

Abstract
Imbalances in gut microbiota and their metabolites have been implicated in osteoporotic disorders. Trimethylamine-n-oxide (TMAO), a metabolite of L-carnitine produced by gut microorganisms and flavin-containing monooxygenase-3, is known to accelerate tissue metabolism and remodeling; however, its role in bone loss remained unexplored. This study investigates the relationship between gut microbiota dysbiosis, TMAO production, and osteoporosis development. We further demonstrate that the loss of beneficial gut microbiota is associated with the development of murine osteoporosis and alterations in the serum metabolome, particularly affecting L-carnitine metabolism. TMAO emerges as a functional metabolite detrimental to bone homeostasis. Notably, transplantation of mouse gut microbiota counteracts obesity- or estrogen deficiency-induced TMAO overproduction and mitigates key features of osteoporosis. Mechanistically, excessive TMAO intake augments bone mass loss by inhibiting bone mineral acquisition and osteogenic differentiation. TMAO activates the PERK and ATF4-dependent disruption of endoplasmic reticulum autophagy and suppresses the folding of ATF5, hindering mitochondrial unfolding protein response (UPRmt) in osteoblasts. Importantly, UPRmt activation by nicotinamide riboside mitigates TMAO-induced inhibition of mineralized matrix biosynthesis by preserving mitochondrial oxidative phosphorylation and mitophagy. Collectively, our findings revealed that gut microbiota dysbiosis leads to TMAO overproduction, impairing ER homeostasis and UPRmt, thereby aggravating osteoblast dysfunction and development of osteoporosis. Our study elucidates the catabolic role of gut microflora-derived TMAO in bone integrity and highlights the therapeutic potential of healthy donor gut microbiota transplantation to alter the progression of osteoporosis.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-024-05501-y.
Keywords: Gut microecosystem, Trimethylamine-n-oxide, ER-phagy, Parkin, OXPHOS, Misfolding
Introduction
Osteoporosis is a leading cause of skeletal fragility related premature death among the elderly population [1]. Metabolic syndromes associated obesity correlate with the development of osteoporotic disorders [2]. For instance, diet-induced obesity inhibits bone formation [3] and over-activates glucocorticoid signaling in osteoblasts [4], thereby exacerbating skeletal tissue deterioration. Additionally, diabetes promotes inflammasome and osteoblast pyroptosis [5], while steering macrophages in adipose tissue into osteoclastogenesis, thereby impairing bone homeostasis [6].
Emerging evidence indicates that gut microorganism dysbiosis and metabolite imbalance deteriorate gut barrier integrity and immune response [7], enhancing osteoclastic resorption [8] and accelerating bone loss [9]. Among these metabolites, trimethylamine n-oxide (TMAO) is an oxidized product of trimethylamine (TMA), which is catalyzed by flavin containing monooxygenase 3 (FMO3) after gut microorganisms convert carnitine into TMA [10]. Elevated plasma TMAO levels are further associated with an increased risk of osteoporotic hip fracture in obese adults [11]. Chronic high-fat diet consumption increases gut oxygen and nitrate availability prompting microflora to produce TMAO [12], which augments oxidative stress for osteoclast formation [13] and tissue inflammation [14].
Endoplasmic reticulum (ER) stress impairs protein folding capacity, leading to the accumulation of unfolded proteins and the activation of the unfolded protein response (UPR) through key regulators such as protein kinase R-link endoplasmic reticulum kinase (PERK), activating transcription factor 4 (ATF4), eukaryotic initiation factor 2 (eIF2α), and CCAAT/enhancer-binding protein homologous protein (CHOP) [15]. Inhibition of UPR reduces osteoclast formation and mitigates osteoporosis development [16]. Furthermore, the mitochondrial UPR (UPRmt), regulated by activating transcription factor 5 (ATF5), is crucial for maintaining proteostasis in the mitochondrial microenvironment, enabling adaptation to extracellular stresses, like obesity and hyperglycemia [17, 18]. Control of UPRmt enhances the osteogenic differentiation capacity of bone-marrow stromal cells in ovariectomized rats [19]. Notably, TMAO is known to activate receptor PERK pathway, disrupting mitochondrial metabolism [20], energy production and redox capacity [21]. However, the specific role of TMAO in osteoblastic activity and osteoporosis development remains poorly understood.
This study aimed to characterize whether gut microbiota affected TMAO production or bone mass loss in high-fat diet-induced obese mice and ovariectomized mice. Additionally, we investigated how TMAO influenced osteoblastic activity and bone homeostasis in osteoporotic bone tissue.
Materials and methods
High-fat diet-induced obesity
With the approval of Institutional Animal Care and Use Committee of Kaohsiung Chang Gung Memorial Hospital (Affidavit #2019091103), male C57BL/6 mice (12 weeks old) were fed high-fat diet (HFD; 60 kcal% fat; D12450K, Research Diet Inc. New Brunswick, NJ) or chow diet (CD; 10 kcal% fat; D1294) together with drinking water ad libitum for 6 consecutive months. All animals were housed in a specific pathogen-free vivarium. Blood glucose levels were quantified using Biochemistry-SP-430 (Arkray Inc., Tokyo, Japan). Animals were euthanatized to dissect bone tissue.
µCT analyses of body adipose and bone mass
Anesthetized mice were µCT scanned (50 kV, 1-mm AI filter, 0.9° rotation, and 2 frames) using 1176 Skyscan system (Bruker, Billerica, MA). Thirty-five-µm pixel size radiographs of whole body were reconstructed into three-dimension images. Muscle and adipose were manually contoured using SKYSCAN® CT-Analysis platform and Micro-CT Mouse Phantom (QRM-70137, Freiburg, Germany) as a reference. Volumes of visceral and subcutaneous fat of the regions of interest (binarization thresholds, 73– 90) were calculated using the software, according to the manufacturer's instructions. In some experiments, femur and tibiae specimens were µCT scanned to capture 300 slices of 9-µm pixel size radiographs, as previously described [22]. Upon calibration with a calcium hydroxyapatite phantom (QRM-70134), bone mineral density (BMD, mg/cm3), trabecular volume (BV/TV, %), trabecular number (Tb.N/mm), trabecular thickness (Tb.Th, mm), trabecular separation (Tb.Sp), structure model index (SMI), cortical BMD, cortical thickness (Ct.Th, mm), and cortical porosity (%) of the specimens were calculated using in-house software.
Biomechanical strength analyses
Biomechanical analyses of femurs were conducted using SHIMADZU Electromechanical Tester (EX-SX, Shimadzu, Kyoto, Japan). Upon placing bone tissue onto a two-jig holder (jig span, 0.5 cm), the middle parts of the specimens were 3-point bended under a load of 50-N, which was displaced at 10 mm/min. The tester's in-house TRAPEZIVMX software was utilized to calculate breaking force (N) and breaking energy (J), which were normalized with the cross-section areas of middle part of the specimens.
16S rRNA sequencing
Fresh feces of mice were harvested upon fasting for 12 h; with all protocols were conducted under sterile conditions. Fecal DNA was isolated using QIAamp PowerFecal DNA Kits (Qiagen, Germantown, MA). To prepare multiplexed SMRTbell Library, a total of 1 ng/μl DNA was used to PCR amplify the V1-V9 regions of full length 16S rRNA genes using specific primers (forward, 5′Phos-GCATCAGRGTTYGATYMTGGCTCAG-3′; reverse, 5′Phos-RGYTACCTTGTTACGACTT-3′). PCR products were purified using AMPure PB beads (100–265-900; PacBio®, Menlo Park, CA); and the SMRTbell library was mixed with sequencing primer v4. Sequel II Binding Kits (102–194-200; PacBio®) were used for primer annealing, and polymerase binding. Gene sequencing was conducted to produce HiFi readouts (predicted accuracy, 30) using PacBio Sequel IIe (circular consensus sequence mode).
Gene readout processing, clustering, and annotation
PacBio Workflow in SMRT Link (minimal prediction accuracy, 0.9 and minimal number passes, 3) was used to process the readouts of Circular Consensus Sequence. In brief, DADA2 (version 1.20) pipeline was utilized for quality filtration, dereplication, chimera removal, and algorithm of amplicon sequence variants (ASVs) from full-length 16S rRNA gene. Annotation of taxonomy classification was conducted using QIIME2 software (version 2021.4) through retrieving NCBI database. Sequence similarities in ASVs against 16S ribosomal RNA database were analyzed using QIIME2 together with MAFFT software. To normalize the sequence depth among specimens, ASVs were rarefied to minimal sequence depth. Bioinformatics analyses were conducted using R package software (version 3.6.0), including ggplot2, factoextra, phyloseq, vegan3d, Rtsne, mixOmics, metagenomeSeq together with Wilcoxon rank-sum test, ALDEx2, and MicroEco. KEGG pathways for functional abundances of bacterial taxa with PICRUSt2, Tax4Fun2, and FAPROTAX (version1.2.4) was used. CPCoA, PCoA, and Heatmap were plotted using TBtools, and ImageGP [23].
UHPLC-MS analyses for serum metabolome
Peripheral blood was drawn using an intracardiac needle. A total of 50 µl serum was mixed with 1000 µl mixture of methanol, acetonitrile, and water (2:2:1 in volume), centrifuged at 10,000 × g for 15 min. Supernatants (10 µl) were eluted through Acquity BEH C18 column (Waters, Milford, MA) with 0.1% formic acid and acetonitrile at 0.25 ml/min for ultrahigh performance liquid chromatography with tandem mass spectrometry (MS) (Orbitrap Elite; Thermo Fischer Scientific. Waltham, MA). MS data acquisition was conducted using positive mode (MS profile resolution, 6000) together with a default data-dependent acquisition (resolution, 15,000), a scan range of 70 – 1000 m/z with the normalized collision energy set to 25. ProteoWizard software was utilized to convert MS data into mzXML format. Data extraction, alignment, and integration was conducted using R package and XCMS software. MS2 bioinformation engine was used for metabolite annotation (cutoff, 0.5) [24].
Fecal microbiota transplantation
Six grams of fresh feces from age-matched CD-fed mice were mixed with 6 ml sterile Ringer's solution. The mixtures were centrifuged (500 × g, 15 min) and fecal microbiota supernatants were harvested for transplantation. Each HFD-fed mouse was transplanted with 0.5 ml fecal microbiota supernatant from CD-fed mice via oral gavage (3 transplantations/week) for 5 consecutive months. In a subset of experiments, 12-week-old female C57BL/6 mice were anesthetized and bilaterally ovariectomized to induce osteoporosis. One week postoperatively, each ovariectomized mouse was transplanted with 0.5 ml fecal microbiota supernatants from aged-matched sham mice via oral gavage for 3 consecutive months (3 transplantations/week).
TMAO treatment
Twelve-week-old male C57BL/6 mice were fed on 0.2% TMAO (317594; Sigma-Aldrich, St Louis, MO) in sterile drinking water and chow diet ad libitum for consecutive 3 months. Control mice received the same diet without TMAO. Animals were euthanatized and peripheral blood and bone tissue were biopsied at the end of study.
ELISA
Serum was mixed with a mixture of methanol/acetonitrile and formaldehyde (15:85) and centrifuged at 14,000 × g for 5 min at 4 °C. The supernatants were eluted through SUPELCO Ascentis C18 column and tandem mass spectrometry to quantify TMA and TMAO. In some experiments, serum TMAO (AMS.E03T0904; Amsbio, Cambridge, MA), L-carnitine (ab83392; Abcam, Cambridge, UK), and FMO3 (MBS9347271; MyBioSource, San Diego, CA) were quantified using specific ELISA kits.
Ex vivo osteogenic and osteoclastogenic differentiation
Bone-marrow mesenchymal cells were isolated from FMT recipient mice or TMAO-fed mice or control mice. A total of 2 × 105 cells/well (24-well plates) were cultured in osteogenic medium (A1007201; Thermal Fisher Scientific) with 10% fetal bovine serum (FBS) for 18 days, as previously described [22]. Mineralized matrices were stained using von Kossa Stain Kits (ab150687; Abcam, Cambridge, UK). Areas of mineralized matrix in 3 random fields of each well from 6 wells of each experiment were measured using Zeiss Image Analysis System. In a subset of experiments, bone-marrow macrophages (5 × 104 cells/well, 48-well plates) were incubated in αMEM with 10% FBS, 20 ng/ml M-CSF and 40 ng/ml RANKL (R&D Systems, Minneapolis, MN) for 10 days [22]. Osteoclasts were stained using tartrate-resistant acid phosphatase staining kits (MK300; Takara Bio Inc., Shiba, Japan). TRAP-stained osteoclasts in 9 random fields from 3 wells were counted.
Histomorphometry and immunohistochemistry
Animals were intraperitoneally injected 50 mg/kg calcein 3 days and 9 days before the end of experiment. Calcein-labeled mineral deposition in the sections of methyl acrylate-embedded bone specimens were evaluated using fluorescence microscopy. Nine random fields of 3 sections of each specimen were selected to calculate mineral apposition rate (MAR, µm/day) and bone formation rate (BFR/BS, µm3/µm2/day), as previously described [25]. Sections of paraffin-embedded bone specimens were stained using alkaline phosphatase and tartrate-resistant acid phosphatase histochemical staining kits and hematoxylin and eosin stain to calculate osteoblast number (Ob.N/mm), osteoclast number (Oc.N/mm), and adipocyte number (Ad.N). In some experiments, sections of paraffine-embedded colon specimens were stained using Periodic Acid-Schiff Stain Kits (ab150680; Abcam, Cambridge, UK) or using primary CLDN-1 (SAB3500438; Sigma-Aldrich, St Louis, MO), TJP-1 (ab276131; Abcam), and IL-17 (ab79056; Abcam) antibodies together with Super Sensitive™ IHC Detection Systems (BioGenex Laboratories; Fremont, CA). Immunostained goblet cells in 9 random fields of 3 different sections of each animal were calculated.
Osteoblast culture
Murine MC3T3-E1 osteoblasts (105 cells/well, 24-well plates) were incubated in osteogenic medium (DMEM with 10% FBS, 50 µg/ml ascorbic acid, 1 mM β-glycerophosphate), supplemented with 50 µM, 100 µM, 200 µM and 300 µM TMAO for 1 day or 18 days. In a subset of experiments, osteoblasts were incubated in osteogenic medium with 200 µM TMAO or 5 µM GSK2606414 (5107; Tocris Bioscience, Bristol, UK) or 2.5 µM nicotinamide riboside (72340; Sigma-Aldrich, St Louis, MO). Mineralized matrices were stained using von Kossa staining kits. Areas of mineralized matrix were calculated from 9 random fields of 3 wells per experiment.
Osteoblast growth and senescence assay
Growth of osteoblasts (2 × 104 cells/well, 96-well plates) were quantified using BrdU Cell Proliferation Assay Kit (#6813; Cell Signaling Technology; Danvers, MA). Senescent cells were stained using Senescence β-Galactosidase Staining Kits (SA-β-gal; #9860; Cell Signaling Technology). SA-β-gal-stained senescent cells in 9 random fields of 3 different wells per experiment were counted.
RT-PCR
Extraction and reverse transcription of total RNA from 106 osteoblasts was conducted using PureLink RNA Mini Kit and High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific), respectively. PCR was conducted using ABI StepOne Plus 96-Well Real-Time PCR Detection System (Thermo Fisher Scientific), with Applied Biosystems™ TaqMan™ Universal PCR Master Mix and specific primers (Supplementary Table 1). The relative fold change in mRNA expression was calculated using the ΔΔCt method, with 18S rRNA as calibrator.
Mitochondrial respiration and ATP production
Seahorse XFe Analyzer together with Seahorse XFp Cell Mito Stress Test Kits (Agilent, Santa Clara, CA) were utilized to quantify mitochondrial respiration capacity. Upon 105 cells were incubated in cartridge with Seahorse XF DMEM Assay Medium, 2 µg/ml oligomycin, 2 µM FCCP, and 2 µM antimycin and rotenone were added to the cartridge to inhibit complex activities. Oxygen consumption rate was calculated automatically. ATP production of 106 osteoblasts were quantified using ATP Assay Kits (ab83355, Abcam), according to the manufacturer's manuals.
Western blotting
Primary antibodies PERK (#3192; Cell Signaling Technology), mTOR (#2983), RPTOR (#2280), FAM134B (#61011), ATF4 (#11815), Mfn2 (#9482), Actin (#4967), phosphorylated PERK (#PA5-102853; Thermal Fisher Scientific), LC3-II (#PA1-16930), ATF5 (#PA5-17988), Opa1 (ab42364, Abcam), and Pierce™ Fast Western Blot Kits (Thermal Fisher Scientific) were utilized for immunoblotting of cell lysates. To characterize aggregated Atf5, 100 µg lysate was ultracentrifuged (125,000 × g, 1 h) to harvest protein aggregates; and proteins from the pellets were separated using SDS-PAGE, followed by Atf5 immunoblotting [22].
Fluorescence microscopy and transmission electron microscopy
Endoplasmic reticulum stress and mitochondria in osteoblasts (102 cells/slide) were stained using ER-Tracker™ (E34251; Thermo Fisher Scientific), MitoTracker™ Green FM (M7514; Thermo Fisher Scientific), respectively. Autophagosome and mitophagosome in cells were stained using CYTO-ID Autophagy Detection Kits (ENZ-51031; Enzo Life Sciences, Farmingdale, NY) and Mitophagy Detection Kits (MD01; Dojindo Laboratories, Tokyo, Japan), respectively. In some experiments, osteoblasts were fixed by 3% glutaraldehyde, post-fixed by 1% OsO4, epoxy resin embedded, and cut into 50-nm sections, which were further coated by gold particles. ER ultrastructure in osteoblasts was investigated using Hitachi SU8229 TEM System.
Statistical analysis
The distribution and the differences in CD and HFD-fed mice or in vehicle and TMAO-fed mice were analyzed using Kolmogorov–Smirnov test and Student t-test, respectively. The differences in 3 or 4 groups were analyzed using ANOVA test and post hoc Bonferroni test. P value < 0.05 is considered significant difference.
Results
Obesity accelerated gut integrity loss and osteoporosis development
We investigated whether diet-induced obesity can alter gut integrity and bone homeostasis. Mice on HFD developed prominent features of obesity, including overweight, hyperglycemia (Fig. 1a), and body adiposis with much higher volumes of visceral and subcutaneous fat (Fig. 1b) than CD-fed animals. Colon in obese mice showed a plethora of atrophy features (Fig. 1c), including decreased colon length and villus height together with goblet cell loss and thus mucin underproduction as evident in periodic acid-Schiff staining (Fig. 1d and 1e). Gut barrier loss and inflammation were also present in obese mice revealing relatively low abundances of tight junction proteins CLDN-1 and TJP-1, but high IL-17 levels in goblet cells (Fig. 1d and e). Moreover, obese animals revealed a very sparse trabecular bone network (Fig. 1f) with decreased trabecular bone mineral density (Tb.BMD), trabecular thickness (Tb.Th), trabecular volume (BV/TV) and trabecular number (Tb.N), respectively, and relatively high trabecular separation (Tb.Sp) and SMI (Fig. 1g). Mineral apposition rate (MAR) and bone formation rate (BFR/BS) in obese mice were less than control animals (Fig. 1h).
Fig. 1.

Body adipose overdevelopment, bone mass loss, and gut atrophy in HFD-induced murine obesity. Increased body weight, blood glucose levels (a) as well as visceral fat and subcutaneous fat development in obese mice (b); scale bar, 1.5 cm. Decreased colon length and villi loss (c and e); scale bar of upper panels, 200 μm; scale bar of lower panels, 100 µm. Losses of PAS-stained mucin, CLDN-1, and TJP1 levels, but increased IL-17 levels in colon goblet cells of obese mice (d and e); scale bar, 15 μm. Sparse trabecular bone network (f); scale bar, 50 μm and decreased trabecular BMD, Tb.Th, BV/TV, Tb.N and increased Tb.Sp and SMI in obese mice (g). Decreased fluorescence calcein-labelling, MAR, and BFR/BS upon HFD consumption (h); scale bar, 20 μm. Mice were fed HFD or CD for 6 months. Data are means ± standard errors calculated from 6 mice. Asterisks (*) stand for significant difference (P < 0.05) analyzed by Kolmogorov–Smirnov test and Student t-test
Obesity induced gut microbiota dysbiosis
Altered gut microflora has been correlated with bone disorders [26]. Colon integrity loss in obese mice prompted us to conduct 16S rRNA sequencing to investigate the correlation between the gut microbiome and bone loss. Principal component analysis revealed distinctively different profiles in the fecal microbiota of obese and control mice, respectively (Fig. 2a). Gut microbiota imbalance was evident in obese mice from decreased α-diversity but increased Firmicute to Bacteroidetes ratios (Fig. 2b). Presence of Lactobacillaceae, Lachnospiraceae, Bacteroidaceae, and Muribaculaceae were indicative of microbial dysbiosis in murine colitis [27]. We identified apparent alterations of these 4 designated bacterial families in obese mice (Fig. 2c). Obesity resulted in a loss of beneficial gut microbiota, revealing a depletion of beneficial microorganism genera (Fig. 2d), including Muriculum, Lactobacillus, Akkermansia, and Ruminococcaceae [28, 29]. Of bacterial species, Bacteroides plebeius and Lactobacillus murinus abundances were reduced, while Lachnospiraceae bacterium abundance was increased by HFD (Fig. 2e). Furthermore, decreased trabecular bone microstructure and loss of bone mass correlated with 20 genera of microorganisms, including Akkermansia, Bacteroides, Ruminococcaceae, Parabacteroides, Lactobacillus, and Blautia in obese mice (Fig. 2f).
Fig. 2.

Gut microbial dysbiosis in HFD-induced obese mice. Principal component analysis showing noticeably different gut microflora profiles (a). Decreased α-diversity and high Firmicute/Bacteroidetes ratios of the gut microflora of obese mice (b). Changes in relative abundances of gut bacterial families (c). Heatmap showing changes in the richness of gut bacterial genera and less beneficial gut microbiome variety in obese mice (d). HFD consumption affected the relative abundances of 3 gut bacterial species (e). Alterations in the gut microbiome correlated with bone mass and trabecular network loss (f). Data are calculated from 5 mice. Asterisks (*) stand for significant difference (P < 0.05) analyzed by Kolmogorov–Smirnov test and Student t-test
Carnitine and TMAO dysmetabolism in obese mice
Gut microorganisms produce metabolites, which may co-regulate bone homeostasis and contribute to osteoporosis development [30]. We characterized the serum metabolome of obese and control mice. Principal component analyses confirmed distinctively different profiles between these two groups (Fig. 3a). Eighty-five metabolites were upregulated; and 187 metabolites were downregulated in obese mice (Fig. 3b). KEGG pathway analyses revealed that these metabolites likely contributed to a plethora of metabolic activities, including glycolysis, lipolysis, tricarboxylic acid cycle, fatty acid metabolism, and carnitine metabolism (Fig. 3c). Fourteen metabolites strongly correlated with the relative abundances of gut microorganisms (Fig. 3d). Of metabolites, L-carnitine is metabolized into trimethylamine (TMA) by these gut microorganisms and flavin containing monooxygenase 3 (FMO3) which oxidizes TMA into trimethylamine n-oxide (TMAO) [12] (Fig. 3e). Moreover, an excess TMAO enhances key osteoclast marker expression and osteoclast formation of murine macrophages [13]. This metabolite was thus selected for subsequent experiments. Of note, obese mice had much higher serum TMAO and FMO3 levels together with lower L-carnitine levels than CD-fed animals. However, serum TMA levels were unaffected upon HFD consumption (Fig. 3f).
Fig. 3.

Altered serum metabolomic landscape in obese mice. Principal component analysis showing distinctive metabolomic profiles (a). Volcanic plot showing 85 and 187 metabolites elevated or reduced, respectively, in obese mice (b). KEGG pathway of metabolomic landscapes, which may contribute to plenty of cellular metabolism (c). Heatmap showing the correlation of serum metabolites and gut microorganisms (d). A drawing scheme depicting L-carnitine conversion into TMA and TMAO by gut microorganisms and FMO3 (e). Increased serum TMAO and FMO3 together with reduced L-carnitine levels in obese mice. Data are calculated from 5 mice. Asterisks (*) stand for significant difference (P < 0.05) analyzed by Kolmogorov–Smirnov test and Student t-test
Transplantation of gut microbiota from CD mice mitigated obesity-mediated TMAO overproduction and bone loss
We then investigated whether fecal microbiota transplantation (FMT) changed TMAO production or bone mass loss in obese mice. Supernatants were harvested from the mixture of sterile Ringer's solution and fresh feces of age-matched CD mice as donors; and were given to HFD mice via oral gavage (3 FMT/week for 5 consecutive months) (Fig. 4a). FMT recipients revealed a reduced body adiposis (Fig. 4b), mucin loss, and IL-17 overproduction in colon goblet cells (Fig. 4c) as compared to vehicle-treated animals. FMT counteracted TMAO overproduction and femur weight loss, while body overweight was unaffected in obese mice (Fig. 4d). The trabecular bone microstructure of FMT recipients further revealed a wider meshed trabecular network than vehicle-treated mice (Fig. 4e). FMT attenuated obesity-mediated loss of Tb.BMD, BV/TV, Tb.Th, and Tb.N (Fig. 4f) and biomechanical properties breaking forces and energy (Fig. 4g); however, cortical BMD or porosity was unaffected. Furthermore, FMT improved bone formation rate (Fig. 4h) and osteoblast number (Ob.N/mm) and repressed obesity-induced marrow adiposis as apparent by a reduced number of adipose cells (Ad.N/mm) (Fig. 4i).
Fig. 4.

Effects of fecal microbiota transplantation (FMT) on body fat formation, gut integrity, and bone mass in obese mice. Schematic depiction of the transplantation procedure of fecal microbiota from CD mice to HFD mice (a). FMT improves body fat volume (b); scale bar, 10 mm, mucin production and IL-17 levels in colon (c); scale bar, 10 μm, and femur weight and serum TMAO levels (d) in obese mice. FMT counteracted obesity-mediated bone loss at a trabecular microstructural level (e); scale bar, 50 μm, and Tb.BMD, Tb.Th, BV/TV, Tb.N (f), breaking force and energy (g). FMT repressed BFR/BS loss (h); scale bar, 20 μm, and osteoblast loss and marrow adiposity (i); scale bar, 20 μm. Data are means ± standard errors calculated from 5 ~ 6 mice. Asterisks (*) stand for significant difference (P < 0.05) from CD-fed group; and hashtags (#) resemble significance from HFD-Veh group analyzed by ANOVA test and post hoc Bonferroni test
FMT attenuated estrogen deficiency-induced osteoporosis
In ovariectomized (OVX) mice as an in vivo model of post-menopausal osteoporosis, we also identified gut microorganism alterations (Fig. 5a) and a decreased abundance of the beneficial bacterial families Bacteroidaceae and Akkermansiaceae (Fig. 5b) together with increased serum TMAO levels. These data prompted us to investigate whether FMT affected estrogen deficiency-mediated bone loss. One week postoperatively, gut microbiota from age-matched sham mice were transplanted into OVX mice for 12 weeks (Fig. 5c). FMT attenuated femur weight loss and serum TMAO overproduction, while body weight was unaffected (Fig. 5d). Bone trabeculae from FMT recipients showed a more interwoven network than vehicle-treated OVX mice (Fig. 5e). Improvement of trabecular bone microarchitecture by FMT further included Tb.BMD, BV/TV, Tb.N, and Tb.Th (Fig. 5f). FMT also reversed cortical bone structure Ct.BMD, Ct.Th, Ct.Vol, and cortical porosity (Fig. 5g). Likewise, FMT resulted in reduced loss of BFR and Ob.N together with fewer marrow adipocyte formation and osteoclast overburden (Fig. 5h) as compared to vehicle-treated animals.
Fig. 5.

FMT-induced bone changes in ovariectomized (OVX) mice. Distinct gut microbial profiles of sham mice and OVX mice (a). Changes in gut bacterial families in sham and OVX mice (b). Schematic illustration of the transplantation procedure of fecal microbiota from aged-matched sham mice to OVX mice (c). FMT suppressed OVX-induced femur weight loss and TMAO overproduction in serum (d). FMT suppressed the loss of trabecular bone (e), Tb.BMD, BV/TV, Tb.N, Tb.Th (f), Ct.BMD, Ct.Th, Ct.Vol and cortical porosity (g) in OVX mice. FMT preserved BFR and Ob.N and repressed marrow adipocyte formation and osteoclast overabundance in OVX mice (h); scale bar, 20 μm. Data are means ± standard errors calculated from 5 to 6 mice. Asterisks (*) stand for significant difference (P < 0.05) from sham group; and hashtags (#) resemble significance from OVX-Veh group analyzed by ANOVA test and post hoc Bonferroni test
TMAO accelerated bone mass loss
To better characterize the role of TMAO in bone turnover, animals were fed chow diet and drinking water with 0.2% TMAO at libitum for 3 consecutive months (Fig. 6a). Water, feed consumption or body weight gain was unaffected throughout the study. Serum TMAO and key bone resorption markers CTX-1 and TRAP5b levels were much higher in TMAO-fed mice as compared to control animals (Fig. 6b). Bone microstructure in TMAO-fed animals was relatively porous (Fig. 6c), revealing low Tb.BMD, Tb.N, BV/TV, and Tb.Th together with high Tb.Sp, and SMI (Fig. 6c); Ct.BMD was also reduced, but Ct.Th was unaffected in these animals (Fig. 6d). Excess TMAO consumption reduced BFR/BS and Ob.N (Fig. 6e) and enhanced Oc.N (Fig. 6f). TMAO intake resulted in a consistent underproduction of mineralized matrix and a decreased expression of key osteogenic markers, like Runx2, collagen 1a1, osteocalcin, and bone alkaline phosphatase together with an increased expression of osteoclastogenic cytokine RANKL expression by bone-marrow mesenchymal cells (Fig. 6g). As a result, osteoclast formation from bone marrow macrophage precursor cells was higher in TMAO-fed mice than in vehicle-fed animals (Fig. 6h). Collectively, our investigations suggested that excess TMAO dysregulated bone formation and resorption to accelerate bone loss.
Fig. 6.

Effects of TMAO intake on bone phenotypes. Schematic illustration of the TMAO feeding regime (a). Increased serum TMAO, bone resorption markers CTX-1, and TRAP5b levels in TMAO-fed mice (b). μCT images showing a sparse trabecular bone microstructure; scale bar, 50 μm. TMAO suppressed Tb.BMD, BV/TV, Tb.N, and Tb.Th and enhanced Tb.Sp. and SMI (c). Ct.BMD was reduced in TMAO-fed mice (d); scale bar, 50 μm. TMAO intake resulted in relatively low BFR/BS and Ob.N (e); scale bar, 20 μm, and high Oc.N in bone tissue (f); scale bar, 20 μm. TMAO inhibited the synthesis of mineralized matrix production as well as osteogenic marker expression, but enhanced RANKL expression of bone-marrow mesenchymal cells (g); scale bar, 5 mm. High osteoclast formation capacity of bone-marrow macrophages in TMAO-fed mice (h); scale bar, 20 μm. Data are means ± standard errors calculated from 5 mice. Asterisks (*) stand for significant difference (P < 0.05) from vehicle group analyzed by Kolmogorov–Smirnov test and Student t-test
TMAO enhanced endoplasmic reticulum stress and senescence in osteoblasts
We further investigated how TMAO can inhibit osteogenic activity during the development of osteoporosis. Interestingly, this metabolite dose-dependently reduced the proliferative and anabolic activity of mouse calvaria osteoblasts, as evident in bromodeoxyuridine uptake (Fig. 7a) and osteogenic marker expression (Fig. 7b). The greatest inhibition was achieved by 200 µM TMAO, which significantly suppressed the production of mineralized matrix and was thus elected for subsequent experiments (Fig. 7c).
Fig. 7.

TMAO regulated endoplasmic reticulum (ER) integrity and senescence in osteoblasts. TMAO dose-dependently suppressed BrdU uptake (a), expression of osteogenic makers Col1a1, Ocn, and Runx2 (b), and von Kossa-stained mineralized nodule production (c); scale bar, 5 mm. TMAO enhanced mRNA expression of PERK and ATF4 as well as protein levels of PERK and phosphorylated PERK, and ATF4 (d). Fluorescent ER tracker staining (scale bar, 10 μm) and disrupted ER ultrastructure (scale bar, 1 μm) in TMAO-treated osteoblasts (e). GSK2606144 counteracted TMAO-induced ER stress and integrity loss. Decreased mRNA expression of Atg4, Atg12, and p62 and protein levels of LC3-II, mTOR, and FAM134B (f) and autophagosome formation (scale bar, 10 μm) and ER-phagic ultrastructure (scale bar, 0.5 μm) (g); scale bar, 10 μm, upon TMAO treatment. Senescence-associated β-galactosidase staining and increased p16, and p21 expression in TMAO-treated cells (h); scale bar, 40 μm. GSK2606414 attenuated TMAO-mediated autophagy loss and senescence. Data are mean ± standard errors calculated from at least three independent experiments, including immunoblotting. Asterisks (*) stand for significant difference (P < 0.05) analyzed by ANOVA test and post hoc Bonferroni test. GSK, GSK2606414; SA-β-gal, senescence association β-galactosidase
Increasing evidence suggests that TMAO binds to endoplasmic reticulum (ER) regulator PERK or enhances cellular senescence [14, 20]. Our data uncovered that TMAO enhanced mRNA expression as well as protein levels of PERK and phosphorylated PERK, together with key ER stress regulator activating transcription factor 4 signaling (Atf4; Fig. 7d and Supplementary Fig. S1 and S2). Furthermore, TMAO-treated osteoblasts were fluorescently stained using ER-Tracker to reveal a disrupted ER ultrastructure (Fig. 7e and Supplementary Fig. S3a). Inactivation of PERK by GSK2606414 mitigated TMAO-mediated PERK as well as Atf4 activation, ER stress, and ER network loss.
Moroever, TMAO inhibited mRNA expression of the autophagic markers Atg4, Atg12, and p62 and protein levels of LC3-II, mTOR and ER-phagy marker FAM134B on protein levels [31] (Fig. 7f and Supplementary Fig. S1 and S2). This metabolite, in turn, repressed autophagic puncta formation and onion ring-like ER-phagic body ultrastructure (Fig. 7g and Supplementary Fig. S3a). As a result, osteoblast senescence was enhanced upon TMAO treatment revealing increased expression of senescence markers p16, p21, and β-galactosidase staining intensity (Fig. 7h). In contrast, GSK2606414 improved TMAO-mediated ER autophagy loss and senescence.
TMAO reduced mitochondrial unfolded protein response, respiration and mitophagy
PERK signaling regulates mitochondrial protein homeostasis and bioenergetics to adapt to extracellular stress [32]. TMAO inhibited plenty of UPRmt regulators, including Atf5, ClpP, Lonp1, Hsp60, and Hsp10 expression (Fig. 8a). It also reduced Atf5 protein levels but enhanced levels of ubiquitinated Atf5 levels (Fig. 8b and Supplementary Fig. S1 and S2). Misfolded proteins usually turn into insoluble aggregates dysregulating protein homeostasis to inhibit intracellular organelle function [33]. Given that TMAO disrupted UPRmt, we thus investigated whether TMAO affected Atf5 folding. Upon ultracentrifugation of cell lysates, aggregated proteins in pellets were characterized using electrophoresis. TMAO enhanced the amounts of protein aggregates corresponding to 150 ~ 15 kDa. Relatively high Atf5 levels corresponding to 31 kDa were also detected in the pellets, suggesting that misfolded Aft5 was increased in these osteoblasts (Fig. 8c and Supplementary Fig. S1 and S2). Of interest, inhibition of PERK by GSK2606414 or gain of UPRmt function by nicotinamide riboside [34] mitigated TMAO-mediated Atf5 ubiquitination and aggregation.
Fig. 8.

TMAO suppressed UPRmt, mitophagy, mitochondrial energetics, and mineralized matrix anabolism. GSK2606144 or nicotinamide riboside counteracted TMAO-induced losses of mRNA expression of UPRmt marker, ATF5, Lonp1, ClpP, Hsp60, and Hsp10 (a) as well as suppression of protein levels of ATF5 and ubiquitinated ATF5 (b) and aggregated Aft5 (c). GSK2606144 or nicotinamide riboside preserved Pink1, Parkin, and LC3-II levels (d) and suppressed TMAO-induced losses of MitoTracker Green-staining of mitochondrial mass and Mitphay Dye/Lyso Dye-staining of mitophagic puncta formation (e); scale bar, 10 μm. GSK2606144 or nicotinamide riboside mitigated TMAO-induced suppression of mitochondrial basal, maximum, ATP-linked oxygen consumption (f), OXPHOS (g), ATP production, proliferation capacity (h), and mineralized matrix synthesis as well as osteogenic marker expression. Data are means ± standard errors calculated from a minimum 3 independent experiments, including immunoblotting. Asterisks (*) stand for significant difference (P < 0.01) analyzed by ANOVA test and post hoc Bonferroni test. GSK, GSK2606414; NR, nicotinamide riboside
Furthermore, TMAO reduced the key mitophagy markers Pink1, Parkin, and LC3-II (Fig. 8d and Supplementary Fig. S1 and S2) and repressed mitochondrial mass and mitophagosome formation, as evident by fluorescent Mtphagy Dye/Lyso Dye labelling (Fig. 8e and Supplementary Fig. S3b). As a result, TMAO inhibited mitochondrial respiration capacity, including basal, maximum, and ATP-linked oxygen consumption rate (Fig. 8f). Mitochondrial OXPHOS (Fig. 8g. and Supplementary Fig. S1 and S2) and ATP production (Fig. 8h) were also repressed upon TMAO incubation. GSK2606414 or nicotinamide riboside also reversed TMAO-mediated inhibition of mitochondrial bioenergetics, proliferation capacity (Fig. 8h), and mineralized extracellular matrix anabolism (Fig. 8i).
Discussion
A plethora of hormonal and biochemical pathways directly or indirectly regulate skeletal tissue and bone turnover, majorly affecting bone homeostasis [35]. Dysbiosis of gut microorganisms correlates with the development of osteoporosis [36], a low bone mass phenotype. An increasing body of evidence has revealed that gut microorganisms produce a great deal of extracellular vesicles or metabolites, which can affect, among others, the osteogenic differentiation of bone-marrow mesenchymal stem cells and bone formation capacity [37]. Our investigations, for the first time, uncovered the novel catabolic actions of gut microflora-derived metabolite TMAO on bone homeostasis during obesity- and estrogen deficiency-induced osteoporosis. We further shed light onto the molecular mechanisms underlying the osteoblast-inhibiting actions of TMAO, which disrupted ER integrity and UPRmt to accelerate senescence and hindered mineralized extracellular matrix underproduction.
Barrier loss and inflammation in the gut microenvironment, like mucin underproduction, tight junction protein loss, and increased IL-17 levels were associated with bone loss in obese animals. Our data are in agreement with other studies showing that gut microecological alteration may interfere with host immune response [38] or brain-gut-bone crosstalk [39] driving the skeleton towards osteoporotic changes. We conducted 16S rRNA sequencing to characterize the gut microbiome landscapes; and correlated gut beneficial microorganisms Akkermansia and Lactobacillus to a plethora of prominent features of the osteoporotic skeleton, including a low BMD, a fragile trabecular network, and an imbalanced bone formation rate. Loss of abundance of Akkermansia in the gut accelerates bone loss in ovariectomized animals [40]; and probiotic lactobacillus supplementation promotes the antioxidative capacity, repressing osteoclast formation and bone loss in ovariectomized mice [41]. Furthermore, our data revealed relatively low abundances of Bacteroides plebeius, Lactobacillus murinus, and Lachnospiraceae bacterium in obesity-induced osteoporotic mice. Dysbiosis of these 3 bacterial species are correlated with rheumatoid arthritis-mediated bone destruction [42], spaceflight-induced bone loss [43] and estrogen deficiency-indced osteoporosis [44]. A reduced loss of bone microarchitecture in obese or ovariectomized mice upon FMT treatment strongly suggests a contribution of gut microbiome to bone integrity. Our data demonstrate the impact of a dysregulation of the gut ecosystem can have on skeletal metabolism in mice with obesity or estrogen deficiency.
Serum metabolomic investigations further identified the dysregulation of the L-carnitine metabolism as another contributor to bone loss, resulting from a gut microorganism dysbiosis. L-carnitine is a cofactor for fatty acid metabolism in the mitochondrial compartment [45]. Decreased carnitine levels are present in patients with low bone mass upon weight control and physiological levels of L-carnitine are advantageous to bone formation [46]. L-carnitine supplementation also attenuates glucocorticoid-induced bone loss and trabecular microstructure destruction [47]. Furthermore, L-carnitine can be modified into TMAO by gut microorganisms and key enzyme FMO3 [10]; and gut microecological dysregulation has been shown to enhance TMAO production in HFD mice [12], which is in line with our results. Loss of serum L-carnitine appears to be a notable feature in explaining the inhibitory effects of obesity on skeletal tissue integrity.
Metabolomic landscapes also showed alterations in other metabolic pathway, like mineral resorption, lipolysis, and fatty acid biosynthesis during bone loss. Lipolysis is important to fuel osteogenic differentiation activity of bone-marrow mesenchymal stem cells at the expense of bone marrow fat [48]. Mitochondrial fatty acid metabolism has been shown to be required to maintain osteoblastic activity and healthy bone mass [49]. The dysregulation of lipid and fatty acid metabolism hinted towards an overall contribution of gut microbiota to the development of fatty bone marrow in the osteoporotic skeleton.
Our investigations uncovered that TMAO is a functional gut microbiota-derived metabolite for obesity and estrogen deficiency-induced osteoporotic changes in the skeleton and thus profoundly interferes with bone mass homeostasis. Specifically, excess TMAO intake accelerated bone loss by disrupting the balance between bone formation and osteoclastic bone resorption. Little is known about how this gut microbiota-derived metabolite affected bone turnover, while only one study has demonstrated that TMAO drives bone-marrow mesenchymal stromal cells toward adipocytes at the cost of bone-forming cells [50]. TMAO usually binds to receptor PERK, which then regulates endoplasmic reticulum homeostasis [20] and mitochondrial activity [21]. We now revealed that TMAO activated PERK-dependent ER stress through disrupting autophagic program and to ultimately augment osteoblast senescence. Not surprisingly, reducing ER stress through PERK inhibition was advantageous to osteoblast survival under TMAO stress. Collectively, these findings conveyed new insights into the suppressive function of TMAO on bone formation.
TMAO appeared to exert context-dependent actions regarding the biosynthesis of a mineralized matrix. In cardiovascular tissues, TMAO is known to enhance osteogenic transcription factor Runx2, consequently leading to calcified extracellular matrix synthesis of vascular smooth muscle cells through activating the NLRP3-dependent inflammasome pathway [51]. TMAO also aggravates mitochondrial stress, promoting an osteogenic response of aortic valve interstitial cells [52]. Not surprisingly, TMAO now inhibited mineralized matrix production in osteogenic cells and in two experimental models of osteoporosis. To the best of our knowledge, our data are the first indication revealing that TMAO disrupts UPRmt by promoting crucial regulator Atf5 misfolding. Additionally, TMAO interrupted a plethora of mitochondrial homeostatic key reactions for energy production, mitophagy, mitochondrial respiration and OXPHOS. All of which are indispensable for mineralized matrix production by osteoblasts [53]. Of note, reversing UPRmt by nicotinamide ribose preserved mitochondrial energetics, steering osteoblasts to produce mineralized extracellular matrices in TMAO stress. Our investigations make clear how this gut microbiota-derived metabolite suppresses mineralized matrix anabolism during osteoporosis.
We cannot exclude the possibility that other gut microbiota-derived metabolites may affect osteoblastic activity and bone mass homeostasis as well. TMAO may influence other mitochondrial metabolic pathways, like glycolysis, TCA cycle or fatty acid biosynthesis to affect osteoblast dysfunction. Our metabolomic studies point towards a complex nature of the gut-microbiota-bone axis. In conclusion, our data show that gut microbiota dysbiosis and dysmetabolism accelerate bone loss. TMAO, among others, is an active gut microbiota-derived metabolite inhibiting osteoblastic activity by enhancing ER stress and Atf5 unfolding in UPRmt. Our data highlight a bone-protective effects of gut microbiota transplantation with potential to slow down osteoporosis (Fig. 9).
Fig. 9.
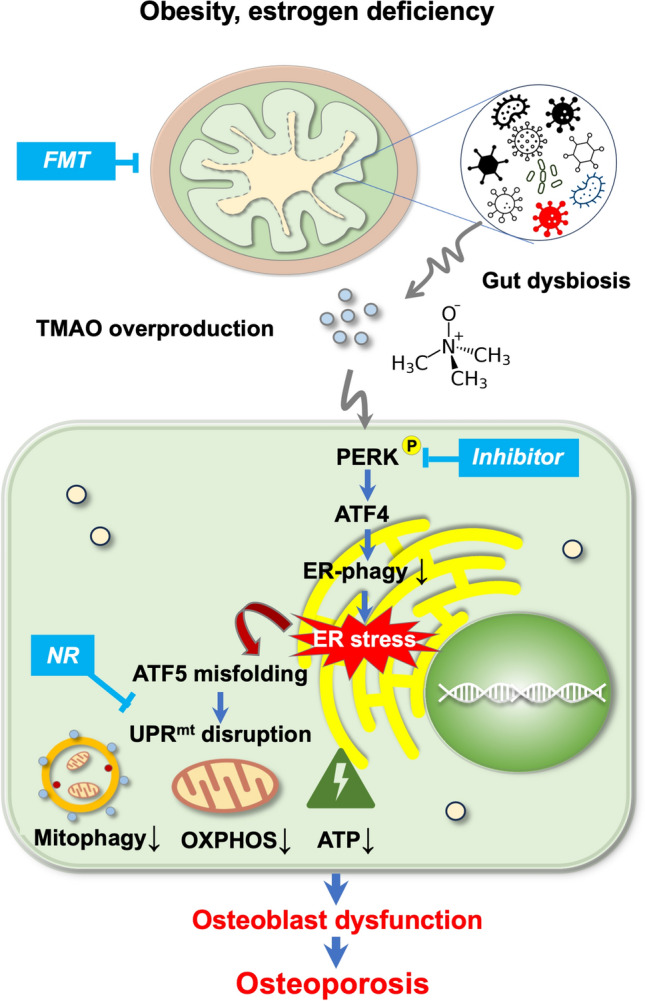
Hypothetical model of how gut microbiota-derived TMAO changes bone integrity. Gut microorganism dysbiosis elevates TMAO production in obese or ovariectomized mice. This metabolite then activates PERK and ATF4 to suppress ER autophagy and thus elevating ER stress, which leads to mitochondrial ATF5 misfolding and UPRmt disruption, together with decreased mitophagy, OXPHOS, and ATP synthesis. This consequently accelerates osteoblast dysfunction, leading to bone loss. FMT (from healthy donors), PERK inhibitor or UPRmt activator mitigates TMAO-induced loss of osteoblastic activity loss and bone mass. FMT, fecal microbiota transplantation; TMAO, trimethylamine-n-oxide; PERK, protein kinase R-link endoplasmic reticulum kinase; ATF4, activating transcription factor 4; ER, endoplasmic reticulum; ATF5, activating transcription factor 5; NR, nicotinamide ribose; UPRmt, mitochondrial unfolding protein response; OXPHOS, oxidative phosphorylation
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work is supported by [NHRI-EX113-11029SI] from National Health Research Institute; [NSTC110-2314-B-182A-MY3] form National Science and Technology Council; [CORPG8M0571] from Chang Gung Memorial Hospital, Taiwan; and BMBF [13XP5206], Germany. Authors also thank Core Laboratory for Gnotobiotic Medicine and Center for Laboratory Animals, Kaohsiung Chang Gung Memorial Hospital, Taiwan for facility support.
Abbreviations
- TMAO
Trimethylamine-n-oxide
- FMO3
Flavin-containing monooxygenase 3
- FMT
Fecal microbiota transplantation
- ER
Endoplasmic reticulum
- PERK
Protein kinase R-like endoplasmic reticulum kinase
- ATF4
Activating transcription factor 4
- UPRmt
Mitochondrial unfolded protein response
- ATF5
Activating transcription factor 5
- OXPHOS
Oxidative phosphorylation
- mTOR
Mammalian target of rapamycin
- RPTOR
Regulatory associated protein of mTOR
- FAM134B
Reticulophagy regulator 1
- Mfn2
Mitofusin 2
- LC3-II
Light chain 3-II
- Opa1
Mitochondrial dynamin like GTPase
- CLDN-1
Claudin 1
- TJP-1
Tight junction protein 1
- Runx2
RUNX family transcription factor 2
- ClpP
Caseinolytic mitochondrial matrix peptidase proteolytic subunit
- Lonp1
Lon peptidase 1
- Hsp60
Heat shock protein 60
- Hsp10
Heat shock protein 10
Author contributions
Study conception and design: Y.-H.L., W.-S.L., R.-W.W., Y.-S.C., S.-L.W., J.-Y.K., S.-Y.W., H. J., F.-S.W.; cell culture analysis: Y.-S.C.; 16S rRNA, metabolome & bioinformation analysis: W.-S.L., Y.-S.C.; in vivo & μCT analysis: Y.-S.C.; histology & immunoblotting: S.-Y.W., S.-L.W.; osteoporosis diagnosis: R.-W.W., J.-Y.K.; Analysis and interpretation of data: Y.-H.L., W.-S.L., W.-R.W., H. J., F.-S.W.; Article drafting and revision: Y.-H.L., W.-S.L., W.-R.W., H. J., F.-S.W.
Funding
This work was supported by [NHRI-EX113-11029SI] from the National Health Research Institute; [NSTC110-2314-B-182A-MY3] form National Science and Technology Council; and [CORPG8M0571] from Chang Gung Memorial Hospital, Taiwan, as well as the BMBF [13XP5206], Germany.
Data availability
Data related to laboratory experimentation, in vitro osteoblast models, 16S rRNA sequencing, and serum metabolome are available on request from the corresponding author.
Declarations
Conflict of interest
Authors have no financial competing interest.
Ethical approval and consent to participate
Animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Kaohsiung Chang Gung Memorial Hospital (IACUC Affidavit #2019091103). This study did not involve human specimens or clinical data.
Consent for publication
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lobo RA, Gompel A (2022) Management of menopause: a view towards prevention. Lancet Diabetes Endocrinol 10:457–470. 10.1016/S2213-8587(21)00269-2 [DOI] [PubMed] [Google Scholar]
- 2.Seimon RV, Wild-Taylor AL, Keating SE et al (2019) Effect of weight loss via severe vs moderate energy restriction on lean mass and body composition among postmenopausal women with obesity: the TEMPO diet randomized clinical trial. JAMA Netw Open 2:e1913733. 10.1001/jamanetworkopen.2019.13733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ali D, Figeac F, Caci A et al (2022) High-fat diet-induced obesity augments the deleterious effects of estrogen deficiency on bone: evidence from ovariectomized mice. Aging Cell 21:e13726. 10.1111/acel.13726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim S, Henneicke H, Cavanagh LL et al (2021) Osteoblastic glucocorticoid signaling exacerbates high-fat-diet- induced bone loss and obesity. Bone Res 9:40. 10.1038/s41413-021-00159-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behera J, Ison J, Voor MJ, Tyagi N (2022) Exercise-linked skeletal irisin ameliorates diabetes-associated osteoporosis by inhibiting the oxidative damage-dependent miR-150-FNDC5/pyroptosis axis. Diabetes 71:2777–2792. 10.2337/db21-0573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai B, Xu J, Li X et al (2022) Macrophages in epididymal adipose tissue secrete osteopontin to regulate bone homeostasis. Nat Commun 13:427. 10.1038/s41467-021-27683-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu M, Pal S, Paterson CW et al (2021) Ovariectomy induces bone loss via microbial-dependent trafficking of intestinal TNF+ T cells and Th17 cells. J Clin Invest 131:e143137. 10.1172/JCI143137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang KL, Mullins BJ, Lejeune A, (2024) Mitigation of osteoclast-mediated arthritic bone remodeling by short chain fatty acids. Arthritis Rheumatol 76:647–659. 10.1002/art.42765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Yang C, Deng Z (2024) Gut microbially produced tryptophan metabolite melatonin ameliorates osteoporosis via modulating SCFA and TMAO metabolism. J Pineal Res 76:e12954. 10.1111/jpi.12954 [DOI] [PubMed] [Google Scholar]
- 10.Schugar RC, Shih DM, Warrier M et al (2017) The TMAO-producing enzyme flavin-containing monooxygenase 3 regulates obesity and the beiging of white adipose tissue. Cell Rep 20:279. 10.1016/j.celrep.2017.06.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elam RE, Bůžková P, Barzilay JI et al (2022) Trimethylamine N-oxide and hip fracture and bone mineral density in older adults: the cardiovascular health study. Bone 161:116431. 10.1016/j.bone.2022.116431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoo W, Zieba JK, Foegeding NJ et al (2021) High-fat diet-induced colonocyte dysfunction escalates microbiota-derived trimethylamine N-oxide. Science 373:813–818. 10.1126/science.aba3683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y, Wang C, Qiu F et al (2024) Trimethylamine-N-oxide promotes osteoclast differentiation and oxidative stress by activating NF-κB pathway. Aging 16:9251–9263. 10.18632/aging.205869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saaoud F, Liu L, Xu K et al (2023) Aorta- and liver-generated TMAO enhances trained immunity for increased inflammation via ER stress/mitochondrial ROS/glycolysis pathways. JCI Insight 8:e158183. 10.1172/jci.insight.158183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo J, Ren R, Sun K et al (2020) PERK controls bone homeostasis through the regulation of osteoclast differentiation and function. Cell Death Dis 11:847. 10.1038/s41419-020-03046-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding ZB, Chen Y, Zheng YR et al (2024) Inhibition of PPP1R15A alleviates osteoporosis via suppressing RANKL-induced osteoclastogenesis. Acta Pharmacol Sin 45:790–802. 10.1038/s41401-023-01209-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C, Li N, Zhang Z et al (2023) The specific mitochondrial unfolded protein response in fast- and slow-twitch muscles of high-fat diet-induced insulin-resistant rats. Front Endocrinol 14:1127524. 10.3389/fendo.2023.1127524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Zhang L, Zhang S et al (2023) ATF5 regulates tubulointerstitial injury in diabetic kidney disease via mitochondrial unfolded protein response. Mol Med 29:57. 10.1186/s10020-023-00651-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Zhang D, Zhang Y et al (2024) Mitochondria-specific antioxidant MitoTEMPO alleviates senescence of bone marrow mesenchymal stem cells in ovariectomized rats. J Cell Physiol. 10.1002/jcp.31323 [DOI] [PubMed] [Google Scholar]
- 20.Chen S, Henderson A, Petriello MC et al (2019) Trimethylamine n-oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab 30:1141-1151.e5. 10.1016/j.cmet.2019.08.021 [DOI] [PubMed] [Google Scholar]
- 21.Dong F, Jiang S, Tang C et al (2022) Trimethylamine N-oxide promotes hyperoxaluria-induced calcium oxalate deposition and kidney injury by activating autophagy. Free Radic Biol Med 179:288–300. 10.1016/j.freeradbiomed.2021.11.010 [DOI] [PubMed] [Google Scholar]
- 22.Lian WS, Ko JY, Chen YS et al (2018) Chaperonin 60 sustains osteoblast autophagy and counteracts glucocorticoid aggravation of osteoporosis by chaperoning RPTOR. Cell Death Dis 9:938. 10.1038/s41419-018-0970-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen C, Chen H, Zhang Y et al (2020) TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant 13:1194–1202. 10.1016/j.molp.2020.06.009 [DOI] [PubMed] [Google Scholar]
- 24.McLean C, Kujawinski EB (2020) AutoTuner: high fidelity and robust parameter selection formetabolomics data processing. Anal Chem 92:5724–5732. 10.1021/acs.analchem.9b04804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dempster DW, Compston JE, Drezner MK et al (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 28:2–17. 10.1002/jbmr.1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo Y, Tong Y, Wu L et al (2023) Alteration of gut microbiota in high-risk individuals for rheumatoid arthritis is associated with disturbed metabolome and initiates arthritis by triggering mucosal immunity imbalance. Arthritis Rheumatol 75:1736–1748. 10.1002/art.42616 [DOI] [PubMed] [Google Scholar]
- 27.Kim MJ, Jung DR, Lee JM et al (2023) Microbial dysbiosis index for assessing colitis status in mouse models: a systematic review and meta-analysis. iScience 27:108657. 10.1016/j.isci.2023.108657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCabe LR, Irwin R, Tekalur A et al (2019) Exercise prevents high fat diet-induced bone loss, marrow adiposity and dysbiosis in male mice. Bone 118:20–31. 10.1016/j.bone.2018.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyamoto J, Igarashi M, Watanabe K et al (2019) Gut microbiota confers host resistance to obesity by metabolizing dietary polyunsaturated fatty acids. Nat Commun 10:4007. 10.1038/s41467-019-11978-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tyagi AM, Yu M, Darby TM et al (2018) The microbial metabolite butyrate stimulates bone formation via T regulatory cell-mediated regulation of WNT10B expression. Immunity 49:1116-1131.e7. 10.1016/j.immuni.2018.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.González A, Covarrubias-Pinto A, Bhaskara RM et al (2023) Ubiquitination regulates ER-phagy and remodelling of endoplasmic reticulum. Nature 618:394–401. 10.1038/s41586-023-06089-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perea V, Baron KR, Dolina V et al (2023) Pharmacologic activation of a compensatory integrated stress response kinase promotes mitochondrial remodeling in PERK-deficient cells. Cell Chem Biol 30:1571-1584.e5. 10.1016/j.chembiol.2023.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu F, Liu Z, Cheng W et al (2023) The PERK branch of the unfolded protein response safeguards protein homeostasis and mesendoderm specification of human pluripotent stem cells. Adv Sci 10:e2303799. 10.1002/advs.202303799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vannini N, Campos V, Girotra M et al (2019) The NAD-booster nicotinamide riboside potently stimulates hematopoiesis through increased mitochondrial clearance. Cell Stem Cell 24:405-418.e7. 10.1016/j.stem.2019.02.012 [DOI] [PubMed] [Google Scholar]
- 35.Foessl I, Dimai HP, Obermayer-Pietsch B (2023) Long-term and sequential treatment for osteoporosis. Nat Rev Endocrinol 19:520–533. 10.1038/s41574-023-00866-9 [DOI] [PubMed] [Google Scholar]
- 36.Ling CW, Miao Z, Xiao ML et al (2021) The association of gut microbiota with osteoporosis is mediated by amino acid metabolism: multiomics in a large cohort. J Clin Endocrinol Metab 106:e3852–e3864. 10.1210/clinem/dgab492 [DOI] [PubMed] [Google Scholar]
- 37.Lin X, Xiao HM, Liu HM et al (2023) Gut microbiota impacts bone via Bacteroides vulgatus-valeric acid-related pathways. Nat Commun 14:6853. 10.1038/s41467-023-42005-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding P, Tan Q, Wei Z et al (2022) Toll-like receptor 9 deficiency induces osteoclastic bone loss via gut microbiota-associated systemic chronic inflammation. Bone Res 10:42. 10.1038/s41413-022-00210-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, Lv M, Liang J et al (2023) Neuropeptide Y-mediated gut microbiota alterations aggravate postmenopausal osteoporosis. Adv Sci 10:e2303015. 10.1002/advs.202303015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu JH, Chen CY, Liu ZZ et al (2021) Extracellular vesicles from child gut microbiota enter into bone to preserve bone mass and strength. Adv Sc 8:2004831. 10.1002/advs.202004831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan Y, Yang J, Zhuge A et al (2022) Gut microbiota modulates osteoclast glutathione synthesis and mitochondrial biogenesis in mice subjected to ovariectomy. Cell Prolif 55:e13194. 10.1111/cpr.13194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng M, Zhao Y, Cui Y et al (2022) Stage-specific roles of microbial dysbiosis and metabolic disorders in rheumatoid arthritis. Ann Rheum Dis 81:1669–1677. 10.1136/ard-2022-222871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bedree JK, Kerns K, Chen T et al (2023) Specific host metabolite and gut microbiome alterations are associated with bone loss during spaceflight. Cell Rep 42:112299. 10.1016/j.celrep.2023.112299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang N, Meng F, Ma S, Fu L (2022) Species-level gut microbiota analysis in ovariectomized osteoporotic rats by Shallow shotgun sequencing. Gene 817:146205. 10.1016/j.gene.2022.146205 [DOI] [PubMed] [Google Scholar]
- 45.Song C, Valeri A, Song F et al (2023) Sexual dimorphism of osteoclast reliance on mitochondrial oxidation of energy substrates in the mouse. JCI Insight 8:e174293. 10.1172/jci.insight.174293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou T, Heianza Y, Chen Y et al (2019) Circulating gut microbiota metabolite trimethylamine N-Oxide (TMAO) and changes in bone density in response to weight loss dets: The POUNDS Lost Trial. Diabetes Care 42:1365–1371. 10.2337/dc19-0134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahmed SA, Abd El Reheem MH, Elbahy DA (2022) l-Carnitine ameliorates the osteoporotic changes and protects against simvastatin induced myotoxicity and hepatotoxicity in glucocorticoid-induced osteoporosis in rats. Biomed Pharmacother 152:113221. 10.1016/j.biopha.2022.113221 [DOI] [PubMed] [Google Scholar]
- 48.Peng H, Hu B, Xie LQ et al (2022) A mechanosensitive lipolytic factor in the bone marrow promotes osteogenesis and lymphopoiesis. Cell Metab 34:1168-1182.e6. 10.1016/j.cmet.2022.05.009 [DOI] [PubMed] [Google Scholar]
- 49.Alekos NS, Kushwaha P, Kim SP et al (2023) Mitochondrial β-oxidation of adipose-derived fatty acids by osteoblasts fuels parathyroid hormone-induced bone formation. JCI Insight 8:e165604. 10.1172/jci.insight.165604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin H, Liu T, Li X et al (2020) The role of gut microbiota metabolite trimethylamine N-oxide in functional impairment of bone marrow mesenchymal stem cells in osteoporosis disease. Ann Transl Med 8: 1009. 10.21037/atm-20-5307. [DOI] [PMC free article] [PubMed]
- 51.Zhang X, Li Y, Yang P et al (2020) Trimethylamine-N-Oxide promotes vascular calcification through activation of NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome and NF-κB (nuclear factor κB) signals. Arterioscler Thromb Vasc Biol 40:751–765. 10.1161/ATVBAHA.119.313414 [DOI] [PubMed] [Google Scholar]
- 52.Li J, Zeng Q, Xiong Z et al (2022) Trimethylamine N-oxide induces osteogenic responses in human aortic valve interstitial cells in vitro and aggravates aortic valve lesions in mice. Cardiovasc Res 118:2018–2030. 10.1093/cvr/cvab243 [DOI] [PubMed] [Google Scholar]
- 53.Hollenberg AM, Smith CO, Shum LC et al (2020) Lactate dehydrogenase inhibition with oxamate exerts bone anabolic effect. J Bone Miner Res 35:2432–2443. 10.1002/jbmr.4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data related to laboratory experimentation, in vitro osteoblast models, 16S rRNA sequencing, and serum metabolome are available on request from the corresponding author.