

Abstract
Objective
Deregulation of the cJUN/AP‐1 and hedgehog/GLI2 signaling pathways has been implicated in fibroblast activation in systemic sclerosis (SSc). However, the consequences of their concomitant up‐regulation are unknown. Here, we tested the hypothesis that mutual amplification of both pathways might drive persistent fibroblast activation.
Methods
Cultured fibroblasts and skin sections of patients with diffuse SSc and healthy volunteers were analyzed. cJUN/AP‐1 signaling and hedgehog/GLI2 signaling were inhibited using knockdown and pharmacologic approaches. Hedgehog signaling was activated in mice by fibroblast‐specific overexpression of constitutively active Smoothened.
Results
cJUN and GLI2 are concomitantly up‐regulated and colocalize in fibroblasts of patients with SSc compared to healthy controls. Activation of hedgehog/GLI2 signaling induces the expression of cJUN in vitro and in vivo, whereas inactivation of GLI2 inhibits cJUN expression. Likewise, inactivation of cJUN impairs the expression of GLI2. This mutual regulation occurs at the level of transcription with binding of cJUN and GLI2 to specific binding motifs. Interference with this mutual amplification of cJUN signaling and GLI2 signaling inhibits fibroblast activation and collagen release: Inhibition of cJUN/AP‐1 signaling ameliorates hedgehog‐induced fibroblast activation and skin fibrosis in SmoACT mice with a reduction of skin thickness of 103% (P = 0.0043) in the treatment group compared to the fibrotic control group. Moreover, combined pharmacologic inhibition of cJUN/AP‐1 and hedgehog/GLI2 exerts additive antifibrotic effects in a model of TGFβ‐driven experimental fibrosis (TBRACT mice).
Conclusion
The transcription factors cJUN and GLI2 reinforce each other's activity to promote fibroblast activation in SSc. Interruption of this crosstalk by combined inhibition of both pathways exerts additive antifibrotic effects at well‐tolerated doses.
INTRODUCTION
Systemic sclerosis (SSc) is a chronic fibrosing connective tissue disorder that affects the skin and various inner organs. 1 , 2 A central hallmark of SSc is the uncontrolled and persistent activation of fibroblasts, which results in the excessive release of extracellular matrix. Myofibroblasts are key effectors of pathologic tissue remodeling and tissue fibrosis. In normal wound healing, myofibroblasts occur transiently to promote repair. In contrast, myofibroblasts persist in the pathologic context of fibrotic diseases such as SSc 1 , 2 and promote chronic fibrotic remodeling. Although the mechanisms of aberrant fibroblast activation are incompletely understood, several core transcription factors of fibrotic tissue remodeling, such as the activator protein 1 (AP‐1) member cJUN or the hedgehog transcription factor GLI2, have been identified. Activation of cJUN and GLI2 are common denominators of fibrotic diseases: Both pathways have been shown to be sufficient and required for the development of fibrosis in preclinical models. 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 Thus, both pathways are potential candidates for targeted antifibrotic therapies. cJUN is centrally involved in homeostatic functions such as cell proliferation and survival, while hedgehog signaling plays a critical role for stem cell revival. 11 , 12 Although small molecule inhibitors are in clinical development (for cJUN/AP‐1) or are already in clinical use (for hedgehog signaling), prolonged inhibition of cJUN/AP‐1 and hedgehog signaling with higher doses of inhibitors in a chronic disease such as SSc might be complicated by dose‐limiting adverse events.
Combinations of synergistic therapies may enable dose reduction of individual drugs, resulting in reduced toxicity but comparable or even improved efficacy. This concept might be particularly critical for targets with major homeostatic functions such as cJUN and GLI2, as low‐grade residual activity is often sufficient to maintain tissue homeostasis, but the achieved extent of inhibition might still be effective in breaking the pathologic signaling output caused by uncontrolled activation in pathologic conditions. First evidence of the mutual interaction of cJUN and GLI2 has been described in keratinocytes, 13 and a previous study demonstrated that cJun induces skin fibrosis through the hedgehog‐mediated multiplication of CD26+, Sca− fibroblasts in a murine, cJun‐driven fibrosis model. 14
The aim of this study was to characterize the mutual interaction of cJUN/AP‐1 and hedgehog/GLI2 signaling in human fibroblasts and different murine models of SSc as a prototypical chronic fibrosing disorder and to test the hypothesis that combination therapies with lower doses of inhibitors of cJUN/AP‐1 and hedgehog/GLI2 signaling may have additive antifibrotic effects and better tolerability than high‐dose monotherapies.
MATERIALS AND METHODS
Patients and fibroblasts
Human dermal fibroblasts were isolated from skin biopsies of 7 patients with SSc and 12 healthy volunteers. All patients fulfilled the 2013 American College of Rheumatology/EULAR criteria for SSc. 15 Four patients were female; three were male. The median age of patients with SSc was 48 years (range: 32–62 years), and their median disease duration was 6.3 years (range: 0.6–23 years). All patients and volunteers signed a consent form approved by the local institutional review board. Patients with active and inactive skin disease were included in the study. Active skin disease was defined as progression of skin fibrosis (increase of modified Rodnan Skin Score >5 within 12 months). 16
Fibroblast‐specific overexpression of Smo (SmoACT ;Col1a2‐Cre‐ER transgenic mice)
To analyze the effects of activated hedgehog signaling in vivo, we generated a mouse model with fibroblast‐specific overexpression of active Smoothened (SmoACT mice). Briefly, Col1a2‐Cre‐ER mice, expressing a tamoxifen‐sensitive Cre recombinase CreERT under the control of a 6 kbp, fibroblast‐specific Col1a2 promoter fragment, 17 were cross‐bred with SmoACT mice that harbor a Cre‐inducible SmoACT mutation (W539L point mutation) to generate SmoACT Col1a2‐Cre‐ER double transgenic mice. Upon induction of recombination by tamoxifen at the age of 4 weeks, these mice overexpress SmoACT selectively in fibroblasts, resulting in increased dermal thickness, myofibroblast counts, and collagen content after 8 weeks. Treatment with the cJUN/AP‐1 inhibitor T5224 was performed in a preventive manner and started 1 week after tamoxifen application at the age of 5 weeks. Control mice were injected with oil, the solvent of T5224.
TBRACT mice
To induce dermal fibrosis by selective activation of transforming growth factor β (TGFβ) signaling, attenuated adenoviruses encoding for a constitutively active TGFβ receptor type I (adTBR) were injected at a concentration of 6.67 × 107 infectious units into the upper back of 4‐week‐old male C57BL/6 mice. Injections of transgene beta‐galactosidase (lacZ) by replication‐deficient adenoviral vectros (adLacZ) served as controls. 18 Injections were performed four times with two weekly intervals. Mice were treated with T5224 (3 or 30 mg/kg/day, oral gavage), GANT‐61 (2 or 20 mg/kg/day, intraperitoneally) or solvents in a preventive manner. The application of T5224 and GANT‐61 was started in parallel with the first injection of adTBR or adLacZ.
Fibroblast‐specific knockdown of Gli2
Mice with conditional alleles of GLI2 (GLI2fl/fl) were cross‐bred with Col1a2‐Cre‐ER mice to generate GLI2fl/fl;Col1a2‐Cre‐ER mice. Cre‐mediated recombination was induced at the age of 4 weeks by injection of tamoxifen (100 mg/kg for 5 days); oil, the solvent of tamoxifen, was used for controls. In addition, GLI2fl/fl;Col1a2‐Cre‐ER mice were injected with attenuated adenoviruses encoding for a constitutively active TGFβ receptor type I (adTBR) as described in the previous section. 3 Injections of adLacZ served to control for the adTBR virus injections. 18 Further methods are described in Supplementary Materials and Methods.
RESULTS
Concurrent up‐regulation of cJUN and GLI2 in fibroblasts in SSc skin
cJUN is the AP‐1 family member with the highest expression levels in SSc fibroblasts, and GLI2 is the predominant hedgehog transcription factor in SSc. 3 , 5 We thus analyzed the expression patterns of cJUN and GLI2 as readouts for activation of both pathways in SSc skin. The expression of cJUN and GLI2 were both significantly increased in patients with SSc with progressive skin fibrosis as compared to matched healthy individuals with increased numbers of cJUN‐ and GLI2‐positive fibroblasts (Figure 1A). Of particular note, the numbers of cJUN‐ and GLI2‐double‐positive fibroblasts were highly increased in SSc, with >50% of SSc fibroblasts coexpressing cJUN and GLI2 compared to 27% of fibroblasts in healthy controls. Confocal microscopy confirmed the concomitant up‐regulation of cJUN and GLI2 in fibroblasts in SSc compared to normal skin (Figure 1B), confirming simultaneous activation of cJUN/AP‐1 and hedgehog/GLI2 signaling in SSc fibroblasts. In addition to fibroblasts, we investigated the expression of cJUN and GLI2 in relation to CD31+ cells and CD45+ cells using imaging mass cytometry (Supplementary Figure S1). We observed a tendency of up‐regulation of cJUN and GLI2 in both CD31+ cells and CD45+ cells; however, the differences were not statically significant and not as pronounced as in fibroblasts. Although the lack of statistically significant differences in expression levels does not exclude functional effects, we thus focused our studies on fibroblasts that demonstrate higher expression levels and statistically significant differences between patients and controls.
Figure 1.
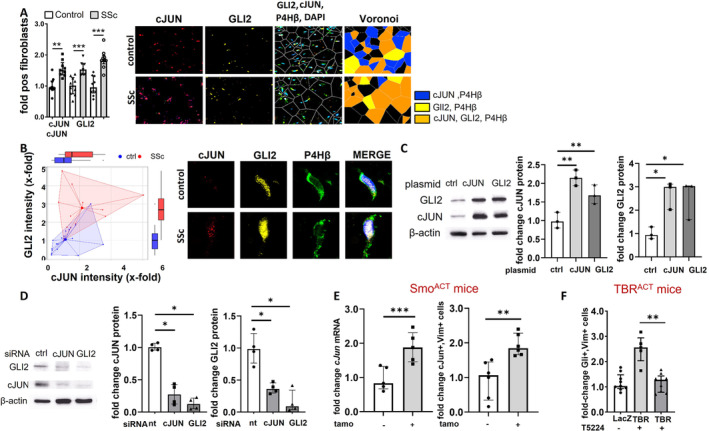
cJUN and GLI2 are concomitantly overexpressed in SSc and induce their mutual expression. (A) Concomitant up‐regulation of cJUN and GLI2 in lesional fibroblasts in SSc versus healthy participants. Representative images of immunofluorescence staining for cJUN (red), GLI2 (yellow), and co‐staining with P4Hβ (green) and DAPI (blue) in the dermis of healthy donors (n = 5; 1–2 technical replicates) and patients with dcSSc (n = 5; 1–2 technical replicates) patients at 1,000‐fold magnification. Voronoi diagrams and quantification of cJUN‐positive fibroblasts, GLI2‐positive fibroblasts, and cJUN/GLI2‐double‐positive fibroblasts are included as bar graphs. (B) Representative fibroblast at 10,000‐fold magnification using confocal microscopy. Quantification: Dots represent individual cells from a total of four patients and four controls. (C,D) Mutual regulation of expression levels of cJUN and GLI2 in fibroblasts upon overexpression (panel C; n = 3 per group) or knockdown by siRNA (panel D; n = 4 per group). (E) Increased expression of cJun in SmoACT mice compared to control mice on mRNA level (n = 5/group) and protein level as assessed by quantification of cJun,Vim+ mesenchymal cells (n = 6/group). (F) Decreased numbers of Gli2‐positive mesenchymal cells in TBRACT mice upon inhibition of cJun/AP‐1 signaling by T5224 (n = 5; 1–2 technical replicates). Data are presented as median with interquartile range. *P < 0.05; **P < 0.01; ***P < 0.001. ctrl, control; dcSSc, diffuse Systemic Sclerosis; LacZ, transgene beta‐galactosidase; mRNA, messenger RNA; nt, nontargeting; siRNA, small interfering RNA; SSc, systemic sclerosis; tamo, tamoxifen; TBR, TGFβ receptor I.
Mutual induction of cJUN and GLI2 signaling in fibroblasts
Next, we aimed to analyze whether GLI2 and cJUN regulate each other. Thus, we overexpressed cJUN and GLI2 in cultured fibroblasts, respectively, and analyzed the expression of GLI2 and cJUN by Western blot: Overexpression of cJUN resulted in the up‐regulation of GLI2, and overexpression of GLI2 induced cJUN expression (Figure 1C). Consistently, we observed a down‐regulation of GLI2 upon knockdown of cJUN and, vice versa, a down‐regulation of cJUN upon GLI2 knockdown (Figure 1D). These results indicate that cJUN and GLI2 mutually regulate the expression of each other. To confirm the mutual regulation of cJun and Gli2 in vivo, we analyzed the expression of cJun and the number of cJun‐positive fibroblasts in a mouse model characterized by the fibroblast‐specific overexpression of a constitutively active Smoothened mutant (SmoACT): Messenger RNA (mRNA) levels of cJun and the number of cJun‐positive mesenchymal cells was significantly increased in SmoACT mice compared to control mice (Figure 1E and Supplementary Figure 2). Other members of the AP‐1 family such as JunB, JunD, FosB, FosC, and Fra2 were not differentially expressed in SmoACT mice (Supplementary Figure 3), highlighting the specific interaction of Gli2 with cJun. Consistently, fibroblast‐specific knockout of Gli2 prevented the induction of cJun in mice overexpressing a constitutively active TGFβ receptor type I (TBRIACT) (Supplementary Figure 4A). In contrast to cJun knockout mice, mice overexpressing cJun under a ubiquitous H2kB promoter do not demonstrate a clinical phenotype, 11 indicating that the overexpression of cJun can be compensated by endogenous mechanisms. Thus, overexpression models may not be suitable options to study the effects of chronic activation of cJun. However, we have previously shown that TGFβ induces the expression of cJUN in SSc fibroblasts. 3 We thus used mice with overexpression of a constitutively active TGFβ receptor 1 (TBRACT mice) upon treatment with adTBR virus to activate cJun/AP‐1 signaling. The number of Gli2‐positive mesenchymal cells was increased in TBRACT mice compared to control mice treated with adLacZ virus (Figure 1F). The inhibition of cJun signaling by the selective AP‐1 inhibitor T5224 6 prevented the increase in the number of Gli2‐positive mesenchymal cells in the skin of TBRACT mice (Figure 1F). Moreover, treatment with T5224 also reduced the levels of Ptch2 mRNA, a prototypical GLI‐target gene, in murine skin of SMOACT mice (Supplementary Figure 4B). These results support the regulatory role of cJun/AP‐1 signaling on Gli2/hedgehog signaling.
Mechanisms of mutual amplification of cJUN and GLI2
We next tested the hypothesis that cJUN and GLI2 may activate the transcription of each other by binding to specific binding motifs in their promoters. We found 12 GLI2 binding sites in the cJUN promotor (−1,000 to +5,000 kb) (Supplementary Figure 4). AP‐1‐dependent reporter assays experimentally confirmed an increase in AP‐1 reporter activity upon stimulation with sonic hedgehog cytokine (SHH).
We also identified seven JUN binding sites in the GLI2 promotor. Moreover, inhibition of cJUN signaling by treatment with T5224 prevented the induction of GLI‐reporter activity by TGFβ in Light2 reporter cells (Supplementary Figure 5). In extension to these experiments, we analyzed DNA binding of GLI2 upon overexpression of cJUN with or without TGFβ stimulation using chromatin immunoprecipitation sequencing (ChIPSeq). We also analyzed DNA binding of cJUN upon GLI2 overexpression by ChIPSeq. Overexpression of cJUN resulted in a 4.6‐fold increase in DNA binding/gene detection of GLI2 (Supplementary Figure 6A). Overexpression of GLI2 had milder effects on DNA binding/gene detection of cJUN, with 1.5‐fold increases (Supplementary Figure 6A), which might be due to higher DNA binding of cJUN in general. Gene ontology term enrichment analysis indicated that DNA binding occurred at genes with relevance to fibrotic tissue remodeling, such as “extracellular matrix organization,” “Notch signaling,” and “vascular smooth muscle cell differentiation” (upon GLI2 overexpression and precipitation for cJUN) and “cytoskeletal organization” and “cell‐cell junction” (upon cJUN overexpression and precipitation for GLI2; Supplementary Figure 6B).
Inhibition of cJUN/AP‐1 signaling ameliorates hedgehog‐induced fibroblast activation and skin fibrosis
Overexpression of constitutively active smoothened (Smo) induced a selective activation of hedgehog signaling with increased mRNA levels of the hedgehog target gene Ptch2 in the skin of mice, without effects on other developmental pathways such as Wnt signaling with its target genes Axin2 and c‐Myc (Figure 2A). The selective activation of hedgehog signaling in SmoACT mice by tamoxifen was sufficient to induce skin fibrosis with increased dermal thickness, accumulation of myofibroblasts, and higher hydroxyproline content compared to control littermates injected with corn oil (Figure 2A) 8 weeks after the application of tamoxifen. Of note, the concomitant inhibition of cJun/AP‐1 signaling using the cJun/AP‐1 inhibitor T5224, which was applied daily starting from the day of the first application of tamoxifen, completely prevented SmoACT‐induced skin fibrosis with dermal thickness, myofibroblast counts, and hydroxyproline content comparable to those in nonfibrotic control mice (Figure 2A). Skin thickening was reduced by 103% (p=0.0043) in SmoACT mice treated with T5224 compared to SmoACT mice treated with corn oil.
Figure 2.
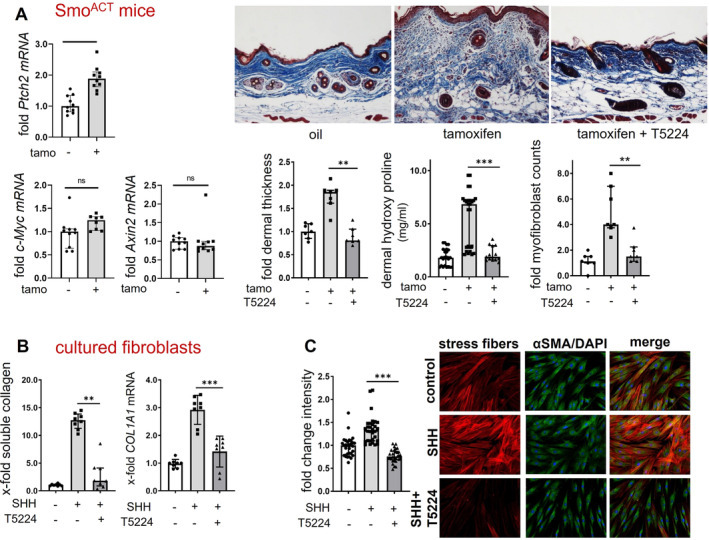
Pharmacologic inhibition of cJUN/AP‐1 ameliorates hedgehog‐induced fibroblast activation and skin fibrosis. (A) Increased mRNA levels of the hedgehog target gene Ptch2 compared to other target genes (n = 4–5, technical duplicates) in SmoACT mice. Effects of treatment with the cJun/AP‐1 inhibitor T5224 on dermal thickness (n = 7/group), dermal hydroxyproline content (n = 7/group; 1–2 technical duplicates), and dermal myofibroblast counts (n = 7/group) in SmoACT mice. Representative images (200‐fold magnification; staining: Masson‐Trichrome) are shown. (B) Treatment with T5224 reduces the release of soluble collagen and the mRNA levels of COL1A1 in cultured fibroblasts stimulated with SHH (n = 4, technical duplicates). (C): T5224 ameliorates the formation of stress fibers in SHH‐stimulated fibroblasts (n = 3 cell lines with 10 random high‐power fields quantified each). Representative images of stress fibers visualized by rhodamine‐phalloidin staining (red), α‐smooth muscle actin (green), and DAPI (blue) are shown at 2,000‐fold magnification. Data are presented as median with interquartile range. *P < 0.05; **P < 0.01; ***P < 0.001. mRNA, messenger RNA; tamo, tamoxifen; SHH, Sonic Hedgehog; αSMA, αSmooth Muscle Actin. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42979/abstract.
Inhibition of cJUN/AP‐1 signaling also blocked GLI2‐induced fibroblast activation and collagen release in human dermal fibroblasts. Incubation of cultured human fibroblasts with recombinant SHH increased the levels of COL1A1 mRNA and the release of collagen protein and induced fibroblast‐to‐myofibroblast transition with increased expression of αSmooth Muscle Actin (αSMA) and of stress fibers (Figure 2B and 2C). These profibrotic effects of SHH were blocked by treatment with cJUN/AP‐1 inhibitor T5224, which returned the levels of collagen and the expression of myofibroblast markers to those of resting fibroblasts (Figure 2B and 2C).
Effects of combined inhibition of cJUN and GLI2 signaling in experimental fibrosis
Selective inhibitors of cJUN and of GLI2 are in advanced clinical development 3 , 5 ; however, long‐term application of these inhibitors, as would be required in chronic fibrotic diseases such as SSc, may be complicated by adverse events. 19 , 20 We thus evaluated whether the combined inhibition of cJun/AP‐1 and Gli2/hedgehog at lower doses may exert additive antifibrotic effects and better tolerability as compared to monotherapies with higher doses of cJun/AP‐1 or Gli2/hedgehog inhibitors.
Treatment with T5224 ameliorated adTBR‐induced fibrosis in a dose‐dependent manner. Dose‐dependent antifibrotic effects were also observed for the Gli2 inhibitor GANT61 (Figure 3). Combination therapy with lower doses of T5224 and of GANT61 were more effective than monotherapies, with the same doses of T5224 or GANT61 with more pronounced reductions in dermal thickening, in myofibroblast counts, and in hydroxyproline content (Figure 3). The antifibrotic effect of the combination therapy of low doses of T5224 and GANT61 was comparable to that of higher doses of T5224 or GANT‐61 (Figure 3).
Figure 3.
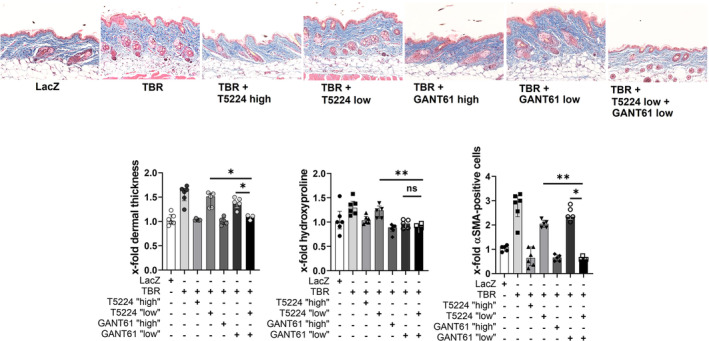
Combined inhibition of cJun/AP‐1 and Gli2/hedgehog signaling exerts additive antifibrotic effects. Treatment of TBRACT mice with T5224 high (30 mg/kg), T5224 low (3 mg/kg), GANT61 high (20 mg/kg), GANT61 low (2 mg/kg/d), or a combination of T5224 low and GANT61 low (n = 3‐6 per group). Representative images of trichrome staining at 200‐fold magnification are shown. Quantification of dermal thickness, hydroxyproline content, and myofibroblasts are shown as bar graphs. Data are presented as median with interquartile range. *P < 0.05; **P < 0.01; ***P < 0.001. LacZ, transgene beta‐galactosidase; αSMA, αSmooth Muscle Actin; TBR, TGFβ receptor I. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42979/abstract.
DISCUSSION
Our study demonstrates that simultaneous induction and amplification of two core pathways of fibrotic tissue remodeling, cJUN/AP‐1 and GLI2/hedgehog signaling, enables fibroblasts to escape their physiologic regulation.
The mutual activation of cJUN/AP‐1 signaling and GLI2/hedgehog signaling is of direct relevance for fibroblast activation and tissue fibrosis. We show that breaking the mutual activation loop by inhibition of cJun in SmoACT mice with hyperactive hedgehog/Gli2 signaling normalizes Gli2/hedgehog‐induced fibroblast activation and tissue fibrosis. Moreover, we also demonstrate that the combined pharmacologic inhibition of cJun and of Gli2 exerts additive antifibrotic effects. Combination therapies are standard regimens in numerous diseases, including rheumatic diseases, as they offer potential for additive efficacy or for dose reduction of individual drugs to allow better tolerability. 21 , 22 However, despite those potential advantages and their widespread use in other diseases, combination therapies are not yet part of the clinical concept in fibrotic disorders. Our study provides first evidence for positive effects of a combination of cJun/AP‐1 inhibitors with Gli2/hedgehog inhibitors in a preclinical model of SSc. Combined inhibition of both pathways demonstrated more potent antifibrotic effects than application of individual inhibitors. The antifibrotic effects of combination therapies with lower doses of inhibitors are comparable to higher doses of monotherapies, but they may not be complicated by their potential adverse events such as increased apoptosis and stem cell depletion, even upon long‐term treatment, as is required for antifibrotic treatment. Indeed, combination therapies of T5224 and GANT‐61 were well‐tolerated in our study, without clinical or histopathologic evidence of obvious toxicity in the preclinical model.
Our study has some limitations: SSc is a complex and heterogenous disease, which is difficult to model in preclinical models. Here, we analyzed the antifibrotic effects of combined inhibition of Gli2/hedgehog signaling and cJun/AP‐1 signaling in a mouse model characterized by the constitutive activation of TGFβ signaling, a central mediator of fibrotic disease. However, other features of SSc such as autoimmunity or inflammation are not predominantly reflected by this model. To address potential additive effects of combined inhibition of Gli2/hedgehog signaling and cJun/AP‐1 signaling on these disease characteristics would require additional models. Moreover, advanced humanized in vitro models such as three‐dimensional vascularized skin grafts have recently become available that may allow us to characterize the combined inhibition of cJUN/AP‐1 and GLI2/hedgehog signaling in an additional setting 23 in the future. Despite these limitations, the concept of mutual dependence of profibrotic pathways and the rationale of their combined inhibition is clearly supported by our study.
In summary, our findings of the mutual activation and perturbation of cJUN/AP‐1 signaling and GLI2/hedgehog in SSc, its role in fibroblast activation and tissue fibrosis, and the demonstration of additive antifibrotic effects of combined inhibition of cJun/AP‐1 signaling and Gli2/hedgehog signaling may have translational implications, as cJUN/AP‐1‐inhibiting drugs are in clinical testing and hedgehog inhibitors are already in clinical use.
AUTHOR CONTRIBUTIONS
All authors contributed to at least one of the following manuscript preparation roles: conceptualization AND/OR methodology, software, investigation, formal analysis, data curation, visualization, and validation AND drafting or reviewing/editing the final draft. As corresponding author, Dr Bergmann confirms that all authors have provided the final approval of the version to be published, and takes responsibility for the affirmations regarding article submission (eg, not under consideration by another journal), the integrity of the data presented, and the statements regarding compliance with institutional review board/Helsinki Declaration requirements.
Supporting information
Disclosure form
Figure S1:
Appendix S1: Supplementary Material
ACKNOWLEDGMENTS
We thank Katja Dreißigacker, Regina Kleinlein, Saskia de Visser van Bloemen, and Wolfgang Espach for excellent technical assistance. Open Access funding enabled and organized by Projekt DEAL.
Supported by the German Research Foundation (grants DI 1537/17‐1, DI 1537/20‐1, DI 1537/22‐1, DI 1537/23‐1, BE 7036/2‐1, and BE 7036/5‐1), Sonderforschungsbereich (SFB) CRC1181 (project A01 and C01) and SFB TR221/ project number 324392634 (B04), German Research Foundation project 441730715 (Leica Stellaris 8 Microscope), German Research Foundation project number 324392634 (Drs Dickel, Liang, and Kunz), and Graduiertenkolleg (GRK) 2740 (B02 to Dr Kunz), German Research Foundation project 424726560 (imaging CyTOF), the Wilhelm‐Sander‐Stiftung (grant 2013.056.1), the Else‐Kröner‐Fresenius‐Foundation (grants 2014_A47 and 2014_A184), the ELAN‐Foundation Erlangen (grant 21‐11‐09‐1‐Bergmann), the Research Committee of the Medical Faculty of the Heinrich‐Heine University Düsseldorf (Forschungskommission ID 2023‐33 to Dr Matei, ID 2022‐18 to Dr Györfi, and ID 2023‐31 to Dr Distler), and the German Federal Ministry of Education and Research (BMBF): MASCARA program, TP 2 (01EC1903A), and iIMMUNE_ACS (01EO2105). Drs Auth and Bergmann's work was supported by NOTICE (RA 2506/7‐21), Clinician Scientist Program of the German Research Foundation. Dr Bergmann's work was supported by the Clinician Scientist Program of IZKF Erlangen. Dr Matei's work was supported by the German Research Foundation (grant MA 9219/2‐1), the Else‐Kröner‐Fresenius‐Foundation (grants 2021_EKEA.03 and 2022_EKMS.02), and The Edith Busch and World Scleroderma Foundation Research Grant Program 2022–2023. The work of Drs Kunz and Dickel was supported by the CompLS program (grant 031L0262C). Dr Kunz's work was supported by the Fraunhofer Cluster of Excellence Immune‐Mediated Diseases. Dr Distler's work was supported by an unrestricted research grant from the Hiller‐Foundation.
Additional supplementary information cited in this article can be found online in the Supporting Information section (https://acrjournals.onlinelibrary.wiley.com/doi/10.1002/art.42979).
Author disclosures are available at https://onlinelibrary.wiley.com/doi/10.1002/art.42979.
REFERENCES
- 1. Distler JHW, Györfi AH, Ramanujam M, et al. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol 2019;15:705–730. [DOI] [PubMed] [Google Scholar]
- 2. Denton CP, Khanna D. Systemic sclerosis. Lancet 2017;390:1685–1699. [DOI] [PubMed] [Google Scholar]
- 3. Liang R, Šumová B, Cordazzo C, et al. The transcription factor GLI2 as a downstream mediator of transforming growth factor‐β‐induced fibroblast activation in SSc. Ann Rheum Dis 2017;76:756–764. [DOI] [PubMed] [Google Scholar]
- 4. Horn A, Palumbo K, Cordazzo C, et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Rheum 2012;64:2724–2733. [DOI] [PubMed] [Google Scholar]
- 5. Avouac J, Palumbo K, Tomcik M, et al. Inhibition of activator protein 1 signaling abrogates transforming growth factor β‐mediated activation of fibroblasts and prevents experimental fibrosis. Arthritis Rheum 2012;64:1642–1652. [DOI] [PubMed] [Google Scholar]
- 6. Zhang F, Hao M, Jin H, et al. Canonical hedgehog signalling regulates hepatic stellate cell‐mediated angiogenesis in liver fibrosis. Br J Pharmacol 2017;174:409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schulien I, Hockenjos B, Schmitt‐Graeff A, et al. The transcription factor c‐Jun/AP‐1 promotes liver fibrosis during non‐alcoholic steatohepatitis by regulating Osteopontin expression. Cell Death Differ 2019;26:1688–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cui L, Chen SY, Lerbs T, et al. Activation of JUN in fibroblasts promotes pro‐fibrotic programme and modulates protective immunity. Nat Commun 2020;11:2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wernig G, Chen SY, Cui L, et al. Unifying mechanism for different fibrotic diseases. Proc Natl Acad Sci U S A 2017;114:4757–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wasson CW, Ross RL, Wells R, et al. Long non‐coding RNA HOTAIR induces GLI2 expression through Notch signalling in systemic sclerosis dermal fibroblasts. Arthritis Res Ther 2020;22:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jochum W, Passegué E, Wagner EF. AP‐1 in mouse development and tumorigenesis. Oncogene 2001;20:2401–2412. [DOI] [PubMed] [Google Scholar]
- 12. Kopinke D, Norris AM, Mukhopadhyay S. Developmental and regenerative paradigms of cilia regulated hedgehog signaling. Semin Cell Dev Biol 2021;110:89–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Laner‐Plamberger S, Kaser A, Paulischta M, et al. Cooperation between GLI and JUN enhances transcription of JUN and selected GLI target genes. Oncogene 2009;28:1639–1651. [DOI] [PubMed] [Google Scholar]
- 14. Lerbs T, Cui L, King ME, et al. CD47 prevents the elimination of diseased fibroblasts in scleroderma. JCI Insight 2020;5:e140458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Ann Rheum Dis 2013;72:1747–1755. [DOI] [PubMed] [Google Scholar]
- 16. Wu W, Jordan S, Graf N, et al. Progressive skin fibrosis is associated with a decline in lung function and worse survival in patients with diffuse cutaneous systemic sclerosis in the European Scleroderma Trials and Research (EUSTAR) cohort. Ann Rheum Dis 2019;78:648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Denton CP, Zheng B, Shiwen X, et al. Activation of a fibroblast‐specific enhancer of the proalpha2(I) collagen gene in tight‐skin mice. Arthritis Rheum 2001;44:712–722. [DOI] [PubMed] [Google Scholar]
- 18. Zehender A, Li YN, Lin NY, et al. TGFβ promotes fibrosis by MYST1‐dependent epigenetic regulation of autophagy. Nat Commun 2021;12:4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grund‐Gröschke S, Stockmaier G, Aberger F. Hedgehog/GLI signaling in tumor immunity—new therapeutic opportunities and clinical implications. Cell Commun Signal 2019;17:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gurzov EN, Bakiri L, Alfaro JM, et al. Targeting c‐Jun and JunB proteins as potential anticancer cell therapy. Oncogene 2008;27:641–652. [DOI] [PubMed] [Google Scholar]
- 21. Melero I, Berman DM, Aznar MA, et al. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer 2015;15:457–472. [DOI] [PubMed] [Google Scholar]
- 22. Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: a review. JAMA 2018;320:1360–1372. [DOI] [PubMed] [Google Scholar]
- 23. Matei AE, Kubánková M, Xu L, et al. Identification of a distinct monocyte‐driven signature in systemic sclerosis using biophysical phenotyping of circulating immune cells. Arthritis Rheumatol 2023:75:768–781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure form
Figure S1:
Appendix S1: Supplementary Material