

Abstract

Understanding the nature of a transition-metal-catalyzed process, including catalyst evolution and the real active species, is rather challenging yet of great importance for the rational design and development of novel catalysts, and this is even more difficult for a bimetallic catalytic system. Pd(0)/carboxylic acid combined system-catalyzed allylic alkylation reaction of alkynes has been used as an atom-economical protocol for the synthesis of allylic products. However, the asymmetric version of this reaction is still rather limited, and the in-depth understanding of the nature of active Pd species is still elusive. Herein we report an enantioselective coupling between readily available aldimine esters and alkynes using a synergistic Cu/Pd catalyst system, affording a diverse set of α-quaternary allyl amino ester derivatives in good yields with excellent enantioselectivities. Mechanistic studies indicated that it is most likely a synergistic asymmetric molecular Cu catalysis with Pd nanoparticle catalysis. The Pd catalyst precursor is transformed to soluble Pd nanoparticles in situ, which are responsible for activating the alkyne to an electrophilic allylic Pd intermediate, while the chiral Cu complex of the aldimine ester enolate provides chiral induction and works in synergy with the Pd nanoparticles.
I. Introduction
Distinguishing between true homogeneous molecular catalysis and soluble or insoluble metal nanoparticle (NP) catalysis is a problem that has attracted much attention in the synthesis community since the key catalytic properties of catalytic activity, selectivity, stability, poisoning, regeneration, and also catalyst optimization are highly dependent on the identity of the active catalyst.1 Various interconversions between metal complexes, clusters, and nanoparticles via leaching and aggregation may occur during the catalytic processes (Scheme 1a, “cocktail” of catalysts).2 For example, ligand-free Pd salts or well-defined Pd complexes containing ligands can form Pd NPs under the reaction conditions, and in some cases, the in situ-formed nanoparticles may induce unexpected catalytic activity or even initiate a new reaction pathway.3,4 Therefore, catalyst evolution and mechanistic insights into the dynamic nature of “cocktail”-type systems are considered of great importance from the perspective of rational catalyst development and optimization.
Scheme 1. Synergistic Homogeneous Asymmetric Cu Catalysis with Pd Nanoparticle Catalysis.

In the last two decades, synergistic bimetallic asymmetric catalysis, wherein two chiral catalysts can work in concert to activate different substrates in two closely related catalytic cycles, has emerged as a powerful approach for reaction engineering (Scheme 1b, left).5 Most commonly, both catalytic cycles are run by homogeneous molecular metal catalysts (M1L*1/M2L*2). In recent years, several elegant relay homogeneous catalysis and nanoparticle catalysis processes have been reported to achieve some challenging transformations, showing the great proficiency of the combination of homogeneous molecular metal catalysis with nanoparticle catalysis in organic synthesis.6 To the best of our knowledge, synergistic homogeneous asymmetric catalysis with nanoparticle catalysis to achieve enantioselective syntheses is still underdeveloped in bimetallic catalysis (Scheme 1b, right).7
Nonproteinogenic α,α-disubstituted α-amino acids (α-AAs) are not only frequently utilized as building blocks in organic synthesis and new drug discovery but also present as structural motifs in numerous biologically active products.8 As a result of their broad utility, the development of efficient methods for the construction of enantiopure α,α-disubstituted α-AAs is highly desirable.9 Among various efficient methodologies, metal-catalyzed asymmetric allylic alkylation of aldimine esters represents an attractive and powerful catalytic process, especially by the elegant asymmetric Cu–Pd bimetallic catalysis.10,11 It is generally believed that both the active Cu and Pd species are molecular in nature, and the chiral Cu complex (Cu(I)/chiral ligand) is responsible for activating the imino ester via formation of a rigid metalated azomethine ylide12 and controlling the stereoselectivity of allylic substitution, while an achiral (or chiral) molecular Pd species forms the active π-allylpalladium intermediate in the reaction. Despite these achievements, the reactions have largely been limited to the use of allylic alcohols or related derivatives as preactivated allylic precursors. In some cases, dienes and allenes are also employed as coupling partners,13,14 which are converted to π-allylmetal intermediates viain situ addition of a metal–H species. Since alkynes are easily accessible and bench-stable synthons in organic synthesis, the employment of alkynes as green electrophilic allyl precursors has attracted great attention in allylic substitution reactions.15−17 Although chiral Rh-catalyzed asymmetric allylic substitution of alkynes with various nucleophiles has achieved great advances,17 the Pd(0)/carboxylic acid combined catalytic system using alkynes as the allylic precursors was largely limited to non-asymmetric reactions,16 and only a few examples18,19 of asymmetric transformations have been reported, mainly based on intramolecular cyclization reactions or α-allylation of carbonyl compounds by cooperative Pd–enamine catalysis. Harsh reaction conditions, such as high temperature (80–100 °C) and acidic conditions, might contribute to the challenges of asymmetric allylic alkylations of alkynes in this Pd(0)/carboxylic acid system. More importantly, an in-depth understanding of the nature of the Pd species involved in such an allylic alkylation process remains elusive so far.
Herein we report our discovery of synergistic homogeneous asymmetric Cu catalysis with Pd nanoparticle catalysis in the allylic alkylation of alkynes with aldimine esters (Scheme 1c). The reaction exhibits broad functional group tolerance, affording a variety of chiral α,α-disubstituted α-amino acid esters in good yields with high enantioselectivities. Data from mechanistic studies, including the induction time measurements, kinetic poisoning experiments, dynamic light scattering (DLS) studies, transmission electron microscopy (TEM) analysis, and filtration experiments, suggested that the active Pd nanoparticles generated in situ from the Pd(0) precursor should be responsible for activating the alkyne to an electrophilic allylic Pd species, which works synergistically with the asymmetric Cu-catalyzed attack of the aldimine ester.
II. Results and Discussion
1. Reaction Development
It has been well-known that aldimine esters can be activated by chiral Cu complexes to form Cu–azomethine ylides, which permits control of the configuration of the generated α-quaternary stereogenic center in allylic alkylation reactions.12 In addition, a combination of Pd(0)/phosphine ligand and a Brønsted acid has been a representative catalyst system for the activation of aryl propynes to electrophilic π-allyl-Pd intermediates.15,16 However, the asymmetric allylic alkylation of aldimine esters with easily accessible alkynes has not been achieved to the best of our knowledge, although it will provide quick access to synthetically valuable α-quaternary allyl amino ester derivatives. Our investigation was initiated using 1-phenyl-1-propyne (1a) and aldimine ester 2a as the model substrates and Pd(PPh3)4 and Cu(MeCN)4PF6/L5 as the bimetallic catalyst system. The reactions were typically run in 2-Me-THF at 100 °C with HOAc and KOAc as the additives. Under these conditions, 3aa was obtained in 74% yield with 93% ee (entry 1, Table 1). Further use of L6–L8 as the chiral ligand for Cu catalysis afforded no better results than that of L5 (entries 2–4 vs 1). Then we tested the combinations of Cu/L5 complex with Pd(dba)2 and different monophosphine ligands (entries 5–8), revealing that Pd(dba)2/L4 gave the best results (97% yield, 93% ee; entry 8). To further improve the enantioselectivity of the reaction, the temperature was lowered to 80 °C, and 3aa was isolated in 83% yield with 95% ee (entry 9). Lastly, control experiments showed that no reaction occurred without Pd(dba)2 or L4 (entries 10 and 11), suggesting that both the Pd source and phosphine ligand were important for the reactivity of the current reaction. While the reaction proceeded with substantially lower reactivity (<10% yield) without Cu salt (entry 12), the reaction in the absence of L5 gave a racemic product 3aa in 65% yield (entry 13). These results demonstrated that the combination of Pd/L4 and Cu/L5 catalysts was crucial for the success of the current asymmetric transformation.
Table 1. Optimization of the Reaction Conditionsa.

| entry | Pd cat. | L for Cu cat. | yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | Pd(PPh3)4 | L5 | 74 | 93 |
| 2 | Pd(PPh3)4 | L6 | 73 | 75 |
| 3 | Pd(PPh3)4 | L7 | 75 | 9 |
| 4 | Pd(PPh3)4 | L8 | 71 | 15 |
| 5 | Pd(dba)2/L1 | L5 | 60 | 90 |
| 6 | Pd(dba)2/L2 | L5 | 60 | 89 |
| 7 | Pd(dba)2/L3 | L5 | 82 | 84 |
| 8 | Pd(dba)2/L4 | L5 | 97 | 93 |
| 9d | Pd(dba)2/L4 | L5 | 88 (83e) | 95 |
| 10d | no L4 | L5 | ND | – |
| 11d | no Pd(dba)2 | L5 | ND | – |
| 12d | L4 | no Cu salt | <10 | 7 |
| 13d | L4 | no L5 | 65 | 0 |
The reaction was carried out using 1a (0.4 mmol), 2a (0.2 mmol), Pd(dba)2 (0.01 mmol, 5 mol %), L1–L4 (0.02 mmol, 10 mol %), Cu(MeCN)4PF6 (0.01 mmol, 5 mol %), L5–L8 (0.012 mmol, 6 mol %), HOAc (0.03 mmol), and KOAc (0.4 mmol) in 2-Me-THF (1.0 mL) at 100 °C for 20 h.
Determined by 1H NMR analyses of the crude products using CH2Br2 as an internal standard. ND = 3aa not detected.
Determined by HPLC on a Daicel OD-H chiral column.
80 °C for 40 h.
Isolated yield.
2. Substrate Scope
With the optimized reaction conditions in hand, we proceeded to explore the substrate scope (Scheme 2a). Propynes containing various monosubstituted aryl groups (1a–l) reacted well with aldimine ester 2a, affording the corresponding products 3aa–la in moderate to good yields (54–89%) with high enantioselectivities (90–95% ee), irrespective of the para (1b–i)/ortho (1j)/meta (1k, 1l) pattern or electron-donating (Me, MeO, Me2N) or electron-withdrawing (F, Cl, CO2Me, CF3) nature of the substituents. The strong electron-donating group −NMe2 may decrease the electrophilicity of the generated π-allylic Pd intermediate, which might account for the slightly lower yield of 3da (54%). The reactions of di- or trisubstituted aryl propynes 1m–o with 2a also worked well, delivering 3ma–oa in 71–87% yield with 85–92% ee. In addition, propynes with thienyl (1p, 1q) and the extended π system (1r) and ferrocenyl (1s) were also suitable substrates, giving the corresponding α,α-disubstituted α-amino acid derivatives 3pa–sa in moderate yields (41–73%) with good enantioselectivities (78–90% ee). The reaction was not restricted to aryl propynes, as the alkyl propynes cyclohexylmethylacetylene and 2-heptyne were also suitable substrates and provided the corresponding products 3ta and 3ua in moderate yields with high enantioselectivities (90 and 92% ee). The relatively weak electrophilicity of the alkyl π-allylic Pd intermediate and the possible side reactions (such as β-H elimination) may lead to the relatively low yields compared to those with aryl propynes.
Scheme 2. (a, b) Substrate Scope, (c) Synthetic Utilization, and (d) Investigation of Alkyne Activation.

Standard conditions: 1 (0.8 mmol), 2 (0.4 mmol), Pd(dba)2 (0.02 mmol, 5 mol %), L4 (0.04 mmol, 10 mol %), Cu(MeCN)4PF6 (0.02 mmol, 5 mol %), L5 (0.024 mmol, 6 mol %), HOAc (0.06 mmol), and KOAc (0.8 mmol) in 2-Me-THF (1.0 mL) at 80 °C for 40 h, then NaBH4 was added to reduce the product. Isolated yields are reported. The ee values were determined by HPLC on a Diacel OD-H chiral column.
Without NaBH4.
Next, we investigated the scope of the reaction with respect to aldimine esters 2 bearing different α-substituents, which were derived from either natural or unnatural α-amino acids (Scheme 2b). The aldimine Schiff bases derived from 2-aminobutyric acid, norleucine, and leucine were suitable coupling partners with 1a, affording the desired products 3ab–ad in 70–83% yield with 76–89% ee. Methionine, aspartic acid and glutamic acid-derived aldimine esters (2e–g) were also readily applicable to this reaction. α-Methylene ether- (2h), α-homobenzyl- (2i), and α-allyl-substituted (2j) aldimine Schiff bases were well-compatible with the protocol, delivering the products 3ah–aj in 65–82% yield with 81–92% ee. Moreover, a cyclic imine was also amenable to the procedure, affording cyclic product 3ak smoothly in 75% yield with 94% ee.
To probe the scalability and utility of the present method, we conducted a gram-scale reaction of 1a with 2a, and the product was further transformed into free amino acid 4 in 71% yield with 93% ee in a one-pot manner (Scheme 2c). Then this densely functionalized amino acid product was proved to be well-amenable to further synthetic manipulations. First, protection of 4 with Boc2O gave a quantitative yield of 5, which on treatment with LiAlH4/KOtBu in THF produced oxazolidinone 6 in 83% yield. In addition, by treatment with Boc2O under DMAP catalysis, 4 was transformed to the corresponding unstable isocyanate, which was converted further to give 7 in >99% yield via the addition of EtNH2.
To prove the possible presence of an allene intermediate in the current alkyne allylic alkylation, the coupling reaction of phenyl allene 8 with aldimine Schiff base 2a was carried out under the standard conditions (Scheme 2d, left). The desired coupling product 3aa was obtained in a lower yield (24%) but still with excellent stereoselectivity (93% ee), suggesting that an allene intermediate might participate in the title reaction. Due to several side reactions of phenyl allene under these conditions, such as dimerization and oligomerization of allene, the yield of 3aa was decreased significantly, suggesting that a low but steady concentration of allene generated in situ from the corresponding alkyne may be optimal for this reaction. Furthermore, a deuterium-labeling experiment with d-1a was conducted (Scheme 2d, right). The observed incorporation of D over all of the positions of the allylic group in d-3aa indicated the involvement of Pd–H in the reaction, and the insertion of Pd–H and β-hydrogen elimination steps was reversible in the catalytic process. These results are consistent with the previous studies of Pd-catalyzed allylic alkylation of alkynes under acidic conditions.15,16 In addition, ligand scrambling was found to be either negligible or absent between Pd(dba)2/L4 and Cu(I)/L5, as revealed by the ligand exchange experiment (for details, see the Supporting Information).
3. Mechanistic Studies
Understanding the nature of the catalytically active species is rather challenging, especially for the current bimetallic catalysis involving two different metal precursors. First, the standard reaction process was measured by taking the yield of 3aa as a function of the reaction time, which exhibited a discontinuity in the course of the reaction, with a slower initial stage being perceptible (Scheme 3a, left). Notably, such a relatively long induction period of about 1.0 h supported the idea that a (sub)nanoparticle nucleation may have occurred in the initiation stage. Indirect evidence for the presence of a NP catalyst can be gained from the mercury drop test.20 In this case, the addition of mercury to a reaction mixture would poison the nanosized catalyst particles by amalgam formation and hence would prevent any product formation thereafter (Scheme 3a, middle). Indeed, when mercury was added into the reaction system (at t = 0 or 20 h), no further product formation was detected. These results demonstrated that nanoparticles or heterogeneous species might be responsible for the catalysis. Besides, poly(4-vinylpyridine) (PVPy) has been used as a selective poison for homogeneous Pd catalysts without inhibiting the activity of heterogeneous Pd catalysts.21 Application of this assay to the model reaction revealed that the catalyst was deactivated immediately upon the addition of PVPy to the reaction mixture at 0 or 20 h (Scheme 3a, right). Taken together, the Hg and PVPy poisoning experiments suggested that the active catalyst was most likely to consist of soluble nanoparticles under the standard conditions.
Scheme 3. Mechanistic Studies.
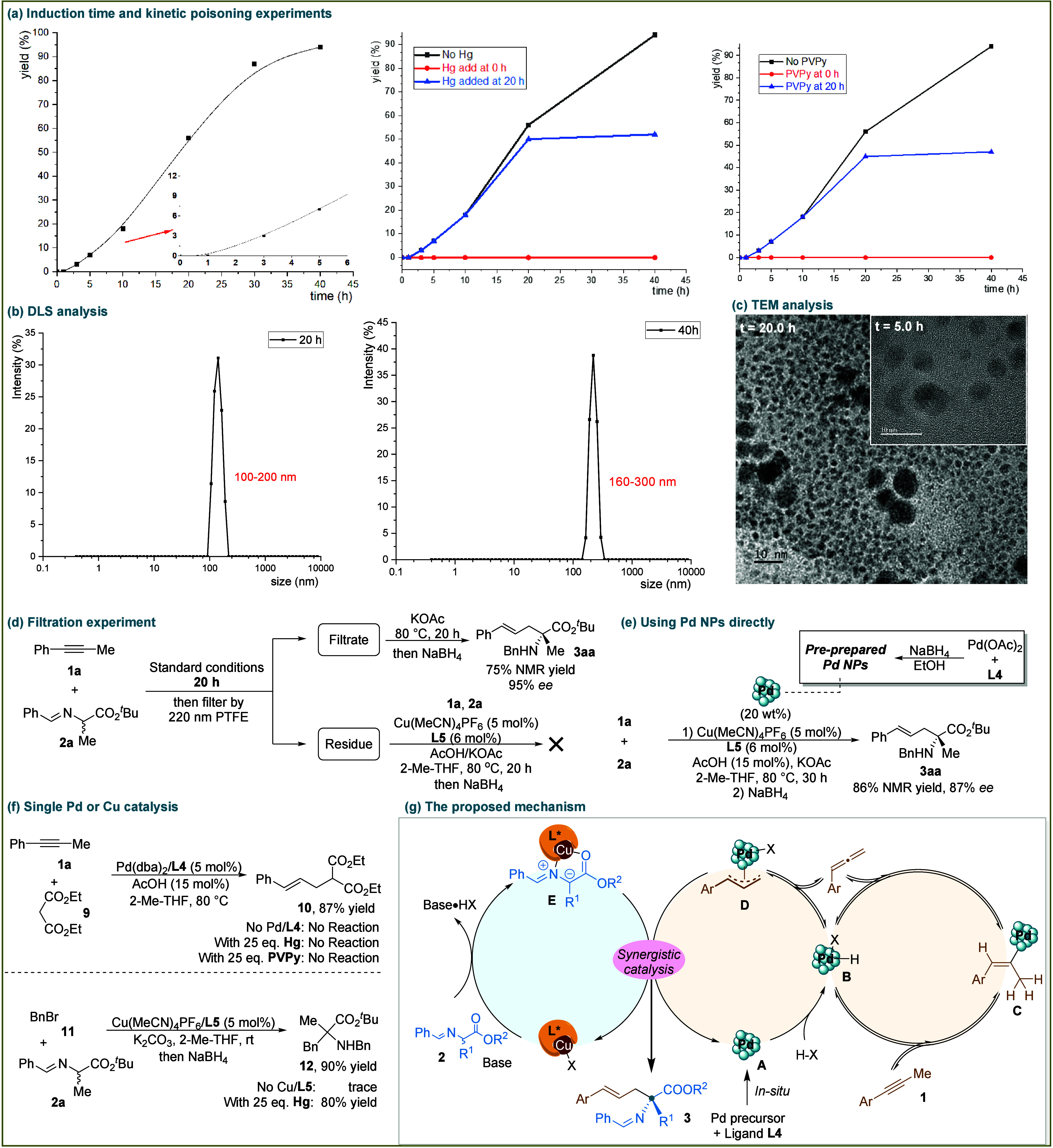
The presence of nanoparticle species was further confirmed by dynamic light scattering experiments. DLS analysis of the reaction mixture revealed the presence of 100–200 nm diameter particles at 20 h after initiation of the reaction (Scheme 3b). After 40 h, particles with sizes ranging from 160 to 300 nm were detected. By contrast, no NPs were observed in the system at the beginning of the reaction (t = 0). On the other hand, it was found by TEM analysis that the samples that were taken at 5.0 and 20.0 h contained nanoparticles (Scheme 3c). By contrast, no NPs were detected by TEM at the initial state (t = 0). The fact that not all samples contained nanoparticles ruled out the possibility of colloid formation following sample withdrawal. Both the DLS data and TEM analysis implicated the formation and continuous growth of NPs in size as the reaction proceeds. Then filtration tests were further carried out to assess whether some heterogeneous metal species may catalyze the current reaction (Scheme 3d). After 20 h, the reaction mixture was filtered through a 200 nm PTFE filter, and the filtrates were further stirred for 20 h at 80 °C with a supplement of KOAc. The reaction still gave a 75% yield of the desired product, suggesting that some catalytically active species were present in the filtrate. In addition, the residue was used as the Pd catalyst instead of Pd(dba)2/L4 for the standard reaction. However, no desired product was obtained, suggesting that the insoluble residue was not active.
Collectively, the long induction time for catalyst activation and the Hg test, DLS, and TEM results all suggested that (sub)nanoparticle nucleation may have occurred from the metal precursors and that either in situ-formed nanoparticles or heterogeneous species are catalytically active. The PVPy test and filtration experiment results revealed that the catalytic activity is associated with soluble metal species. All of these results indicated that some soluble nanoparticles are catalytically active and most likely to be responsible for the current asymmetric catalysis.
To clarify whether the NPs are either Pd or Cu (or both) in nature is challenging. To this end, further control experiments were carried out. L4-coordinated Pd NPs were independently prepared by the reduction of a mixture of Pd(OAc)2 and L4 by NaBH4 (Scheme 3e).22 Under otherwise standard conditions, the reaction of 1a and 2a using these preprepared Pd NPs instead of Pd(dba)2/L4 also occurred smoothly. Notably, no detectable induction period was observed (for details, see the Supporting Information), further supporting that the in situ formation of Pd NPs via aggregation of particles from the Pd precursor is essential and that the in situ-formed Pd NPs are most likely the catalytically active species in the title reaction.23 Furthermore, a Pd-catalyzed allylic alkylation of alkyne 1a with 9 was carried out in the presence of HOAc, and it was found that both Hg and PVPy inhibited the activity (Scheme 3f), suggesting that soluble Pd NPs might be active for the reaction. In addition, NPs were also detected in the reaction mixture by TEM analysis (see the Supporting Information). Cu(I)/chiral ligand-catalyzed asymmetric reactions using aldimine esters as the nucleophiles have been well-developed, and the obtained high and reproducible enantioselectivities are generally consistent with homogeneous molecular catalysis. A Cu/L5-catalyzed substitution reaction of 10 and 2a was taken as an example. The reaction occurred smoothly (80% yield) in the presence of excess Hg, and no NPs were detected in the reaction mixture by TEM analysis. These results indicated that in the title reaction, Pd/L4 is likely to form soluble Pd NPs while Cu/L5 might be molecular species to keep the whole catalytic system still homogeneous. The reproducibility of the high enantioselectivities of the title reaction also argues for a molecular nature in the chirality control process. Therefore, the current asymmetric reaction is most likely a synergistic process of soluble Pd NP catalysis with homogeneous chiral Cu catalysis, although we cannot rule out the potential for the involvement of Cu NPs or mixed Pd/Cu NPs.
Based on the literature and our results, we propose a possible Pd/Cu dual-catalyzed pathway (Scheme 3g). The in situ-generated Pd NPs A activate the alkyne by Pd–H insertion and β-H elimination to afford the allene intermediate. Insertion of the Pd–H species B into the allene produced the key π-allyl species bound with Pd NPs (D). Meanwhile, the in situ-formed α-substituted N-metalated azomethine ylide E, which was activated by the chiral copper(I) complex, could result in a linearly selective allylic alkylation process that permits control of the configuration of the generated α-quaternary stereogenic center. The reaction exhibited high atom and step economy since the in situ-formed electrophilic π-allyl-Pd species omitted the need to install leaving groups on the allylic reagents.
III. Summary and Conclusions
In summary, we have developed a synergistic Pd/Cu-catalyzed asymmetric allylic alkylation of alkynes with aldimine esters, affording a diverse set of α-quaternary allylated amino esters in good yields with excellent enantioselectivities (up to 95% ee). Notably, mechanistic studies suggested that the Pd precursor together with a phosphine ligand is likely to form soluble Pd nanoparticles to activate the alkyne to an allylic Pd species, which can act in synergy with chiral Cu-catalyzed asymmetric allylic alkylation to form the chiral product. This work demonstrates the remarkable power of synergistic homogeneous molecular catalysis with nanoparticle catalysis to access difficult or otherwise unattainable transformations and will stimulate future work for the development of mixed homogeneous molecular catalysis and nanoparticle catalysis.
Acknowledgments
The authors acknowledge financial support from the National Key R&D Program of China (2022YFA1503200), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDB0610000), the National Natural Science Foundation of China (92256303, 22171278, and 21821002), the Shanghai Science and Technology Committee (23ZR1482400), the Natural Science Foundation of Ningbo (2023J034), and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c09983.
Experimental procedures, complete characterization data, and copies of 1H, 13C, 19F, and 31P NMR spectra (PDF)
Author Contributions
# Y.L. and H.C. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews, see:; a Widegren J. A.; Finke R. G. A Review of Soluble Transition-Metal Nanoclusters as Arene Hydrogenation Catalysts. J. Mol. Catal. A: Chem. 2003, 191, 187–207. 10.1016/S1381-1169(02)00125-5. [DOI] [Google Scholar]; b Widegren J. A.; Finke R. G. A Review of the Problem of Distinguishing True Homogeneous Catalysis from Soluble or Other Metal-Particle Heterogeneous Catalysis under Reducing Conditions. J. Mol. Catal. A: Chem. 2003, 198, 317–341. 10.1016/S1381-1169(02)00728-8. [DOI] [Google Scholar]; c Dyson P. J. Arene Hydrogenation by Homogeneous Catalysts: Fact or Fiction?. Dalton Trans. 2003, 15, 2964–2974. 10.1039/b303250g. [DOI] [Google Scholar]; d Ott L. S.; Finke R. G. Transition-Metal Nanocluster Stabili-zation for Catalysis: A Critical Review of Ranking Methods and Putative Stabilizers. Coord. Chem. Rev. 2007, 251, 1075–1100. 10.1016/j.ccr.2006.08.016. [DOI] [Google Scholar]; e Narayanan R.; Tabor C.; El-Sayed M. A. Can the Observed Changes in the Size or Shape of a Colloidal Nanocatalyst Reveal the Nanocatalysis Mechanism Type: Homogeneous or Heterogeneous?. Top. Catal. 2008, 48, 60–74. 10.1007/s11244-008-9057-4. [DOI] [Google Scholar]; f Crabtree R. H. Resolving Heterogeneity Problems and Impurity Artifacts in Operationally Homogeneous Transition Metal Catalysts. Chem. Rev. 2012, 112, 1536–1554. 10.1021/cr2002905. [DOI] [PubMed] [Google Scholar]; g Schmidt A. F.; Kurokhtina A. A. Distinguishing between the Homogeneous and Heterogeneous Mechanisms of Catalysis in the Mizoroki-Heck and Suzuki-Miyaura Reactions: Problems and Prospects. Kinet. Catal. 2012, 53, 714–730. 10.1134/S0023158412060109. [DOI] [Google Scholar]; h Liu L.; Meira D. M.; Arenal R.; Concepcion P.; Puga A. V.; Corma A. Determination of the evolution of heterogeneous single metal atoms and nanoclusters under reaction conditions: which are the working catalytic sites?. ACS. Catal. 2019, 9, 10626–10639. 10.1021/acscatal.9b04214. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Gärtner D.; Sandl S.; Jacobi von Wangelin A. Homogeneous vs. heterogeneous: mechanistic insights into iron group metal-catalyzed reductions from poisoning experiments. Catal. Sci. Technol. 2020, 10, 3502–3514. 10.1039/D0CY00644K. [DOI] [Google Scholar]
- a Ananikov V. P.; Beletskaya I. P. Toward the Ideal Catalyst: From Atomic Centers to a “Cocktail” of Catalysts. Organometallics 2012, 31, 1595–1604. 10.1021/om201120n. [DOI] [Google Scholar]; b Eremin D. B.; Ananikov V. P. Understanding active species in catalytic transformations: From molecular catalysis to nanoparticles, leaching, “Cocktails” of catalysts and dynamic systems. Coord. Chem. Rev. 2017, 346, 2–19. 10.1016/j.ccr.2016.12.021. [DOI] [Google Scholar]; c Prima D. O.; Kulikovskaya N. S.; Galushko A. S.; Mironenko R. M.; Ananikov V. P. Transition metal ‘cocktail’-type catalysis. Curr. Opin. Green Sustainable Chem. 2021, 31, 100502. 10.1016/j.cogsc.2021.100502. [DOI] [Google Scholar]
- For selected reviews, see:; a Phan N. T. S.; Van Der Sluys M.; Jones C. W. On the Nature of the Active Species in Palladium Catalyzed Mizoroki-Heck and Suzuki-Miyaura Couplings Homogeneous or Heterogeneous Catalysis, A Critical Review. Adv. Synth. Catal. 2006, 348, 609–679. 10.1002/adsc.200505473. [DOI] [Google Scholar]; b de Vries J. G. A unifying mechanism for all high-temperature Heck reactions. The role of palladium colloids and anionic species. Dalton Trans. 2006, 421–429. 10.1039/B506276B. [DOI] [PubMed] [Google Scholar]; c Trzeciak A. M.; Ziółkowski J. J. Monomolecular, nanosized and heterogenized palladium catalysts for the Heck reaction. Coord. Chem. Rev. 2007, 251, 1281–1293. 10.1016/j.ccr.2006.11.013. [DOI] [Google Scholar]; d Balanta A.; Godard C.; Claver C. Pd nanoparticles for C-C coupling reactions. Chem. Soc. Rev. 2011, 40, 4973–4985. 10.1039/c1cs15195a. [DOI] [PubMed] [Google Scholar]; e Kashin A. S.; Ananikov V. P. Catalytic C-C and C-Heteroatom Bond Formation Reactions: In Situ Generated or Preformed Catalysts? Complicated Mechanistic Picture Behind Well-Known Experimental Procedures. J. Org. Chem. 2013, 78, 11117–11125. 10.1021/jo402038p. [DOI] [PubMed] [Google Scholar]; f Deraedt C.; Astruc D. Homeopathic” Palladium Nanoparticle Catalysis of Cross Carbon-Carbon Coupling Reactions. Acc. Chem. Res. 2014, 47, 494–503. 10.1021/ar400168s. [DOI] [PubMed] [Google Scholar]; g Richter C.; Schaepe K.; Glorius F.; Ravoo B. J. Tailor-Made N-Heterocyclic Carbenes for Nanoparticle Stabilization. Chem. Commun. 2014, 50, 3204–3207. 10.1039/c4cc00654b. [DOI] [PubMed] [Google Scholar]; h Polynski M. V.; Ananikov V. P. Modeling Key Pathways Proposed for the Formation and Evolution of “Cocktail”-Type Systems in Pd-Catalyzed Reactions Involving ArX Reagents. ACS Catal. 2019, 9, 3991–4005. 10.1021/acscatal.9b00207. [DOI] [Google Scholar]; i Trzeciak A. M.; Augustyniak A. W. The Role of Palladium Nanoparticles in Catalytic C-C Cross-Coupling Reactions. Coord. Chem. Rev. 2019, 384, 1–20. 10.1016/j.ccr.2019.01.008. [DOI] [Google Scholar]; j Jeddi N.; Scott N. W. J.; Fairlamb I. J. S. Well-Defined Pdn Clusters for Cross-Coupling and Hydrogenation Catalysis: New Opportunities for Catalyst Design. ACS Catal. 2022, 12, 11615–11638. 10.1021/acscatal.2c03345. [DOI] [Google Scholar]
- For selected examples, see:; a Reetz M. T.; Westermann E. Phosphane-Free Palladium-Catalyzed Coupling Reactions: The Decisive Role of Pd Nanoparticles. Angew. Chem., Int. Ed. 2000, 39, 165–168. . [DOI] [PubMed] [Google Scholar]; b de Vries A. H. M.; Mulders J. M. C. A.; Mommers J. H. M.; Henderickx H. J. W.; de Vries J. G. Homeopathic Ligand-Free Palladium as a Catalyst in the Heck Reaction. A Comparison with a Palladacycle. Org. Lett. 2003, 5, 3285–3288. 10.1021/ol035184b. [DOI] [PubMed] [Google Scholar]; c Reetz M. T.; de Vries J. G. Ligand-free Heck reactions using low Pd-loading. Chem. Commun. 2004, 1559–1563. 10.1039/b406719n. [DOI] [PubMed] [Google Scholar]; d Thathagar M. B.; ten Elshof J. E.; Rothenberg G. Pd Nanoclusters in C-C Coupling Reactions: Proof of Leaching. Angew. Chem., Int. Ed. 2006, 45, 2886–2890. 10.1002/anie.200504321. [DOI] [PubMed] [Google Scholar]; e Ellis P. J.; Fairlamb I. J. S.; Hackett S. F. J.; Wilson K.; Lee A. F. Evidence for the Surface-Catalyzed Suzuki-Miyaura Reaction over Palladium Nanoparticles: An Operando XAS Study. Angew. Chem., Int. Ed. 2010, 49, 1820–1824. 10.1002/anie.200906675. [DOI] [PubMed] [Google Scholar]; f Pun D.; Diao T.; Stahl S. S. Aerobic Dehydrogenation of Cyclohexanone to Phenol Catalyzed by Pd(TFA)2/2-Dimethylaminopyridine: Evidence for the Role of Pd Nanoparticles. J. Am. Chem. Soc. 2013, 135, 8213–8221. 10.1021/ja403165u. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Leyva-Pérez A.; Oliver-Meseguer J.; Rubio-Marqués P.; Corma A. Water-Stabilized Three-and Four-Atom Palladium Clusters as Highly Active Catalytic Species in Ligand-Free C-C Cross-Coupling Reactions. Angew. Chem., Int. Ed. 2013, 52, 11554–11559. 10.1002/anie.201303188. [DOI] [PubMed] [Google Scholar]; h Baumann C. G.; De Ornellas S.; Reeds J. P.; Storr T. E.; Williams T. J.; Fairlamb I. J. S. Formation and propagation of well-defined Pd nanoparticles (PdNPs) during C-H bond functionalization of heteroarenes: are nanoparticles a moribund form of Pd or an active catalytic species?. Tetrahedron 2014, 70, 6174–6187. 10.1016/j.tet.2014.06.002. [DOI] [Google Scholar]; i Fricke C.; Sherborne G. J.; Funes-Ardoiz I.; Senol E.; Guven S.; Schoenebeck F. Orthogonal Nanoparticle Catalysis with Organogermanes. Angew. Chem., Int. Ed. 2019, 58, 17788–17795. 10.1002/anie.201910060. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Fernández E. a.; Rivero-Crespo M. A.; Domínguez I.; RubioMarqués P.; Oliver-Meseguer J.; Liu L.; Cabrero-Antonino M.; Gavara R.; Hernández-Garrido J. C.; Boronat M.; Leyva-Pérez A.; Corma A. Base-controlled Heck, Suzuki, and Sonogashira reactions catalyzed by ligand-free platinum or palladium single atom and sub-nanometer clusters. J. Am. Chem. Soc. 2019, 141, 1928–1940. 10.1021/jacs.8b07884. [DOI] [PubMed] [Google Scholar]; k Uno K.; Itoh T.; Sato H.; Lloyd-Jones G. C.; Orito Y. Direct Observation of Palladium Leaching from Pd/C by a Simple Method: X-ray Absorption Spectroscopy of Heterogeneous Mixtures. ACS Omega 2023, 8, 21787–21792. 10.1021/acsomega.3c01343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reviews of synergistic bimetallic asymmetric catalysis, see:; a Park J.; Hong S. Cooperative bimetallic catalysis in asymmetric transformations. Chem. Soc. Rev. 2012, 41, 6931–6943. 10.1039/c2cs35129c. [DOI] [PubMed] [Google Scholar]; b Pye D. R.; Mankad N. P. Bimetallic catalysis for C-C and C-X coupling reactions. Chem. Sci. 2017, 8, 1705–1718. 10.1039/C6SC05556G. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kim U B.; Jung D. J.; Jeon H. J.; Rathwell K.; Lee S.-g. Synergistic Dual Transition Metal Catalysis. Chem. Rev. 2020, 120, 13382–13433. 10.1021/acs.chemrev.0c00245. [DOI] [PubMed] [Google Scholar]; d Martínez S.; Veth L.; Lainer B.; Dydio P. Challenges and Opportunities in Multicatalysis. ACS Catal. 2021, 11, 3891–3915. 10.1021/acscatal.0c05725. [DOI] [Google Scholar]; e Huo X.; Li G.; Wang X.; Zhang W. Bimetallic Catalysis in Stereodivergent Synthesis. Angew. Chem., Int. Ed. 2022, 61, e202210086. 10.1002/anie.202210086. [DOI] [PubMed] [Google Scholar]; f Wei L.; Wang C.-J. Asymmetric transformations enabled by synergistic dual transition-metal catalysis. Chem. Catal. 2023, 3, 100455. 10.1016/j.checat.2022.10.031. [DOI] [Google Scholar]; g Ackerman-Biegasiewicz L. K. G.; Kariofillis S. K.; Weix D. J. Multimetallic-Catalyzed C-C Bond-Forming Reactions: From Serendipity to Strategy. J. Am. Chem. Soc. 2023, 145, 6596–6614. 10.1021/jacs.2c08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Alonso J. M.; Paz Muñoz M. Evidence of Hybrid Homogeneous-Heterogeneous Catalysis in a Pt/Au Heterobimetallic System. ChemCatChem 2018, 10, 2646–2654. 10.1002/cctc.201800076. [DOI] [Google Scholar]; b Cui M.; Qian Q.; Zhang J.; Wang Y.; Asare Bediako B. B.; Liu H.; Han B. Liquid fuel synthesis via CO2 hydrogenation by coupling homogeneous and heterogeneous catalysis. Chem 2021, 7, 726–737. 10.1016/j.chempr.2020.12.005. [DOI] [Google Scholar]; c Wu H.; Yang J.; Peters B. B. C.; Massaro L.; Zheng J.; Andersson P. G. Asymmetric Full Saturation of Vinylarenes with Cooperative Homogeneous and Heterogeneous Rhodium Catalysis. J. Am. Chem. Soc. 2021, 143, 20377–20383. 10.1021/jacs.1c09975. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang M.; Zhao Z.; Li C.; Li H.; Liu J.; Yang Q. Synergy of metal nanoparticles and organometallic complex in NAD(P)H regeneration via relay hydrogenation. Nat. Commun. 2022, 13, 5699. 10.1038/s41467-022-33312-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Li X.; Hao W.; Yi N.; He Y.-M.; Fan Q.-H. Asymmetric Transfer Hydrogenation of Naphthol and Phenol Derivatives with Cooperative Heterogeneous and Homogeneous Catalysis. CCS Chem. 2023, 5, 2277–2289. 10.31635/ccschem.023.202303007. [DOI] [Google Scholar]
- For selected reviews of asymmetric nanoparticle catalysis, see:; a Yasukawa T.; Miyamura H.; Kobayashi S. Chiral metal nanoparticle-catalyzed asymmetric C-C bond formation reactions. Chem. Soc. Rev. 2014, 43, 1450–1461. 10.1039/C3CS60298B. [DOI] [PubMed] [Google Scholar]; b Yasukawa T.; Miyamura H.; Kobayashi S. Chiral Ligand-Modified Metal Nanoparticles as Unique Catalysts for Asymmetric C-C Bond-Forming Reactions: How Are Active Species Generated?. ACS Catal. 2016, 6, 7979–7988. 10.1021/acscatal.6b02446. [DOI] [Google Scholar]; c Savitha G.; Saha R.; Sekar G. Bimetallic chiral nanoparticles as catalysts for asymmetric synthesis. Tetrahedron Lett. 2016, 57, 5168–5178. 10.1016/j.tetlet.2016.10.011. [DOI] [Google Scholar]; d Schrader I.; Neumann S.; Šulce A.; Schmidt F.; Azov V.; Kunz S. Asymmetric Heterogeneous Catalysis: Transfer of Molecular Principles to Nanoparticles by Ligand Functionalization. ACS Catal. 2017, 7, 3979–3987. 10.1021/acscatal.7b00422. [DOI] [Google Scholar]; e Mallat T.; Orglmeister E.; Baiker A. Asymmetric Catalysis at Chiral Metal Surfaces. Chem. Rev. 2007, 107, 4863–4890. 10.1021/cr0683663. [DOI] [PubMed] [Google Scholar]
- a Ohfune Y.; Shinada T. Enantio- and Diastereoselective Construction of α, α-Disubstituted α-Amino Acids for the Synthesis of Biologically Active Compounds. Eur. J. Org. Chem. 2005, 2005, 5127–5143. 10.1002/ejoc.200500434. [DOI] [Google Scholar]; b Ager D. J.Synthesis of Unnatural/Nonproteinogenic α-Amino Acids. In Amino Acids, Peptides and Proteins in Organic Chemistry, Volume 1: Origins and Synthesis of Amino Acids; Hughes A. B., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp 495–526. [Google Scholar]; c Unnatural Amino Acids: Methods and Protocols; Pollegioni L., Servi S., Eds.; Springer: New York, 2012. [Google Scholar]
- For selected reviews of the synthesis of α-AAs, see:; a Vogt H.; Bräse S. Recent Approaches towards the Asymmetric Synthesis of α, α-Disubstituted α-Amino Acids. Org. Biomol. Chem. 2007, 5, 406–430. 10.1039/B611091F. [DOI] [PubMed] [Google Scholar]; b Mizota I.; Shimizu M. Umpolung Reactions of α-Imino Esters: Useful Methods for the Preparation of α-Amino Acid Frameworks. Chem. Rec. 2016, 16, 688–702. 10.1002/tcr.201500267. [DOI] [PubMed] [Google Scholar]; c Jiang H.; Jin Y.; Lin J. New Progress in Asymmetric Synthesis of Quaternary α-Amino Acids. Mini-Rev. Org. Chem. 2017, 14, 434–447. 10.2174/1570193X14666170420121255. [DOI] [Google Scholar]; d Dickstein J. S.; Kozlowski M. C. Organometal Additions to α-Iminoesters: N-Alkylation via Umpolung. Chem. Soc. Rev. 2008, 37, 1166–1173. 10.1039/b709139g. [DOI] [PubMed] [Google Scholar]; e Carloni A.; Porzi G.; Sandri S. Stereoselective Synthesis of Uncommon α, α′-Dialkyl-α-Aminoacids. Part 1. Tetrahedron: Asymmetry 1998, 9, 2987–2998. 10.1016/S0957-4166(98)00272-9. [DOI] [Google Scholar]; f Bera K.; Namboothiri I. N. N. Asymmetric Synthesis of Quaternary α-Amino Acids and Their Phosphonate Analogues. Asian J. Org. Chem. 2014, 3, 1234–1260. 10.1002/ajoc.201402178. [DOI] [Google Scholar]
- a Fu J.; Huo X.; Li B.; Zhang W. Cooperative bimetallic catalysis in asymmetric allylic substitution. Org. Biomol. Chem. 2017, 15, 9747–9759. 10.1039/C7OB02476B. [DOI] [PubMed] [Google Scholar]; b Wu Y.; Huo X.; Zhang W. Synergistic Pd/Cu Catalysis in Organic Synthesis. Chem.—Eur. J. 2020, 26, 4895–4916. 10.1002/chem.201904495. [DOI] [PubMed] [Google Scholar]
- For selected examples, see:; a Wei L.; Xu S.-M.; Zhu Q.; Che C.; Wang C.-J. Synergistic Cu/Pd Catalysis for Enantioselective Allylic Alkylation of Aldimine Esters: Access to α, α-Disubstituted α-Amino Acids. Angew. Chem., Int. Ed. 2017, 56, 12312–12316. 10.1002/anie.201707019. [DOI] [PubMed] [Google Scholar]; b Huo X.; He R.; Fu J.; Zhang J.; Yang G.; Zhang W. Stereoselective and Site-Specific Allylic Alkylation of Amino Acids and Small Peptides via a Pd/Cu Dual Catalysis. J. Am. Chem. Soc. 2017, 139, 9819–9822. 10.1021/jacs.7b05460. [DOI] [PubMed] [Google Scholar]; c He R.; Huo X.; Zhao L.; Wang F.; Jiang L.; Liao J.; Zhang W. Stereodivergent Pd/Cu Catalysis for the Dynamic Kinetic Asymmetric Transformation of Racemic Unsymmetrical 1,3-Disubstituted Allyl Acetates. J. Am. Chem. Soc. 2020, 142, 8097–8103. 10.1021/jacs.0c02150. [DOI] [PubMed] [Google Scholar]; d Xiao L.; Chang X.; Xu H.; Xiong Q.; Dang Y.; Wang C.-J. Cooperative Catalyst-Enabled Regio- and Stereodivergent Synthesis of α-Quaternary α-Amino Acids via Asymmetric Allylic Alkylation of Aldimine Esters with Racemic Allylic Alcohols. Angew. Chem., Int. Ed. 2022, 61, e202212948. 10.1002/anie.202212948. [DOI] [PubMed] [Google Scholar]; e Li B.; Xu H.; Dang Y.; Houk K. N. Dispersion and Steric Effects on Enantio-/Diastereoselectivities in Synergistic Dual Transition-Metal Catalysis. J. Am. Chem. Soc. 2022, 144, 1971–1985. 10.1021/jacs.1c12664. [DOI] [PubMed] [Google Scholar]
- For reviews of Cu-catalyzed asymmetric transformations of aldimine esters, see:; a Coldham I.; Hufton R. Intramolecular Dipolar Cycloaddition Reactions of Azomethine Ylides. Chem. Rev. 2005, 105, 2765–2810. 10.1021/cr040004c. [DOI] [PubMed] [Google Scholar]; b Narayan R.; Potowski M.; Jia Z.-J.; Antonchick P.; Waldmann H. Catalytic Enantioselective 1,3-Dipolar Cycloadditions of Azomethine Ylides for Biology-Oriented Synthesis. Acc. Chem. Res. 2014, 47, 1296–1310. 10.1021/ar400286b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wei L.; Xiao L.; Hu Y.; Wang Z.; Tao H.; Wang C.-J. Recent Advances in Metallated Azomethine Ylides for the Synthesis of Chiral Unnatural α-Amino Acids. Chin. J. Org. Chem. 2019, 39, 2119–2130. 10.6023/cjoc201904060. [DOI] [Google Scholar]; d Wei L.; Chang X.; Wang C.-J. Catalytic Asymmetric Reactions with N-Metallated Azomethine Ylides. Acc. Chem. Res. 2020, 53, 1084–1100. 10.1021/acs.accounts.0c00113. [DOI] [PubMed] [Google Scholar]
- For selected reviews, see:; a Li G.; Huo X.; Jiang X.; Zhang W. Asymmetric synthesis of allylic compounds via hydrofunctionalisation and difunctionalisation of dienes, allenes, and alkynes. Chem. Soc. Rev. 2020, 49, 2060–2118. 10.1039/C9CS00400A. [DOI] [PubMed] [Google Scholar]; b Wang H.; Zhang Q.; Zi W. Synergistic Catalysis Involving Palladium for Stereodivergent Csp3–Csp3 Coupling Reactions. Acc. Chem. Res. 2024, 57, 468–488. 10.1021/acs.accounts.3c00639. [DOI] [PubMed] [Google Scholar]; c Patil N. T.; Kavthe R. D.; Shinde V. S. Transition metal-catalyzed addition of C-, N- and O-nucleophiles to unactivated C-C multiple bonds. Tetrahedron 2012, 68, 8079–8146. 10.1016/j.tet.2012.05.125. [DOI] [Google Scholar]
- For selected examples, see:; a Zhang Q.; Yu H.; Shen L.; Tang T.; Dong D.; Chai W.; Zi W. Stereodivergent Coupling of 1,3-Dienes with Aldimine Esters Enabled by Synergistic Pd and Cu Catalysis. J. Am. Chem. Soc. 2019, 141, 14554–14559. 10.1021/jacs.9b07600. [DOI] [PubMed] [Google Scholar]; b Zhu M.; Zhang Q.; Zi W. Diastereodivergent Synthesis of β-Amino Alcohols by Dual-Metal-Catalyzed Coupling of Alkoxyallenes with Aldimine Esters. Angew. Chem., Int. Ed. 2021, 60, 6545–6552. 10.1002/anie.202014510. [DOI] [PubMed] [Google Scholar]; c Chai W.; Guo B.; Zhang Q.; Zi W. Cooperative Pd/Cu-Catalyzed Diastereodivergent Coupling of Allenamides and Aldimine Esters to Access the Mannich-type Motifs. Chem. Catal. 2022, 2, 1428–1439. 10.1016/j.checat.2022.04.006. [DOI] [Google Scholar]; d Xia J.; Hirai T.; Katayama S.; Nagae H.; Zhang W.; Mashima K. Mechanistic Study of Ni and Cu Dual Catalyst for Asymmetric C-C Bond Formation; Asymmetric Coupling of 1,3-Dienes with C-nucleophiles to Construct Vicinal Stereocenters. ACS Catal. 2021, 11, 6643–6655. 10.1021/acscatal.1c01626. [DOI] [Google Scholar]
- a Koschker P.; Breit B. Branching Out: Rhodium-Catalyzed Allylation with Alkynes and Allenes. Acc. Chem. Res. 2016, 49, 1524–1536. 10.1021/acs.accounts.6b00252. [DOI] [PubMed] [Google Scholar]; b Haydl A. M.; Breit B.; Liang T.; Krische M. J. Alkynes as Electrophilic or Nucleophilic Allylmetal Precursors in Transition-Metal Catalysis. Angew. Chem., Int. Ed. 2017, 56, 11312–11325. 10.1002/anie.201704248. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cera G.; Maestri G. Palladium/Brønsted Acid Catalysis for Hydrofunctionalizations of Alkynes: From Tsuji-Trost Allylations to Stereoselective Methodologies. ChemCatChem 2022, 14, e202200295. 10.1002/cctc.202200295. [DOI] [Google Scholar]
- For selected examples of Pd-catalyzed allylic substitution using propynes, see:; a Trost B. M.; Schmidt T. A simple synthesis of dienones via isomerization of alkynones effected by palladium catalysts. J. Am. Chem. Soc. 1988, 110, 2301–2303. 10.1021/ja00215a051. [DOI] [Google Scholar]; b Trost B. M.; Brieden W.; Baringhaus K. H. Intermolecular Additions and Cycloisomerizations by a Pd-Catalyzed Sequence of an Intramolecular Redox Reaction and an Addition. Angew. Chem., Int. Ed. Engl. 1992, 31, 1335–1336. 10.1002/anie.199213351. [DOI] [Google Scholar]; c Kadota I.; Shibuya A.; Gyoung Y. S.; Yamamoto Y. Palladium/Acetic Acid Catalyzed Allylation of Some Pronucleophiles with Simple Alkynes. J. Am. Chem. Soc. 1998, 120, 10262–10263. 10.1021/ja981299c. [DOI] [Google Scholar]; d Patil N. T.; Nawaz Khan F.; Yamamoto Y. MicrowaveEnhanced Pd(0)/Acetic Acid Catalyzed Allylation Reactions of C, N, and O-Pronucleophiles with Alkynes. Tetrahedron Lett. 2004, 45, 8497–8499. 10.1016/j.tetlet.2004.09.099. [DOI] [Google Scholar]; e Bajracharya G. B.; Huo Z.; Yamamoto Y. Intramolecular Hydroamination of Alkynes Catalyzed by Pd(PPh3)4/Triphenyl-phosphine under Neutral Conditions. J. Org. Chem. 2005, 70, 4883–4886. 10.1021/jo050412w. [DOI] [PubMed] [Google Scholar]; f Yao S.; Liu J.; Yang Z.; Gui Q.; Chen X.; Tan Z.; Li P. Palladium-Catalyzed Allylation of α-Nitroacetates with Propynes. Syn. Commun. 2014, 44, 3165–3172. 10.1080/00397911.2014.928939. [DOI] [Google Scholar]; g Gao S.; Wu Z.; Fang X.; Lin A.; Yao H. Palladium-Catalyzed Dearomative Allylic Alkylation of Indoles with Alkynes To Synthesize Indolenines with C3-Quarternary Centers. Org. Lett. 2016, 18, 3906–3909. 10.1021/acs.orglett.6b01947. [DOI] [PubMed] [Google Scholar]; h Lu C.-J.; Chen H.; Chen D.-K.; Wang H.; Yang Z.-P.; Gao J.; Jin H. Palladium-catalyzed allylation of sulfonyl hydrazides with alkynes to synthesize allylic arylsulfones. Org. Biomol. Chem. 2016, 14, 10833–10839. 10.1039/C6OB01929C. [DOI] [PubMed] [Google Scholar]; i Fang X.; Zeng Y.; Li Q.; Wu Z.; Yao H.; Lin A. Redox-Neutral Atom-Economic Pd(0)-Catalyzed Dearomatization of β-Naphthols with Alkynes toward Naphthalenones. Org. Lett. 2018, 20, 2530–2533. 10.1021/acs.orglett.8b00662. [DOI] [PubMed] [Google Scholar]; j Ren W.; Zuo Q.-M.; Niu Y.-N.; Yang S.-D. Palladium-NHC-Catalyzed Allylic Alkylation of Pronucleophiles with Alkynes. Org. Lett. 2019, 21, 7956–7960. 10.1021/acs.orglett.9b02937. [DOI] [PubMed] [Google Scholar]; k Zheng P.; Wang C.; Chen Y.-C.; Dong G.-B. Pd-Catalyzed Intramolecular α-Allylic Alkylation of Ketones with Alkynes: Rapid and Stereodivergent Construction of [3.2.1] Bicycles. ACS Catal. 2019, 9, 5515–5521. 10.1021/acscatal.9b00997. [DOI] [Google Scholar]; l Lu C.-J.; Yu X.; Chen D.-K.; Wang H.; Song Q.-B.; Gao J.-R. Palladium-catalyzed allylation of aminophenol with alkynes to construct C-N bonds. Org. Biomol. Chem. 2019, 17, 3545–3551. 10.1039/C9OB00333A. [DOI] [PubMed] [Google Scholar]; m Yang C.; Zhang K.; Wu Z.; Yao H.; Lin A. Cooperative Palladium/Proline-Catalyzed Direct α-Allylic Alkylation of Ketones with Alkynes. Org. Lett. 2016, 18, 5332–5335. 10.1021/acs.orglett.6b02649. [DOI] [PubMed] [Google Scholar]; n Patil N. T.; Song D.; Yamamoto Y. Palladium-Catalyzed Allylation of Pronucleophiles with Alkynes at 50 °C - Remarkable Effect of 2-(Dicyclohexylphosphanyl)-2′-(dimethylamino)biphenyl as Ligand. Eur. J. Org. Chem. 2006, 2006, 4211–4213. 10.1002/ejoc.200600337. [DOI] [Google Scholar]
- For selected examples of Rh-catalyzed allylic substitution using propynes, see:; a Lumbroso A.; Koschker P.; Vautravers N. R.; Breit B. Redox Neutral Atom Economic Rhodium-Catalyzed Coupling of Terminal Alkynes with Carboxylic Acids Toward Branched Allylic Esters. J. Am. Chem. Soc. 2011, 133, 2386–2389. 10.1021/ja1108613. [DOI] [PubMed] [Google Scholar]; b Gellrich U.; Meißner A.; Steffani A.; Kähny M.; Drexler H.; Heller D.; Plattner D. A.; Breit B. Mechanistic Investigations of the Rhodium Catalyzed Propargylic C-H Activation. J. Am. Chem. Soc. 2014, 136, 1097–1104. 10.1021/ja411204d. [DOI] [PubMed] [Google Scholar]; c Koschker P.; Kähny M.; Breit B. Enantioselective RedoxNeutral Rh-Catalyzed Coupling of Terminal Alkynes with Carboxylic Acids Toward Branched Allylic Esters. J. Am. Chem. Soc. 2015, 137, 3131–3137. 10.1021/jacs.5b01131. [DOI] [PubMed] [Google Scholar]; d Chen Q.-A.; Chen Z.; Dong V. M. Rhodium-Catalyzed Enantioselective Hydroamination of Alkynes with Indolines. J. Am. Chem. Soc. 2015, 137, 8392–8395. 10.1021/jacs.5b05200. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Cruz A. F.; Dong V. M. Stereo-divergent Coupling of Aldehydes and Alkynes via Synergistic Catalysis Using Rh and Jacobsen’s Amine. J. Am. Chem. Soc. 2017, 139, 1029–1032. 10.1021/jacs.6b10680. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zheng W.-F.; Xu Q.-J.; Kang Q. Rhodium/Lewis Acid Catalyzed Regioselective Addition of 1,3-Dicarbonyl Compounds to Internal Alkynes. Organometallics 2017, 36, 2323–2330. 10.1021/acs.organomet.7b00284. [DOI] [Google Scholar]; g Xie L.; Yang H.; Ma M.; Xing D. Rhodium-Catalyzed Branched and Enantioselective Direct α-Allylic Alkylation of Simple Ketones with Alkynes. Org. Lett. 2020, 22, 2007–2011. 10.1021/acs.orglett.0c00375. [DOI] [PubMed] [Google Scholar]; h Ji D.-W.; Yang F.; Chen B.-Z.; Min X.-T.; Kuai C.-S.; Hu Y.-C.; Chen Q.-A. Rhodium-catalyzed regio- and enantioselective allylic alkylation of pyrazol-5-ones with alkynes. Chem. Commun. 2020, 56, 8468–8471. 10.1039/D0CC04002A. [DOI] [PubMed] [Google Scholar]; i Davison T. R.; Parker P. D.; Hou X.; Chung P. C.; Augustine S. A.; Dong V. M. Enantioselective Addition of α-Nitroesters to Alkynes. Angew. Chem., Int. Ed. 2021, 60, 4599–4603. 10.1002/anie.202014015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For asymmetric intramolecular cyclization, see:; a Patil N. T.; Lutete L. M.; Wu H.; Pahadi N. K.; Gridnev I. D.; Yamamoto Y. Palladium Catalyzed Intramolecular Asymmetric Hydroamination, Hydro-alkoxylation, and Hydrocarbonation of Alkynes. J. Org. Chem. 2006, 71, 4270–4279. 10.1021/jo0603835. [DOI] [PubMed] [Google Scholar]; b Patil N. T.; Wu H.; Yamamoto Y. A Route to 2-Substituted Tetrahydroquinolines via Palladium-Catalyzed Intramolecular Hydroamination of Anilino-alkynes. J. Org. Chem. 2007, 72, 6577–6579. 10.1021/jo0708137. [DOI] [PubMed] [Google Scholar]; c Narsireddy M.; Yamamoto Y. Catalytic Asymmetric Intramolecular Hydroamination of Alkynes in the Presence of a Catalyst System Consisting of Pd(0)-Methyl Norphos (or Tolyl Renorphos)-Benzoic Acid. J. Org. Chem. 2008, 73, 9698–9709. 10.1021/jo801785r. [DOI] [PubMed] [Google Scholar]
- a Lee J. T. D.; Zhao Y. Direct Enantioselective α-Allylation of Unfunctionalized Cyclic Ketones with Alkynes through Pd-Amine Cooperative Catalysis. Chem.—Eur. J. 2018, 24, 9520–9524. 10.1002/chem.201802273. [DOI] [PubMed] [Google Scholar]; b Su Y.-L.; Li L.-L.; Zhou X.-L.; Dai Z.-Y.; Wang P.-S.; Gong L.-Z. Asymmetric α-Allylation of Aldehydes with Alkynes by Integrating Chiral Hydridopalladium and Enamine Catalysis. Org. Lett. 2018, 20, 2403–2406. 10.1021/acs.orglett.8b00740. [DOI] [PubMed] [Google Scholar]; c Tang M.-Q.; Yang Z.-J.; He Z. T. Asymmetric formal sp2-hydrocarbonations of dienes and alkynes via palladium hydride catalysis. Nat. Commun. 2023, 14, 6303. 10.1038/s41467-023-42160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chernyshev V. M.; Astakhov A. V.; Chikunov I. E.; Tyurin R. V.; Eremin D. B.; Ranny G. S.; Khrustalev V. N.; Ananikov V. P. Pd and Pt Catalyst Poisoning in the Study of Reaction Mechanisms: What Does the Mercury Test Mean for Catalysis?. ACS Catal. 2019, 9, 2984–2995. 10.1021/acscatal.8b03683. [DOI] [Google Scholar]; b Chagunda C. I.; Fisher T.; Schierling M.; McIndoe J. S. Poisonous Truth about the Mercury Drop Test: The Effect of Elemental Mercury on Pd(0) and Pd(II)ArX Intermediates. Organometallics 2023, 42, 2938–2945. 10.1021/acs.organomet.3c00340. [DOI] [Google Scholar]
- Richardson J. M.; Jones C. W. Poly(4-vinylpyridine) and Quadrapure TUas Selective Poisons for Soluble Catalytic Species in Palladium-Catalyzed Coupling Reactions - Application to Leaching from Polymer-Entrapped Palladium. Adv. Synth. Catal. 2006, 348, 1207–1216. 10.1002/adsc.200606021. [DOI] [Google Scholar]
- Landge V. G.; Grant A. J.; Fu Y.; Rabon A. M.; Payton J. L.; Young M. C. Palladium-Catalyzed γ, γ’-Diarylation of Free Alkenyl Amines. J. Am. Chem. Soc. 2021, 143, 10352–10360. 10.1021/jacs.1c04261. [DOI] [PubMed] [Google Scholar]
- In the case of Pd2(dba)3 precursors, PdNP formation is a significant issue. See:; a Zalesskiy S. S.; Ananikov V. P. Pd2(dba)3 as a Precursor of Soluble Metal Complexes and Nanoparticles: Determination of Palladium Active Species for Catalysis and Synthesis. Organometallics 2012, 31, 2302–2309. 10.1021/om201217r. [DOI] [Google Scholar]; b Kapdi A. R.; Whitwood A. C.; Williamson D. C.; Lynam J. M.; Burns M. J.; Williams T. J.; Reay A. J.; Holmes J.; Fairlamb I. J. S. The Elusive Structure of Pd2(dba)3. Examination by Isotopic Labeling, NMR Spectroscopy, and X-ray Diffraction Analysis: Synthesis and Characterization of Pd2(dba-Z)3 Complexes. J. Am. Chem. Soc. 2013, 135, 8388–8399. 10.1021/ja403259c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.