

Abstract
RNA-based agents (siRNA, miRNA, and mRNA) can selectively manipulate gene expression/proteins and are set to revolutionize a variety of disease treatments. Nanoparticle (NP) platforms have been developed to deliver functional mRNA or siRNA inside cells to overcome their inherent limitations. Recent studies have focused on siRNA to knock down proteins causing drug resistance or mRNA technology to introduce tumor suppressors. However, cancer needs multitargeted approaches to selectively manipulate multiple gene expressions/proteins. In this proof-of-concept study, we developed NPs containing Luc-mRNA and siRNA-GFP as model agents ((M+S)-NPs) and showed that NPs can simultaneously deliver functional mRNA and siRNA and impact the expression of two genes/proteins in vitro. Additionally, after in vivo administration, (M+S)-NPs successfully knocked down GFP while introducing luciferase into a TNBC mouse model, indicating that our NPs have the potential to develop RNA-based anticancer therapeutics. These studies pave the way to develop RNA-based, multitargeted approaches for complex diseases like cancer.
Keywords: nanoparticles, siRNA, mRNA, codelivery, gene, protein, restoration and knockdown
Introduction
RNA-based therapies have the capacity to selectively manipulate gene expressions and hold the potential to revolutionize current therapeutic strategies for various diseases, including cancer.1−3 RNA-based agents such as siRNA, miRNA, and mRNA can downregulate, augment, or correct specific gene products that are otherwise undruggable with small molecules.4,5 mRNA agents, typically over 2000 nucleotides long, include conventional mRNA, self-amplifying mRNA (saRNA), trans-amplifying mRNA (taRNA), and circular mRNA (circRNA).6 These mRNA agents can be designed to carry optimized genetic information and be translated for the production of encoded proteins without integrating into the host genome.1,6−8 mRNA technology offers greater flexibility for targeting various diseases, including cancer, cardiovascular diseases, as well as vaccines.6,8,9 In cancer treatment, mRNA applications are multifaceted. They can encode tumor suppressors to inhibit cancer cell proliferation, tumor antigens to trigger immune responses, and chimeric antigen receptors (CARs) or T cell receptors (TCRs) for T cell therapies.6,10 While mRNA therapies can produce specific proteins in the target cells, RNA interference (RNAi), a natural defense mechanism against exogenous genes, can specifically knock down target gene expression.11,12 RNAi agents, such as siRNA and miRNA, modulate target genes by mediating targeted mRNA degradation (siRNA and miRNA) or mRNA translation repression (miRNA). Both siRNA and miRNA-based RNAi therapies have shown significant potential in cancer therapies, specifically in knocking down/modulating the expression of genes/proteins involved in drug resistance and cancer stem cell (CSC) enrichment, inducing cell death, and sensitizing cancer cells to chemotherapeutic drugs.13,14
Despite their potential, mRNA and RNAi therapies face several challenges in clinical applications.3 RNA agents, such as mRNA, siRNA, and miRNA, possess undesirable physicochemical and pharmacological properties, are susceptible to degradation and unwanted immune reactions, and must be effectively delivered into the target cells.2,3,14 Therefore, sophisticated delivery platforms are essential for developing safe and effective nucleic acid-based therapies.15 Nanotherapeutic strategies offer several advantages over conventional therapies. Nanoparticle-based drug delivery platforms can encapsulate various therapeutic agents irrespective of their physicochemical properties, protect them from degradation, and deliver them into the cells.16 However, delivering RNA agents is more challenging than traditional therapeutic agents due to the large size of mRNA (over 2000 nucleotides) and the phosphate backbone common to all RNA agents, which complicates their encapsulation in NPs.7,11,15,16 Additionally, NPs must escape the endosome and release functional RNA agents into the cytosol to be effective. To overcome these challenges, advanced NP platforms will be required.2,11 In this context, recent lipid NPs have shown success in delivering either mRNA or siRNA, with examples including ONPATTRO (patisiran), an siRNA-containing NP currently in the clinic for treating hereditary amyloidogenic transthyretin (TTR) amyloidosis, and the Moderna Spikevax and Pfizer-BioNTech COVID-19 vaccines, which are NPs containing mRNA encoding the SARS-CoV-2 spike glycoprotein.8,17 Despite these successes, strategies targeting a single protein or cytokine are insufficient for complex diseases like cancer and cardiovascular diseases, the leading causes of mortality worldwide.18
Triple-negative breast cancer (TNBC) is a highly heterogeneous disease that accounts for a disproportionate number of breast cancer-related deaths.19 Effective treatment strategies must address significant challenges related to drug resistance, cancer stem cell enrichment, and tumorigenesis, which represent critical unmet medical needs.20,21 Multiple genes and proteins are implicated in tumor progression and metastasis, including mutations in tumor suppressor genes.22,23 We recently showed that TNBC stem cells exist in two phenotypically distinct, yet interconvertible, epithelial-like and mesenchymal-like populations, regulated by Wnt and YAP signaling pathways.24 We demonstrated that simultaneous inhibition of these pathways is essential for developing effective therapy.20,23,24 Additionally, using clinically relevant TNBC patient-derived xenograft (PDX) models, we also showed that nanotherapies targeting multiple signaling pathways involved in CSC enhancement, in combination with chemotherapy, can arrest PDX tumor growth, suppress CSCs, and diminish tumor regeneration.20,25,26 In complex diseases like TNBC, CSCs and tumor cells can modulate tumor microenvironments and phenotypical states or enter dormancy to develop resistance to single-agent therapies. Simultaneous targeting of multiple pathways can eliminate both bulk tumor cells and CSCs, ultimately leading to better patient outcomes. In this context, restoring tumor suppressor genes via mRNA to control tumor growth while silencing genes/proteins involved in drug resistance and tumorigenesis using RNAi presents a potent therapeutic strategy for TNBCs.
To date, a variety of NP systems have been developed to encapsulate and deliver two or more drugs with diverse physicochemical properties, including hydrophobic, hydrophilic, small molecules, proteins/peptides, and RNAi agents.14,20,27,28 These systems are designed to provide different release kinetics, such as spontaneous, controlled, stimuli-responsive, or sequential release.29,30 However, the codelivery NPs containing both mRNA and siRNA to simultaneously restore and knock down the expression of two different genes have not yet been reported in the literature. Here, we developed a codelivery 2-in-1 NP system containing both mRNA and siRNA to simultaneously restore and knock down the expressions of two distinct genes. Compared to delivering two agents separately as single-agent NPs or in separate vehicles, combining them in a single 2-in-1 NP system ensures synchronized pharmacological action at both tissue and cellular levels.14,18,25 Since the biophysicochemical properties of NPs critically influence their nanobio interactions, endosomal escape, and intracellular delivery, encapsulating both agents in a single NP (2-in-1) platform maximizes their codelivery within the tumor microenvironments. This approach ensures that both agents are effectively delivered into the cells and remain functional within the cellular context. As a proof-of-concept, we used Luc-mRNA and siRNA against GFP to develop our NPs, studying their effects in vitro and in vivo using GFP+ cells as a model. Our results demonstrated that NPs successfully encapsulated both mRNA and siRNA, protected them from degradation, delivered functional agents into the cells, and simultaneously and effectively knocked down and restored the target gene expression in both in vitro and in vivo settings.
Results
Development and Characterization of Codelivery NPs
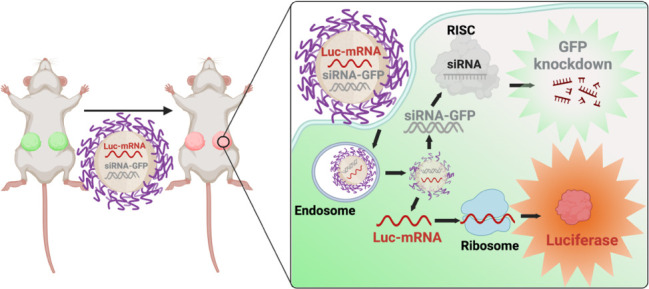
For this proof-of-concept study, we first developed single and dual RNA agents containing NPs via a self-assembling process using a PLGA10K–PEG5K polymer. PLGA–PEG polymeric NPs were chosen due to their biodegradability, biocompatibility, stability, scalability, and versatility in various drug delivery applications.31 To address the hydrophilic nature of RNA’s phosphate backbone and facilitate encapsulation within the NPs, we used PEI-C14 as a cationic lipid (Figures S1 and S2). Both single and dual-drug NPs were synthesized using the nanoprecipitation method by blending siRNA and/or mRNA with PEI-C14, followed by the addition of PLGA10K–PEG5K, and dropwise addition to nuclease-free water.20 The resulting NPs were Luc-mRNA containing (M)-NPs, siRNA-GFP containing (S)-NPs, and Luc-mRNA + siRNA-GFP containing (M+S)-NPs (Figure 1). NPs were instantly formed with PEI-C14:siRNA and PEI-C14:mRNA complexes embedded in the PLGA hydrophobic core stabilized by a PEG shell.32
Figure 1.

Schematic diagram of NP development (upper panel) and their in vitro and in vivo evaluation (lower panel).
After synthesis, NPs were collected and purified using centrifugal filters and characterized for their physicochemical properties. Hydrodynamic size and surface charge were measured by dynamic light scattering (DLS). All three NPs have uniform sizes ranging between 50 and 60 nm, with low PDI of 0.15–0.28 (Figure S4) and surface charges between −0.2 and −20 mV (Figure 2A,B,E). TEM characterization of NPs showed all three NP formulations have a spherical shape, and the actual sizes are 40–50 nm, which are lower than DLS sizes as expected (Figure 2C). The % of encapsulation efficiency (EE%) of siRNA, measured using Cy5-labeled siRNA, was approximately 92% and 85% for single and dual drug NPs, respectively. EE of Luc-mRNA, determined by RiboGreen assay, was 100% and 91% for single and dual drug NPs, respectively.33 We then studied the serum stability of our NPs by incubating them for 6 h at different serum concentrations (up to 10%). Results indicated that NPs remained stable in the presence of serum proteins, as there were no significant changes in NP sizes before and after incubation (Figure 2D).34
Figure 2.
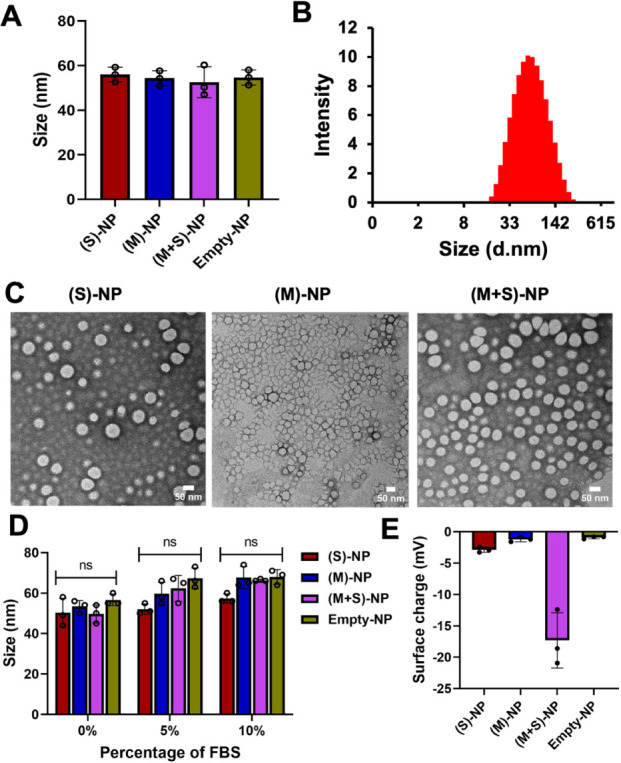
Physicochemical characterization of nanoparticles. (A) Size of single- and dual-drug NPs, along with control-NPs, measured by diluting 20 μL of NPs in 1 mL of sterile water using dynamic light scattering, n = 3. (B) Size distribution intensity of (M+S)-NPs, measured by DLS. (C) Size and morphology of single and dual-drug NPs by transmission electron microscopy (scale bar: 50 nm). (D) Stability of all the NPs in different percentages of FBS; NPs incubated for 6 h and size was measured; n = 3 (ns: no significant difference). (E) Surface of single and dual-drug NPs, along with control-NPs, measured by diluting 20 μL of NPs in 1 mL using dynamic light scattering n = 3.
Co-Delivery NPs Capable of Protecting and Delivering Both Functional mRNA and siRNA In vitro
To evaluate our NP’s capacity to escape endosomes and deliver functional mRNA and/or siRNA, we performed a flow cytometry study. For this purpose, we developed NPs loaded with Cy5-labeled siRNA to mimic siRNA-GFP and EGFP mRNA as model mRNA (Cy5-siRNA+EGFP mRNA)-NPs). The codelivery NPs’ size, surface charge, and stability are similar to those of (M+S)-NPs (S3A–D). HT1080 cells were treated with (Cy5-siRNA+EGFP-mRNA)-NPs and control NPs for 48 h, and the NP delivery capacity was analyzed by flow cytometry. The Cy5 label could track siRNA in NPs, while GFP expression from the cells revealed functional mRNA delivery.35,36
After 48 h of treatment, we observed a significant percentage of cells associated with Cy5 (100%) (Figures 3B,C and S5–S7) and GFP (25%) (Figures 3D–F and S5–S7) signals, demonstrating that NPs can deliver agents intracellularly and are functional. Despite some Cy5 signal bleeding into the GFP channel, there was a 3-fold increase in the GFP signal in the cells. These results indicate that the NPs successfully entered the cells and delivered both mRNA and siRNA.
Figure 3.

Cellular interactions of codelivery NPs. (A) Model representation of NP treatment to HT1080 cells; Cy5 cellular entry and expression of EGFP. (B,E) Representative flow cytometry histogram showing Cy5 and GFP expression in HT1080 cells 48 h post-treatment with Cy5-siRNA+EGFP-mRNA-NPs (1 nmol + 0.016 nmol) or control-NPs. (C,D) Percentage of Cy5 and GFP in HT1080 cells (control: nontreated) quantified by flow cytometry 48 h post-treatment with Cy5-siRNA+EGFP-mRNA-NPs (1 nmol + 0.016 nmol) or control-NPs (10 μM); n = 3 (data represent means ± SD, ****p < 0.0001). (F) Fluorescence microscopy images of HT1080 cells after 48 h of treatment with dual-drug NPs or empty NPs (scale bar: 100 μm).
Next, we explored whether delivered mRNA and siRNA were functional and simultaneously processed by the cellular machinery to perform their intended functions. To achieve this, we developed monoclonal cell lines of MDA-MB-231 and HT1080 that stably express GFP, termed MDA-MB-231-GFP+ and HT1080-GFP+ throughout the manuscript. These cell lines were created by retroviral transduction and subsequent ring cloning. Before we explored the transfection capacities of dual agent (M+S)-NPs, we studied single agent (M)-NPs and (S)-NPs. To this end, HT1080 and MDA-MB-231 cells were treated with (M)-NPs, and their corresponding GFP+ cell lines were treated with (S)-NPs for 48 h, and luminescence (luciferase RLU) and flow cytometry assays were performed, respectively. Our results showed that, when compared to empty NPs, single agent (M)-NPs and (S)-NPs were successful in delivering functional agents in both cell lines, as shown in Figures 4A–E and S8–S11.
Figure 4.

Single-drug NP’s gene restoration and knockdown, and simultaneous gene restoration and knockdown mediated by codelivery dual-drug NPs using mRNA and siRNA in two cell lines. (A,B) Single drug (M)-NPs (0.016 nmol) and empty-NPs (10 μM) treated with HT1080 and MDA-MB-231 cells, and 48 h post-treatment with luciferase (RLU) measured by a plate reader, n = 3 (data represent means ± SD, *p < 0.05). (C) Single-drug (S)-NPs (1 nmol) and empty-NPs (10 μM) treated to GFP+ cells (HT1080 and MDA-MB-231). (D,E) Count of GFP knockdown was analyzed by flow cytometry histogram; no GFP cells were used as a positive control. (F) GFP+ cells (HT1080 and MDA-MB-231) were treated with (M+S)-NPs (∼60 nm size) coloaded with siRNA-GFP (1 nmol) and Luc mRNA (0.016 nmol) for 48 h. (G,H) GFP knockdown count was analyzed via flow cytometry; no GFP cells were used as positive control. (I) Luciferase expression was measured in GFP+ cells by luminescence, n = 3 (data represent means ± SD, *p < 0.05, **p < 0.01).
To evaluate the transfection efficiency of our codelivery NPs, we treated both GFP+ cells with (M+S)-NPs and control NPs for 48 h. We assessed the simultaneous delivery of Luc-mRNA and siRNA-GFP by (M+S)-NPs and their effects on the cells. Flow cytometry was used to analyze GFP knockdown induced by siRNA-GFP from (M+S)-NPs, while a luciferase assay was performed to measure the expression of the luciferase gene by Luc-mRNA delivered via the same (M+S)-NPs. The results indicated that our (M+S)-NPs successfully escaped endosomes and simultaneously knocked down GFP expression with siRNA-GFP while introducing the expression of the luciferase gene via Luc-mRNA (Figures 4F–I and S8–S11). Compared to empty NPs, (M+S)-NPs produced a strong bioluminescence signal in both cell lines, confirming the successful delivery of Luc-mRNA by codelivery NPs. Additionally, the mRNA transfection efficiency was higher in HT1080-GFP+ than in MDA-MB-231-GFP+ cell lines. As expected, (M+S)-NPs delivered siRNA-GFP and also successfully knocked down GFP in both cell types, as evidenced by flow cytometry.
Overall, our in vitro studies with GFP+ cells showed that (M+S)-NPs can successfully escape the endosomes, simultaneously knocking down GFP expression and introducing luciferase gene expression into the cells.
Co-Delivery NPs Can Express Target Genes within TNBC Tumors While Simultaneously Silencing Another Gene In Vivo
To assess the translation of our (M+S)-NPs’ in vitro effects into an in vivo setting, we studied their efficacy using a TNBC in vivo model. As a proof of concept, MDA-MB-231-GFP+ cells were implanted into the mammary gland of NSG mice to produce GFP+ TNBC tumors. Once tumors reached a mean diameter of 1 cm, mice were randomized into two cohorts and treated with (M+S)-NPs and empty NPs for 24 h. Luciferase induction was examined using an in vivo imaging system (IVIS) at 0 and 20 h post-treatment and with ex vivo bioluminescence at 24 h. GFP knockdown was measured using flow cytometry 24 h of treatment. At 0 h post-treatment, no luminescence was observed in either the control groups or (M+S)-NPs treated groups. However, at 20 h post-treatment, a strong luciferase bioluminescence was detected in the tumors of mice treated with (M+S)-NPs, while those treated with empty NPs showed no luminescence signal (Figure 5B). Subsequently, after 24 h treatment, tumors were extracted, single-cell suspensions were prepared, and luciferase bioluminescence and GFP were quantified using bioluminescence and flow cytometry, respectively. Compared to the control treatments, tumor cells treated with (M+S)-NPs exhibited a significantly strong (>300-fold) luciferase signal (Figure 5C). In addition, (M+S)-NPs achieved approximately 20% knockdown of GFP expression in tumor cells (Figure 5D,E). These results unequivocally demonstrate that following administration, (M+S)-NPs can penetrate the tumor cells and deliver both mRNA and siRNA, enabling simultaneous knockdown of the target gene product while inducing the expression of another protein.
Figure 5.
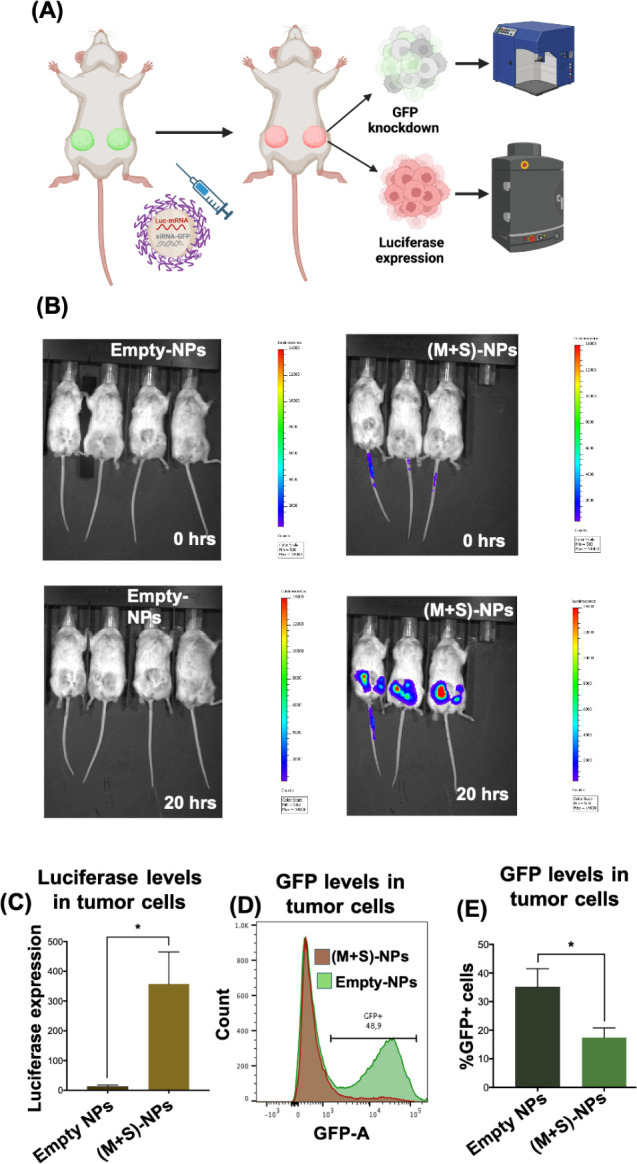
In vivo, codelivery NPs achieve simultaneous functional restoration and gene knockdown using mRNA and siRNA. GFP+ cells (MDA-MB-231) implanted into the mammary gland of NSG mice were treated via intratumoral injection (IT) with (M+S)-NPs coloaded with siRNA-GFP and Luc mRNA ((2.5 nmol siRNA-GFP + 0.4 nmol Luc mRNA)/mouse) for 24 h (A). Luciferase expression of Luc mRNA was measured using IVIS (B). GFP knockdown and luciferase expression were further analyzed via flow cytometry after dissociation of tumor into single cells (C,D), and the percentage of GFP+ cells was summarized (E). n = 4 for control-NPs and n = 3 for (M+S)-NPs; *p < 0.05.
Discussion
RNAi and mRNA technologies hold immense potential to treat a variety of diseases by targeting genes that are undruggable for traditional therapies.4 They offer versatile mechanisms for modulating gene and protein expression, including downregulation, augmentation, or correction. However, the full potential of RNA-based therapies remains unrealized due to inherent limitations, such as stability and unfavorable PK/PD properties. Effective delivery of RNA agents into cells is crucial to their treatment efficacy. In this context, advancements in NP-based drug delivery platforms have paved the way for novel NP systems capable of successfully delivering either siRNA or mRNA. Notably, current clinical successes, such as patisiran, a siRNA-based therapy for polyneuropathy in people with hereditary transthyretin-mediated amyloidosis, and mRNA-based COVID-19 vaccines, have accelerated RNA-based drug development for a variety of therapies.8,17
In cancer, siRNA agents, alone or in combination with chemotherapeutic agents, are predominantly explored for their ability to silence proteins implicated in drug resistance.13,14 Conversely, mRNA therapies are primarily studied for their potential to restore tumor suppressor gene/protein expressions, or to produce cytokines/antigens to improve antitumor immune response.6 However, cancer is a multifaceted disease, and genomic studies have highlighted that several factors are involved in cancer progression and survival.14,25 Additionally, single-target therapies for cancer often encounter challenges such as drug resistance and tumorigenesis.26
In TNBC, conventional chemotherapeutic treatments are frequently associated with drug resistance, the development of cancer stem cells, tumorigenesis, and adverse side effects. Our previous research has shown that multitargeted approaches can effectively control tumor growth and diminish CSC enrichment using clinically relevant tumor models.20,25 In light of these findings, we reasoned that knocking down or restoring a single target gene may have limitations in effectively treating cancer, necessitating multitargeted approaches. To address this, we developed (M+S)-NPs using siRNA-GFP and Luc-mRNA as model agents to simultaneously restore one gene while silencing the other. This approach represents a promising strategy for overcoming the challenges associated with single-target therapies and advancing the treatment of complex diseases like cancer.
Our results showed that (M+S)-NPs effectively encapsulated both agents, protected them from degradation, and successfully delivered them inside the cells. Notably, despite the size difference between siRNA (20–21 nucleotides) and Luc-mRNA (>2000 nucleotides), the NPs efficiently encapsulated both within a single particle. The encapsulation efficiencies are comparable to those reported for single-agent formulations in the literature. Additionally, our NP’s sizes are around 60 nm comparable to our previous NPs known to accumulate in tumors after intravenous administration.25 In vitro and in vivo characterization of (M+S)-NPs confirmed their ability to effectively enter cells, escape endosomes, and release their cargo in the cytosol. Additionally, (M+S)-NPs delivered siRNA-GFP and Luc-mRNA were functional and effective, as evidenced by a significant decrease in GFP expression and an increase in the Luciferase signal in both in vitro and in vivo studies (Figures 4 and 5). The significant GFP knockdown and luciferase expression achieved with (M+S)-NPs are comparable to those reported in current nanoparticle systems carrying single agents.36,37 Within the cells, the two RNA agents are processed differently. While mRNA is recruited into ribosomes for translation, siRNA is incorporated into the multiprotein RNA-induced silencing complex (RISC), which recognizes and cleaves complementary mRNA.5,12 Our NP results showed that these processes did not interfere with each other and operated independently. However, further studies are needed to understand the kinetics of this complex and any complementary interactions.
Diseases like cancer and cardiovascular diseases are complex and require multitargeted approaches. Our proof-of-concept study demonstrated the feasibility of precisely modulating the expression of multiple genes simultaneously. Drug resistance, CSC enhancement, and tumorigenesis are the primary challenges in current TNBC therapies. Our in vivo studies using the TNBC cell line model suggest that we could induce tumor suppressor gene expressions such as PTEN, P53, or tumor antigens for immunotherapy while simultaneously knocking down the genes/proteins involved in drug resistance/CSC developments.36,38 Although GFP and luciferase are not directly interconnected, several signaling pathway crosstalks, such as MAPK/ERK and PI3K-Akt, Wnt and NF-kB, and JAK/STAT and RAS/MAPK, have been reported in tumors. Our approach can selectively enhance or interfere with these cross-talks to improve therapeutic outcomes. Additionally, our study suggests that we can synergistically promote antitumoral factors while minimizing protumoral factors via mRNA introduction, RNA interference, translational inhibition, and/or translational repression. Overall, the studies presented here will pave the way for new therapeutic strategies for complex diseases such as cancer, with significant implications for drug development.
Conclusions
RNA-based strategies, such as RNAi and mRNA technologies, hold significant promise in treating major diseases, including cancer. Current studies primarily focus on introducing tumor suppressors using mRNA or knocking down proteins that cause drug resistance with siRNA. However, cancer’s complexity necessitates multitargeted approaches. To address this, we developed codelivery NPs using Luc-mRNA and siRNA-GFP as model agents. Our NPs were efficiently transfected and successfully functioned in two different cancer cell lines in vitro. When administered to a TNBC mouse model created using the MDA-MB-231 GFP+ cell line, the codelivery NPs efficiently knocked down GFP while simultaneously introducing luciferase. This proof-of-concept has significant implications in developing RNA-based multitargeted therapies for complex diseases like cancer.
Materials and Methods
Materials and Reagents
mPEG–PLGA was procured from PolySciTech. Polyethylenimine (PEI) and 1,2- epoxytetradecane were purchased from Sigma-Aldrich. EGFP-mRNA and Luc-mRNA were purchased from GenScript Biotech. GFP-siRNA was purchased from Integrated DNA Technologies. Cy5-siRNA universal negative control was purchased from Sigma-Aldrich. Centrifuge filters were obtained from Pall Corporation. DMSO solvent was obtained from Fisher Scientific. Copper grids were obtained from Ted Pella Inc. UranyLess EM stain was procured from Electron Microscopy Sciences. RiboGreen assay reagent and luciferase reagent kit were purchased from Thermo Fisher Scientific. Dulbecco’s modified Eagle’s medium was purchased from Wisent Bioproducts, and trypsin for cell splitting was obtained from Corning Inc.
Nanoparticle Synthesis and Characterization
All NPs were synthesized using the nanoprecipitation method by mixing PLGA–PEG, PEI-C14, and mRNA/siRNAs in appropriate ratios. To mRNA (0.08 nmol) or siRNA (4 nmol) in 100 μL of sterile water, PEI-C14 lipid (0.1 mM (0.2 mM for dual drug NPs)) was added with 500 μL of DMSO and mixed gently. Subsequently, a solution of mPEG–PLGA (0.2 mM (0.4 mM for dual drug NPs)) in 500 μL DMF was added dropwise. After 10 min of spinning, 2 mL sterile water was added to the NP solution and spinning was continued for 2 h. For empty NP synthesis, a similar concentration of PEI-C14 and mPEG–PLGA was used without mRNA or siRNA. Then, NP solutions were concentrated for two rounds for 10 min. The resulting NPs were characterized for size and surface charge and stored at 4 °C. The particle size distribution and morphological appearance of NPs were examined under transmission electron microscopy. Briefly, 10 μL of the sample was spread onto a Cu grid (300 mesh) for 30 s and allowed to stain using 10 μL of UranyLess. The grid was dried before visualizing under a JEM-1400Plus transmission electron microscope operated at 120 kV. The images were acquired on FIJI ImageJ software. Also, NPs were tested for stability by incubating them for 6 h in 5% or 10% fetal bovine serum (FBS) at 37 °C and then measuring the size. The efficiency (EE%) of mRNA in the NPs was measured using the RiboGreen (Thermo Fisher Scientific) assay as previously described.1 Briefly, single- or dual-drug NPs (15 μL) and 1× TE buffer (235 μL) were mixed as one solution. From this NP solution, three different volumes were taken in a replicate manner in a 96-well black well plate. Next, 1% Triton X-100 buffer was added to each sample. RNA standard solutions were prepared in 1× TE buffer in the same plate. After 10–15 min, fluorescent RiboGreen reagent was added to NP solutions and RNA standard solutions. The resulting solution’s fluorescence intensity was read at an excitation of 490 nm and an emission of 520 nm, using the plate reader BioTek Synergy Neo2 Hybrid Multimode. To measure the amount of mRNA and determine the encapsulation efficiency, we employed a standard curve.
For the siRNA encapsulation efficiency (EE%), the Cy5-labeled siRNA fluorescence intensity was measured. Briefly, in a black 96-well plate, Cy5-siRNA standard solutions (six concentrations) were placed with increasing concentrations. Each Cy5-siRNA concentration’s volume was made up to 100 μL with DMSO. In the same plate, Cy5-siRNA-based single- or dual-drug NPs (50 μL) and DMSO (50 μL) were mixed in other wells in a replicate manner. Fluorescence intensity was read at an excitation of 650 nm and an emission of 670 nm, using the plate reader BioTek Synergy Neo2 Hybrid Multimode. A standard curve was made to determine the amount of siRNA and the efficiency of encapsulation.
In Vitro Studies
The MDA-MB-231 breast cancer cell line and HT1080 cell line were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), and both cell lines were maintained in DMEM media supplemented with 10% FBS and 1% penicillin/streptomycin.
To generate the GFP+ MDA-MB-231, and GFP+ HT1080 monoclonal cell lines, retroviral vectors encoding GFP were produced by cotransfection of HEK293T cells (ATCC) with MLV LTR-GFP, Gag-Pol, and a plasmid encoding VSV-G (all kind gifts of Dr. James Cunningham, Brigham and Women’s Hospital) using the JetPrime (Polyplus-transfection) reagent following the manufacturer’s protocol. Supernatants containing the retroviral pseudotypes were filtered (0.45 μm) and used to transduce MDA-MB-231 and HT1080 cells in the presence of Polybrene (3 μg/mL). 48 to 72 h later, transduced cells were seeded at very low density in 10 cm-dishes. Cells were incubated at 37 °C until the generation of colonies, which were selected based on GFP expression as visualized using the ZOE imager (Bio-Rad) and picked using cloning cylinders.
GFP Knockdown
GFP+ cells were seeded in a 12-well plate at 50 000 cells per well. At 60% confluency, cells were treated with (S)-NP (1 nmol) or (M+S)-NP (1 nmol + 0.016 nmol) and incubated for 48 h. Then, the cells were trypsinized and centrifuged. The cell pellet was resuspended with a 1× FACS buffer. GFP knockdown was assessed quantitatively using the BD LSRFortessa flow cytometer, and data were analyzed with FlowJo software.
(Cy5-siRNA+EGFP-mRNA)-NP Co-Delivery to HT1080 Cells
In a 12-well plate, HT1080 cells were seeded with a density of 50 000 cells per well. After 1 day, cells were treated with (Cy5-siRNA+EGFP-mRNA)-NP (1 nmol+0.016 nmol) and incubated for 48 h. GFP production in live cell imaging was performed by fluorescence microscopy using the ZOE Fluorescent Cell Imager (Bio-Rad, CA, USA). Subsequently, cells were trypsinized and centrifuged. The cell pellet was resuspended in a FACS buffer. The percentage of GFP production and Cy5 cellular entry were assessed quantitatively using the BD LSRFortessa flow cytometer, and data were analyzed with FlowJo software.
Luciferase Assay
In a white 96-well plate, at a density of 7 × 103 cells per well (GFP+ or GFP−), the cells were seeded in triplicate 1 day before treatment. Single (0.016 nmol) or dual-drug NPs (1 nmol + 0.016 nmol) were added to cells with fresh media and incubation was continued for 48 h. Then, the medium was removed and each well was added with 50 μL of 1× luciferase lysis buffer and the cells were lysed by a single freeze–thaw cycle. Subsequently, 50 μL of luciferin reagent was added to each well and luminescence reading was initiated using the BioTek Synergy Neo2 Hybrid Multimode Reader.
In Vivo Studies
The animal studies were approved by the Animal Care and Use Committee at the University of Ottawa (protocol # BMIe-4035). The MDA-MB-231-GFP+ breast cancer cells were mixed in a 1:1 ratio with Matrigel and injected under aseptic conditions into the mammary fat pads (2 × 106 cells per fat pad) of NSG mice. When the tumor reached a mean diameter of approximately 1 cm, tumor-bearing NSG mice were treated via intratumoral injection (IT) of 50 μL (M+S)-NPs ((2.5 nmol siRNA-GFP + 0.4 nmol Luc mRNA)/mouse) or empty NPs. After 20 h postinjection, relative bioluminescent intensity in tumors and different organs in mice was quantified using the PerkinElmer IVIS Spectrum In Vivo Imaging System (IVIS). Following IVIS, mice were humanely euthanized 24 h postinjection. The tumors were harvested and minced using a scalpel and incubated in DMEM media containing collagenase/hyaluronidase (STEMCELL Technologies, #07912) at 37 °C for 60 min. Afterward the solution was passed through a 40 μm nylon mesh for the creation of a single-cell solution. GFP knockdown was analyzed via flow cytometry, and luciferase expression was assessed by bioluminescence. n = 4 for control empty-NPs and n = 3 for mRNA/siRNA NPs.
Flow Cytometry
Dissociated cancer cells were filtered through a 40 μm strainer and suspended in PBS supplemented with 2% FBS and 2 mM EDTA. 1 μL portion of mouse IgG (1 mg/mL) was added and incubated at 4 °C for 10 min. Afterward, the cells were resuspended in 1× binding buffer (eBioscience, San Diego, CA, USA) and incubated with Annexin-V (eBioscience) for 15 min at room temperature. The cells were then washed twice and 7-aminoactinomycin D (7-AAD, eBioscience, San Diego, CA) was added to exclude dead cells. Flow cytometry was performed on a Cyan-ADP 9 or the BD LSRFortessa. Data were analyzed with FlowJo software (Ashland, OR, USA).
Statistical analysis
Data are represented as means ± standard deviation (SD) or standard error (SE). Statistical significance was determined using ANOVA or student’s t-test wherever appropriate and reported as (*) for p < 0.05, (**) for p < 0.01, (***) for p < 0.001, (****) for p < 0.0001, and n for no significant difference. Unless otherwise stated, experiments have a minimum of three biological repeats.
Acknowledgments
The authors would like to thank Ardeshir Ariana for help with flow cytometry and Vera Tang from the uOttawa Flow Cytometry and Virometry Core for technical support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnanoscienceau.4c00040.
Synthesis and characterization of PEI-C14; Cy5-siRNA+EGFP-mRNA-NPs characterization; PDI of all NPs; flow cytometry analysis of Cy5 and EGFP expression in HT1080 cells; flow cytometry analysis of GFP knockdown in GFP+ cells; percentage (quantitative graph) of GFP knockdown in GFP+ cells with single and dual-drug NPs; luciferase analysis with single-drug and dual-drug NPs in GFP+ and GFP– cells, respectively (PDF)
Author Contributions
○ S.M. and S.E.S. contributed equally to the manuscript. All authors were involved in data collection, analysis, and final editing.
This work was supported by a Canadian Institutes of Health Research Project Grant, PJT 175177 (L.W. and S.G.), National Research Council, Canada, Cell and Gene Therapy (CGT) Challenge program grant, CGT-504–1 (S.G., U.I., and L.W.), and Natural Sciences and Engineering Research Council RGPIN-2019–05220 (L.W.). M.C. is a Tier II Canada Research Chair in Molecular Virology and Antiviral Therapeutics. M.C. is a recipient of an Ontario Ministry of Research, Innovation and Science Early Researcher Award. S.E., E.D., M.K., and R.D. are Canada Graduate Scholarship recipients. BioRender was used in creating some of the illustrations.
The animal studies were approved on 12/20/2023 by the Animal Care and Use Committee (ACUC) at the University of Ottawa (protocol # BMIe-4035).
The authors declare no competing financial interest.
Supplementary Material
References
- Huang X.; Kong N.; Zhang X.; Cao Y.; Langer R.; Tao W. The landscape of mRNA nanomedicine. Nat. Med. 2022, 28 (11), 2273–2287. 10.1038/s41591-022-02061-1. [DOI] [PubMed] [Google Scholar]
- Mendes B. B.; Conniot J.; Avital A.; Yao D.; Jiang X.; Zhou X.; Sharf-Pauker N.; Xiao Y.; Adir O.; Liang H.. et al. Nanodelivery of nucleic acids. Nat. Rev. Methods Primers, 2022, 2, (1), . 10.1038/s43586-022-00104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; Witzigmann D.; Thomson S. B.; Chen S.; Leavitt B. R.; Cullis P. R.; van der Meel R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16 (6), 630–643. 10.1038/s41565-021-00898-0. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R. The druggable genome. Nat. Rev. Drug Discovery 2002, 1 (9), 727–730. 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Qin S.; Tang X.; Chen Y.; Chen K.; Fan N.; Xiao W.; Zheng Q.; Li G.; Teng Y.; Wu M.; et al. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduct. Targeted Ther. 2022, 7 (1), 166. 10.1038/s41392-022-01007-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Shi Q.; Huang X.; Koo S.; Kong N.; Tao W. mRNA-based cancer therapeutics. Nat. Rev. Cancer 2023, 23 (8), 526–543. 10.1038/s41568-023-00586-2. [DOI] [PubMed] [Google Scholar]
- Hou X.; Zaks T.; Langer R.; Dong Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6 (12), 1078–1094. 10.1038/s41578-021-00358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschmann M. D.; Carrasco M. J.; Alishetty S.; Paige M.; Alameh M. G.; Weissman D. Nanomaterial Delivery Systems for mRNA Vaccines. Vaccines 2021, 9 (1), 65. 10.3390/vaccines9010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Tang M.; Ho W.; Teng Y.; Chen Q.; Bu L.; Xu X.; Zhang X. Q. Modulating Plaque Inflammation via Targeted mRNA Nanoparticles for the Treatment of Atherosclerosis. ACS Nano 2023, 17 (18), 17721–17739. 10.1021/acsnano.3c00958. [DOI] [PubMed] [Google Scholar]
- Parayath N. N.; Stephan S. B.; Koehne A. L.; Nelson P. S.; Stephan M. T. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 2020, 11 (1), 6080. 10.1038/s41467-020-19486-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; Witzigmann D.; Chen S.; Cullis P. R.; van der Meel R. Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Acc. Chem. Res. 2019, 52 (9), 2435–2444. 10.1021/acs.accounts.9b00368. [DOI] [PubMed] [Google Scholar]
- Hu B.; Zhong L.; Weng Y.; Peng L.; Huang Y.; Zhao Y.; Liang X. J. Therapeutic siRNA: State of the art. Signal Transduct. Targeted Ther. 2020, 5 (1), 101. 10.1038/s41392-020-0207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully M. A.; Wilhelm R.; Wilkins D. E.; Day E. S. Membrane-Cloaked Nanoparticles for RNA Interference of beta-Catenin in Triple-Negative Breast Cancer. ACS Biomater. Sci. Eng. 2024, 10 (3), 1355–1363. 10.1021/acsbiomaterials.4c00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadde S. Multi-drug delivery nanocarriers for combination therapy. MedChemcomm 2015, 6 (11), 1916–1929. 10.1039/C5MD00365B. [DOI] [Google Scholar]
- Paunovska K.; Loughrey D.; Dahlman J. E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23 (5), 265–280. 10.1038/s41576-021-00439-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon W.; Kingston B. R.; Ouyang B.; Ngo W.; Chan W. C. W. A framework for designing delivery systems. Nat. Nanotechnol. 2020, 15 (10), 819–829. 10.1038/s41565-020-0759-5. [DOI] [PubMed] [Google Scholar]
- Akinc A.; Maier M. A.; Manoharan M.; Fitzgerald K.; Jayaraman M.; Barros S.; Ansell S.; Du X.; Hope M. J.; Madden T. D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14 (12), 1084–1087. 10.1038/s41565-019-0591-y. [DOI] [PubMed] [Google Scholar]
- Shi J.; Kantoff P. W.; Wooster R.; Farokhzad O. C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17 (1), 20–37. 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis C.; Ma J.; Bryan L.; Jemal A. Breast cancer statistics, 2013. Ca-Cancer J. Clin. 2014, 64 (1), 52–62. 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]
- El-Sahli S.; Hua K.; Sulaiman A.; Chambers J.; Li L.; Farah E.; McGarry S.; Liu D.; Zheng P.; Lee S. H.; et al. A triple-drug nanotherapy to target breast cancer cells, cancer stem cells, and tumor vasculature. Cell Death Dis. 2021, 12 (1), 8. 10.1038/s41419-020-03308-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent R.; Trudeau M.; Pritchard K. I.; Hanna W. M.; Kahn H. K.; Sawka C. A.; Lickley L. A.; Rawlinson E.; Sun P.; Narod S. A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- Chai C.; Wu H. H.; Abuetabh Y.; Sergi C.; Leng R. Regulation of the tumor suppressor PTEN in triple-negative breast cancer. Cancer Lett. 2022, 527, 41–48. 10.1016/j.canlet.2021.12.003. [DOI] [PubMed] [Google Scholar]
- Sulaiman A.; McGarry S.; Lam K. M.; El-Sahli S.; Chambers J.; Kaczmarek S.; Li L.; Addison C.; Dimitroulakos J.; Arnaout A.; et al. Co-inhibition of mTORC1, HDAC and ESR1alpha retards the growth of triple-negative breast cancer and suppresses cancer stem cells. Cell Death Dis. 2018, 9 (8), 815. 10.1038/s41419-018-0811-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulaiman A.; McGarry S.; Li L.; Jia D.; Ooi S.; Addison C.; Dimitroulakos J.; Arnaout A.; Nessim C.; Yao Z.; et al. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol. Oncol. 2018, 12 (4), 423–440. 10.1002/1878-0261.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulaiman A.; McGarry S.; El-Sahli S.; Li L.; Chambers J.; Phan A.; Al-Kadi E.; Kahiel Z.; Farah E.; Ji G.; et al. Nanoparticles Loaded with Wnt and YAP/Mevalonate Inhibitors in Combination with Paclitaxel Stop the Growth of TNBC Patient-Derived Xenografts and Diminish Tumorigenesis. Adv. Ther. 2020, 3 (11), 2000123. 10.1002/adtp.202000123. [DOI] [Google Scholar]
- Sulaiman A.; McGarry S.; El-Sahli S.; Li L.; Chambers J.; Phan A.; Cote M.; Cron G. O.; Alain T.; Le Y.; et al. Co-targeting Bulk Tumor and CSCs in Clinically Translatable TNBC Patient-Derived Xenografts via Combination Nanotherapy. Mol. Cancer Ther. 2019, 18 (10), 1755–1764. 10.1158/1535-7163.MCT-18-0873. [DOI] [PubMed] [Google Scholar]
- Detappe A.; Nguyen H. V.; Jiang Y.; Agius M. P.; Wang W.; Mathieu C.; Su N. K.; Kristufek S. L.; Lundberg D. J.; Bhagchandani S.; et al. Molecular bottlebrush prodrugs as mono- and triplex combination therapies for multiple myeloma. Nat. Nanotechnol. 2023, 18 (2), 184–192. 10.1038/s41565-022-01310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel Y.; Manturthi S.; Tiwari S.; Gahunia E.; Courtemanche A.; Gandelman M.; Côté M.; Gadde S. Development of Pro-resolving and Pro-efferocytic Nanoparticles for Atherosclerosis Therapy. ACS Pharmacol. Transl. Sci. 2024, 7, 3086. 10.1021/acsptsci.4c00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Xu Y.; Xiao H.; Xiao Z.; Guo Y.; Cheng D.; Shuai X. Core-Shell Distinct Nanodrug Showing On-Demand Sequential Drug Release To Act on Multiple Cell Types for Synergistic Anticancer Therapy. ACS Nano 2019, 13 (6), 7036–7049. 10.1021/acsnano.9b02149. [DOI] [PubMed] [Google Scholar]
- Ling X.; Chen X.; Riddell I. A.; Tao W.; Wang J.; Hollett G.; Lippard S. J.; Farokhzad O. C.; Shi J.; Wu J. Glutathione-Scavenging Poly(disulfide amide) Nanoparticles for the Effective Delivery of Pt(IV) Prodrugs and Reversal of Cisplatin Resistance. Nano Lett. 2018, 18 (7), 4618–4625. 10.1021/acs.nanolett.8b01924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamaly N.; Yameen B.; Wu J.; Farokhzad O. C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116 (4), 2602–2663. 10.1021/acs.chemrev.5b00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Xie K.; Zhang X. Q.; Pridgen E. M.; Park G. Y.; Cui D. S.; Shi J.; Wu J.; Kantoff P. W.; Lippard S. J.; et al. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (46), 18638–18643. 10.1073/pnas.1303958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley R. S.; Kashyap M. V.; Billingsley M. M.; White B.; Alameh M. G.; Bose S. K.; Zoltick P. W.; Li H.; Zhang R.; Cheng A. Y.; et al. Ionizable lipid nanoparticles for in utero mRNA delivery. Sci. Adv. 2021, 7, 3. 10.1126/sciadv.aba1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. K. T.; Kahiel Z.; LeBlond N. D.; Ghorbani P.; Farah E.; Al-Awosi R.; Cote M.; Gadde S.; Fullerton M. D. Characterization of Redox-Responsive LXR-Activating Nanoparticle Formulations in Primary Mouse Macrophages. Molecules 2019, 24 (20), 20. 10.3390/molecules24203751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Xu Y.; Solis L. M.; Tao W.; Wang L.; Behrens C.; Xu X.; Zhao L.; Liu D.; Wu J.; et al. Long-circulating siRNA nanoparticles for validating Prohibitin1-targeted non-small cell lung cancer treatment. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (25), 7779–7784. 10.1073/pnas.1505629112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. A.; Xu Y.; Tao W.; Ubellacker J. M.; Lim M.; Aum D.; Lee G. Y.; Zhou K.; Zope H.; Yu M.; et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2 (11), 850–864. 10.1038/s41551-018-0284-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Wu J.; Liu Y.; Yu M.; Zhao L.; Zhu X.; Bhasin S.; Li Q.; Ha E.; Shi J.; et al. Ultra-pH-Responsive and Tumor-Penetrating Nanoplatform for Targeted siRNA Delivery with Robust Anti-Cancer Efficacy. Angew. Chem., Int. Ed. 2016, 55 (25), 7091–7094. 10.1002/anie.201601273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong N.; Tao W.; Ling X.; Wang J.; Xiao Y.; Shi S.; Ji X.; Shajii A.; Gan S. T.; Kim N. Y.; et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 2019, 11, 523. 10.1126/scitranslmed.aaw1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.