

Abstract
Background
Lynch syndrome (LS) is an autosomal‐dominant, hereditary cancer predisposition syndrome caused by pathogenic variants (PVs) in one of the mismatch‐repair genes MLH1, MSH2/EPCAM, MSH6, or PMS2. Individuals who have MLH1 PVs have high lifetime risks of colorectal cancer (CRC) and endometrial cancer (EC). There is controversy regarding whether a younger age at diagnosis (or anticipation) occurs in MLH1‐associated LS. The objective of this study was to assess anticipation in families with MLH1‐associated LS by using statistical models while controlling for potential confounders.
Methods
Data from 31 families with MLH1 PVs were obtained from an academic registry. Wilcoxon signed‐rank tests on parent–child‐pairs as well as parametric Weibull and semiparametric Cox proportional hazards and Cox mixed‐effects models were used to calculate hazard ratios or to compare mean ages at CRC/EC diagnosis by generation. Models were also corrected for ascertainment bias and birth‐cohort effects.
Results
A trend toward younger ages at diagnosis of CRC/EC in successive generations, ranging from 3.2 to 15.7 years, was observed in MLH1 PV carrier families. A greater hazard for cancer in younger generations was not precluded by the inclusion of birth cohorts in the model. Individuals who had MLH1 variants with no Mlh1 activity were at a 78% greater hazard for CRC/EC than those who retained Mlh1 activity.
Conclusions
The current results demonstrated evidence in support of anticipation in families with MLH1‐associated LS across all statistical models. Mutational effects on Mlh1 activity influenced the hazard for CRC/EC. Screening based on the youngest age of cancer diagnosis in MLH1‐LS families is recommended.
Keywords: anticipation, birth cohorts, generations, Lynch syndrome, Mlh1 activity
Short abstract
Anticipation in MLH1‐associated Lynch syndrome was not precluded by the inclusion of birth cohorts in the analyses. The effects of MLH1 mutations on Mlh1 activity influence age of diagnosis.
INTRODUCTION
Lynch syndrome (LS) is an autosomal‐dominant, hereditary cancer predisposition syndrome due to a pathogenic variant (PV) in one of the mismatch‐repair (MMR) genes MLH1, MSH2/EPCAM, MSH6, or PMS2 1 and manifests with increased lifetime risks of colorectal cancer (CRC) and endometrial cancer (EC) among other less common cancer types. 2 Of the variants in LS, 43% have been reported in MLH1, 40% have been reported in MSH2, and the remaining 17% have been reported in MSH6 and PMS2. Penetrance estimates for CRC and EC in LS vary by gene, variant, sex, and geographic location. 3
The tendency for cancer to develop at an earlier age in successive generations within a family was first reported in 1913 by Aldred Scott Warthin. 4 This defines the phenomenon of anticipation in which the age at onset of a disorder is reduced and/or the severity of the phenotype is increased in successive generations. 5 Anticipation has been observed in familial melanoma; Li Fraumeni syndrome; breast, ovarian, and pancreatic cancers; and von Hippel–Lindau syndrome. 6 Statistical models for estimating the effects of anticipation on cancer range from a simple comparison of the mean or median ages at the onset of cancer in parent–child pairs (PCPs) to more complex models that follow randomized distributions with the inclusion of covariates. Several types of biases, such as ascertainment bias, birth‐cohort effects, and right truncation effects, can potentially influence the estimation of anticipation. 7
In LS, studies on anticipation have indicated decreases in the age of onset in successive generations by 1–2 years for most MMR genes and up to 7 years for those with PMS2 PVs. 6 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 These anticipation effects reportedly disappeared after correcting for birth cohort, 7 , 13 , 16 except in one study. 17 The presence and magnitude of anticipation effects in LS in these studies have depended on the type of model used, the choice of covariates included in the model, and the methods used to correct for bias. 18
Given the mixed results of anticipation effects in LS, the objective of this study was to estimate genetic anticipation in families with MLH1 PVs. Families with MLH1‐associated LS were selected because of their higher incidence and strong association with CRC and EC. We applied three statistical models to examine the effects of generation on age at first diagnosis of CRC or EC while correcting for ascertainment bias and birth‐cohort effects.
MATERIALS AND METHODS
Study cohort
Families with confirmed MLH1 PVs or likely PVs that included at least two first‐degree relatives with cancer were identified in a prospective, Institutional Review Board‐approved registry collected from 1992 through 2021 at The University of Chicago. Pedigree data collected on these families included age at diagnosis of cancer, year of birth, age and sex of family members, genetic test results, and site of cancer (Figure 1A).
FIGURE 1.
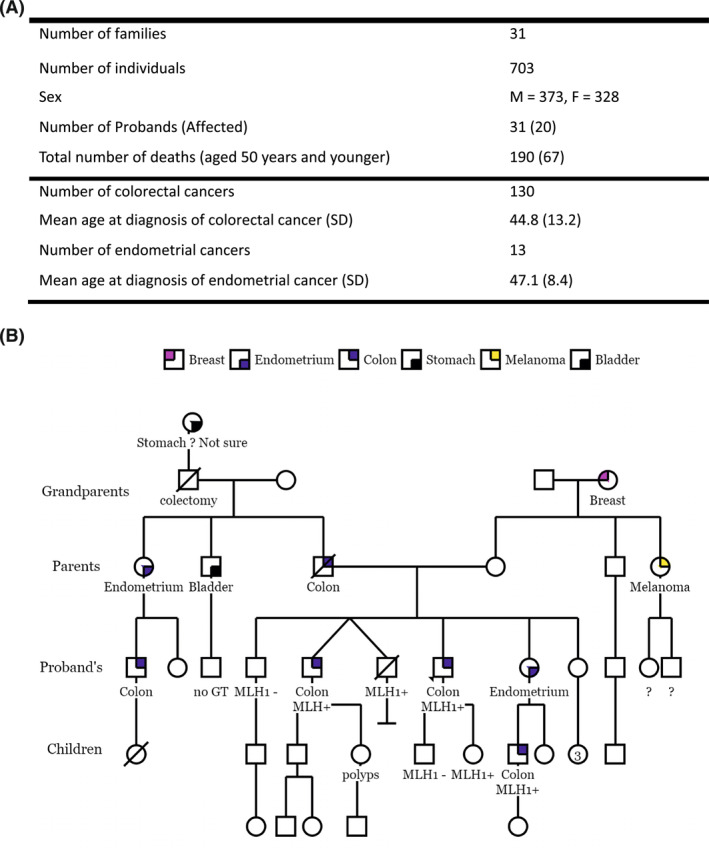
Descriptive statistics of (A) the study cohort and (B) an example pedigree depicting the creation of generations with respect to the proband. Individuals who did not fall into one of the four generations shown or who had cancers other than colorectal or endometrial were not included in the analysis. F indicates female; GT, genetic test; M, male; MLH1−, negative for MLH1 mutation 1; MLH1+, positive for MLH1 mutation 1; SD, standard deviation.
Generations and birth cohorts
The proband was the individual indicated as such on the pedigree. Generations were created with respect to the proband for each family as the proband's parents, grandparents, and children (Figure 1B). Probands and their siblings or cousins were designated as the proband's generation, henceforth referred to as probands to distinguish these from the proband from a ped. PCPs were created with individuals who had both a parent and their child diagnosed with CRC or EC. Birth cohorts were created at 20‐year and 10‐year intervals starting with those born before 1920 through 2000. Birth cohorts were also created for affected PCPs who had children born before and after 1945. Assuming that proactive screening of unaffected family members from age 25 years began with publication of the Amsterdam I criteria 19 in 1991, two birth cohorts of those born before and after 1966 were also created.
For individuals with missing age information, it was assumed that both parents of the proband were born in the same year, that the years of birth differed by 25 years in each generation, and that the year of birth of siblings was the same. 20 For individuals who had died and were reported as affected with an unknown age of diagnosis, the age of death was considered as the age of diagnosis. 20 Year of birth, if not available, was calculated from the last age reported on the pedigree and the year the pedigree was recorded or updated.
Variant effects and Mlh1 proficiency
Germline MLH1 PVs were known in 28 of 31 families and were recorded as MLH1‐positive in the remaining three families. Variants were classified based on their effect on the protein as a single‐residue variation (SRV), in which only a single amino acid residue in the Mlh1 protein was altered; a termination (T) effect if the variant resulted in a longer or shorter form of protein because of displacement of the termination codon; and a splicing effect (SP), in which the variant was known through in‐silico or functional studies to cause in‐frame exon skipping, resulting in an altered transcript (see Supporting Information S1). Variants were also classified as Mlh1‐proficient or Mlh1‐deficient based on published literature on functional studies (see Supporting Information S1). Mlh1 was considered MMR‐proficient if it demonstrated an MMR level activity above that of controls. Variants were also categorized into truncating and nontruncating as in a previous study. 21 Variants that could not be classified unambiguously were excluded from the analyses.
Statistical analysis
Differences in ages of diagnosis between PCPs were determined using the Wilcoxon pairwise signed‐rank test (see Supporting Information S2). The study outcome variable in multivariate regression models was age at first diagnosis of CRC/EC. Follow‐up was defined as the time elapsed from birth until the first CRC/EC diagnosis or censoring. An individual was censored either at the age of last contact with the family, as recorded in the pedigree, or at the age of death. The primary predictor variable of interest in multivariate regression models was the categorical variable generation, with sex, birth cohort, and the effect of MLH1 mutation on protein as covariates. Hazard ratios (HRs) for CRC/EC between generations were determined using nonparametric Cox proportional‐hazard models with (Cox mixed‐effects) and without (Cox‐PH) inclusion of families as random effects. Multivariate parametric regression with Weibull models was used to estimate the mean age of diagnosis for each generation. Cox‐PH and Weibull models were run both with and without the inclusion of probands. Survival analyses were also performed with pre‐1945 and post‐1945, pre‐1966 and post‐1966, and 10‐year birth cohorts as covariates in Cox‐PH models (see Supporting Information S1). The effect of Mlh1 protein activity (see Supporting Information S1) on age at diagnosis of CRC/EC was examined through univariate Cox‐PH regression. Mutation effects were included as a covariate to assess their relevance to the effect of generation on age of diagnosis (see Supporting Information S1). Mutational probabilities were added as weights to all statistical models. One‐way analysis of variance was used when the Cox‐PH model indicated a poor fit to the data. Statistical significance was set at 95% confidence intervals (CIs; α = .05) for all models. Values of p < .001, p < .01, p < .05, p < .1, and p ≥ 0.1 were considered as very strong, strong, moderate, trend, and insufficient evidence, respectively. 22
RESULTS
Our data comprised a total of 703 members from 31 families that had at least one member ascertained to carry a PV in MLH1 through genetic testing. A description of the cohort demographics is provided in Figure 1A. All generations except children had a lower mean age of diagnosis compared with their parent's generation (Table 1). Median ages of diagnosis of CRC/EC in generations did not show any specific trend. A consistent decrease in mean and median ages of diagnosis was observed from older to younger birth cohorts in individuals born after 1920 (Table 1).
TABLE 1.
Distribution by age of diagnosis weighted by mutation probabilities for individuals diagnosed with colorectal or endometrial cancer by generation, birth cohort, mismatch repair proficiency, and mismatch repair deficiency.
| Total no. | No. affected (%) | Age of diagnosis: Mean ± SD, years | Age of diagnosis: Median [IQR], years | |
|---|---|---|---|---|
| Generation, N = 668 | ||||
| Grandparents | 129 | 21 (16.3) | 52.6 ± 14.0 | 48 [23] |
| Parents | 193 | 48 (24.9) | 46.3 ± 12.2 | 45 [15] |
| Probands | 218 | 53 (24.3) | 40.1 ± 11.9 | 40 [15] |
| Children | 128 | 17 (13.2) | 42.1 ± 8.4 | 40 [13] |
| Cohort, N = 656 | ||||
| <1920 | 82 | 19 (23.2) | 45.5 ± 11.0 | 42 [10] |
| 1920–1939 | 140 | 38 (27.1) | 53.1 ± 13.5 | 50 [13] |
| 1940–1959 | 173 | 51 (29.0) | 42.9 ± 10.3 | 44 [13] |
| 1960–1979 | 164 | 26 (15.9) | 39.6 ± 6.9 | 39 [10] |
| 1980–2000 | 97 | 7 (7.2) | 25.7 ± 6.4 | 24 [8] |
| MMR‐proficient, N = 158 | ||||
| Grandparents | 31 | 5 (16.1) | 58.5 ± 20.7 | 42 [26] |
| Parents | 46 | 7 (15.2) | 46.0 ± 12.6 | 45 [16] |
| Probands | 56 | 3 (5.3) | 36.6 ± 15.4 | 31 [26] |
| Children | 23 | 0 (0.0) | — | — |
| MMR‐deficient, N = 310 | ||||
| Grandparents | 50 | 5 (10.0) | 52.3 ± 10.5 | 47 [3] |
| Parents | 89 | 26 (29.2) | 45.3 ± 10.7 | 44 [13] |
| Probands | 86 | 28 (32.5) | 42.9 ± 13.6 | 40 [13] |
| Children | 56 | 12 (21.4) | 43.9 ± 8.1 | 39.5 [9] |
Abbreviations: IQR, interquartile range; MMR, mismatch repair; SD, standard deviation.
There were 62 PCPs in which both the parent and a child had been diagnosed with CRC or EC. The pseudomedian of differences (see Supporting Information S2) between ages at diagnosis of the children compared with their parents (age P − age C) among the PCPs was 6 years and was statistically significant (p < .01; paired, two‐sided Wilcoxon signed‐rank test; Table 3). When separated into birth cohorts, parents were found to be diagnosed at a median of approximately 10 years later compared with their affected children when children were born after 1945 (Table 2). The exclusion of probands showed a similar trend with a median increase of approximately 11 years in age at diagnosis of parents compared with their children born after 1945, but with lower statistical significance (Table 2).
TABLE 3.
Estimated mean age at onset of cancer for each generation from parametric Weibull distribution models with and without the inclusion of probands and adjusted for birth cohort, sex, and MLH1 variant effect.
| Generation | Without probands, N = 411 | With probands, N = 438 | ||
|---|---|---|---|---|
| Age of diagnosis: Mean ± SE, years | 95% CI | Age of diagnosis: Mean ± SE, years | 95% CI | |
| Grandparents | 78.2 ± 8.3 | 61.8–94.5 | 75.0 ± 7.8 | 59.6–90.3 |
| Parents | 62.5 ± 3.4 | 55.9–69.1 | 62.5 ± 3.3 | 56.0–69.0 |
| Probands | 59.3 ± 3.6 | 52.3–66.4 | 55.7 ± 2.7 | 50.5–61.0 |
| Children | 44.4 ± 3.9 | 36.7–52.0 | 45.4 ± 3.9 | 37.6–53.1 |
Abbreviations: CI, confidence interval; SE, standard error.
TABLE 2.
(Pseudo)‐median of differences in age at diagnosis of affected parents and their affected children with the Wilcoxon signed‐rank test, n = 20 probands.
| P‐C pairs | No. without probands | Pseudomedian, P‐C pairs (95% CI) | p | No. with probands | Pseudomedian, P‐C pairs (95% CI) | p |
|---|---|---|---|---|---|---|
| All P–C pairs | 54 | 5.49 (0.50–11.00) | .03 | 62 | 6.00 (2.00–10.99) | < .01 |
| Child born <1945 | 25 | 4.7*e−5 (4.99, 6.00) | .94 | 26 | 1.50 (−4.49, 7.50) | .66 |
| Child born >1945 | 29 | 10.99 (2.50–21.00) | .12 | 36 | 9.99 (3.99–17.50) | < .01 |
Abbreviations: CI, confidence interval; P–C pairs, parent–child pairs.
Predicted mean ages of onset in different generations calculated from multivariate Weibull accelerated failure time models indicated that children, probands, and parents were diagnosed at an average of 10.3, 6.8, and 12.5 years earlier, respectively, compared with their previous generations (Table 3). When probands were excluded, children, probands, and parents showed a decline in age of diagnosis by 14.9, 3.2, and 15.7 years, respectively, compared with their previous generations (Table 3). These predicted mean ages of onset were estimated accounting for censoring, whereas the mean ages at diagnosis in Table 2 were calculated only among patients who were diagnosed with CRC or EC.
Cox‐PH models with or without probands showed a trend toward a greater hazard at any given age in younger generations (Figure 2). Probands were at 54% greater hazard for CRC/EC than their grandparents, similar to the hazard among parents, and at 150% lower hazard compared with children when probands were excluded from the multivariate Cox‐PH model (Figure 2A). With the inclusion of probands, probands were at 60% greater hazard than grandparents, at 33% greater hazard than parents (p = .06; 95% CI, 0.44–1.01), and at 53% lower hazard than children (p = .14; 95% CI, 0.87–2.7; Figure 2B). The cumulative incidence of CRC/EC predicted by the Cox‐PH model stratified by generation indicated that the median age at diagnosis of cancer was approximately 44, 46, 55, and 72 years in children, probands, parents, and grandparents respectively (Figure 3). The cumulative incidence of CRC/EC in children was lower before and greater after age 40 years compared with the incidence in probands (Figure 3).
FIGURE 2.
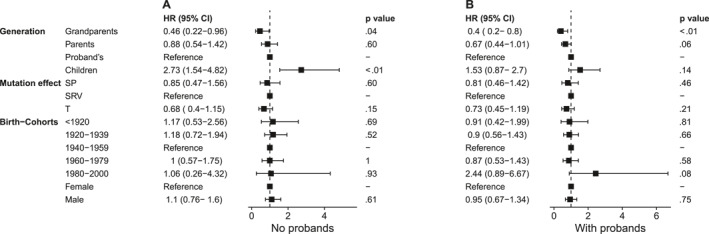
Forest plots showing HRs for CRC/EC as estimated by the Cox‐PH models (A) without probands and (B) with probands. CI indicates confidence interval; Cox‐PH, Cox proportional hazards; CRC/EC, colorectal and/or endometrial cancers; HR, hazard ration; SP, splicing effect; SRV, single residue variation; T, termination effect.
FIGURE 3.
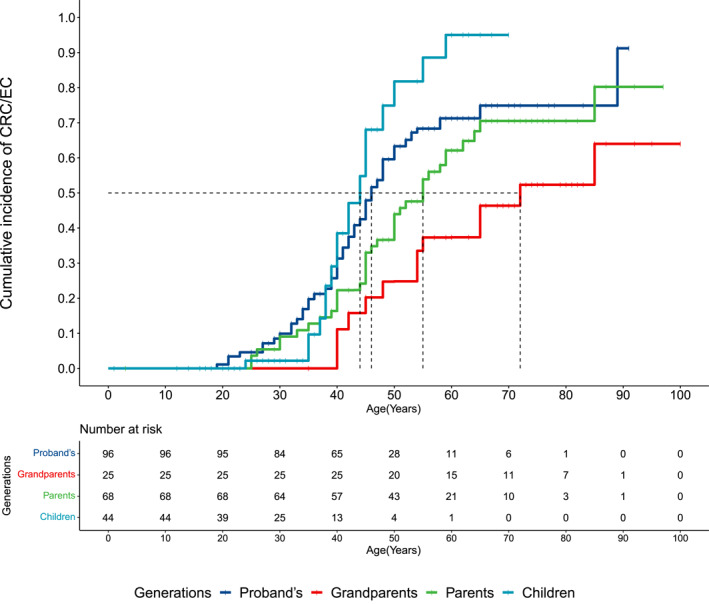
Cumulative incidence of CRC/EC by age as predicted by the multivariate COX‐PH model stratified by generation, probands included. The median age at diagnosis for each generation is indicated. Censoring is represented by vertical bars. Cox‐PH indicates Cox proportional hazards; CRC/EC, colorectal and endometrial cancers.
The hazard for CRC/EC with different mutation effects appeared slightly lower in SP and T mutation effects compared with SRV effects, but the difference was statistically insignificant (Figure 2). There were no significant differences in the hazard for truncating variants compared with nontruncating variants (HR, 0.9; p = .67; 95% CI, 0.56–1.45). MMR activity was known in 19 families, of which seven had mutations that were MMR‐proficient. MMR deficiency showed a 78% increased hazard for CRC/EC compared with variants that retained MMR proficiency (HR, 1.78; p = .03; 95% CI, 1.05–3.04) through univariate Cox‐PH analysis. Mean ages of diagnosis weighted by mutation probabilities decreased by 12.5 and 9.4 years between grandparents, parents, and probands in the MMR‐proficient group (Table 1), with no CRC/EC in children. In the MMR‐deficient group, the corresponding decrease in mean age of diagnosis were 7.0 years and 2.4 years between grandparents, parents and probands, while it increased by a year in children compared to probands. Cox‐PH analysis in the MMR‐deficient group showed no significant differences in HRs over generations except for grandparents (see Supporting Information S1). The Cox‐PH analysis model for the MMR proficient group was a poor fit (likelihood ratio test, 0.72; degrees of freedom [df] = 3; p = .80). However, an analysis of variance indicated a significant effect of generation on age at diagnosis in the MMR‐proficient group (df = 3; F = 6.91; p < .01), whereas it was not significant in the MMR‐deficient group (df = 3; F = 1.62; p = .18).
HRs between males and females were not significant in any of the Cox models (Figure 2). HRs from multivariate Cox mixed‐effects regression with families included as random effects were similar to HRs in the Cox‐PH model (see Supporting Information S1).
HRs in birth cohorts in Cox‐PH models did not follow a specific trend, with only the 1980–2000 birth cohort showing a 144% greater hazard than the 1940–1959 cohort when probands were included (Figure 2B). HRs between birth cohorts were statistically insignificant in Cox‐PH models with 10‐year, pre‐1945 and post‐1945, or pre‐1966 and post‐1966 birth cohorts (see Supporting Information S1).
DISCUSSION
The results from our study show evidence of anticipation in families with MLH1‐associated LS. At any given age, the hazard for CRC/EC was consistently lower in older generations across all models, and this effect was not precluded by the inclusion of birth cohorts. Younger mean ages at diagnosis (Weibull model) for younger generations aligned with their increase in hazard (Cox‐PH model). We also observed that mutations causing MMR deficiency resulted in a greater hazard for CRC/EC compared with those that retained MMR activity.
Our results are in agreement with previous studies of MLH1‐associated LS that reported anticipation effects ranging from 2.8 8 , 9 to 6 years 17 or an increased hazard of cancer in subsequent generations. 6 Our study augments previously observed effects of birth cohorts by demonstrating dependence of anticipation effects on the criteria for defining birth cohorts. For example, in comparing PCPs in our study, children born before 1945 had similar ages of cancer onset compared with their parents, whereas those born after 1945 were diagnosed approximately 10 years earlier compared with their parents. In the general population, a birth‐cohort effect for CRC is hypothesized to be caused by exposures, such as sedentary lifestyle, obesity, alcohol intake, and consumption of processed meat, among others, that began in the 1950s 23 , 24 and may explain the earlier onset of CRC in post‐1945 birth cohorts in the current study. Studies similar to ours that reported the year of birth as a likely explanation of anticipation in LS 7 , 16 did not report the HRs associated with birth cohorts, which, in our study, were observed to be of negligible effect size and statistically insignificant. Another study reporting evidence against genetic anticipation in familial CRC also suggested that anticipation could not be explained by a secular trend. 13 This was evident in our models also, none of which showed a trend or significant difference in ages of diagnosis among birth cohorts irrespective of how they were defined (see Supporting Information S1). Birth‐cohort effects are implicated in an increase in the worldwide incidence of cancer, 25 and well established risk factors for CRC have been associated with early onset CRC and later onset CRC to similar degrees, 26 which manifest as an increase in incidence but not a younger age at diagnosis. Several new risk factors for CRCs, such as exposures during fetal development and the gut microbiome, have continued to emerge. 25 , 27 The chronological occurrences of this array of risk factors is highly variable, making the categorization of birth cohorts by secular effects challenging. The occurrence of mutations in MMR genes in patients with pediatric cancer, in whom exposure to environmental effects is limited, also favors genetic rather than environmental effects in anticipation. 28 , 29 , 30 , 31 , 32 , 33
Ascertainment bias can overestimate disease risk and penetrance in clinical genetic variant–cancer association studies 34 because of an overrepresentation of young onset cases in probands and right truncation effects, 35 and is corrected by excluding probands from analysis. Age at cancer diagnosis of affected family members was reported during family history‐taking irrespective of whether individuals were seen at clinic, and all family members with CRC/EC were included in the analysis. This prevents ascertainment bias, so that the exclusion of probands is not necessary. Also, 11 of the 31 probands in our study were not affected by CRC/EC and did not contribute to younger ages of diagnosis. The exclusion of probands from the regression models unnecessarily discards reliable data with high mutation probabilities and may be less accurate than models with probands included.
It is interesting that the cumulative incidence of cancer in the children's generation is lower than that of probands as well as parents at ages younger than 40 years. A possible explanation is screening followed by family members who carry the familial MLH1 mutation. Screening through colonoscopy or sigmoidoscopy has been shown to reduce the risk and incidence of CRC. 36 , 37 Early detection of adenomas followed by polypectomy would prevent the transition to adenocarcinoma, postponing the onset of CRC/EC to an older age, which possibly explains lower incidence in younger individuals of the children's generation. Early detection of an existing CRC would lead to early diagnosis, which, in our data, aligns with the rapid increase in incidence of CRC/EC in children older than 40 years. This also indicates that more frequent screening after age 35 years may be desirable.
To our knowledge, this is the first study that has modeled mutation effects on Mlh1 protein as a covariate in a Cox‐PH model to study anticipation effects in pedigree data. A previous study that stratified LS cancer risks by gene and variant type found significantly older median ages at the onset (lower hazard) of EC for MLH1‐truncating variants compared with splice‐site variants and large rearrangements. 38 However, a recent, prospective LS database study that examined the effect of type of variant in greater than 5000 carriers of MMR PVs found no difference in penetrance between truncating and missense/aberrant splicing variants in MLH1 and MSH2. 21 Lastella et al. 39 demonstrated that missense variants in MLH1 can have in‐vivo effects different from those predicted by in‐silico analysis, so that variant types may not always be correlated with Mlh1 protein activity. An international study has proposed the presence of risk modifiers in LS that may not be variant‐specific, and the authors have proposed estimating penetrance according to the effect of a variant on protein function. 3 Our categorization of MLH1 variants as SRV, SP, and T was based on published functional and in‐silico evidence and augmented generational effects on age of diagnosis. Admittedly, some of these categories may approximate, but cannot replace, Mlh1 activity determined through functional assays.
Our study provides evidence for the influence of Mlh1 protein activity on age at onset of CRC/EC. Differences in the proportion of affected individuals and mean ages at diagnosis of CRC/EC over generations were notable between the MMR‐proficient and MMR‐deficient groups. Pathogenicity because of MLH1 mutations is attributed to protein instability as well as catalytic dysfunction, 40 and an MLH1 variant that produces active protein may still be pathogenic if its expression is below a certain threshold. 41 Also, the expression of immune‐related genes has been identified as distinct in cancer‐free patients with LS and those with CRC. 42 A substantial number of LS cases are associated with genomic rearrangements, susceptibility to which may be determined by Alu density in MLH1. 43 This complex interplay of MLH1 cis‐acting factors, epistasis, and the level of Mlh1 activity may be an essential part of the mechanism of anticipation, but further studies are needed to confirm this hypothesis. Statistical models with larger pedigree and functional data grouped by Mlh1 activity will help discern the role of the Mlh1 activity level in anticipation.
To the best of our knowledge, this is the first regression analysis in MLH1‐associated LS that includes mutation effects as a covariate while adjusting for ascertainment bias and birth‐cohort effects. Despite the applied corrections, the possibility of other uncontrolled biases and skewness because of calculated ages for older generations cannot be excluded. Our study also limited cancer diagnoses to CRC or EC, which are strongly associated with MLH1 mutations, while excluding other cancers known to be associated with MLH1, such as gastric, bladder, pancreatic, prostate, and brain cancers. 44 , 45 Inclusion of these cancer types in anticipation studies would present a more realistic picture of generational differences in ages of diagnosis of MLH1‐associated cancers. Another limitation was the insufficient number of affected individuals with MMR proficiency. Future studies on a larger number of families with MLH1‐associated LS caused by MMR‐proficient mutations are needed to discern its effect on age of diagnosis.
In conclusion, we report evidence in support of an anticipation effect in families with MLH1‐associated LS that cannot be explained by birth‐cohort effects. Our study supports screening for CRC/EC 2 to 10 years earlier than the youngest age of diagnosis of MLH1‐associated LS in the family. We noted that variant types, when defined by their effects on Mlh1 protein activity, constitute a contributing factor to alterations in cancer risk. Risk assessment for CRC/EC in families with MLH1‐associated LS should factor in information on MMR proficiency or deficiency when available.
AUTHOR CONTRIBUTIONS
Arti S. Pandey: Conceptualization, methodology, data curation, software, investigation, validation, formal analysis, resources, visualization, writing–original draft, and writing–review and editing. Christine Drogan: Data curation, resources, and writing–review and editing. Dezheng Huo: Methodology, investigation, validation, formal analysis, supervision, resources, writing–review and editing, and software. Kristen Postula: Methodology and writing–review and editing. Shreshtha M. Garg: Project administration and supervision. Sonia S. Kupfer: Methodology, resources, supervision, validation, investigation, project administration, writing–review and editing, and conceptualization.
CONFLICT OF INTEREST STATEMENT
The authors declared no conflicts of interest.
Supporting information
Supporting Information S1
Supporting Information S2
Pandey AS, Drogan C, Huo D, Postula K, Garg SM, Kupfer SS. Anticipation in families with MLH1‐associated Lynch syndrome. Cancer. 2025;e35589. doi: 10.1002/cncr.35589
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article because no new data were created or analyzed in this study.
REFERENCES
- 1. Biller LH, Syngal S, Yurgelun MB. Recent advances in Lynch syndrome. Fam Cancer. 2019;18(2):211‐219. doi: 10.1007/s10689-018-00117-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Valle L, Vilar E, Tavtigian SV, Stoffel EM. Genetic predisposition to colorectal cancer: syndromes, genes, classification of genetic variants and implications for precision medicine. J Pathol. 2019;247(5):574‐588. doi: 10.1002/path.5229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. International Mismatch Repair Consortium . Variation in the risk of colorectal cancer in families with Lynch syndrome: a retrospective cohort study. Lancet Oncol. 2021;22(7):1014‐1022. doi: 10.1016/S1470-2045(21)00189-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Warthin AS. The further study of a cancer family. J Cancer Res. 1925;9(2):279‐286. doi: 10.1158/jcr.1925.279 [DOI] [Google Scholar]
- 5. Strachan T, Read AP . Human Molecular Genetics. 2nd ed. Wiley‐Liss; 1999. [Google Scholar]
- 6. von Salomé J, Boonstra PS, Karimi M, et al. Genetic anticipation in Swedish Lynch syndrome families. PLoS Genet. 2017;13(10):e1007012. doi: 10.1371/journal.pgen.1007012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ten Broeke SW, Rodriguez‐Girondo M, Suerink M, et al. The apparent genetic anticipation in PMS2‐associated Lynch syndrome families is explained by birth‐cohort effect. Cancer Epidemiol Biomarkers Prev. 2019;28(6):1010‐1014. doi: 10.1158/1055-9965.EPI-18-0576 [DOI] [PubMed] [Google Scholar]
- 8. Larsen K, Petersen J, Bernstein I, Nilbert M. A parametric model for analyzing anticipation in genetically predisposed families. Stat Appl Genet Mol Biol. 2009;8(1):Article26‐11. doi: 10.2202/1544-6115.1424 [DOI] [PubMed] [Google Scholar]
- 9. Nilbert M, Timshel S, Bernstein I, Larsen K. Role for genetic anticipation in Lynch syndrome. J Clin Oncol. 2009;27(3):360‐364. doi: 10.1200/JCO.2008.16.1281 [DOI] [PubMed] [Google Scholar]
- 10. Ponti G, Ruini C, Tomasi A. Mismatch repair gene deficiency and genetic anticipation in Lynch syndrome: myth or reality? Dis Colon Rectum. 2015;58(1):141‐142. doi: 10.1097/DCR.0000000000000275 [DOI] [PubMed] [Google Scholar]
- 11. Stella A, Surdo NC, Lastella P, et al. Germline novel MSH2 deletions and a founder MSH2 deletion associated with anticipation effects in HNPCC. Clin Genet. 2007;71(2):130‐139. doi: 10.1111/j.1399-0004.2007.00745.x [DOI] [PubMed] [Google Scholar]
- 12. Stupart D, Goldberg P, Algar U, Vorster A, Ramesar R. No evidence of genetic anticipation in a large family with Lynch syndrome. Fam Cancer. 2014;13(1):29‐34. doi: 10.1007/s10689-013-9669-0 [DOI] [PubMed] [Google Scholar]
- 13. Tsai YY, Petersen GM, Booker SV, Bacon JA, Hamilton SR, Giardiello FM. Evidence against genetic anticipation in familial colorectal cancer. Genet Epidemiol. 1997;14(4):435‐446. doi: [DOI] [PubMed] [Google Scholar]
- 14. Vasen HF, Taal BG, Griffioen G, et al. Clinical heterogeneity of familial colorectal cancer and its influence on screening protocols. Gut. 1994;35(9):1262‐1266. doi: 10.1136/gut.35.9.1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Westphalen AA, Russell AM, Buser M, et al. Evidence for genetic anticipation in hereditary non‐polyposis colorectal cancer. Hum Genet. 2005;116(6):461‐465. doi: 10.1007/s00439-005-1272-5 [DOI] [PubMed] [Google Scholar]
- 16. Voskuil DW, Vasen HF, Kampman E, van't Veer P. Colorectal cancer risk in HNPCC families: development during lifetime and in successive generations. National Collaborative Group on HNPCC. Int J Cancer. 1997;72(2):205‐209. doi: [DOI] [PubMed] [Google Scholar]
- 17. Boonstra PS, Mukherjee B, Taylor JM, Nilbert M, Moreno V, Gruber SB. Bayesian modeling for genetic anticipation in presence of mutational heterogeneity: a case study in Lynch syndrome. Biometrics. 2011;67(4):1627‐1637. doi: 10.1111/j.1541-0420.2011.01607.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bozzao C, Lastella P, Stella A. Anticipation in Lynch syndrome: where we are where we go. Curr Genomics. 2011;12(7):451‐465. doi: 10.2174/138920211797904070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non‐Polyposis Colorectal Cancer (ICG‐HNPCC). Dis Colon Rectum. 1991;34(5):424‐425. doi: 10.1007/BF02053699 [DOI] [PubMed] [Google Scholar]
- 20. Ten Broeke SW, van der Klift HM, Tops CMJ, et al. Cancer risks for PMS2‐associated Lynch syndrome. J Clin Oncol. 2018;36(29):2961‐2968. doi: 10.1200/JCO.2018.78.4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dominguez‐Valentin M, Plazzer JP, Sampson JR, et al. No difference in penetrance between truncating and missense/aberrant splicing pathogenic variants in MLH1 and MSH2: a prospective Lynch syndrome database study. J Clin Med. 2021;10(13):2856. doi: 10.3390/jcm10132856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ganesh S, Cave V. P‐values, p‐values everywhere. N Z Vet J. 2018;66(2):55‐56. doi: 10.1080/00480169.2018.1415604 [DOI] [PubMed] [Google Scholar]
- 23. Ugai T, Sasamoto N, Lee HY, et al. Is early‐onset cancer an emerging global epidemic? Current evidence and future implications. Nat Rev Clin Oncol. 2022;19(10):656‐673. doi: 10.1038/s41571-022-00672-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Akimoto N, Ugai T, Zhong R, et al. Rising incidence of early‐onset colorectal cancer—a call to action. Nat Rev Clin Oncol. 2021;18(4):230‐243. doi: 10.1038/s41571-020-00445-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. World Cancer Research Fund/American Institute for Cancer Research . Diet, Nutrition, Physical Activity and Colorectal Cancer. Continuous Update Project Expert Report. World Cancer Research Fund Network; 2018. Accessed March, 2024. https://www.wcrf.org/wp‐content/uploads/2021/02/Colorectal‐cancer‐report.pdf [Google Scholar]
- 26. Archambault AN, Lin Y, Jeon J, et al. Nongenetic determinants of risk for early‐onset colorectal cancer. JNCI Cancer Spectr. 2021;5(3):pkab029. doi: 10.1093/jncics/pkab029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gupta S, May FP, Kupfer SS, Murphy CC. Birth cohort colorectal cancer (CRC): implications for research and practice. Clin Gastroenterol Hepatol. 2023;22(3):455‐469.e7. doi: 10.1016/j.cgh.2023.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Self C, Suttman A, Wolfe Schneider K, Hoffman L. Lynch syndrome: further defining the pediatric spectrum. Cancer Genet. 2021;258‐259:37‐40. doi: 10.1016/j.cancergen.2021.07.002 [DOI] [PubMed] [Google Scholar]
- 29. Scollon S, Eldomery MK, Reuther J, et al. Clinical and molecular features of pediatric cancer patients with Lynch syndrome. Pediatr Blood Cancer. 2022;69(11):e29859. doi: 10.1002/pbc.29859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stewart M, McCormick M, Windreich RM, Munro C, Meade J. A mismatched syndrome: a five‐year‐old girl with very‐high‐risk leukemia and Lynch syndrome. Pediatr Blood Cancer. 2023;70(11):e30660. doi: 10.1002/pbc.30660 [DOI] [PubMed] [Google Scholar]
- 31. Tua‐Caraccia R, Livingston A, Routh JC. Recurrent papillary bladder tumors in a boy with Lynch syndrome. Urology. 2023;181:133‐135. doi: 10.1016/j.urology.2023.06.016 [DOI] [PubMed] [Google Scholar]
- 32. MacArthur TA, Ongie LJ, Lanpher BC, Ishitani MB. Pediatric manifestations of Lynch syndrome: a single center experience. J Pediatr Surg Case Rep. 2022;86:102431. doi: 10.1016/j.epsc.2022.102431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liepert M, Brundler MA, Galante GJ. A rare presentation of pediatric Lynch syndrome presenting with recurrent adenomatous polyps. JPGN Rep. 2023;4(4):e354. doi: 10.1097/PG9.0000000000000354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ranola JMO, Tsai GJ, Shirts BH. Exploring the effect of ascertainment bias on genetic studies that use clinical pedigrees. Eur J Hum Genet. 2019;27(12):1800‐1807. doi: 10.1038/s41431-019-0467-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guindalini RS, Song A, Fackenthal JD, Olopade OI, Huo D. Genetic anticipation in BRCA1/BRCA2 families after controlling for ascertainment bias and cohort effect. Cancer. 2016;122(12):1913‐1920. doi: 10.1002/cncr.29972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bretthauer M, Loberg M, Wieszczy P, et al. Effect of colonoscopy screening on risks of colorectal cancer and related death. N Engl J Med. 2022;387(17):1547‐1556. doi: 10.1056/NEJMoa2208375 [DOI] [PubMed] [Google Scholar]
- 37. Holme O, Schoen RE, Senore C, et al. Effectiveness of flexible sigmoidoscopy screening in men and women and different age groups: pooled analysis of randomised trials. BMJ. 2017;356:i6673. doi: 10.1136/bmj.i6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ryan NAJ, Morris J, Green K, et al. Association of mismatch repair mutation with age at cancer onset in Lynch syndrome: implications for stratified surveillance strategies. JAMA Oncol. 2017;3(12):1702‐1706. doi: 10.1001/jamaoncol.2017.0619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lastella P, Surdo NC, Resta N, Guanti G, Stella A. In silico and in vivo splicing analysis of MLH1 and MSH2 missense mutations shows exon‐ and tissue‐specific effects. BMC Genom. 2006;7(1):243. doi: 10.1186/1471-2164-7-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mahdouani M, Ben Ahmed S, Hmila F, et al. Functional characterization of MLH1 missense variants unveils mechanisms of pathogenicity and clarifies role in cancer. PLoS One. 2022;17(12):e0278283. doi: 10.1371/journal.pone.0278283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hinrichsen I, Brieger A, Trojan J, Zeuzem S, Nilbert M, Plotz G. Expression defect size among unclassified MLH1 variants determines pathogenicity in Lynch syndrome diagnosis. Clin Cancer Res. 2013;19(9):2432‐2441. doi: 10.1158/1078-0432.CCR-12-3299 [DOI] [PubMed] [Google Scholar]
- 42. Bohaumilitzky L, Kluck K, Huneburg R, et al. The different immune profiles of normal colonic mucosa in cancer‐free Lynch syndrome carriers and Lynch syndrome colorectal cancer patients. Gastroenterology. 2022;162(3):907‐919.e10. doi: 10.1053/j.gastro.2021.11.029 [DOI] [PubMed] [Google Scholar]
- 43. Li L, McVety S, Younan R, et al. Distinct patterns of germ‐line deletions in MLH1 and MSH2: the implication of Alu repetitive element in the genetic etiology of Lynch syndrome (HNPCC). Hum Mutat. 2006;27(4):388. doi: 10.1002/humu.9417 [DOI] [PubMed] [Google Scholar]
- 44. Engel C, Loeffler M, Steinke V, et al. Risks of less common cancers in proven mutation carriers with lynch syndrome. J Clin Oncol. 2012;30(35):4409‐4415. doi: 10.1200/JCO.2012.43.2278 [DOI] [PubMed] [Google Scholar]
- 45. Watson P, Vasen HFA, Mecklin JP, et al. The risk of extra‐colonic, extra‐endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123(2):444‐449. doi: 10.1002/ijc.23508 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1
Supporting Information S2
Data Availability Statement
Data sharing is not applicable to this article because no new data were created or analyzed in this study.