

Abstract
Meningiomas are the most common primary central nervous system tumor. Clinical trials have failed to support effective medical treatments, despite initially promising animal studies. A key issue could be that available experimental models fail to mimic the clinical situation. Hence, there is a need for meningioma models with high translational value for understanding pathophysiology and tests of possible medical treatments. Resemblance between models and clinical meningiomas should be optimized with respect to morphology, immunohistochemistry and epigenetic factors, which we aimed to do. Third passage primary patient-derived benign meningiomas were implanted intracranially in athymic nude rats. The animals were euthanized after three months. We found intra- and intertumoral variability in terms of tumor take rate (79.5% for superficially implanted cells and 25% for deeply implanted cells) and xenograft sizes. There were close resemblance between primary tumors and xenografts in morphology and immunohistochemistry. Furthermore, we performed DNA-methylation using the EPIC 850 K array on three pairs of primary tumors and xenografts. Copy number variation profiles and correlation plots on CpGs showed a high degree of similarities between primary tumors and corresponding xenografts. On differential methylation analysis, most probes were insignificant (866,074), 25 were hypermethylated, and 382 were hypomethylated, where no significant differentially methylated regions were revealed.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-83456-7.
Keywords: Meningioma, Xenograft, Orthotopic tumor model, In vivo, DNA methylation profiling
Subject terms: Cancer models, Experimental models of disease, Preclinical research
Introduction
Meningiomas are the most common primary central nervous system tumor, accounting for approximately 40% of these tumors1. They originate from arachnoid cap cells associated with dura mater or choroid plexus. They are predominately benign (80%), atypical (18%), and rarely malignant (1.5%)1. Surgery remains the main treatment of symptomatic meningiomas2, unfortunately gross total resection is only possible in two-thirds of skull base meningiomas and is rarely achieved in for instance cavernous sinus and petro-clival meningiomas3. Recurrences are common and can be intractable, further highlighting the need for better understanding of pathophysiology, and thereby valid model systems to increase knowledge and test non-surgical treatments.
Despite more than 30 completed trials and several ongoing trials, effective medical compounds for refractory or recurrent meningiomas are still lacking4. Several pharmaceuticals have shown potential in meningioma animal trials5–14. However in general, the translatability of small animal models to human clinical cancer trials is low, with a translation success rate of less than 8%15. Many meningioma animal trials are performed on commercially available/established cell lines such as IOMM-Lee16, which do not mimic meningioma properties and clinical scenarios. This emphasizes the need to reassess the in vivo models used for understanding pathophysiological processes and pharmaceutical testing.
Meningiomas display heterogenous histological features, with 15 distinct subtypes identified17,18and a wide range of genetic and epigenetic profiles9,19–25. Furthermore, meningiomas originate from both mesoderm and neural crest26with varying gene expression depending on location27,28. This contributes to the challenges in translating research findings to human conditions, and the animal models used for pharmaceutical testing should ideally reflect this heterogeneity of meningiomas as a group. Among numerous meningioma animal models16, the orthotopic xenograft model with tumor tissue/cells remains the closest representation of human tumors in vivo despite the complete or partial lack of recipient immune system. Only a few studies have successfully established an orthotopic meningioma model with primary meningioma cells with preserved and overall comparable immunohistochemical profiles5,29–33. However, only two research groups have published successful use of benign meningioma cells in mice models with reported tumor take rates of 54–100%5,29,30.
Establishing suitable animal disease models is crucial in translational medical science, and rats and mice have emerged as the preferred animals. Mice have long been preferred due to small size, cost-effectiveness, and flexibility in gene editing, which are useful factors in meningioma models16. However, rats are closer to humans physiologically, morphologically, and genetically and should be ideal for biomedical and clinical research34.
The main objective of our study was to establish the first orthotopic meningioma rat model based on histologically verified primary benign meningiomas. We aimed to: (1) Explore tumor take rates in a superficial implantation model at the convexity and a deep implantation model at the skull base, (2) Analyze morphological and immunohistochemical differences and similarities between primary tumors and corresponding xenografts, and, (3) Compare epigenetic profiles using DNA methylation.
Materials and methods
Approvals and declarations
The study was approved by the Regional Committees on Health Research Ethics for Southern Denmark (S-20190105). The study was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki and in compliance to regional guidelines and regulations. Animal experiments were approved by the Danish Animal Experiments Inspectorate (2019-15-0201-00195) and all experiments were conducted in accordance to local guidelines, national regulations and to those of the Danish 3R-Center. This study was reported in compliance with ARRIVE 2.0 The Recommended Set35.
Patient inclusion and meningioma tissue handling
Patients ≥ 18 years with an MRI-suspected meningioma were eligible for inclusion. Informed consent was obtained from all subjects prior to surgery and extraction of primary tumor material. The meningioma diagnosis was confirmed in accordance with guidelines17 prior to implantation into animals. Samples were obtained during surgery and transferred to Dulbecco’s Eagle Medium (DMEM AQ) (D0819, Sigma, Merck KgaA, Germany) with additions of 10% fetal bovine serum (FBS) (10500064, Thermo Fisher Scientific Inc., MA, USA), 1% Penicillin/Streptomycin (15140122, Thermo Fisher Scientific Inc., MA, USA), 4 mM GlutaMax (35050038, Thermo Fisher Scientific Inc., MA, USA), 1 mM Sodium Pyruvate (11360039, Thermo Fisher Scientific Inc., MA, USA), and Progesterone 0.025 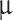 g/ml (P7556, Sigma, Merck KgaA, Germany). Primary tumor samples were washed twice using PBS (10140169, Thermo Fisher Scientific Inc., MA, USA). Tumors were mechanically dissociated to pieces of approximately 1 mm3 and resuspended in the same medium as described above and transferred to T75 cell culture flasks (75 cm2). Cells were incubated at 37 C
g/ml (P7556, Sigma, Merck KgaA, Germany). Primary tumor samples were washed twice using PBS (10140169, Thermo Fisher Scientific Inc., MA, USA). Tumors were mechanically dissociated to pieces of approximately 1 mm3 and resuspended in the same medium as described above and transferred to T75 cell culture flasks (75 cm2). Cells were incubated at 37 C /5% CO2 in a humidified atmosphere. The medium was changed 2–3 times each week and cells were trypsinized and passaged at 70% confluence using TrypLExpress (12605028, Thermo Fisher Scientific Inc., MA, USA). Third-passage cells were used for implantation. Remaining cells were embedded in fibrin clots using plasma and thrombin in a 1:1 ratio and subsequently paraffin-embedded for further histological and immunohistochemical analyses.
/5% CO2 in a humidified atmosphere. The medium was changed 2–3 times each week and cells were trypsinized and passaged at 70% confluence using TrypLExpress (12605028, Thermo Fisher Scientific Inc., MA, USA). Third-passage cells were used for implantation. Remaining cells were embedded in fibrin clots using plasma and thrombin in a 1:1 ratio and subsequently paraffin-embedded for further histological and immunohistochemical analyses.
Animals
We used 80 Crl: NIH-Fox1rnu – athymic, female, nude rats homozygous, 4–6 weeks old (5–7 at implantation) (Charles River Laboratories, Germany) with mean weight of 167 g (SD 20.6). The animals were housed at the animal facility of the University of Southern Denmark in the specific pathogen-free section with humidity of 53%, temperature of 22 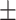 1C
1C , and a 12-hour light/darkness cycle. Animals were kept in Type IV cages with four animals per cage and access to food (Altromin 1324, Brogaarden, Denmark) and water ad libitum. Animals were weighed and inspected for well-being and neurological symptoms twice a week during the entire course of the experiments in accordance with animal approvals. Predetermined exclusion criteria included humane endpoints: weight loss > 20% compared to animals of the same breed and similar age and sex, visible pain, or debilitating neurological symptoms. Animals with untimely death would be excluded from final analyses. See Supplemental Material (SM1A) for an overview of all animals used in the study.
, and a 12-hour light/darkness cycle. Animals were kept in Type IV cages with four animals per cage and access to food (Altromin 1324, Brogaarden, Denmark) and water ad libitum. Animals were weighed and inspected for well-being and neurological symptoms twice a week during the entire course of the experiments in accordance with animal approvals. Predetermined exclusion criteria included humane endpoints: weight loss > 20% compared to animals of the same breed and similar age and sex, visible pain, or debilitating neurological symptoms. Animals with untimely death would be excluded from final analyses. See Supplemental Material (SM1A) for an overview of all animals used in the study.
In vivo design and surgical implantation
Pilot studies with various cell numbers, concentrations, and implantation methods were conducted to determine the following approach. This included sacrificing two animals randomly selected (one from each group from the first round of animals) to assess tumor volume and localization due to previous experiments with no tumor formation.
With respect to the significant risk of publication bias36, a resource equation was used to determine number of animals as a means of sample size assessment37and was cross-referenced with previous successful studies using benign meningioma cells5,29,30.
Third-passage cells were harvested (trypsinized and centrifuged at 180 G for 5 minutes) and resuspended in medium at a concentration of 105 cells/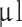 . Before surgery, animals were administered Temgesic (buprenorfin) (2CARE4 Aps, Denmark) in a mix of 0.2 mg/1.0 g food in a dose of 0.4 mg/kg per animal. Animals were anesthetized subcutaneously with Hypnorm (Skanderborg Apotek (Skanderborg Pharmacy), Denmark) (Fentanyl, 0.315 mg/ml; Fluanisone 10 mg/ml) and Midazolam (Hameln Pharma Aps, Denmark) (5 mg/ml) in a 1:1 ratio matched with same volume sterile volume in a dose of 0.3 ml/100 g bodyweight. Animals were placed in a small animal stereotaxic frame (David Kopf Instruments, CA, USA). For experimental setup, see Fig. 1A. A 0.5–1 cm midline incision was performed, and bregma was identified. A burr hole was placed 2 mm to the right and 1 mm anterior to bregma after local administration of 0.1 ml Lidocaine 10 mg/ml. Drilling was performed carefully to ensure a tight fit around the needle to avoid backflow of cells during and after implantation. 10
. Before surgery, animals were administered Temgesic (buprenorfin) (2CARE4 Aps, Denmark) in a mix of 0.2 mg/1.0 g food in a dose of 0.4 mg/kg per animal. Animals were anesthetized subcutaneously with Hypnorm (Skanderborg Apotek (Skanderborg Pharmacy), Denmark) (Fentanyl, 0.315 mg/ml; Fluanisone 10 mg/ml) and Midazolam (Hameln Pharma Aps, Denmark) (5 mg/ml) in a 1:1 ratio matched with same volume sterile volume in a dose of 0.3 ml/100 g bodyweight. Animals were placed in a small animal stereotaxic frame (David Kopf Instruments, CA, USA). For experimental setup, see Fig. 1A. A 0.5–1 cm midline incision was performed, and bregma was identified. A burr hole was placed 2 mm to the right and 1 mm anterior to bregma after local administration of 0.1 ml Lidocaine 10 mg/ml. Drilling was performed carefully to ensure a tight fit around the needle to avoid backflow of cells during and after implantation. 10 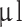 of cell suspension (7.5 × 104−105 cells/
of cell suspension (7.5 × 104−105 cells/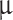 l) was injected using a customized 26G blunt-tipped, 10
l) was injected using a customized 26G blunt-tipped, 10 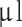 Hamilton syringe (Hamilton Company, NV, USA). The needle was kept in place for 1 min before injecting the cell suspension (1
Hamilton syringe (Hamilton Company, NV, USA). The needle was kept in place for 1 min before injecting the cell suspension (1 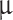 l/30 seconds). Following injection/implantation, the needle was kept in place for 1 min before slow retraction (1mm/30 seconds). The burr hole was sealed with bone wax, and the skin was sutured with absorbable suture 5−0. Post-surgery anesthetics comprised 0.5 mg/100 g subcutaneous Carprofen (Rimayl) (Zoetis Animal Health ApS, NJ, USA). Animals were randomly selected to receive either superficial implantation at the convexity (2 mm deep from the top of the bone) or deep implantations at the skull base (which was found at 7.6 mm (SD 0.88)). Animals were euthanized 3 months after implantation using 200 mg/kg IV pentobarbital (Exagon® Vet., 400 mg/ml) (Salfarm Danmark A/S, Denmark). Animal handlers were blinded to the implantation site.
l/30 seconds). Following injection/implantation, the needle was kept in place for 1 min before slow retraction (1mm/30 seconds). The burr hole was sealed with bone wax, and the skin was sutured with absorbable suture 5−0. Post-surgery anesthetics comprised 0.5 mg/100 g subcutaneous Carprofen (Rimayl) (Zoetis Animal Health ApS, NJ, USA). Animals were randomly selected to receive either superficial implantation at the convexity (2 mm deep from the top of the bone) or deep implantations at the skull base (which was found at 7.6 mm (SD 0.88)). Animals were euthanized 3 months after implantation using 200 mg/kg IV pentobarbital (Exagon® Vet., 400 mg/ml) (Salfarm Danmark A/S, Denmark). Animal handlers were blinded to the implantation site.
Fig. 1.

Methods. (A) Experimental setup with the small-animal stereotactic frame and microscope. The middle image displays an animal fixed with three fixed points: two pins in the ear canals and one clamp on the nose. The last image (to the right) is a microsurgical view of the syringe in the skull of an animal through the burr hole. (B) Whole-brain extraction post-mortem, and preparation for histological and immunohistochemical analyses.
Histology and immunohistochemistry
Tumor samples from patients were obtained during surgery were immediately fixed in 4% neutral-buffered formaldehyde for 24 h before being paraffin-embedded. After euthanasia, whole rat brains were extracted within 10 min post-mortem and fixed in 4% neutral-buffered formaldehyde for 24 h. Brain sections of 1 mm were hand-cut using a guide (Fig. 1B) and were then paraffin-embedded. All paraffin-embedded samples (primary tumor, rat brain sections, and third-passage cells in fibrin clots) were cut into 3 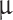 m sections on a microtome. Sections were stained with Hematoxylin (CS701, Dako, Denmark)/Eosin (CS700, Dako, Denmark) (HE) and bluing buffer (CS702, Dako, Denmark) on a Dako Coverstainer (Dako, Denmark). After heat-induced-epitope retrieval (protocol depending on antibody), tumor sections were stained with the following antibodies: epithelial membrane antigen (EMA) (Clone E29, 1:100, 20 min/32oC, Dako, Denmark), somatostatin receptor type 2 (SSTR2) (Clone EP149, 1:400, 20 min/32oC, Cell Marque, CA, USA), progesterone receptor (PR) (Clone 1E2, ready-to-use (RTU), 12 min/36oC, Ventana Medical Systems, Inc., AZ, USA), KI67 (Clone 30 − 9, RTU, 16 min/36oC, Ventana Medical Systems, Inc., AZ, USA), glial fibrillary acidic protein (GFAP) (Catalog Z0334, 1:4000, 24 min/36oC, Dako, Denmark) and Nestin (Clone 196908, Catalog MAB1259, 1:5000, 16 min/36oC, R&D Systems, MN, USA). EMA and SSTR2 stainings were performed on the Omnis Platform (Dako, Denmark), and PR, KI67, GFAP and Nestin stainings were performed on the BenchMark Ultra Platform (Ventana Medical Systems, Inc., AZ, USA). Only tumor-bearing animals based on HE assessment received selected immunohistochemical stainings as described above.
m sections on a microtome. Sections were stained with Hematoxylin (CS701, Dako, Denmark)/Eosin (CS700, Dako, Denmark) (HE) and bluing buffer (CS702, Dako, Denmark) on a Dako Coverstainer (Dako, Denmark). After heat-induced-epitope retrieval (protocol depending on antibody), tumor sections were stained with the following antibodies: epithelial membrane antigen (EMA) (Clone E29, 1:100, 20 min/32oC, Dako, Denmark), somatostatin receptor type 2 (SSTR2) (Clone EP149, 1:400, 20 min/32oC, Cell Marque, CA, USA), progesterone receptor (PR) (Clone 1E2, ready-to-use (RTU), 12 min/36oC, Ventana Medical Systems, Inc., AZ, USA), KI67 (Clone 30 − 9, RTU, 16 min/36oC, Ventana Medical Systems, Inc., AZ, USA), glial fibrillary acidic protein (GFAP) (Catalog Z0334, 1:4000, 24 min/36oC, Dako, Denmark) and Nestin (Clone 196908, Catalog MAB1259, 1:5000, 16 min/36oC, R&D Systems, MN, USA). EMA and SSTR2 stainings were performed on the Omnis Platform (Dako, Denmark), and PR, KI67, GFAP and Nestin stainings were performed on the BenchMark Ultra Platform (Ventana Medical Systems, Inc., AZ, USA). Only tumor-bearing animals based on HE assessment received selected immunohistochemical stainings as described above.
The NanoZoomer 2.0 digital image scanner (Hamamatsu Photonics, Japan) was used to digitize stained slides for subsequent semiquantitative assessment by two neuropathologists (MWN and JKP). We included tumors (positive tumor take rate) displaying specific meningioma morphology using HE staining. To corroborate the diagnosis and facilitate comparison with primary tumors, immunohistochemistry was performed in selected cases. Animals identified with diffuse meningiomatosis and those with insufficient tumor formation were excluded. For examples of the smallest included tumors and excluded cases of meningiomatosis and gliosis, see Supplemental Material 3 (SM3).
To estimate size, we used coronal histological sections and applied the trapezoidal rule, a linear approximation method. This method calculates the volume by taking into account the average lesion area and the distance38. NanoZoomer 2.0 digital image scanner’s Freehand Region tool was used to delineate tumors on the HE staining to achieve individual areas (see Supplemental Material (SM4) for examples). Tumors (xenografts) spanning multiple sections were delineated separately. Tumor volume was calculated by adding the tumor area of each 1 mm section. During assessment, the neuropathologists were blinded to one another in terms of the subtype of meningioma and the scoring of immunohistochemical staining (0, + 1, +2, + 3), where 0 was no staining, + 1 was weak staining, + 2 was moderate staining, and + 3 was strong staining. Assessors were considered in agreement if their scores differed by no more than one grade (e.g., 1 and 1, or 2 and 3). When scores differed by one grade, the result was reported as a range (see results). All disagreements (>1 grade scoring e.g. 1 and 3) were resolved through a consensus meeting between the neuropathologists. All KI67 results for primary tumors, brain slices, and implanted cells were discussed prior to final scoring. Individual and consensus scorings are available in SM1A.
DNA methylation protocol and analyses
Formalin-fixed paraffin-embedded (FFPE) primary tumor pieces were cut into two or three 10 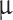 m sections and subjected to DNA extraction using QIAamp DNA FFPE Tissue Kit (QIAGEN, Germany) as described in Supplemental Material (SM2). FFPE brains from tumor-bearing animals were macro-dissected with a 15-blade scalpel to divide brain from tumor. Depending on tumor sizes, tumor tissue from more than one animal (same primary tumor) was pooled to ensure sufficient tissue for analyses.
m sections and subjected to DNA extraction using QIAamp DNA FFPE Tissue Kit (QIAGEN, Germany) as described in Supplemental Material (SM2). FFPE brains from tumor-bearing animals were macro-dissected with a 15-blade scalpel to divide brain from tumor. Depending on tumor sizes, tumor tissue from more than one animal (same primary tumor) was pooled to ensure sufficient tissue for analyses.
Primary tumors and corresponding xenografts underwent DNA extraction and subsequent DNA methylation profiling on the same EPIC 850k chip to ensure uniformity. A full stepwise protocol is available in Supplemental Material (SM2). In brief, Quality Check of DNA was performed using the Infinium HD FFPE Methylation Assay (Illumina inc., CA, USA). Bisulfite conversion was performed using EZ DNA Methylation kit (Zymo Research Corp. CA, USA). Restoration of DNA was performed by first breaking down DNA using Infinium HD Assay Kit FFPE – Restore Kit (Illumina inc., CA, USA) and ZR-96 DNA Clean and Concentrator kit (Zymo Research Corp. CA, USA). DNA was amplified using Infinium HD FFPE Methylation Assay amplification protocol (Illumina inc., CA, USA). Methylation data were collected on the iScan System (Illumina inc., CA, USA). IDAT files were uploaded to the DNA methylation-based CNS classifier39 (https://www.molecularneuropathology.org/mnp/), and the Brain Tumor Classifier (v12.5) was used to assess classification and copy number variation.
Statistical analysis was performed using RStudio Pro 2022.12.0 + 353.pro20 based on R version 4.1.2 (R-Tools Technology Inc., ON, Canada). DNA methylation data was processed using a modified version of the minfi package. The pfilter and dasen function from the wateRmelon package were used to filter samples and CpG sites with bad quality and subsequent normalization. Probes association with sex chromosomes or SNPs or reported cross-reactivity were filtered using the rmSNPandCH function from the DMRcate package. The 2,000 most variable CpGs were selected based on the highest standard deviation across all samples. Heatmaps were generated using the ComplexHeatmap package on b-values. We conducted a pvclust bootstrap analysis with 200 resampling iterations on our dataset to evaluate the stability and reliability of the clustering results. This involved generating 200 bootstrap samples from the original data and performing unsupervised hierarchical clustering on each sample to assess the robustness of the obtained clusters, with the pvclust package providing statistical support through approximately unbiased (AU) p-values. Differentially methylated positions (DMPs) (differentially methylation CpG sites) were identified using moderated t-test as implemented in the limma package and using the Benjamini-Hochberg method for adjusted p-values<0.05 for multiple testing correction on M-values. Differentially methylated regions (DMRs) were calculated using the DMRcate method. Finally, we performed correlation plots for the primary tumor and corresponding xenograft focusing on the CpGs relevant for the v12.5 BrainClassifier. Finally, a functional gene enrichment analysis of DMPs, focusing on potential gene overlaps, was performed using the Molecular Signatures Database (MsigDB) through Gene Set Enrichment Analysis (GSEA) developed by the Broad Institute40 with a cut-off of FDR (False Descriptive Rate) q-value < 0.05.
Statistical considerations
Other descriptive statistics (mean, SD, median) were performed using GraphPad Prism 9.5.1 (GraphPad Software, MA, USA). Mann Whitney U test was performed on non-parametric data.
Results
Patient samples
All primary tumor samples were WHO-grade 1 benign PR-positive meningiomas based on the WHO classification of central nervous system tumors 5th edition from 202117. The four patients (referred to as Meningioma Odense (MO) patients) were women aged 63–80 years) (Table 1).
Table 1.
Sample characteristics.
| Sample | Age | Sex | Location | WHO-Grade | Tumor subtype |
|---|---|---|---|---|---|
| MO-4 | 63 | Female | Sphenoid wing | 1 - Benign | Translational |
| MO-12 | 72 | Female | Convexity | 1 - Benign | Microcystic |
| MO-25 | 64 | Female | Falx | 1 - Benign | Transitional |
| MO-27 | 80 | Female | Olfactory | 1 - Benign | Fibrous |
Validation of tumor cell implantation and animal well-being throughout experiments
Two animals were sacrificed after one week to assess the implantation method of MO-04 experiments. There was no definite tumor formation for either skull base or convexity implantations but clear cell clustering, scarce cytoplasmatic staining reaction for EMA, and negative nuclei staining reaction for PR (Fig. 2). Based on this we proceeded with the method.
Fig. 2.

Implantation validation. Validation of cell implantation after one week in two animals – one from each group of MO-4. The skull base implantation showed diffusely aligned tumor cells along the cannula tract, scarce cytoplasmatic staining reaction for EMA, and negative nuclei staining reaction for PR. There were no cells present on the ventral side of the brain (injection site). The convexity implantation 2 mm below the top of the skull showed viable cells but again with scarce cytoplasmatic staining reaction for EMA and negative nuclear staining for PR alongside a cystic formation probably due to a perioperative lesion. OV: Overview, HE: Hematoxylin and Eosin, EMA: Epithelial membrane antigen, PR: Progesterone receptor. Black bars 2,5 mm for OV and 100 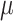 m for HE, EMA and PR.
m for HE, EMA and PR.
One animal died suddenly and unexpectedly after 16 days unrelated to surgery or tumor load. None of the remaining animals met humane endpoints (pain, neurological symptoms, > 20% weight loss) after 3 months. The remaining 77 animals were used for final analyses.
Tumor take rate and tumor size in convexity and skull base implantations
In general, we found a tendency towards higher tumor take rate in all convexity xenografts compared to all skull base xenografts (median 79.5% (IQR 40.0–89.8%) vs. 25.0% (IQR 14.4–45.0%); P-value = 0.125). Tumor take rate did not differ statistically within the individual samples (skull base vs. convexity) (Table 2) but showed a tendency towards higher tumor take rate for tumors in the convexity group. The xenografts varied in size both inter- and intratumorally without statistical significance (Table 2). Overall, there was no statistical difference (P-value = 0.45) in convexity vs. skull base tumors, but there was a clear tendency towards the larger tumors being in the superficial group. The median tumor volume for all 38 xenografts was 0.22 mm3 (IQR 0.13–1.02) (Fig. 3). The full overview of individual tumor sizes is available in Supplemental Material (SM1B).
Table 2.
Overview of tumor subtype, tumor take rate, and tumor size. Data are median and IQR (interquartile range). Mann-Whitney U unpaired tests were performed on both tumor take rate (TTR) and tumor size, comparing deep vs. superficial implantation. *Not significant. **Microcystic elements in larger tumors (2/5 tumors too small to ascertain). ***Largely with transitional elements, 60% of tumors displayed primarily meningothelial subtype. ****Fibrous subtype was less apparent – Meningothelial pattern in primary tumor present in most of the samples (9/14). NA: Not applicable.
| Sample | Xenograft subtype match | Skull base xenografts | Convexity xenografts | P-values | |||
|---|---|---|---|---|---|---|---|
| Tumor take rate | Tumor sizes mm3 (median (IQR)) |
Tumor take rate | Tumor sizes mm3 (median (IQR)) |
TTR | Size | ||
| MO-4 | Transitional | 1/8 (12.5%) | 0.059 (NA) | 8/9 (89%) | 0.26 (0.12–1.41) | 0.003 | NA |
| MO-12 | Microcystic** | 2/10 (20%) | 0.018 (0.015–0.021) | 3/10 (30%) | 0.17 (0.048–0.27) | 0.99* | 0.20* |
| MO-25 | Transitional/meningothelial*** | 3/10 (30%) | 0.26 (0.19–0.31) | 7/10 (70%) | 0.18 (0.13–0.56) | 0.18* | 0.38* |
| MO-27 | Meningothelial/Fibrous**** | 5/10 (50%) | 0.34 (0.19–1.08) | 9/10 (90%) | 2.42 (0.17–2.65) | 0.14* | 0.61* |
Fig. 3.

Xenograft sizes. Box plots with min, max, 25% percentile, 75% percentile, and median volumes of all xenografts. MO-X-D: Deep implantations at the skull base and MO-X-S: Convexity implantations below skull surface. MO-12 had the smallest size with a deep median size of 0.018 mm3, and the MO-27 tumors were much larger with a median of 2.42 mm3.
Morphological comparisons
Based on HE staining, all xenografts retained the morphological features of the corresponding primary tumors (Fig. 4) with key traits of each individual subtypes and only small differences. Fibrous/collagenous features in one transitional tumor (MO-25) and one fibrous tumor (MO-27) translated to a lesser degree. The microcysts seen in the MO-12 primary tumor did translate, but not to all xenografts. Larger images with notes of features are available in Supplemental Material (SM7).
Fig. 4.

Histology and immunohistochemistry of primary tumors (P) and corresponding xenografts (X). Morpholog: Assessed on HE sections, MO-4 and MO-25 primary tumors were classified as transitional due to the presence of meningothelial (lobulated architecture, clusters of syncytial cells, and meningothelial whorls) and fibroblastic features with collagen. MO-4 xenografts retained transitional features in all samples, and MO-25 retained traits in 60%. The transitional subtype can also contain psammoma bodies as seen in MO-25, which translated to the xenografts albeit in a lesser degree in comparison to the primary tumor. MO-27 primary tumor was classified as fibrous as it displayed bundles of collagen and maintained spindle cell morphology with few to no meningothelial nests or whorls. The xenografts had clear fibrous morphological traits in 4/14 cases but were dominated by the meningothelial subset, similarly to the transitional tumors. The MO-12 primary tumor was classified as microcystic because cells had elongated processes and vacuolated cytoplasm that resembles microcysts, which was also found especially in the largest (3/5) of the xenografts. Immunohistochemistry: All but one xenografted tumor showed consistent EMA staining. PR expression in xenografts was very heterogeneous within the primary tumors and between xenografts. Xenografted MO-4, MO-12, MO-25, and MO-27 regained PR in 89%, 40%, 20%, and 50% of animals, respectively, with a lower staining intensity in general. KI67 ranges (1–10%) were generally comparable between primary tumors and xenografts. SSTR2 displayed similar consistent staining scoring for MO-4. MO-12 and MO-25 showed a lower degree of staining intensity and also negative xenografts (Table 3). MO-27 showed well-matched high grade + 3 SSTR2 staining for the majority of xenografts. All xenografts showed similar negative (varying degree of unspecific) GFAP staining. OV: Overview, HE: Hematoxylin and Eosin, EMA: Epithelial membrane antigen, PR: Progesterone receptor, SSTR2: Somatostatin receptor 2, GFAP: Glial fibrillary acidic protein. Black bars: OV-X: 2,5 mm. Remaining slides: 100 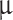 m. Magnetic resonance images are T1-weighted with contrast.
m. Magnetic resonance images are T1-weighted with contrast.
Immunohistochemical comparisons between primary tumors, xenografts, and implanted cells
Overall, primary tumors and corresponding xenografts displayed similar immunohistochemical profiles with minor differences (Tables 3and Fig. 4). The cytoplasmatic stainings of EMA, SSTR2, and GFAP all showed consistent staining from primary tumor to xenografts. The third-passage cells showed lower staining intensity for EMA, comparable staining for SSTR, and the same negative unspecific staining for GFAP compared to primary tumors and xenografts (Tables 3and Fig. 5). All primary tumors displayed positive PR. In vitro third-passage cells lost their PR expression upon culturing, and PR was regained in 20–89% of all xenografts. KI67 displayed comparable values for primary tumors and xenografts (1–10%). In general, KI67 was higher in the third-passage cells (10–30%). Further evaluations of the immunohistochemistry of individual xenografts are available in Supplemental Material SM1A, and examples of scoring are available in SM8 (primary tumor and xenografts) and SM9 (third-passage cells). Finally, we stained primary, implanted third-passage cells and xenografts for Nestin – a stem cell related biomarker. Primary and xenografts showed similar perivascular/endothelial staining. Third-passage cells used for implantation showed positive staining in few cells (Fig. 6).
Table 3.
Overview of immunohistochemical staining scores. Primary tumors and corresponding xenograft stainings were scored in a semiquantitative manner using the grades 0, + 1, +2, + 3, where 0 was no staining, + 1 was weak staining, + 2 was moderate staining, and + 3 was strong staining. Scoring values presented from min to max for both assessors to display the range. This also applied to KI67. EMA: Epithelial membrane antigen; PR: Progesterone receptor; SSTR2: Somatostatin receptor 2; GFAP: Glial fibrillary acidic protein. *KI67 was scored with a general score, but the highest score is shown in parenthesis if there were areas with a distinctly higher score. Representable images of general and highest scores (if applicable) are available in Supplemental Material (SM8 and 9). **GFAP slides display varying degrees of unspecific stainings (US) (negative) as is known especially for polyclonal GFAP antibody.
| Sample | Type | EMA | PR | SSTR2 | KI67* | GFAP** |
|---|---|---|---|---|---|---|
| MO-4 | Primary tumor | + 2–3 | + 1–2 | + 2 | 5% (10%) | US |
| Xenografts | + 3 (3/3) |
+ 1–2 (8/9) 0 (1/9) |
+ 2 (3/3) | 5% (10%) | US (1/1) | |
| Implanted cells | + 1 | 0 | + 1–2 | 30% | US | |
| MO-12 | Primary tumor | + 1 | + 2–3 | + 2–3 | 2% (5%) | US |
| Xenografts |
+ 1–2 (4/5) 0 (1/5) |
+ 1 (2/5) 0 (3/5) |
+ 1–2 (2/3) 0 (1/3) |
1–2% (5%) | US (3/3) | |
| Implanted cells | + 1–2 | 0 | + 2–3 | 20% | US | |
| MO-25 | Primary tumor | + 3 | + 1–2 | + 3 | 10% (20%) | US |
| Xenografts | + 2–3 (5/5) |
+ 1 (2/10) 0 (8/10) |
+ 1 (1/3) 0 (2/3) |
2–5% | US (3/3) | |
| Implanted cells | + 1–2 | 0 | + 2 | 10% | US | |
| MO-27 | Primary tumor | + 2 | + 3 | + 3 | 3–5% | US |
| Xenografts | + 1–3 (4/4) |
+ 1–2 (6/12) 0 (6/12) |
+ 3 (3/4) 0 (1/4) |
1–5% (10–40%) | US (3/3) | |
| Implanted cells | + 1 | 0 | + 3 | 20% | US |
Fig. 5.

Immunohistochemistry of third-passage cells. HE panels show single cells. EMA showed consistent positive cytoplasmatic staining around the cells albeit lower scoring in comparison to primary tumors. None of the samples displayed positive PR
in vitro. All cell lines showed high KI67 (10–30%). SSTR2 showed consistent staining and scoring, and GFAP showed (negative) unspecific staining for all samples. For further evaluation see Table 3. OV: Overview, HE: Hematoxylin and Eosin, EMA: Epithelial membrane antigen, PR: Progesterone receptor, SSTR2: Somatostatin receptor 2, GFAP: Glial fibrillary acidic protein. Black bars: 100 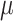 m.
m.
Fig. 6.

Nestin staining for cells with stem-like features. All primary tumors exhibit perivascular/endothelial Nestin staining, which is also observed with a similar pattern in xenografts. As illustrated in higher magnification (insert), Nestin-positive cells, although fewer in number, could also be observed in the third-passage cells used for implantation.
Epigenetic analyses
One sample, MO-12, was ineligible for DNA extraction due to small xenografts. At least 100 ng of DNA per sample were needed for optimal quality, which was met for all samples included in the epigenetic analyses. The bisulfite conversion was efficient for all samples. The full data sheet on parameters is available as Supplemental Material (SM5).
DNA methylation profiling using EPIC 850 K was performed to explore and compare primary tumors and in vivo xenografts. All primary tumors and corresponding xenografts matched overall as benign, but not with subclasses using the BrainClassifier v12.5 (Table 4). Copy number variation profiles showed similarities between primary tumors and xenografts (Fig. 7). To further explore the methylome, a heatmap was created using unsupervised hierarchical clustering. This analysis focused on the 2,000 most variable CpGs and showed MO-27 primary tumor and xenograft in the same edge; MO-4 and MO-25 primary tumors were placed together, as were the MO-4 and MO-25 xenografts (Fig. 8A). Correlation plots revealed high correlation between primary tumor and corresponding xenografts (Fig. 8B).
Table 4.
BrainClassifier v12.5 results. Using the BrainClassifier v12.5, all primary tumors were identified as benign meningiomas (0.99 (SD: 0.0)). All in vivo xenografts had an overall best match with benign meningiomas (0.79 (0.22)). None of the xenografts had their best subclass match with the corresponding primary tumors. Full reports are available in Supplemental Material (SM6).
| Sample | Origin | Overall match | Benign subclass 1 |
Benign subclass 2 | Benign subclass 3 |
|---|---|---|---|---|---|
| MO-04 | Primary tumor | 0.99 | 0.01 | 0.01 | 0.96 |
| Xenografts | 0.86 | 0.61 | 0.05 | 0.17 | |
| MO-25 | Primary tumor | 0.99 | 0.99 | 0.00 | 0.00 |
| Xenografts | 0.54 | 0.02 | 0.00 | 0.50 | |
| MO-27 | Primary tumor | 0.99 | 0.00 | 0.76 | 0.21 |
| Xenografts | 0.96 | 0.00 | 0.04 | 0.92 |
Fig. 7.

Copy number variation (CNV) profiles. Primary tumors on the left and in vivo xenografts on the right for each of the three analyzed samples. MO-04 displayed gain of chromosome 3 and loss of chromosome 22 with NF2 and SMARCB1 loss. MO-25 CNV showed similar profiles as MO-4 but without gain of chromosome 3. MO-27 displayed gain of chromosome 1q on the primary tumor and had a tendency towards the same in the xenograft, but it was difficult to assess due to the noisy profile.
Fig. 8.

Heatmap and correlation plots. (A) The heatmap contains the 2000 most variant CpG sites (unsupervised). It displays primary tumors (Purple bars) and corresponding xenografts (Green bars). The unsupervised clustering was performed with 200x bootstrap. It placed MO-27 primary tumor and xenograft together in one branch and pooled the remaining tumors and xenografts together. Here, however, the primary tumors (edge 1) and xenografts (edge 2) were more alike based on the top 2000 most variant CpG sites. The bootstrap shows robust and significant values for Edge 1 and 2 AU/BP (approximately unbiased/bootstrap probability) 100/100 and robust, but not significant for edge 4 and 3 AU/BP 90/95 and 89/95 respectively. (B) Correlation plots for the three sample pairs focusing on the CpGs relevant for the v12.5 BrainClassifier showed high correlations (MO-4 (R = 0.86), MO-25 (R = 0.82), and MO-27 (R = 0.93)).
Differential DNA methylation analysis on grouped primary tumors and xenografts only revealed small differences on a differentially methylated position (DMP or differing CpG) level. Most probes (866,074) were insignificant, 25 were hypermethylated, and 382 were hypomethylated. There was no single significant differentially methylated region.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was performed on the genes involved in the 407 DMPs. There were no genes related to DMRs; however, we found two significant overlaps using DMPs in the Reactome pathways: post-translational protein modification with 18 overlapping genes (FDR q-value: 0.007) and cellular responses to stimuli with 12 overlapping genes (FDR q-value: 0.04). The full GSEA analysis can be found in the Supplemental Material (SM10).
Discussion
In this study, we established benign meningioma xenografts in a rat model using primary cell cultures at third passage from four different benign meningiomas. There was a tendency for higher tumor take rates in convexity models compared to skull base models. In accordance with comparable previous studies29,30, we found close morphological and immunohistochemical resemblances between the primary meningiomas and the corresponding xenografted tumors, with only minor differences. Original expression of PR was lost in primary cultures but was regained in many xenografts, while EMA, SSTR, and GFAP remained largely consistent from primary tumor to in vitro conditions to rat xenografts. Furthermore, copy number variations were retained, and the analysis of DNA methylation profiling patterns revealed a close resemblance between primary tumors and xenografts, underscoring the usability of this in vivo model for future preclinical meningioma studies.
Meningioma models with human xenografts in immunocompromised mice have been used since the 1980s16but not previously in rats. Besides having a closer resemblance to humans physiologically and morphologically, the rat genome (2.75 gigabases) is also closer to the human genome (2.9 gigabases) than the mice genome (2.6 gigabases)41. Rats have a size advantage over mice (up to 10-fold) enabling easier microsurgery and larger blood draws and tissue volumes, and are preferred in neurobehavioral studies34. Their size may make rats preferable for studies where a focal treatment is intended. We also found that our xenografted tumor volumes were larger than those reported in a similar mouse study. Specifically, we obtained much larger tumors, with a median tumor volume of 0.22 mm3 (range 0.015 mm3 to 3.6 mm3), compared to Friedrich et al.29, who reported tumor volumes ranging from 0.004 mm3 to 0.224 mm3, with the majority (18/27 xenografts) being smaller than 0.05 mm3.
We observed an overall tumor take rate of 80% when implanting tumor cells superficially at the convexity. Our findings align with previous successful studies reporting comparable superficial tumor take rates ranging from 54 to 100%5,29,30when using a similar number of benign cells. Using a tenth of the number of cells has shown to be unsuccessful16,31. In contrast to convexity implantation, injections into the temporal cranial base failed in 75% of the animals, where McCutcheon et al.30 showed a tumor take rate of 85%. We hypothesize that a higher tumor take rate could have been achieved for the deep implantations if the cells had been injected just below the cortex, avoiding penetration of the brain surface at the skull base. This would mean that no cells would be dispersed over a larger area and would remain close proximity with cell-cell contact, which facilitates communication and growth42.
Meningiomas exhibit substantial intertumoral morphological heterogeneity43. Overall, the histological subtypes observed in primary tumors translated well in the xenografts for all tumors, with no significant transformations detected (SM7). In xenografts derived from translational and fibrous subtypes, some of the collagenous tissue diminished, and the tumor morphology shifted towards a more meningothelial appearance consistent with previous studies5,29,44. Retention of morphological features in meningioma models has also been described in previous publications30,45,46.
Three immunohistochemical markers typical of meningioma, EMA, SSTR2A, and PR were retained in xenografts, but not fully. EMA is normally expressed in 90–95% of meningiomas47, SSTR2A in > 95%47,48, and PR in over 70%49,50. The four primary tumors in our study expressed all three. The membrane/cytoplasmatic stainings of our third-passage cells (EMA and SSTR2) were comparable to primary tumor. Less so for EMA than SSTR2, where SSTR2 showed a more consistent staining in comparison to the primary tumor. The differences might be due to monolayer cell conditions in vitro compared to in situ/in vivo samples. EMA was retained in almost all our xenografts (94%, i.e. 16/17) with comparable staining intensity. Friedrich et al.29 reported positive EMA expression in fewer xenografts (72%, i.e. 18/25) with lower staining intensity compared to primary tumors, explained by microenvironmental influence. As with McCutcheon et al.30, we used third-passage cells while Friedrich et al.5,29, who achieved the highest published tumor take rate using benign cells, attributed their success to the use of vital cells in passage six. Passage number affects a cell line’s characteristics over time51. Cell lines at high passage numbers experience altered morphology, response to stimuli, growth rates, protein expression, and transfection efficiency compared to lower passage cells. This could also explain the lower EMA expression in the study by Friedrich et al.29. Despite the low cell passage number in our study, however, SSTR2 expression was more varied in our xenografts with an overall expression of 69%. We attribute this to microenvironmental changes although we cannot specify this further. PR also displayed varying results. PRs are known to be elusive in vitro in both monolayer52and spheroid cultures53, but 3D spheroid cultures have a greater chance of retaining PR expression54, which we also found in our in vitro experiments (spheroid data not shown here). Interestingly, many of our xenografts regained PR expression. A similar pattern of PR expression in vitro and in vivo has previously been shown in an immortalized primary tumor cell line55. To our knowledge, the underlying mechanisms are currently unknown, but it is possible that in vitro conditions do not adequately support PR expression, possibly due to the absence of specific factors, or that meningioma cells lose the ability to respond to these factors despite their presence53. The current study is the first to report in vivo PR expression in an orthotopic meningioma model using primary tumors (without immortalization).
An important aspect of a model is the proliferative index, which should accurately represent the parent tumor’s growth rate. As we show here, monolayer cultures of benign meningiomas can experience a surge in KI67 due to the favorable in vitro environment in comparison to the primary tumors54. As in Friedrich et al.29, we found the proliferative indexes to return to comparable levels after implantation in the more representative microenvironment to human conditions (orthotopic implantation) for the benign cells. It is not uncommon for proliferative indexes to rise due to positive selection through the cell passages for primary cells31,44, which is why low passaged cells are preferred.
Passaging cells through multiple passages can lead to clonal selection, resulting in a more homogenous cell population. To assess the presence of cells with stem cell-like features, we stained the samples with Nestin—a class IV intermediate filament protein and a recognized stem cell-related biomarker56,57. Benign meningiomas typically exhibit low Nestin immunoreactivity, except in perivascular regions58, where higher staining intensity is associated with higher malignancy grades57. In all four samples, including both primary tumors and xenografts, staining was consistently observed in the perivascular/endothelial regions. Interestingly, cells from the third passage, used for implantation, exhibited a few Nestin-positive cells. This suggests the presence of cells with stem cell-like features within the cell population, hinting a heterogeneous pool of cells being implanted into the animals. Stem-like cells serve as a reservoir for self-renewal and differentiation into various cell types59, contributing to the xenografts complexity mirroring that of the primary tumors.
Meningiomas as a group exhibit a wide range of copy number alterations, with benign displaying fewer alterations than malignant meningiomas23. Comparative molecular assessments between primary tumors and xenografts have been limited. Only Zhang et al.31 have conducted such assessments on two pairs (WHO II and III – WHO 2016 classification60) using RNA sequencing to assess DNA copy numbers and genes. The xenografts retained the copy number alterations of the primary tumors, which we also show. One xenograft sample displayed an especially noisy copy number profile, despite good DNA quality and bisulfite conversion. It is most likely that the noisy xenograft copy number profiles were due to rodent DNA, which can lead to unspecific binding/cross-reactivity of probes. Notably, the PDXNET and EurOPDX consortia found robustness of copy number alterations and reported no enrichment of cancer-related genes in xenografts in a mouse host61.
Epigenetically, we also found a close resemblance using DNA methylation analysis with only 407 out of 866,074 probes differing between primary tumors and xenografts. We could not identify any specific differentially methylated regions (DMRs) between primary tumors and xenografts, indicating conservation of the methylation signatures and, accordingly, the absence of potential functional/biological significant regions. We hypothesize that the small changes (the 407 differing CpGs or DMPs) were most likely introduced by the changing microenvironment, interacting foreign/rodent DNA, and unspecific binding/cross-reactivity of probes - contamination from the host animal. The GSEA could provide some insight into this. Despite having no DMRs meaning the findings may not be biologically significant, there were two significant gene overlaps using the DMPs. The Reactome pathway post-translational protein modifications introduce structural changes in existing proteins, enabling them to participate in multiple biological processes at both histone and non-histone levels. These modifications are also associated with T-cell regulation and interaction with the tumor microenvironment62. Hence, this could explain the small but significant changes in the DMPs due to the new microenvironment and the lack of T-cells and their involvement. This is further emphasized by the related Reactome pathway: cellular response to stimuli, which is also associated with changes in the microenvironment and immune system due to changes in cellular stress and response to stimuli that occur in patient-derived models, with changing stromal cells from human to animal over the passages of cells63. However, this remains suggestive since there is no overall DMR. Overall, these observations underscore the complexity of tumor microenvironments and the critical role of T-cells and post-translational modifications in shaping the molecular landscape, even in the absence of clear DMRs.
The unsupervised hierarchical clustering found MO-27 to be in the same cluster, which is emphasized by the high correlation (0.93). The remaining samples were clustered as xenografts together and primary tumors together. The mix of rodent DNA (from the genetically identical rats) might have caused this clustering, and hence the similar CpGs affected in the groups. This also suggests no biological differences between the two groups. Unfortunately, there is no way to assess the content rodent DNA using a microarray as this is based on hybridizing DNA to predesigned probes and not sequencing. It has previously been shown that xenografts retain similar genomic profiles despite the presence of rodent DNA64. The EPIC bead chip (850 K array) covers > 850,000 sites and although it is robust, there will always be some probes that measure differently even in identical replicates (congruency > 95%65).
Using the BrainClassifier v12.5, the methylation subclasses remained “benign meningioma”, but B1, B2, and B3 identities were not preserved from primary tumors to xenografts. The BrainClassifier does not allow us to identify which CpGs the classifier focusses on in the specific classes. However, the similar percentages of CpGs when using the classifier (0.06%) and when assessing all 850 K CpGs (0.05%) show that the methylation patterns are equally distributed.
Although we could not identify any specific differentially methylated regions, there seems to be distinct differences, although possibly not biologically significant, when human tissue is transferred to T-cell deficient animals. Based on the present study, we cannot completely exclude actual biological significance, which will have to be explored in future studies. Given the rather complex nature of DNA methylation changes, differences in single CpGs rather than larger regions are uninformative and are unlikely to lead to differences in gene expression.
A final note: We believe it is essential when establishing tumor models to assess the likeness of a xenograft animal model to the original patient-derived tumor prior to its use. Genetic and epigenetic profiling can validate these similarities and elucidate differences, thereby strengthen study outcomes and translation of results from clinic to laboratory and back.
Limitations
There are several limitations to this study. We performed histology and immunohistochemistry on FFPE brain tissue as shown in Fig. 1, where we could potentially have induced artefacts and possibly unintentionally removed tumor tissue thus decreasing size and possibly tumor take rate. Furthermore, we macro-dissected tumors from the rodent brain for epigenetic analyses, which—despite our care—may have included some rodent brain tissue. A model-related limitation was the range of cells injected at 7.5 × 104−105 cells/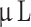 . We intended to inject 10
. We intended to inject 10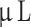 of 105 cells/
of 105 cells/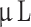 , but the heterogeneity of the cells due to the low cell passage at the time of implantation meant that some of the cell suspensions were too viscous and had to be diluted. However, there was no correlation between lower number of cells injected and lower tumor take rate. Finally, xenografting human tumor cells in immunocompromised animals decreases the translatability overall and renders the model unusable for testing immunotherapies. As our model only lacks T-cells, the innate immune system is still intact.
, but the heterogeneity of the cells due to the low cell passage at the time of implantation meant that some of the cell suspensions were too viscous and had to be diluted. However, there was no correlation between lower number of cells injected and lower tumor take rate. Finally, xenografting human tumor cells in immunocompromised animals decreases the translatability overall and renders the model unusable for testing immunotherapies. As our model only lacks T-cells, the innate immune system is still intact.
Conclusion
Personalized/targeted therapy is an evolving field in meningiomas and emphasizes the need for ‘personalized’ meningioma tumor models that accurately represent the heterogeneity of meningiomas as a group. The rat model presented here uses approximately one million third-passage benign primary patient-derived tumor cells and proved to be a viable and consistent approach, especially at the convexity. It closely mirrors corresponding primary tumors morphologically, immunohistochemically, and epigenetically, with only minor differences observed. The epigenetic differences were specific to the level of CpGs, with no biologically relevant differentially methylated regions identified. Moreover, our xenograft model has an incomplete immune system that lacks the adaptive aspect but retains the innate system. Investigations are needed into how the animal innate immune system influences the tumor in further tests of the model’s validity and usability.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgements
We would address a special thanks to biomedical laboratory technician Helle Wohlleben for invaluable assistance in preparing samples and performing sections and immunohistochemical stainings and also biomedical laboratory technician Mirjeta Krasniqi for helping with performing EPIC 850k on all samples. Finally, we would like to thank Claire Gudex, MD, MPH, MBChB, University of Southern Denmark, for final proofreading.
Author contributions
MSA: first draft, conceptualization, in vitro and in vivo experimental parts, methodology, statistics, figures, and tables and analyses and interpretation of all data; BH: conceptualization, in vivo experimental part, methodology; JKP and MW: assessment of primary tumors and xenograft morphology and immunohistochemical staining, PJ and MM: DNA methylation analyses and interpretation, BBO: methodology and in vitro study part. FRP: conceptualization, methodology. All authors performed review, writing, editing, and approved the final draft.
Funding
MSA received funding from Max Th. Harding Larsen Foundation, Overlægerådet (Odense University Hospital), and Familien Hede Nielsens Foundation. FRP and MSA received funding from Innovation foundation Denmark. None of the funders had any part in the design, analyses, or reporting of the study.
Data availability
Most data generated or analysed during this study are included in this published article (and its Supplementary Information files). The remaining datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. The digital histology and immunohistochemistry files and the IDAT files generated and analyzed are not publicly available due to large file sizes, but they are available from the corresponding author on reasonable request.
Declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was approved by the Regional Committees on Health Research Ethics for Southern Denmark (S-20190105). All participants signed consent forms. The animal protocol was submitted to and approved (29th August 2019) by the Danish Animal Experiments Inspectorate (2019-15-0201-00195). The full approved protocol (in Danish) can be provided upon reasonable request.
Consent for publication
All patients involved in the project have signed a regionally approved consent form.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ostrom, Q. T. et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro Oncol.24(Suppl 5), v1–v95 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldbrunner, R. et al. EANO guideline on the diagnosis and management of meningiomas. Neuro Oncol.23(11), 1821–1834 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voss, K. M. et al. The Simpson grading in meningioma surgery: does the tumor location influence the prognostic value? J. Neurooncol. 133(3), 641–651 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Shahbandi, A., Shah, D. S., Hadley, C. C. & Patel, A. J. The Role of Pharmacotherapy in Treatment of Meningioma: A Systematic Review. Cancers (Basel) ;15(2). (2023). [DOI] [PMC free article] [PubMed]
- 5.Friedrich, S., Schwabe, K., Grote, M., Krauss, J. K. & Nakamura, M. Effect of systemic celecoxib on human meningioma after intracranial transplantation into nude mice. Acta Neurochir. (Wien). 155(1), 173–182 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Ragel, B. T. et al. Calcium channel antagonists augment hydroxyurea- and ru486-induced inhibition of meningioma growth in vivo and in vitro. Neurosurgery59(5), 1109–1120 (2006). discussion 20 – 1. [DOI] [PubMed] [Google Scholar]
- 7.Angus, S. P. et al. EPH receptor signaling as a novel therapeutic target in NF2-deficient meningioma. Neuro Oncol.20(9), 1185–1196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horbinski, C. et al. The effects of palbociclib in combination with radiation in preclinical models of aggressive meningioma. Neurooncol Adv.3(1), vdab085 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choudhury, A. et al. Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities. Nat. Genet.54(5), 649–659 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakano, T. et al. Eribulin prolongs survival in an orthotopic xenograft mouse model of malignant meningioma. Cancer Sci.113(2), 697–708 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pachow, D. et al. mTORC1 inhibitors suppress meningioma growth in mouse models. Clin. Cancer Res.19(5), 1180–1189 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Chen, X., Tian, F., Lun, P. & Feng, Y. Curcumin Inhibits HGF-Induced EMT by Regulating c-MET-Dependent PI3K/Akt/mTOR Signaling Pathways in Meningioma. Evid. Based Complement. Alternat Med.2021, 5574555 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jungwirth, G. et al. KIF11 inhibitors filanesib and ispinesib inhibit meningioma growth in vitro and in vivo. Cancer Lett.506, 1–10 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Yu, T. et al. Receptor-Tyrosine Kinase Inhibitor Ponatinib Inhibits Meningioma Growth In Vitro and In Vivo. Cancers (Basel) ;13(23). (2021). [DOI] [PMC free article] [PubMed]
- 15.Mak, I. W., Evaniew, N. & Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. Am. J. Transl Res.6(2), 114–118 (2014). [PMC free article] [PubMed] [Google Scholar]
- 16.Andersen, M. S. et al. Meningioma animal models: a systematic review and meta-analysis. J. Transl Med.21(1), 764 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Louis, D. N. et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol.23(8), 1231–1251 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhat, A. R., Wani, M. A., Kirmani, A. R. & Ramzan, A. U. Histological-subtypes and anatomical location correlated in meningeal brain tumors (meningiomas). J. Neurosci. Rural Pract.5(3), 244–249 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nigim, F., Wakimoto, H., Kasper, E. M., Ackermans, L. & Temel, Y. Emerging Medical Treatments for Meningioma in the Molecular Era. Biomedicines ;6(3). (2018). [DOI] [PMC free article] [PubMed]
- 20.Bi, W. L., Mei, Y., Agarwalla, P. K., Beroukhim, R. & Dunn, I. F. Genomic and Epigenomic Landscape in Meningioma. Neurosurg. Clin. N Am.27(2), 167–179 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Suppiah, S. et al. Molecular and translational advances in meningiomas. Neuro Oncol.21(Suppl 1), i4–i17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahm, F. et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol.18(5), 682–694 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Driver, J. et al. A molecularly integrated grade for meningioma. Neuro Oncol.24(5), 796–808 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang, J. Z., Nassiri, F., Mawrin, C. & Zadeh, G. Genomic Landscape of Meningiomas. Adv. Exp. Med. Biol.1416, 137–158 (2023). [DOI] [PubMed] [Google Scholar]
- 25.Wang, J. Z., Nassiri, F., Aldape, K., von Deimling, A. & Sahm, F. The Epigenetic Landscape of Meningiomas. Adv. Exp. Med. Biol.1416, 175–188 (2023). [DOI] [PubMed] [Google Scholar]
- 26.Dasgupta, K. & Jeong, J. Developmental biology of the meninges. Genesis57(5), e23288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang, J. Z. et al. Meningioma: International Consortium on Meningiomas (ICOM) consensus review on scientific advances & treatment paradigms for clinicians, researchers, and patients. Neuro Oncol. (2024). [DOI] [PMC free article] [PubMed]
- 28.Soni, N. et al. Meningioma: Molecular Updates from the 2021 WHO Classification of CNS Tumors and Imaging Correlates. AJNR Am. J. Neuroradiol. (2024). [DOI] [PubMed]
- 29.Friedrich, S. et al. Comparative morphological and immunohistochemical study of human meningioma after intracranial transplantation into nude mice. J. Neurosci. Methods. 205(1), 1–9 (2012). [DOI] [PubMed] [Google Scholar]
- 30.McCutcheon, I. E. et al. Intracranial injection of human meningioma cells in athymic mice: an orthotopic model for meningioma growth. J. Neurosurg.92(2), 306–314 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Zhang, H. et al. Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Primary and Recurrent Meningioma. Cancers (Basel) ;12(6). (2020). [DOI] [PMC free article] [PubMed]
- 32.Linsler, S., Muller, S. J., Muller, A., Senger, S. & Oertel, J. M. Fluorescence image-guided resection of intracranial meningioma: an experimental in vivo study on nude mice. Ann. Anat.237, 151752 (2021). [DOI] [PubMed] [Google Scholar]
- 33.Hu, D., Wang, X., Mao, Y. & Zhou, L. Identification of CD105 (endoglin)-positive stem-like cells in rhabdoid meningioma. J. Neurooncol. 106(3), 505–517 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Jacob, H. J. The rat: a model used in biomedical research. Methods Mol. Biol.597, 1–11 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Percie du Sert, N. et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol.18(7), e3000411 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wieschowski, S. et al. Publication rates in animal research. Extent and characteristics of published and non-published animal studies followed up at two German university medical centres. PLoS One. 14(11), e0223758 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Festing, M. F. & Altman, D. G. Guidelines for the design and statistical analysis of experiments using laboratory animals. ILAR J.43(4), 244–258 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Park, H. J. et al. Semi-automated method for estimating lesion volumes. J. Neurosci. Methods. 213(1), 76–83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capper, D. et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol.136(2), 181–210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102(43), 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibbs, R. A. et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature428(6982), 493–521 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Schwager, S. C., Taufalele, P. V. & Reinhart-King, C. A. Cell-Cell Mechanical Communication in Cancer. Cell. Mol. Bioeng.12(1), 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raghunathan, A. & Giannini, C. Histopathology of Meningiomas. Adv. Exp. Med. Biol.1416, 35–45 (2023). [DOI] [PubMed] [Google Scholar]
- 44.Ragel, B. T. et al. A comparison of the cell lines used in meningioma research. Surg. Neurol.70(3), 295–307 (2008). discussion. [DOI] [PubMed] [Google Scholar]
- 45.Malham, G. M., Thomsen, R. J., Synek, B. J. & Baguley, B. C. Establishment of primary human meningiomas as subcutaneous xenografts in mice. Br. J. Neurosurg.15(4), 328–334 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Matsuda, Y. et al. Antitumor effects of antiprogesterones on human meningioma cells in vitro and in vivo. J. Neurosurg.80(3), 527–534 (1994). [DOI] [PubMed] [Google Scholar]
- 47.Boulagnon-Rombi, C. et al. Immunohistochemical Approach to the Differential Diagnosis of Meningiomas and Their Mimics. J. Neuropathol. Exp. Neurol.76(4), 289–298 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Silva, C. B. et al. Expression of somatostatin receptors (SSTR1-SSTR5) in meningiomas and its clinicopathological significance. Int. J. Clin. Exp. Pathol.8(10), 13185–13192 (2015). [PMC free article] [PubMed] [Google Scholar]
- 49.Agopiantz, M., Carnot, M., Denis, C., Martin, E. & Gauchotte, G. Hormone Receptor Expression in Meningiomas: A Systematic Review. Cancers (Basel) ;15(3). (2023). [DOI] [PMC free article] [PubMed]
- 50.Baxter, D. S., Orrego, A., Rosenfeld, J. V. & Mathiesen, T. An audit of immunohistochemical marker patterns in meningioma. J. Clin. Neurosci.21(3), 421–426 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Briske-Anderson, M. J., Finley, J. W. & Newman, S. M. The influence of culture time and passage number on the morphological and physiological development of Caco-2 cells. Proc. Soc. Exp. Biol. Med.214(3), 248–257 (1997). [DOI] [PubMed] [Google Scholar]
- 52.Waelti, E. R. & Markwalder, T. M. Endocrine manipulation of meningiomas with medroxyprogesterone acetate. Effect of MPA on growth of primary meningioma cells in monolayer tissue culture. Surg. Neurol.31(2), 96–100 (1989). [DOI] [PubMed] [Google Scholar]
- 53.Blankenstein, M. A. et al. Occurrence, regulation, and significance of progesterone receptors in human meningioma. Steroids65(10–11), 795–800 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Tonn, J. C. et al. Inverse correlation of cell proliferation and expression of progesterone receptors in tumor spheroids and monolayer cultures of human meningiomas. Neurosurgery41(5), 1152–1159 (1997). [DOI] [PubMed] [Google Scholar]
- 55.Cargioli, T. G. et al. Establishment of an in vivo meningioma model with human telomerase reverse transcriptase. Neurosurgery. 60(4), 750–759; discussion 9–60. (2007). [DOI] [PubMed]
- 56.Alamir, H. et al. In situ characterization of stem cells-like biomarkers in meningiomas. Cancer Cell. Int.18, 77 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiao, Z. Y., Chen, X. J., Pan, Q., Yang, Q. Z. & Li, K. Z. Expression of Nestin, CD133 and Sox2 in Meningiomas. Turk. Neurosurg.28(6), 910–914 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Petricevic, J. et al. Expression of nestin, mesothelin and epithelial membrane antigen (EMA) in developing and adult human meninges and meningiomas. Acta Histochem.113(7), 703–711 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Rich, J. N. Cancer stem cells: understanding tumor hierarchy and heterogeneity. Med. (Baltim).95(1 Suppl 1), S2–S7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Louis, D. N. et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol.131(6), 803–820 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Woo, X. Y. et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat. Genet.53(1), 86–99 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li, W., Li, F., Zhang, X., Lin, H-K. & Xu, C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal. Transduct. Target. Therapy. 6(1), 422 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu, Y. et al. Patient-derived xenograft models in cancer therapy: technologies and applications. Signal. Transduct. Target. Ther.8(1), 160 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rossello, F. J. et al. Next-generation sequence analysis of cancer xenograft models. PLoS One. 8(9), e74432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol.17(1), 208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Most data generated or analysed during this study are included in this published article (and its Supplementary Information files). The remaining datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request. The digital histology and immunohistochemistry files and the IDAT files generated and analyzed are not publicly available due to large file sizes, but they are available from the corresponding author on reasonable request.