

Abstract
Covalent chemistry is a versatile approach for expanding the ligandability of the human proteome. Activity-based protein profiling (ABPP) can infer the specific residues modified by electrophilic compounds through competition with broadly reactive probes. The extent to which such residue-directed platforms, however, fully assess the protein targets of electrophilic compounds in cells remains unclear. Here, we evaluate a complementary protein-directed ABPP method that identifies proteins showing stereoselective reactivity with alkynylated, chiral electrophilic compounds—termed stereoprobes. Integration of protein- and cysteine-directed data from cancer cells treated with tryptoline acrylamide stereoprobes revealed generally well-correlated ligandability maps and highlighted features, such as protein size and the proteotypicity of cysteine-containing peptides, that explain gaps in each ABPP platform. In total, we identified stereoprobe binding events for >300 structurally and functionally diverse proteins, including compounds that stereoselectively and site-specifically disrupt MAD2L1BP interactions with the spindle assembly checkpoint complex leading to delayed mitotic exit in cancer cells.
Graphical Abstract
Introduction
Advances in our understanding of the genetic basis of human disease offer unprecedented opportunities for precision medicine1. Nonetheless, many genotype-phenotype relationships lack mechanistic understanding and remain therapeutically unactionable due, in large part, to a dearth of functional assays and selective pharmacological tools for studying disease-relevant proteins. Several innovative strategies have emerged to address this gap, including approaches to discover small molecule binders to proteins that may otherwise lack screening-compatible functional assays2. Such “binding-first” methods include fragment-based ligand discovery3–7, DNA-encoded libraries8–10, and chemical proteomics11, 12. Among these approaches, chemical proteomics has the advantage of assaying small molecules for interactions with many proteins in parallel directly in native biological systems, which can circumvent the need to recombinantly express and purify proteins for ligand discovery. Chemical proteomics has shown particular utility in the discovery and optimization of chemical probes and drugs that operated by a covalent mechanism11–13.
Covalent chemistry often leverages electrophilic small molecules to modify nucleophilic amino acid residues in proteins. Advantages of covalent chemistry include – i) enhanced interactions with shallow pockets in proteins that are challenging to target by solely reversible binding; ii) improved selectivity by targeting isotype-restricted nucleophilic amino acids within sets of related proteins; and iii) sustained pharmacological activity that is dependent on protein turnover rather than the pharmacokinetics of small molecules14, 15. Original covalent probes and drugs were mainly discovered serendipitously or by the incorporation of electrophilic groups into established reversible ligands, as has been shown for kinase inhibitors16–18. More recently, the intentional pursuit of covalent chemical probes, either by targeting specific disease-relevant nucleophilic residues (e.g., G12C-KRAS)19, 20 or by globally profiling the reactivity of structurally diverse electrophilic small molecules in cells12, 21–26 has begun to showcase the broad potential of covalent chemistry to expand the small-molecule interactions (or ‘ligandability’) of the human proteome.
Large-scale investigations of electrophilic small molecule-protein interactions frequently leverage the activity-based protein profiling (ABPP) technology, which infers the identity of nucleophilic residues modified by electrophilic compounds in biological systems through competition with broadly reactive amino acid-directed probes24, 27. For instance, we and others have used cysteine-directed iodoacetamide (IA) probes with alkyne or (desthio)biotin (DTB) affinity handles22, 27, 28 in competitive ABPP experiments that map cysteines engaged by electrophilic fragments21, 29, natural products30, and diversity-oriented synthesis-inspired collections of stereochemically defined compounds (‘stereoprobes’)22, 31–33. These cysteine-directed ABPP experiments have discovered covalent ligands that produce functional effects through modification of cysteine residues on diverse proteins, including DNA/RNA-binding proteins34, 35, metabolic enzymes and protein kinases36, 37, scaffolding/adaptor proteins38, and E3 ligases32, 39–41. Nonetheless, in certain instances, cysteine-directed ABPP has fallen short of mapping biologically relevant protein targets of electrophilic compounds in human cells31 due to gaps in its coverage of ligandable cysteines. Such gaps may reflect, for instance, cysteines residing on non-proteotypic peptides that fail to be detected by conventional liquid chromatography-mass spectrometry (LC-MS) protocols42.
Understanding the proportion of ligandable cysteines that evade detection in original cysteine-directed ABPP platforms is important to minimize false-negative outcomes (i.e., electrophilic small molecule-cysteine interactions that are overlooked) and to better understand the proteome-wide selectivity of covalent chemical probes. Here, we describe a multi-tiered chemical proteomic strategy to address this question by evaluating a focused library of tryptoline acrylamide stereoprobes22, 32, 33 for their protein reactivity in cells by two complementary approaches: 1) cysteine-directed ABPP, wherein hit compounds are defined as those that stereoselectively block cysteine reactivity with an IA-DTB probe; and 2) protein-directed ABPP43, 44, wherein hit compounds are defined as those that block the stereoselective enrichment of proteins by stereochemically matched alkyne-modified probes. We show that each approach has distinct attributes and, when deployed together, generates near-comprehensive maps of covalent liganding events for electrophilic small molecules in a biological system. Additionally, our data indicate that tryptoline acrylamides, combined with controls that account for their stereoselective (inactive enantiomeric compounds) and site-specific (cysteine mutant proteins) reactivity in cells, can serve as useful tools for initial biology experiments even at early stages in their maturation as chemical probes.
Results
Alkyne stereoprobe design and initial characterization
Previous cysteine-directed ABPP studies of tryptoline acrylamide stereoprobes have identified stereoselectively liganded cysteines on a diverse array of proteins22, 31, 33. We felt that this scope of proteomic reactivity would provide for a rigorous comparison of cysteine- and protein-directed ABPP platforms. We accordingly synthesized five sets of alkyne-functionalized tryptoline acrylamide stereoprobes, each consisting of four stereoisomers (20 total compounds in black; Fig. 1a). By placing the alkyne handle at different positions on the tryptoline core, we hoped to account for potential effects of this group on proteomic interactions in comparison to the unmodified tryptoline acrylamide stereoprobes to be used as competitors in ABPP experiments (eight total compounds in red; Fig. 1a).
Figure 1. Alkyne stereoprobes for mapping electrophilic small molecule-protein interactions in cells.
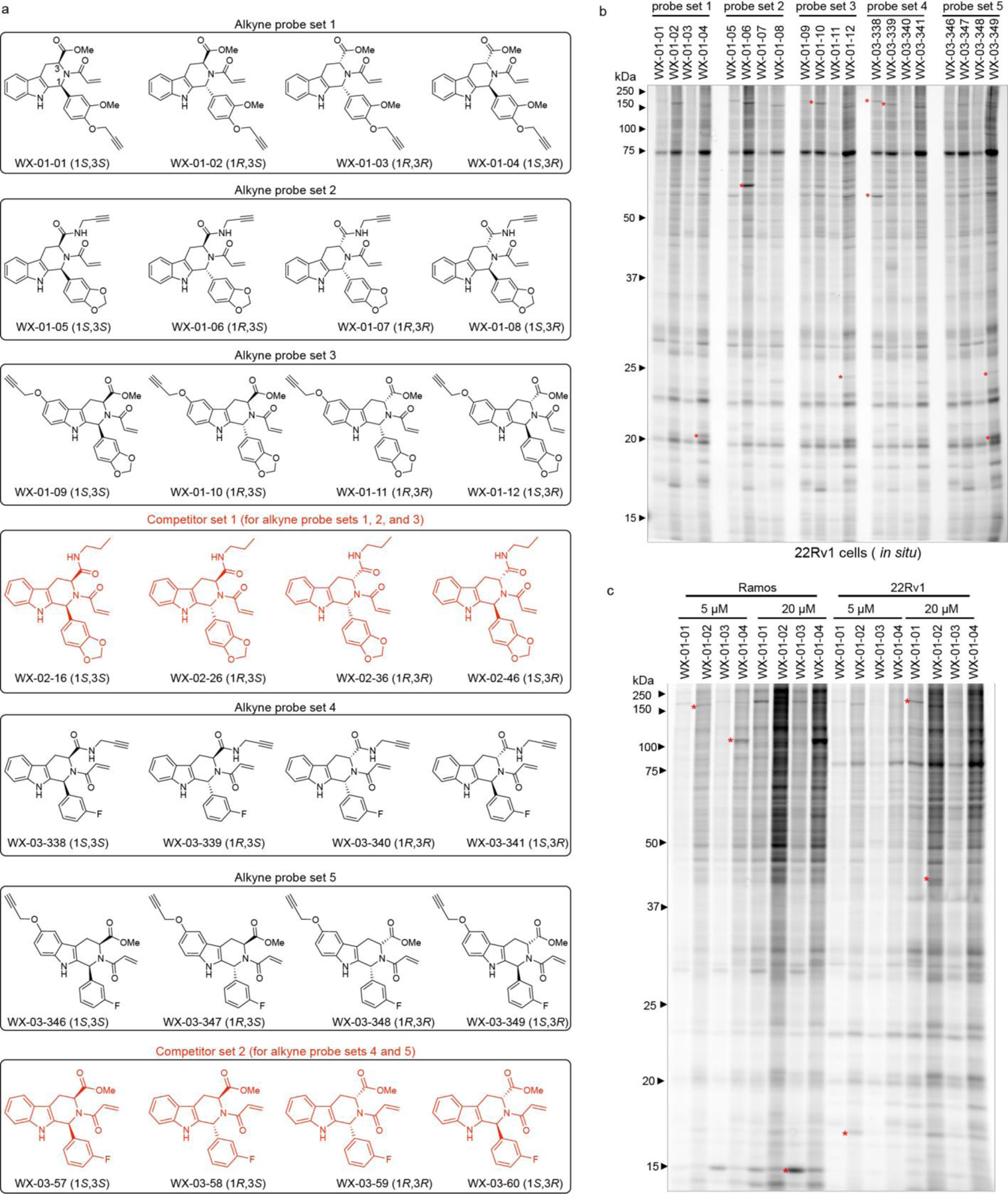
a, Structures of alkynylated tryptoline acrylamide stereoprobes used in this study (black) and the corresponding non-alkyne stereoprobes (red) used as competitors in ABPP experiments. b, In situ reactivity of alkyne stereoprobes (5 μM, 1 h) in 22Rv1 cells as determined by gel-ABPP where stereoprobe-reactive proteins are detected by CuAAC conjugation to an azide-rhodamine reporter group, SDS-PAGE, and in-gel fluorescence scanning. c, Concentration-dependent in situ reactivity of alkyne stereoprobe set 1 in Ramos and 22Rv1 cells (tested at 5 μM and 20 μM, 1 h). Red asterisks mark examples of stereoselective stereoprobe-protein interactions. b, c, data are from a single experiment representative of two independent experiments.
We first assessed the overall proteomic reactivity of alkyne stereoprobes by gel-ABPP in two human cancer cell lines – an adherent prostate carcinoma epithelial cell line (22Rv1) and a suspension B lymphoid cell line (Ramos). These cell lines display distinct gene expression profiles45 that we felt would provide complementary portraits of protein ligandability. Cells were treated with 5 or 20 μM of each stereoprobe for 1 h, lysed, and stereoprobe-reactive proteins visualized by Cu(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC)46, 47 conjugation to a rhodamine reporter tag, SDS-PAGE, and in-gel fluorescence scanning48. Each alkyne stereoprobe produced clear concentration-dependent proteomic reactivity (Fig. 1b, c and Extended Data Fig. 1a), with trans 1R, 3S and 1S, 3R stereoisomers generally showing greater reactivity (Fig. 1b, c and Extended Data Fig. 1a), a property that was also observed in cell lysates (Extended Data Fig. 1b) and with glutathione (Supplementary Table 1). Even with the limited resolution afforded by SDS-PAGE, several stereoselective stereoprobe-protein interactions were detected (red asterisks, Fig. 1b, c and Extended Data Fig. 1a, b).
Protein-directed ABPP with alkyne stereoprobes
We next performed MS analysis of initial protein-directed ABPP experiments of Ramos cells treated with a representative set of alkyne stereoprobes (probe set 2; 5 or 20 μM, 3 h). Following cell lysis, stereoprobe-reactive proteins were conjugated to azide-biotin by CuAAC, isolated with streptavidin beads, digested with trypsin, labeled by tandem mass tagging (TMT), and identified (MS1/MS2 analysis) and quantified (MS3 analysis) by multiplexed (TMT10plex) MS (Extended Data Fig. 2a). Proteins showing >3-fold enrichment by one stereoprobe compared to its enantiomer were assigned as stereoselective targets. More than 150 proteins were stereoselectively enriched at 5 and/or 20 μM, and we noted that these proteins tended to show higher stereoenrichment ratios in cells treated with 5 μM of the stereoprobes (Extended Data Fig. 2b). This result could indicate that, at 20 μM, the stereoprobes begin to show low-level labeling of multiple cysteines on proteins, which could erode at the stereoenrichment values associated with specific engagement of individual cysteines. We therefore performed future protein-directed ABPP experiments with 5 μM of alkyne stereoprobes.
We next determined the stoichiometry of tryptoline acrylamide-protein interactions using competitive protein-directed ABPP experiments, where cells were first treated with DMSO or non-alkyne competitor stereoprobes (20 μM; WX-02-16/26/36/46 or WX-03-57/58/59/60; Fig. 1a) for 2 h, followed by treatment with corresponding alkyne stereoprobes (5 μM; Fig. 1a) for 1 h, and processed and analysed by multiplexed (TMT16plex) MS (Fig. 2a). Proteins were designated as being stereoselectively liganded if they met the following criteria: 1) >3-fold enantioselective enrichment with at least one pair of alkyne stereoprobe enantiomers; and 2) >50% blockade of this enantioselective enrichment by the corresponding competitor stereoprobe.
Figure 2. Protein-directed ABPP platform for mapping stereoselectively liganded proteins in human cells.
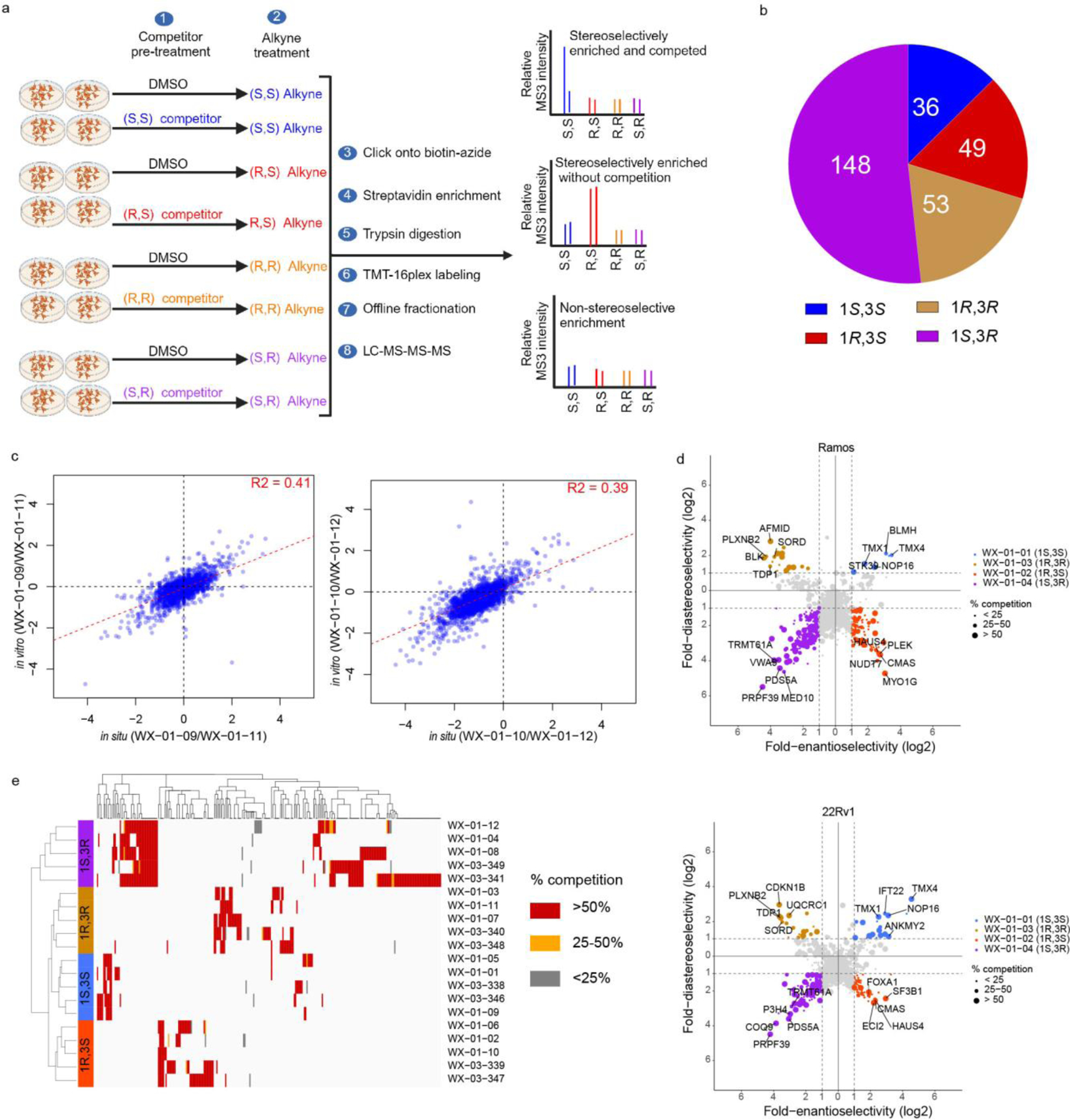
a, Workflow for protein-directed ABPP experiments where the stereoselective enrichment of proteins by alkyne stereoprobes and blockade of this enrichment by corresponding non-alkyne competitor stereoprobes are determined by multiplexed (tandem mass tagging, TMT16plex) MS-based proteomics. Created with BioRender.com. b, Pie chart showing total number of proteins stereoselectively liganded in situ by each stereoconfiguration of tryptoline acrylamide stereoprobes. Note that this number is >271 because 14 proteins show cross reactivity with more than one stereoconfiguration. c, Correlation plots comparing in situ (5 μM, 1 h) and in vitro (10 μM, 1 h) enantioselective protein enrichment values from protein-directed ABPP experiments performed in Ramos cells treated with stereoprobe set 3. d, Quadrant plots highlighting stereoselectively liganded proteins for each stereoconfiguration of alkyne stereoprobe set 1 in Ramos (top) and 22Rv1(bottom) cells. Enantioselectivity (x-axis) is the ratio of enrichment for one stereoisomer vs its enantiomer, and diastereoselectivity (y-axis) is the ratio of enrichment of one stereoisomer vs the average of its two diastereomers. e, Unsupervised clustering of the full stereoprobe interaction profiles for each stereoselectively liganded protein, where clustering was based on the magnitude of competitor stereoprobe blockade of stereoselective enrichment observed for each liganded protein. White spaces represent protein-stereoprobe interactions that did not qualify as stereoselectively enriched by the indicated alkyne stereoprobe. Protein-directed ABPP data represent average values from 2–4 biological replicates per stereoprobe, per cell line.
In total, 271 proteins were stereoselectively liganded in protein-directed ABPP experiments performed in Ramos and 22Rv1 cells with five sets of alkyne stereoprobes (Fig. 2b and Supplementary Dataset 1). Each stereochemical configuration of the tryptoline acrylamide core liganded a distinct set of proteins, with the (1S, 3R) stereoisomer engaging the greatest number of proteins (Fig. 2b). The (1S, 3R) stereoisomer also stereoselectively engaged a greater number of proteins in cell lysates (Extended Data Fig. 2c). We more generally observed well-aligned stereoprobe reactivity profiles in cells (in situ) versus cell lysates (in vitro) (Fig. 2c), although there were exceptional proteins that were stereoselectively liganded only in cells or lysates (Extended Data Fig. 2d). Despite the greater overall reactivity of the trans (1R, 3S) and (1S, 3R) stereoprobes, most of the proteins enantioselectively liganded by the cis (1R, 3R) and (1S, 3S) stereoprobes did not cross react with the trans stereoprobes (Extended Data Fig. 2e), underscoring the importance of the absolute stereoconfiguration of the tryptoline core for stereoprobe-protein interactions.
We found that quadrant plots offered a way to visualize stereoselectively liganded proteins, where the positions of proteins on the x- and y-axes reflect enantioselective and diastereoselective enrichment, respectively, and the size of the dot represents the degree of competitive blockade of this enrichment by the corresponding non-alkyne stereoprobe for the designated quadrant (Fig. 2d). This presentation format also highlighted stereoselectively liganded proteins expressed by only one cell type (e.g., immune-restricted proteins PLEK and MYO1G in Ramos cells or the prostate lineage transcription factor FOXA149 in 22Rv1 cells; Fig. 2d and Extended Data Fig. 3). Finally, hierarchical clustering revealed similar target profiles for alkyne stereoprobes of the same stereochemistry, while also highlighting proteins that were engaged by only a single alkyne stereoprobe (Fig. 2e).
Having established an initial map of proteins stereoselectively liganded by tryptoine acrylamide stereoprobes, we next aimed to understand how these protein-directed ABPP results correlated with, and complemented data generated by cysteine-directed ABPP.
Integration of cysteine- and protein-directed ABPP data
Cysteine-directed ABPP experiments were performed with five sets of alkyne stereoprobes and two sets of non-alkyne competitor stereoprobes (Fig. 1a) in Ramos and 22Rv1 cells following described protocols50, where each multiplexed (TMT10plex) experiment compared in duplicate all four stereoisomers of a given stereoprobe set alongside a DMSO control (20 μM compound, 3 h; Extended Data Fig. 4a). Cysteines were assigned as stereoselectively liganded if they showed a >66.7% decrease in IA-DTB reactivity in cells treated with a stereoprobe, and this decrease in IA-DTB reactivity was at least 2.5-fold greater than that observed for the enantiomeric stereoprobe. In total, >38,000 cysteines on >9,000 proteins were quantified, of which 238 cysteines on 217 proteins were stereoselectively liganded (Fig. 3a and Supplementary Dataset 1). As was observed in the protein-directed ABPP experiments (Fig. 2b and Extended Data Fig. 2c), each stereochemical configuration of the tryptoline acrylamide core liganded a distinct set of cysteines, and the (1S, 3R) stereoisomer engaged the greatest number of cysteines (Extended Data Fig. 4b).
Figure 3. Integrated protein- and cysteine-directed ABPP.
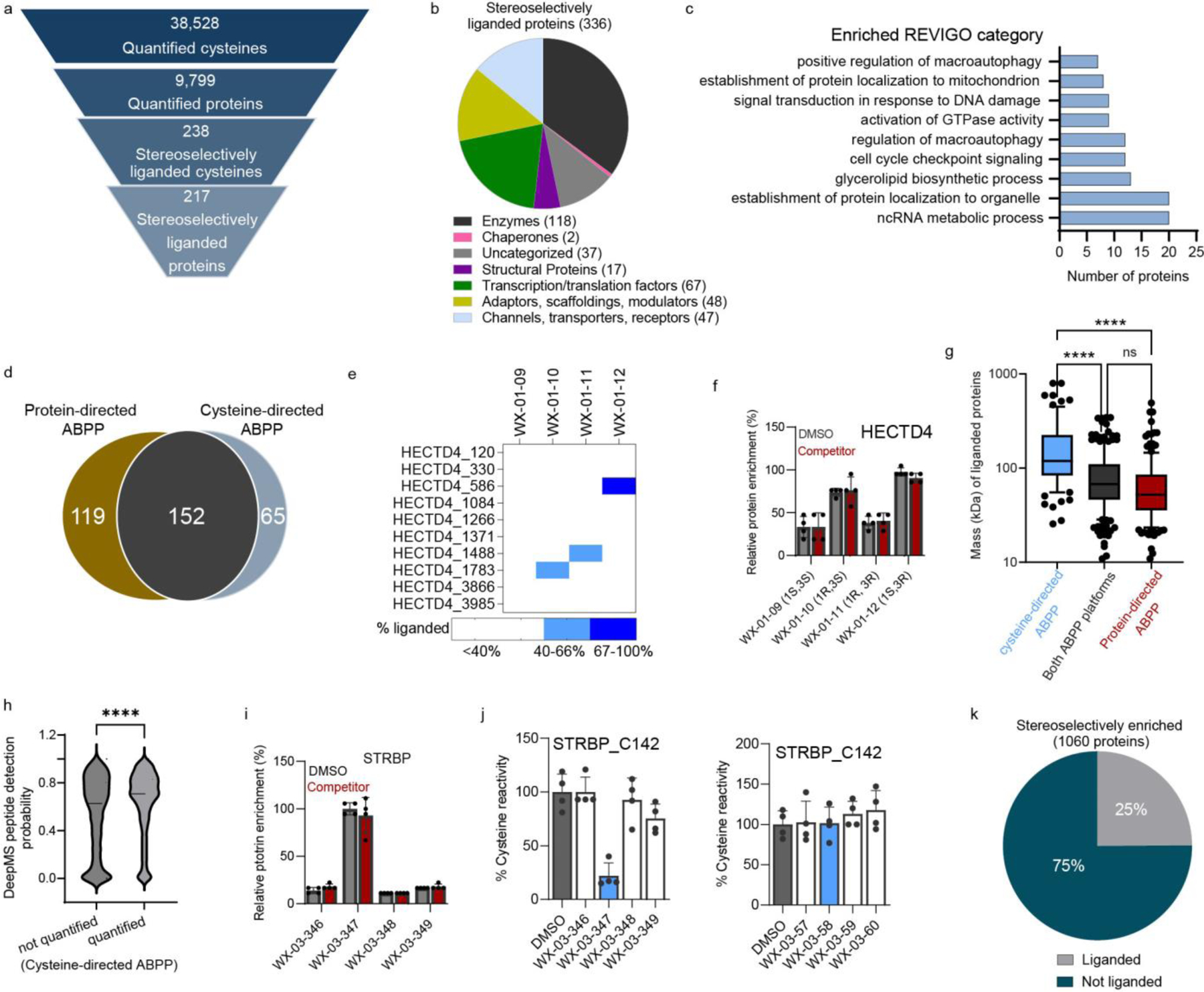
a, Total numbers and stereoselectively liganded fractions of proteins/cysteines quantified by cysteine-directed ABPP in Ramos and 22Rv1 cells. b, Functional class distribution of stereoselectively liganded proteins assigned by Gene Ontology (Panther) and KEGG Brite databases. c, GO-term enrichment analysis for stereoselectively liganded proteins. d, Venn diagram showing fraction of stereoselectively liganded proteins mapped by cysteine- and/or protein-directed ABPP. e, Heatmap showing multiple cysteines (C586, C1488, C1783) in HECTD4 displaying distinct stereoselective liganding profiles in cysteine-directed ABPP experiments. f, Bar graph showing lack of enantioselective enrichment of HECTD4 in protein-directed ABPP experiments. g, Box and Whisker plot showing the relationship between protein size and likelihood of being assigned as liganded in cysteine- or protein-directed ABPP experiments. (Ordinary one-way ANOVA with Tukey’s multiple comparison, ***adjusted p value < 0.0001). Cysteine-directed ABPP: minimum 25.57, maximum 796.4, median 119.0, 25% percentile 83.41, 75% percentile 224.5, bounds 10%-90% percentile (55.23–448.7); Both ABPP platforms: minimum 10.89, maximum 343.5, median 67.50, 25% percentile 46.34, 75% percentile 109.9, bounds 10%-90% percentile (28.46–192.2); protein-directed ABPP: minimum 10.89, maximum 491.9, median 52.02, 25% percentile 35.59, 75% percentile 85.15, bounds 10%-90% percentile (23.37–145.8). h, Violin plot showing MS detection probability (DeepMS score) for cysteine-containing tryptic peptides from stereoselectively liganded proteins that were quantified (right) or not quantified (left) in cysteine-directed ABPP experiments (unpaired two-tailed t-test, **** p=0.0001). i, Protein-directed ABPP data showing stereoselective enrichment of STRBP by WX-03-347 without blockade of this enrichment by WX-03-58. j, Cysteine-directed ABPP data showing stereoselective liganding of STRBP_C142 by WX-03-347 (left), but not WX-03-58 (right). For f, i, j data represent average values ± SD of four independent experiments. k, Pie chart displaying fraction of stereoselectively enriched proteins in protein-directed ABPP experiments that showed (gray; liganded) or did not show (teal; not liganded) blockade of enrichment by non-alkyne competitor stereoprobes or engagement by alkyne and/or competitor stereoprobes in cysteine-directed ABPP experiments. For g and h, data represent all the proteomic datasets generated in this study (n=4 biological replicates for each stereoprobe per ABPP experiment, per cell line).
Integration of cysteine- and protein-directed ABPP data generated a total of 336 stereoselectively liganded proteins from a wide range of functional protein classes, including enzymes, channels/transporters, transcription/translation factors, and adaptor/scaffolding proteins (Fig. 3b and Supplementary Dataset 1). About 40% (134) of these liganded proteins show essentiality (common or strongly selective) in the Cancer Dependency Map51 (Extended Data Fig. 4c). GO pathway analysis further revealed an enrichment of stereoselectively liganded proteins in processes involving RNA metabolism, autophagy, and cell cycle (Fig. 3c).
Nearly half of the stereoselectively liganded proteins (152 total) were identified by both ABPP platforms, with the remaining proteins distributed between those identified exclusively by cysteine- (65 total) or protein- (119 total) directed ABPP (Fig. 3d). In contemplating why some proteins might be identified as stereoselectively liganded by cysteine-, but not protein-directed ABPP, we noted examples of large proteins, like HECTD4 or PRKDC, that harbored multiple cysteines each liganded by a stereoprobe of distinct stereochemistry (Fig. 3e and Extended Data Fig. 4d, e). While such site-specific differences in stereoselective liganding can be resolved by cysteine-directed ABPP, they would be expected to confound stereoselective enrichment profiles in protein-directed ABPP experiments, and HECTD4 and PRKDC accordingly did not show enantioselective enrichment by protein-directed ABPP (Fig. 3f and Extended Data Fig. 4d, e). Indeed, we observed an overall correlation between the size of proteins harboring stereoselectively liganded cysteines and their likelihood of being assigned as ligandable exclusively by cysteine-directed ABPP (Fig. 3g). While these results indicate that larger proteins may prove generally challenging to interpret by protein-directed ABPP, we did find examples of proteins >2000 amino acids in length that were assigned as stereoselectively liganded by this method (e.g., PIKFYVE; Extended Data Fig. 4f).
When considering reasons why stereoprobe liganding events might be observed by protein-, but not cysteine-directed ABPP, we surmised that the proteotypicity of tryptic peptides containing liganded cysteines could be a contributory factor. Proteotypicity refers to the likelihood that a given peptide will be identified in MS-based proteomics experiments and can be affected by physicochemical properties such as tryptic peptide length, hydrophobicity, and isoelectric point42. Consistent with this hypothesis, we found that tryptic peptides quantified in cysteine-directed ABPP experiments had higher predicted proteotypicity (DeepMS Peptide probability score for detection52) compared to tryptic peptides that were not quantified (Fig. 3h). Protein-directed ABPP, on the other hand, can better overcome challenges with peptide proteotypicity by quantifying multiple tryptic peptides per covalently liganded protein. We additionally found that predicted proteotypic peptides not quantified in our cysteine-directed ABPP experiments were enriched in peptides showing greater reactivity with the IA-DTB probe in denatured proteomes (Extended Data Fig. 4g), suggesting that some proteotypic peptides may fail to be quantified in cysteine-directed ABPP experiments performed in native proteomes because they contain buried cysteines that are inaccessible to chemical probes53.
We also observed another category of stereoprobe liganding events that was exclusively identified by protein-directed ABPP due to interactions dependent on the alkyne modification itself. In these cases, as exemplified by STRBP (Fig. 3i, j) and FXR1 (Extended Data Fig. 4h, i), stereoselective enrichment was observed without competition in protein-directed ABPP experiments (Fig. 3i and Extended Data Fig. 4h), while cysteine-directed ABPP experiments revealed stereoselective liganding of a cysteine by the alkyne stereoprobe, but not its non-alkyne counterpart (Fig. 3j, Extended Data Fig. 4i, and Supplementary Dataset 1). Finally, integration of protein- and cysteine-directed ABPP data facilitated interpretation of other categories of stereoprobe-protein interactions, including: i) proteins that were stereoselectively enriched, but not competed, and for which liganding of a cysteine by the alkyne stereoprobe was not observed (Fig. 3k and Supplementary Dataset 1); and 2) proteins that were uniformly enriched and competed by all stereoisomers of one or more stereoprobe sets (73 total proteins in this category; Extended Data Fig. 4j and Supplementary Dataset 1). We interpret the first category as mostly reflecting low-stoichiometry, but nonetheless stereoselective stereoprobe-protein interactions, and the second category as a less frequent group of liganded cysteines residing in pockets that non-stereoselectively bind each of the stereoprobes.
Characterization of stereoprobe targets
We next aimed to verify and characterize a representative set of stereoprobe-protein interactions. We initially focused on confirming covalent liganding events mapped by both cysteine- and protein-directed ABPP. We selected liganded proteins with diverse structural and functional annotations, including a pleckstrin homology (PH) domain-containing protein (PLEK), an iron-sulfur cluster scaffolding protein (NFU1), integral membrane proteins (TMX1, TMX4), and various classes of enzymes (TYMS (metabolic enzyme); LIMK1 (kinase); STAMBP (deubiquitinase)). Each protein and the corresponding mutant(s) where the liganded cysteine was converted to alanine were expressed as Flag epitope-tagged proteins by transient transfection in HEK293T cells. The transiently transfected cells were subsequently treated for 1 h with alkyne stereoprobes (5 μM, except where noted), and analyzed by gel-ABPP. We also performed competitive gel-ABPP experiments, where transfected cells were pretreated with non-alkyne competitor stereoprobes (20 μM, 1 h, except where noted) prior to exposure to alkyne stereoprobes. For each recombinantly expressed protein, we confirmed stereoselective and site-specific engagement by the preferred alkyne stereoprobe, as well as competition of this engagement by the preferred competitor stereoprobe (Extended Data Fig. 5–7 and Supplementary Discussion).
We next investigated a representative set of stereoprobe-protein interactions exclusively mapped by protein-directed ABPP. These interactions presented an additional technical challenge, as we needed to experimentally deduce the cysteines liganded by the stereoprobes. The expectation that stereoselectively liganded cysteines should be absent in the tryptic peptide maps of proteins enriched by stereoprobes assisted in elucidating some stereoprobe-cysteine interactions. For instance, the poorly characterized protein C15orf57 (or CCDC32) was stereoselectively enriched and competed by (1S, 3R) stereoprobes (Fig. 4a), and only one of the four cysteines in this protein (C111) was absent from the tryptic peptide maps from protein-directed ABPP experiments (Extended Data Fig. 8a). We confirmed stereoselective labeling of recombinant WT-C15orf57, but not the C111A mutant, by the (1S, 3R) alkyne WX-01-12 (Fig. 4b), as well as stereoselective blockade of this labeling by pre-treatment with the non-alkyne competitor WX-02-46 (Extended Data. Fig. 8b).
Figure 4. Characterization of stereoprobe-protein interactions.
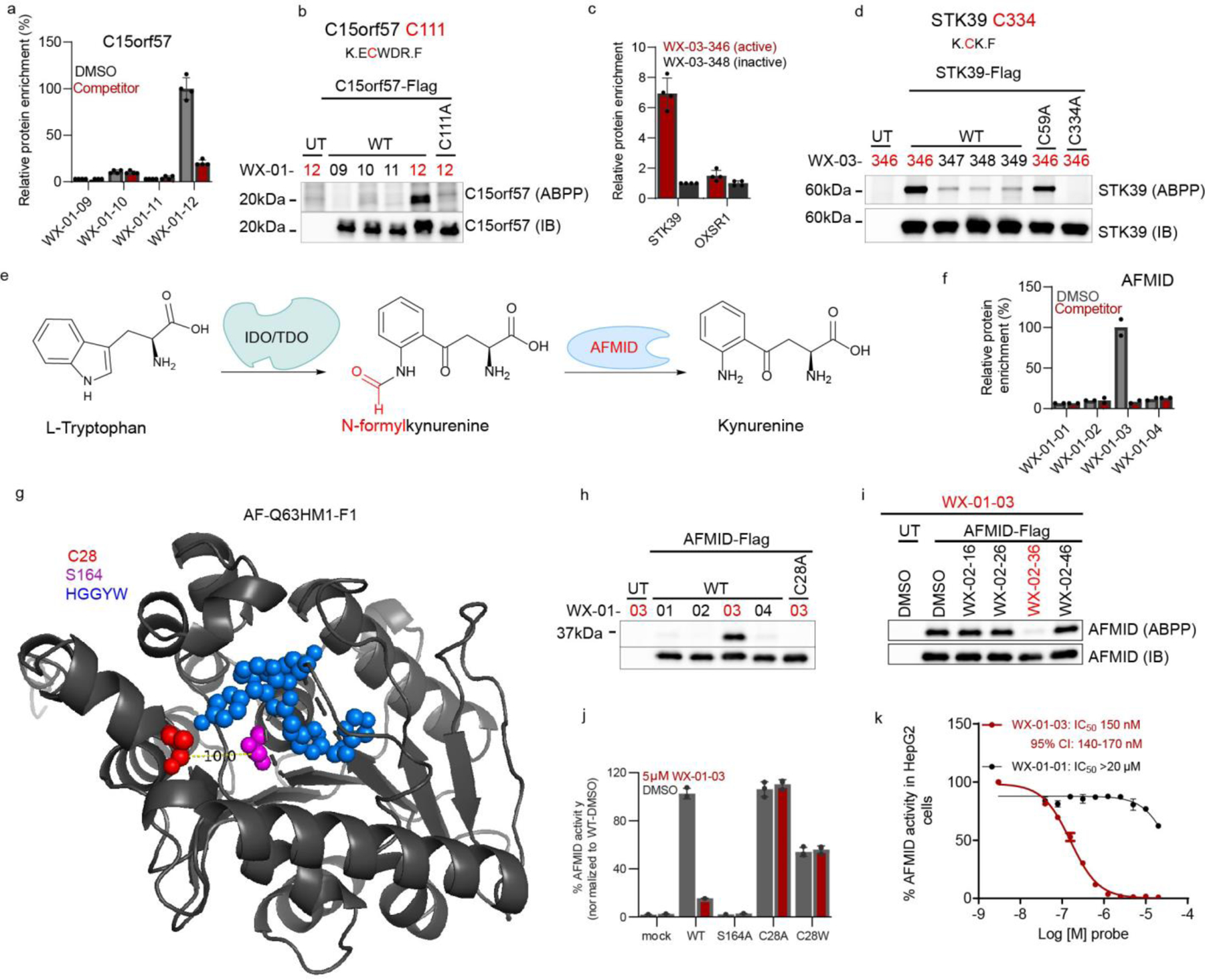
a, Protein-directed ABPP data showing stereoselective enrichment of C15orf57 by WX-01-12 and blockade of this enrichment by WX-02-46 in Ramos cells. b, Gel-ABPP data demonstrating stereoselective engagement of recombinant WT-C15orf57, but not C111A-C15orf57 by WX-01-12 (5 μM, 1 h). Data represent in situ stereoprobe treatment of HEK293T cells transiently expressing Flag-tagged C15orf57 proteins. ABPP signals were measured by CuAAC conjugation to rhodamine-azide tag followed by SDS-PAGE and in-gel fluorescence scanning. Shown above the gel is the C111-containing tryptic peptide. c, Protein-directed ABPP data showing enantioselective enrichment of STK39, but not paralog OXSR1, by WX-03-346 in Ramos cells. d, Gel-ABPP data demonstrating engagement of WT- and C59A-STK39, but not C334A-STK39, by WX-03-346 (1 μM, 1 h). Shown above the gel is the C334-containing tryptic peptide. e, Function of AFMID in the tryptophan-kynurenine metabolic pathway. IDO, indoleamine 2, 3-dioxygenase; TDO, tryptophan 2, 3-dioxygenase. f, Protein-directed ABPP data showing stereoselective enrichment of AFMID by WX-01-03 and blockade of this enrichment by WX-02-36 in Ramos cells. Data represent average values from 2 biological replicates. g, AlphaFold model of AFMID showing location of C28 (red), catalytic serine S164 (purple), and the HGGYW oxyanion motif (blue). h, i, Gel-ABPP data demonstrating stereoselective engagement of recombinant WT-AFMID, but not C28A-AFMID by WX-01-03 (5 μM, 1 h) (h) and blockade of this engagement by WX-02-36 (20 μM, 1 h) (i). j, N-formylamidase activity of indicated AFMID proteins and effects of WX-01-03 (5 μM, 1 h pre-treatment). AFMID variants were expressed in HEK293T cells, and lysates incubated with N-formylkynurenine (500 μM, 30 min) and the kynurenine product measured by LC-MS/MS. k, Concentration-dependent and stereoselective inhibition of endogenous AFMID activity in HepG2 cells by WX-01-03 (3 h). AFMID activity measured as described in j (2 h incubation). For d, h, i experiments were performed as described in Fig. 4b, and data are representative of at least two independent experiments; IB = anti-Flag immunoblot, UT = untransfected cells. For j, k, data represent mean values ± SD for three independent biological experiments. For a, c, data represent mean values ± SD for four independent biological experiments.
Integrating the tryptic peptide maps of stereoprobe targets with knowledge of paralog reactivity also proved helpful for identifying liganded cysteines. For instance, the protein kinase STK39, but not its closely related paralog OXSR1 (74% sequence identity), was stereoselectively enriched and competed by a subset of (1S, 3S) stereoprobes (Fig. 4c). Among the cysteines absent from the stereoprobe-enriched tryptic peptide map of STK39, only two residues (C59 and C334) were unique to STK39 compared to OXSR1. Mutagenesis of C334, but not C59, blocked stereoprobe reactivity with STK39 (Fig. 4d). We also confirmed by gel-ABPP that recombinant STK39 reactivity with alkyne WX-03-346 was stereoselectivity blocked by non-alkyne competitor WX-03-57 (Extended Data Fig. 8c). A crystal structure of STK39 indicates that C334 is distal to the ATP-binding site of the kinase located in region that is generally well conserved in OXSR154 (Extended Data Fig. 8d, e), suggesting that compounds engaging this residue may have allosteric, rather than orthosteric potential. Of note, C15orf57_C111 and STK39_C334 are both found on small (non-proteotypic) tryptic peptides (2–5 amino acids in length; Fig. 4b, d), potentially explaining why they were not quantified in cysteine-directed ABPP experiments.
Taken together, our experiments offer a roadmap for identifying liganded cysteines that may elude detection in cysteine-directed ABPP experiments due to, for instance, their presence on non-proteotypic peptides. In the Supplementary Discussion and Extended Data Fig. 8f–l, we also describe another likely rare, but intriguing source of stereoprobe targets that are exclusively mapped by protein-directed ABPP – proteins reacting with tryptoline acrylamides at non-cysteine residues.
Stereoprobes inhibit the kynurenine pathway enzyme AFMID
The stereoprobe-liganded protein arylformamidase (AFMID) is a serine hydrolase that converts N-formylkynurenine to kynurenine (Fig. 4e) and plays an important role in the tryptophan-kynurenine metabolic pathway that is implicated in immunological/inflammatory disorders55,56 and host response to infectious agents57,58. To our knowledge, inhibitors of AFMID are lacking.
Protein-directed ABPP experiments identified AFMID as being stereoselectively liganded by the (1R, 3R) alkyne/competitor stereoprobe pair WX-01-03/WX-01-36 (Fig. 4f). We did not detect a stereoselectively liganded cysteine for AFMID in cysteine-directed ABPP experiments, but an AlphaFold59, 60 structural model suggested that C28 was proximal to the active-site serine (S164) (Fig. 4g), and docking studies supported a productive binding mode for WX-01-03 compared to the inactive enantiomer WX-01-01 in the AFMID active site (Extended Data Fig. 9a). We found that recombinant WT-AFMID, but not the C28A-AFMID mutant stereoselectively reacted with WX-01-03 in HEK293T cells (Fig. 4h), and this interaction was stereoselectively blocked by pre-treatment with WX-02-36 (Fig. 4h).
WX-01-03 also stereoselectively inhibited the hydrolytic conversion of N-formylkynurenine to kynurenine61, 62 catalyzed by recombinant WT-AFMID (Fig. 4j). In contrast, the C28A-AFMID mutant was insensitive to WX-01-03, and a C28W-AFMID mutant showed partial loss in activity that was not further attenuated by WX-01-03 (Fig. 4j and Extended Data Fig. 9b). As anticipated, an S164A AFMID mutant displayed negligible catalytic activity (Fig. 4j). WX-01-03, but not WX-01-01, further blocked the reactivity of WT-AFMID with a serine-hydrolase directed fluorophosphonate activity-based probe63 (Extended Data Fig. 9c).We finally found that WX-01-03 stereoselectively blocked the catalytic activity of endogenous AFMID with an IC50 value of 150 nM as measured in the human hepatocellular carcinoma line HepG2 (Fig. 4k and Extended Data Fig. 9d). The genetic disruption of AFMID similarly eliminated the N-formylkynurenine hydrolysis activity in HepG2 cells (Extended Data Fig. 9e).
We noted that, in many other mammalian organisms, including mouse, C28 in AFMID is replaced by a serine (Extended Data Fig. 9f). Considering the importance of mouse models for studying immune-relevant metabolic pathways, we generated a S26C mutant of mouse AFMID and found that it was stereoselectively engaged (Extended Data Fig. 9g) and inhibited (Extended Data Fig. 9h) by WX-01-03 with an IC50 value of 1.8 μM. In contrast WT mouse AFMID was insensitive to WX-01-03 (Extended Data Fig. 9g, h). These results, taken together, indicate that tryptoline acrylamides act as stereoselective and site-specific inhibitors of human AFMID by engaging a non-catalytic, active site-proximal cysteine, and that mouse AFMID can be engineered by site-directed mutagenesis to display sensitivity to these compounds.
Stereoprobes disrupt MAD2L1BP protein complexes in cells
We next sought to understand if the tryptoline acrylamide stereoprobes could be used to study the function of a non-enzymatic adaptor or scaffolding protein, as such proteins have been historically challenging for chemical probe development. This line of experimental inquiry was additionally important because covalent liganding events on such non-enzymatic proteins discovered by chemical proteomics are often of unclear functional impact (e.g., occurring at cryptic or allosteric pockets). If stereoprobe hits could prove useful for assessing the biological relevance of newly discovered ligandable pockets, this information would help to prioritize functional stereoprobe-protein interactions for future medicinal chemistry optimization.
Cysteine- and protein-directed ABPP identified a stereoselective and site-specific interaction between (1S, 3R) tryptoline acrylamide stereoprobes and C186 of MAD2L1BP (or p31comet) (Figure 5a, b), an adaptor protein that negatively regulates the spindle assembly checkpoint (SAC) complex responsible for ensuring proper chromosome segregation during mitosis64–66. MAD2L1BP executes its functions by binding to the SAC complex member MAD2L1 and converting this protein from an active to an inactive state67. Interestingly, a crystal structure of the MAD2L1BP-MAD2L1 complex indicated that C186 of MAD2L1BP is proximal to the interface for binding MAD2L1, suggesting that covalent ligands targeting this cysteine might affect MAD2L1BP-MAD2L1 interactions.
Figure 5. Stereoprobes block MAD2L1BP interactions with the spindle assembly checkpoint (SAC) complex.
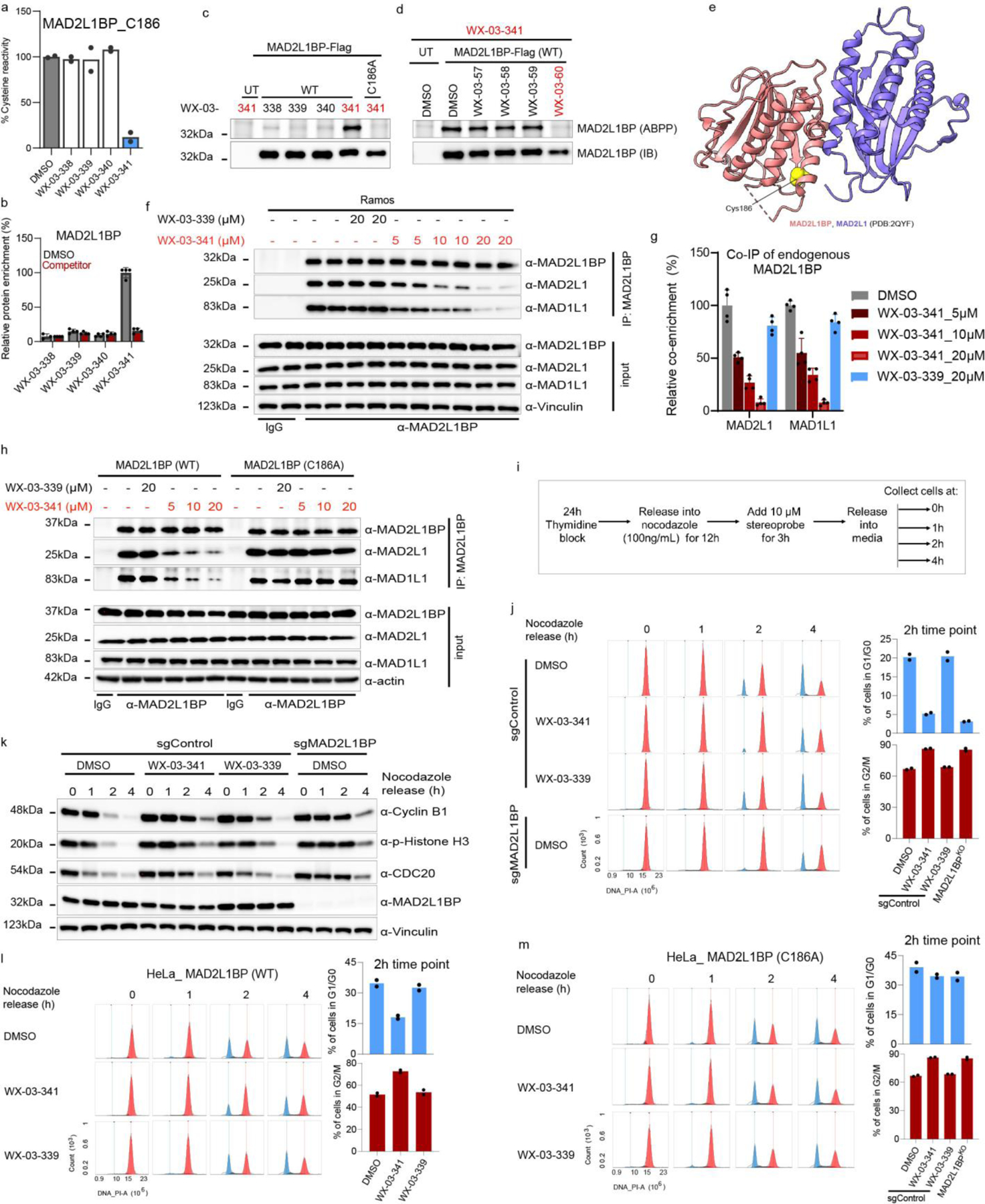
a, b, Cysteine- (a) and protein- (b) directed ABPP data showing stereoselective engagement of MAD2L1BP (C186) in Ramos cells by WX-03-341 (a) and blockade of WX-03-341 enrichment by WX-03-60 (b) in Ramos cells. Data represent mean values of two (a) or mean values ± SD of four (b) biological replicates. c, d, Gel-ABPP data demonstrating stereoselective engagement of recombinant WT-MAD2L1BP, but not the C186A_MAD2L1BP mutant by WX-03-341 (5 μM, 1 h) (c), and blockade of this engagement (d) by WX-03-60 (1 h, 20 μM pre-treatment) in transfected HEK293T cells. e, Crystal structure of MAD2L1BP (pink)-MAD2L1(purple) complex showing location of C186 (yellow). f, Western blots of anti-MAD2L1BP co-immunoprecipitations (Co-IPs) showing stereoselective disruption of MAD2L1BP-MAD2L1/MAD1L1 interactions by WX-03-341 (5–20 μM, 3 h) versus WX-03-339 (20 μM, 3 h) in Ramos cells. g, Quantification of blotting signals from f. MAD2L1 and MAD1L1 signals were normalized to MAD2L1BP and then to DMSO signals. Data represent mean values ± SD of four biological replicates. h, Western blots of anti-MAD2L1BP co-immunoprecipitations (Co-IPs) showing that disruption of MAD2L1BP-MAD2L1/MAD1L1 interactions is C186 dependent. MAD2L1BP-knockout HeLa cells (sgMAD2L1BP cells) stably transduced with WT- or C186A-MAD2L1BP mutant were treated as described in f. i, Workflow for cell cycle synchronization and monitoring of G2/M transition into G0/G1 in HeLa cells. j, Cell cycle histogram (cell count (y-axis)) versus DNA content (propidium iodide (PI) signal) (x-axis)) showing delayed mitotic exit (red) into G1 (blue) in sgMAD2L1BP and WX-03-341-treated sgControl cells compared to DMSO- or WX-03-339-treated sgControl cells (see Extended Data Fig. 10c for flow cytometry gating information). k, Immunoblotting of cells in j for mitotic markers cyclin B1, phosphorylated histone H3 (ser10), and CDC20. l, m, Cell cycle histogram showing that WX-03-341 effects on mitotic exit are preserved in sgMAD2L1BP HeLa cells ectopically expressing WT- (l), but not C186A-MAD2L1BP (m). For c, d, h j, k, l and m, data are representatives of two independent experiments; IB = anti-Flag immunoblot, UT= untransfected cells. For j, l, and m, bar graphs are means of two biological replicates at 2 h.
We first confirmed by gel-ABPP that WX-03-341 stereoselectively reacted with recombinant WT-MAD2L1BP, but not a C126A mutant, expressed in HEK293T cells (Fig.5c), and that the WT-MAD2L1BP-WX-03-341 interaction was stereoselectively blocked by pre-treatment with WX-03-60 (Fig. 5d). We next performed co-immunoprecipitation (co-IP) experiments of endogenous MAD2L1BP from Ramos and K562 cells treated with DMSO, active stereoprobe WX-03-341 (5, 10, or 20 μM, 3 h), and the inactive enantiomer WX-03-339 (20 μM, 3 h). Western blotting of the co-IPs revealed that WX-03-341 blocked the interactions of MAD2L1BP with MAD2L1, as well as with an additional SAC member MAD1L1, in both cell lines, while WX-03-339 had negligible effects (Fig. 5f, g and Extended Data Fig. 10a). We repeated these co-IP experiments in HeLa cells expressing recombinant WT- or C186A-MAD2L1BP against a backdrop of genetic deletion of endogenous MAD2L1BP by CRISPR/Cas9 technology (sgMAD2L1BP HeLa cells; Extended Data Fig. 10b), which revealed that WX-03-341 disrupted WT-MAD2L1BP, but not C186A-MAD2L1BP, interactions with MAD2L1 and MAD1L1 (Fig.5h).
To evaluate the functional effect of stereoprobe disruption of MAD2L1BP interactions with members of the SAC complex, we measured the transition of HeLa cells from mitosis to G1 using the workflow summarized in Fig.5i68. Briefly, HeLa cells were synchronized to G1 phase by blocking thymidine synthase with thymidine for 24 h, then washed and released into nocodazole (an inhibitor of microtubule polymerization and mitotic spindle formation) for 12 h, to activate the SAC. Cells were further treated with DMSO or stereoprobes (10 μM) for 3 h. Mitotic cells (round and detached from the plate) were collected and, after nocodazole removal by washing, released into fresh media, and collected at 0, 1, 2 and 4 h time points. The transition from mitosis (M) into G1 phase of the cell cycle was assessed by measuring DNA content (2N for G1/G0 and 4N for G2/M cells) by flow cytometry of propidium iodide-stained cells. Previous studies have shown that human disease-associated truncation mutants of MAD2L1BP that impair interactions with MAD2L1 result in a delay in the G2/M to G1/G0 transition68, and we observed a similar outcome for sgMAD2L1BP HeLa cells, as reflected by the very limited entry of these cells into the G1/G0 phase at the 2 h time point after nocodazole removal (Fig. 5j). Notably, WX-03-341, but not WX-03-339, also delayed mitotic exit into the G1 phase of the cell cycle to a similar degree as observed in sgMAD2L1BP cells (Fig. 5j). Immunoblotting further revealed the slower degradation of mitosis-specific markers such as cyclin B1, phosphorylated histone H3, and CDC20 in WX-03-341-treated (and sgMAD2L1BP) cells compared to cells treated with DMSO or the inactive enantiomer WX-03-339 (Fig. 5k). Finally, we found that the stereoselective effects of WX-03-341 on delayed mitotic exit were also site-specific in that they were preserved in sgMAD2L1BP HeLa cells ectopically expressing WT-MAD2LBP (Fig. 5l), but not C186A-MAD2L1BP (Fig. 5m).
In summary, we discovered that (1S, 3R) tryptoline acrylamides stereoselectively and site-specifically engage C186 of MAD2L1BP, resulting in the blockade of the interaction of this protein with members of the SAC complex and delayed cell exit from mitosis.
Discussion
By generating global portraits of small molecule-protein interactions in native biological systems, ABPP has enriched our understanding of covalent chemistry as a means to expand the druggable proteome. An ideal ABPP platform would, in a single experiment, identify each amino acid residue on each protein that reacts with an electrophilic compound in cells and quantify the extent of these reactions (i.e., the stoichiometry of residue modification). However, technical limitations in MS-based proteomics prevent the full realization of this goal, and the number of covalent protein binding events that may be overlooked by current ABPP protocols remains an open and important question. Here, we show that a multi-tiered chemical proteomic approach integrating residue (cysteine)- and protein-directed ABPP generates enriched maps of electrophilic small molecule-protein interactions in human cells.
Our results indicate that gaps in the proteomic coverage of cysteine-directed ABPP originate in large part from covalent reactions occurring with cysteines on non-proteotypic peptides. These reactions, especially those that occur with clear SAR (e.g., stereoselectivity) and high stoichiometry, can frequently be identified by protein-directed ABPP. On the other hand, we are less confident about the compatibility of protein-directed ABPP for screening higher-reactivity electrophilic fragments21, 29, where the engagement of multiple cysteines on the same protein may yield complicated SARs. While protein-directed ABPP does not generally identify the residues liganded by electrophilic compounds in each protein, we have provided guidance for how to deduce this information by, for instance, examining the tryptic peptide maps of alkyne probe-enriched proteins for the absence of cysteine-containing peptides (see Supplementary Discussion) and by comparing cysteine conservation in paralogous proteins that either share or do not share ligandability profiles. Then, mutagenesis of candidate cysteines can be used to infer the site of liganding by electrophilic compounds. It is also possible that the cysteines engaged by alkyne stereoprobes may be directly mapped by MS-based proteomics25, 69, 70, at least in cases where potentially complicating factors such as suppression of peptide ionization by the covalently adducted stereoprobe and/or concurrent probe and peptide fragmentation events can be overcome71.
While we do not anticipate many covalent small molecule-protein interactions will be overlooked by integrated cysteine- and protein-directed ABPP, we can imagine rare cases of liganded cysteines on non-proteotypic peptides from very large proteins continuing to present technical challenges. Alternative protease digestion can provide complementary proteotypic peptide maps to those generated by trypsin72 which should increase coverage of cysteine-directed ABPP. We additionally cannot exclude that some cysteines may show low intrinsic reactivity with the IA-DTB probe used herein, and other broad-spectrum, cysteine-reactive probes may accordingly be employed as alternative73–75.
Our work, along with other recent studies22, 76, 77, underscore how stereochemically defined compounds can facilitate the discovery of specific small molecule-protein binding events in chemical proteomic experiments. Stereoselectivity may not only assist in prioritizing high, but also lower stoichiometry stereoprobe-protein interactions for future study. As has been shown in previous studies, stereoselective small molecule-protein interactions, regardless of their fractional occupancy, frequently occur at functional sites22, 31, 34, 77, and low-stoichiometry covalent compounds may accordingly find utility as tools to screen nascent ligandable pockets in proteins for higher affinity small-molecule binders. Finally, we only screened two human cell lines in this study with a single class of stereoprobes, and the evaluation of additional cell types and cell states with more structurally diverse stereoprobe libraries should identify additional covalent liganding events across the proteome, including those occurring on proteins restricted in expression to specific cell lineages or dictated by differential post-translational modifications, biomolecular associations, and/or localization (rather than mere expression) of proteins. Each of these opportunities for expanding the ligandable proteome should benefit from the rich maps of covalent small molecule-protein interactions afforded by integrated cysteine- and protein-directed ABPP.
Conclusion
From a modest-sized library of tryptoline acrylamide stereoprobes, we discovered >300 stereoselectively liganded proteins from diverse structural and functional classes. In each instance, a low-μM (IC50 < 20 μM) chemical tool, and an associated target engagement assay, are now available for initial biological studies. However, we believe it is also important to clarify the types of biology experiments that may be performed with the tryptoline acrylamide-protein interactions reported herein, given their generally limited potency and selectivity. As summarized in the workflow in Figure 6, we posit that the nascent stage of maturation of the tryptoline acrylamides as chemical probes can be counterbalanced, at least in part, by leveraging their stereoselectivity and site-specificity to provide key controls (inactive enantiomers and stereoprobe-resistant cysteine mutant proteins, respectively) for interpreting on-target pharmacological effects in biological studies, as shown previously22, 31, 34, 53 and herein for proteins like AFMID and MAD2L1BP. We further emphasize the value of acute assays that can measure stereoprobe effects on proximal biochemical or cellular readouts of protein function in <24 h, as longer-term experiments, or those that record downstream consequences of protein perturbation, may require compounds of greater potency and selectivity to minimize general side effects associated with electrophilic stress78.
Figure 6. Schematic for mapping and functionally evaluating stereoprobe-protein interactions in cells.
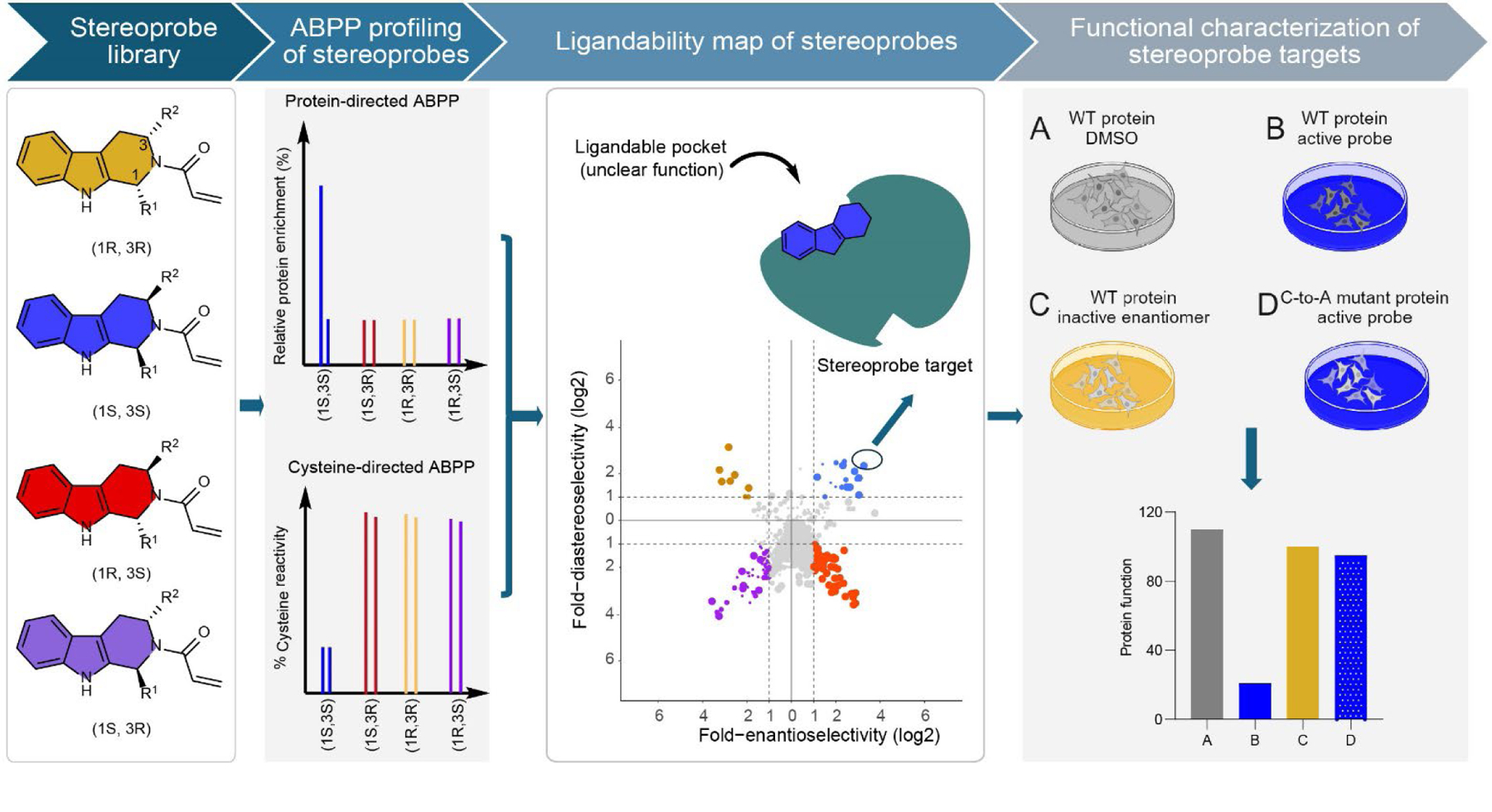
From left to right, tryptoline acrylamide stereoprobes are screened in human cells by both cysteine- and protein-directed ABPP to generate extensive maps of covalent ligandability, from which proteins showing stereoselective and site-specific interactions with the tryptoline acrylamides can be selected for further biological investigation. In these biology experiments, a tryptoline acrylamide is considered to have on-target functional effects if these activities are not observed in control cells treated with the inactive enantiomer or expressing the stereoprobe-resistant cysteine-to-alanine (C-to-A) mutant protein. Overall, this approach allows for the discovery of stereoselective and site-specific covalent ligand-protein interactions in cells and initial evaluation of the functional consequences of these interactions, providing useful information to guide future efforts to develop more advanced chemical probes for newly discovered ligandable pockets. The top right panel was created with BioRender.com.
Even if the initial types of biology experiments performable with tryptoline acrylamide-protein interactions are limited in scope, they can still provide important evidence of functionality for newly discovered ligandable pockets in proteins. Consider, for instance, the (1S, 3R) tryptoline acrylamide interactions with C186 of MAD2L1BP. By leveraging the stereoselectivity and site specificity of stereoprobes, we were able to conduct well-controlled biology experiments that demonstrated these compounds both block MAD2L1BP-MAD2L1/MAD1L1 interactions and delay mitotic exit in cancer cells. Future efforts to improve the potency and selectivity of covalent ligands for MAD2L1BP_C186 can now be pursued with confidence that these compounds bind a functional site on the protein based on the biological studies performed with the initial stereoprobe hits. If some of the stereoprobe interactions described herein are found to lack direct functional effects on proteins, the ligands may serve as starting points for the design of heterobifunctional compounds that mediate the degradation of proteins of interest79. We also call attention to the potential for the stereoprobes to function in high-throughput screening assays of larger compound libraries80, opening up the possibility to identify structurally distinct (and even reversible) chemistries that bind the ligandable pockets initially mapped by chemical proteomics.
Methods
Research ethics and regulations
All experiments were performed in compliance with protocols approved by The Scripps Research Institute Institutional Review Board.
Cell lines and cell culture
All cell lines were obtained from American Type Culture Collection (ATCC). 22Rv1 (ATCC, CRL-2505™), Ramos (ATCC, CRL-1596™), HEK293T (ATCC, CRL-3216™), HeLa (ATTC, CCL-2™), K562 (ATCC, CCL-243™) , and SW480 (ATCC, CCL-228™) cells were grown in RPMI (22Rv1, Ramos, and K562), or DMEM (HEK293T, SW480, HeLa), supplemented with 10% fetal bovine serum (FBS), 2 mM L-alanyl-L-glutamine (GlutaMAX, 22Rv1 only) or 2 mM L-glutamine, penicillin (100 U/mL), and streptomycin (100 μg/mL), in a humidified, 37 °C/5% CO2 tissue culture incubator.
Reagents
Additional reagents, source and catalog numbers are found in Supplementary Table 2 and 3 in the supplementary information.
Gel-ABPP for proteome-wide reactivity
-In situ reactivity:
Ramos (5 mL of 3 million cells/mL) or 22Rv1 (3 mL of 1 million/mL seeded in 6 cm dish overnight) cells were treated with 5 or 20 μM alkyne probes for 1 h. Cells were collected and washed 3x with chilled DPBS. Cell pellets were resuspended in 200 μL of cold DPBS and lysed by sonication (2x15 pulses, 10% power output). Total protein content of whole cell lysates was measured using Pierce™ BCA Protein Assay Kit. Samples were normalized to 1 mg/mL and 50 μL treated with 6 μL of click mix (45 μL of 1.7 mM TBTA in 4:1 t-BuOH:DMSO, 15 μL of 50 mM CuSO4 in H2O, 15 μL of 1.25 mM rhodamine-PEG-Azide in DMSO, 15 μL of freshly prepared 50 mM TCEP in DPBS) for 1 h at RT with vigorous vortexing every 20 min. Click reaction was quenched by the addition of 18 μL of 4x SDS gel loading buffer and samples resolved on 10% SDS-PAGE and imaged by in gel fluorescent scanning using BioRad imager with Image Lab software version 6.1.
-In vitro reactivity:
Cell lysates (50 μL) were treated with 1 μL of 50x alkyne probe for 1 h at RT and analyzed as above, following click reaction.
Glutathione (GSH) reactivity assay
In brief, GSH was diluted to a final concentration of 50 μM in buffer consisting of 0.1M Tris pH 8.8, 30% acetonitrile. In triplicate, 100 μL of the GSH solution was added to a clear 384 well plate (Greiner 781101). Stereoprobes (5 μL of 10mM) were then added to the GSH solution to achieve a final probe concentration of 500 μM and the reaction incubated for 2 h and 6 h timepoints at RT. Ellman’s reagent (5 μL of 100 mM) was then added to the plate and absorbance read at 440 nm. The concentration of GSH remaining was derived from a standard curve and observed rate (kobs/[I]) was calculated assuming pseudo first-order reaction kinetics from the following equations: d[GSH]/dt = −k*[GSH], [GSH]t = [GSH]t0*e−kt.
Protein-directed ABPP
-In situ treatment and sample processing:
Ramos cells (suspension cells:12.5 mL of 3 million cells/mL— seeded 3 h prior to treatment) or 22Rv1 cells (adherent: 20 mL of 1 million/mL in 15 cm dish—seeded 24 h prior to treatment) were treated with DMSO or 20 μM of the non-alkyne competitor stereoprobes (WX-02-16/26/36/46 or WX-03-57/58/59/60) for 2 h. Cells were further treated with 5 μM of stereochemically matched alkyne probes for 1 h. Cells were washed 3x with chilled DPBS, and immediately processed or stored at −80 °C. For non-competitive protein-directed ABPP, cells were treated with 5 or 20 μM of the alkyne stereoprobe only, for 3 h. Cell pellets were resuspended in 500 μL of cold DPBS and lysed by sonication (2x15 pulses, 10% power output). Total protein content of whole cell lysates was measured using Pierce™ BCA Protein Assay Kit. Samples were normalized to 2 mg/mL and 500 μL (1 mg of proteome) treated with 55 μL of click mix (30 μL of 1.7 mM TBTA in 4:1 t-BuOH:DMSO, 10 μL of 50 mM CuSO4 in H2O, 5 μL of 10 mM Biotin-PEG4-azide (BroadPharm, cat# BP-22119) in DMSO, 10 μL of freshly prepared 50 mM TCEP in DPBS) for 1 h at RT with vigorous vortexing every 20 min. Proteins were precipitated out of solution by the addition of chilled methanol (600 μL), chloroform (200 μL) and water (100 μL), followed by vigorous vortexing and centrifugation at 16,000 × g for 10 min, to create a disk. Without disrupting the protein disk, both top and bottom layers were aspirated, and the protein disk resonicated in 500 μL of methanol and centrifuged at 16,000 × g for 10 min. After complete aspiration of the methanol, protein pellets were resuspended in 500 μL of freshly made 8 M urea in DPBS, followed by the addition of 10 μL of 10% SDS and probe sonicated to clarity. Samples were reduced with 25 μL of 200 mM DTT at 65 °C for 15 min, followed by alkylation with 25 μL of 400 mM Iodoacetamide at 37 °C for 30 min. Samples were quenched with 130 μL of 10% SDS, transferred to 15 mL tube and the total volume brought up to 6 mL with DPBS (0.2% final SDS). Washed streptavidin beads (Thermo cat # 20353; 100 μL 50% slurry/sample) was then added and probed labeled protein enriched for 1.5 h at RT with rotation. After incubation, beads were pelleted (2 min x 2000 g) and washed with 0.2% SDS in DPBS (2 x 10 mL), DPBS (1 x 5 mL, then transferred to protein low-bind eppendorf safe-lock tube), HPLC water (2 x 1 mL), and 200 mM EPPS (1x 1 mL), at RT. Enriched proteins were digested on-bead overnight with 200 μL of trypsin mix (2 M urea, 1 mM CaCl2, 10 μg/mL trypsin, 200 mM EPPS, pH 8.0). Beads were spun down, supernatant collected and 100 μL of acetonitrile (30% final) added, followed by 6 μL of 20 mg/mL (in dry acetonitrile) of the corresponding TMT16plex tag (for competitive protein-directed ABPP or TMT10plex for non-competitive protein-directed ABPP) for 1.5 h at RT with vortexing every 30 min. TMT labeling was quenched by the addition of hydroxylamine (6 μL 5% solution in H2O) and incubated for 15 min at RT. Samples were then acidified with 20 μL 100% formic acid, combined and SpeedVac to dryness. Samples were desalted with Sep-Pak column and then high pH fractionated into 10 fractions, using Peptide Desalting Spin Columns (as described below).
-In vitro treatment and sample processing:
For in vitro treatment, 500 μL (1 mg) of proteome were treated with 5 μL of 100x probe for 1 h at RT and processed for MS-analysis as described for in situ treatment above.
-Data processing:
Enrichment ratios (probe vs probe) were calculated for each peptide-spectra match by dividing each TMT reporter ion intensity by the sum intensity for all the channels. Peptide-spectra matches were then grouped based on protein ID and, excluding peptides with summed reporter ion intensities < 10,000, coefficient of variation of > 0.5, and < 2 distinct peptides. Replicate channels were grouped across each experiment, and average values were computed for each protein. A variability metric was also computed across replicate channels, which equaled the ratio of median absolute deviation to average and was expressed in percentage. A protein was considered enantioselectively liganded if the variability corresponding to the alkyne probe leading to highest enrichment did not exceed 20%, and at least one of the following additional criteria were met: (i) the average enrichment by the alkyne probe was > 3-fold that of its enantiomer and > 2-fold the enrichment observed following treatment with a stereochemically matched non-alkyne competitor; (ii) the average enrichment by the alkyne probe was > 3-fold that of its enantiomer, and at least one site in the protein was deemed liganded only by the alkyne probe (not by non-alkyne competitors) in cysteine-directed ABPP experiments; (iii) the average enrichment by the alkyne probe was > 2-fold that of its enantiomer and > 2-fold the enrichment observed following treatment with a stereochemically matched non-alkyne competitor, and at least one site in the protein was deemed liganded (by any probe) in cysteine-directed ABPP experiments (vide infra). Additionally, for all proteins determined to be stereoselectively liganded by protein-directed ABPP, we confirmed that the preferred alkyne stereoprobe accounted for > 33% of the total enrichment signal across the corresponding set of four alkyne stereoprobes.
Cysteine-directed ABPP
In situ treatment and sample processing:
Cysteine-directed ABPP was carried out as previously reported22, 50 with slight modifications. In summary, Ramos cells (suspension cells: 12.5 mL of 3 million cells/mL—seeded 3 h prior to treatment) or 22Rv1 cells (adherent: 20 mL of 1 million/mL in 15 cm dish—seeded 24 h prior to treatment) were treated with DMSO or 20 μM of the stereoprobes for 3 h. Cells were washed 3x with chilled DPBS, and immediately processed or stored at −80 °C. Cell pellets were resuspended in 500 μL of cold DPBS and lysed by sonication (2x15 pulses, 10% power output). Total protein content of whole cell lysates was measured using Pierce™ BCA Protein Assay Kit. Samples were normalized to 2 mg/mL and 500 μL (1mg of proteome) treated with 5 μL of 10 mM iodoacetamide desthiobiotin (IA-DTB, in DMSO) for 1 h at room temperature with occasional vortexing. Proteins were precipitated out of solution by the addition of chilled HPLC grade methanol (600 μL), chloroform (200 μL) and water (100 μL), followed by vigorous vortexing and centrifugation at 16,000 x g for 10 min, to create a disk. Without disrupting the protein disk, both top and bottom layers were aspirated, and the protein disk washed with 1 mL cold methanol and centrifuged at 16,000 × g for 10 min. The pellets were allowed to air dry (just enough get rid of methanol droplets), and then resuspended in 90 μL of denaturing/reducing buffer (9 M urea, 10 mM DTT, 50 mM triethylammonium bicarbonate (TEAB) pH 8.5). Samples were reduced by heating at 65 °C for 20 min, followed by the addition of 10 μL (500 mM) iodoacetamide for 30 min, at 37 °C to cap free cysteines. Samples were then centrifuged at maximum speed (16,000 × g for 2 min) to pellet any insoluble precipitate and probe sonicated once more to ensure complete resuspension, and then diluted with 300 μL 50 mM TEAB pH 8.5 to reach a final urea concentration of 2 M. Trypsin (4 μL of 0.25 μg/μL in trypsin resuspension buffer with 25 mM CaCl2) was added to each sample and digested at 37°C overnight. Digested samples were then diluted with 300 μL wash buffer (50 mM TEAB pH 8.5, 150 mM NaCl, 0.2% NP-40) containing streptavidin-agarose beads (50 μL of 50% slurry/sample) and were rotated at room temperature for 2 h. Samples were centrifuged (2,000 × g, 2 min), and the entire content transferred to BioSpin columns and washed (3x1 mL wash buffer, 3x1 mL DPBS, 3x1 mL water). Enriched peptides were eluted from beads with 300 μL 50% acetonitrile with 0.1% formic acid and speedVac to dryness. IA-DTB labeled and enriched peptides were resuspended in 100 μL EPPS buffer (200 mM, pH 8.0) with 30% acetonitrile, vortexed, and water bath sonicated. Samples were TMT labeled by the addition of 3 μL of 20 mg/mL (in dry acetonitrile) of corresponding TMT10plex tag, vortexed, and incubated at room temperature for 1.5 h. TMT labeling was quenched with the addition of hydroxylamine (5 μL 5% solution in H2O) and incubated for 15 min at room temperature. Samples were then acidified with 5 μL formic acid, combined and dried using SpeedVac. Samples were desalted with Sep-Pak and then high pH fractionated with HPLC (as described below) into 96-well plate and recombined into12 fractions total.
Cysteine-directed ABPP of denatured proteome
Cysteine-directed ABPP of denatured proteome was performed as previously reported53. Briefly, cell lysates from Ramos or 22Rv1 cells (500 μL of 2 mg/mL) was added to empty 1.5 mL low bind tubes (native sample) and kept on ice. For denatured samples, cell lysates were added to tubes containing 240 mg of urea (8 M final concentration) and boiled at 65°C for 15 min. Both native and denatured samples were equilibrated to RT and then treated with 5 μL of 10 mM IA-DTB at RT for 1 h. Proteins were precipitated out of solution and processed for proteomic analysis as described in the cysteine-directed ABPP section above.
-Data processing:
Cysteine engagement ratios (DMSO vs compound) were calculated for each peptide-spectra match by dividing each TMT reporter ion intensity by the average intensity for the DMSO channels. Peptide-spectra matches were then grouped based on protein ID and residue number (e.g., NFU1 C210), excluding peptides with summed reporter ion intensities for the DMSO channels < 10,000, coefficient of variation for DMSO channels > 0.5. Replicate channels were grouped across each experiment, and average values were computed for each cysteine site. A variability metric was also computed across replicate channels, which equaled the ratio of median absolute deviation to average and was expressed in percentage. A cysteine site was considered enantioselectively liganded if the variability corresponding to the probe leading to highest blockade of iodoacetamide-desthiobiotin (IA-DTB) did not exceed 20%, and at least one of the following additional criteria were met: (i) the average IA-DTB blockade by the probe was > 66.7% and > 2.5-fold that of its enantiomer, and either (a) the same probe led to < 25% IA-DTB blockade of at least one other cysteine in the same protein, or (b) the same cysteine site was deemed liganded in another cysteine-directed ABPP experiment in this study; (ii) the average IA-DTB blockade by a given probe was > 50% and > 1.5-fold that of its enantiomer, and the protein was deemed enantioselectively liganded by protein-directed ABPP in this study (vide supra). Finally, we also required at least one additional unchanging cysteine on the parent protein harboring a stereoselectively liganded cysteine ( except for cases where only one cysteine was quantified and we also observed evidence of ligandability in protein-directed ABPP experiments) to avoid misinterpreting protein expression changes as cysteine liganding events.
Offline fractionation
-High pH spin column fractionation:
High pH fractionation was carried out as previously reported22, 50 using Peptide Desalting Spin Columns (Thermo 89852). Samples (protein-directed ABPP samples) were resuspended in 300 μL of buffer A (5% acetonitrile, 0.1% formic acid) by water bath sonication and bound to the spin columns. Bound peptides were then washed 2x with water, 1x with 5% acetonitrile in 10 mM NH4HCO3, and eluted into 30 fractions with increasing gradient of acetonitrile. Every 10th fraction was combined (e.g., 1, 10, and 30) and SpeedVac to dryness. Each of the resulting 10 fractions were resuspended in buffer A (5% acetonitrile, 0.1% formic acid) and analyzed by mass spectrometry.
-HPLC fractionation:
Samples (cysteine-directed ABPP samples) were resuspended in 500 μL buffer A and fractionated with Agilent HPLC into a 96 deep-well plate containing 20 μL of 20% formic acid to acidify the eluting peptides, as previously reported31 The peptides were eluted onto a capillary column (ZORBAX 300Extend-C18, 3.5 μm) and separated at a flow rate of 0.5 mL/min using the following gradient: 100% buffer A from 0–2 min, 0%–13% buffer B from 2–3 min, 13%–42% buffer B from 3–60 min, 42%–100% buffer B from 60–61 min, 100% buffer B from 61–65 min, 100%–0% buffer B from 65–66 min, 100% buffer A from 66–75 min, 0%–13% buffer B from 75–78 min, 13%–80% buffer B from 78–80 min, 80% buffer B from 80–85 min, 100% buffer A from 86–91 min, 0%–13% buffer B from 91–94 min, 13%–80% buffer B from 94–96 min, 80% buffer B from 96–101 min, and 80%–0% buffer B from 101–102 min (buffer A: 10 mM aqueous NH4HCO3; buffer B: acetonitrile). The plates were evaporated to dryness using SpeedVac and peptides resuspended in 80% acetonitrile, with 0.1% formic acid and combined to a total of 12 fractions (e.g., fraction1= well 1A+ 1B…1H, fraction 2= well 2A+2B….2H) (3x300 μL/column). Samples were SpeedVac to dryness and the resulting 12 fractions were re-suspended in buffer A (5% acetonitrile, 0.1% formic acid) and analyzed by mass spectrometry.
TMT liquid chromatography-mass-spectrometry (LC-MS) analysis
Samples were analyzed by liquid chromatography tandem mass-spectrometry using an Orbitrap Fusion mass spectrometer (Thermo Scientific) coupled to an UltiMate 3000 Series Rapid Separation LC system and autosampler (Thermo Scientific Dionex), as previously reported22, 50, and data acquired with Thermo Scientific Xcalibur software version 2.2. The peptides were eluted onto a capillary column (75 μm inner diameter fused silica, packed with C18 (Waters, Acquity BEH C18, 1.7 μm, 25 cm)) or an EASY-Spray HPLC column (Thermo ES902, ES903) using an Acclaim PepMap 100 (Thermo 164535) loading column, and separated at a flow rate of 0.25 μL/min. Data was acquired using an MS3-based TMT method on Orbitrap Fusion or Orbitrap Eclipse Tribrid Mass Spectrometers. Briefly, the scan sequence began with an MS1 master scan (Orbitrap analysis, resolution 120,000, 400−1700 m/z, RF lens 60%, automatic gain control [AGC] target 2E5, maximum injection time 50 ms, centroid mode) with dynamic exclusion enabled (repeat count 1, duration 15 s). The top ten precursors were then selected for MS2/MS3 analysis. MS2 analysis consisted of: quadrupole isolation (isolation window 0.7) of precursor ion followed by collision-induced dissociation (CID) in the ion trap (AGC 1.8E4, normalized collision energy 35%, maximum injection time 120 ms). Following the acquisition of each MS2 spectrum, synchronous precursor selection (SPS) enabled the selection of up to 10 MS2 fragment ions for MS3 analysis. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap (collision energy 55%, AGC 1.5E5, maximum injection time 120 ms, resolution was 50,000). For MS3 analysis, we used charge state–dependent isolation windows. For charge state z = 2, the MS isolation window was set at 1.2; for z = 3–6, the MS isolation window was set at 0.7. Raw files were uploaded to Integrated Proteomics Pipeline (IP2, version 6.0.2) available at (http://ip2.scripps.edu/ip2/mainMenu.html) and MS2 and MS3 files extracted from the raw files using RAW Converter (version 1.1.0.22, available at http://fields.scripps.edu/rawconv/) and searched using the ProLuCID algorithm using a reverse concatenated, non-redundant variant of the Human UniProt database (release 2016–07). Cysteine residues were searched with a static modification for carboxyamidomethylation (+57.02146 Da). A dynamic modification for IA-DTB labeling (+398.25292 Da) was included with a maximum number of 2 differential modifications per peptide. N-termini and lysine residues were also searched with a static modification corresponding to the TMT tag (+229.1629 Da for 10plex and +304.2071 Da for 16plex). Peptides were required to be at least 6 amino acids long. ProLuCID data was filtered through DTASelect (version 2.0) to achieve a peptide false-positive rate below 1%. The MS3-based peptide quantification was performed with reporter ion mass tolerance set to 20 ppm with Integrated Proteomics Pipeline (IP2).
Lysine-directed ABBP
Lysine-directed ABPP was carried out in the same way as cysteine directed ABPP, with the following modifications: (1) NHS-DTB (30 μM final) was used in place of IA-DTB and (2) a dynamic modification for NHS-DTB labeling (−33.04175 Da) was used in place of IA-DTB labeling (+398.25292 Da) during the search.
Cloning and mutagenesis
All full-length plasmids were obtained from either OriGene in pCMV6 vector with C-terminal Myc-DDK (FLAG) epitope tag or from GenScript in pcDNA3.1-C-(k) DYK (FLAG), as shown in Supplementary Table 3. Mutagenesis was carried out using Q5® Site-Directed Mutagenesis Kit (New England BioLabs, E0554S), using primers shown in Supplementary Table 3.
Gel-ABPP with recombinant proteins
HEK293T cells (3x105) were seeded in 6-well plate overnight and transfected with 1–2 μg of FLAG-epitope tag plasmids (depending on difficulty of expression) using PEI at a ratio of 1:3 (DNA: PEI), for 48 h. Cells were treated with alkyne probe only, for 1 h or with competitor probe for 1 h, followed by alkyne probe for a further 1 h, processed and analyzed by in gel fluorescent scanning as described above.
Cell proliferation assay (pH-dependent compound toxicity)
SW480 cells were cultured in standard DMEM and then switched to NaHCO3-free DMEM supplemented with 22 mM NaHCO3/22 mM NaCl (pH 7.4), 5.5 mM NaHCO3/38.5 mM NaCl (pH 6.9), or 2.75 mM NaHCO3/41.25 mM NaCl (pH 6.6), 10% FBS and pen/strep for 3 days, to acclimatized to the pH conditions. Cells were then seeded in the corresponding pH medium at 5000 cells per well (50 μL of 1x105 cells/mL) in 96-well flat bottom white wall plates. After 24 h, 50 μL of the corresponding pH medium containing DMSO or 2x compound dilutions (from 1000x DMSO stocks) were added to the wells and cultured for 72 h. Plates were brought to RT and 50 μL of CellTiter-Glo® (Promega, G7570) added to each well and vigorously shaken with a microplate orbital shaker for 20 sec and incubated on a gentle rotating platform for 30 min at RT. Luminescence readings were taken with CLARIOstar (BMG Labtech) plate reader version 5.4. After background subtraction, each treatment was normalized to DMSO control at the corresponding pH and graphed with GraphPad PRISM software version 9.5. Data represents mean values ±SD for three independent experiments, each of which was set up in triplicates.
LIMK1 NanoBRET target engagement assay
LIMK1 NanoBRET target engagement assay was carried out in HEK293T cells transiently transfected with WT or C349A LIMK1-NanoLuc® Fusion Vector (Promega, NV3391) using the NanoBRET™ TE Intracellular Kinase Assay kit, following the manufacturer’s protocol (Promega, N2640). Briefly, HEK293T cells were trypsinized and resuspended in assay medium (Opti-MEM without phenol red, 1% FBS) to a density of 2x105 cells/mL. DNA mixtures (10 μg/mL) were prepared in 1 mL of serum free Opti-MEM at a ratio of 9 μg/mL of Transfection Carrier DNA (Promega, E4881) and 1 μg/mL of LIMK1-NanoLuc plasmid. FuGENE HD reagent (30 μL) was then added to the DNA mixture, mixed by inversion, and incubated at RT for 20 min to form lipid-DNA complexes. Lipid–DNA complexes were then mixed with the HEK293T cell suspension at a 1:20 ratio and 100 μL of the final mixture added to white 96-well tissue culture plates (Corning/Falcon, 353377). Transfected cells were incubated in a humidified, 37 °C/5% CO2 tissue culture incubator for 20 h. A 100x solution of K-10 NanoBRET tracer in 100% DMSO was diluted in Tracer Dilution Buffer to generate 20x (10 μM) Complete NanoBRET Tracer Reagent. The Complete NanoBRET Tracer Reagent (5 μL/well of the 20x stock) was added to the transfected cells and mixed by shaking at 900 rpm on an orbital shaker. Test compounds (1000x stock in DMSO) were diluted to 10x final concentration in Opti-MEM reduced serum medium, no phenol red and 10 μL added to the cells containing 1x NanoBRET Tracer Reagent. Plates were thoroughly mixed for 15 sec at 900 rpm and incubated in a humidified, 37 °C/5% CO2 tissue culture incubator for 3 h. Assay plates were brought to RT and 50 μL of 3x complete substrate plus inhibitor solution added to each well and incubated at RT for 2 min. Each condition was set up in triplicate and untransfected cells were used as background control. Donor (450 nm) and acceptor (610 nm) BRET signals were measured using a CLARIOstar microplate reader (BMG Labtech), version 5.4. Background correction was performed by subtracting average BRET ratio in the absence of LIMK1 from BRET ratio of each sample and milliBRET (mBu) ratios calculated as follows: ((Acceptorsample/Donorsample) – (Acceptoruntransfected control/Donor untransfected control)) × 1,000. Results were normalized to DMSO control and graphed using GraphPad PRISM v.9.5.
LIMK1 Nanoluc immunoblot
Following transfection and treatment with stereoprobes (in 6-well plate) as described above in the NanoBRET section, cells were washed 2x with chilled DPBS and lysed with RIPA buffer supplemented with complete protease and phosphatase inhibitor cocktail. After protein quantification and normalization, samples were boiled in SDS loading buffer and immunoblotted with either mouse anti-Nanoluc (Promega, cat# N7000) or anti-actin (Cell signaling technology, cat# 4967), followed by anti-mouse HRP, and visualization with luminol reagent, using BioRad imager with Image Lab software version 6.1.
AFMID enzyme assay
AFMID activity was assayed in HepG2 cells (endogenous AFMID) or HEK293T cells over expressing AFMID (recombinant AFMID). For in situ treatment, HepG2 cells (1x106) were seeded in 6-well plates overnight and treated with compounds for 3 h. Cell pellets were collected, washed with DPBS, and lysed by sonication (3x 8pulses, 10% output) in assay buffer (50 mM Na2HPO4, 1 mM EDTA). Samples were spun at 16,000 x g for 10 min, and supernatant collected for enzyme assay. The concentration of soluble proteome was measured by Pierce™ BCA Protein Assay Kit and samples normalized to 1 mg/mL for endogenous and 0.025 mg/mL for recombinant AFMID. AFMID activity was initiated by the addition of 1 μL of 25 mM N-formylkynurenine (Millipore Sigma, cat# PHR8908) in DMSO (500 μM final)62 to 50 μL of the lysate in Eppendorf tubes (LC-MS) or in half-area clear bottom 96-well plate (absorbance measurement) and the reaction allowed to proceed for 30 min (recombinant AFMID) or for 2 h (endogenous AFMID) at RT. For LC-MS analysis, reaction was quenched with 150 μL methanol containing internal standard (5 μg/mL L-Kynurenine D6: Cambridge Isotope Laboratories Inc, cat# DLM-7842). Samples were vigorously vortexed, chilled at −80°C for 30 min, and centrifuged at 16,000 × g for 10 min at 4 °C to pellet precipitated proteins. Supernatant (100 μL) was transferred to an LC-MS/MS vial for metabolomic analysis. For in vitro compound treatment, 50 μL of 1 mg/mL (HepG2 soluble lysate) or 0.025 mg/mL of HEK29T cells overexpressing AFMID (WT or mutants) were treated with DMSO or compound for 1h prior to enzyme assay (2 h for endogenous AFMID and 30 min for recombinant AFMID). For Absorbance measurement, absorbance readings were taken at 365nm on a CLARIOstar microplate reader, version 5.4.
LC-MS/MS measurement of kynurenine
Metabolomic profiling of kynurenine was achieved in positive mode by LC/MS–based multiple reaction monitoring (MRM) methods with Agilent Technologies 6460 Triple Quad coupled to Agilent 1290 Infinity LC HPLC. Samples were injected onto an Agilent Eclipse XDB-C18 (4.6mm x 150mm x5 μm) reverse-phase analytical column with a flow rate of 0.25 mL/min. The column was eluted isocratically with 5% mobile phase A (10 mM ammonium formate with 0.1% formic acid in water) for 0.5 min followed by a linear gradient to 40% mobile phase B (methanol with 0.1% formic acid) over 20 min. The following ESI parameters were used for MS analysis: drying gas temperature, 350°C; drying gas flow, 9 L/min; nebulizer pressure, 45 Ψ; sheath gas temperature, 375°C; sheath gas flow, 12 l/min; fragmentor voltage, 100 V; and capillary voltage, 3.5 kV. The MRM transitions m/z for the targeted analytes were 209.1→146 for kynurenine, 215.6→ 152 for Kynurenine D6 (internal standard) and 237→136 for N-formylkynurenine as reported by others61, 62. MRM data was acquired with Agilent MassHunter Workstation LC/MS Acquisition Console version 07.02, and data analyzed with Agilent MassHunter Quantitative Analysis software version 10.0.707.0. For each treatment condition, the kynurenine signal was divided by that of the internal standard and then expressed as a percentage of the DMSO control.
Generation of MAD2L1BP and AFMID CRISPR/Cas9 knockout cells
Stable knock-out cells were generated by transduction of cells with LentiCRISPR v2-Blast carrying sgControl, sgAFMID or sgMAD2L1BP. Briefly, sgRNAs (sgControl-01_sense5’- CACCGGAACCTACGGGCTACGATACG; sgControl-02_sense5’- CACCGACGGAGGCTAAGCGTCCACCGGGCCGACTGGACCAAGCATG) CACCGGACAATCATGGTGAAAGCGG; sgMAD2L1BP-01_sense5’- CACCGAAGAGACTGCATGGTACCAG; sgMAD2L1BP-02_ sense5’_CACCGACTTGAGACAAGCTCTACGC; sgMAD2L1BP-03_ sense5’- CACCGCAGGAAATGCCAACAAGCCC; sgAFMID01_sense5’- CACCGGCCACTCCCTGTGCCGTCAG; sgAFMID02_sense5’- CACCGAGTCGGACAACCCATCGGCT; and sgAFMID03_sense5’- CACCGGGCCGACTGGACCAAGCATG ) were annealed and cloned into LentiCRISPR v2 Blast using BsmBI-v2 (NEB Golden Gate Assembly Kit, NEB #E1602). To generate lentivirus, 2 million Lenti-X 293T cells (Takara) were seeded in 10 cm dishes overnight, in 10 mL of antibiotic free DMEM. CRISPR v2-Blast-sgRNA, lentiviral packaging vector (pCMV-dR8.91) and envelope vector (VSV-G) were mixed at 6:6:1 ratio in OPTI-MEM media, and 3 μg of PEI (1mg/mL, Polysciences) added per μg of total plasmids. The DNA: PEI complex was incubated at RT for 15 min and the complex added dropwise to the Lenti-X 293T cells. Media was replaced with fresh DMEM (Corning) with 30% FBS plus pen-strep and 2mM Glutamine, 24h post transfection. The virus was collected at 48h post-transfection and filtered through a 0.45 μm syringe filter (Millipore) to eliminate floating cells. For transduction, 1 million HeLa or HepG2 cells were mixed with 500 μL of viral supernatant in 3 mL supplemented with 8 μg/mL polybrene in 6-well plates. Cells were spin-infected at 930xg and 30 °C for 1 h and incubated for 24 h at 37 °C. Selection was initiated with 10 μg/mL blasticidin for one-week. Selected pools were characterized by immunoblots (MAD2L1BP) or enzyme assay (AFMID) and used for knock-out cell line experiments.
Generation of MAD2L1BP stable cells
Full length MAD2L1BP (WT and C186) carrying C-terminal Myc and Flag tags with silent mutation in the PAM NGG sequence (Q40Q) were cloned into pLEX307 (Adgene plasmid# 41392) vector by Gateway cloning. Viral supernatants were generated as described above and MAD2L1BP CRISPR KO cells (sgMAD2L1BP_01) transduced and selected with 10 μg/mL of puromycin for one-week. Selected stable pools were evaluated by immunoblot with anti-MAD2L1BP antibody (1:500dilution).
Co-immunoprecipitation of MAD2L1BP
Ramos, K562 cells (40 mL of 3 million cells/mL, per condition) or HeLa cells stably expressing MAD2L1BP-Flag-Myc (4million cells in 15cm dish overnight) were treated with DMSO or stereoprobes (5, 10 and 20 μM) for 3h. Cells were collected and washed 2x with cold DPBP. Pellets were either analyzed immediately or frozen at −80°C. Ice-thawed pellets were resuspended in 1 mL of IP lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 0.5% NP-40) supplemented with EDTA-free complete protease inhibitor and phosphoSTOP. Lysing was achieved by rotating the samples at 4°C for 1h. Lysate was clarified by spinning at 16,000xg for 10 min and supernatant assayed for total protein using BCA reagent. Lysate (4mg for endogenous protein and 1.5mg for recombinant protein) were mixed with 5 μg of rabbit polyclonal MAD2L1BP antibody (Proteintech, cat# 15344-1-AP) and incubated at 4°C overnight with rotation. Control samples were enriched with an equal amount of normal rabbit IgG (Cell signaling cat# 2729). To each sample was added 50 μL of prewashed protein A magnetic bead (ThermoFisher Scientific, cat# 88846) for a further 3 h at 4°C with rotation. Samples were washed 3x with IP wash buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 0.2% NP-40), 1x with DPBS and 1x with 50 mM EPPS, pH 8.0. Enriched proteins were eluted off the bead by boiling with 60 μL of 8M urea at 65°C for 10 min and supernatant collected with a magnetic stand into new tubes. Beads were rinsed with 60 μL of 200 mM EPPS, pH 8.0 and combined. Part of the eluate (20 μL) was boiled with equal volume of 1x SDS loading buffer for immunoblot and the remaining 100 μL reduced with 5 μL of 200 mM DTT for 15 min at 65°C and alkylated with 5 μL of 400 mM iodoacetamide for 30 min at 37°C. Samples were further diluted with 100 μL of 200 mM EPPS, pH 8.0 containing 0.02 μg/μL trypsin, 4 mM CaCl2 (For a final concentration of 2 mM urea, 2μg trypsin, and 2 mM CaCl2) and digested overnight at 37°C. Acetonitrile (100 μL) was added to each sample and TMT labeled as described in the protein-directed ABPP section. Combined samples were offline fractionated and combined into three fractions. Further processing and analysis by mass spectrometry followed the protein-directed ABPP protocol described above. Input lysate or immunoprecipitated samples were resolved on 4–20% SDS-PAGE, blotted onto nitrocellulose membrane, blocked with 5% milk in TBST for 1h at RT, immunoprobed with anti-MAD2L1BP (1:500 dilution), anti-MAD1L1 (1:1000 dilution), anti-MAD2L1 (1:1000 dilution), anti-actin HRP (1: 4000 dilution) or anti-vinculin HRP (1:1000 dilution) overnight at 4°C. Membranes were washed 5x5minutes with TBST and incubated in the corresponding anti-mouse or or anti-Rabbit HRP conjugated secondary antibody for 1h at RT and washed 5x5minutes. Membranes were developed with ECL western blot substrate (Thermofisher Scientific, cat# PI32106) and images taken with BioRad Image Lab version 6.1.
Flow cytometry and immunoblot measurement of cell cycle progression.
HeLa (2million cells in 15cm dish; 3 plates per condition to get enough cells for flow and western blot) were seeded in DMEM overnight. Cells were synchronized to G1/Go phase with 2 mM thymidine for 24h. Thymidine synchronized cells were washed 2x with warm DPBS and released into fresh media containing 100 ng/mL of nocodazole for 12 h. DMSO or stereoprobes were added to the cells and cultured for a further 3h. Mitotic round off cells were collected into tubes and washed 3x with warm DPBS and aliquot taken out for T0 time point. The remaining cells were resuspended in DMEM, cultured and aliquoted into individual plate for 1, 2 and 4 h. At each time point, cells were collected by trypsinization (both suspension and adherent cells were collected), washed, and resuspended in 500 μL of cold DPBS and 100 μL taken out and fixed with 900 μL of 80% ice cold ethanol on ice for 1h or overnight at −20°C. The remaining 400 μL were spun down and pellets saved at −80°C for immunoblot of mitotic markers: cyclin B1 (1:1000), phosphorylated histone (ser10) H3 (1:2000) and CDC20 (1:1000), as described above, following whole cell lysate extraction with RIPA buffer containing protease and phosphatase inhibitors. For flow cytometry, fixed cells were spun at 1000xg for 10 min and cell pellets transferred into round bottom 96 well plate with 200 μL of 1x permeabilization buffer (ThermoFisher Scientific, cat # 00-8333-56) in DPBS. Cells were washed two more times with 1x permeabilization buffer by spinning at 300xg for 3min, flipping to decant the supernatant and resuspending the pellet in fresh buffer. Cells were resuspended in 65 μL of PBS containing 2x RNase A (100 μg/mL) for 5min, followed by the addition of 65 μL of 2X FACS buffer (1% FBS, 2 mM EDTA) with propidium iodide for a final concentration of 50 μg/mL and incubated at RT for 30 min. Data were acquired on ACEA NovoCyte using B615 channel (PE/Texas Red) at a slow flow rate of 14 μL/min. Instrument was set to analyze 100 μL or 15,000 cells in the final gate (Extended Data Fig. 10c). Cell cycle results were fitted with NovoExpress software using Dean-Jett-Fox (DJF) model.
For immunoblot of mitotic markers, cells were lysed by sonication in IP lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 0.5% NP-40) supplemented with EDTA-free complete protease inhibitor and phosphoSTOP. Clarified lysate were boiled with SDS loading buffer, resolved by SDS-PAGE and immunoblotted with the respective antibody.
Statistics and Reproducibility
Statistics were only performed on experiments with a minimum of three biological replicates. All other experiments with representative data shown were replicated at least twice with similar results. Graphing and statistics were performed with GraphPad Prism 9.5.0. Specific test used and p-values are included in the associated figure legends.
Extended Data
Extended Data Fig. 1. Gel-ABPP of alkyne stereoprobes in human cancer cells.

a, In situ reactivity of alkyne stereoprobes (5 μM, 1 h) in Ramos cells as determined by gel-ABPP. b, In vitro reactivity of probe set 1 (5 or 20 μM, 1 h) in Ramos and 22Rv1 cell lysate. Red asterisks mark examples of stereoselective stereoprobe-protein interactions. a, b, data are from a single experiment representative of at least two independent experiments.
Extended Data Fig. 2. Protein-directed ABPP platform for mapping stereoselectively liganded proteins in human cells.
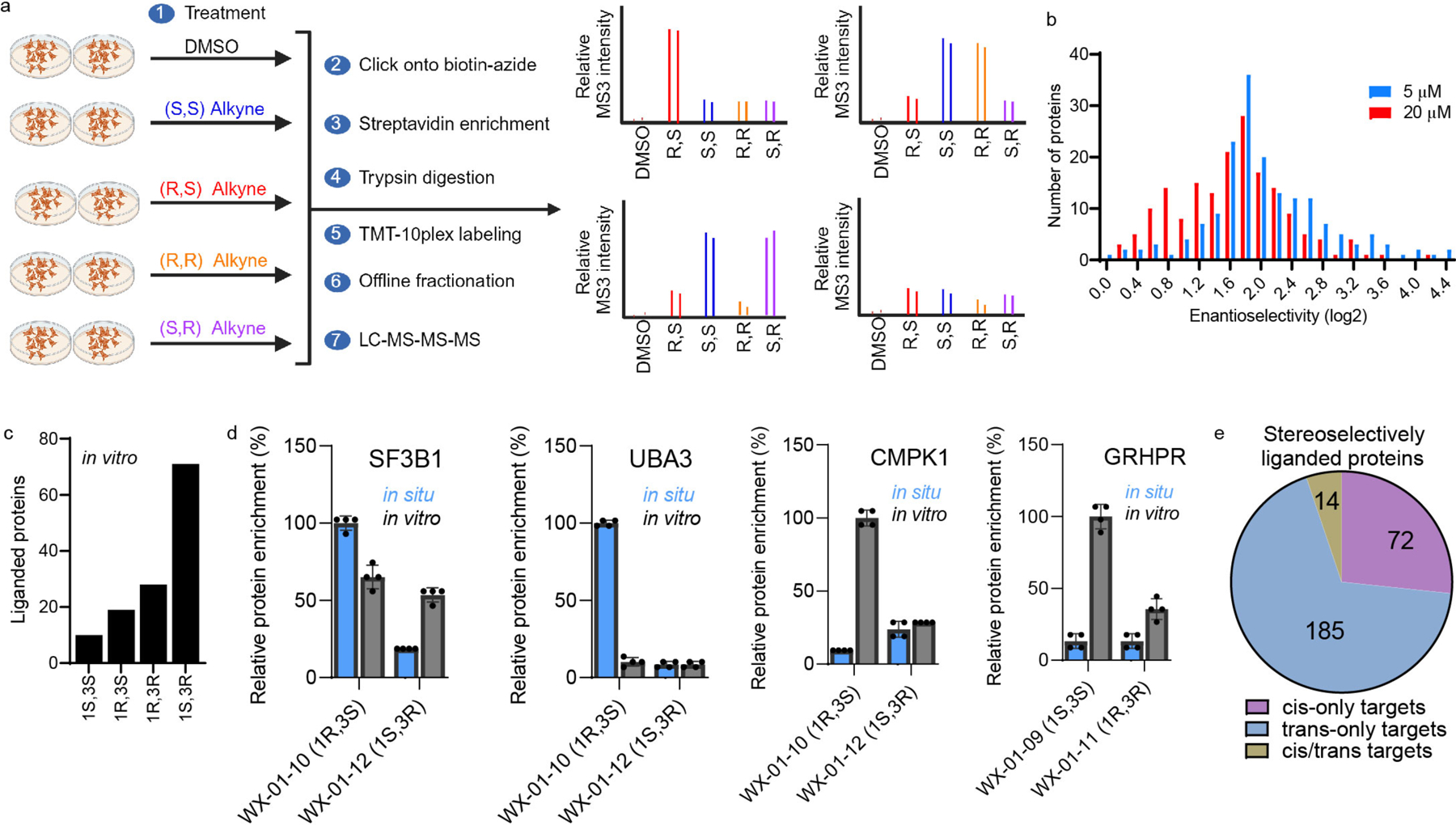
a, Workflow for protein-directed ABPP experiments where the stereoselective enrichment of proteins by alkyne stereoprobes is determined by multiplexed (tandem mass tagging, TMT10plex) MS-based proteomics. Image was created with BioRender.com. b, Comparison of enantioselective enrichment values in protein-directed ABPP experiments in Ramos cells treated with alkyne stereoprobe set 2 (WX-01-05/06/07/08) at 5 vs 20 μM (3 h). Proteins shown are: 1) designated as stereoselective targets in either the 5 or 20 μM data sets (> 3-fold enantioselective enrichment); and 2) quantified in both the 5 and 20 μM data sets. c, Number of proteins stereoselectively liganded by each stereoconfiguration of stereoprobe set 3 (WX-01-09/10/11/12; 10 μM, 1 h) in vitro. d, Examples of proteins showing preferential stereoselective enrichment by stereoprobe set 3 in situ (SF3B1, UBA3) or in vitro (CMPK1, GRHPR). In situ conditions: 5 μM stereoprobe, 1 h; in vitro conditions: 10 μM stereorpobe, 1 h. Data represent mean values ± SD for four independent experiments (n=4). e, Pie chart showing fraction of proteins that were enantioselectively liganded by cis (1R, 3R and 1S, 3S) stereoprobes (cis-only), trans (1R, 3S and 1S, 3R) stereoprobes (trans-only), or both cis and trans stereoprobes (cis/trans).
Extended Data Fig. 3. Stereoprobe-protein interaction maps from protein-directed ABPP experiments in human cancer cells.
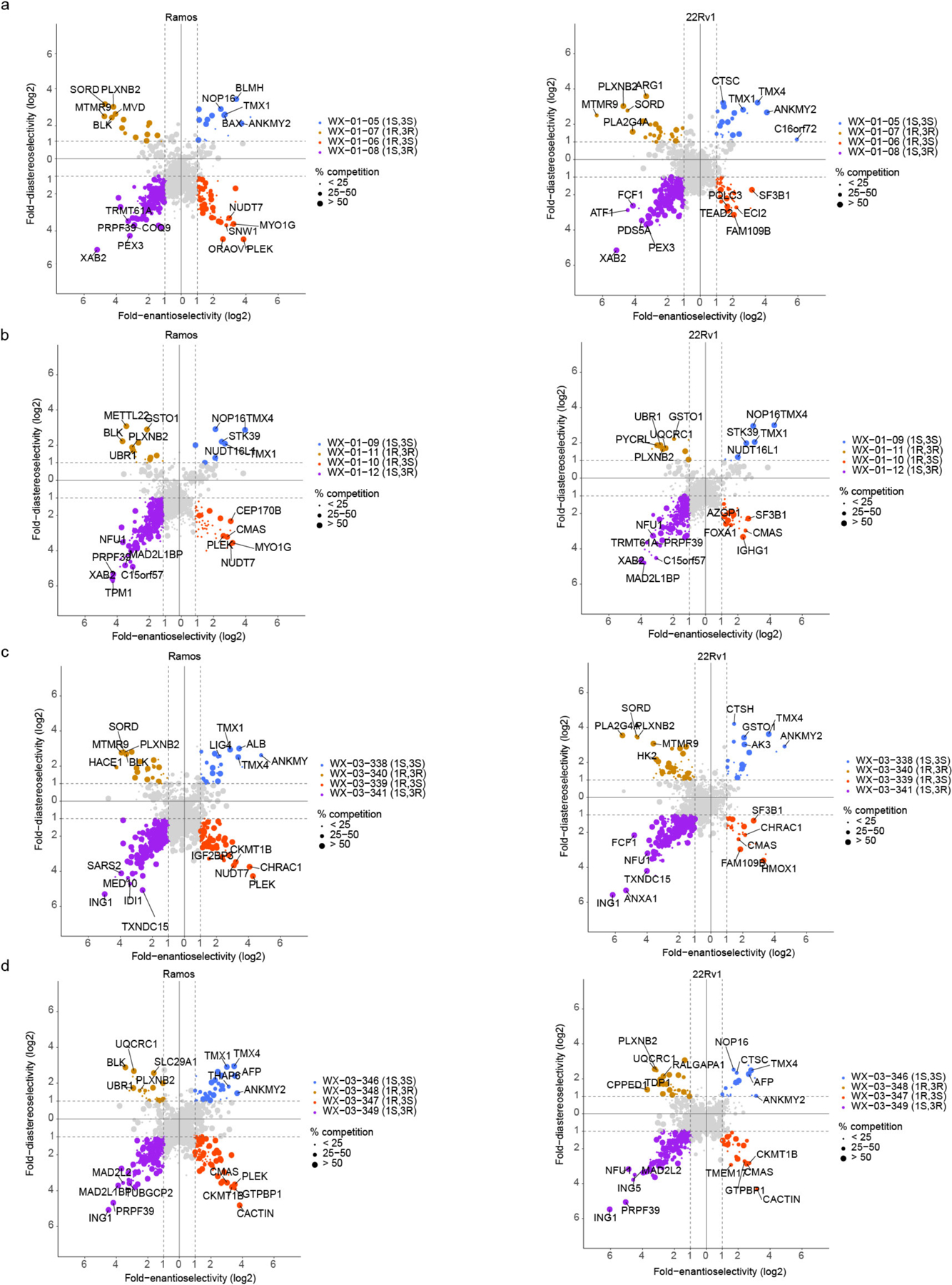
a-d, Quadrant plots highlighting stereoselectively liganded proteins for each stereoconfiguration of alkyne stereoprobe sets 2–5 in Ramos (left) and 22Rv1(right) cells. Enantioselectivity (x-axis) is the ratio of enrichment for one stereoisomer vs its enantiomer, and diastereoselectivity (y-axis) is the ratio of enrichment of one stereoisomer vs the average of its two diastereomers. Data represent mean values for four independent experiments per stereoprobe, per cell line.
Extended Data Fig. 4. Integrated protein- and cysteine-directed.
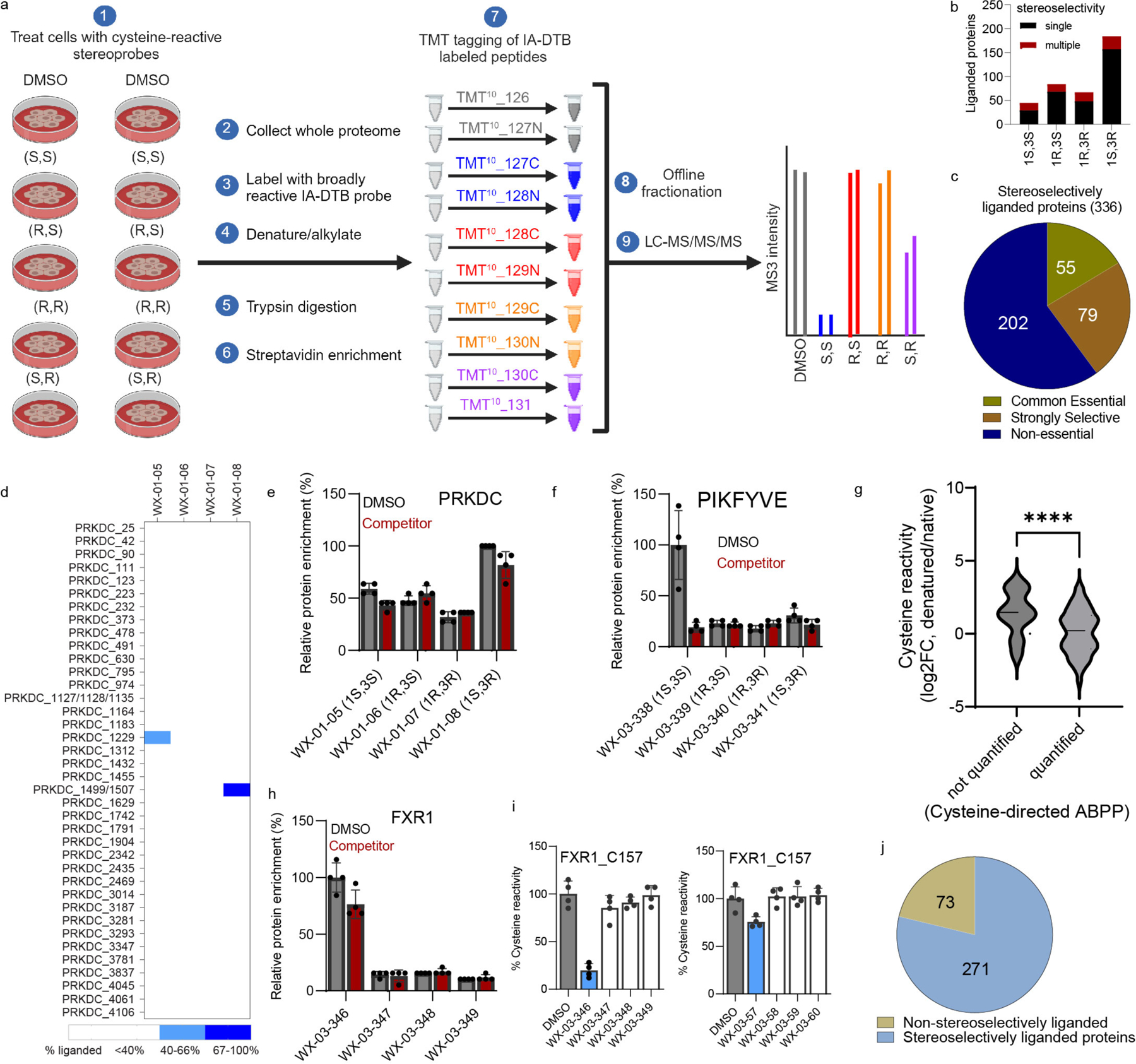
a, Workflow for cysteine-directed ABPP experiments where stereoprobe reactivity with cysteines is determined by multiplexed (tandem mass tagging, TMT10plex) MS-based proteomics, as described previously22. The workflow was created with BioRender.com. b, Number of proteins stereoselectively liganded by one (single; black) versus multiple (red) stereoprobe core configurations. c, Proportion of stereoselectively liganded proteins showing essentiality in the Cancer Dependency Map. d, Heatmap of quantified cysteines in PRKDC showing two cysteines (C1229 and C1499/C1507) displaying distinct stereoselective liganding profiles in cysteine-directed ABPP experiments in Ramos cells. e, Protein-directed ABPP data showing lack of enantioselective enrichment for PRKDC. f, Protein-directed ABPP data showing stereoselective enrichment of PIKFYVE by alkyne WX-03-338 in 22Rv1 cells and blockade of this enrichment by WX-03-57. g, Violin plot showing native-vs-denatured reactivity ratios for cysteine-containing tryptic peptides from stereoselectively liganded proteins with good predicted proteotypicity (DeepMS probability > 0.5) that were either quantified or not quantified in cysteine-directed ABPP experiments performed in native cell proteomes. n=3 biological replicate/cell line; unpaired two-tailed t-test, **** p=0.0001. h, Protein-directed ABPP data showing stereoselective enrichment of FXR1 by WX-03-346 without blockade of this enrichment by WX-03-57. i, Cysteine-directed ABPP data showing greater stereoselective liganding of FXR1_C157 by WX-03-346 (left) versus WX-03-57 (right). j, Pie chart showing fraction of proteins liganded in a stereoselective (blue) or non-stereoselective (green) manner in protein-directed ABPP experiments. For e, f, h, and i, data represent mean values ± SD of n=4 biological replicates.
Extended Data Fig. 5. Characterization of stereoprobe-protein interactions.
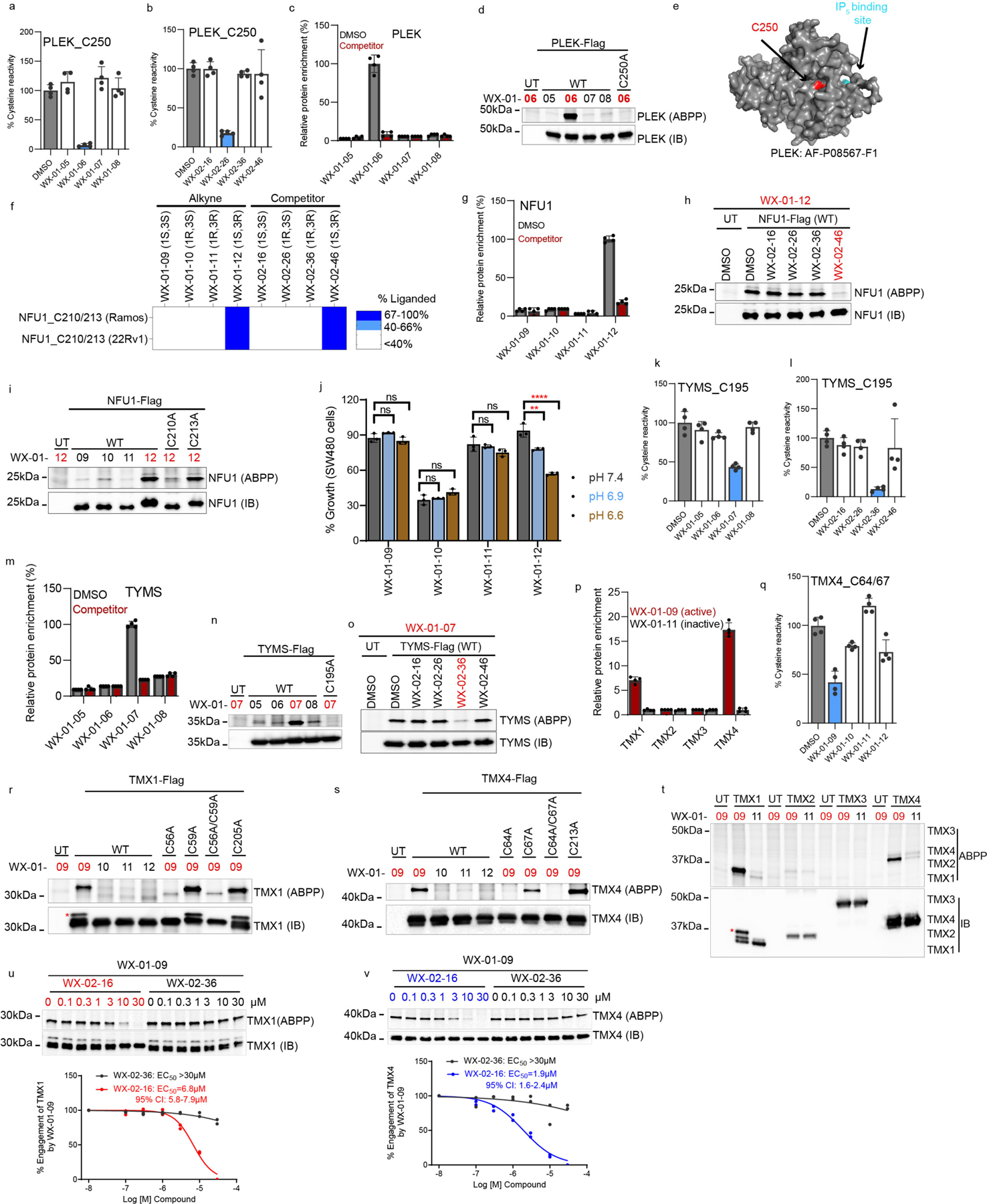
a, b, Cysteine-directed ABPP data showing stereoselective liganding of PLEK_C250 in Ramos cells by WX-01-06 (a) and WX-02-26 (b) in Ramos cells. c, Protein-directed ABPP data showing stereoselective enrichment of PLEK by WX-01-06 and blockade of this enrichment by WX-02-26. d, Gel-ABPP data demonstrating stereoselective engagement of recombinant WT-PLEK, but not a C250A-PLEK mutant by WX-01-06 (5 μM, 1 h). e, AlphaFold-predicted structure of PLEK showing location of C250 (red) relative to the IP5 binding pocket (blue). f, Cysteine-directed ABPP data showing stereoselective liganding of C210/213 of NFU1 by WX-01-12 and WX-02-46. g, Protein-directed ABPP data showing stereoselective enrichment of NFU1 by WX-01-12 and blockade of this enrichment by WX-02-46. h, Competitive gel-ABPP data showing stereoselective blockade of WX-01-12 reactivity with recombinant WT-NFU1 by WX-02-46 (20 μM, 1 h pre-treatment). i, Gel-ABPP data demonstrating engagement of WT-NFU1 and the C213A-NFU1 mutant, but not the C210A-NFU1 mutant, by WX-01-12. j, CellTiter-Glo data showing pH-dependent impairment in SW480 cell growth by WX-01-12 (5 μM, 72 h). Data are mean values ± SD from three independent experiments. One-way ANOVA with Dunnett’s multiple comparison, **p=0.0023, ***p<0.0001. k, l, Cysteine-directed ABPP data showing stereoselective liganding of TYMS_C195 by WX-01-07 (k) and WX-02-36 (l) in Ramos cells. m, Protein-directed ABPP data showing stereoselective enrichment of recombinant TYMS by WX-01-07 (5 μM, 1 h) and blockade of this enrichment by WX-02-36 (20 μM, 1 h pre-treatment). n, o, Gel-ABPP data showing stereoselective engagement of WT-TYMS, but not a C195-TYMS mutant by WX-01-07 (n) and stereoselective blockade of this engagement by WX-02-36 (o). p, Bar graph showing enantioselective enrichment of TMX1/4, but not TMX2/3 by WX-01-09 from protein-directed ABPP experiments in Ramos cells. For each TMX protein, the signal intensity in WX-01-11-treated cells was set to a value of 1. q, Cysteine-directed ABPP data showing stereoselective liganding of TMX4_C64/67 by WX-01-09 in Ramos cells. r, s, Gel-ABPP data demonstrating engagement of recombinant WT-TMX1 and C59A- and C205A-TMX1 mutants, but not C56A- or C56A/C59A TMX1 mutants (r), and recombinant WT-TMX4 and C67A- and C213A-TMX4 mutants, but not C64A- or C64A/C67A-TMX4 mutants (s) by WX-01-09 (5 μM, 1 h). t, Gel-ABPP confirming stereoselective engagement of recombinant TMX1 and TMX4, but not TMX2 and TMX3, by WX-01-09. u, v, Competitive gel-ABPP data showing concentration-dependent and enantioselective blockade of WX-01-09 (5 μM, 1 h) engagement of TMX1 (u) and TMX4 (v) by WX-02-16 (1 h pre-treatment). Top, representative gel-ABPP data; bottom, quantification of gel-ABPP. Data represent 2 biological replicates. The red asterisk in r represents alkyne liganded and rhodamine tagged species of TMX1 (this corresponds to the signal seen in gel-ABPP above the IB). Proteomic data presented in a-c, g, k- m, and p-q are mean values ± SD of n=4 biological replicates. For d, h, i, n-o, r- t experiments were performed in transfected HEK293T cells as described in Fig. 4b, and data are from a single experiment representative of two experiments; IB = anti-Flag immunoblot, UT= untransfected cells.
Extended Data Fig. 6. Characterization of tryptoline acrylamide stereoprobe-LIMK1 interactions.
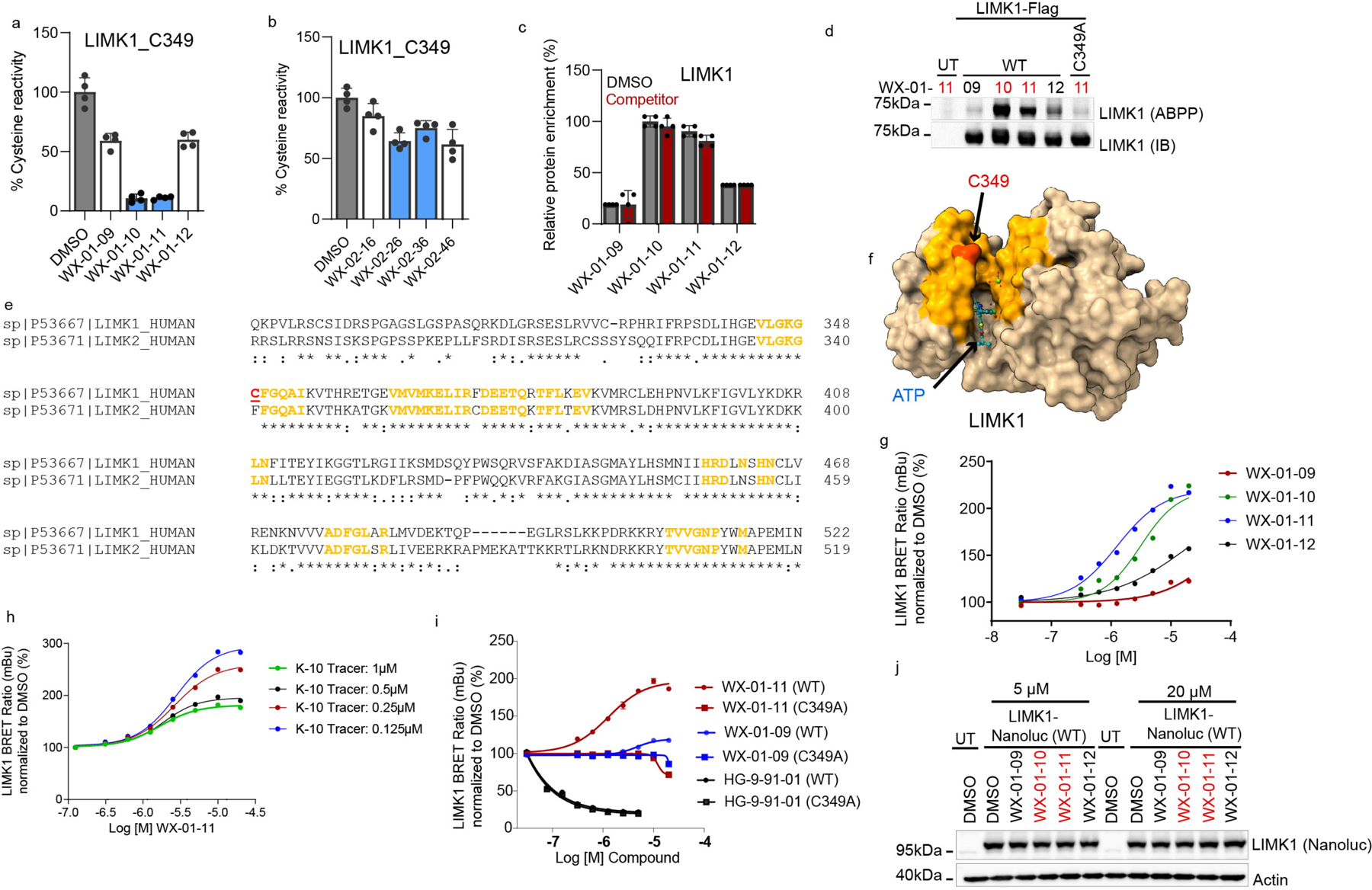
a, Cysteine-directed ABPP data showing enantioselective liganding of C349 of LIMK1 by alkyne stereoprobes WX-01-10 and WX-01-11 in Ramos cells. b, Cysteine-directed ABPP data showing lack of engagement of LIMK1_C349 by WX-02-26 and WX-02-36. c, Protein-directed ABPP data showing enantioselective enrichment of LIMK1 by WX-01-10 and WX-01-11 that is not blocked by WX-02-26 and WX-02-36, respectively. d, Gel-ABPP data demonstrating engagement of recombinant WT-LIMK1, but not the C349A-LIMK1 mutant by WX-01-11 (5 μM, 1 h). Experiments were performed in transfected HEK293T cells as described in Fig. 4b. e, Sequence alignment of LIMK1 and LIMK2 showing conserved residues (yellow) that are proximal (<15 Å) to the LIMK1-restricted stereoprobe-liganded cysteine C349 (red). f, Crystal structure of LIMK1 (PDB: 8AAU) showing C349 (red) in a pocket adjacent to the ATP (blue) binding site. Highlighted in yellow are residues conserved between LIMK1 and LIMK2 located within 15 Å of LIMK1 C349. g, Enantioselective and concentration-dependent enhancement of BRET signal in an LIMK1 NanoBRET kinase assay by WX-01-10 and WX-01-11 compared to their respective enantiomers WX-01-12 and WX-01-09. Data were generated in HEK293T cells transiently expressing LIMK1-nanoLuciferase fusion protein, where cells were treated with 0.5 μM of the NanoBRET K-10 tracer and different concentrations of stereoprobes for 3 h (data represent mean values for one experiment setup in triplicates). h, NanoBRET kinase assay showing that WX-01-11 increases signals for LIMK1 with the general kinase NanoBRET probe K-10, with the largest effect observed at lower concentrations of the NanoBRET probe. Data represent mean values for one experiment setup in triplicates. i, Enantioselective increase in BRET signal by WX-01-11 in WT- but not C349A-LIMK1 mutant cells. The ATP-binding pocket kinase inhibitor HG-9-91-01 decreased NanoBRET signals for both WT- and C349A-LIMK1 mutant (data represent mean values ± SD, n=3 independent experiments). j, Immunoblot of LIMK1-Nanoluc protein expressed in HEK293T cells showing that alkyne stereoprobes do not affect LIMK1 expression under conditions where they increase NanoBRET signals. For a-c, data represent mean values ± SD for n=4 biological replicates. For d, g-h, j data are from a single experiment representative of two independent experiments with similar results. For d, j, IB = anti-Flag immunoblot, and UT= untransfected cells.
Extended Data Fig. 7. Stereoselective liganding of a conserved cysteine in deubiquitinase paralogs STAMBP and STAMBPL1.
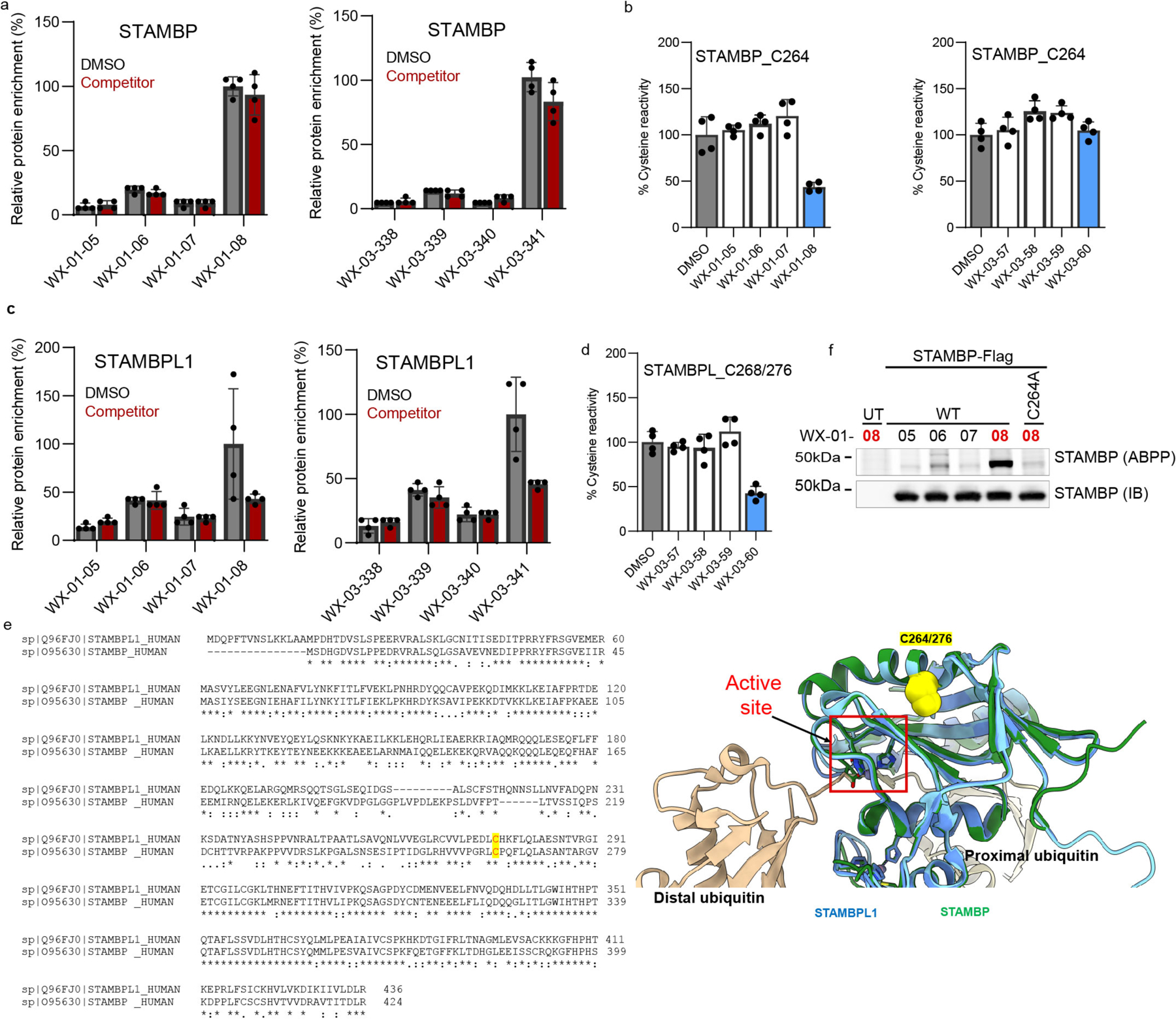
a, Protein-directed ABPP data showing stereoselective enrichment of STAMBP by WX-01-08 and WX-03-341 without blockade of this enrichment by the corresponding competitors WX-02-46 and WX-03-60, respectively. b, Cysteine-directed ABPP data showing stereoselective liganding of STAMBP_C264 by WX-01-08, but not WX-03-60. c, Protein-directed ABPP data showing stereoselective enrichment of STAMBPL1 by WX-01-08 and WX-03-341 and blockade of this enrichment by the corresponding competitor stereoprobes WX-02-46 and WX-03-60, respectively. d, Cysteine-directed ABPP data showing stereoselective liganding of STAMBPL1_C266/C276 by WX-03-60. For a-d, data represent mean values ± SD, n=4 biological replicates. e, Left, sequence alignment of STAMBP and STAMBPL1 showing conservation of the stereoprobe-liganded cysteine (C264 in STAMBP and inferred as C276 in STAMBPL1) highlighted in yellow. Right, overlay of the crystal structures of STAMBP (green, PDB: 3RZV) and STAMBPL1 (blue, PDB: 2ZNV) showing location of the conserved cysteines C264/276 (yellow) with respect to the deubiquitinase active site). f, Gel-ABPP data (performed as in Fig. 4c) showing stereoselective liganding of recombinant WT-STAMBP, but not a C264A STAMBP mutant by WX-01-08 (5 μM, 1 h). Experiments were performed in transfected HEK293T cells as described in Fig. 4b. Data are from a single experiment representative of two independent experiments. IB = anti-Flag immunoblot, and UT= untransfected cells.
Extended Data Fig. 8. Leveraging tryptic peptide maps from protein-directed ABPP experiments to deduce stereoselectively liganded residues.
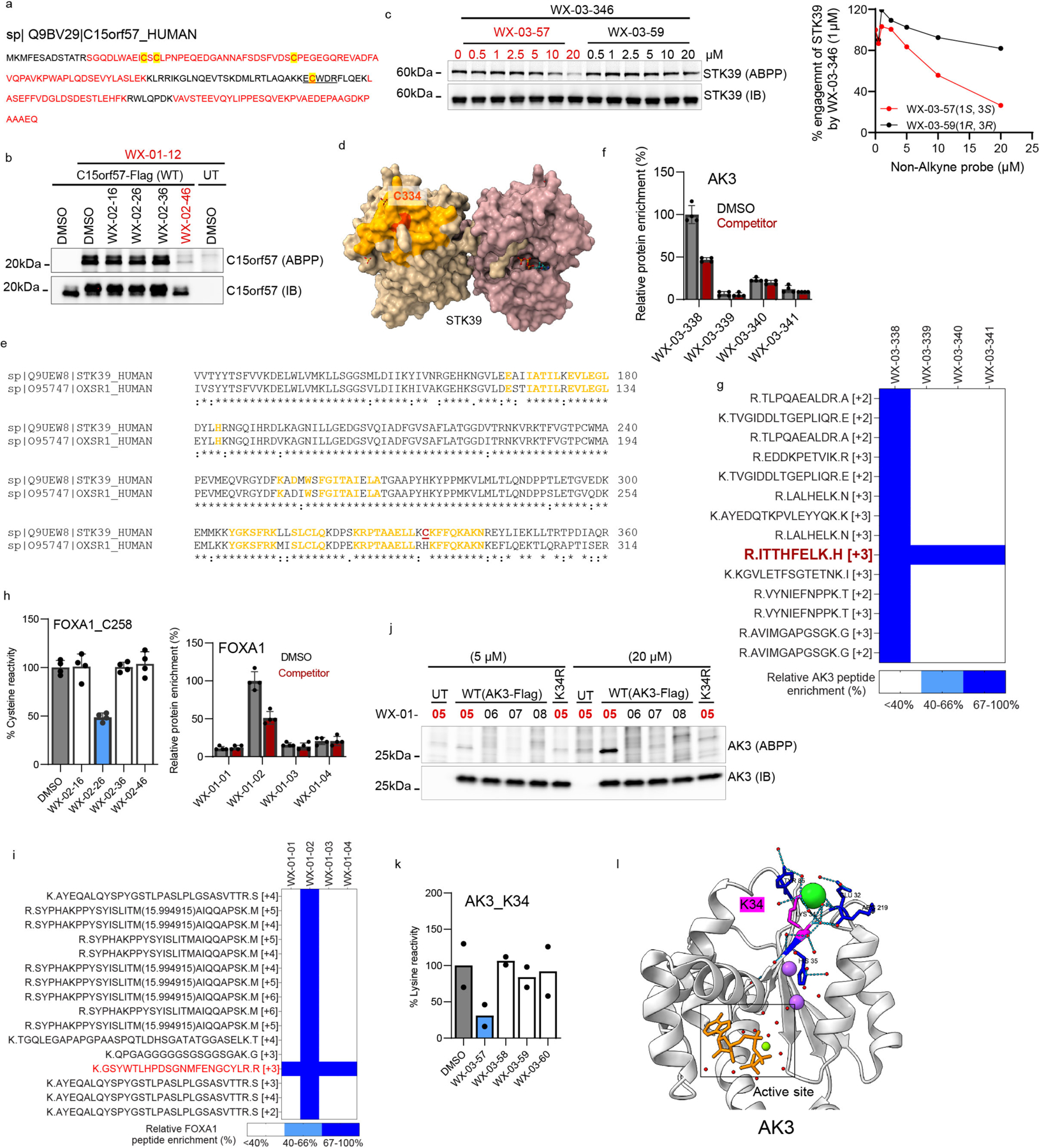
a, Sequence of C15orf57 showing the three cysteines (yellow highlight), peptides that were quantified in protein-directed ABPP (red), and the tryptic peptide containing the liganded cysteine, C111 (underlined). b, Competitive gel-ABPP data showing stereoselective blockade of WX-01-12 (5 μM, 1 h) engagement of C15orf57 by WX-02-46 (20 μM, 1 h pre-treatment). c, Competitive gel-ABPP data showing concentration-dependent, enantioselective blockade of WX-03-346 (1 μM, 1 h) engagement of STK39 by WX-03-59 (1 h pre-treatment) (left) and quantitation of these data (right). d, e, Crystal structure of STK39 (PDB: 5D9H) showing location of stereoprobe-liganded cysteine C334 (red) distal to the ATP pocket and highlighted in yellow are residues around C334 (< 15 Å) that are conserved between STK39 and paralog OXSR1 (yellow-highlighted residues also shown in the sequence alignment in e). f, Protein-directed ABPP data showing stereoselective enrichment of AK3 by WX-03-338 and blockade of this enrichment by WX-03-57 in 22Rv1 cells. Data represent mean values ± SD, n=4 biological replicates. g, Tryptic peptide map of AK3 from protein-directed ABPP experiments showing stereoselective enrichment of all quantified AK3 peptides by WX-03-338 except for the peptide containing K34 (red). In the heat map display, tryptic peptide signal intensities were normalized to 100% for the WX-03-338 treatment group. h, Left, cysteine-directed ABPP data showing stereoselective liganding of FOXA1_C258 by WX-02-26 in 22Rv1 cells. Right, protein-directed ABPP data showing stereoselective enrichment of FOXA1 by WX-01-02 and blockade of this enrichment by WX-02-26 in 22Rv1 cells. Data represent mean values ± SD, n=4 biological replicates. i, Tryptic peptide map of FOXA1 from protein-directed ABPP experiments showing stereoselective enrichment of all quantified FOXA1 peptides by WX-01-02 except for the peptide containing C258 (red). In the heat map display, tryptic peptide signal intensities were normalized to 100% for the WX-01-02 treatment group. j, Gel-ABPP data demonstrating stereoselective engagement of recombinant WT-AK3, but not the K34R-AK3 mutant by WX-01-05. Experiments were performed in transfected HEK293T cells as described in Fig. 4b. k, Lysine-directed ABPP showing stereoselective liganding of AK3_K34 by WX-03-57. Data represent mean and individual values from two biological replicates. l, Crystal structure of AK3 (PDB: 6ZJD) showing distal location of K34 relative to the enzyme active site. For b, c, and j, data are from a single experiment representative of at least two independent experiments; IB = anti-Flag immunoblot, UT= untransfected cells.
Extended Data Fig. 9. Stereoselective and site-specific covalent ligands that inhibit the kynurenine biosynthetic enzyme AFMID.
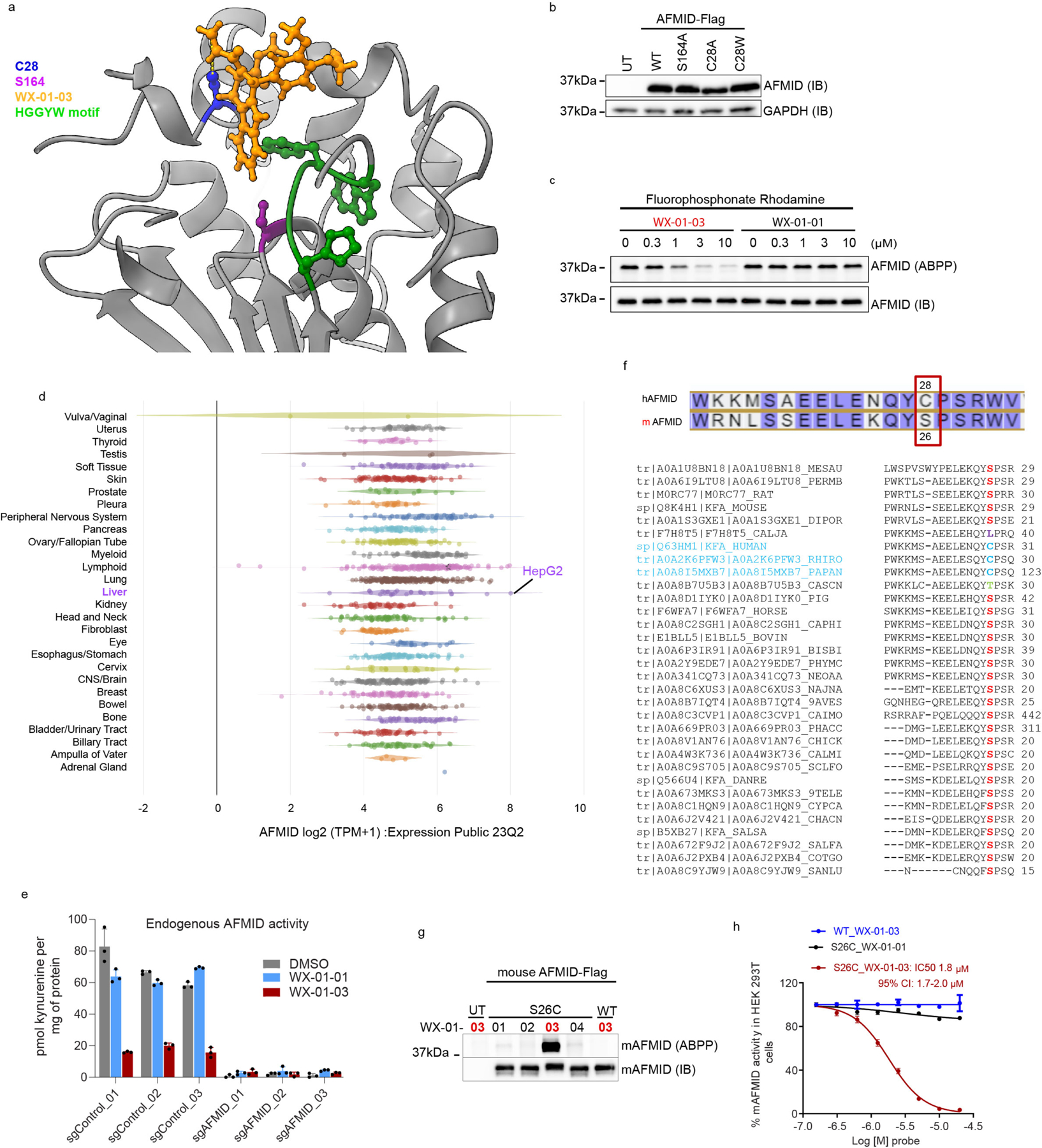
a, Molecular docking showing the preferred non-covalent binding pose for WX-01-03 to C28 of AFMID AlphaFold structure (AF-Q63HM1-F1). Highlighted is C28 (blue) of AFMID in which the sulfur atom and the beta carbon of WX-01-03 acrylamide group are 5.92 Å apart. Also highlighted is the catalytic serine, S164 (purple) and the HGGYW motif (green) that forms the oxyanion hole during substrate cleavage. b, Anti-Flag immunoblot showing expression of recombinant WT-, S164A-, C28A-, and C28W-AFMID in HEK293T cells used in enzyme assay in Fig. 4j; UT= untransfected cells, IB=immunoblot. Data is from a single experiment. c, Competitive gel-ABPP data showing enantioselective blockade of fluorophosphonate-rhodamine (1 μM, 1 h) reactivity with WT-AFMID by WX-01-03 (1 h pre-treatment). Data is representative of two independent experiments. d, Public RNAseq data showing relative mRNA expression of AFMID in cell lines of different lineages (data source: https://depmap.org/portal/, 23Q2 release). Highlighted in purple is the liver cell line HepG2 showing high AFMID expression. e, AFMID activity in HepG2 cells stably infected with three different CRISPR/Cas9 control or AFMID guide (sg) RNAs. Cells were treated with DMSO or 0.5 μM stereoprobes (3 h) and AFMID activity measured as described in Fig.4j. f, Alignment of AFMID sequences (https://www.uniprot.org/) from different organisms showing that the residue corresponding to human AFMID_C28 is substituted for serine in mouse (top panel) other species (bottom panel). g, Gel-ABPP data showing stereoselective engagement of a recombinant mouse S26C-AFMID, but not WT-AFMID, by WX-01-03 (5 μM, 1 h). h, Concentration-dependent and stereoselective inhibition of mouse S26C-AFMID, but not WT-AFMID activity, by WX-01-03. For e, h, Data represent average values ± SD, n=3 biological replicates.
Extended Data Fig. 10. Stereoprobes block MAD2L1BP interactions with the spindle assembly checkpoint (SAC) complex.
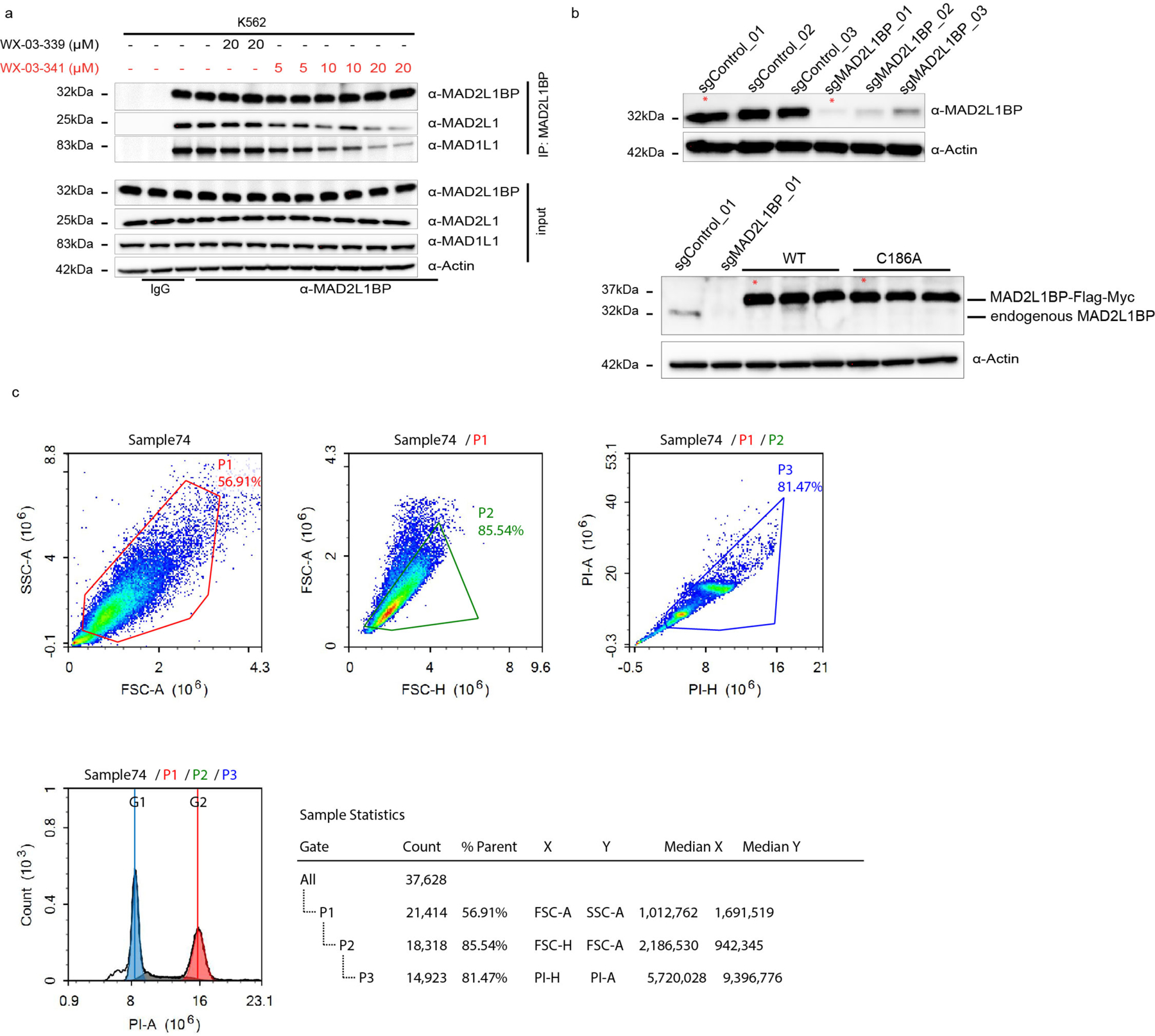
a, Western blots of anti-MAD2L1BP co-immunoprecipitations (Co-IPs) showing stereoselective disruption of MAD2L1BP-MAD2L1/MAD1L1 interactions by WX-03-341 (5–20 μM, 3 h) versus WX-03-339 (20 μM, 3 h) in K562 cells. b, HeLa cells showing CRISPR/Cas9 knockout of MAD2L1BP (top panel, first row) and the reintroduction of WT or C186A-MAD2L1BP into sgMAD2L1BP_01 cells (bottom panel, first row). Red asterisk shows sgControl, sgMAD2L1BP, WT-MAD2L1BP and C186A-MAD2L1BP cell populations used for functional studies in Fig. 5h–m. c, Example of data collection and gating out of cell debris and aggregates on Novocyte for DNA content by PI staining. Gating was strategized to remove irregular events as follows; cell debris were gated out based on side scatter area (SSC-A) and forward scatter area (FSC-A) signal (top panel, left plot), cell aggregates were removed based on forward scatter height (FSC-H) and area (FSC-A) signal (top panel, middle blot). Further removal of aggregates and debris from propidium iodide (PI) positive cells was by PI area (PI-A) and PI height (PI-H) (top panel, right plot). Bottom panel plot shows cell cycle fitting based on the final gate (P3) and table shows the statistics and number of cells in each gate.
Supplementary Material
Acknowledgments
This work was supported by the NIH (U19 AI142784, R35 CA231991) and delivered as part of the eDyNAmiC team supported by the Cancer Grand Challenges partnership funded by Cancer Research UK (CGCATF-2021/100012 + CGCATF-2021/100021) and the National Cancer Institute (OT2CA278688 + OT2CA278692). HHMI Hanna H Gray Fellowship (E.N., GT15176), Jane Coffin Childs Memorial Fellowship (K.E.D.), and Vividion Therapeutics. The authors thank Dr. Xuedong Liu and Dr. Bing Chen (WuXi AppTec) for small-molecule synthesis. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing Interests
G.M.S., T. N., and P.A. are employees of Vividion Therapeutics, and B.F.C. is a founder and member of the Board of Directors of Vividion Therapeutics. The remaining authors declare no competing interests.
Code Availability
Custom codes used for data analysis are available at https://github.com/cravattlab/njomen.
Peer review Information:
Nature Chemistry thanks John Pezacki, Zhengqiu Li and the other, anonymous, reviewer for their contribution to the peer review of this work.
Editorial Summary:
The ligandability of the human proteome can be expanded using covalent chemistry. A multi-tiered chemical proteomic strategy now provides in-depth maps of tryptoline acrylamide-protein interactions in cancer cells. This platform afforded the discovery of stereoselective covalent ligands for hundreds of human proteins, including compounds that disrupt protein-protein interactions regulating the cell cycle.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE81 partner repository with the dataset identifier PXD042541. Raw proteomic files were searched using the ProLuCID algorithm using a reverse concatenated, non-redundant variant of the Human UniProt database (release 2016-07). Processed proteomic data are provided in Supplementary Dataset1. Source Data are provided as Source Data files and all other data associated with the manuscript can be found in the supplementary information.
References
- 1.Chakravarty D; Solit DB, Clinical cancer genomic profiling. Nat Rev Genet 2021, 22 (8), 483–501. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber SL, A Chemical Biology View of Bioactive Small Molecules and a Binder-Based Approach to Connect Biology to Precision Medicines. Isr J Chem 2019, 59 (1–2), 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang ZZ; Shi XX; Huang GY; Hao GF; Yang GF, Fragment-based drug discovery supports drugging 'undruggable' protein-protein interactions. Trends Biochem Sci 2023, 48 (6), 539–552. [DOI] [PubMed] [Google Scholar]
- 4.Denis JD; Hall RJ; Murray CW; Heightman TD; Rees DC, Fragment-based drug discovery: opportunities for organic synthesis. RSC Med Chem 2020, 12 (3), 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundquist KP; Panchal V; Gotfredsen CH; Brenk R; Clausen MH, Fragment-Based Drug Discovery for RNA Targets. ChemMedChem 2021, 16 (17), 2588–2603. [DOI] [PubMed] [Google Scholar]
- 6.Erlanson DA; Davis BJ; Jahnke W, Fragment-Based Drug Discovery: Advancing Fragments in the Absence of Crystal Structures. Cell Chem Biol 2019, 26 (1), 9–15. [DOI] [PubMed] [Google Scholar]
- 7.Bon M; Bilsland A; Bower J; McAulay K, Fragment-based drug discovery-the importance of high-quality molecule libraries. Mol Oncol 2022, 16 (21), 3761–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen X; Wu X; Jin R; Lu X, Privileged heterocycles for DNA-encoded library design and hit-to-lead optimization. Eur J Med Chem 2023, 248, 115079. [DOI] [PubMed] [Google Scholar]
- 9.Shi B; Zhou Y; Huang Y; Zhang J; Li X, Recent advances on the encoding and selection methods of DNA-encoded chemical library. Bioorg Med Chem Lett 2017, 27 (3), 361–369. [DOI] [PubMed] [Google Scholar]
- 10.Peterson AA; Liu DR, Small-molecule discovery through DNA-encoded libraries. Nat Rev Drug Discov 2023, 22 (9), 699–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan WC; Sharifzadeh S; Buhrlage SJ; Marto JA, Chemoproteomic methods for covalent drug discovery. Chem Soc Rev 2021, 50 (15), 8361–8381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spradlin JN; Zhang E; Nomura DK, Reimagining Druggability Using Chemoproteomic Platforms. Acc Chem Res 2021, 54 (7), 1801–1813. [DOI] [PubMed] [Google Scholar]
- 13.Cuesta A; Taunton J, Lysine-Targeted Inhibitors and Chemoproteomic Probes. Annu Rev Biochem 2019, 88, 365–381. [DOI] [PubMed] [Google Scholar]
- 14.Singh J; Petter RC; Baillie TA; Whitty A, The resurgence of covalent drugs. Nat Rev Drug Discov 2011, 10 (4), 307–17. [DOI] [PubMed] [Google Scholar]
- 15.Boike L; Henning NJ; Nomura DK, Advances in covalent drug discovery. Nature Reviews Drug Discovery 2022, 21 (12), 881–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cross DA; Ashton SE; Ghiorghiu S; Eberlein C; Nebhan CA; Spitzler PJ; Orme JP; Finlay MR; Ward RA; Mellor MJ; Hughes G; Rahi A; Jacobs VN; Red Brewer M; Ichihara E; Sun J; Jin H; Ballard P; Al-Kadhimi K; Rowlinson R; Klinowska T; Richmond GH; Cantarini M; Kim DW; Ranson MR; Pao W, AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014, 4 (9), 1046–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang ML; Rule S; Martin P; Goy A; Auer R; Kahl BS; Jurczak W; Advani RH; Romaguera JE; Williams ME; Barrientos JC; Chmielowska E; Radford J; Stilgenbauer S; Dreyling M; Jedrzejczak WW; Johnson P; Spurgeon SE; Li L; Zhang L; Newberry K; Ou Z; Cheng N; Fang B; McGreivy J; Clow F; Buggy JJ; Chang BY; Beaupre DM; Kunkel LA; Blum KA, Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013, 369 (6), 507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdeldayem A; Raouf YS; Constantinescu SN; Moriggl R; Gunning PT, Advances in covalent kinase inhibitors. Chem Soc Rev 2020, 49 (9), 2617–2687. [DOI] [PubMed] [Google Scholar]
- 19.Ostrem JM; Peters U; Sos ML; Wells JA; Shokat KM, K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503 (7477), 548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong DS; Fakih MG; Strickler JH; Desai J; Durm GA; Shapiro GI; Falchook GS; Price TJ; Sacher A; Denlinger CS; Bang YJ; Dy GK; Krauss JC; Kuboki Y; Kuo JC; Coveler AL; Park K; Kim TW; Barlesi F; Munster PN; Ramalingam SS; Burns TF; Meric-Bernstam F; Henary H; Ngang J; Ngarmchamnanrith G; Kim J; Houk BE; Canon J; Lipford JR; Friberg G; Lito P; Govindan R; Li BT, KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med 2020, 383 (13), 1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Backus KM; Correia BE; Lum KM; Forli S; Horning BD; González-Páez GE; Chatterjee S; Lanning BR; Teijaro JR; Olson AJ; Wolan DW; Cravatt BF, Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534 (7608), 570–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vinogradova EV; Zhang X; Remillard D; Lazar DC; Suciu RM; Wang Y; Bianco G; Yamashita Y; Crowley VM; Schafroth MA; Yokoyama M; Konrad DB; Lum KM; Simon GM; Kemper EK; Lazear MR; Yin S; Blewett MM; Dix MM; Nguyen N; Shokhirev MN; Chin EN; Lairson LL; Melillo B; Schreiber SL; Forli S; Teijaro JR; Cravatt BF, An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 2020, 182 (4), 1009–1026 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abbasov ME; Kavanagh ME; Ichu TA; Lazear MR; Tao Y; Crowley VM; Am Ende CW; Hacker SM; Ho J; Dix MM; Suciu R; Hayward MM; Kiessling LL; Cravatt BF, A proteome-wide atlas of lysine-reactive chemistry. Nat Chem 2021, 13 (11), 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C; Weerapana E; Blewett MM; Cravatt BF, A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods 2014, 11 (1), 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y; Craven GB; Kamber RA; Cuesta A; Zhersh S; Moroz YS; Bassik MC; Taunton J, Direct mapping of ligandable tyrosines and lysines in cells with chiral sulfonyl fluoride probes. Nat Chem 2023, 15 (11), 1616–1625. [DOI] [PubMed] [Google Scholar]
- 26.Maurais AJ; Weerapana E, Reactive-cysteine profiling for drug discovery. Curr Opin Chem Biol 2019, 50, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weerapana E; Wang C; Simon GM; Richter F; Khare S; Dillon MB; Bachovchin DA; Mowen K; Baker D; Cravatt BF, Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 468 (7325), 790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gygi SP; Rist B; Gerber SA; Turecek F; Gelb MH; Aebersold R, Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol 1999, 17 (10), 994–9. [DOI] [PubMed] [Google Scholar]
- 29.Kuljanin M; Mitchell DC; Schweppe DK; Gikandi AS; Nusinow DP; Bulloch NJ; Vinogradova EV; Wilson DL; Kool ET; Mancias JD; Cravatt BF; Gygi SP, Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nat Biotechnol 2021, 39 (5), 630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grossman EA; Ward CC; Spradlin JN; Bateman LA; Huffman TR; Miyamoto DK; Kleinman JI; Nomura DK, Covalent Ligand Discovery against Druggable Hotspots Targeted by Anti-cancer Natural Products. Cell Chem Biol 2017, 24 (11), 1368–1376.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazear MR; Remsberg JR; Jaeger MG; Rothamel K; Her HL; DeMeester KE; Njomen E; Hogg SJ; Rahman J; Whitby LR; Won SJ; Schafroth MA; Ogasawara D; Yokoyama M; Lindsey GL; Li H; Germain J; Barbas S; Vaughan J; Hanigan TW; Vartabedian VF; Reinhardt CJ; Dix MM; Koo SJ; Heo I; Teijaro JR; Simon GM; Ghosh B; Abdel-Wahab O; Ahn K; Saghatelian A; Melillo B; Schreiber SL; Yeo GW; Cravatt BF, Proteomic discovery of chemical probes that perturb protein complexes in human cells. Mol Cell 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao Y; Remillard D; Vinogradova EV; Yokoyama M; Banchenko S; Schwefel D; Melillo B; Schreiber SL; Zhang X; Cravatt BF, Targeted Protein Degradation by Electrophilic PROTACs that Stereoselectively and Site-Specifically Engage DCAF1. J Am Chem Soc 2022, 144 (40), 18688–18699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feldman HC; Merlini E; Guijas C; DeMeester KE; Njomen E; Kozina EM; Yokoyama M; Vinogradova E; Reardon HT; Melillo B; Schreiber SL; Loreto A; Blankman JL; Cravatt BF, Selective inhibitors of SARM1 targeting an allosteric cysteine in the autoregulatory ARM domain. Proc Natl Acad Sci U S A 2022, 119 (35), e2208457119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kathman SG; Koo SJ; Lindsey GL; Her HL; Blue SM; Li H; Jaensch S; Remsberg JR; Ahn K; Yeo GW; Ghosh B; Cravatt BF, Remodeling oncogenic transcriptomes by small molecules targeting NONO. Nat Chem Biol 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boike L; Cioffi AG; Majewski FC; Co J; Henning NJ; Jones MD; Liu G; McKenna JM; Tallarico JA; Schirle M; Nomura DK, Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC. Cell Chem Biol 2021, 28 (1), 4–13.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kavanagh ME; Horning BD; Khattri R; Roy N; Lu JP; Whitby LR; Ye E; Brannon JC; Parker A; Chick JM; Eissler CL; Wong AJ; Rodriguez JL; Rodiles S; Masuda K; Teijaro JR; Simon GM; Patricelli MP; Cravatt BF, Selective inhibitors of JAK1 targeting an isoform-restricted allosteric cysteine. Nat Chem Biol 2022, 18 (12), 1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darabedian N; Ji W; Fan M; Lin S; Seo HS; Vinogradova EV; Yaron TM; Mills EL; Xiao H; Senkane K; Huntsman EM; Johnson JL; Che J; Cantley LC; Cravatt BF; Dhe-Paganon S; Stegmaier K; Zhang T; Gray NS; Chouchani ET, Depletion of creatine phosphagen energetics with a covalent creatine kinase inhibitor. Nat Chem Biol 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bar-Peled L; Kemper EK; Suciu RM; Vinogradova EV; Backus KM; Horning BD; Paul TA; Ichu TA; Svensson RU; Olucha J; Chang MW; Kok BP; Zhu Z; Ihle NT; Dix MM; Jiang P; Hayward MM; Saez E; Shaw RJ; Cravatt BF, Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 2017, 171 (3), 696–709.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X; Crowley VM; Wucherpfennig TG; Dix MM; Cravatt BF, Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol 2019, 15 (7), 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X; Luukkonen LM; Eissler CL; Crowley VM; Yamashita Y; Schafroth MA; Kikuchi S; Weinstein DS; Symons KT; Nordin BE; Rodriguez JL; Wucherpfennig TG; Bauer LG; Dix MM; Stamos D; Kinsella TM; Simon GM; Baltgalvis KA; Cravatt BF, DCAF11 Supports Targeted Protein Degradation by Electrophilic Proteolysis-Targeting Chimeras. J Am Chem Soc 2021, 143 (13), 5141–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward CC; Kleinman JI; Brittain SM; Lee PS; Chung CYS; Kim K; Petri Y; Thomas JR; Tallarico JA; McKenna JM; Schirle M; Nomura DK, Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem Biol 2019, 14 (11), 2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao Z; Chang C; Yang J; Zhu Y; Fu Y, AP3: An Advanced Proteotypic Peptide Predictor for Targeted Proteomics by Incorporating Peptide Digestibility. Anal Chem 2019, 91 (13), 8705–8711. [DOI] [PubMed] [Google Scholar]
- 43.Lanning BR; Whitby LR; Dix MM; Douhan J; Gilbert AM; Hett EC; Johnson TO; Joslyn C; Kath JC; Niessen S; Roberts LR; Schnute ME; Wang C; Hulce JJ; Wei B; Whiteley LO; Hayward MM; Cravatt BF, A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol 2014, 10 (9), 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niessen S; Dix MM; Barbas S; Potter ZE; Lu S; Brodsky O; Planken S; Behenna D; Almaden C; Gajiwala KS; Ryan K; Ferre R; Lazear MR; Hayward MM; Kath JC; Cravatt BF, Proteome-wide Map of Targets of T790M-EGFR-Directed Covalent Inhibitors. Cell Chem Biol 2017, 24 (11), 1388–1400.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warren A; Chen Y; Jones A; Shibue T; Hahn WC; Boehm JS; Vazquez F; Tsherniak A; McFarland JM, Global computational alignment of tumor and cell line transcriptional profiles. Nat Commun 2021, 12 (1), 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rostovtsev VV; Green LG; Fokin VV; Sharpless KB, A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew Chem Int Ed Engl 2002, 41 (14), 2596–9. [DOI] [PubMed] [Google Scholar]
- 47.Tornoe CW; Christensen C; Meldal M, Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 2002, 67 (9), 3057–64. [DOI] [PubMed] [Google Scholar]
- 48.Speers AE; Adam GC; Cravatt BF, Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc 2003, 125 (16), 4686–7. [DOI] [PubMed] [Google Scholar]
- 49.Teng M; Zhou S; Cai C; Lupien M; He HH, Pioneer of prostate cancer: past, present and the future of FOXA1. Protein Cell 2021, 12 (1), 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vinogradova EV; Cravatt BF, Multiplexed proteomic profiling of cysteine reactivity and ligandability in human T cells. STAR Protoc 2021, 2 (2), 100458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsherniak A; Vazquez F; Montgomery PG; Weir BA; Kryukov G; Cowley GS; Gill S; Harrington WF; Pantel S; Krill-Burger JM; Meyers RM; Ali L; Goodale A; Lee Y; Jiang G; Hsiao J; Gerath WFJ; Howell S; Merkel E; Ghandi M; Garraway LA; Root DE; Golub TR; Boehm JS; Hahn WC, Defining a Cancer Dependency Map. Cell 2017, 170 (3), 564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Serrano G; Guruceaga E; Segura V, DeepMSPeptide: peptide detectability prediction using deep learning. Bioinformatics 2020, 36 (4), 1279–1280. [DOI] [PubMed] [Google Scholar]
- 53.Li H; Ma T; Remsberg JR; Won SJ; DeMeester KE; Njomen E; Ogasawara D; Zhao KT; Huang TP; Lu B; Simon GM; Melillo B; Schreiber SL; Lykke-Andersen J; Liu DR; Cravatt BF, Assigning functionality to cysteines by base editing of cancer dependency genes. Nat Chem Biol 2023, 19 (11), 1320–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor C. A. t.; Juang YC; Earnest S; Sengupta S; Goldsmith EJ; Cobb MH, Domain-Swapping Switch Point in Ste20 Protein Kinase SPAK. Biochemistry 2015, 54 (32), 5063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davis I; Liu A, What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Rev Neurother 2015, 15 (7), 719–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tan L; Yu JT; Tan L, The kynurenine pathway in neurodegenerative diseases: mechanistic and therapeutic considerations. J Neurol Sci 2012, 323 (1–2), 1–8. [DOI] [PubMed] [Google Scholar]
- 57.Dewulf JP; Martin M; Marie S; Oguz F; Belkhir L; De Greef J; Yombi JC; Wittebole X; Laterre PF; Jadoul M; Gatto L; Bommer GT; Morelle J, Urine metabolomics links dysregulation of the tryptophan-kynurenine pathway to inflammation and severity of COVID-19. Sci Rep 2022, 12 (1), 9959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas T; Stefanoni D; Reisz JA; Nemkov T; Bertolone L; Francis RO; Hudson KE; Zimring JC; Hansen KC; Hod EA; Spitalnik SL; D'Alessandro A, COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020, 5 (14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varadi M; Anyango S; Deshpande M; Nair S; Natassia C; Yordanova G; Yuan D; Stroe O; Wood G; Laydon A; Žídek A; Green T; Tunyasuvunakool K; Petersen S; Jumper J; Clancy E; Green R; Vora A; Lutfi M; Figurnov M; Cowie A; Hobbs N; Kohli P; Kleywegt G; Birney E; Hassabis D; Velankar S, AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 2022, 50 (D1), D439–d444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu LJ; Li XF; Hu JQ; Ni XJ; Lu HY; Wang JJ; Huang XN; Lin CX; Shang DW; Wen YG, A Simple HPLC-MS/MS Method for Determination of Tryptophan, Kynurenine and Kynurenic Acid in Human Serum and its Potential for Monitoring Antidepressant Therapy. J Anal Toxicol 2017, 41 (1), 37–44. [DOI] [PubMed] [Google Scholar]
- 62.Dobrovolsky VN; Bowyer JF; Pabarcus MK; Heflich RH; Williams LD; Doerge DR; Arvidsson B; Bergquist J; Casida JE, Effect of arylformamidase (kynurenine formamidase) gene inactivation in mice on enzymatic activity, kynurenine pathway metabolites and phenotype. Biochim Biophys Acta 2005, 1724 (1–2), 163–72. [DOI] [PubMed] [Google Scholar]
- 63.Bachovchin DA; Ji T; Li W; Simon GM; Blankman JL; Adibekian A; Hoover H; Niessen S; Cravatt BF, Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc Natl Acad Sci U S A 2010, 107 (49), 20941–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Westhorpe FG; Tighe A; Lara-Gonzalez P; Taylor SS, p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci 2011, 124 (Pt 22), 3905–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hagan RS; Manak MS; Buch HK; Meier MG; Meraldi P; Shah JV; Sorger PK, p31(comet) acts to ensure timely spindle checkpoint silencing subsequent to kinetochore attachment. Mol Biol Cell 2011, 22 (22), 4236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lok TM; Wang Y; Xu WK; Xie S; Ma HT; Poon RYC, Mitotic slippage is determined by p31(comet) and the weakening of the spindle-assembly checkpoint. Oncogene 2020, 39 (13), 2819–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang M; Li B; Tomchick DR; Machius M; Rizo J; Yu H; Luo X, p31comet blocks Mad2 activation through structural mimicry. Cell 2007, 131 (4), 744–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abdel-Salam GMH; Hellmuth S; Gradhand E; Käseberg S; Winter J; Pabst AS; Eid MM; Thiele H; Nürnberg P; Budde BS; Toliat MR; Brecht IB; Schroeder C; Gschwind A; Ossowski S; Häuser F; Rossmann H; Abdel-Hamid MS; Hegazy I; Mohamed AG; Schneider DT; Bertoli-Avella A; Bauer P; Pearring JN; Pfundt R; Hoischen A; Gilissen C; Strand D; Zechner U; Tashkandi SA; Faqeih EA; Stemmann O; Strand S; Bolz HJ, Biallelic MAD2L1BP (p31comet) mutation is associated with mosaic aneuploidy and juvenile granulosa cell tumors. JCI Insight 2023, 8 (22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Browne CM; Jiang B; Ficarro SB; Doctor ZM; Johnson JL; Card JD; Sivakumaren SC; Alexander WM; Yaron TM; Murphy CJ; Kwiatkowski NP; Zhang T; Cantley LC; Gray NS; Marto JA, A Chemoproteomic Strategy for Direct and Proteome-Wide Covalent Inhibitor Target-Site Identification. J Am Chem Soc 2019, 141 (1), 191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wozniak JM; Li W; Governa P; Chen LY; Jadhav A; Dongre A; Forli S; Parker CG, Enhanced mapping of small-molecule binding sites in cells. Nat Chem Biol 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mons E; Kim RQ; Mulder MPC, Technologies for Direct Detection of Covalent Protein-Drug Adducts. Pharmaceuticals (Basel) 2023, 16 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Giansanti P; Tsiatsiani L; Low TY; Heck AJ, Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat Protoc 2016, 11 (5), 993–1006. [DOI] [PubMed] [Google Scholar]
- 73.Abegg D; Tomanik M; Qiu N; Pechalrieu D; Shuster A; Commare B; Togni A; Herzon SB; Adibekian A, Chemoproteomic Profiling by Cysteine Fluoroalkylation Reveals Myrocin G as an Inhibitor of the Nonhomologous End Joining DNA Repair Pathway. J Am Chem Soc 2021, 143 (48), 20332–20342. [DOI] [PubMed] [Google Scholar]
- 74.Tessier R; Nandi RK; Dwyer BG; Abegg D; Sornay C; Ceballos J; Erb S; Cianférani S; Wagner A; Chaubet G; Adibekian A; Waser J, Ethynylation of Cysteine Residues: From Peptides to Proteins in Vitro and in Living Cells. Angew Chem Int Ed Engl 2020, 59 (27), 10961–10970. [DOI] [PubMed] [Google Scholar]
- 75.Motiwala HF; Kuo YH; Stinger BL; Palfey BA; Martin BR, Tunable Heteroaromatic Sulfones Enhance in-Cell Cysteine Profiling. J Am Chem Soc 2020, 142 (4), 1801–1810. [DOI] [PubMed] [Google Scholar]
- 76.Cuesta A; Wan X; Burlingame AL; Taunton J, Ligand Conformational Bias Drives Enantioselective Modification of a Surface-Exposed Lysine on Hsp90. J Am Chem Soc 2020, 142 (7), 3392–3400. [DOI] [PubMed] [Google Scholar]
- 77.Wang Y; Dix MM; Bianco G; Remsberg JR; Lee HY; Kalocsay M; Gygi SP; Forli S; Vite G; Lawrence RM; Parker CG; Cravatt BF, Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat Chem 2019, 11 (12), 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jacobs AT; Marnett LJ, Systems analysis of protein modification and cellular responses induced by electrophile stress. Acc Chem Res 2010, 43 (5), 673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Békés M; Langley DR; Crews CM, PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov 2022, 21 (3), 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Y; Liu Z; Hirschi M; Brodsky O; Johnson E; Won SJ; Nagata A; Petroski MD; Majmudar JD; Niessen S; VanArsdale T; Gilbert AM; Hayward MM; Stewart AE; Nager AR; Melillo B; Cravatt B, Expanding the ligandable proteome by paralog hopping with covalent probes. bioRxiv 2024. [DOI] [PubMed] [Google Scholar]
- 81.Perez-Riverol Y; Bai J; Bandla C; García-Seisdedos D; Hewapathirana S; Kamatchinathan S; Kundu DJ; Prakash A; Frericks-Zipper A; Eisenacher M; Walzer M; Wang S; Brazma A; Vizcaíno JA, The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 2022, 50 (D1), D543–d552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE81 partner repository with the dataset identifier PXD042541. Raw proteomic files were searched using the ProLuCID algorithm using a reverse concatenated, non-redundant variant of the Human UniProt database (release 2016-07). Processed proteomic data are provided in Supplementary Dataset1. Source Data are provided as Source Data files and all other data associated with the manuscript can be found in the supplementary information.