

Abstract

The synthesis of coumarin– and flavonoid–chalcone hybrids via Pd-catalyzed Heck-type coupling of arene diazonium salts and 8-allylcoumarins and −flavonoids is reported. The β-hydride elimination step proceeds with high regioselectivity if an OMOM-substituent is present at the position C7, adjacent to the allyl group. A selective allylic oxidation of the coupling products was accomplished using DDQ in the presence of silica to furnish the chalcones.
Introduction
In medicinal chemistry hybrid molecules are defined as compounds with two or more pharmacologically active molecular entities that are linked through covalent bonds. The different pharmacophores can either bind to the same target, independently to different targets or simultaneously to two or more targets in close proximity.1 For example, trioxaquines are antimalarial drugs in which a 1,2,4-trioxane inspired by artemisinin is linked to a chloroquine moiety. The activity of these hybrids against chloroquine resistant Plasmodium falciparum strains is significantly higher than the activity of the single molecules.1 Many hybrid molecules based on both natural product and synthetic pharmacophores have been developed since the original disclosure of the concept and investigated for numerous anti-infective activities, anti-inflammatory activity and anticancer activity.2 Although some natural products are already hybrid molecules in themselves, it has been reasoned that the synthetic linkage of natural products can further broaden the chemical space of pharmacologically active compounds.3 A natural product scaffold that has been incorporated in many synthetic hybrid structures is the chalcone moiety. Chalcones are naturally occurring 1,3-diarylpropenones, a subgroup of diarylpropanoids, that are mostly found in plants.4 They are biosynthetically derived from coumaryl-CoA and three units of malonyl-CoA and are biosynthetic precursors of flavanones, flavones and isoflavones (Figure 1). For this reason, chalcones accumulate only in a limited number of plants, which, however, play a prominent role in traditional medicine.4
Figure 1.

Benzopyran–chalcone hybrids and their bioactivities.
Bioactivities that have been reported for naturally occurring chalcones range from anti-infective activities (antibiotic, antiviral, antifungal, antiprotozoal activity) over cytotoxic5 to enzyme inhibiting activity (e.g., inhibition of tyrosinase6) and cardio- and neuroprotective effects.4,7 By using strategies such as bioisosterism, pro-drug development and molecular hybridization chalcones have been identified as attractive entry points for the design and development of new lead structures and drugs.8 Hybrids of chalcones and other natural product (e.g., quinolines, artemisinin and coumarins) and synthetic (e.g., azoles and ferrocenes) scaffolds have inter alia been tested as anticancer9−12 and antimalarial13 agents. An example is the synthetic coumarin–chalcone hybrid 1,14 which was named S009-131 and proposed as a promising candidate for the treatment of cervical cancer as it inhibits proliferation of HeLa and C33A cancer cells by apoptosis.15 Compound S009-131 (1) triggers DNA damage through binding to the minor groove via hydrogen bonds and stabilizes the tumor suppressor protein p53.16 Other coumarin–chalcone hybrids were successfully tested for their radical scavenging ability (e.g., 2)17 toward reactive oxygen species (ROS),17−20 for their antibacterial21 and trypanocidal activities (e.g., 3),22 and as selective inhibitors of monoamine oxidase B (MAO-B).23 The pyranochalcone xanthohumol C (4) is a naturally occurring chromene–chalcone hybrid isolated from hops that shows neuroprotective activity,24 and desmosflavans A and B (5)25 are diastereomeric plant metabolites with a flavane–chalcone hybrid structure (Figure 1).
Most syntheses of coumarin–chalcone hybrids8 rely on classical carbonyl condensation methods, such as Claisen–Schmidt-condensation,14 Knoevenagel-condensation/cyclization sequences18,22 or the Friedel–Crafts acylation of coumarins and subsequent Claisen–Schmidt-condensation.17 The development of alternative approaches would open up the opportunity to use other starting materials and reaction conditions and access substitution patterns that can not, or only with difficulties, be realized through conventional carbonyl condensation chemistry.
We26−28 and others29−34 have investigated the Pd-catalyzed Heck-type arylation using arene diazonium salts as electrophilic coupling partners, commonly referred to as Matsuda–Heck reaction,35 for several years.36−38 This reaction allows the regio- and stereoselective arylation of olefins at ambient temperature under ligand-free conditions in the absence of strong bases or even under base-free conditions. A remarkable feature is the possibility to introduce electron-rich arenes, including unprotected phenols,39 which is particularly important for the synthesis of chalcones and other diarylpropanoids. With a view to the synthesis of diarylpropanoid–coumarin and diarylpropanoid–flavonoid hybrids with the general structure 9 we investigated the Matsuda–Heck-arylation of 8-allylbenzopyranones 6 (i.e., coumarins, flavanones, flavones and isoflavones) and arene diazonium salts 7. After oxidative addition of the electrophilic coupling partner 7 to the Pd(0) catalyst and migratory insertion into the C–C-double bond a cationic Pd–II-σ-complex 8 is formed, which undergoes β-hydride elimination either with Hβ to the product 9β or with Hβ′ to the product 9β′. The Heck-coupling products 9 will then be converted to chalcone–coumarin or chalcone–flavonoid hybrids 10 through allylic oxidation, a reaction that might also be associated with a regioselectivity issue (Scheme 1).
Scheme 1. Possible Products of Matsuda–Heck Reactions for 8-Allylbenzopyranones 6.

In this work, we investigate whether the regioselectivity issues described above can be controlled, either by choosing appropriate reaction conditions or by varying the substituent R adjacent to the olefin, and whether the sequence of Pd-catalyzed arylation and allylic oxidation opens up a synthetically useful route to chalcone hybrids.
Results and Discussion
Optimization of Conditions for the Coupling of Arene Diazonium Salts with Allylarenes
As an entry point toward benzopyrane–chalcone hybrids we identified the MOM-protected 8-allylcoumarin 11a as an olefinic coupling partner. This compound was available from previous projects in our group directed at the synthesis of prenylated coumarin natural products from plants.40 We knew from our earlier experience with Matsuda–Heck reactions that different types of olefins require a specific optimization of reaction conditions, but that the number of variables is fortunately limited: most Matsuda–Heck reactions with electron-deficient alkenes (e.g., acrylates) work well in alcohols (preferably methanol),27 whereas for electron-rich alkenes, such as enolethers, better results are obtained in acetonitrile.28 For related arene substituted alkenes (styrenes, allylbenzene) THF41 and N,N-dimethylacetamide42 have been used as solvents, which were therefore also included in this study. The second important variable is the presence or absence of a base. In those cases where basic conditions are preferable sodium acetate is normally added to the reaction mixture.
For optimization purposes we have used in this and earlier studies the 4-methoxyphenyl diazonium salt 7f, because the symmetry of the 4-methoxyphenyl substituent simplifies NMR-spectroscopic analysis of crude reaction mixtures and therefore allows rates of conversion to be estimated. The experiments summarized in Table 1 revealed that for electron-neutral allylbenzene-type olefins such as 11a, basic conditions are mandatory in all solvents. In the absence of a base (entries 2, 4, 6 and 8) 1H NMR spectra of the reaction solutions pointed at a decomposition of the starting material without formation of any defined product. In these cases, no characteristic signals of either 11a or 12af were observed, and in all cases the MOM-ether was cleaved. Under basic conditions (addition of 3 equiv of NaOAc to the reaction mixture) in DMA (entry 7) only unreacted starting material 11a was observed in the 1H NMR spectrum, whereas in THF (entry 5) conversion to the coupling product 12af was ca. 30%. Synthetically useful rates of conversion were only observed in methanol (entry 1) and in acetonitrile (entry 3). In the latter solvent conversion was quantitative.
Table 1. Screening of Conditions for the Pd-Catalyzed Arylation of 11a.

| entry | solvent | Base (n equiv) | Conversion to 12af (isolated yield)a |
|---|---|---|---|
| 1 | methanol | NaOAc (3.0) | >80b (50) |
| 2 | methanol | – | dec.c |
| 3 | acetonitrile | NaOAc (3.0) | quant. (79) |
| 4 | acetonitrile | – | dec.c |
| 5 | THF | NaOAc (3.0) | <30%b |
| 6 | THF | – | <10%d |
| 7 | DMAe | NaOAc (3.0) | <5b |
| 8 | DMAe | – | dec.c |
Conversion was determined by TLC and 1H NMR spectroscopy of the reaction solution.
Starting material 11 detected in 1H NMR spectrum.
dec: decomposition; no signals of 12af or 11a visible in 1H NMR spectrum.
Signals for 11a with MOM-ether cleavage observed in 1H NMR spectrum.
DMA: N,N-dimethylacetamide.
Regardless of the solvent used, we found that this particular Matsuda–Heck reaction is rather slow compared to many other Pd-catalyzed couplings with arene diazonium salts that were previously investigated in our group. For this reason we routinely used a reaction time of 16 h and employed the diazonium salt in slight excess. Under the optimized conditions (entry 3; acetonitrile as solvent, addition of NaOAc as a base) we obtained the coumarin–phenylpropanoid hybrid 12af after chromatographic purification as a single isomer in 79% yield.
Representative Structure Elucidation of Coupling Product 12af
1H NMR-spectra of the crude reaction mixtures of 11a and 7f (Table 1, entries 1 and 3) revealed that 12af was the predominant product, but that small amounts of other products were also formed. These may arise from decomposition reactions of the diazonium salt, which is used in excess, because additional signals are mainly observed in the aromatic region. The NMR spectra of the reaction mixtures did not point at the formation of any regio- or stereoisomers of the predominant coupling product 12af, because we observed only the signals for the E-configured C=C-double bond of 12af in the olefinic region of the 1H NMR spectrum. Chromatographic purification led to the isolation of the main product as a single isomer with an E-configured double bond, based on a 3J value of 15.8 Hz. It was, however, not possible to assign the position of the double bond based on standard 1D-NMR-spectra alone. Therefore, a full signal assignment based on the 2D-experiments H,H–COSY, HSQC, HMBC and NOESY was performed. Indicative for the assigned structure of 12af, pointing at a β- rather than a β′-selective hydride elimination, are four HMBC interactions. The HMBC experiment is most sensitive for C–H-couplings across three, and to some extent across two bonds. We observed strong cross peaks between the allylic hydrogen atoms (position H1′) and quaternary carbons C7, C8 and C8a located in the coumarin part of the hybrid molecule. A fourth HMBC interaction was observed between the olefinic hydrogen H3′ and carbon C2″ of the 4-methoxyphenyl ring that was introduced by the Pd-catalyzed arylation. Other HMBC interactions (not shown in Figure 2) between the central position of the propanoid chain (H2′) and C8 of the coumarin part and C1″ of the 4-methoxyphenyl substituent are visible but are not suitable to distinguish between the possible regioisomers. This structural assignment was corroborated by two NOE-interactions between the H2″-protons of the 4-methoxyphenyl ring and both olefinic protons in the propene bridge (H2′ and H3′) (Figure 2).
Figure 2.
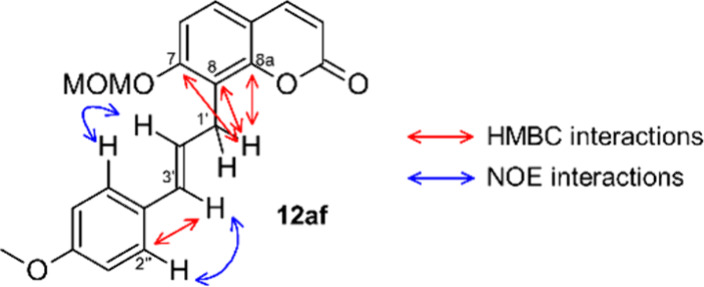
HMBC and NOE interactions in 12af.
Scope and Limitations of the Matsuda–Heck Coupling Reactions with 8-Allylcoumarin 11a
We applied the optimized conditions for the coupling of 11a with 7f to 21 other ortho-, meta-, and para-substituted arene diazonium salts 7 (Table 2). With few exceptions the coupling products 12 were obtained in synthetically useful yields and in all cases as single isomers. We repeated the structure elucidation and full signal assignment process based on 2D-NMR experiments, as described in the preceding paragraph for 12af, for coupling products 12ah, 12ai, 12ap and 12at. With this selection a broad range of electronic and steric substituent effects at the electrophilic coupling partners 7 was covered, from strongly electron donating (para-OCH3, 12af) over moderately electron donating (para-CH3, 12ah) to strongly electron withdrawing (para-CO2Et, 12ai). Compounds 12ap (meta-OCH3), 12at (ortho-OCH3) and 12au (ortho-CO2CH3) result from arene diazonium salts with varying degrees of steric hindrance and electron densities at the reactive site. We reasoned that both electronic and steric effects of the substituents might affect the course of the β-hydride elimination step. This would remain undetected by routine 1H and 13C NMR spectra, as the expected chemical shift values, multiplicities and coupling constants for the propene bridges in 9β and 9β′ are expected to be very similar. For coupling products 12ah, 12ai, 12ap and 12at the same HMBC- and NOE-interactions were observed as for 12af, which indicates that the β-hydride elimination step is not affected by steric or electronic substituent effects, but gives mainly 9β-isomers irrespective of the arene diazonium salt used. Only in the cases of 7j (entry 10; para-NO2, complex mixture of products), 7s (entry 19; ortho-Br, incomplete and sluggish conversion), and 7v (entry 22; ortho-CH3, no conversion) no coupling products 12 could be isolated. In the cases of 7s and 7v significant amounts of unreacted starting material 11a were recovered. The failure of the coupling reaction with most ortho-substituted arene diazonium salts might be explained by steric hindrance.
Table 2. Synthesis of Coupling Products 12: Scope and Limitations.

| entry | 7 | R1 | R2 | R3 | 12 | yield (%) |
|---|---|---|---|---|---|---|
| 1 | 7a | H | H | H | 12aa | 74 |
| 2 | 7b | F | H | H | 12ab | 67 |
| 3 | 7c | Cl | H | H | 12ac | 73 |
| 4 | 7d | Br | H | H | 12ad | 75 |
| 5 | 7e | I | H | H | 12ae | 61 |
| 6 | 7f | OCH3 | H | H | 12af | 79 |
| 7 | 7g | OH | H | H | 12ag | 53 |
| 8 | 7h | CH3 | H | H | 12ah | 71 |
| 9 | 7i | CO2Et | H | H | 12ai | 79 |
| 10 | 7j | NO2 | H | H | 12aj | <5a |
| 11 | 7k | CF3 | H | H | 12ak | 49 |
| 12 | 7l | H | F | H | 12al | 73 |
| 13 | 7m | H | Cl | H | 12am | 72 |
| 14 | 7n | H | Br | H | 12an | 50 |
| 15 | 7o | H | I | H | 12ao | 24 |
| 16 | 7p | H | OCH3 | H | 12ap | 93 |
| 17 | 7q | H | CH3 | H | 12aq | 75 |
| 18 | 7r | H | CO2CH3 | H | 12ar | 69 |
| 19 | 7s | H | H | Br | 12as | <5b |
| 20 | 7t | H | H | OCH3 | 12at | 64 |
| 21 | 7u | H | H | CO2CH3 | 12au | 75 |
| 22 | 7v | H | H | CH3 | 12av | <5b |
Complex mixture of products.
Low conversion (<5%); unreacted starting material recovered.
Mechanistic Rationale for the Observed Regioselectivity
Electronically nonbiased olefins, such as allylarene 11a, normally give mixtures of regioisomeric coupling products in Heck-type arylations, because most catalyst systems are unable to distinguish between the electronically very similar β-hydrogens (designated as Hβ and Hβ′ in Scheme 1).43 A solution to this problem was proposed by Sigman and co-workers, who found that a cationic bimetallic Pd–Cu-catalyst system equipped with a strong σ-donor ligand yields styrene-type coupling products in oxidative Heck-coupling reactions with very high regioselectivity. In this study, arene boronic acids were used as arylating agents.44 The authors reasoned that this catalyst system is sensitive to subtle differences in C–Hβ and C–Hβ′ bond strengths, which are caused by the electronic nature of the substituents at the arene boronic acid. In a follow-up study, Sigman and co-workers investigated the Heck-type coupling of the nonbiased olefin allylbenzene with various arene diazonium salts bearing electron-donating or electron-withdrawing substituents.42 It was found that only with the donor solvent N,N-dimethylacetamide (DMA) high selectivity in the β-hydride elimination step could be accomplished, and that strongly electron-donating substituents at the arene diazonium salt favor the formation of the double bond in conjugation to the newly introduced arene. This selectivity can be explained by a relatively higher hydridic character of the β-hydrogen adjacent to the aryl substituent coming from the diazonium salt.
Our results obtained for the Pd-catalyzed arylation of 11 with arene diazonium salts show some notable differences: a) contrary to the results from the Sigman group, DMA is an unsuitable solvent, but acetonitrile in combination with a base results in high conversions, irrespective of the electronic nature of the diazonium salt (Table 1). This solvent, however, gave poor yields and selectivity in the arylation of allylbenzene, as described by Sigman and co-workers;42 b) as outlined in the previous section, no significant and coherent effects of the electronic nature of the substituents at the diazonium salt on yield and selectivity were observed for the Pd-catalyzed couplings of 11. In all cases, the same regioisomers 12 were obtained, which points at a strongly preferred elimination of Hβ irrespective of the electronic nature of the arylating agent.
The observed selectivity can be explained if a coordination of the cationic and electrophilic Pd atom in Pd-σ-complex 8 (Scheme 1) is assumed. Selectivity control of the β-hydride elimination step through chelation with O- and N-donor groups of the substrate has previously been suggested by White and co-workers to rationalize unusually high levels of regioselectivity in oxidative Heck reactions.45 In our case, chelation might occur through the O-MOM group or through the ring oxygen and/or carbonyl oxygen of the coumarin heterocycle. In both cases the β-hydrogens Hβ will be located at an exo-CH2-group that is connected to the chelate complex via a freely rotatable C–C-bond, which will allow a synperiplanar orientation of Pd and Hβ. On the other hand, for both chelate complexes the β-hydrogens Hβ′ will be located at an endocyclic C atom. This should result in limited conformational flexibility, which will make synperiplanar conformations with the Pd atom less likely (Figure 3).
Figure 3.

Proposed Pd-σ-alkyl chelates.
To test the hypothesis that chelation of the cationic Pd-center contributes to the regioselectivity of the β-hydride elimination step, we first investigated the Matsuda–Heck coupling of 8-allylcoumarin (11b),46 that lacks a coordinating substituent at C-7, with various diazonium salts (7a,d,f,g,h,i,n,r) under our standard conditions (Scheme 2).
Scheme 2. Arylation Reactions without a Chelating OMOM Group.

For all coupling reactions of 11b the regioisomer resulting from hydride elimination of Hβ (β-12b#) is the major product. However, in contrast to the coupling products of 11a, notable amounts of the β′-isomer, in which the double bond is in conjugation with the coumarin part, were detected. The β- and β′-isomers were obtained as inseparable mixtures after chromatography, and the product ratio was determined from the 1H NMR spectrum by integration of the signals for the CH2-group. The ratio of regioisomers (β:β′) is in the range of 3:1 to 4:1 in all cases. All coupling reactions of 11b proceeded rather sluggishly with the formation of several inseparable and unidentified byproducts, which prevented the isolation of pure coupling products and determining reliable yields. We conclude that coordination of the OMOM-group to the electrophilic Pd substantially contributes to the observed selectivity of the β-hydride elimination, but that a coordination to the ring-oxygen and probably the carbonyl oxygen is also a notable contributor to the observed regioselectivity.
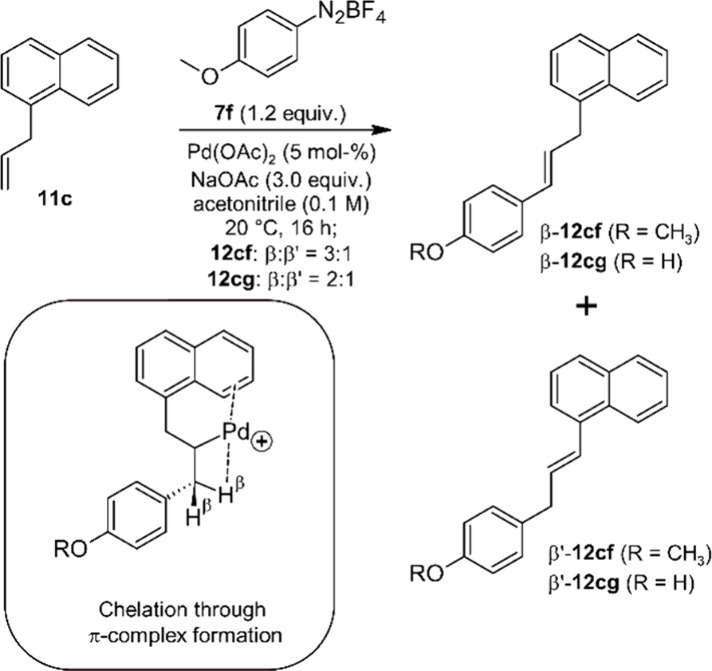
In order to further reduce possible coordinating sites at the allylarene substrate while maintaining the general steric situation of a coumarin, we investigated the regioselectivity of the coupling reaction of 1-allylnaphthalene (11c) and diazonium salts 7f and 7g (Scheme 3). The two regioisomers β-12cf and β′-12-cf were isolated as an inseparable mixture in a ratio (β:β′ = 3:1) very similar to that observed for the coupling reaction of 8-allylcoumarin (11b) with the same diazonium salt 7f. A similar result was obtained for the coupling reaction of 1-allylnaphthalene (11c) with 4-phenoldiazonium salt 7g (ratio β-12cg: β′-12cg = 2:1). These observations suggest that the π-system of the annulated benzene ring coordinates to the electrophilic Pd similar to a ring- and/or carbonyl-oxygen of a coumarin. The major product β-12cf was identified by comparing its NMR data with literature data reported for the same compound, but synthesized independently.47 As for 8-allylcoumarin (11b), both coupling reactions investigated with 11c proceed sluggishly and with incomplete conversion, and with formation of other byproducts that could not be separated from products 12cf and 12cg, respectively. Therefore, a yield can again not be reported for these coupling products, but the ratio of β- and β′-isomer could be determined from the 1H NMR-spectrum through integration of the signals for the CH2-protons. In the particular case of 12cf we also observed a doublet (J = 15.5 Hz) of the C=C-double bond of β′-12cf, which is shifted by 0.5 ppm downfield from the corresponding proton of β-12cf. Integration of these protons confirms the ratio obtained through integration of the CH2-groups.
Scheme 3. Arylation of 1-Allylnaphthalene (11c) with 7f and 7g.
With the aim to rule out any bias of the β-hydride elimination step caused by chelation, we investigated the Heck coupling of 5-allyl-1,2,3,4-tetrahydronaphthalene 11d under our standard conditions. Model substrate 11d was synthesized in four steps from commercially available naphthalene-1-yl-acetic acid (see Supporting Information for details). We assume that the steric situation in test substrate 11d is similar to that of 11b and 11c, but that a chelation of the electrophilic Pd in the transition state of the β-hydride elimination step is not possible (Scheme 4).
Scheme 4. Arylation of 5-Allyl-1,2,3,4-tetrahydronaphthalene (11d) and 7f.
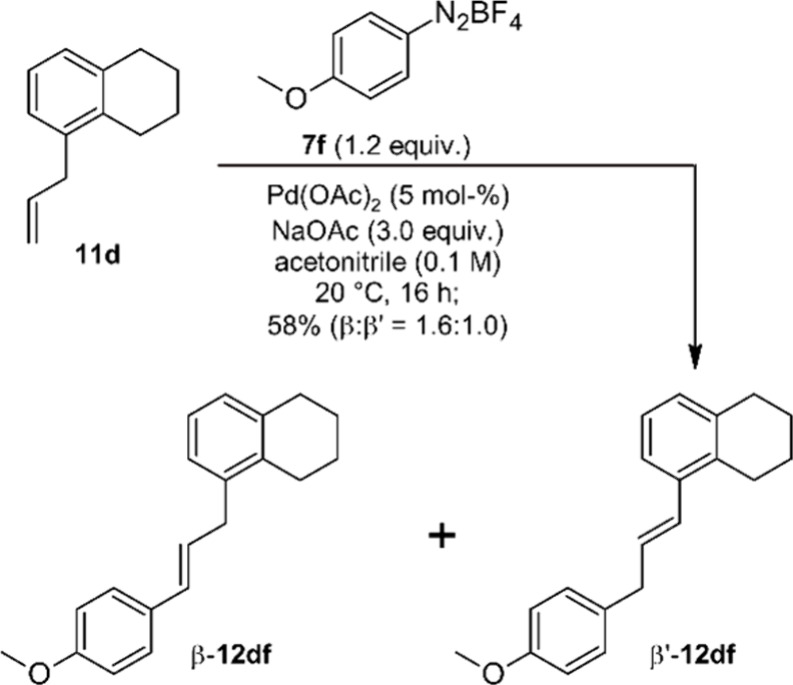
We noticed that under the standard conditions conversion of 11d remained incomplete. Analysis of the product mixture by GC-MS revealed that five isomeric coupling products were formed, and 1H NMR spectroscopy indicates that β-12df and β′-12df are the main products. These isomers are present in a ratio of 1.6:1.0. The slight preference of β-12df is most likely caused by steric effects.
Allylic Oxidation of Heck-Coupling Products 12
Numerous stoichiometric and catalytic methods for allylic and benzylic oxidation reactions are available. These methods48 and their applications in natural product synthesis49 have been reviewed. To identify a suitable method for the envisaged allylic oxidation and optimize the reaction conditions for the task at hand, Heck coupling product 12af was chosen as a substrate (Table 3). We first investigated Ru-catalyzed protocols. This chemistry, pioneered by Murahashi and co-workers,50 relies on the use of catalytic amounts of a Ru source, e.g. [Ru(PPh3)2Cl2],51 in combination with an oxidant such as tert-butyl hydroperoxide. During our investigations into a tandem ring-closing metathesis/allylic oxidation sequence52 we discovered that Ru-based metathesis catalysts, such as the first generation Grubbs’ catalyst A,53 are also capable of catalyzing allylic oxidations if tert-butyl hydroperoxide is used as an oxidant. For the present task, however, Ru-catalyzed protocols were unsuitable as conversion to the chalcone remained below 5%, regardless of the precatalyst and the reaction time and temperatures (entries 1–6). A similar protocol that uses catalytic amounts of CrO3 was also unsuccessful (entry 7),54 as was the use of pyridinium chloro chromate (PCC) in large excess (entry 8).55 With a catalytic amount of PCC and tert-BuOOH as oxidant, however, some conversion to the desired chalcone oxidation product could be detected (entry 9).54 No product formation was detected with SeO2- (entry 10)56,57 and CuI-catalyzed (entry 11)58 methods, whereas CuBr as a catalyst led to some conversion (entry 12).58 Eventually, a suitable method was identified when the reagent combination of dichlorodicyano benzoquinone (DDQ) and silica under microwave irradiation was tested (entry 13).59 This method had been developed by Sinha and co-workers, who originally used ultrasonication to promote the reaction,60,61 but later found that microwave irradiation gives higher yields of the oxidation products.59 The authors found that the addition of silica notably increases the yield and they reason that this is due to a protonation of the oxidant DDQ.60
Table 3. Optimization of Allylic Oxidation Conditions.

| entry | Conditionsa | Yield of 13af (%) |
|---|---|---|
| 1 | [RuPPh3)2Cl2] (5 mol %); tert-BuOOH (4 equiv), benzene, 40 °C, 3 h51 | <5b |
| 2 | A (5 mol %); tert-BuOOH (4 equiv), benzene, 40 °C, 3 h52 | <5b |
| 3 | [RuPPh3)2Cl2] (5 mol %); tert-BuOOH (8 equiv), benzene, 40 °C, 16 h51 | <5b |
| 4 | A (5 mol %); tert-BuOOH (8 equiv), benzene, 40 °C, 16 h52 | <5b |
| 5 | [RuPPh3)2Cl2] (5 mol %); tert-BuOOH (8 equiv), benzene, 60 °C, 16 h51 | <5b |
| 6 | A (5 mol %); tert-BuOOH (8 equiv), benzene, 60 °C, 16 h52 | <5b |
| 7 | CrO3 (5 mol %); tert-BuOOH (7 equiv), C6H5CF3, 20 °C, 16 h54 | <5b |
| 8 | Pyridinium chlorochromate (25 equiv), 4 Å-molecular sieves, benzene, 80 °C, 16 h55 | <5b |
| 9 | Pyridinium chlorochromate (10 mol %), tert-BuOOH (4 equiv), C6H5CF3, 20 °C, 96 h54 | <20c |
| 10 | SeO2 (5 mol %), PIDA (5 equiv), dioxane, 100 °C, 16 h56,57 | <5b |
| 11 | CuI (2.6 mol %), tert-BuOOH (6 equiv), benzene, 70 °C, 24 h.58 | <5b |
| 12 | CuBr (2.0 mol %), tert-BuOOH (6 equiv), acetonitrile, 55 °C, 24 h58 | <20c |
| 13 | DDQ, silica, dioxane, microwave irradiation @90 °C, 25 min59 | 78d |
Method and conditions adapted from this reference.
Conversion <5% (TLC), unreacted starting material recovered.
Conversion <20% (1H NMR-spectroscopy).
Isolated along with ca. 20% of 14af.
With these conditions we were able to isolate the chalcone–coumarin hybrid 13af in 78% yield. Minor quantities of a second, slightly more polar product were obtained in impure form, contaminated with small amounts of 13af. This side product was identified as the deprotected oxidation product 14af. Cleavage of acetals, e.g. THP ethers, in the presence of catalytic amounts of DDQ has been reported,62,63 although in this case the acidic silica might also be responsible for the observed partial MOM-ether cleavage.
The allylic oxidation step can proceed either through a radical or a hydride abstraction. In both cases a resonance stabilized intermediate (either an allyl radical or a carbenium ion) will be formed, which can then react with or without net-transposition of the C–C-double bond to the oxidation product. Thus, not only 13af, but also its regioisomer, with the carbonyl group bound to the coumarin part of the hybrid structure, are possible oxidation products. We obtained just one regioisomer with very high selectivity, but similar to the Heck coupling products 12 the structure could not be unambiguously assigned solely based on 1D-NMR spectra. For this reason, a full signal assignment based on the 2D-NMR methods H,H–COSY, HSQC, HMBC and NOESY was performed. While no significant NOE-interactions could be observed for protons H1′ and H2′, HMBC interactions between H1′ and C7 and C8a, between H2′ and C1″, and between H2″ and C3′ (the chalcone carbonyl carbon) clearly point at the structure shown for 13af in Figure 4.
Figure 4.
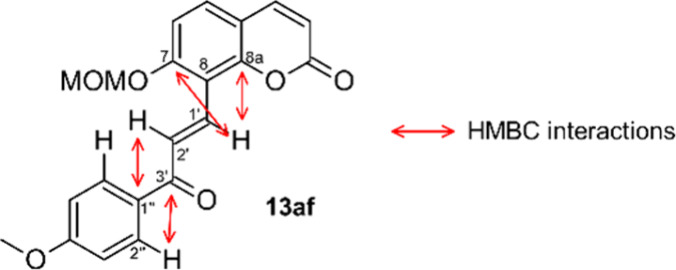
HMBC interactions in 13af.
The optimized allylic oxidation conditions (Table 3, entry 13) were first applied to the other C7-OMOM-substituted Matsuda–Heck coupling products 12a#. In most cases the allylic oxidation proceeds with high regioselectivity (Table 4). For the oxidation products of 12aa (enry 1), 12am (entry 12), 12aq (entry 16), 12ar (entry 17) and 12au (entry 19) the complete signal assignment process outlined above was repeated to check whether the regioselectivity of the allylic oxidation is affected by the substitution pattern of the aromatic substituent bound at C3′. In all cases single regioisomers with a C1′=C2′-double bond and a carbonyl group at C3′-position, as for 13af (entry 6), were obtained. While we observed cleavage of the MOM-group only to a small extent for this example and for the para-iodo-substituted derivative 13ae, concomitant deprotection occurred quantitatively during the oxidation of the meta-bromo-substituted compound 12an, the meta-carbomethoxy-substituted starting material 12ar, and the ortho-carbomethoxy-substituted precursor 12au. These reactions furnished exclusively phenols 14an (entry 13), 14ar (entry 17) and 14au (entry 19). Quantitative decomposition was observed during oxidation of phenol-substituted compound 12ag (entry 7), and partial decomposition for iodo-substituted coupling product 12ao (entry 14) and para-methyl-substituted coupling product 12ah (entry 8). In these cases the coumarin–chalcone hybrids were obtained as inseparable mixtures with other unidentified reaction products.
Table 4. Allylic Oxidation of 12a#: Scope and Limitations.

| entry | 12 | R1 | R2 | R3 | Product | yield (%) |
|---|---|---|---|---|---|---|
| 1 | 12aa | H | H | H | 13aa | 56 |
| 2 | 12ab | F | H | H | 13ab | 85 |
| 3 | 12ac | Cl | H | H | 13ac | 47 |
| 4 | 12ad | Br | H | H | 13ad | 43 |
| 5 | 12ae | I | H | H | 13ae | 29a |
| 6 | 12af | OCH3 | H | H | 13af | 78a |
| 7 | 12ag | OH | H | H | 13ag | <5b |
| 8 | 12ah | CH3 | H | H | 13ah | n.d.c |
| 9 | 12ai | CO2Et | H | H | 13ai | 36 |
| 10 | 12ak | CF3 | H | H | 13ak | 22 |
| 11 | 12al | H | F | H | 13al | 33 |
| 12 | 12am | H | Cl | H | 13am | 60 |
| 13 | 12an | H | Br | H | 14an | 26 |
| 14 | 12ao | H | I | H | 13ao | n.d.c |
| 15 | 12ap | H | OCH3 | H | 13ap | 24 |
| 16 | 12aq | H | CH3 | H | 13aq | 37 |
| 17 | 12ar | H | CO2CH3 | H | 14ar | 48 |
| 18 | 12at | H | H | OCH3 | 13at | 56 |
| 19 | 12au | H | H | CO2CH3 | 14au | 92 |
Unprotected phenols 14ae (16%) and 14af (ca. 10%) were isolated as byproducts.
Decomposition of starting material.
n.d.: not determined; oxidation products 13 were detected by NMR but could not be isolated in pure form due to inseparable byproducts.
Upon careful inspection of the NMR-spectra of oxidation products 14 (free phenols) we noted some significant differences compared to the spectra of chalcones 13 (MOM-ethers). In particular, an unusually high δ(13C) value of ca. 167 ppm was detected for carbon atoms C7 in all oxidation products 14. This prompted us to double check the assigned chalcone structure through a full signal assignment based on 2D-NMR-experiments. To our surprise, we did not detect the characteristic HMBC-correlations that are indicative for the C1′=C2′-double bond present in MOM-ethers 13, but found instead HMBC-interactions that point at a double bond located between positions C2′ and C3′, as shown in Figure 5 for the representative example 14au. It is not clear why the regioselectivity of the allylic oxidation is reversed in these cases, but it appears likely that cleavage of the MOM-ether occurs prior to the allylic oxidation. The phenol thus generated at C7 then directs the oxidant to the C1′-position, probably through hydrogen bond formation. In contrast, the selective C3′-oxidation observed in MOM-ethers 13 could originate from steric hindrance.
Figure 5.
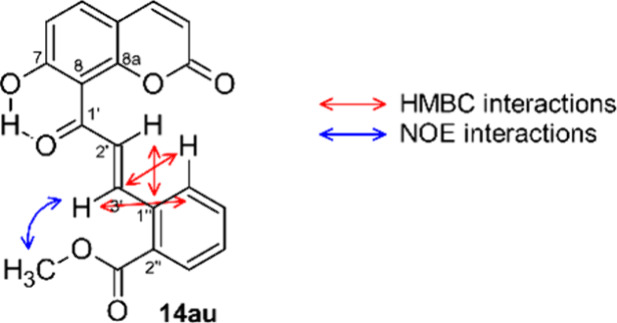
HMBC interactions in 14au.
Application to Flavanone–Chalcone Hybrids
The Matsuda–Heck-arylation/allylic oxidation sequence for the synthesis of coumarin–chalcone hybrids can be applied to other natural product scaffolds. We first investigated the synthesis of flavanone–chalcone hybrids 18, which started from 8-allylflavanones 15. Flavanones 15a and 15c are known compounds that were previously used by us for the synthesis of prenylated flavanones.64 Flavanone 15b was synthesized through an analogous sequence of steps (see Supporting Information for details). By using the standard conditions from Table 1, entry 3, three coupling products 16af, 16bf and 16cf were obtained from 8-allylflavanones 15a-c and diazonium salt 7f in yields comparable to those obtained for the coupling reactions of 8-allylcoumarin 11a (Scheme 5). All products 16 were obtained as single regioisomers with a C2′=C3′-double bond, which was proven by 2D-NMR methods via a line of reasoning analogous to that described for the coumarin coupling products 12 (see Supporting Information for a detailed chart). In the case of 16cf the assigned structure (and hence the selectivity of the Matsuda–Heck-reaction) was independently corroborated by single-crystal X-ray analysis (Figure 6).
Scheme 5. Synthesis of Flavanone–Chalcone Hybrids 18.

Figure 6.
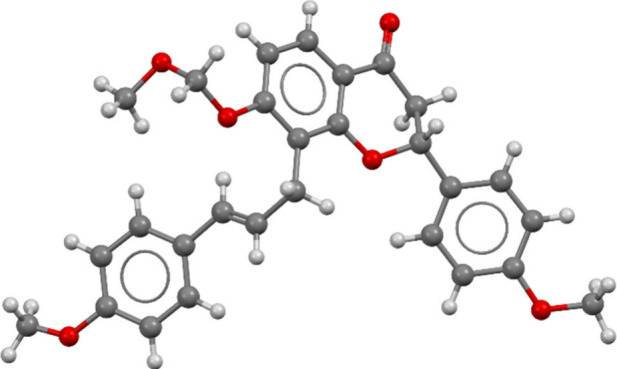
Molecular structure of coupling product 16cf. Displacement ellipsoids are shown at the 50% probability level.
All coupling products 16 underwent the allylic oxidation, using the DDQ-silica reagent under microwave irradiation, in fair yields and high regioselectivity to furnish MOM-protected flavanone-chalcone hybrids 17. Cleavage of the MOM-ether was accomplished with methanol in the presence of acid to furnish the phenolic hybrid compounds 18 (Scheme 5).
Application to Isoflavone–Chalcone Hybrids
For the synthesis of an isoflavone–chalcone hybrid with the chalcone connected to the isoflavone-A-ring we started from 8-allyl isoflavone 19. This is an intermediate en route to the prenylated isoflavone natural product 7-methoxyebenosin and was previously synthesized by us via an oxidative flavanone rearrangement.65 Matsuda–Heck arylation under the established standard conditions furnished 20 as a single regioisomer, and oxidation with DDQ-silica gave the isoflavone–chalcone hybrid 21 (Scheme 6). As discussed above for the coumarin– and flavanone–chalcone hybrids, structure elucidation is based on 2D-NMR-experiments, in particular HMBC (see Supporting Information for details).
Scheme 6. Synthesis of Isoflavone-A-Ring-Chalcone-Hybrid 21.
We also investigated the application of our approach to the synthesis of an isoflavone-chalcone hybrid with the chalcone moiety attached to the B-ring (Scheme 7). To this end isoflavone 22, an intermediate in our synthesis of the natural product 5-deoxy-3′-prenylbiochanin A66 was used as a starting material and reacted with diazonium salt 7f under the standard conditions. Compared to other allylarenes from this study that underwent coupling reactions with high β-selectivity, 22 is sterically less congested and lacks a second coordinating group adjacent to the allyl chain. Nevertheless, coupling product 23 was obtained in fair yield and high β-selectivity. Allylic oxidation under the established standard conditions proceeded with full conversion of the starting material, but resulted in the formation of an inseparable mixture of isomers. This observation suggests that a synthetically useful regioselectivity of the allylic oxidation step requires a strong steric and/or chelating bias, and that the presence of just one adjacent substituent is not sufficient in this regard.
Scheme 7. Attempted Synthesis of an Isoflavone-B-Ring-Chalcone-Hybrid 23.
Conclusions
In summary, we showed that electronically nonbiased olefins, such as allyl benzenes, undergo Pd-catalyzed arylation reactions in high regioselectivity if two potentially coordinating substituents are present at the positions adjacent to the allyl group. We reason that the regioselectivity of the β-hydride elimination step arises from a chelation of the Pd-σ-complex, that forces one β-hydrogen into an orientation that is geometrically unfavorable for the β-H-elimination. The subsequent allylic oxidation also occurs in high selectivity, but with transposition of the C=C-double bond. Most likely steric reasons are responsible for the observed regioselectivity of this step. The sequence of Pd-catalyzed arylation with arene diazonium salts and allylic oxidation was applied in the synthesis of various chalcone hybrids.
Experimental Section
General Methods
All experiments were conducted in dry reaction vessels under an atmosphere of dry nitrogen. Solvents were purified by standard procedures. Unless otherwise stated, reaction mixtures were heated with silicon oil baths. Microwave reactions were carried out in sealed vials in an Anton-Paar-monowave 300 or Anton-Paar-monowave 400 reactor (monowave, maximum power 850 W, temperature of reaction mixture and temperature–time profile were monitored by IR-sensor, vial volume 10 mL). 1H NMR spectra were obtained at 400 MHz in CDCl3 with CHCl3 (δ = 7.26 ppm) as an internal standard. Coupling constants are given in Hz. 13C{1H} NMR spectra were recorded at 100 MHz in CDCl3 with CDCl3 (δ = 77.1 ppm) as an internal standard. Whenever the solubility of the sample was insufficient in CDCl3, it was replaced by either acetone-d6 (acetone-d5 as internal standard for 1H NMR spectroscopy, δ = 2.05 ppm, acetone-d6 as internal standard for 13C{1H} NMR spectroscopy, δ = 29.9 ppm) or DMSO-d6 (DMSO-d5 as internal standard for 1H NMR spectroscopy, δ = 2.50 ppm, DMSO-d6 as internal standard for 13C{1H} NMR spectroscopy, δ = 39.5 ppm). Structural assignments were made with additional information from gNOESY, gCOSY, gHSQC, and gHMBC experiments. Signal assignments refer to the numbering scheme shown in Figure 1. IR spectra were recorded as ATR-FTIR spectra. Wavenumbers are given in cm–1. The peak intensities are defined as strong (s), medium (m) or weak (w). Low and high resolution mass spectra were obtained by EI-TOF or ESI-TOF. Chromatographic purification of compounds was accomplished using the dry column vacuum chromatography (DCVC) method as described in the literature.67
Caution: arene diazonium salts with tetrafluoroborate counterions are in most cases very stable. We have never experienced any safety incidents while working with these reagents, but there have been reports describing explosions or violent decomposition reactions caused by some arene diazonium tetrafluoroborates. We urgently recommend that operators familiarize themselves with the potential hazards and recommended safety measures as e.g. suggested by Firth and Fairlamb68 and by Correia and co-workers.69
General Procedure for the Coupling of Allylbenzopyranones and Arene Diazonium Salts
To a solution of the respective arene diazonium salt 7 (1.20 mmol, 1.20 equiv) in acetonitrile (10 mL, 0.1 M in olefin 11) was added Pd(OAc)2 (11.2 mg, 5.0 mol %). The mixture was stirred at 20 °C for 5 min, and NaOAc (246 mg, 3.00 mmol, 3.00 equiv) and the respective allylbenzopyranone 11 (1.00 mmol, 1.00 equiv) were added. The mixture was stirred at 20 °C for 16 h, filtered through a short silica-Celite pad which was washed with MTBE (20 mL). All volatiles were evaporated, and the residue was purified by flash chromatography on silica, using hexanes–ethyl acetate mixtures of increasing polarity as eluent, to furnish the coupling products.
7-(Methoxymethoxy)-8-[(2E)-3-phenylprop-2-en-1-yl]-2H-chromen-2-one (12aa)
Obtained from 7a (58 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 60 mg (0.19 mmol, 74%); mp 105–106 °C; IR (ATR) ν 2906 (w), 1714 (s), 1601 (s), 1492 (m), 1246 (s), 1018 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 9.5 Hz, 1H), 7.33–7.29 (m, 3H), 7.25 (t, J = 7.4 Hz, 2H), 7.16 (tt, J = 7.3, 1.3 Hz, 1H), 7.07 (d, J = 8.7 Hz, 1H), 6.51 (d, J = 15.8 Hz, 1H), 6.34 (dt, J = 15.8, 6.6 Hz, 1H), 6.27 (d, J = 9.5 Hz, 1H), 5.31 (s, 2H), 3.78 (d, J = 6.5 Hz, 2H), 3.49 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.2, 158.0, 153.1, 143.8, 137.6, 131.0, 128.5, 127.1, 127.0, 126.7, 126.1, 117.0, 113.7, 113.6, 110.6, 94.4, 56.5, 26.3; HRMS (EI) m/z [M]+ calcd for C20H18O4 322.1205, found 322.1215.
8-[(2E)-3-(4-Fluorophenyl)prop-2-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (12ab)
Obtained from 7b (63 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 57 mg (0.17 mmol, 67%); mp 110–112 °C; IR (ATR) ν 2930 (w), 1722 (s), 1605 (s), 1508 (m), 1116 (m), 1058 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 9.5 Hz, 1H), 7.31 (d, J = 8.7 Hz, 1H), 7.26 (dd, J = 8.5, 5.9 Hz, 2H), 7.07 (d, J = 8.7 Hz, 1H), 6.93 (t, J = 8.5 Hz, 2H), 6.47 (d, J = 15.8 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 6.25 (dt, J = 15.9, 6.6 Hz, 1H), 5.31 (s, 2H), 3.76 (dd, J = 6.6, 0.8 Hz, 2H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 162.0 (d, J = 245.9 Hz), 161.1, 158.0, 153.1, 143.9, 133.8 (d, J = 3.4 Hz), 129.8, 127.6 (d, J = 7.9 Hz), 126.8, 126.6 (d, J = 2.3 Hz), 117.0, 115.4 (d, J = 21.5 Hz), 113.8, 113.7, 110.6, 94.5, 56.5, 26.3; HRMS (EI) m/z [M+] calcd for C20H17O4F 340.1111, found 340.1114.
8-[(2E)-3-(4-Chlorophenyl)prop-2-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (12ac)
Obtained from 7c (68 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 65 mg (0.18 mmol, 73%); mp 108–110 °C; IR (ATR) ν 2957 (w), 1723 (s), 1606 (s), 1491 (m), 1116 (m), 1058 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 9.5 Hz, 1H), 7.31 (d, J = 8.7 Hz, 1H), 7.25–7.18 (4H), 7.08 (d, J = 8.6 Hz, 1H), 6.44 (d, J = 15.8 Hz, 1H), 6.31 (dt, J = 15.8, 6.4 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 5.31 (s, 2H), 3.77 (dd, J = 6.4, 0.7 Hz, 2H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.3, 158.0, 153.1, 143.8, 136.2, 132.7, 129.8, 128.7, 127.8, 127.4, 126.9, 116.8, 113.8, 113.8, 110.6, 94.4, 56.5, 26.4; HRMS (EI) m/z [M+] calcd for C20H17O435Cl 356.0815, found 356.0805.
8-[(2E)-3-(4-Bromophenyl)prop-2-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (12ad)
Obtained from 7d (81 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 76 mg (0.19 mmol, 75%); mp 100–101 °C; IR (ATR) ν 2956 (w), 1722 (s), 1606 (s), 1488 (m), 1115 (m), 1054 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 9.5 Hz, 1H), 7.36 (d, J = 8.5 Hz, 2H), 7.32 (d, J = 8.7 Hz, 1H), 7.16 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 8.7 Hz, 1H), 6.43 (d, J = 15.9 Hz, 1H), 6.33 (dt, J = 15.7, 6.1 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 5.31 (s, 2H), 3.76 (d, J = 6.0 Hz, 2H), 3.47 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.2, 158.0, 153.1, 143.8, 136.6, 131.6, 129.8, 127.9, 127.7, 126.9, 120.7, 116.7, 113.8, 113.7, 110.6, 94.4, 56.5, 26.4; HRMS (EI) m/z [M+] calcd for C20H17O479Br 400.0310, found 400.0311.
8-[(2E)-3-(4-Iodophenyl)prop-2-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (12ae)
Obtained from 7e (95 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 68 mg (0.15 mmol, 61%); mp 122–124 °C; IR (ATR) ν 2955 (w), 1720 (s), 1605 (s), 1483 (m), 1115 (m), 1059 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 9.5 Hz, 1H), 7.56 (d, J = 8.4 Hz, 2H), 7.32 (d, J = 8.7 Hz, 1H), 7.07 (d, J = 8.7 Hz, 1H), 7.04 (d, J = 8.4 Hz, 2H), 6.41 (d, J = 15.9 Hz, 1H), 6.34 (dt, J = 15.9, 5.5 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 5.31 (s, 2H), 3.76 (d, J = 5.5 Hz, 1H), 3.47 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.3, 158.0, 153.1, 143.9, 137.6, 137.2, 130.0, 128.1, 128.0, 126.9, 116.7, 113.9, 113.8, 110.6, 94.4, 92.2, 56.5, 26.4; HRMS (EI) m/z [M+] calcd for C20H17O4127I 448.0172, found 448.0158.
7-(Methoxymethoxy)-8-[(2E)-3-(4-methoxyphenyl)prop-2-en-1-yl]-2H-chromen-2-one (12af)
Obtained from 7f (67 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 70 mg (0.20 mmol, 79%); mp 100–105 °C; IR (ATR) ν 2956 (w), 1727 (s), 1607 (s), 1511 (m), 1247 (s), 1060 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 9.5 Hz, 1H, H4), 7.29 (d, J = 8.7 Hz, 1H, H5), 7.23 (d, J = 8.7 Hz, 2H, H2″/H6″), 7.23 (d, J = 8.7 Hz, 2H), 7.06 (d, J = 8.7 Hz, 1H, H6), 6.78 (d, J = 8.8 Hz, 2H), 6.45 (d, J = 15.8 Hz, 1H, H3′), 6.26 (d, J = 9.5 Hz, 1H, H3), 6.20 (dt, J = 15.7, 6.7 Hz, 1H, H2′), 5.30 (s, 2H, −OCH2O−), 3.77 (s, 3H), 3.76 (dd, J = 6.6, 1.2 Hz, 1H, H1′), 3.48 (s, 3H, −OCH2OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 161.3 (C2), 158.8 (C4″), 158.0 (C7), 153.0 (C8a), 143.8 (C4), 130.5 (C1″), 130.3 (C3′), 127.2 (C2″/C6″), 126.6 (C5), 124.8 (C2′), 117.3 (C8), 113.9 (C3″/C5″), 113.7 (C4a), 113.6 (C3), 110.6 (C6), 94.4 (−OCH2OCH3), 56.5 (−OCH2OCH3), 55.3 (C4″–OCH3), 26.3 (C1′); HRMS (EI) m/z [M+] calcd for C21H20O5 352.1311, found 352.1325.
8-[(2E)-3-(4-Hydroxyphenyl)prop-2-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (12ag)
Obtained from 7g (250 mg, 1.20 mmol) and 11a (246 mg, 1.00 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 179 mg (0.53 mmol, 53%); mp 105–107 °C; IR (ATR) ν 3346 (w), 2932 (w), 1698 (s), 1604 (s), 1512 (m), 1247 (m), 1065 (m), 835 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 9.5 Hz, 1H), 7.38 (d, J = 8.7 Hz, 1H), 7.17 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 8.6 Hz, 1H), 6.73 (d, J = 8.5 Hz, 2H), 6.42 (d, J = 15.8 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 6.17 (dt, J = 15.8, 6.7 Hz, 1H), 5.31 (s, 2H), 3.72 (dd, J = 6.7, 1.0 Hz, 2H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 162.2, 158.1, 155.5, 153.0, 144.4, 130.5, 130.1, 127.4, 126.8, 124.2, 117.5, 115.5, 113.8, 113.4, 110.8, 94.4, 56.6, 26.3; HRMS (EI) m/z [M+] calcd for C20H18O5 338.1154, found 338.1132.
7-(Methoxymethoxy)-8-[(2E)-3-(4-methylphenyl)prop-2-en-1-yl]-2H-chromen-2-one (12ah)
Obtained from 7h (62 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 60 mg (0.18 mmol, 71%); mp 119–121 °C; IR (ATR) ν 2920 (w), 1715 (s), 1601 (s), 1492 (m), 1246 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 9.5 Hz, 1H, H4), 7.30 (d, J = 8.7 Hz, 1H, H5), 7.20 (d, J = 8.1 Hz, 2H, H2″), 7.07 (d, J = 8.8 Hz, 1H, H6), 7.05 (d, J = 8.1 Hz, 2H, H3″), 6.47 (d, J = 15.8 Hz, 1H, H3′), 6.29 (dt, J = 15.7, 6.6 Hz, 1H, H2′), 6.27 (d, J = 9.5 Hz, 1H, H3), 5.31 (s, 2H, −OCH2O−), 3.77 (dd, J = 6.7, 0.8 Hz, 2H, H1′), 3.48 (s, 3H, −OCH3), 2.29 (s, 3H, −C4″CH3); 13C{1H} NMR (100 MHz, CDCl3) δ 161.2 (C2), 158.0 (C7), 153.1 (C8a), 143.8 C4), 136.8 (C4″), 134.9 (C1″), 130.9 (C3′), 129.2 (C3″), 126.7 (C5), 126.0 (C2″), 125.9 (C2′), 117.3 (C8), 113.7 (C4a), 113.6 (C3), 110.6 (C6), 94.5 (−OCH2OCH3), 56.5 (−OCH2OCH3), 26.3 (C1′), 21.2 (C4″–CH3); HRMS (EI) m/z [M+] calcd for C21H20O4 336.1362, found 336.1354.
Ethyl 4-{(1E)-3-[7-(Methoxymethoxy)-2-oxo-2H-chromen-8-yl]prop-1-en-1-yl}benzoate (12ai)
Obtained from 7i (79 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 78 mg (0.20 mmol, 79%); mp 75–77 °C; IR (ATR) ν 2980 (w), 1710 (s), 1605 (s), 1272 (s), 1107 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 8.4 Hz, 2H, H3″) 7.64 (d, J = 9.5 Hz, 1H, H4), 7.34 (d, J = 8.3 Hz, 2H, H2″) 7.32 (d, J = 8.6 Hz, 1H, H5), 7.08 (d, J = 8.6 Hz, 1H, H6), 6.52 (d, J = 15.8 Hz, 1H, H3′), 6.46 (dt, J = 15.8, 5.6 Hz, 1H, H2′), 6.28 (d, J = 9.5 Hz, 1H, H3), 5.31 (s, 2H, −OCH2O−), 4.34 (q, J = 7.1 Hz, 2H, −CO2CH2CH3), 3.80 (d, J = 5.5 Hz, 2H, H1′), 3.47 (s, 3H, −OCH3), 1.37 (t, J = 7.1 Hz, 3H, −CO2CH2CH3); 13C{1H} NMR (100 MHz, CDCl3): δ = 166.6 (−CO2CH2CH3), 161.2 (C2), 158.0 (C7), 153.1 (C8a), 143.8 (C4), 142.1 (C1″), 130.2 (C3′), 130.0 (C3″), 130.0 (C2′), 128.9 (C4″), 126.9 (C5), 126.0 (C2″), 116.5 (C8), 113.8 (C4a), 113.7 (C3), 110.6 (C6), 94.4 (−OCH2OCH3), 60.9 (−CO2CH2CH3), 56.5 (−OCH2OCH3), 26.5 (C1′), 14.4 (−CO2CH2CH3); HRMS (EI) m/z [M+] calcd for C23H22O6 394.1416, found 394.1408.
7-(Methoxymethoxy)-8-{(2E)-3-[4-(trifluoromethyl)phenyl]prop-2-en-1-yl}-2H-chromen-2-one (12ak)
Obtained from 7k (78 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 48 mg (0.12 mmol, 49%); mp 128–130 °C; IR (ATR) ν 2934 (w), 1725 (s), 1607 (s), 1323 (s), 1066 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 9.5 Hz, 1H), 7.49 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.7 Hz, 1H), 7.09 (d, J = 8.7 Hz, 1H), 6.52 (d, J = 15.9 Hz, 1 H), 6.44 (dt, J = 15.9, 6.0 Hz, 1H), 6.29 (d, J = 9.5 Hz, 1H), 5.32 (s, 2H), 3.80 (d, J = 6.0 Hz, 2H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.2, 158.0, 153.2, 143.8, 141.2, 129.9, 129.8, 128.9 (q, J = 32.0 Hz), 127.0, 126.3, 125.5 (q, J = 3.9 Hz), 124.4 (q, J = 271.0 Hz), 116.5, 113.8, 113.8, 110.6, 94.5, 56.5, 26.4; HRMS (EI) m/z [M+] calcd for C21H17O4F3 390.1079, found 390.1072.
8-[(2E)-3-(3-Fluorophenyl)prop-2-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (12al)
Obtained from 7l (63 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 62 mg (0.18 mmol, 73%); mp 105–107 °C; IR (ATR) ν 2906 (w), 1715 (s), 1602 (s), 1490 (m), 1246 (s), 1019 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 9.5 Hz, 1H), 7.32 (d, J = 8.7 Hz, 1H), 7.20 (td, J = 7.9, 6.1 Hz, 1H), 7.08 (d, J = 8.6 Hz, 1H), 7.05 (d, J = 7.2 Hz, 1H), 6.99 (dm, J = 9.5 Hz, 1H), 6.84 (td, J = 8.5, 2.5 Hz, 1H), 6.46 (d, J = 15.9 Hz, 1H), 6.35 (dt, J = 15.9, 6.2 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 5.31 (s, 2H), 3.78 (d, J = 6.2 Hz, 1H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 163.2 (d, J = 244.9 Hz), 161.2, 158.0, 153.1, 143.8, 140.1 (d, J = 7.8 Hz), 130.0, 129.9 (d, J = 7.5 Hz), 128.5, 126.9, 122.1 (d, J = 2.6 Hz), 116.7, 113.9 (d, J = 21.2 Hz), 113.8, 113.7, 112.6 (d, J = 21.7 Hz), 110.6, 94.5, 56.5, 26.3; HRMS (EI) m/z [M+] calcd for C20H17O4F 340.1111, found 340.1103.
8-[(2E)-3-(3-Chlorophenyl)prop-2-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (12am)
Obtained from 7m (226 mg, 1.20 mmol) and 11a (246 mg, 1.00 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 258 mg (0.72 mmol, 72%); mp 83–85 °C; IR (ATR) ν 1714 (s), 1603 (s), 1247 (s), 1119 (s), 1066 (s), 1022 (s), 966 (s), 836 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 9.5 Hz, 1H), 7.32 (d, J = 8.7 Hz, 1H), 7.29–7.27 (m, 1H), 7.19–7.10 (m, 3H), 7.08 (d, J = 8.7 Hz, 1H), 6.44 (d, J = 15.9 Hz, 1H), 6.34 (dt, J = 15.9, 6.2 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 5.31 (s, 2H), 3.78 (d, J = 6.2 Hz, 1H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.3, 158.0, 153.1, 143.8, 139.6, 134.5, 129.8, 129.7, 128.7, 127.1, 126.9, 126.1, 124.4, 116.7, 113.8, 113.8, 110.6, 94.5, 56.6, 26.3; HRMS (ESI) m/z [M + H]+ calcd for C20H18O4Cl 357.0894, found 357.0902.
8-[(2E)-3-(3-Bromophenyl)prop-2-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (12an)
Obtained from 7n (81 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a red-brown solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 50 mg (0.12 mmol, 50%); mp 106–108 °C; IR (ATR) ν 2907 (w), 1710 (s), 1603 (s), 1494 (m), 1245 (s), 1058 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 9.5 Hz, 1H), 7.43 (s(br), 1H), 7.32 (d, J = 8.7 Hz, 1H), 7.27 (d, J = 7.2 Hz, 1H), 7.21 (d, J = 8.1 Hz, 1H), 7.12 (d, J = 7.8 Hz, 1H), 7.08 (d, J = 8.7 Hz, 1H), 6.42 (d, J = 15.9 Hz, 1H), 6.33 (dt, J = 15.8, 5.7 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 5.31 (s, 2H), 3.77 (d, J = 5.7 Hz, 2H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.2, 158.0, 153.1, 143.8, 139.8, 130.0, 129.9, 129.6, 129.0, 128.7, 126.9, 124.8, 122.7, 116.6, 113.7, 113.7, 110.6, 94.4, 56.5, 26.3; HRMS (EI) m/z [M+] calcd for C20H17O4[79]Br 400.0310, found 400.0314.
8-[(2E)-3-(3-Iodophenyl)prop-2-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (12ao)
Obtained from 7o (318 mg, 1.20 mmol) and 11a (246 mg, 1.00 mmol) as a red-brown solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 108 mg (0.24 mmol, 24%); mp 129–132 °C; IR (ATR) ν 2961 (w), 1709 (s), 1603 (s), 1244 (s), 1115 (s), 1056 (s), 1036 (s), 964 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 9.5 Hz, 1H), 7.64 (t, J = 1.5 Hz, 1H), 7.48 (dm, J = 7.8 Hz, 1H), 7.31 (d, J = 8.7 Hz, 1H), 7.25 (dm, J = 7.8 Hz, 1H), 7.08 (d, J = 8.7 Hz, 1H), 6.97 (t, J = 7.8 Hz, 1H), 6.38 (d, J = 15.9 Hz, 1H), 6.32 (dt, J = 15.8, 5.7 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 5.31 (s, 2H), 3.77 (d, J = 5.7 Hz, 1H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.2, 158.0, 153.1, 143.8, 139.9, 135.9, 135.0, 130.2, 129.5, 128.6, 126.9, 125.4, 116.6, 113.8, 113.8, 110.6, 94.7, 94.4, 56.6, 26.3; HRMS (ESI) m/z [M + H]+ calcd for C20H18O4I 449.0244, found 449.0241.
7-(Methoxymethoxy)-8-[(2E)-3-(3-methoxyphenyl)prop-2-en-1-yl]-2H-chromen-2-one (12ap)
Obtained from 7p (67 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 82 mg (0.23 mmol, 93%); mp 73–75 °C; IR (ATR) ν 2936 (w), 1721 (s), 1604 (s), 1491 (m), 1245 (s), 1152 (s), 1115 (s), 1040 (s), 832 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 9.5 Hz, 1H, H4), 7.31 (d, J = 8.6 Hz, 1H, H5), 7.16 (dd, J = 8.2, 7.7 Hz, 1H, H5″), 7.07 (d, J = 8.7 Hz, 1H, H6), 6.90 (d, J = 7.7 Hz, 1H, H6″), 6.85–6.83 (m, 1H, H2″), 6.72 (dd, J = 8.2, 1.6 Hz, 1H, H4″), 6.47 (d, J = 15.8 Hz, 1H, H3′), 6.33 (dt, J = 15.8, 6.5 Hz, 1H, H2′), 6.28 (d, J = 9.5 Hz, 1H, H3), 5.31 (s, 2H, −OCH2O−), 3.78 (d, J = 6.5 Hz, 2H, H1′), 3.77 (s, 3H, −OCH3), 3.48 (s, 3H, −OCH2OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 161.3 (C2), 159.8 (C3″), 158.0 (C7), 153.1 (C8a), 143.8 (C4), 139.1 (C1″), 130.9 (C3′), 129.5 (C5″), 127.4 (C2′), 126.8 (C5), 118.8 (C6″), 117.0 (C8), 113.8 (C4a), 113.7 (C3), 112.9 (C4″), 111.4 (C2″), 110.6 (C6), 94.4 (−OCH2OCH3), 56.5 (−OCH3), 55.3 (−OCH2OCH3), 26.3 (C1′); HRMS (EI): m/z [M+] calcd for C21H20O5 352.1311, found 352.1309.
7-(Methoxymethoxy)-8-[(2E)-3-(3-methylphenyl)prop-2-en-1-yl]-2H-chromen-2-one (12aq)
Obtained from 7q (62 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 63 mg (0.19 mmol, 75%); mp 95–96 °C; IR (ATR) ν 2907 (w), 1714 (s), 1602 (s), 1413 (m), 1246 (s), 1024 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 9.5 Hz, 1H), 7.31 (d, J = 8.7 Hz, 1H), 7.16–7.08 (3H), 7.07 (d, J = 8.7 Hz, 1H), 6.98 (d, J = 7.0 Hz, 1H), 6.47 (d, J = 15.8 Hz, 1H), 6.32 (dt, J = 15.7, 6.5 Hz, 1H), 6.28 (d, J = 9.5 Hz, 1H), 5.31 (s, 2H), 3.77 (d, J = 6.5 Hz, 2H), 3.49 (s, 3H), 2.29 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 161.3, 158.0, 153.1, 143.8, 138.1, 137.6, 131.1, 128.5, 127.9, 126.9, 126.8, 126.7, 123.3, 117.2, 113.8, 113.7, 110.6, 94.5, 56.5, 26.4, 21.5; HRMS (EI) m/z [M+] calcd for C21H20O4 336.1362, found 336.1365.
Methyl 3-{(1E)-3-[7-(Methoxymethoxy)-2-oxo-2H-chromen-8-yl]prop-1-en-1-yl}benzoate (12ar)
Obtained from 7r (79 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 66 mg (0.17 mmol, 69%); mp 110–112 °C; IR (ATR) ν 2955 (w), 1714 (s), 1604 (s), 1432 (m), 1246 (s), 1025 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.95 (t, J = 1.7 Hz, 1H), 7.82 (dt, J = 7.7, 1.2 Hz, 1H), 7.64 (d, J = 9.5 Hz, 1H), 7.49 (dt, J = 7.8, 1.4 Hz, 1H), 7.32 (t, J = 7.8 Hz, 1H), 7.31 (d, J = 8.6 Hz, 1H), 7.09 (d, J = 8.7 Hz, 1H), 6.52 (d, J = 15.9 Hz, 1H), 6.42 (dt, J = 15.8, 6.3 Hz, 1H), 6.29 (d, J = 9.5 Hz, 1H), 5.32 (s, 2H), 3.89 (s, 3H), 3.80 (d, J = 6.3 Hz, 2H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 167.2, 161.3, 158.0, 153.2, 143.9, 138.0, 130.6, 130.5, 130.0, 128.6, 128.5, 128.2, 127.3, 126.9, 116.8, 113.8, 113.8, 110.6, 94.5, 56.6, 52.2, 26.4; HRMS (EI) m/z [M+] calcd for C22H20O6 380.1260, found 380.1264.
7-(Methoxymethoxy)-8-[(2E)-3-(2-methoxyphenyl)prop-2-en-1-yl]-2H-chromen-2-one (12at)
Obtained from 7t (67 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a yellowish oil; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 56 mg (0.16 mmol, 64%); IR (ATR) ν 2961 (w), 1721 (s), 1605 (s), 1488 (m), 1243 (s), 1053 (s), 1019 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 9.6 Hz, 1H, H4), 7.35 (dd, J = 7.6, 1.8 Hz, 1H, H6″), 7.28 (d, J = 8.6 Hz, 1H, H5), 7.14 (td, J = 7.6, 1.8 Hz, 1H, H4″), 7.06 (d, J = 8.6 Hz, 1H, H6), 6.86 (d, J = 15.9 Hz, 1H, H3′), 6.84 (t, J = 7.5 Hz, 1H, H5″), 6.80 (d, J = 8.1 Hz, 1H, H3″), 6.34 (dt, J = 15.8, 6.9 Hz, 1H, H2′), 6.25 (d, J = 9.5 Hz, 1H, H3), 5.30 (s, 2H, −OCH2O−), 3.80 (d, J = 6.9 Hz, 2H, H1′), 3.79 (s, 3H, −OCH3), 3.49 (s, 3H, −OCH2OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 161.3 (C2), 158.0 (C7), 156.4 (C2″), 153.0 (C8a), 143.8 (C4), 128.1 (C4″), 127.5 (C2′), 126.6 (C1″), 126.6 (C6″), 126.5 (C5), 125.9 (C3′), 120.6 (C5″), 117.3 (C8), 113.7 (C4a), 113.5 (C3), 110.8 (C3″), 110.6 (C6), 94.3 (−OCH2OCH3), 56.4 (−OCH2OCH3), 55.4 (−OCH3), 26.8 (C1′); HRMS (EI) m/z [M+] calcd for C21H20O5 352.1311, found 352.1302.
Methyl 2-{(1E)-3-[7-(Methoxymethoxy)-2-oxo-2H-chromen-8-yl]prop-1-en-1-yl}benzoate (12au)
Obtained from 7u (75 mg, 0.30 mmol) and 11a (61 mg, 0.25 mmol) as a colorless oil; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 71 mg (0.19 mmol, 75%); IR (ATR) ν 2951 (w), 1716 (s), 1605 (s), 1433 (w), 1245 (s), 1115 (s), 1058 (s), 832 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.79 (dd, J = 7.9, 1.1 Hz, 1H), 7.64 (d, J = 9.5 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.38 (td, J = 7.6, 1.1 Hz, 1H), 7.31 (d, J = 8.6 Hz, 1H), 7.24–7.17 (m, 2H), 7.09 (d, J = 8.6 Hz, 1H), 6.30–6.21 (m, 3H), 5.31 (s, 2H), 3.83 (s, 3H), 3.82 (dd, J = 6.3, 1.3 Hz, 2H), 3.48 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 168.1, 161.3, 158.3, 153.1, 143.8, 139.2, 132.0, 130.4, 129.9, 129.7, 128.5, 127.3, 126.8, 126.8, 117.0, 113.7, 113.7, 110.8, 94.6, 56.5, 52.0, 26.6; HRMS (EI) m/z [M+] calcd for C22H20O6 380.1260, found 380.1271.
(E)-7-(Methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-8-(3-(4-methoxyphenyl)allyl) chroman-4-one (16af)
Obtained from 7f (270 mg, 1.20 mmol) and 15a (390 mg, 1.00 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 372 mg (0.76 mmol, 76%); mp 105–107 °C; IR (ATR) ν 2886 (w), 1689 (s), 1595 (m), 1510 (s), 1244 (s), 1150 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.9 Hz, 1H, H5), 7.40 (d, J = 8.6 Hz, 2H, H2‴), 7.21 (d, J = 8.7 Hz, 2H, H2″), 7.08 (d, J = 8.6 Hz, 2H, H3‴), 6.84 (d, J = 8.9 Hz, 1H, H6), 6.81 (d, J = 8.7 Hz, 2H, H3″), 6.34 (d, J = 15.8 Hz, 1H, H3′), 6.13 (dt, J = 15.8, 6.7 Hz, 1H, H2′), 5.44 (dd, J = 12.8, 3.0 Hz, 1H, H2), 5.29 (s, 2H, C7–OCH2–OCH3), 5.21 (s, 2H, C4‴–OCH2–OCH3), 3.79 (s, 3H, −OCH3), 3.59–3.53 (m, 2H, H1′), 3.50 (s, 3H, C4‴–OCH2–OCH3), 3.48 (s, 3H, C7–OCH2–OCH3), 3.02 (dd, J = 16.8, 12.8 Hz, 1H, H3ax), 2.85 (dd, J = 16.8, 3.0 Hz, 1H, H3eq); 13C{1H} NMR (100 MHz, CDCl3) δ 191.6 (C4), 161.0 (C7), 160.7 (C8a), 158.8 (C4″), 157.5 (C4‴), 132.6 (C1‴), 130.7 (C1″), 129.9 (C3′), 127.6 (C2‴), 127.2 (C2″), 126.6 (C5), 125.5 (C2′), 117.2 (C8), 116.5 (C3‴), 116.0 (C4a), 114.0 (C3″), 107.9 (C6), 94.5 (C4‴–OCH2–OCH3), 94.2 (C7–OCH2–OCH3), 79.3 (C1), 56.5 (C7–OCH2–OCH3), 56.2 (C7–OCH2–OCH3), 55.4 (C4″–OCH3), 44.3 (C3), 26.7 (C2′); HRMS (EI) m/z [M+] calcd for C29H30O7 490.1992, found 490.1984.
(E)-7-Methoxy-2-(4-(methoxymethoxy)phenyl)-8-(3-(4-methoxyphenyl)allyl)chroman-4-one (16bf)
Obtained from 7f (270 mg, 1.20 mmol) and 15b (355 mg, 1.00 mmol) as a yellow paste; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 295 mg (0.64 mmol, 64%); IR (ATR) ν 2902 (w), 2837 (w), 1681 (s), 1595 (s), 1510 (s), 1243 (s), 1108 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 8.8 Hz, 1H), 7.40 (d, J = 8.7 Hz, 2H), 7.22 (d, J = 8.7 Hz, 2H), 7.08 (d, J = 8.7 Hz, 2H), 6.81 (d, J = 8.7 Hz, 2H), 6.65 (d, J = 8.8 Hz, 1H), 6.33 (d, J = 15.9 Hz, 1H), 6.12 (dt, J = 15.9, 6.7 Hz, 1H), 5.43 (dd, J = 12.8, 3.1 Hz, 1H), 5.20 (s, 2H), 3.92 (s, 3H), 3.79 (s, 3H), 3.55–3.51 (m, 2H), 3.50 (s, 3H), 3.01 (dd, J = 16.9, 12.8 Hz, 1H), 2.84 (dd, J = 16.9, 3.1 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 191.6, 163.4, 160.5, 158.8, 157.4, 132.7, 130.7, 129.9, 127.6, 127.2, 126.8, 125.6, 116.5, 116.4, 115.5, 114.0, 105.0, 94.5, 79.2, 56.2, 56.1, 55.4, 44.3, 26.5; HRMS (EI) m/z [M+] calcd for C28H28O6 460.1886, found 460.1870.
(E)-7-(Methoxymethoxy)-2-(4-methoxyphenyl)-8-(3-(4-methoxyphenyl)allyl)chroman-4-one (16cf)
Obtained from 7f (270 mg, 1.20 mmol) and 15c (355 mg, 1.00 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 275 mg (0.60 mmol, 60%). Suitable crystals for single crystal X-ray diffraction analysis were obtained by slowly evaporating the solvent from a solution of 16cf in a hexanes–ethyl acetate mixture: mp 101–102 °C; IR (ATR) ν 2892 (w), 1688 (m), 1588 (s), 1510 (s), 1250 (s), 1032 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.9 Hz, 1H), 7.40 (d, J = 8.5 Hz, 2H), 7.21 (d, J = 8.7 Hz, 2H), 6.94 (d, J = 8.7 Hz, 2H), 6.83 (d, J = 8.9 Hz, 1H), 6.81 (d, J = 8.5 Hz, 2H), 6.33 (d, J = 15.7 Hz, 1H), 6.13 (dt, J = 15.7, 6.7 Hz, 1H), 5.44 (dd, J = 12.7, 3.1 Hz, 1H), 5.28 (s, 2H), 3.83 (s, 3H), 3.79 (s, 3H), 3.58–3.54 (m, 2H), 3.47 (s, 3H), 3.03 (dd, J = 16.9, 12.7 Hz, 1H), 2.86 (dd, J = 16.9, 3.1 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 191.7, 161.0, 160.7, 159.9, 158.8, 131.4, 130.7, 129.9, 127.7, 127.2, 126.6, 125.5, 117.2, 116.0, 114.2, 114.0, 107.8, 94.2, 79.3, 56.5, 55.5, 55.4, 44.2, 26.7; HRMS (EI): m/z [M+] calcd for C28H28O6 460.1886, found 460.1893.
7-Methoxy-3-(4-methoxyphenyl)-8-[(2E)-3-(4-methoxyphenyl)prop-2-en-1-yl]-4H-chromen-4-one (20)
Obtained from 7f (270 mg, 1.20 mmol) and 19 (324 mg, 1.00 mmol) as a colorless solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 305 mg (0.71 mmol, 71%); mp 123–124 °C; IR (ATR) ν 1651 (s), 1605 (s), 1588 (s), 1511 (s), 1250 (s), 1176 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.22 (d, J = 8.9 Hz, 1H, H5), 8.00 (s, 1H, H2), 7.51 (d, J = 8.7 Hz, 2H, H2‴), 7.26 (d, J = 8.7 Hz, 2H, H2″), 7.05 (d, J = 8.9 Hz, 1H, H6), 6.97 (d, J = 8.7 Hz, 2H, H3‴), 6.81 (d, J = 8.7 Hz, 2H, H3″), 6.38 (d, J = 15.8 Hz, 1H, H3′), 6.20 (dt, J = 15.8, 6.5 Hz, 1H, H2′), 3.98 (s, 3H, C7–OCH3), 3.84 (s, 3H, C4‴–OCH3), 3.78 (s, 3H, C4″–OCH3), 3.75 (dd, J = 6.5, 1.0 Hz, 2H, H1′); 13C{1H} NMR (100 MHz, CDCl3) δ 176.5 (C4), 161.2 (C7), 159.6 (C4‴), 158.9 (C4″), 155.3 (C8a), 152.5 (C2), 130.4 (C1″), 130.2 (C2‴), 130.1 (C3′), 127.3 (C2″), 125.9 (C5), 125.0 (C2′), 124.5 (C1‴), 124.3 (C3), 118.7 (C4a), 116.1 (C8), 114.1 (C3‴), 114.0 (C3″), 109.2 (C6), 56.4 (C7–OCH3), 55.4 (C4‴–OCH3), 55.4 (C4″–OCH3), 26.3 (C1′); HRMS (EI) m/z [M+] calcd for C27H24O5 428.1624, found 428.1628.
7-(Methoxymethoxy)-3-{4-methoxy-3-[(2E)-3-(4-methoxyphenyl)-prop-2-en-1-yl]phenyl}-4H-chromen-4-one (23)
Obtained from 7f (270 mg, 1.20 mmol) and 22 (353 mg, 1.00 mmol) as an amorphous colorless solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 224 mg (0.52 mmol, 52%); IR (ATR) ν 2922 (w), 1641 (s), 1606 (s), 1241 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 9.5 Hz, 1H, H5), 7.91 (s, 1H, H2), 7.44 (dd, J = 8.6, 2.0 Hz, 1H, H6′), 7.35 (d, J = 2.0 Hz, 1H, H2′), 7.28 (d, J = 8.6 Hz, 2H, H2‴), 7.09–7.04 (m, 2H, H6, H8), 6.94 (d, J = 8.6 Hz, 1H, H5′), 6.82 (d, J = 8.6 Hz, 2H, H3‴), 6.40 (d, J = 15.8 Hz, 1H, H3″), 6.25 (dt, J = 15.8, 6.8 Hz, 1H, H2″), 5.27 (s, 2H, −OCH2O−), 3.88 (s, 3H, C4′–OCH3), 3.78 (s, 3H, C4‴–OCH3), 3.55 (d, J = 6.8 Hz, 2H, H1″), 3.51 (s, 3H, −OCH2OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 176.1 (C4), 161.5 (C7), 158.8 (C4‴), 157.8 (C8a), 157.5 (C4′), 152.4 (C2), 130.7 (C1‴), 130.5 (C2′), 130.3 (C3″), 129.2 (C3′), 128.3 (C6′), 128.0 (C5), 127.3 (C2‴), 126.7 (C2″), 125.2 (C3), 124.1 (C1′), 119.4 (C4a), 115.5 (C6 or C8), 114.0 (C3‴), 110.6 (C5′), 103.2 (C6 or C8), 94.5 (−OCH2OCH3), 56.5 (−OCH2OCH3), 55.7 (C4′–OCH3) 55.4 (C4‴–OCH3), 33.6 (C1″); HRMS (ESI): m/z [M + H]+ calcd for C28H27O6 459.1808, found 459.1814.
General Procedure for the Allylic Oxidation of Matsuda–Heck Coupling Products
Arylallyl chromanones 12 (0.25 mmol, 1.00 equiv), DDQ (136 mg, 0.60 mmol, 2.40 equiv) and silica (100 mg) were suspended in 1,4-dioxane (5 mL, 0.05 M in 12) in a vessel suitable for microwave irradiation. The vessel was sealed, and the mixture was heated to 90 °C under microwave irradiation for 25 min. After cooling to ambient temperature, it was filtered through a silica-Celite pad and washed with MTBE (150 mL). The solvent was removed under reduced pressure. The residue was purified by flash chromatography on silica, using hexanes–MTBE mixtures of increasing polarity as eluent. Attempts for upscaling to a 1 mmol scale were unsuccessful due to limitations in vial size, and because the initial substrate concentration of 0.05 M could not be increased.
7-(Methoxymethoxy)-8-[(1E)-3-oxo-3-phenylprop-1-en-1-yl]-2H-chromen-2-one (13aa)
Obtained from 12aa (81 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 47 mg (0.14 mmol, 56%); mp 141–144 °C; IR (ATR) ν 2909 (w), 1726 (s), 1661 (m), 1588 (s), 1115 (m), 1044 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 16.2 Hz, 1H), 8.22 (d, J = 16.1 Hz, 1H), 8.10 (dm, J = 8.0 Hz, 2H, H2″), 7.67 (d, J = 9.5 Hz, 1H, H4), 7.59 (tm, J = 7.3 Hz, 1H, H4″), 7.51 (tm, J = 7.5 Hz, 2H, H3″), 7.43 (d, J = 8.7 Hz, 1H, H5), 7.15 (d, J = 8.7 Hz, 1H, H6), 6.34 (d, J = 9.5 Hz, 1H, H3), 5.37 (s, 2H, −OCH2O−), 3.53 (s, 3H, −OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 191.2 (C3′), 160.1 (C2), 159.6 (C7), 154.2 (C8a), 143.8 (C4), 138.3 (C1″), 133.0 (C4″), 132.5 (C1′), 130.1 (C5), 128.8 (C2″), 128.8 (C3″), 127.8 (C2′), 114.0 (C3), 113.8 (C4a), 113.1 (C8), 111.0 (C6), 95.0 (−OCH2OCH3), 56.9 (−OCH2OCH3); HRMS (ESI) m/z [M + H]+ calcd for C20H17O5 337.1071, found 337.1068.
8-[(1E)-3-(4-Fluorophenyl)-3-oxoprop-1-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (13ab)
Obtained from 12ab (85 mg, 0.25 mmol) as a colorless solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 75 mg (0.21 mmol, 85%); mp 158–160 °C; IR (ATR) ν 1729 (s), 1663 (m), 1596 (s), 1276 (m), 1240 (m), 1154 (s), 1116 (s), 1045 (s), 835 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J = 16.1 Hz, 1H), 8.22 (d, J = 16.1 Hz, 1H), 8.17–8.10 (m, 2H), 7.68 (d, J = 9.5 Hz, 1H), 7.44 (d, J = 8.8 Hz, 1H), 7.23–7.14 (m, 2H), 6.35 (d, J = 9.5 Hz, 1H), 5.37 (s, 2H), 3.53 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 189.5, 165.8 (d, J = 254.3 Hz), 160.1, 159.6, 154.1, 143.8, 134.6 (d, J = 3.0 Hz), 132.6, 131.4 (d, J = 9.7 Hz), 130.2, 127.2, 115.9 (d, J = 21.8 Hz), 113.9, 113.7, 112.8, 111.0, 95.0, 56.9; HRMS (ESI) m/z [M + H]+ calcd for C20H16O5F 355.0976, found 355.0970.
8-[(1E)-3-(4-Chlorophenyl)-3-oxoprop-1-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (13ac)
Obtained from 12ac (91 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 44 mg (0.12 mmol, 47%); mp 156–160 °C; IR (ATR) ν 1720 (s), 1662 (s), 1593 (s), 1554 (s), 1082 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 16.1 Hz, 1H), 8.21 (d, J = 16.1 Hz, 1H), 8.03 (d, J = 8.6 Hz, 2H), 7.67 (d, J = 9.5 Hz, 1H), 7.49 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 8.7 Hz, 1H), 7.16 (d, J = 8.7 Hz, 1H), 6.34 (d, J = 9.5 Hz, 1H), 5.37 (s, 2H), 3.53 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 190.0, 160.1, 159.6, 154.2, 143.8, 139.4, 136.6, 133.0, 130.3, 130.2, 129.1, 127.2, 114.0, 113.8, 112.9, 111.1, 95.0, 56.9; HRMS (ESI) m/z [M + H]+ calcd for C20H16O5Cl 371.0681, found 371.0674.
8-[(1E)-3-(4-Bromophenyl)-3-oxoprop-1-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (13ad)
Obtained from 12ad (100 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 45 mg (0.11 mmol, 43%); mp 166–169 °C; IR (ATR) ν 2973 (w), 1713 (s), 1592 (m), 1361 (m), 1221 (s), 1166 (w) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 16.3 Hz, 1H), 8.22 (d, J = 16.3 Hz, 1H), 7.96 (dm, J = 8.5 Hz, 2H), 7.67 (d, J = 9.5 Hz, 1H), 7.66 (dm, J = 8.6 Hz, 2H), 7.44 (d, J = 8.8 Hz, 1H), 7.16 (d, J = 8.8 Hz, 1H), 6.34 (d, J = 9.5 Hz, 1H), 5.37 (s, 2H), 3.53 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 190.1, 160.1, 159.6, 154.2, 143.8, 137.1, 133.0, 132.1, 130.4, 130.3, 128.2, 127.0, 114.0, 113.8, 112.9, 111.1, 95.0, 56.9; HRMS (ESI) m/z [M + H]+ calcd for C20H16O5Br 415.0181, found 415.0158.
8-[(1E)-3-(4-Iodophenyl)-3-oxoprop-1-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (13ae)
Obtained from 12ae (112 mg, 0.25 mmol) as an off white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 34 mg (0.07 mmol, 29%); mp 150–154 °C. A second fraction contained 7-hydroxy-8-[(2E)-3-(4-iodophenyl)prop-2-enoyl]-2H-chromen-2-one (14ae): off-white solid; yield: (17 mg, 0.04 mmol, 16%); mp 220–222 °C. Analytical data for13ae: IR (ATR) ν 2926 (w), 1730 (s), 1661 (m), 1591 (s), 1557 (m), 1277 (m), 1245 (m), 1116 (m), 1047 (s), 1005 (m), 830 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 16.2 Hz, 1H, H1′), 8.21 (d, J = 16.2 Hz, 1H, H2′), 7.88 (dm, J = 8.6 Hz, 2H, H3″), 7.80 (dm, J = 8.6 Hz, 2H, H2″), 7.67 (d, J = 9.5 Hz, 1H, H4), 7.44 (d, J = 8.7 Hz, 1H, H5), 7.15 (d, J = 8.7 Hz, 1H, H6), 6.34 (d, J = 9.5 Hz, 1H, H3), 5.37 (s, 2H, −OCH2O−), 3.53 (s, 3H, −OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 190.5 (C3′), 160.1 (C2), 159.6 (C7), 154.2 (C8a), 143.8 (C4), 138.1 (C3″), 137.6 (C1″), 133.0 (C2′), 130.3 (C5), 130.2 (C2″), 127.1 (C1′), 114.0 (C3), 113.8 (C4a), 112.9 (C8), 111.1 (C6), 101.0 (C4″), 95.0 (−OCH2OCH3), 56.9 (−OCH2OCH3); HRMS (ESI) m/z [M + H]+ calcd for C20H16O5I 463.0037, found 463.0043. Analytical data for14ae: IR (ATR) ν 1730 (s), 1631 (s), 1599 (s), 1544 (s), 1584 (s), 1351 (s), 1193 (s), 1117 (m), 833 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 13.88 (s, 1H, -OH), 8.28 (d, J = 15.5 Hz, 1H, H2′), 7.87 (d, J = 15.5 Hz, 1H, H3′), 7.79 (dm, J = 8.4 Hz, 2H, H3″), 7.66 (d, J = 9.5 Hz, 1H, H4), 7.54 (d, J = 8.8 Hz, 1H, H5), 7.46 (dm, J = 8.4 Hz, 2H, H2″), 6.94 (d, J = 8.8 Hz, 1H, H6), 6.31 (d, J = 9.5 Hz, 1H, H3); 13C{1H} NMR (100 MHz, CDCl3): δ = 193.2 (C1′), 167.9 (C7), 159.5 (C2), 155.7 (C8a), 144.9 (C3′), 144.4 (C4), 138.5 (C3″), 134.8 (C5), 134.4 (C1″), 130.5 (C2″), 126.8 (C2′), 115.9 (C6), 112.3 (C3), 111.3 (C4a), 109.8 (C8), 97.8 (C4″); HRMS (ESI) m/z [M + H]+ calcd for C18H12O4127I 418.9775, found 418.9781.
7-(Methoxymethoxy)-8-[(1E)-3-(4-methoxyphenyl)-3-oxoprop-1-en-1-yl]-2H-chromen-2-one (13af)
Obtained from 12af (88 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 71 mg (0.19 mmol, 78%); mp 140–143 °C. A second, slightly more polar fraction consisted of 7-hydroxy-8-[(1E)-3-(4-methoxyphenyl)-3-oxoprop-1-en-1-yl]-2H-chromen-2-one (14af), contaminated with ca. 10% of 13af. Analytical data for13af: IR (ATR) ν 1726 (s), 1655 (m), 1588 (s), 1453 (m), 1243 (m), 1167 (s), 1045 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 16.0 Hz, 1H, H2′), 8.19 (d, J = 16.1 Hz, 1H, H1′), 8.11 (dm, J = 8.5 Hz, 2H, H2″), 7.67 (d, J = 9.5 Hz, 1H, H4), 7.42 (d, J = 8.7 Hz, 1H, H5), 7.14 (d, J = 8.8 Hz, 1H, H6), 6.99 (dm, J = 8.5 Hz, 2H, H3″), 6.33 (d, J = 9.5 Hz, 1H, H3), 5.36 (s, 2H, −OCH2O−), 3.89 (s, 3H, −OCH3), 3.52 (s, 3H, −OCH2OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 189.6 (C3′), 163.6 (C4″), 160.2 (C2), 159.5 (C7), 154.1 (C8a), 143.8 (C4), 131.6 (C1′), 131.3 (C1″), 131.2 (C2″), 129.9 (C5), 127.8 (C2′), 114.1 (C3″), 114.0 (C3), 113.8 (C4a), 113.3 (C8), 111.1 (C6), 95.0 (−OCH2OCH3), 56.9 (−OCH2OCH3), 55.6 (−OCH3); HRMS (ESI) m/z [M + H]+ calcd for C21H19O6 367.1176, found 367.1171. Selected analytical data of the minor byproduct14af(obtained from a sample contaminated with13af): 1H NMR (400 MHz, acetone-d6) δ 8.41 (d, J = 16.0 Hz, 1H), 8.28 (d, J = 16.1 Hz, 1H), 8.09 (dm, J = 8.5 Hz, 2H), 7.93 (d, J = 9.5 Hz, 1H), 7.58 (d, J = 9.5 Hz, 1H), 7.09 (dm, J = 8.6 Hz, 2H), 7.06 (d, J = 8.6 Hz, 1H), 6.27 (d, J = 9.5 Hz, 1H), 3.92 (s, 3H).
Ethyl 4-{(2E)-3-[7-(Methoxymethoxy)-2-oxo-2H-chromen-8-yl]prop-2-enoyl}benzoate (13ai)
Obtained from 12ai (99 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 36 mg (0.09 mmol, 36%); mp 115–118 °C; IR (ATR) ν 2925 (w), 1713 (m), 1663 (m), 1608 (m), 1583 (s), 1275 (s), 1045 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 16.4 Hz, 1H), 8.23 (d, J = 16.4 Hz, 1H), 8.19 (dm, J = 8.4 Hz, 2H), 8.12 (dm, J = 8.4 Hz, 2H), 7.67 (d, J = 9.5 Hz, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.15 (d, J = 8.7 Hz, 1H), 6.34 (d, J = 9.5 Hz, 1H), 5.37 (s, 2H), 4.41 (q, J = 7.1 Hz, 2H), 3.53 (s, 3H), 1.42 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 190.8, 166.0, 160.0, 159.7, 154.2, 143.7, 141.6, 134.1, 133.3, 130.4, 130.0, 128.6, 127.5, 114.1, 113.8, 112.9, 111.0, 95.0, 61.5, 56.9, 14.4; HRMS (ESI) m/z [M + H]+ calcd for C23H21O7 409.1287, found 409.1263.
7-(Methoxymethoxy)-8-{(1E)-3-oxo-3-[4-(trifluoromethyl)phenyl]prop-1-en-1-yl}-2H-chromen-2-one (13ak)
Obtained from 12ak (98 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 22 mg (0.05 mmol, 22%); mp 100–104 °C; IR (ATR) ν 1725 (s), 1666 (m), 1591 (s), 1554 (m), 1325 (m), 1287 (m), 1165 (m), 1106 (s), 1066 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.31 (d, J = 16.1 Hz, 1H), 8.26 (d, J = 16.1 Hz, 1H), 8.19 (d, J = 8.5 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H), 7.68 (d, J = 9.5 Hz, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.17 (d, J = 8.8 Hz, 1H), 6.36 (d, J = 9.5 Hz, 1H), 5.38 (s, 2H), 3.54 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 190.3, 160.0, 159.7, 154.3, 143.8, 141.2, 134.4, 133.7, 130.5, 129.1, 127.1, 125.9 (q, J = 3.9 Hz), 124.0 (d, J = 272.3 Hz), 114.1, 113.8, 112.8, 111.1, 95.1, 56.9; HRMS (ESI) m/z [M + H]+ calcd for C21H16O5F3 405.0944, found 405.0947.
8-[(1E)-3-(3-Fluorophenyl)-3-oxoprop-1-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (13al)
Obtained from 12al (85 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 29 mg (0.08 mmol, 33%); mp 92–95 °C; IR (ATR) ν 1732 (s), 1664 (m), 1583 (s), 1246 (s), 1115 (m), 1047 (s), 728 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.27–8.22 (m, 2H), 7.88 (dm, J = 7.6 Hz, 1H), 7.76 (dm, J = 9.5 Hz, 1H), 7.67 (d, J = 9.5 Hz, 1H), 7.50 (td, J = 8.0, 5.6 Hz, 1H), 7.45 (d, J = 8.7 Hz, 1H), 7.28 (td, J = 8.3, 2.7 Hz, 1H), 7.16 (d, J = 8.6 Hz, 1H), 6.35 (d, J = 9.6 Hz, 1H), 5.38 (s, 2H), 3.54 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 190.0, 163.1 (d, J = 248.0 Hz), 160.1, 159.6, 154.2, 143.8, 140.5 (d, J = 6.4 Hz), 133.2, 130.5 (d, J = 8.1 Hz), 130.4, 127.2, 124.6 (d, J = 3.1 Hz), 120.0 (d, J = 21.7 Hz), 115.5 (d, J = 23.0 Hz), 114.0, 113.8, 112.9, 111.0, 95.0, 56.9; HRMS (ESI) m/z [M + H]+ calcd for C20H16O5F 355.0976, found 355.0969.
8-[(1E)-3-(3-Chlorophenyl)-3-oxoprop-1-en-1-yl]-7-(methoxymethoxy)-2H-chromen-2-one (13am)
Obtained from 12am (89 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 56 mg (0.15 mmol, 60%); mp 127–130 °C; IR (ATR) ν 1726 (s), 1663 (m), 1596 (s), 1585 (s), 1291 (s), 1204 (s), 1048 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.24–8.21 (m, 2H, H1′, H2′), 8.04 (t, J = 1.8 Hz, 1H, H2″), 7.96 (dt, J = 7.7, 1.2 Hz, 1H, H6″), 7.66 (d, J = 9.5 Hz, 1H, H4), 7.55 (dm, J = 7.9 Hz, 1H, H4″), 7.46 (t, J = 7.8 Hz, 1H, H5″), 7.44 (d, J = 8.8 Hz, 1H, H5), 7.16 (d, J = 8.8 Hz, 1H, H6), 6.34 (d, J = 9.5 Hz, 1H, H3), 5.37 (s, 2H, −OCH2O−), 3.53 (s, 3H, −OCH2OCH3); 13C{1H} NMR (101 MHz, CDCl3) δ 190.0 (C3′), 160.0 (C2), 159.6 (C7), 154.2 (C8a), 143.7 (C4), 139.9, 135.1, 133.3 (C1′), 132.9 (C4″), 130.4 (C5), 130.2 (C5″), 128.9 (C2″), 127.3 (C2′), 126.9 (C6″), 114.1 (C3), 113.8 (C4a), 112.9 (C8), 111.0 (C6), 95.0 (−OCH2OCH3), 56.9 (−OCH3); HRMS (ESI) m/z [M + H]+ calcd for C20H16O5Cl 371.0681, found 371.0677.
8-[(2E)-3-(3-Bromophenyl)prop-2-enoyl]-7-hydroxy-2H-chromen-2-one (14an)
Obtained from 12an (100 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 24 mg (0.06 mmol, 26%); mp 178–181 °C; IR (ATR) ν 1740 (s), 1631 (s), 1599 (s), 1548 (s), 1242 (s), 1190 (s), 1110 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 13.78 (s, 1H, -OH), 8.24 (d, J = 15.5 Hz, 1H, H2′), 7.85 (d, J = 15.5 Hz, 1H, H3′), 7.83 (s, 1H, H2″), 7.70 (dm, J = 7.8 Hz, 1H, H6″), 7.66 (d, J = 9.5 Hz, 1H, H4), 7.56 (dm, J = 7.7 Hz, 1H, H4″), 7.54 (d, J = 8.7 Hz, 1H, H5), 7.33 (t, J = 7.9 Hz, 1H, H5″), 6.94 (d, J = 8.7 Hz, 1H, H6), 6.31 (d, J = 9.5 Hz, 1H, H3); 13C{1H} NMR (101 MHz, CDCl3) δ 193.2 (C1′), 167.8 (C7), 159.4 (C2), 155.7 (C8a), 144.4 (C4), 144.1 (C3′), 137.1 (C1″), 134.9 (C5), 133.8 (C4″), 132.3 (C2″), 130.8 (C5″), 127.6 (C2′), 127.1 (C6″), 123.3 (C3″), 115.9 (C6), 112.3 (C3), 111.3 (C4a), 109.8 (C8); HRMS (ESI) m/z [M + H]+ calcd for C18H11O479Br 370.9913, found 370.9916.
7-(Methoxymethoxy)-8-[(1E)-3-(3-methoxyphenyl)-3-oxoprop-1-en-1-yl]-2H-chromen-2-one (13ap)
Obtained from 12ap (88 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 21 mg (0.06 mmol, 24%); mp 107–110 °C; IR (ATR) ν 2943 (w), 1723 (s), 1661 (m), 1604 (m), 1581 (s), 1446 (m), 1286 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.30 (d, J = 16.2 Hz, 1H), 8.20 (d, J = 16.2 Hz, 1H), 7.73–7.55 (m, 3H), 7.47–7.37 (m, 2H), 7.17–7.10 (m, 2H), 6.33 (d, J = 9.5 Hz, 1H), 5.36 (s, 2H), 3.90 (s, 3H), 3.53 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 191.0, 160.1, 160.0, 159.7, 154.2, 143.8, 139.7, 132.5, 130.1, 129.8, 127.9, 121.4, 120.0, 114.0, 113.8, 113.1, 112.7, 111.0, 95.0, 56.9, 55.6; HRMS (ESI) m/z [M + H]+ calcd for C21H19O6 367.1182, found 367.1183.
7-(Methoxymethoxy)-8-[(1E)-3-(3-methylphenyl)-3-oxoprop-1-en-1-yl]-2H-chromen-2-one (13aq)
Obtained from 12aq (84 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 32 mg (0.09 mmol, 37%); mp 151–154 °C; IR (ATR) ν 2922 (w), 1730 (m), 1661 (m), 1591 (s), 1277 (m), 1245 (s), 1116 (s), 1046 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 16.2 Hz, 1H, H2′), 8.20 (d, J = 16.2 Hz, 1H, H1′), 7.93–7.86 (m, 2H, H2″-H6″), 7.67 (d, J = 9.5 Hz, 1H, H4), 7.43 (d, J = 8.7 Hz, 1H, H5), 7.44–7.38 (m, 2H, H2″-H6″), 7.16 (d, J = 8.8 Hz, 1H, H6), 6.34 (d, J = 9.6 Hz, 1H, H3), 5.37 (s, 2H, −OCH2O−), 3.54 (s, 3H, −OCH2OCH3), 2.46 (s, 3H, −CH3); 13C{1H} NMR (100 MHz, CDCl3) δ 191.6 (C3′), 160.2 (C2), 159.6 (C7), 154.1 (C8a), 143.8 (C4), 138.6 (C3″), 138.4 (C1″), 133.8 (C2″, C4″-C6″), 132.3 (C1′), 130.0 (C5), 129.4 (C2″, C4″-C6″), 128.7 (C2″, C4″-C6″), 128.3 (C2′), 126.1 (C2″, C4″-C6″), 114.1 (C3), 113.8 (C4a), 113.3 (C8), 111.0 (C6), 95.0 (−OCH2OCH3), 56.9 (−OCH2OCH3), 21.6 (−CH3); HRMS (ESI) m/z [M + H]+ calcd for C21H19O5 351.1227, found 351.1220.
Methyl 3-[(1E)-3-(7-hydroxy-2-oxo-2H-chromen-8-yl)-3-oxoprop-1-en-1-yl]benzoate (14ar)
Obtained from 12ar (95 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 42 mg (0.12 mmol, 48%); mp 187–190 °C; IR (ATR) ν 2954 (w), 2924 (w), 1723 (s), 1634 (m), 1598 (s), 1346 (m), 1190 (m), 1113 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 13.82 (s, 1H, – OH), 8.35 (t, J = 1.7 Hz, 1H, H2″), 8.32 (d, J = 15.6 Hz, 1H, H2′), 8.09 (dt, J = 7.8, 1.4 Hz, 1H, H4″), 7.97 (d, J = 15.4 Hz, 1H, H3′), 7.97 (dm, J = 7.9 Hz, 1H, H6″), 7.66 (d, J = 9.5 Hz, 1H, H4), 7.54 (t, J = 7.7 Hz, 1H, H5″), 7.54 (d, J = 8.7 Hz, 1H, H5), 6.94 (d, J = 8.7 Hz, 1H, H6), 6.31 (d, J = 9.5 Hz, 1H, H3), 3.96 (s, 3H, −OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 193.2 (C1′), 167.9 (C7), 166.6 (−CO2CH3), 159.4 (C2), 155.7 (C8a), 144.7 (C3′), 144.4 (C4), 135.3 (C1″), 134.8 (C5), 132.5 (C6″), 131.9 (C4″), 131.2 (C3″), 130.8 (C2″), 129.4 (C5″), 127.4 (C2′), 115.9 (C6), 112.3 (C3), 111.3 (C4a), 109.8 (C8), 52.5 (−OCH3); HRMS (ESI) m/z [M + H]+ calcd for C20H14O6 351.0863, found 351.0898.
7-(Methoxymethoxy)-8-[(1E)-3-(3-methylphenyl)-3-oxoprop-1-en-1-yl]-2H-chromen-2-one (13at)
Obtained from 12at (88 mg, 0.25 mmol) as an off-white solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 50 mg (0.14 mmol, 56%); mp 146–148 °C; IR (ATR) ν 2839 (w), 1725 (s), 1653 (m), 1597 (m), 1581 (s), 1438 (m), 1245 (s), 1045 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 16.3 Hz, 1H, H2′), 8.12 (d, J = 16.2 Hz, 1H, H1′), 7.73 (dd, J = 7.7, 1.8 Hz, 1H, H6″), 7.64 (d, J = 9.5 Hz, 1H, H4), 7.49 (ddd, J = 8.5, 7.5, 1.7 Hz, 1H, H4″), 7.39 (d, J = 8.7 Hz, 1H, H5), 7.13 (d, J = 8.7 Hz, 1H, H6), 7.06–7.00 (m, 2H, H3″, H5″), 6.29 (d, J = 9.5 Hz, 1H, H3), 5.34 (s, 2H, −OCH2O−), 3.99 (s, 3H, −OCH2OCH3), 3.51 (s, 3H, −OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 193.1 (C3′), 160.2 (C2), 159.5 (C7), 158.9 (C2″), 154.1 (C8a), 143.8 (C4), 133.4 (C4″), 132.5 (C2′), 131.0 (C6″), 130.7 (C1′), 129.8 (C5), 129.2 (C1″), 120.8 (C5″), 113.9 (C3), 113.8 (C4a), 113.4 (C8), 111.9 (C3″), 111.0 (C6), 94.9 (−OCH2O−), 56.8 (−OCH2OCH3), 56.0 (−OCH3); HRMS (ESI) m/z [M + H]+ calcd for C21H19O6 367.1182, found 367.1175.
Methyl 2-[(1E)-3-(7-Hydroxy-2-oxo-2H-chromen-8-yl)-3-oxoprop-1-en-1-yl]benzoate (14au)
Obtained from 12au (96 mg, 0.25 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 80 mg (0.23 mmol, 92%); mp 214–218 °C; IR (ATR) ν 2959 (w), 1723 (s), 1713 (s), 1599 (s), 1235 (m), 1266 (m), 1195 (m), 1116 (s), 1031 (m) cm–1; 1H NMR (400 MHz, CDCl3) δ 13.78 (s, 1H, -OH), 8.73 (d, J = 15.5 Hz, 1H, H3′), 8.05 (d, J = 15.5 Hz, 1H, H2′), 8.01–7.97 (m, 2H, H3″, H6″), 7.66 (d, J = 9.5 Hz, 1H, H4), 7.64 (t, J = 7.5 Hz, 1H, H5″), 7.53 (d, J = 8.9 Hz, 1H, H5), 7.48 (t, J = 7.8 Hz, 1H, H4″), 6.94 (d, J = 8.8 Hz, 1H, H6), 6.29 (d, J = 9.5 Hz, 1H, H3), 3.96 (s, 3H, −OCH3); 13C{1H} NMR (101 MHz, CDCl3) δ 193.6 (C1′), 167.7 (C7), 167.3 (-CO2CH3), 159.6 (C2), 155.7 (C8a), 144.8 (C3′), 144.4 (C4), 136.8 (C1″), 134.7 (C5), 133.0 (C5″), 130.9 (C3″), 130.5 (C2″), 130.0 (C4″), 128.9 (C2′), 128.7 (C6″), 115.9 (C6), 112.2 (C3), 111.2 (C4a), 109.8 (C8), 52.6 (−OCH3); HRMS (ESI) m/z [M + H]+ calcd for C20H15O6 351.0863, found 351.0862.
(E)-7-(Methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-8-(3-(4-methoxyphenyl)-3-oxoprop-1-en-1-yl)chroman-4-one (17af)
Obtained from 16af (240 mg, 0.49 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 120 mg, (0.24 mmol, 49%); mp 143–144 °C; IR (ATR) ν 2839 (w), 1673 (m), 1654 (m), 1578 (s), 1256 (m), 1026 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 16.0 Hz, 1H, H1′), 8.06 (d, J = 16.0 Hz, 1H, H2′), 7.96 (d, J = 8.9 Hz, 1H, H5), 7.75 (d, J = 9.0 Hz, 2H, H2″), 7.51 (d, J = 8.6 Hz, 2H, H2‴), 7.16 (d, J = 8.6 Hz, 2H, H3‴), 6.91 (d, J = 8.9 Hz, 1H, H6), 6.84 (d, J = 9.0 Hz, 2H, H3″), 5.53 (dd, J = 13.6, 2.8 Hz, 1H, H2), 5.35 (s, 2H, C7–OCH2OCH3), 5.24 (s, 2H, C4‴–OCH2OCH3), 3.86 (s, 3H, C4″–OCH3), 3.52 (s, 3H, C7–OCH2OCH3), 3.52 (s, 3H, C4‴–OCH2OCH3), 3.16 (dd, J = 16.8, 13.6 Hz, 1H, H3ax), 2.89 (dd, J = 16.8, 2.8 Hz, 1H, H3eq); 13C{1H} NMR (100 MHz, CDCl3) δ 190.8 (C4), 189.3 (C3′), 163.4 (C4″), 162.7 (C7), 162.4 (C8a), 158.0 (C4‴), 133.0 (C1′), 131.8 (C1‴), 131.4 (C1″), 130.9 (C2″), 130.1 (C5), 128.2 (C2‴), 125.8 (C2′), 116.8 (C3‴), 116.0 (C4a), 113.8 (C3″), 113.5 (8), 108.2 (C6), 94.7 (C7–OCH2OCH3) 94.5 (C4‴–OCH2OCH3), 80.4 (C2), 56.9 (C7–OCH2OCH3), 56.3 (C4‴–OCH2OCH3), 55.5 (C4″–OCH3), 43.8 (C3); HRMS (EI) m/z [M+] calcd for C29H28O8 504.1784, found 504.1782.
(E)-7-Methoxy-2-(4-(methoxymethoxy)phenyl)-8-(3-(4-methoxyphenyl)-3-oxoprop-1-en-1-yl)chroman-4-one (17bf)
Obtained from 16bf (230 mg, 0.50 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 85 mg (0.18 mmol, 36%); mp 198–199 °C; IR (ATR) ν 2936 (w), 2837 (w), 1676 (m), 1650 (m), 1601 (s), 1580 (s), 1230 (s), 1081 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 15.9 Hz, 1H), 8.02 (d, J = 15.9 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H), 7.76 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 8.6 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 6.68 (d, J = 8.8 Hz, 1H), 5.52 (dd, J = 13.5, 2.8 Hz, 1H), 5.24 (s, 2H), 3.98 (s, 3H), 3.85 (s, 3H), 3.52 (s, 3H), 3.15 (dd, J = 16.9, 13.5 Hz, 1H), 2.88 (dd, J = 16.9, 2.8 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 190.9, 189.4, 165.0, 163.3, 162.3, 157.9, 133.0, 131.9, 131.5, 130.9, 130.3, 128.1, 125.6, 116.7, 115.4, 113.7, 112.7, 105.1, 94.5, 80.3, 56.4, 56.3, 55.5, 43.8; HRMS (EI) m/z [M+] calcd for C28H26O7 474.1679, found 474.1676.
(E)-7-(Methoxymethoxy)-2-(4-methoxyphenyl)-8-(3-(4-methoxyphenyl)-3-oxoprop-1-en-1-yl)chroman-4-one (17cf)
Obtained from 16cf (137 mg, 0.30 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 104 mg (0.22 mmol, 73%); mp 188–189 °C; IR (ATR) ν 2839 (w), 1674 (m), 1655 (m), 1578 (s), 1256 (m), 1027 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.16 (d, J = 16.0 Hz, 1H), 8.04 (d, J = 16.0 Hz, 1H), 7.95 (d, J = 8.9 Hz, 1H), 7.70 (d, J = 8.8 Hz, 2H), 7.51 (d, J = 8.6 Hz, 2H), 7.03 (d, J = 8.6 Hz, 2H), 6.91 (d, J = 8.9 Hz, 1H), 6.79 (d, J = 8.8 Hz, 2H), 5.52 (dd, J = 13.7, 2.7 Hz, 1H), 5.35 (s, 2H), 3.88 (s, 3H), 3.86 (s, 3H), 3.52 (s, 3H), 3.19 (dd, J = 16.9, 13.7 Hz, 1H), 2.89 (dd, J = 16.9, 2.7 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3) δ 190.9, 189.3, 163.3, 162.7, 162.4, 160.3, 132.9, 131.5, 130.9, 130.6, 130.1, 128.4, 125.8, 116.0, 114.5, 113.7, 113.5, 108.2, 94.7, 80.5, 56.9, 55.5, 55.5, 43.6; HRMS (EI) m/z [M+] calcd for C28H26O7 474.1679, found 474.1692.
7-Methoxy-3-(4-methoxyphenyl)-8-[(1E)-3-(4-methoxyphenyl)-3-oxoprop-1-en-1-yl]-4H-chromen-4-one (21)
Obtained from 20 (180 mg, 0.42 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (9:1 (v/v) to 5:1 (v/v)); yield: 96 mg (0.21 mmol, 50%); mp 177–178 °C; IR (ATR) ν 1660 (s), 1594 (s), 1513 (m), 1417 (m), 1296 (s), 1172 (s) cm–1; 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 9.0 Hz, 1H, H5), 8.24 (d, J = 16.0 Hz, 1H, H1′), 8.07 (d, J = 16.0 Hz, 1H, H2′), 8.06 (d, J = 8.7 Hz, 2H, H2″), 8.03 (s, 1H, H2), 7.52 (d, J = 8.7 Hz, 2H, H2‴), 7.06 (d, J = 9.0 Hz, 1H, H6), 6.98 (d, J = 8.7 Hz, 2H, H3″ or H3‴), 6.97 (d, J = 8.7 Hz, 2H, H3″ or H3‴), 4.06 (s, 3H, C7–OCH3), 3.88 (s, 3H, C4″–OCH3), 3.84 (s, 3H, C4‴–OCH3); 13C{1H} NMR (100 MHz, CDCl3) δ 189.6 (C3′), 175.9 (C4), 163.5 (C4″), 162.9 (C7), 159.8 (C4‴), 156.0 (C8a), 152.1 (C2), 132.1 (C1′), 131.3 (C1″), 131.0 (C2″), 130.2 (C2‴), 129.3 (C5), 126.8 (C2′), 124.8 (C3), 123.9 (C1‴), 118.7 (C4a), 114.1 (C3″ or C3‴), 114.0 (C3″ or C3‴), 112.4 (C8), 109.3 (C6), 56.6 (C7–OCH3), 55.6 (C4″–OCH3), 55.4 (C4‴–OCH3); HRMS (EI) m/z [M+] calcd for C27H22O6 442.1416, found 442.1410.
General Procedure for the Cleavage of MOM-Protecting Groups
To a solution of the respective MOM-ether 17 (0.5 mmol, 1.0 equiv) in methanol (10 mL, 0.05 M in 17) was added aqueous HCl (4 M, 1.5 mmol, 3.0 equiv per MOM-group) and the mixture was heated at 60 °C for 2 h. It was then cooled to ambient temperature, diluted with water (20 mL), and extracted with ethyl acetate (three times 20 mL). The combined organic extracts were dried with MgSO4 and filtered, and all volatiles were evaporated in vacuo. The residue was purified by column chromatography on silica, using hexanes–ethyl acetate mixtures as eluent.
(E)-7-Hydroxy-2-(4-hydroxyphenyl)-8-(3-(4-methoxyphenyl)-3-oxoprop-1-en-1-yl)chroman-4-one (18af)
Obtained from 17af (75 mg, 0.15 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (3:1 (v/v) to 1:1 (v/v)); yield: 40 mg (0.10 mmol, 67%); mp 170–171 °C; IR (ATR) ν 3270 (w), 2972 (w), 1657 (m), 1601 (s), 1576 (s), 1548 (s), 1234 (s), 1165 (s) cm–1; 1H NMR (400 MHz, DMSO-d6) δ 11.52 (s,1H), 9.79 (s, 1H), 8.01 (d, J = 15.9 Hz, 1H), 7.96 (d, J = 15.9 Hz, 1H), 7.75 (d, J = 8.8 Hz, 1H), 7.60 (d, J = 8.7 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 8.4 Hz, 2H), 6.71 (d, J = 8.8 Hz, 1H), 5.63 (dd, J = 13.8, 2.6 Hz, 1H), 3.85 (s, 3H), 3.35 (dd, J = 16.8, 13.8 Hz, 1H), 2.71 (dd, J = 16.8, 2.6 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 190.3, 187.7, 164.2, 163.0, 162.8, 158.1, 133.1, 130.7, 130.2, 129.7, 128.9, 128.9, 123.5, 115.4, 113.9, 113.7, 109.9, 109.9, 79.9, 55.5, 42.2; HRMS (ESI) m/z [M + H]+ calcd for C25H21O6 417.1338, found 417.1348.
(E)-2-(4-Hydroxyphenyl)-7-methoxy-8-(3-(4-methoxyphenyl)-3-oxoprop-1-en-1-yl)chroman-4-one (18bf)
Obtained from 17bf (40 mg, 0.08 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (3:1 (v/v) to 1:1 (v/v)); yield: 34 mg (0.08 mmol, quant.); mp 214–215 °C; IR (ATR) ν 3279 (w), 2832 (w), 1682 (m), 1588 (s), 1552 (m), 1231 (s), 1162 (s) cm–1; 1H NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 7.98 (d, J = 16.0 Hz, 1H), 7.92 (d, J = 16.0 Hz, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 8.0 Hz, 2H), 6.98–6.83 (m, 4H), 6.89 (d, J = 8.9 Hz, 1H), 5.62 (dd, J = 13.8, 2.7 Hz, 1H), 3.98 (s, 3H), 3.84 (s, 3H), 3.37 (dd, J = 16.7, 13.8 Hz, 1H), 2.74 (dd, J = 16.7, 2.7 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 190.7, 187.6, 164.2, 163.0, 161.9, 158.2, 132.3, 130.6, 130.3 (2C), 130.1, 128.9 (2C), 128.8, 124.4, 115.5 (2C), 115.0, 113.9 (2C), 111.2, 105.4, 80.1, 56.8, 55.5, 42.3; HRMS (ESI) m/z [M + H]+ calcd for C26H23O6 431.1495, found 431.1475.
(E)-7-Hydroxy-2-(4-methoxyphenyl)-8-(3-(4-methoxyphenyl)-3-oxoprop-1-en-1-yl)chroman-4-one (18cf)
Obtained from 17cf (70 mg, 0.15 mmol) as a yellow solid; eluent for chromatography: hexanes–ethyl acetate mixtures of increasing polarity (3:1 (v/v) to 1:1 (v/v)); yield: 35 mg (0.08 mmol, 54%); mp 186–187 °C; IR (ATR) ν 3120 (w), 2931 (w), 1687 (m), 1636 (m), 1595 (s), 1438 (s), 1254 (s), 1230 (s), 1160 (s) cm–1; 1H NMR (400 MHz, DMSO-d6) δ 7.98 (s, 1H), 7.74 (d, J = 8.6 Hz, 1H), 7.61 (d, J = 8.7 Hz, 2H), 7.60 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 6.70 (d, J = 8.6 Hz, 1H), 5.67 (dd, J = 13.6, 2.8 Hz, 1H), 3.83 (s, 3H), 3.83 (s, 3H), 3.32 (dd, J = 16.8, 13.6 Hz, 1H), 2.72 (dd, J = 16.8, 2.8 Hz, 1H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 190.3, 187.9, 164.3, 163.0, 162.7, 159.8, 133.2, 130.8, 130.8, 130.3, 129.7, 128.2, 123.6, 114.2, 113.9, 113.7, 110.1, 110.0, 79.8, 55.5, 55.4, 42.2; HRMS (ESI) m/z [M + H]+ calcd for C26H23O6 431.1495, found 431.1500.
Acknowledgments
We thank Evonik Oxeno for generous donations of solvents and Umicore (Hanau, Germany) for generous donations of precious metal catalysts.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.4c02520.
Crystallographic details and refinement data for compound 16cf; synthesis procedures, characterization data and copies of spectra for test substrate 11d and all intermediates; synthesis procedures, characterization data and copies of spectra for 15b and its precursor; copies of spectra for all products 12, 13, 14, 16, 17, 18, 20, 21 and 23 (PDF)
FAIR data, including the primary NMR FID files, for compounds 11d, 12aa–12ai, 12ak–12ar, 12at, 12au, 13aa–13am, 13ap, 13aq, 13at, 14ae, 14af, 14an, 14ar, 14au, 15b, 16af–16cf, 17af–17cf, 18af–18cf, 20, 21, 23, SI-2, SI-3 and SI-7 (ZIP)
The authors declare no competing financial interest.
Supplementary Material
References
- Meunier B. Hybrid Molecules with a Dual Mode of Action: Dream or Reality?. Acc. Chem. Res. 2008, 41, 69–77. 10.1021/ar7000843. [DOI] [PubMed] [Google Scholar]
- Shaveta; Mishra S.; Singh P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. 10.1016/j.ejmech.2016.08.039. [DOI] [PubMed] [Google Scholar]
- Tietze L. F.; Bell H. P.; Chandrasekhar S. Natural Product Hybrids as New Leads for Drug Discovery. Angew. Chem., Int. Ed. 2003, 42, 3996–4028. 10.1002/anie.200200553. [DOI] [PubMed] [Google Scholar]
- Rozmer Z.; Perjési P. Naturally occurring chalcones and their biological activities. Phytochem. Rev. 2016, 15, 87–120. 10.1007/s11101-014-9387-8. [DOI] [Google Scholar]
- Russell A. E.; Gines B. R. Chalcones: Potential Chemotherapeutic Compounds and Educational Tools for Closing the Loop in STEM. Acc. Chem. Res. 2023, 56, 1256–1262. 10.1021/acs.accounts.2c00583. [DOI] [PubMed] [Google Scholar]
- Obaid R. J.; Mughal E. U.; Naeem N.; Sadiq A.; Alsantali R. I.; Jassas R. S.; Moussa Z.; Ahmed S. A. Natural and synthetic flavonoid derivatives as new potential tyrosinase inhibitors: a systematic review. RSC Adv. 2021, 11, 22159–22198. 10.1039/D1RA03196A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang C.; Zhang W.; Sheng C.; Zhang W.; Xing C.; Miao Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes M. N.; Muratov E. N.; Pereira M.; Peixoto J. C.; Rosseto L. P.; Cravo P. V. L.; Andrade C. H.; Neves B. J. Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules 2017, 22, 1210. 10.3390/molecules22081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F.; Huang G.; Xiao J. Chalcone hybrids as potential anticancer agents: Current development, mechanism of action, and structure-activity relationship. Med. Res. Rev. 2020, 40, 2049–2084. 10.1002/med.21698. [DOI] [PubMed] [Google Scholar]
- Mohamed M. F. A.; Abuo-Rahma G. E.-D. A. Molecular targets and anticancer activity of quinoline–chalcone hybrids: literature review. RSC Adv. 2020, 10, 31139–31155. 10.1039/D0RA05594H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leechaisit R.; Mahalapbutr P.; Boonsri P.; Karnchanapandh K.; Rungrotmongkol T.; Prachayasittikul V.; Prachayasittikul S.; Ruchirawat S.; Prachayasittikul V.; Pingaew R. Discovery of Novel Naphthoquinone–Chalcone Hybrids as Potent FGFR1 Tyrosine Kinase Inhibitors: Synthesis, Biological Evaluation, and Molecular Modeling. ACS Omega 2023, 8, 32593–32605. 10.1021/acsomega.3c03176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koley M.; Han J.; Soloshonok V. A.; Mojumder S.; Javahershenas R.; Makarem A. Latest developments in coumarin-based anticancer agents: mechanism of action and structure–activity relationship studies. RSC Med. Chem. 2024, 15, 10–54. 10.1039/D3MD00511A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng P.; Yang L.; Huang X.; Wang X.; Gong M. Chalcone hybrids and their antimalarial activity. Arch. Pharm. 2020, 353, 1900350 10.1002/ardp.201900350. [DOI] [PubMed] [Google Scholar]
- Sashidhara K. V.; Kumar A.; Kumar M.; Sarkar J.; Sinha S. Synthesis and in vitro evaluation of novel coumarin–chalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 7205–7211. 10.1016/j.bmcl.2010.10.116. [DOI] [PubMed] [Google Scholar]
- Singh N.; Sarkar J.; Sashidhara K. V.; Ali S.; Sinha S. Anti-tumour activity of a novel coumarin–chalcone hybrid is mediated through intrinsic apoptotic pathway by inducing PUMA and altering Bax/Bcl-2 ratio. Apoptosis 2014, 19, 1017–1028. 10.1007/s10495-014-0975-2. [DOI] [PubMed] [Google Scholar]
- Ashraf R.; Hamidullah; Hasanain M.; Pandey P.; Maheshwari M.; Singh L. R.; Siddiqui M. Q.; Konwar R.; Sashidhara K. V.; Sarkar J. Coumarin-chalcone hybrid instigates DNA damage by minor groove binding and stabilizes p53 through post translational modifications. Sci. Rep. 2017, 7, 45287. 10.1038/srep45287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi G.-L.; Liu Z.-Q. Coumarin moiety can enhance abilities of chalcones to inhibit DNA oxidation and to scavenge radicals. Tetrahedron 2014, 70, 8397–8404. 10.1016/j.tet.2014.08.063. [DOI] [Google Scholar]
- Pérez-Cruz F.; Vazquez-Rodriguez S.; Matos M. J.; Herrera-Morales A.; Villamena F. A.; Das A.; Gopalakrishnan B.; Olea-Azar C.; Santana L.; Uriarte E. Synthesis and Electrochemical and Biological Studies of Novel Coumarin–Chalcone Hybrid Compounds. J. Med. Chem. 2013, 56, 6136–6145. 10.1021/jm400546y. [DOI] [PubMed] [Google Scholar]
- Mazzone G.; Galano A.; Alvarez-Idaboy J. R.; Russo N. Coumarin–Chalcone Hybrids as Peroxyl Radical Scavengers: Kinetics and Mechanisms. J. Chem. Inf. Model. 2016, 56, 662–670. 10.1021/acs.jcim.6b00006. [DOI] [PubMed] [Google Scholar]
- Xue Y.; Liu Y.; Luo Q.; Wang H.; Chen R.; Liu Y.; Li Y. Antiradical Activity and Mechanism of Coumarin–Chalcone Hybrids: Theoretical Insights. J. Phys. Chem. A 2018, 122, 8520–8529. 10.1021/acs.jpca.8b06787. [DOI] [PubMed] [Google Scholar]
- Hamdi N.; Fischmeister C.; Puerta M. C.; Valerga P. A rapid access to new coumarinyl chalcone and substituted chromeno[4,3-c]pyrazol-4(1H)-ones and their antibacterial and DPPH radical scavenging activities. Med. Chem. Res. 2011, 20, 522–530. 10.1007/s00044-010-9326-1. [DOI] [Google Scholar]
- Vazquez-Rodriguez S.; Figueroa-Guíñez R.; Matos M. J.; Santana L.; Uriarte E.; Lapier M.; Maya J. D.; Olea-Azar C. Synthesis of coumarin–chalcone hybrids and evaluation of their antioxidant and trypanocidal properties. MedChemComm 2013, 4, 993–1000. 10.1039/c3md00025g. [DOI] [Google Scholar]
- Moya-Alvarado G.; Yañez O.; Morales N.; González-González A.; Areche C.; Núñez M. T.; Fierro A.; García-Beltrán O. Coumarin-Chalcone Hybrids as Inhibitors of MAO-B: Biological Activity and In Silico Studies. Molecules 2021, 26, 2430. 10.3390/molecules26092430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urmann C.; Bieler L.; Priglinger E.; Aigner L.; Couillard-Despres S.; Riepl H. M. Neuroregenerative Potential of Prenyl- and Pyranochalcones: A Structure–Activity Study. J. Nat. Prod. 2021, 84, 2675–2682. 10.1021/acs.jnatprod.1c00505. [DOI] [PubMed] [Google Scholar]
- Bajgai S. P.; Prachyawarakorn V.; Mahidol C.; Ruchirawat S.; Kittakoop P. Hybrid flavan-chalcones, aromatase and lipoxygenase inhibitors, from Desmos cochinchinensis. Phytochemistry 2011, 72, 2062–2067. 10.1016/j.phytochem.2011.07.002. [DOI] [PubMed] [Google Scholar]
- Riemer N.; Riemer M.; Krüger M.; Clarkson G. J.; Shipman M.; Schmidt B. Synthesis of Arylidene-β-lactams via exo-Selective Matsuda-Heck Arylation of Methylene-β-lactams. J. Org. Chem. 2021, 86, 8786–8796. 10.1021/acs.joc.1c00638. [DOI] [PubMed] [Google Scholar]
- Krause A.; Sperlich E.; Schmidt B. Matsuda–Heck arylation of itaconates: a versatile approach to heterocycles from a renewable resource. Org. Biomol. Chem. 2021, 19, 4292–4302. 10.1039/D1OB00392E. [DOI] [PubMed] [Google Scholar]
- Otte F.; Schmidt B. Matsuda-Heck Arylation of Glycals for the stereoselective Synthesis of Aryl C-Glycosides. J. Org. Chem. 2019, 84, 14816–14829. 10.1021/acs.joc.9b02410. [DOI] [PubMed] [Google Scholar]
- de Oliveira J. M.; Angnes R. A.; Khan I. U.; Polo E. C.; Heerdt G.; Servilha B. M.; Menezes da Silva V. H.; Braga A. A. C.; Correia C. R. D. Enantioselective, Noncovalent, Substrate-Directable Heck–Matsuda and Oxidative Heck Arylations of Unactivated Five-Membered Carbocyclic Olefins. Chem.—Eur. J. 2018, 24, 11738–11747. 10.1002/chem.201801910. [DOI] [PubMed] [Google Scholar]
- Polo E. C.; Wang M. F.; Angnes R. A.; Braga A. A. C.; Correia C. R. D. Enantioselective Heck Arylation of Acyclic Alkenol Aryl Ethers: Synthetic Applications and DFT Investigation of the Stereoselectivity. Adv. Synth. Catal. 2020, 362, 884–892. 10.1002/adsc.201901471. [DOI] [Google Scholar]
- Oger N.; Felpin F.-X. Heterogeneous Palladium Catalysts for Suzuki–Miyaura Coupling Reactions Involving Aryl Diazonium Salts. ChemCatChem. 2016, 8, 1998–2009. 10.1002/cctc.201600134. [DOI] [Google Scholar]
- Trusova M. E.; Rodriguez-Zubiri M.; Kutonova K. V.; Jung N.; Brase S.; Felpin F.-X.; Postnikov P. S. Ultra-fast Suzuki and Heck reactions for the synthesis of styrenes and stilbenes using arenediazonium salts as super-electrophiles. Org. Chem. Front. 2018, 5, 41–45. 10.1039/C7QO00750G. [DOI] [Google Scholar]
- Matelienė L.; Knaup J.; von Horsten F.; Gu X.; Brunner H. Benign Arylations of Dimethyl Itaconate via Heck–Matsuda Reaction Utilizing in-Situ Generated Palladium on Aluminum Oxide. Eur. J. Org. Chem. 2020, 2020, 127–135. 10.1002/ejoc.201901708. [DOI] [Google Scholar]
- Li Z.; Tong R. Asymmetric Total Syntheses of (−)-Hedycoropyrans A and B. J. Org. Chem. 2017, 82, 1127–1135. 10.1021/acs.joc.6b02738. [DOI] [PubMed] [Google Scholar]
- Kikukawa K.; Matsuda T. Reaction of diazonium salts with transition metals. 1. Arylation of olefins with arene diazonium salts catalyzed by zero valent palladium. Chem. Lett. 1977, 6, 159–162. 10.1246/cl.1977.159. [DOI] [Google Scholar]
- Roglans A.; Pla-Quintana A.; Moreno-Mañas M. Diazonium Salts as Substrates in Palladium-Catalyzed Cross-Coupling Reactions. Chem. Rev. 2006, 106, 4622–4643. 10.1021/cr0509861. [DOI] [PubMed] [Google Scholar]
- Felpin F.-X.; Nassar-Hardy L.; Le Callonnec F.; Fouquet E. Recent Advances in the Heck-Matsuda Reaction in Heterocyclic Chemistry. Tetrahedron 2011, 67, 2815–2831. 10.1016/j.tet.2011.02.051. [DOI] [Google Scholar]
- Taylor J. G.; Moro A. V.; Correia C. R. D. Evolution and Synthetic Applications of the Heck–Matsuda Reaction: The Return of Arenediazonium Salts to Prominence. Eur. J. Org. Chem. 2011, 2011, 1403–1428. 10.1002/ejoc.201001620. [DOI] [Google Scholar]
- Schmidt B.; Hölter F.; Berger R.; Jessel S. Mizoroki-Heck reactions with 4-phenol diazonium salts. Adv. Synth. Catal. 2010, 352, 2463–2473. 10.1002/adsc.201000493. [DOI] [Google Scholar]
- Schultze C.; Schmidt B. Prenylcoumarins in One or Two Steps by a Microwave-Promoted Tandem Claisen Rearrangement/Wittig Olefination/Cyclization Sequence. J. Org. Chem. 2018, 83, 5210–5224. 10.1021/acs.joc.8b00667. [DOI] [PubMed] [Google Scholar]
- Andrus M. B.; Song C.; Zhang J. Q. Palladium-imidazolium carbene catalyzed Mizoroki-Heck coupling with aryl diazonium ions. Org. Lett. 2002, 4, 2079–2082. 10.1021/ol025961s. [DOI] [PubMed] [Google Scholar]
- Werner E. W.; Sigman M. S. Operationally Simple and Highly (E)-Styrenyl-Selective Heck Reactions of Electronically Nonbiased Olefins. J. Am. Chem. Soc. 2011, 133, 9692–9695. 10.1021/ja203164p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigman M. S.; Werner E. W. Imparting Catalyst Control upon Classical Palladium-Catalyzed Alkenyl C–H Bond Functionalization Reactions. Acc. Chem. Res. 2012, 45, 874–884. 10.1021/ar200236v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner E. W.; Sigman M. S. A Highly Selective and General Palladium Catalyst for the Oxidative Heck Reaction of Electronically Nonbiased Olefins. J. Am. Chem. Soc. 2010, 132, 13981–13983. 10.1021/ja1060998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcamp J. H.; Brucks A. P.; White M. C. A General and Highly Selective Chelate-Controlled Intermolecular Oxidative Heck Reaction. J. Am. Chem. Soc. 2008, 130, 11270–11271. 10.1021/ja804120r. [DOI] [PubMed] [Google Scholar]
- Schmidt B.; Riemer M. Synthesis of Allyl- and Prenylcoumarins via Microwave-promoted Tandem Claisen-Rearrangement/Wittig-Olefination. Synthesis 2015, 48, 141–149. 10.1055/s-0035-1560501. [DOI] [Google Scholar]
- Yang H.; Sun P.; Zhu Y.; Yan H.; Lu L.; Qu X.; Li T.; Mao J. Copper-catalyzed decarboxylative C(sp2)–C(sp3) coupling reactions via radical mechanism. Chem. Commun. 2012, 48, 7847–7849. 10.1039/c2cc33203e. [DOI] [PubMed] [Google Scholar]
- Weidmann V.; Maison W. Allylic oxidations of olefins to enones. Synthesis 2013, 45, 2201–2221. 10.1055/s-0033-1338491. [DOI] [Google Scholar]
- Nakamura A.; Nakada M. Allylic Oxidations in Natural Product Synthesis. Synthesis 2013, 45, 1421–1451. 10.1055/s-0033-1338426. [DOI] [Google Scholar]
- Murahashi S.-I.; Komiya N.. Oxidation Reactions. In Ruthenium in Organic Synthesis; Murahashi S.-I., Ed.; Wiley-VCH: Weinheim, 2004; pp 53–93. [Google Scholar]
- Murahashi S.-I.; Komiya N.; Oda Y.; Kuwabara T.; Naota T. Ruthenium-Catalyzed Oxidation of Alkanes with tert-Butyl Hydroperoxide and Peracetic Acid. J. Org. Chem. 2000, 65, 9186–9193. 10.1021/jo001348f. [DOI] [PubMed] [Google Scholar]
- Schmidt B.; Krehl S. A single precatalyst Tandem RCM-allylic oxidation sequence. Chem. Commun. 2011, 47, 5879–5881. 10.1039/c1cc11347j. [DOI] [PubMed] [Google Scholar]
- Schwab P.; Grubbs R. H.; Ziller J. W. Synthesis and applications of RuCl2(=CHR′)(PR3)2: the influence of the alkylidene moiety on metathesis activity. J. Am. Chem. Soc. 1996, 118, 100–110. 10.1021/ja952676d. [DOI] [Google Scholar]
- Fousteris M. A.; Koutsourea A. I.; Nikolaropoulos S. S.; Riahi A.; Muzart J. Improved chromium-catalyzed allylic oxidation of Δ5-steroids with t-butyl hydroperoxide. J. Mol. Catal. A: Chem. 2006, 250, 70–74. 10.1016/j.molcata.2006.01.031. [DOI] [Google Scholar]
- Parish E. J.; Wei T.-Y. Allylic Oxidation of Δ5-Steroids with Pyridinium Chlorochromate (PCC) and Pyridinium Dichromate (PDC). Synth. Commun. 1987, 17, 1227–1233. 10.1080/00397918708063974. [DOI] [Google Scholar]
- Umbreit M. A.; Sharpless K. B. Allylic oxidation of olefins by catalytic and stoichiometric selenium dioxide with tert-butyl hydroperoxide. J. Am. Chem. Soc. 1977, 99, 5526–5528. 10.1021/ja00458a072. [DOI] [Google Scholar]
- Barton D. H. R.; Crich D. Oxidation of olefins with 2-pyridineseleninic anhydride. Tetrahedron 1985, 41, 4359–4364. 10.1016/S0040-4020(01)97207-2. [DOI] [Google Scholar]
- Salvador J. A. R.; Sáe Melo M. L.; Campos Neves A. S. Copper-catalysed allylic oxidation of Δ5-steroids by t-butyl hydroperoxide. Tetrahedron Lett. 1997, 38, 119–122. 10.1016/S0040-4039(96)02231-9. [DOI] [Google Scholar]
- Sharma A.; Sharma N.; Shard A.; Kumar R.; Mohanakrishnan D.; Saima; Sinha A. K.; Sahal D. Tandem allylic oxidation–condensation/esterification catalyzed by silica gel: an expeditious approach towards antimalarial diaryldienones and enones from natural methoxylated phenylpropenes. Org. Biomol. Chem. 2011, 9, 5211–5219. 10.1039/c1ob05293d. [DOI] [PubMed] [Google Scholar]
- Joshi B. P.; Sharma A.; Sinha A. K. Ultrasound-assisted convenient synthesis of hypolipidemic active natural methoxylated (E)-arylalkenes and arylalkanones. Tetrahedron 2005, 61, 3075–3080. 10.1016/j.tet.2005.01.071. [DOI] [Google Scholar]
- Joshi B. P.; Sharma A.; Sinha A. K. Efficient one-pot, two-step synthesis of (E)-cinnmaldehydes by dehydrogenation–oxidation of arylpropanes using DDQ under ultrasonic irradiation. Tetrahedron 2006, 62, 2590–2593. 10.1016/j.tet.2005.12.028. [DOI] [Google Scholar]
- Tanemura K.; Suzuki T.; Horaguchi T. Deprotection of Acetals and Silyl Ethers Using Some π-Acceptors. Bull. Chem. Soc. Jpn. 1994, 67, 290–292. 10.1246/bcsj.67.290. [DOI] [Google Scholar]
- Raina S.; Singh V. K. DDQ as a Mild and Efficient Catalyst for Deprotection of Tetrahydropyranyl Ethers. Synth. Commun. 1995, 25, 2395–2400. 10.1080/00397919508015442. [DOI] [Google Scholar]
- Schultze C.; Foß S.; Schmidt B. 8-Prenylflavanones via Microwave Promoted Tandem Claisen Rearrangement/6-endo-trig Cyclization and Cross Metathesis. Eur. J. Org. Chem. 2020, 2020, 7373–7384. 10.1002/ejoc.202001378. [DOI] [Google Scholar]
- Kwesiga G.; Sperlich E.; Schmidt B. Scope and Applications of 2,3-Oxidative Aryl Rearrangements for the Synthesis of Isoflavone Natural Products. J. Org. Chem. 2021, 86, 10699–10712. 10.1021/acs.joc.1c01375. [DOI] [PubMed] [Google Scholar]
- Kwesiga G.; Kelling A.; Kersting S.; Sperlich E.; von Nickisch-Rosenegk M.; Schmidt B. Total Syntheses of Prenylated Isoflavones from Erythrina sacleuxii and Their Antibacterial Activity: 5-Deoxy-3′-prenylbiochanin A and Erysubin F. J. Nat. Prod. 2020, 83, 3445–3453. 10.1021/acs.jnatprod.0c00932. [DOI] [PubMed] [Google Scholar]
- Pedersen D. S.; Rosenbohm C. Dry Column Vacuum Chromatography. Synthesis 2001, 2001, 2431–2434. 10.1055/s-2001-18722. [DOI] [Google Scholar]
- Firth J. D.; Fairlamb I. J. S. A Need for Caution in the Preparation and Application of Synthetically Versatile Aryl Diazonium Tetrafluoroborate Salts. Org. Lett. 2020, 22, 7057–7059. 10.1021/acs.orglett.0c02685. [DOI] [PubMed] [Google Scholar]
- Souza E. L. S. d.; Chorro T. H. D.; Correia C. R. D. Thermal analysis of arenediazonium tetrafluoroborate salts: Stability and hazardous evaluation. Process Saf. Environ. 2023, 177, 69–81. 10.1016/j.psep.2023.06.082. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.