

Abstract
CHD6, a member of the chromodomain helicase DNA-binding protein family, has been implicated in various diseases and tumors. However, its precise binding model of CHD6 on regulatory functional genes remains poorly understood. In this study, we discovered sharp peaks of CHD6, as the first member of CHD family for housekeeping process, binding only to the promoter region of genes in the C4-2 cell line. These genes, with conserved sharp CHD6 peaks across tumor cells, likely represent housekeeping genes ADNP and GOLGA5. Genes with sharp CHD6 peaks exhibit stable and low expression levels, sharing epigenetic features similar to housekeeping genes. Furthermore, this regulatory model also exists in both HEK293 cells and cardiomyocytes. Overall, the results of this study demonstrate that CHD6 binds to the promoter regions of housekeeping genes, regulating their histone modifications, chromatin structure, and gene expression.
Keywords: MT: Bioinformatics, CHD6, housekeeping genes, histone modifications, epigenetics, chromatin remodeling
Graphical abstract
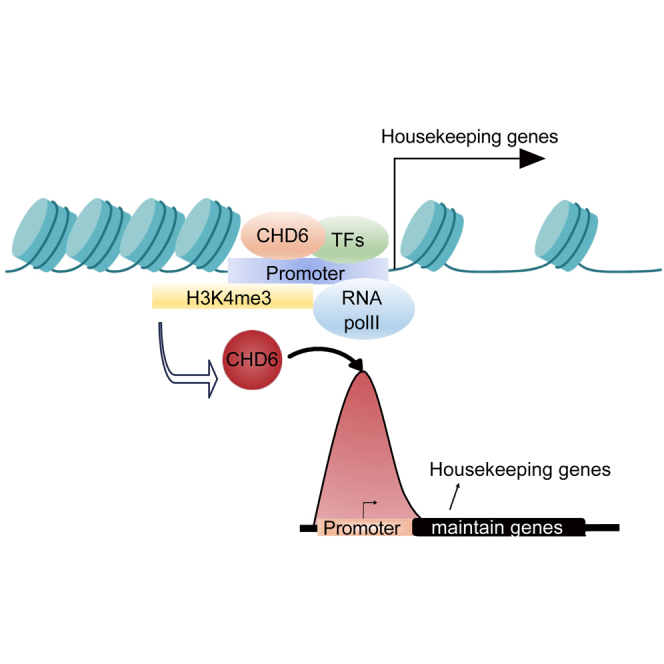
Zhao and colleagues demonstrated that CHD6 binds specifically to the promoter regions of the housekeeping genes in both normal and cancer cells through multi-omics analysis. This study is the first to characterize the unique binding patterns of CHD6, providing insights into its role in gene regulation.
Introduction
Prostate cancer (PC) is one of the most widespread malignancies in men worldwide, with an estimated 1,466,718 new cases diagnosed globally in 2024.1 Despite multiple available treatment options, including active surveillance, chemotherapy, radiation therapy, hormone therapy, surgery, and cryotherapy, PC remains incurable.2 As time progresses, this disease continues to develop resistance to various conventional treatment approaches.3 Castration-resistant prostate cancer (CRPC) is considered incurable.4 Thus, there is an urgent need to identify more molecular biomarkers for CRPC. In addition, investigating the molecular mechanisms underlying CRPC progression is critical and may help provide new therapeutic targets. Chromatin remodelers are pivotal in regulating chromatin accessibility and nucleosome positioning on genomic DNA, which is essential for all DNA-dependent biological processes.5,6 Recent research has indicated that chromatin remodelers are involved in both human cancer and neurological disorders, offering insights into new mechanisms and potential therapeutic avenues.7,8,9
As a member of the chromatin remodeling factor family, the chromodomain helicase DNA-binding protein (CHD) family includes nine members, CHD1–CHD9.10 These members share double chromodomains for binding specifically modified histones11,12,13 and an SNF2-like ATP-dependent helicase domain that alters chromatin packaging.14 They are considered crucial factors in establishing the nucleosome landscape essential for oncogenes (OGs), tumor suppressor genes (TSGs), and housekeeping (HK) genes, thereby controlling fundamental processes, including transcription, proliferation, and epigenetic perturbation.15 The emerging dysregulation of CHD in various human cancers emphasizes the vital significance of chromatin dynamics in tumorigenesis.16,17,18,19,20 CHD6, belonging to the subfamily III of CHD, is expressed ubiquitously in mammalian tissues. CHD6 plays a crucial role in and has associations with multiple diseases. Previous reports have highlighted the significant involvement of CHD6 in DNA damage and repair and autophagy.21,22 CHD6 collaborates with TCF4 to positively regulate TMEM65 gene expression, thereby promoting colorectal cancer development and metastasis.23 Meanwhile, CHD6 binds on chromatin to evict nucleosomes from promoters and gene bodies for transcriptional activation in PC.24 HK, developmental, and OG transcriptional programs are highly regulated to maintain cell identity and function. The involvement of CHD6 in the regulation of HK genes and how it impacts the chromatin structure of these genes remains unclear, despite CHD6 being a cancer driver gene.
HK genes are often assumed to be stably expressed and essential for the maintenance of basal cellular processes, which ensures the essential functions for cell survival across various tissues or organisms.25,26 However, evidence indicates that HK genes may vary in expression levels under diverse experimental conditions.27,28,29 Despite their common use as internal reference genes for gene expression assessment, changes in their expression levels have been associated with cancer development.30 A study focused on prostate tumorigenesis revealed that HK genes were more likely differentially expressed, hinting at their potential role in driving cancer development. Another study suggested that the variable expression levels of HK genes can act as diagnostic and prognostic indicators in lung cancers.31 While cancer development induced by HK genes expressions has been reported previously, the mechanisms by which chromatin remodelers regulate their expression levels and affect their epigenetic modifications remain poorly understood.
In this study, to gain insights into how the binding models of CHD6 influence the expression levels and epigenetic modifications of distinct functional genes, we conducted a comprehensive analysis using multi-omics approaches on prostate cancer cells. We found that genes associated with sharp CHD6 peaks are enriched in HK genes rather than cancer-related genes. It is interesting that this model is consistent across normal human cells.
Results
Sharp enrichment of CHD6 at promoter region of HK genes in cancer cells
Our previous study demonstrated that CHD6 binds on chromatin to evict nucleosomes from promoters and gene bodies, which leads to the transcriptional activation of oncogenic pathways.24 However, previous research has not clearly elucidated how the distinct binding regions of CHD6 influence transcriptional programs. To assess the direct regulatory regions of CHD6, we analyzed the distribution of all peaks across the genome, from transcription start site (TSS) to transcription termination site (TTS), using CHD6 chromatin immunoprecipitation sequencing (ChIP-seq). Among the total peaks, a notable enrichment of promoters and gene bodies is shown in a pie chart (Figure S1A). To gain further insight into how CHD6 regulates gene expression levels, we recently observed the CHD6 signal and found that it bound to the promoter region (±1 kb of TSS) of several well-known HK genes,25 such as ADNP and GOLGA5, in the C4-2 cell line (Figure 1A). By contrast, CHD6 covered low-density genetic regions, from promoters to gene bodies, on OGs such as FGFR3 and CBX8 (Figure 1A). This observation motivated us to perform a systematic analysis of the regions (promoters and gene bodies) associated with each CHD6 peak. We observed a subset of high-density CHD6 peaks on relatively narrow promoters (defined as sharp CHD6 peaks) and a subset of low-density CHD6 peaks that were exceptionally wide (defined as broad CHD6 peaks), spanning both the promoter and gene body regions (Figure 1B). We found no overlap between the sharp and broad peaks, which suggests potentially distinct mechanisms for these two groups of peaks. To conduct a quantitative comparison of sharp and broad CHD6 peaks, we retrieved 1,000 genes associated with sharp and broad CHD6 peaks based on the total signal of CHD6 and randomly chose another 1,000 genes as control (Figure 1C). Distinct CHD6 binding signals were found only in the promoter region on sharp CHD6 peaks, whereas the entire gene exhibited CHD6 binding signals on broad CHD6 peaks (Figure 1D). As expected, the signal density of the sharp CHD6 peaks was higher than that of the broad and control CHD6 peaks in the promoter region.
Figure 1.

Sharp CHD6 peaks in the C4-2 cell line
(A) Density of CHD6 in the housekeeping (HK) genes ANDP and GOLGA5 and the oncogenes (OGs) FGFR3 and CBX8. (B) Definition of genes with sharp and broad CHD6 peaks. (C) Heatmap of CHD6. Each row represents a gene region from −1 to +1 kb with respect to the gene. Top: the top 1,000 sharp CHD6 peaks; center: the top 1,000 broad CHD6 peaks; bottom: 1,000 random genes. (D) Average ChIP-seq signal value of CHD6 plotted around groups. (E) KEGG pathway enrichment of genes with sharp, broad, and control peaks. (F) Enrichment p values (y axis) of HK genes, OGs, and tumor suppressor genes (TSGs) in the different groups. (G) The percentage of gene expression. (H) Expression levels of the HK genes ANDP and GOLGA5 in tumor and normal cells. For (F), p values determined using Fisher's exact test. For (G), p values determined using one-tailed Wilcoxon test. ∗p < 0.05; ∗∗p <0.01; ∗∗∗p < 0.001.
We used Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis to characterize enriched functions of the genes associated with the three groups of CHD6 peaks. Endocytosis32 (hsa04144) and autophagy33 (hsa04140) were explicitly enriched in the group with sharp CHD6 peaks but not in the groups with broad or control CHD6 peaks (Figure 1E). This observation suggests that sharp CHD6 peaks might be associated with genes maintaining the HK process. By contrast, the Wnt signaling pathway34 (hsa04310) is uniquely enriched in broad CHD6 peaks (Figure 1E). Pathways in cancer (hsa05200), referring to well-curated signaling networks involved in cancer development, were also significantly enriched in broad CHD6 peaks but not in the groups with sharp or control peaks (Figure 1E). To further confirm the association between sharp CHD6 peaks and the HK process, we collected high-confidence HK genes,25 TSGs, and OGs (https://cancer.sanger.ac.uk/cosmic). We confirmed that this enrichment preference for HK genes was not due to bias in our driver gene collection, as TSGs and OGs showed similarly significant enrichment for the KEGG pathway in endocytosis (hsa04144) (Figure S1B), and OGs showed similarly significant enrichment for the KEGG pathway in autophagy animal (hsa04140) (Figure S1B). Surprisingly, the CHD6 group with the sharp peaks in the promoter region was enriched in HK genes but not in OG or TSG genes (Figure 1F).
We next analyzed the percentage of gene expression across different groups using the genotype-tissue expression (GTEx) tissue dataset and found that genes with sharp CHD6 peaks showed a universal expression compared with other genes (Figure 1G). We also used the single-cell RNA-seq (scRNA-seq) of 21,762 cells from 6 patients with CRPC using the 10X Genomics platform (Table S1). Analysis of the scRNA-seq data using CopyKAT resulted in the identification of aneuploid tumor and diploid normal cells in all six patients (Figure S1C). We found that genes with sharp CHD6 peaks, such as ANDP and GOLGA5, were expressed in all cells (Figure 1H), whereas genes with broad CHD6 peaks, such as FGFR3 and CBX8, were expressed in only a few tumor cells (Figure S1D). Taking these data together, we conclude that sharp CHD6 peaks in the C4-2 cell line are strongly and uniquely associated with HK genes.
Sharp CHD6 genes have epigenetic signatures similar to those of HK genes
The enrichment level of histone H3 lysine 4 trimethylation (H3K4me3) at the initiation site of transcription, a hallmark of active gene promoters, is closely associated with gene expression level.35,36 The latest research findings have indicated that H3K4me3 regulates RNA polymerase II (RNA Pol II) promoter-proximal pause-release and transcriptional elongation.37,38 To understand the potential mechanisms underlying the sharp CHD6 peaks of genes, we mapped the ChIP-seq data of H3K4me3 and RNA Pol II in C4-2 cell line in three groups. The H3K4me3 and RNA Pol II ChIP-seq signals were more enriched in promoter regions of the genes with sharp CHD6 peaks than in those of the other genes (Figures 2A and 2B). This pattern of narrow H3K4me3 has been identified in the promoter regions of HK genes in several studies.39,40,41 Furthermore, we observed that broad H3K4me3 (spanning over 4 kb) was enriched in the genes with broad CHD6 peaks but not in those with sharp peaks (Figure S2A). Given that the pausing index is the promoter-to-gene body ratio of RNA Pol II ChIP-seq density, a higher pausing index indicates more poised RNA Pol II and less elongation. Genes with sharp CHD6 peaks exhibited a higher RNA Pol II pausing index than other genes (Figure 2C), indicating that genes with sharp CHD6 peaks are in a state of promoter-proximal pausing. A previous study has shown that the promoter pausing of RNA Pol II plays a critical role in regulating HK genes.42 Consistent with this observation, the elongation-associated histone mark trimethylation of histone H3 at lysine 36 (H3K36me3)43,44 showed weak signals in the genes with sharp CHD6 peaks (Figure S2B). In addition, we demonstrated that sharp CHD6 peaks were associated with expression levels significantly lower than those of genes with broad CHD6 peaks (Figure 2D). These results indicate that genes with sharp CHD6 exhibit epigenetic characteristics that are more similar to those of HK genes.
Figure 2.

Epigenetic features of genes with sharp CHD6 peaks
(A) Average ChIP-seq signal value of H3K4me3 plotted around groups. (B) Average ChIP-seq signal value of RNA Pol II plotted around groups. (C) RNA Pol II pausing index plotted against sharp, broad, and control CHD6 peaks. (D) Boxplots showing gene expression levels (y axis) of the genes (n = 1,000 for each group), with the center line indicating the median. (E) Average ChIP-seq signal value of H3K27ac plotted around groups. (F) Boxplot showing CGI proportion (y axis) of groups. p values determined using one-tailed Wilcoxon test. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Common enhancers are generally believed to be involved in the transcription of HK genes.45,46 Acetylation of histone H3 at lysine 27 (H3K27ac), an enhancer-associated mark, showed lower enrichment in the genes with sharp CHD6 peaks than in those with broad peaks (Figure 2E). Multiple adjacent enhancer elements can be clustered into super-enhancers (SEs), which are the major drivers of transcriptional activation.47 In our analysis of ChIP-seq data on H3K27ac, we identified 1,121 SE genes. Similarly, genes with sharp CHD6 peaks did not display significant enrichment in SE genes, whereas the genes with broad peaks showed significant enrichment (Figure S2C). Another enhancer-associated mark, H3K4me1, also showed a lower signal in the genes with sharp CHD6 peaks than in those with broad peaks (Figure S2D), which aligns with the H3K27ac results. Moreover, using assay for transposase accessible chromatin sequencing to evaluate genome-wide chromatin accessibility, we observed that the genes with sharp CHD6 peaks exhibited lower chromatin accessibility than the genes with broad CHD6 peaks (Figure S2E).
H3K4me3 displayed a robust correlation with the boundaries of promoter-linked CpG islands (CGIs), and HK genes tended to have high CpG density.48,49 Therefore, we compared the proportion of CGIs in the promoters of sharp and broad peaks and found a significantly higher proportion of CGIs on the genes with sharp CHD6 peaks than on the genes with broad and control peaks (Figure 2F). The results further revealed that the genes with sharp CHD6 peaks, such as HK genes, are enriched with CGIs in the promoter regions.
Genes with sharp CHD6 peaks show low but stable expression levels
To assess gene expression stability, we calculated the coefficient of variation (CV) and expression levels of genes across different groups using the GTEx tissue dataset. The CV of the genes with sharp CHD6 peaks was significantly lower than that of other genes (Figure 3A). Moreover, we observed significant decreases in the expression levels of genes with sharp CHD6 peaks compared with those with broad CHD6 peaks (Figure S3A). These expression patterns resembled those of HK genes, which are characterized by low but stable expression levels.50 Therefore, our findings suggest that genes with sharp CHD6 peaks and HK genes share similar expression characteristics.
Figure 3.

Expression levels of genes with sharp CHD6 peaks
(A) Coefficient of variation around groups. (B) Average MNase-seq read density plotted around genes. (C) Average ChIP-seq signal value of H3K9me3 plotted around genes. (D) Average ChIP-seq signal value of H3K27me3 plotted around genes. (E) The logFC (y axis) change in the genes with sharp and broad CHD6 peaks after CHD6 knockdown. (F) Nucleosome density of genes with sharp CHD6 peaks in both the C4-2 control cells and the CHD6 knockdown cells. (G) Comparison of gene expression levels between normal and cancer cell lines. p values determined using one-tailed Wilcoxon test. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Given that CHD6 is an ATPase-dependent nucleosome remodeler that influences gene expression levels, we reasoned that CHD6 depletion might alter the occupancy of nucleosome at CHD6 binding sites. To map nucleosome occupancy on chromatin, we used micrococcal nuclease digestion with deep sequencing (MNase-seq) in both the C4-2 control and CHD6 knockdown cells. The occupancy of nucleosomes was higher in the genes with sharp peaks than in those with broad CHD6 peaks (Figure 3B) in the C4-2 control cells. The nucleosome occupancy was increased significantly across the promoters of sharp CHD6 peaks compared with the promoters of broad CHD6 peaks (Figure S3B). High nucleosome occupancy is often associated with gene silencing and can affect gene expression levels, thereby influencing the cell function.51 H3K9me3 and H3K27me3, as common repressive histone modifications,52,53 exhibited higher signals in gene bodies with sharp CHD6 peaks than in those with broad CHD6 peaks (Figures 3C and 3D). To further confirm the impact of nucleosome density on the expression levels of genes characterized by different CHD6 binding patterns, we performed fold change (FC) analyses of gene expression levels between CHD6 knockdown and control cells. The results revealed that CHD6 knockdown led to the downregulation of the expression levels of genes with broad peaks and to minimal or no change in the expression levels of genes with sharp peaks (Figure 3E). Moreover, nucleosome occupancy at CHD6 binding sites with sharp peaks showed no change in response to CHD6 knockdown (Figure 3F), while significant alterations were observed across broad CHD6 peaks in promoter and gene body regions (Figure S3C). These observations suggest that CHD6 may regulate the density of nucleosomes at promoters, thereby controlling the low but stable expression levels of genes associated with sharp CHD6 peaks.
To further understand sharp CHD6 peaks beyond the C4-2 cell line, we collected RNA-seq data from normal (RWPE1, BPH1, and PrEC) and cancer cell lines (VCaP, LNCaP, 22RV1, PC3, and DU145) associated with the prostate. A significant decrease in expression level was observed in the genes with sharp CHD6 peaks compared with normal cell lines in most cancer cell lines (Figure 3G). Conversely, gene expression levels significantly increased in the genes with broad CHD6 peaks within the cancer cell lines compared with the normal cell lines (Figure S3D). Previous studies have suggested that the expression levels of HK genes undergo changes during the transition from normal to tumor cells.30,31 Taking these data together, we conclude that genes characterized by sharp CHD6 peaks sustain low but stable expression levels, showing an expression pattern that is similar to that of HK genes.
Enriched motif of HK transcription factor in the genes with sharp CHD6 peaks
To examine the preferred binding motifs associated with distinct binding patterns of CHD6 and to elucidate the mechanisms involved in gene expression regulation, we performed a motif analysis on the regions associated with sharp and broad CHD6 peaks. We found that sharp CHD6 peaks were strongly enriched with the TFE3, USF2, USF1, and ETS1 motifs (Figure 4A), whereas broad CHD6 peaks were enriched with the ZNF711 and ZFX motifs (Figure 4B). TFE3 transcription factors belong to the microphthalmia family and are master regulators of organelle signaling, metabolism, and stress adaptation.54 Previous studies have indicated that the functional domain of the TFE3 gene typically fuses with the promoter region of HK genes, which is a critical event for the occurrence of Xp11.2-translocation renal cell carcinoma.55 USF2 and USF1 transcription factors, as determined by separate analyses using weight matrix collections of vertebrate transcription factor binding sites from TRANSFAC and PROMO, were predicted binding motifs that are overrepresented in HK gene promoters.56 A previous study showed that ZNF711 may play a subordinate role in ZFX, which acts as a transcriptional activator in various types of human tumors.57 These results indicate that the expression levels of genes with sharp CHD6 peaks are regulated by HK transcription factors.
Figure 4.

Characterization of sharp CHD6 peaks motifs and transcription factors
(A) Volcano plots of motif enrichment scores (percentage enrichment/percentage background) of the sharp CHD6 peaks compared with the log10 (p value). (B) Volcano plots of motif enrichment scores of the broad CHD6 peaks compared with the log10 (p value). (C) Distribution of ETS1 binding sites. (D) Venn diagram showing the overlap between the sharp CHD6 peaks and ETS1 binding peaks.
To gain further support for the results that genes with sharp CHD6 peaks are regulated by the HK transcription factor, we collected ChIP-seq for ETS1 in the DU145 cell line. The distribution of all peaks represented the binding of ETS1 across the entire genome, indicating the regions where ETS1 interacted with the DNA. Among the total peaks, a notable enrichment in promoter regions is shown in the pie chart in Figure 4C. Consistent with our hypothesis, a striking 76% of the sharp CHD6 peaks were found to coincide with the regions where ETS1 binds in the promoter regions (Figure 4D). ETS1 and GABPA, both members of the ETS family transcription factor, were found to redundantly occupy promoters of HK genes in T cells.58 The above studies indirectly suggested that the genes with sharp CHD6 peaks are associated with HK programs.
Sharp CHD6 marks HK genes in normal cells
Based on the results of the analysis of the genes with sharp CHD6 peaks in cancer cells, we supposed that sharp CHD6 peaks might also exist in normal cells. Therefore, we tested this hypothesis in two normal cells, HEK293 and cardiomyocytes. We observed remarkably consistent binding patterns of CHD6 in the normal cells resembling those observed in the cancer cells, which were characterized by high-density CHD6 peaks predominantly located on promoters, exhibiting narrow profiles. By contrast, the low-density CHD6 peaks were notably broader, spanning both promoter and gene body regions (Figure 5A). KEGG enrichment analysis revealed that pathways associated with mitophagy (hsa04137), autophagy (hsa04140) and apoptosis (hsa04210) were significantly enriched in the genes with sharp CHD6 peaks, but not in those with broad peaks or the control peaks (Figure 5B). The CHD6 peaks displaying sharp profiles in the promoter regions were found to be specifically enriched in HK genes, while lacking enrichment in OGs or TSGs (Figure 5C).
Figure 5.

Characterization of genes with sharp CHD6 peaks in normal cells
(A) Average signal value of CHD6 peaks plotted around different genes in normal cells. (B) KEGG pathway enrichment of genes with sharp, broad, and control peaks in HEK293 and cardiomyocytes. (C) Enrichment p values (y axis) of the HK genes in HEK293 and cardiomyocytes. p values determined using Fisher's test exact test.
To determine whether the CHD6 binding pattern is conserved across different cell types, we identified 253 overlapping sharp CHD6 genes between HEK293 cells and cardiomyocytes (Table S2). These overlapping genes were significantly enriched in HK genes (Figure S4A). In addition, gene functional enrichment analysis using DAVID revealed strong associations with apoptosis and autophagy pathways (Figure S4B), which are both linked to HK processes. These findings provide compelling evidence that the observed pattern of sharp CHD6 peaks is closely linked to the HK regulatory network, a phenomenon consistently observed in both cancer and normal cells.
Discussion
Chromatin remodelers are transcription regulators and play crucial roles in developmental processes.59 Recent studies have indicated that developmental and HK gene transcription were regulated by distinct chromatin remodelers, highlighting the connection between chromatin structure and functional properties.60 In this study, we have identified genes that characterize exclusive CHD6 binding at their promoter regions, namely sharp CHD6 peaks, which are preferentially enriched at HK genes within the C4-2 cell line. Our findings also show CHD6 binding in both the promoter and gene body regions of the genes, which suggests their potential as OGs, consistent with the findings of previous research.24 The discovery of sharp CHD6 peaks, enriched at promoter regions of HK genes, implies the presence of a specialized regulatory mechanism that ensures the maintenance of essential cellular functions. Moreover, our results underscore the significance of CHD6 in prostate cancer progression, as evidenced by the differential expression levels of genes associated with sharp CHD6 peaks in cancer compared with normal prostate cells. Overall, our findings shed light on the role of CHD6 in modulating the expression levels of HK genes, thereby influencing fundamental cellular processes and potentially contributing to the rapid proliferation of cancer cells.
Different from broad CHD6 peaks, sharp CHD6 peaks are an indicator of decreased transcription elongation and enhancer inactivity. The distinct chromatin structure associated with sharp CHD6 peaks, which are characterized by increased nucleosome density and repressive histone modifications, suggests a mechanism for gene silencing and decreased transcriptional activity. In addition, the absence of significant enrichment in broad H3K4me3 modifications and SEs, which are characteristic of broad peaks, in sharp CHD6 peaks further supports the result of diminished enhancer activity. The lower levels of chromatin accessibility observed in sharp CHD6 peaks indicate a reduced activation state that may influence transcription machinery and gene expression.
Our study provides valuable insights into the regulatory role of CHD6 in PC, but our analysis focused primarily on coding genes, overlooking the potential regulatory roles of CHD6 in non-coding regions. In addition, beyond PC, we demonstrated the presence of a sharp CHD6 signature in the HK in normal cells. In future studies, we can further examine the potential regulatory role of CHD6 in noncoding regions and investigate its universality in other cancer types and normal tissues. This comprehensive investigation will deepen our understanding of how CHD6 regulates gene expression and tumor development.
In summary, our study emphasizes the role of sharp CHD6 peaks in maintaining the expression levels of HK genes, which are essential for sustaining life. Moreover, our findings elucidate the distinct CHD6 binding models involved in shaping the chromatin structure and participating in various transcriptional programs. Overall, these findings deepen our understanding of the molecular mechanisms driven by CHD6, shedding light on essential cellular processes and their role in cancer or disease development. Importantly, they provide a foundation for future therapeutic interventions targeting chromatin remodelers in cancer treatment.
Materials and methods
Cell line and cell culture
The cell line HEK293 was purchased from the American Type Culture Collection and grown in DMEM complete medium with 10% fetal bovine serum. The cells were incubated at 37°C with 5% CO2 and continuously cultured for less than 2 months. Cell lines were mycoplasma negative in routine tests.
Cleavage under targets and tagmentation
The cleavage under targets and tagmentation (CUT&Tag) assay (N259-YH01, Novoprotein Scientific) was performed in accordance with the manufacturer’s instructions. Concanavalin A-conjugated magnetic beads were activated with binding buffer and incubated with washed cells. The cell-bead complex was incubated with antibodies, washed, and subjected to tagmentation with hyperactive pA-Tn5 transposase. After tagmentation, DNA was purified and libraries were prepared using the NovoNGS CUT&Tag 4.0 High-Sensitivity Kit. The libraries were then quantified and amplified. Sequencing was performed using Illumina NovaSeq PE150.
ChIP-seq analysis
ChIP-seq raw reads were mapped to the human genome version hg19 using Bowtie version 1.2.2 with default parameter values.61 We then submitted the mapped reads to the Dpeak function in DANPOS version 2.2.2 (https://sites.google.com/site/danposdoc/) to calculate the ChIP-seq signal (read density) at each base pair of the genome,62 subtract background (input) signal, normalize read number, and define individual enrichment peaks. The Dpeak stored the signal value at each base pair in a Wiggle format file, which we next converted to bigWig format using the tool wigToBigWig (https://www.encodeproject.org/software/wigtobigwig/). The Dpeak also stored individual feature values for each enrichment peak of the ChIP-seq signal. These feature values include peak width, height, and total signal. To calculate signal value at each base pair across each gene, we used the Profile function in DANPOS version 2.2.2. The Profile function in DANPOS 2.2.2 was also used to calculate the average ChIP-seq signal at each gene group. The colocalization of genomic loci was investigated with the Integrative Genomics Viewer.
CUT&Tag analysis
Approximately 1 × 105 cells were used for CHD6 (Bethyl, A301-221A) CUT&Tag assay. The raw sequencing image data were examined by the Illumina NovaSeq analysis pipeline. Before read mapping, clean reads were obtained from the raw reads by removing the adaptor sequences. The clean reads were then aligned to the unmasked human genome version hg19 using Bowtie2 version 2.4.563 and further analyzed by DANPOS version 2.2.2 (https://sites.google.com/site/danposdoc/).
RNA-seq and scRNA-seq analysis
RNA-seq raw reads were mapped to human genome version hg19 using TopHat version 2.1.1 with default parameter values.61 The expression value (number of raw reads) for each gene was determined by the software HTSeq version 0.9.1 with default parameter values.61 Normalized (the trimmed mean of M-values method) expression values and differentially expressed genes were determined by edgeR version 3.10.5 run with an R version 3.2.1. We used the tool bedGraphToBigWig (https://www.encodeproject.org/software/bedgraphtobigwig/) to generate a bigWig file that contains RNA-seq signal (read density) at each base pair across the genome.
The scRNA-seq data on patients with PC were accessed in the NCBI GEO database under accession number GSE137829 and analyzed by Seurat (version 4.2.1) and an R toolkit (https://github.com/satijalab/seurat), using the software R (version 4.3.1). CopyKAT R package64 is utilized to calculate single-cell copy-number profiles from 10× single-cell RNA data and predict tumor and normal cells.
MNase-seq analysis
MNase-seq raw reads were mapped to the human genome version hg19 using Bowtie version 1.2.2 with default parameter values.61 We then submitted the mapped reads to the Dpos function (parameter: -smooth_width 0 -c 50000000 -u 1 -pheight 1e−200) in DANPOS version 2.2.2 (https://sites.google.com/site/danposdoc/) to calculate the MNase-seq signal (read density) at each base pair of the genome. The pipeline to observe the MNase-seq read density is the same as that described above for ChIP-seq. The Profile function in DANPOS version 2.2.2 was also used to calculate the average MNase-seq signal at each gene group.
Calculation of the RNA Pol II pausing index
We defined the pausing region as the region from 50 bp upstream to 300 bp downstream of the TSS and defined the elongation region as the region from 300 bp downstream of the TSS to 1 kb downstream of the TTS. We also normalized the read counts by the total number of mapped reads, presenting the results as reads per million. We then calculated the pausing index as the pausing-to-elongation ratio of RNA Pol II ChIP-seq read density.
Identification of SEs
SEs were identified using the ROSE tool.65 Briefly, individual enhancers within 12.5 kb of one another were stitched together to form a single larger enhancer domain. Stitched enhancer domains were then ranked for input-normalized ChIP-seq occupancy of CHD6. The point on the x axis at which the tangent to a scaled graph with the x and y axes ranging from 0 to 1 that had a slope of 1 was used as a cutoff, above which stitched enhancers were classified as SEs.
Motif analysis
Motif enrichment analysis was performed using the findMotifsGenome.pl program in the HOMER software suite.66
Function enrichment analysis
Gene Ontology and KEGG pathway analyses were performed using the DAVID database version 6.8 (https://david.ncifcrf.gov). Pathways with p values smaller than 0.05 were defined as significantly enriched. Enrichment levels for HK genes, tumor suppressors, and OGs were defined based on Fisher’s exact tests.
Statistical analysis
For bar plots and boxplots, p values were calculated using Wilcoxon’s test and Fisher’s exact test. Differences were considered significant when the p value was <0.05 (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
Data and code availability
The CUT&Tag sequencing data have been deposited to GEO (https://www.ncbi.nlm.nih.gov/geo/), with the accession number GSE264397. The RNA-seq, scRNA-seq, MNase-seq, H3K4me3, H3K4me1, H3K27ac, H3K9me3, and H3K27me3 ChIP-seq data for the cell types of prostate tissue were downloaded from the GEO database project website (https://www.ncbi.nlm.nih.gov/geo/). Table S1 contains all data information used in this study. All data are available in the article and its supplemental information.
Acknowledgments
This work was supported by the National Key R&D Program of China (2021YFF1201100), the National Natural Science Foundation of China (32270603) and the Fundamental Research Funds for the Central Universities (BMU2021YJ057).
Author contributions
D.Z. and L.B.: conceptualization, resources, supervision, funding acquisition, methodology, and writing – review & editing. S.H.: data curation, data analysis, and writing – review & editing. Z.R. and C.W.: investigation, methodology, and software. B.-Y.S.: investigation and software. Y.L. and L.H.: methodology, validation, and experiments. All authors reviewed the results and approved the final version of the manuscript.
Declaration of interests
The authors declare no conflicts of interest that pertain to this work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102397.
Supplemental information
References
- 1.Bernal A., Bechler A.J., Mohan K., Rizzino A., Mathew G. The Current Therapeutic Landscape for Metastatic Prostate Cancer. Pharmaceuticals. 2024;17 doi: 10.3390/ph17030351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sekhoacha M., Riet K., Motloung P., Gumenku L., Adegoke A., Mashele S. Prostate Cancer Review: Genetics, Diagnosis, Treatment Options, and Alternative Approaches. Molecules. 2022;27 doi: 10.3390/molecules27175730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braga-Basaria M., Muller D.C., Carducci M.A., Dobs A.S., Basaria S. Lipoprotein profile in men with prostate cancer undergoing androgen deprivation therapy. Int. J. Impot. Res. 2006;18:494–498. doi: 10.1038/sj.ijir.3901471. [DOI] [PubMed] [Google Scholar]
- 4.Rebello R.J., Oing C., Knudsen K.E., Loeb S., Johnson D.C., Reiter R.E., Gillessen S., Van der Kwast T., Bristow R.G. Prostate cancer. Nat. Rev. Dis. Prim. 2021;7:9. doi: 10.1038/s41572-020-00243-0. [DOI] [PubMed] [Google Scholar]
- 5.Tyagi M., Imam N., Verma K., Patel A.K. Chromatin remodelers: We are the drivers. Nucleus. 2016;7:388–404. doi: 10.1080/19491034.2016.1211217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reyes A.A., Marcum R.D., He Y. Structure and Function of Chromatin Remodelers. J. Mol. Biol. 2021;433 doi: 10.1016/j.jmb.2021.166929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Centore R.C., Sandoval G.J., Soares L.M.M., Kadoch C., Chan H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020;36:936–950. doi: 10.1016/j.tig.2020.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Hankey W., Chen Z., Wang Q. Shaping Chromatin States in Prostate Cancer by Pioneer Transcription Factors. Cancer Res. 2020;80:2427–2436. doi: 10.1158/0008-5472.CAN-19-3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H., Gigi L., Zhao D. CHD1, a multifaceted epigenetic remodeler in prostate cancer. Front. Oncol. 2023;13 doi: 10.3389/fonc.2023.1123362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marfella C.G.A., Imbalzano A.N. The Chd family of chromatin remodelers. Mutat. Res. 2007;618:30–40. doi: 10.1016/j.mrfmmm.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sims R.J., Chen C.F., Santos-Rosa H., Kouzarides T., Patel S.S., Reinberg D. Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J. Biol. Chem. 2005;280:41789–41792. doi: 10.1074/jbc.C500395200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flanagan J.F., Mi L.Z., Chruszcz M., Cymborowski M., Clines K.L., Kim Y., Minor W., Rastinejad F., Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 13.Flanagan J.F., Blus B.J., Kim D., Clines K.L., Rastinejad F., Khorasanizadeh S. Molecular implications of evolutionary differences in CHD double chromodomains. J. Mol. Biol. 2007;369:334–342. doi: 10.1016/j.jmb.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hauk G., Bowman G.D. Structural insights into regulation and action of SWI2/SNF2 ATPases. Curr. Opin. Struct. Biol. 2011;21:719–727. doi: 10.1016/j.sbi.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mills A.A. The Chromodomain Helicase DNA-Binding Chromatin Remodelers: Family Traits that Protect from and Promote Cancer. Csh Perspect Med. 2017;7 doi: 10.1101/cshperspect.a026450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colbert L.E., Petrova A.V., Fisher S.B., Pantazides B.G., Madden M.Z., Hardy C.W., Warren M.D., Pan Y., Nagaraju G.P., Liu E.A., et al. CHD7 Expression Predicts Survival Outcomes in Patients with Resected Pancreatic Cancer. Cancer Res. 2014;74:2677–2687. doi: 10.1158/0008-5472.CAN-13-1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim M.S., Chung N.G., Kang M.R., Yoo N.J., Lee S.H. Genetic and expressional alterations of genes in gastric and colorectal cancers. Histopathology. 2011;58:660–668. doi: 10.1111/j.1365-2559.2011.03819.x. [DOI] [PubMed] [Google Scholar]
- 18.Caldon C.E., Sergio C.M., Schütte J., Boersma M.N., Sutherland R.L., Carroll J.S., Musgrove E.A. Estrogen regulation of cyclin E2 requires cyclin D1 but not c-Myc. Mol. Cell Biol. 2009;29:4623–4639. doi: 10.1128/MCB.00269-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bagchi A., Papazoglu C., Wu Y., Capurso D., Brodt M., Francis D., Bredel M., Vogel H., Mills A.A. is a tumor suppressor at human. Cell. 2007;128:459–475. doi: 10.1016/j.cell.2006.11.052. [DOI] [PubMed] [Google Scholar]
- 20.March H.N., Rust A.G., Wright N.A., ten Hoeve J., de Ridder J., Eldridge M., van der Weyden L., Berns A., Gadiot J., Uren A., et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat. Genet. 2011;43:1202–1209. doi: 10.1038/ng.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore S., Berger N.D., Luijsterburg M.S., Piett C.G., Stanley F.K.T., Schräder C.U., Fang S., Chan J.A., Schriemer D.C., Nagel Z.D., et al. The CHD6 chromatin remodeler is an oxidative DNA damage response factor. Nat. Commun. 2019;10 doi: 10.1038/s41467-018-08111-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kargapolova Y., Rehimi R., Kayserili H., Brühl J., Sofiadis K., Zirkel A., Palikyras S., Mizi A., Li Y., Yigit G., et al. Overarching control of autophagy and DNA damage response by CHD6 revealed by modeling a rare human pathology. Nat. Commun. 2021;12 doi: 10.1038/s41467-021-23327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B., Liu Q., Wen W., Gao H., Wei W., Tang A., Qin B., Lyu H., Meng X., Li K., et al. The chromatin remodeler CHD6 promotes colorectal cancer development by regulating TMEM65-mediated mitochondrial dynamics via EGF and Wnt signaling. Cell Discov. 2022;8 doi: 10.1038/s41421-022-00478-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao D., Zhang M., Huang S., Liu Q., Zhu S., Li Y., Jiang W., Kiss D.L., Cao Q., Zhang L., Chen K. CHD6 promotes broad nucleosome eviction for transcriptional activation in prostate cancer cells. Nucleic Acids Res. 2022;50:12186–12201. doi: 10.1093/nar/gkac1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenberg E., Levanon E.Y. Human housekeeping genes, revisited. Trends Genet. 2013;29:569–574. doi: 10.1016/j.tig.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 26.Zhu J., He F., Hu S., Yu J. On the nature of human housekeeping genes. Trends Genet. 2008;24:481–484. doi: 10.1016/j.tig.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 27.de Kok J.B., Roelofs R.W., Giesendorf B.A., Pennings J.L., Waas E.T., Feuth T., Swinkels D.W., Span P.N. Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab. Invest. 2005;85:154–159. doi: 10.1038/labinvest.3700208. [DOI] [PubMed] [Google Scholar]
- 28.Lee P.D., Sladek R., Greenwood C.M.T., Hudson T.J. Control genes and variability: Absence of ubiquitous reference transcripts in diverse mammalian expression studies. Genome Res. 2002;12:292–297. doi: 10.1101/gr.217802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barber R.D., Harmer D.W., Coleman R.A., Clark B.J. GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genom. 2005;21:389–395. doi: 10.1152/physiolgenomics.00025.2005. [DOI] [PubMed] [Google Scholar]
- 30.Byun J., Logothetis C.J., Gorlov I.P. Housekeeping genes in prostate tumorigenesis. Int. J. Cancer. 2009;125:2603–2608. doi: 10.1002/ijc.24680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang Y.C., Ding Y., Dong L., Zhu L.J., Jensen R.V., Hsiao L.L. Differential expression patterns of housekeeping genes increase diagnostic and prognostic value in lung cancer. PeerJ. 2018;6 doi: 10.7717/peerj.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gundu C., Arruri V.K., Yadav P., Navik U., Kumar A., Amalkar V.S., Vikram A., Gaddam R.R. Dynamin-Independent Mechanisms of Endocytosis and Receptor Trafficking. Cells. 2022;11 doi: 10.3390/cells11162557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marx V. Autophagy: eat thyself, sustain thyself. Nat. Methods. 2015;12:1121–1125. doi: 10.1038/nmeth.3661. [DOI] [PubMed] [Google Scholar]
- 34.Mo Y., Wang Y., Zhang L., Yang L., Zhou M., Li X., Li Y., Li G., Zeng Z., Xiong W., et al. The role of Wnt signaling pathway in tumor metabolic reprogramming. J. Cancer. 2019;10:3789–3797. doi: 10.7150/jca.31166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lauberth S.M., Nakayama T., Wu X., Ferris A.L., Tang Z., Hughes S.H., Roeder R.G. H3K4me3 Interactions with TAF3 Regulate Preinitiation Complex Assembly and Selective Gene Activation. Cell. 2013;152:1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu S., Song A., Peng L., Tang N., Qiao Z., Wang Z., Lan F., Chen F.X. H3K4me2/3 modulate the stability of RNA polymerase II pausing. Cell Res. 2023;33:403–406. doi: 10.1038/s41422-023-00794-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H., Fan Z., Shliaha P.V., Miele M., Hendrickson R.C., Jiang X., Helin K. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature. 2023;615:339–348. doi: 10.1038/s41586-023-05780-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z., Shi L., Dawany N., Kelsen J., Petri M.A., Sullivan K.E. H3K4 tri-methylation breadth at transcription start sites impacts the transcriptome of systemic lupus erythematosus. Clin. Epigenet. 2016;8:14. doi: 10.1186/s13148-016-0179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins B.E., Sweatt J.D., Greer C.B. Broad domains of histone 3 lysine 4 trimethylation are associated with transcriptional activation in CA1 neurons of the hippocampus during memory formation. Neurobiol. Learn. Mem. 2019;161:149–157. doi: 10.1016/j.nlm.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park S., Kim G.W., Kwon S.H., Lee J.S. Broad domains of histone H3 lysine 4 trimethylation in transcriptional regulation and disease. FEBS J. 2020;287:2891–2902. doi: 10.1111/febs.15219. [DOI] [PubMed] [Google Scholar]
- 42.Sayed D., He M., Yang Z., Lin L., Abdellatif M. Transcriptional regulation patterns revealed by high resolution chromatin immunoprecipitation during cardiac hypertrophy. J. Biol. Chem. 2013;288:2546–2558. doi: 10.1074/jbc.M112.429449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao C., Fan T., Tian H., Zheng Y., Zhou Z., Li S., Li C., He J. H3K36 trimethylation-mediated biological functions in cancer. Clin. Epigenet. 2021;13:199. doi: 10.1186/s13148-021-01187-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu S., Li J., Ji G., Ng Z.L., Siew J., Lo W.N., Ye Y., Chew Y.Y., Long Y.C., Zhang W., et al. Npac Is A Co-factor of Histone H3K36me3 and Regulates Transcriptional Elongation in Mouse Embryonic Stem Cells. Dev. Reprod. Biol. 2022;20:110–128. doi: 10.1016/j.gpb.2020.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu I., Landsman D. Clustered and diverse transcription factor binding underlies cell type specificity of enhancers for housekeeping genes. Genome Res. 2023;33:1662–1672. doi: 10.1101/gr.278130.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beacon T.H., Delcuve G.P., López C., Nardocci G., Kovalchuk I., van Wijnen A.J., Davie J.R. The dynamic broad epigenetic (H3K4me3, H3K27ac) domain as a mark of essential genes. Clin. Epigenet. 2021;13:138. doi: 10.1186/s13148-021-01126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hughes A.L., Kelley J.R., Klose R.J. Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation. Bba-Gene Regul Mech. 2020;1863 doi: 10.1016/j.bbagrm.2020.194567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian H., He Y., Xue Y., Gao Y.Q. Expression regulation of genes is linked to their CpG density distributions around transcription start sites. Life Sci. Alliance. 2022;5 doi: 10.26508/lsa.202101302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joshi C.J., Ke W., Drangowska-Way A., O'Rourke E.J., Lewis N.E. What are housekeeping genes? PLoS Comput. Biol. 2022;18 doi: 10.1371/journal.pcbi.1010295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hesson L.B., Sloane M.A., Wong J.W., Nunez A.C., Srivastava S., Ng B., Hawkins N.J., Bourke M.J., Ward R.L. Altered promoter nucleosome positioning is an early event in gene silencing. Epigenetics. 2014;9:1422–1430. doi: 10.4161/15592294.2014.970077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim J., Kim H. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J. 2012;53:232–239. doi: 10.1093/ilar.53.3-4.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou C., Halstead M.M., Bonnet-Garnier A., Schultz R.M., Ross P.J. Histone remodeling reflects conserved mechanisms of bovine and human preimplantation development. EMBO Rep. 2023;24 doi: 10.15252/embr.202255726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slade L., Pulinilkunnil T. The MiTF/TFE Family of Transcription Factors: Master Regulators of Organelle Signaling, Metabolism, and Stress Adaptation. Mol. Cancer Res. 2017;15:1637–1643. doi: 10.1158/1541-7786.MCR-17-0320. [DOI] [PubMed] [Google Scholar]
- 55.Zhuang W., Dong X., Wang B., Liu N., Guo H., Zhang C., Gan W. NRF-1 directly regulates TFE3 and promotes the proliferation of renal cancer cells. Oncol. Lett. 2021;22:679. doi: 10.3892/ol.2021.12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farré D., Bellora N., Mularoni L., Messeguer X., Albà M.M. Housekeeping genes tend to show reduced upstream sequence conservation. Genome Biol. 2007;8:1–10. doi: 10.1186/gb-2007-8-7-r140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rhie S.K., Yao L., Luo Z., Witt H., Schreiner S., Guo Y., Perez A.A., Farnham P.J. ZFX acts as a transcriptional activator in multiple types of human tumors by binding downstream from transcription start sites at the majority of CpG island promoters. Genome Res. 2018;28:310–320. doi: 10.1101/gr.228809.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hollenhorst P.C., Chandler K.J., Poulsen R.L., Johnson W.E., Speck N.A., Graves B.J. DNA specificity determinants associate with distinct transcription factor functions. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang F.L., Li D.Q. Targeting Chromatin-Remodeling Factors in Cancer Cells: Promising Molecules in Cancer Therapy. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms232112815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hendy O., Serebreni L., Bergauer K., Muerdter F., Huber L., Nemcko F., Stark A. Developmental and housekeeping transcriptional programs in require distinct chromatin remodelers. Mol. Cell. 2022;82:3598-+. doi: 10.1016/j.molcel.2022.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen K., Xi Y., Pan X., Li Z., Kaestner K., Tyler J., Dent S., He X., Li W. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013;23:341–351. doi: 10.1101/gr.142067.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gao R., Bai S., Henderson Y.C., Lin Y., Schalck A., Yan Y., Kumar T., Hu M., Sei E., Davis A., et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat. Biotechnol. 2021;39:599–608. doi: 10.1038/s41587-020-00795-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The CUT&Tag sequencing data have been deposited to GEO (https://www.ncbi.nlm.nih.gov/geo/), with the accession number GSE264397. The RNA-seq, scRNA-seq, MNase-seq, H3K4me3, H3K4me1, H3K27ac, H3K9me3, and H3K27me3 ChIP-seq data for the cell types of prostate tissue were downloaded from the GEO database project website (https://www.ncbi.nlm.nih.gov/geo/). Table S1 contains all data information used in this study. All data are available in the article and its supplemental information.