.47–6.43 (m, 2H), 6.35 (s, 1H), 2.30 (s, 3H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 143.0, 137.7, 136.6, 134.0, 130.5, 129.6, 128.9, 128.5, 128.3, 128.2, 128.1, 127.1, 125.8, 121.1, 119.9, 117.7, 113.0, 44.9, 21.6 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H27N4, 491.2236; found, 491.2228.
1,1′-((3-Fluorophenyl)methylene)bis(3-(pyridin-2-yl)imidazo[1,5-a]pyridine) (3q)
Compound 3q was obtained as a green semisolid (93 mg, 73%). 1H NMR (400 MHz, CDCl3, δ): 9.91–9.89 (m, 2H), 8.61–8.59 (m, 2H), 8.30 (d, J = 8.15 Hz, 2H), 7.72 (dt, J = 7.80, 1.56 Hz, 2H), 7.61–7.59 (m, 2H), 7.23–7.19 (m, 1H), 7.18–7.11 (m, 4H), 6.91–6.87 (m, 1H), 6.77–6.73 (m, 2H), 6.70–6.66 (m, 2H), 6.36 (s, 1H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 151.3, 148.1, 136.4, 133.8 (d, J = 32.18 Hz), 130.2, 129.5 (d, J = 8.19 Hz), 125.9, 124.5, 122.1, 121.4, 119.5, 118.7, 115.8 (d, J = 21.35 Hz), 113.6, 113.2 (d, J = 21.28 Hz), 44.6 ppm; 19F NMR (377 MHz, CDCl3): δ −113.5 to −113.6 (m) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C31H22N6F, 497.1890; found, 497.1890.
1,1′-((3-Fluorophenyl)methylene)bis(3-(3-fluorophenyl)imidazo[1,5-a]pyridine) (3r)
Compound 3r was obtained as a green solid (94 mg, 75%), mp = 124–126 °C. 1H NMR (400 MHz, CDCl3, δ): 7.59–7.53 (m, 4H), 7.49 (t, J = 7.70 Hz, 1H), 7.28–7.23 (m, 2H), 7.19 (d, J = 9.19 Hz, 2H), 7.13–7.02 (m, 5H), 6.98–6.88 (m, 2H), 6.58 (s, 1H), 6.47–6.44 (m, 2H), 6.35 (t, J = 6.70 Hz, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 161.3, 158.8, 132.8, 132.7 (d, J = 3.31 Hz), 132.2, 131.0 (d, J = 3.81 Hz), 130.8 (d, J = 8.27 Hz), 129.8 (d, J = 14.08 Hz), 129.0, 128.2 (d, J = 8.12 Hz), 124.9 (d, J = 3.45 Hz), 124.0 (d, J = 3.53 Hz), 122.3 (d, J = 6.77 Hz), 118.9, 118.4 (2C), 118.3, 116.1 (d, J = 21.44 Hz), 115.3 (d, J = 22.02 Hz), 112.8, 37.9 (d, J = 3.49 Hz) ppm; 19F NMR (377 MHz, CDCl3): δ −110.7 to −110.8, 116.7–116.7 (m) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H22N4F3, 531.1797; found, 531.1799.
1,1′-((4-Fluorophenyl)methylene)bis(3-phenylimidazo[1,5-a]pyridine) (3s)
Compound 3s was obtained as a green semisolid (99 mg, 78%). 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 7.26 Hz, 2H), 7.78–7.76 (m, 4H), 7.50–7.44 (m, 8H), 7.42–7.38 (m, 2H), 6.99–6.94 (m, 2H), 6.59–6.55 (m, 2H), 6.50–6.46 (m, 2H), 6.38 (s, 1H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 162.2, 160.4, 148.4, 138.8, 136.7, 133.6, 130.4 (d, J = 7.85 Hz), 129.0, 128.6 (d, J = 3.46 Hz), 128.3, 121.2, 119.7, 118.1, 115.0 (d, J = 21.03 Hz), 113.2, 43.9 ppm; 19F NMR (377 MHz, CDCl3): δ −117.2 (s) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H22N4F, 495.1985; found, 495.1981.
1,1′-((4-Fluorophenyl)methylene)bis(3-(pyridin-2-yl)imidazo[1,5-a]pyridine) (3t)
Compound 3t was obtained as a green semisolid (97 mg, 76%). 1H NMR (400 MHz, CDCl3, δ): 9.90 (d, J = 7.22 Hz, 2H), 8.60 (dd, J = 4.81, 0.71 Hz, 2H), 8.30 (d, J = 8.12 Hz, 2H), 7.72 (dt, J = 7.79, 1.65 Hz, 2H), 7.56 (d, J = 8.90 Hz, 2H), 7.39–7.35 (m, 2H), 7.16–7.13 (m, 2H), 6.95 (t, J = 8.69 Hz, 2H), 6.76–6.72 (m, 2H), 6.69–6.66 (m, 2H), 6.36 (s, 1H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 162.8, 160.3, 151.2, 148.1, 138.6, 136.4, 133.9 (d, J = 18.46 Hz), 130.3, 130.2 (d, J = 12.50 Hz), 125.9, 122.1, 121.4, 119.4, 118.7, 114.9 (d, J = 20.99 Hz), 113.6, 44.1 ppm; 19F NMR (377 MHz, CDCl3): δ −117.3 (s) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C31H22N6F, 497.1890; found, 497.1886.
1,1′-((3-Fluorophenyl)methylene)bis(3-(2-fluorophenyl)imidazo[1,5-a]pyridine) (3u)
Compound 3u was obtained as a green solid (92 mg, 74%), mp = 137–139 °C. 1H NMR (400 MHz, CDCl3, δ): 8.16 (d, J = 7.33 Hz, 2H), 7.84 (d, J = 7.55 Hz, 2H), 7.72 (d, J = 9.19 Hz, 2H), 7.58–7.53 (m, 4H), 7.51–7.48 (m, 2H), 7.46–7.41 (m, 2H), 7.12–7.07 (m, 2H), 6.65–6.61 (m, 2H), 6.56–6.53 (m, 2H), 6.48 (s, 1H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 164.3 (d, J = 11.78 Hz), 163.8, 161.8 (d, J = 11.83 Hz), 161.4, 135.5, 133.1, 130.6 (d, J = 8.61 Hz), 130.1 (d, J = 7.58 Hz), 129.7 (d, J = 8.27 Hz), 129.0, 125.8, 124.6, 123.7 (d, J = 2.90 Hz), 121.1, 120.3 (d, J = 21.31 Hz), 119.7, 118.8, 117.0 (d, J = 23.32 Hz), 115.7 (t, J = 21.08 Hz), 115.3 (d, J = 23.02 Hz), 113.9, 113.4 (d, J = 21.31 Hz), 43.7 ppm; 19F NMR (377 MHz, CDCl3): δ −111.8 (s), −112.5 to −112.5 (m), −113.2 (s) ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H22N4F3, 531.1797; found, 531.1799.
1,1′-(Thiophen-3-ylmethylene)bis(3-phenylimidazo[1,5-a]pyridine) (3v)
Compound 3v was obtained as a green semisolid (91 mg, 73%). 1H NMR (400 MHz, CDCl3): δ 8.18 (dd, J = 7.22, 0.75 Hz, 2H), 7.80 (d, J = 7.43 Hz, 4H), 7.58 (d, J = 9.22 Hz, 2H), 7.53–7.49 (m, 4H), 7.43–7.39 (m, 2H), 7.29–7.25 (m, 2H), 7.19 (dd, J = 4.80, 1.30 Hz, 1H), 6.59 (t, J = 7.60 Hz, 2H), 6.50–6.47 (m, 2H), 6.38 (s, 1H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 143.8, 136.6, 133.9, 130.6, 129.0, 128.9, 128.4, 128.3, 128.2, 125.1, 122.0, 121.1, 119.7, 117.8, 113.0, 40.9 ppm; HRMS (ESI) m/z: calcd for C31H23N4S [M + H]+, 483.1643; found, 483.1644.
1,1′-((5-Nitrothiophen-3-yl)methylene)bis(3-phenylimidazo[1,5-a]pyridine) (3w)
Compound 3w was obtained as a green semisolid (93 mg, 68%). 1H NMR (400 MHz, CDCl3, δ): 8.20 (d, J = 7.24 Hz, 2H), 7.98–7.98 (m, 1H), 7.79–7.77 (m, 4H), 7.66 (d, J = 9.24 Hz, 2H), 7.54–7.50 (m, 5H), 7.45–7.43 (m, 2H), 6.67–6.63 (m, 2H), 6.54–6.50 (m, 2H), 6.26 (s, 1H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 144.2, 137.0, 132.1, 130.4, 130.3, 129.3, 129.1, 128.7, 128.4, 128.2, 121.4, 119.2, 118.6, 113.4, 40.8 ppm; HRMS (ESI) m/z: calcd for C31H22N5O2S [M + H]+, 528.1494; found, 528.1490.
4-Methyl-2-phenyl-5-((3-phenylimidazo[1,5-a]pyridin-1-yl)methyl)-2,4-dihydro-3H-pyrazol-3-one (3x)
Compound 3x was obtained as an orange semisolid (63 mg, 64%). 1H NMR (400 MHz, CDCl3, δ): 8.10 (d, J = 7.21 Hz, 1H), 7.76 (d, J = 7.71 Hz, 2H), 7.64 (d, J = 7.32 Hz, 2H), 7.43 (d, J = 8.35 Hz, 3H), 7.39 (d, J = 7.23 Hz, 1H), 7.31 (t, J = 7.96 Hz, 2H), 7.11 (t, J = 7.39 Hz, 1H), 6.63 (t, J = 7.70 Hz, 1H), 6.45 (t, J = 6.31 Hz, 1H), 3.93 (s, 2H), 2.27 (s, 3H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ = 174.8, 163.2, 137.9, 136.9, 129.8, 129.2, 128.9, 128.7 (2C), 127.9, 125.5, 124.9, 121.3, 119.0, 118.7, 118.1, 113.3, 64.5, 15.1 ppm; HRMS (ESI) m/z: calcd for C24H21N4O [M + H]+, 381.1715; found, 381.1723.
3-(3-Fluorophenyl)-1-((3-fluorophenyl)(3-(pyridin-2-yl)imidazo[1,5-a]pyridin-1-yl)methyl)imidazo[1,5-a]pyridine (3y)
Compound 3y was obtained as a green solid (94 mg, 71%), mp = 128–130 °C. 1H NMR (400 MHz, CDCl3): δ 9.88 (tt, J = 7.20, 1.06 Hz, 1H), 8.59 (qd, J = 4.88, 0.88 Hz, 1H), 8.30 (dd, J = 8.12, 0.93 Hz, 1H), 7.73–7.68 (m, 3H), 7.57 (dt, J = 7.77, 1.46 Hz, 1H), 7.44–7.40 (m, 3H), 7.28–7.24 (m, 1H), 7.22–7.17 (m, 2H), 7.15–7.12 (m, 1H), 7.10–7.01 (m, 2H), 6.73–6.65 (m, 3H), 6.64–6.59 (m, 1H), 6.51 (dt, J = 6.78, 1.20 Hz, 1H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 162.1, 161.2, 159.6, 158.8, 151.3, 148.1, 136.4, 134.0, 132.9 (d, J = 9.71 Hz), 132.7 (2C), 132.1, 130.8 (d, J = 3.89 Hz), 130.7, 130.1, 129.9 (d, J = 14.19 Hz), 129.0, 128.2 (d, J = 8.12 Hz), 125.8, 124.9 (d, J = 3.17 Hz), 123.9 (d, J = 3.21 Hz), 122.3, 122.3 (d, J = 3.93 Hz), 121.3, 119.5, 119.1, 118.3 (d, J = 6.39 Hz), 116.1 (d, J = 21.49 Hz), 115.3 (d, J = 22.07 Hz), 113.5, 112.8, 38.0 (d, J = 3.07 Hz) ppm; 19F NMR (377 MHz, CDCl3): δ −110.7 to −110.8, −116.3 to −116.3 (m) ppm; HRMS (ESI) m/z: calcd for C32H22N5F2 [M + H]+, 514.1843; found, 514.1842.
Bis(3-phenylimidazo[1,5-a]pyridin-1-yl)methane-d2(3z)
Compound 3z was obtained as a dark colored semi solid (60 mg, 58%); 1H NMR (400 MHz, CDCl3, δ): 8.17 (d, J = 7.40 Hz, 2H), 7.79 (d, J = 7.17 Hz, 4H), 7.55–7.49 (m, 6H), 7.42 (d, J = 7.40 Hz, 2H), 6.61–6.57 (m, 2H), 6.49–6.46 (m, 2H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 137.1 (t, J = 25.0 Hz), 131.5, 129.4 (t, J = 15.18 Hz), 128.8 (d, J = 2.92 Hz), 128.7, 128.4 (d, J = 25.83 Hz), 127.9, 121.1 (d, J = 13.16 Hz), 119.0 (d, J = 28.03 Hz), 118.0 (d, J = 11.44 Hz), 113.4 (d, J = 5.69 Hz) ppm.
N-(Methoxymethyl)-2-nitrobenzamide (5a)
Compound 5a was obtained as a white solid (53 mg, 84%), mp = 170–172 °C. 1H NMR (400 MHz, CDCl3, δ): 8.08 (d, J = 8.01 Hz, 1H), 7.70 (t, J = 7.34 Hz, 1H), 7.61 (t, J = 7.75 Hz, 1H), 7.55 (d, J = 7.58 Hz, 1H), 6.65 (s, 1H), 4.88 (d, J = 6.69 Hz, 2H), 3.50 (s, 3H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 167.3, 146.5, 133.9, 132.7, 130.9, 128.6, 124.8, 71.9, 56.7 ppm; HRMS (ESI) m/z: calcd for C9H10N2O4Na [M + Na]+, 233.0538; found, 233.0531.
N-(Methoxy(phenyl)methyl)-2-nitrobenzamide (5b)
Compound 5b was obtained as a white solid (79 mg, 92%), mp = 84–86 °C. 1H NMR (400 MHz, CDCl3 and DMSO-d6, δ): 8.44 (s, 1H), 7.14 (s, 1H), 6.83 (s, 1H), 6.74–6.65 (m, 4H), 6.49 (s, 3H), 5.37 (d, J = 8.75 Hz, 1H), 2.70 (s, 3H) ppm; 13C{1H} NMR (101 MHz, CDCl3 and DMSO-d6, δ): 165.7, 145.4, 1381, 132.3, 131.5, 129.2, 127.9, 126.9 (2C), 125.0, 122.8, 80.2, 54.5 ppm; HRMS (ESI) m/z: calcd for C15H14N2O4 [M + Na]+, 309.0851; found, 309.0844.
N-((4-Chlorophenyl)(methoxy)methyl)-2-nitrobenzamide (5c)
Compound 5c was obtained as a white solid (84 mg, 88%), mp = 90–92 °C. 1H NMR (400 MHz, CDCl3, δ): 8.07 (dd, J = 8.17, 0.91 Hz, 1H), 7.67 (dt, J = 7.46, 1.22 Hz, 1H), 7.59 (dt, J = 7.86, 1.39 Hz, 1H), 7.50 (dd, J = 7.50, 1.33 Hz, 1H), 7.46 (d, J = 8.43 Hz, 2H), 7.35–7.33 (m, 2H), 6.39 (s, 1H), 6.30 (d, J = 9.41 Hz, 1H), 3.65 (s, 3H) ppm; 13C{1H} NMR (101 MHz, CDCl3): δ 166.8, 146.4, 137.5, 134.6, 133.9, 132.5, 130.9, 128.9, 128.5, 127.4, 124.8, 81.2, 56.7 ppm; HRMS (ESI) m/z: calcd for C15H13N2O4NaCl [M + Na]+, 343.0462; found, 343.0448.
2,4,5-Triphenyl-1H-imidazole (7a)
Compound 7a was obtained as a white solid (60 mg, 85%), mp = 276–278 °C. 1H NMR (400 MHz, pyridine-d5, δ): 8.49 (s, 2H), 7.89 (s, 3H), 7.49 (s, 3H), 7.37–7.31 (m, 8H) ppm; 13C{1H} NMR (101 MHz, pyridine-d5, δ): 147.5, 132.2, 129.5, 129.1, 128.9, 127.8, 126.4 ppm; HRMS (ESI) m/z: calcd. for C21H17N2 [M + H]+, 297.1392; found, 297.1379.
2-(4-Chlorophenyl)-4,5-diphenyl-1H-imidazole (7b)
Compound 7b was obtained as a white solid (63 mg, 80%), mp = 260–262 °C. 1H NMR (400 MHz, pyridine-d5, δ): 8.39 (d, J = 8.50 Hz, 3H), 7.89 (s, 1H), 7.51 (d, J = 8.50 Hz, 3H), 7.40–7.36 (m, 5H), 7.33–7.30 (m, 3H) ppm; 13C{1H} NMR (101 MHz, pyridine-d5, δ): 146.3, 134.3, 130.8, 129.6, 129.1, 128.9, 127.8 ppm; HRMS (ESI) m/z: calcd for C21H16N2Cl [M + H]+, 331.1001; found, 331.1002.
Acknowledgments
This research was generously supported by CSIR HCP-23 project. S.M. thanks CSIR for SRF fellowship and AcSIR for Ph.D support.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c03823.
Experimental procedures, characterization, and copies of 1H and 13C NMR spectra for all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Gensch T.; Hopkinson M. N.; Glorius F.; Wencel-Delord J. Mild metal-catalyzed C–H activation: examples and concepts. Chem. Soc. Rev. 2016, 45, 2900–2936. 10.1039/C6CS00075D. [DOI] [PubMed] [Google Scholar]; b Altus K. M.; Love J. A. The continuum of carbon–hydrogen (C–H) activation mechanisms and terminology. Commun. Chem. 2021, 4, 173. 10.1038/s42004-021-00611-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dalton T.; Faber T.; Glorius F. C–H activation: toward sustainability and applications. ACS Cent. Sci. 2021, 7, 245–261. 10.1021/acscentsci.0c01413. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Roy S.; Panja S.; Sahoo S. R.; Chatterjee S.; Maiti D. Enroute sustainability: metal free C–H bond functionalisation. Chem. Soc. Rev. 2023, 52, 2391–2479. 10.1039/D0CS01466D. [DOI] [PubMed] [Google Scholar]
- a Renno G.; Cardano F.; Volpi G.; Barolo C.; Viscardi G.; Fin A. Imidazo [1,5-a] pyridine-Based Fluorescent Probes: A Photophysical Investigation in Liposome Models. Molecules 2022, 27, 3856. 10.3390/molecules27123856. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Volpi G.; Rabezzana R. Imidazo [1, 5-a] pyridine derivatives: useful, luminescent andversatile scaffolds for different applications. New J. Chem. 2021, 45, 5737–5743. 10.1039/D1NJ00322D. [DOI] [Google Scholar]; c Colombo G.; Ardizzoia G. A.; Brenna S. Imidazo[1,5-a]pyridine-based derivatives as highly fluorescent dyes. Inorg. Chim. Acta 2022, 535, 12849. 10.1016/j.ica.2022.120849. [DOI] [Google Scholar]
- a Bagdi A. K.; Santra S.; Monir K.; Hajra A. Synthesis of imidazo[1,2-a]pyridines: a decade update. Chem. Commun. 2015, 51, 1555–1575. 10.1039/C4CC08495K. [DOI] [PubMed] [Google Scholar]; b Ramana Reddy M.; Darapaneni C. M.; Patil R. D.; Kumari H. Recent synthetic methodologies for imidazo [1, 5-a] pyridines and related heterocycles. Org. Biomol. Chem. 2022, 20, 3440–3468. 10.1039/d2ob00386d. [DOI] [PubMed] [Google Scholar]
- a Al-Jumaili M. H.; Hamad A. A.; Hashem H. E.; Hussein A. D.; Muhaidi M. J.; Ahmed M. A.; Albanaa A. H.; Siddique F.; Bakr E. A. Comprehensive review on the Bis-heterocyclic compounds and their anticancer efficacy. J. Mol. Struct. 2023, 1271, 133970. 10.1016/j.molstruc.2022.133970. [DOI] [Google Scholar]; b Kaveti B.; Rentería- Gómez A. ´.; Unnamatla M. V.; Gámez-Montaño R.. Synthesis of bis-heterocycles type spacer containing 1, 5-disubstituted-1H-tetrazoles. In 21st International Electronic Conference on Synthetic Organic Chemistry, 2017; Vol. 1, pp 1–6.; c Soural M.; Bouillon I.; Krchnak V. Combinatorial libraries of bis-heterocyclic compounds with skeletal diversity. J. Comb. Chem. 2008, 10, 923–933. 10.1021/cc8001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bag D.; Sawant S. D. Gold(I)-Catalyzed Cycloisomerization–Indole Addition Cascade: Synthesis of 3 (2 H)-Furanone-IncorporatedUnsymmetrical 3,3′-Bis (indolyl) methanes. Org. Lett. 2022, 24, 4930–4934. 10.1021/acs.orglett.2c01845. [DOI] [PubMed] [Google Scholar]; b Kour J.; Khajuria P.; Verma P. K.; Kapoor N.; Kumar A.; Sawant S. D. Selective Synthesis of Bis-Heterocycles via Mono-and Di-Selenylation of Pyrazoles and Other Heteroarenes. ACS Omega 2022, 7 (15), 13000–13009. 10.1021/acsomega.2c00323. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kour J.; Venkateswarlu V.; Verma P. K.; Hussain Y.; Dubey G.; Bharatam P. V.; Sahoo S. C.; Sawant S. D. Oxone-DMSO triggered methylene insertion and C (sp2)–C(sp3)-H–C(sp2) bond formation to access functional bis-heterocycles. J. Org. Chem. 2020, 85 (7), 4951–4962. 10.1021/acs.joc.9b03477. [DOI] [PubMed] [Google Scholar]
- a Parameswaran P.; Majik M.; Praveen P. Bis (indolyl) methane alkaloids: isolation, bioactivity, and syntheses. Synthesis 2015, 47, 1827–1837. 10.1055/s-0034-1380415. [DOI] [Google Scholar]; b Liu X.; Ma S.; Toy P. H. Halogen bond-catalyzed Friedel–Crafts reactions of aldehydes and ketones using a bidentate halogen bond donor catalyst: synthesis of symmetrical bis (indolyl) methanes. Org. Lett. 2019, 21, 9212–9216. 10.1021/acs.orglett.9b03578. [DOI] [PubMed] [Google Scholar]; c Bandgar B. P.; Shaikh K. A. Molecular iodine-catalyzed efficient and highly rapid synthesis of bis(indolyl)methanes under mild conditions. Tetrahedron Lett. 2003, 44, 1959–1961. 10.1016/S0040-4039(03)00032-7. [DOI] [Google Scholar]; d Chen D.; Yu L.; Wang P. G. Lewis acid-catalyzed reactions in protic media. Lanthanide-catalyzed reactions of indoles with aldehydes or ketones. Tetrahedron Lett. 1996, 37, 4467–4470. 10.1016/0040-4039(96)00958-6. [DOI] [Google Scholar]
- a Phuc B. V.; Nguyen N. T.; Van N. T. H.; Nguyen T. L.; Nguyen V. H.; Tran C. M.; Nguyen H.; Nguyen M. T.; Hung T. Q.; Dang T. T. Facile iodine-promoted synthesis of bis(1-imidazo[1,5-a]pyridyl)arylmethanes and exploration of applications. Chem. Commun. 2023, 59, 1947–1950. 10.1039/D2CC05419A. [DOI] [PubMed] [Google Scholar]; b Murai T.; Nagaya E.; Shibahara F.; Maruyama T. Imidazo[1,5-a] pyridine-1-ylalkylalcohols: synthesis via intramolecular cyclization of N-thioacyl 1, 2-aminoalcohols and their silyl ethers and molecular structures. Org. Biomol. Chem. 2012, 10, 4943. 10.1039/c2ob25438g. [DOI] [PubMed] [Google Scholar]; c Tahara S.; Shibahara F.; Maruyama T.; Murai T. Iodine-mediated cyclization of N-thioacyl-1-(2-pyridyl)-1, 2-aminoalcohols and their subsequent condensation leading to the formation of novel bis(1-imidazo[1,5-a]pyridyl)arylmethanes. Chem. Commun. 2009, 7009–7011. 10.1039/b910172a. [DOI] [PubMed] [Google Scholar]
- a Liu P.; Shen Z.; Yuan Y.; Sun P. Synthesis of symmetrical methylene-bridged imidazoheterocycles using DMSO as methylene source under metal-free conditions. Org. Biomol. Chem. 2016, 14, 6523–6530. 10.1039/C6OB00977H. [DOI] [PubMed] [Google Scholar]; b Mondal S.; Samanta S.; Santra S.; Bagdi A. K.; Hajra A. N. N-Dimethylformamide as a Methylenating Reagent: Synthesis of Heterodiarylmethanes via Copper-Catalyzed Coupling between Imidazo[1,2-a]pyridines and Indoles/N, N-Dimethylaniline. Adv. Synth. Catal. 2016, 358, 3633. 10.1002/adsc.201600674. [DOI] [Google Scholar]; c Modi A.; Ali W.; Patel B. K. N. N-Dimethylacetamide (DMA) as a Methylene Synthon for Regioselective Linkage of Imidazo[1,2-a]pyridine. Adv. Synth. Catal. 2016, 358, 2100–2107. 10.1002/adsc.201600067. [DOI] [Google Scholar]; d Kaswan P.; Nandwana N. K.; DeBoef B.; Kumar A. Vanadyl Acetylacetonate Catalyzed Methylenation of Imidazo[1,2-a]pyridines by Using Dimethylacetamide as a Methylene Source: Direct Access to Bis(imidazo[1,2-a]pyridin-3-yl)methanes. Adv. Synth. Catal. 2016, 358, 2108–2115. 10.1002/adsc.201600225. [DOI] [Google Scholar]; e Kumar R.; Rawat D.; Adimurthy S. Polyethylene Glycol (PEG-400) as Methylene Spacer and Green Solvent for the Synthesis of Heterodiarylmethanes under Metal-Free Conditions. Eur. J. Org Chem. 2020, 2020, 3499–3507. 10.1002/ejoc.202000467. [DOI] [Google Scholar]
- Mahajan S.; Sawant S. D. Iodine/TBHP-Mediated One-Pot Multicomponent Protocol for Tandem C–N and C–S Bond Formation To Access Sulfenylimidazo[1,5-a]pyridines via C–H Functionalization. J. Org. Chem. 2022, 87, 11387–11398. 10.1021/acs.joc.2c00890. [DOI] [PubMed] [Google Scholar]
- a Shibahara F.; Shibata Y.; Murai T. Imidazo[1,5-a] pyridinylidenes as π-Accepting NHC Ligands in Catalysis. Chem. Lett. 2021, 50, 1892–1900. 10.1246/cl.210461. [DOI] [Google Scholar]; b Volpi G. Luminescent Imidazo[1,5-a]pyridine Scaffold: Synthetic Heterocyclization Strategies-Overview and Promising Applications. Asian J. Org. Chem. 2022, 11, e202200171 10.1002/ajoc.202200171. [DOI] [Google Scholar]; c Colombo G.; Cinco A.; Ardizzoia G. A.; Brenna S. Long-alkyl chain functionalized imidazo[1,5-a]pyridine derivatives as blue emissive dyes. Colorants 2023, 2, 179–193. 10.3390/colorants2020012. [DOI] [Google Scholar]; d Weber M. D.; Garino C.; Volpi G.; Casamassa E.; Milanesio M.; Barolo C.; Costa R. D. Origin of a counterintuitive yellow light-emitting electrochemical cell based on a blue-emitting heteroleptic copper (I) complex. Dalton Trans. 2016, 45, 8984–8993. 10.1039/C6DT00970K. [DOI] [PubMed] [Google Scholar]; e Priyanga S.; Khamrang T.; Velusamy M.; Karthi S.; Ashokkumar B.; Mayilmurugan R. Coordination geometry-induced optical imaging of L-cysteine in cancer cells using imidazopyridine-based copper (II) complexes. Dalton Trans. 2019, 48, 1489–1503. 10.1039/C8DT04634D. [DOI] [PubMed] [Google Scholar]
- a Halli J.; Hofman K.; Beisel T.; Manolikakes G. Synthesis of N-Acyl-N, O-acetals from Aldehydes, Amides and Alcohols. Eur. J. Org Chem. 2015, 2015, 4624–4627. 10.1002/ejoc.201500655. [DOI] [Google Scholar]; b Tan B. Y. H.; Teo Y. C. Efficient cobalt-catalyzed C–N cross-coupling reaction between benzamide and aryl iodide in water. Org. Biomol. Chem. 2014, 12, 7478–7481. 10.1039/C4OB01483A. [DOI] [PubMed] [Google Scholar]; c Panda N.; Mothkuri R.; Nayak D. K. Copper-Catalyzed Regioselective Synthesis of N-Aryl Amides from Aldoximes and Aryl Halides. Eur. J. Org Chem. 2014, 2014, 1602–1605. 10.1002/ejoc.201301868. [DOI] [Google Scholar]
- a Sohail M.; Bilal M.; Maqbool T.; Rasool N.; Ammar M.; Mahmood S.; Malik A.; Zubair M.; Abbas Ashraf G. Iron-catalyzed synthesis of N-heterocycles via intermolecular and intramolecular cyclization reactions: A review. Arab. J. Chem. 2022, 15, 104095. 10.1016/j.arabjc.2022.104095. [DOI] [Google Scholar]; b Hosseini S.; Kiasat A. R.; Farhadi A. Fe3O4@ SiO2/Bipyridinium Nanocomposite as a Magnetic and Recyclable Heterogeneous Catalyst for the Synthesis of Highly Substituted Imidazoles Via Multi-Component Condensation Strategy. Polycyclic Aromat. Compd. 2021, 41, 762–771. 10.1080/10406638.2019.1616306. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information