

Abstract
Employing electrochemistry for the selective functionalization of liquid alkanes allows for sustainable and efficient production of high-value chemicals. However, the large potentials required for C(sp3)-H bond functionalization and low water solubility of such alkanes make it challenging. Here we discover that a Pt/IrOx electrocatalyst with optimized Cl binding energy enables selective generation of Cl free radicals for C-H chlorination of alkanes. For instance, we achieve monochlorination of cyclohexane with a current up to 5 A, Faradaic efficiency (FE) up to 95% and stable performance over 100 h in aqueous KCl electrolyte. We further demonstrate that our system can directly utilize concentrated seawater derived from a solar evaporation reverse osmosis process, achieving a FE of 93.8% towards chlorocyclohexane at a current of 1 A. By coupling to a photovoltaic module, we showcase solar-driven production of chlorocyclohexane using concentrated seawater in a membrane electrode assembly cell without any external bias. Our findings constitute a sustainable pathway towards renewable energy driven chemicals manufacture using abundant feedstock at industrially relevant rates.
Subject terms: Electrocatalysis, Electrochemistry, Materials for energy and catalysis
Large potentials required for C(sp3)-H bond activation and low water solubility make electrochemical functionalization of alkanes challenging. Here, the authors report that Pt/IrOx enables selective generation of Cl free radicals for chlorination of cyclohexane at industrially relevant rates.
Introduction
The functionalization of alkyl C(sp3)-H bonds is crucial for the synthesis of a broad range of fine chemicals with important applications in the medical, agrochemical and electronics industries1–4. For instance, halogenation to form alkyl C(sp3)-Cl is typically employed as the first functionalization step for alkanes. The resulting organohalides produced are versatile building blocks which are highly amenable towards the formation of C-C or C-heteroatom (e.g., C-N, C-S) bonds through synthetic chemistry5,6. Such halogenation reactions are highly crucial in the modern chemicals industry, being involved in the production routes of more than 50% of industrial chemicals and about 20% of pharmaceutical small molecule drugs7. As a result, 75 million tons of Cl2 are generated annually for the production of chlorinated chemicals, which is costly and energy-intensive7. Moreover, the use of the Cl2 reactant is limited to only 50% due to formation of unreactive HCl in the reaction pathway. In the US alone, ~1090 million kilograms of HCl acid waste is generated annually from the production of chlorinated solvents8, which then requires a chlorine recycling process or large amounts of NaOH for neutralization.
An important organohalide is chlorocyclohexane, which is widely used as a precursor for the manufacture of pesticides, pharmaceuticals, and rubber anti-scorching agents9,10. This can be directly produced through the formation of Cl free radicals from Cl2 under photothermal conditions, which then react with cyclohexane to form chlorocyclohexane11,12. This results in the formation of one mole of HCl acid waste for every mole of chlorocyclohexane produced. Hence, the toxic reactant/byproduct and low reactant atomic efficiency make this process non-ideal from a green chemistry standpoint. Alternatively, chlorocyclohexane can also be manufactured through a two-step thermochemical process starting with cyclohexane (Fig. 1a). In the first step, cyclohexane is partially oxidized at high temperature and pressure (150–200 °C and 10–14 atm) into cyclohexanol13,14. Next, cyclohexanol is reacted with HCl to generate chlorocyclohexane at a temperature of 80–100 °C15–17. Although this process has a higher atom economy, toxic and expensive reactants are still used, and two steps are required as opposed to only a single step. Furthermore, the HCl feed is not completely utilized, which results in diluted HCl waste that needs to be either recycled or neutralized.
Fig. 1. Renewable energy powered electrosynthesis of chlorocyclohexane.

a Schematic of thermochemical process involving Cl2 or HCl as the reactant. b Schematic of our proposed green electrochemical process for the complete solar-driven one-step conversion of cyclohexane to chlorocyclohexane and co-production of green H2, using concentrated seawater as the electrolyte. The concentrated seawater is obtained through a solar evaporation reverse osmosis process.
There has therefore been strong interest in developing processes for single-step alkane halogenation using mild reagents (e.g. KCl, NaCl) under ambient conditions with minimal waste generation. This has benefits including lower costs as well as greater simplicity and safety of operation18. Motivated by this need, recent studies have shown that one-step halogenation is possible with mild reagents. For example, Zhou and co-workers showed that a Mn metal-organic framework could catalyze C-H halogenation using NaClO in an acetone/water mixture at room temperature19,20. Photoelectrochemical C-H halogenation was also demonstrated with a TiO2 photoelectrode, whereby Cl free radicals could be generated from Cl− in the electrolyte. These Cl free radicals then react with cyclohexane to produce chlorocyclohexane21. Although these previous studies have made important progress, production rates need to be significantly increased for industrial relevance and techno-economic viability. For instance, photoelectrochemical systems have constraints on the maximum current density that can be attained, which tend to limit production rates22–24.
We reason that electrochemical systems25–28 could potentially solve these issues, with advantages including (1) greener production using mild reagents, (2) industrially relevant production rates, (3) single step halogenation under ambient conditions, (4) direct utilization of renewable electricity, (5) co-generation of green H2 gas and (6) utilization of seawater (Fig. 1b). For instance, Lei and co-workers developed a system for electrochemical C-H halogenation27 using HX/NaX (X represents the halogenation element, Cl or Br) as the reagent and a mixture of N,N-dimethylformamide (10.5 ml) and H2O (0.5 ml) as the solvent. With a C electrode at an applied current of 12 mA, product yields of up to 81% could be obtained in their experiments.
However, significant advances are still required to realize electrochemical C-H halogenation at high current density and Faradaic efficiency (FE), and this is mainly hampered by two important challenges. Firstly, C-H functionalization imposes large positive overpotentials, which can lead to side reactions such as the oxygen evolution reaction (OER), chlorine evolution reaction (CER) and even uncontrolled overoxidation of cyclohexane into CO229–31. Secondly, liquid alkanes such as cyclohexane have limited solubility if aqueous electrolytes are employed (0.006% in water)32. Hence, if the C-H halogenation process is mediated on the electrode surface, the system can rapidly become reactant mass transfer-limited, making it difficult to achieve a high FE when large current densities are applied.
Here we discover that performing electrolysis in Cl− containing cyclohexane/water emulsions allows for the selective production of chlorocyclohexane at high current densities (Fig. 2). Key to achieving this is maintaining rapid agitation of the emulsion and designing electrocatalysts which display a preference for the formation of Cl free radicals as opposed to Cl2 evolution. Importantly, these Cl free radicals enable cyclohexane to chlorocyclohexane conversion to take place in the bulk of the electrolyte, allowing for mass transport limitations to be overcome. This is enabled by designing a Pt/IrOx electrocatalyst with an optimized Cl binding energy. As a result of this strategy, we achieve cyclohexane to chlorocyclohexane conversion at high currents of up to 5 A (1 A/cm2), with a FE of up to 95% and stable operation over a 100 h period, achieving a yield of 1668 mmol of chlorocyclohexane. We also demonstrate that similar performance can be attained using concentrated seawater derived from a solar evaporation reverse osmosis process. Finally, solar-driven chlorocyclohexane production in a membrane electrode assembly (MEA) system is also achieved by coupling with a photovoltaic module.
Fig. 2. Chlorination of cyclohexane under ambient conditions.
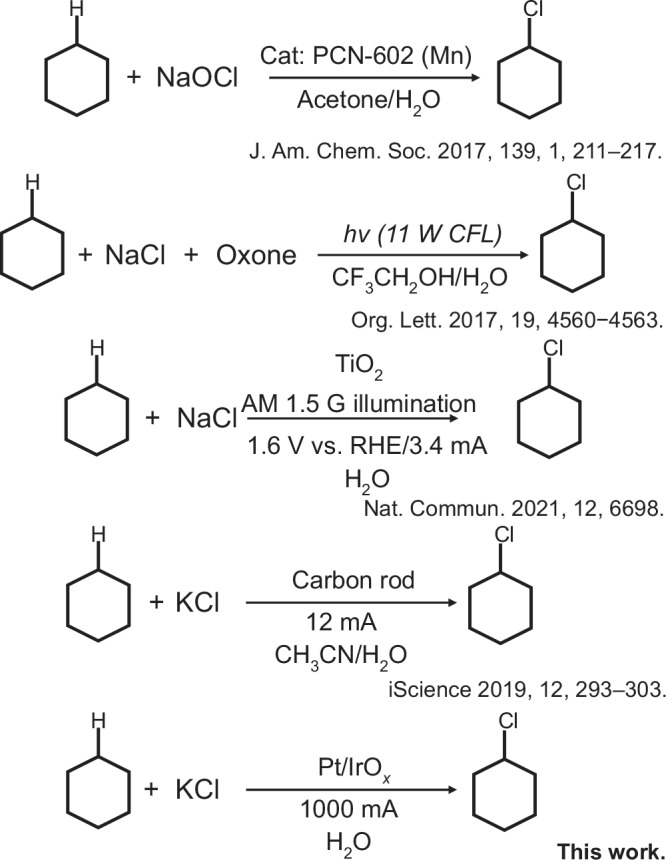
Comparison with the prior literature and brief description of reaction conditions.
Results
Mechanism of chlorocyclohexane electrosynthesis
We first sought to develop a working system for the electrosynthesis of chlorocyclohexane. For this, we performed bulk electrolysis experiments with a simple H-type cell (Fig. S1), using 5 ml of cyclohexane added to 45 ml of 1.0 M KCl as the electrolyte. As cyclohexane has low solubility in aqueous media, it separates out as a top organic layer. Hence, we employed continuous and rapid mechanical stirring (500 rpm) to create and maintain an emulsion for facilitating the reaction (Supplementary Movie 1). The stirring is stopped at the end of the reaction, which causes the insoluble chlorocyclohexane and cyclohexane to separate out and form a top organic layer again. This top organic layer can be easily collected using a separating funnel, for product analysis and quantification using Fourier-transform infrared spectroscopy (FTIR) and gas chromatography mass spectrometry (GCMS) respectively. A schematic that illustrates each of the steps involved in the process is shown in Fig. S2.
Our initial trials involved applying a constant current of 300 mA with different acid stable anode materials (C, Pt, Ti and IrOx) to evaluate the chlorocyclohexane FE for each case. The results (Fig. S3-S5) indicate that monochlorinated chlorocyclohexane can indeed be generated by this process and is the sole product detected for all cases. In addition, the overpotential required and chlorocyclohexane FE obtained are observed to be dependent on the electrode material used, with the best results obtained for IrOx. Specifically, an FE of 20.4%, 15.8%, 28.4% and 45.3% was obtained for C, Ti, Pt and IrOx respectively (Fig. S6). Due to its higher FE, we selected IrOx as the electrode of choice for our further investigations (geometric area 5 cm2).
To understand the reasons for the higher FE obtained with the IrOx electrode, we firstly consider the plausible reaction mechanisms for chlorocyclohexane formation. It was previously proposed that Cl2 evolution on metal oxide surfaces involves the Volmer-Heyrovksy mechanism33,34. The first Volmer step involves a single electron oxidation of Cl− to form surface adsorbed *Cl (Cl− → *Cl + e−). In the subsequent Heyrovsky step, Cl2 is generated through another electron transfer via an Eley-Rideal mechanism involving *Cl and Cl− (*Cl + Cl− → Cl2 + e−). This generated Cl2 then undergoes disproportionation in the aqueous media to form HOCl, which could potentially serve as the active species19,35 for cyclohexane monochlorination. Another possibility is the generation of Cl free radicals from Cl− oxidation (Cl− → Cl• + e−)36–38. From here, cyclohexane would be transformed into chlorocyclohexane through a series of free radical substitution reactions39,40. Based on these two possibilities, we employed a suite of mechanism studies to identify the C-H halogenation pathway.
We firstly performed a control experiment where Cl2 gas was produced via electrolysis and directly purged into a separate cyclohexane solution (Fig. S7a). In this case, no chlorocyclohexane formation was detected (Fig. S7b), confirming that Cl2 is unable to directly react with cyclohexane. To investigate the role of HOCl as the active species, we then carried out Cl2 evolution with 1 M KCl as the electrolyte at 1000 mA for 30 min (without cyclohexane). This creates an electrolyte solution that is saturated with HOCl. 5 ml of cyclohexane was then added to this HOCl saturated solution and stirred at 500 rpm for 30 min, without any applied current. Similarly, chlorocyclohexane was also not detected (Fig. S8), which rules out the possibility of HOCl as the active species.
Following this, we conducted experiments where we added 0.1 mM of 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) into the 1 M KCl electrolyte as a free radical trap21,29. The current density was then set to 200 mA/cm2 with the IrOx electrode for a duration of 4 min. Using electron paramagnetic resonance (EPR) spectroscopy (Fig. S9), we detected the presence of DMPO-Cl, which provides evidence for the presence of electrogenerated Cl free radicals21,29. Another set of experiments was performed with 2,2,6,6-tetramethylpiperidine oxide (TEMPO) added to the electrolyte, which also functions as a free radical trap41. When electrolysis was carried out with IrOx, we found that the chlorocyclohexane FE at 800 mA (160 mA/cm2) decreases from 80.7% to 59.4% when 0.01 M of TEMPO was used. Doubling the TEMPO concentration to 0.02 M results in a further decrease in the FE to 32.9% (Fig. 3a and S10), which further supports the idea that C-H halogenation is mediated by Cl free radicals.
Fig. 3. Proposed mechanism for electrosynthesis of chlorocyclohexane.

a GCMS analysis of samples obtained with electrolyte containing 0.01 M or 0.02 M of TEMPO at a current of 800 mA. b EPR signal of the electrolyte after different reaction times with a current of 400 mA using IrOx. Normalized Faradaic efficiency of (c) O2 (m/z = 32) and (d) Cl2 (m/z = 69.9) collected when different electrolytes are employed using IrOx at a current of 400 mA. e Illustration of the competing Cl2 evolution and Cl free radical mediated C-H functionalization pathways. Source data are provided as a Source Data file.
We then carried out EPR spectroscopy tests where we found that the TEMPO signal gradually decreases with increasing reaction time (Fig. 3b). This is due to trapping of electrogenerated Cl free radicals to form TEMPO-Cl, which we detected through electrospray ionization–high-resolution mass spectrometry (ESI-MS) measurements42 (Fig. S11). Similar EPR spectroscopy experiments were also performed with the C anode, where we observed longer extinction times for the TEMPO signal, even though a same TEMPO concentration and current was employed (Fig. S12). These results strongly imply that Cl free radical formation competes with Cl2 evolution and is dependent on the electrode material used. In this case, IrOx has a higher propensity to generate Cl free radicals than C, which results in a higher FE towards chlorocyclohexane. This hypothesis is illustrated in Fig. 3e, where the generation of Cl free radicals leads to chlorocyclohexane formation. On the other hand, if Cl2 evolution occurs instead, chlorocyclohexane is unable to be generated.
To understand this competition between Cl free radical formation and Cl2 evolution, we performed online differential electrochemical mass spectroscopy (DEMS) experiments with IrOx, tracking signals related to Cl2 (m/z = 69.9) and O2 (m/z = 32) (Fig. S13). Electrolysis in 1.0 M KCl electrolyte was carried out at a current of 400 mA, where only Cl2 generation was observed (Fig. 3d). The lack of any O2 signal shows that OER does not occur under these electrolysis conditions (Fig. 3c). Next, when cyclohexane was added to the electrolyte, a much lower Cl2 signal was observed (Fig. 3d), which we attribute to consumption of Cl free radicals to form chlorocyclohexane. On the other hand, when C was used instead, we continued to observe a strong Cl2 signal (albeit slightly lower) even when cyclohexane was present in the electrolyte (Fig. S14). These experiments indicate that C has a stronger preference for Cl2 evolution, which impedes the generation of Cl free radicals needed for C-H halogenation.
We also performed DEMS experiments with KOH added into the electrolyte, where only an O2 signal was observed indicating that OER has become dominant on IrOx. Through iodometric titration experiments, we detected the presence of unreacted ClO− which accounts for an FE of about 12.4% (Fig. S15), with the balance going to O2 evolution (Fig. 3c). When we attempted to perform C-H halogenation with alkaline electrolyte, we were unable to detect any chlorocyclohexane product. Hence, we postulate that a high concentration of OH− in the electrolyte may result in the balance of the equation (Cl ↔ Cl• ↔ Cl2 + H2O ↔ HCl + HOCl) moving to the right. Hence, the formation of Cl free radicals is suppressed, which prevents the formation of chlorocyclohexane. On the other hand, chlorocyclohexane could still be generated with 1.0 M HCl acidic electrolyte (Fig. S16).
Linear sweep voltammetry (LSV) experiments (Fig. S17) were also carried out in the low current density regime to analyze the Tafel slopes for the C, PtOx, and IrOx anodes in 1.0 M KCl electrolyte with and without the presence of cyclohexane. For these experiments, a rotating disk electrode (RDE) system was employed for better control and uniformity of hydrodynamic conditions43. For all rotation rates employed, we found that the Tafel slopes for C, PtOx and IrOx are all quite similar with and without cyclohexane (Fig. S18). Hence, there is no change in the rate determining step with the addition of cyclohexane. This is consistent with cyclohexane chlorination being a free radical mediated pathway occurring in the electrolyte solution, which does not influence electron transfer on the electrode surface.
We also reasoned that a significant amount of C-H halogenation should be occurring in the bulk of the electrolyte since the low solubility of cyclohexane in water (0.006%) should quickly lead to mass transport limitations. To understand this, we evaluated the boundary layer thickness of our H-cell by determining the limiting current density44 for the reduction of 0.1 mM K3Fe(CN)6 at various stirring rates (Fig. S19). At the highest stirring rate of 500 rpm, we found the boundary layer thickness to be 2.87 µm, with a limiting current density of only 22.6 mA/cm2. Comparatively at an identical stirring rate, we could perform chlorination of cyclohexane at ≥200 mA/cm2 without suffering from mass transport issues. Hence, this indicates that the chlorination of cyclohexane cannot be limited to only the near electrode surface and boundary layer. Notably, the lifetime of Cl radicals in aqueous solutions is ~5 µs45,46, hence these radicals should in principle be able to leave the boundary layer and react with cyclohexane in the bulk electrolyte.
We also considered whether the formation of chlorocyclohexane could also occur through a chain21 mechanism (Fig. S20) rather than the nonchain mechanism47 that we depicted in Fig. 3e. To investigate this, we firstly performed electrolysis at 200 mA/cm2 for durations of 5, 10, or 15 min using 1 M HCl as the electrolyte without any cyclohexane added. This yields pretreated electrolytes containing Cl2 with concentrations of 0.03, 0.057, and 0.082 M for 5, 10, and 15 min respectively (Fig. S21). We then added cyclohexane to these solutions and performed another electrolysis at 200 mA/cm2 for another 5 min (two-step). In a separate experiment, we performed electrolysis at 200 mA/cm2 for 5 min with 1 M HCl as the electrolyte, with cyclohexane added (one-step). Importantly, if the chain mechanism is operative, then we would expect a larger amount of chlorocyclohexane to be generated in the two-step experiment, due to the presence of Cl2 reactant in the electrolyte. For the two-step case, we obtained conversion yields of 2.93%, 2.94%, and 2.90% for pretreatment electrolysis times of 5, 10, and 15 min respectively (Fig. S22). As for the one-step case, this yielded a very similar conversion yield of 2.92%, which indicates that the nonchain mechanism is the dominant pathway in our system.
To investigate the effect of the emulsion on the functionalization chemistry, we first varied the amount of cyclohexane reactant (2, 3, 5 and 8 ml), whilst keeping the amount of 1 M KCl electrolyte constant at 45 ml. In all cases, a constant current of 1000 mA was applied for 30 min. For the lower cyclohexane amounts, we started to notice non-negligible amounts of dichlorocyclohexane formation (Fig. S23), with an FE of 22.8% and 9.3% for 2 ml and 3 ml of cyclohexane respectively. There was also a concomitant decrease in the chlorocylohexane FE from 89.1% with 5 ml cyclohexane to 65.9% and 78.8% with 2 ml and 3 ml cyclohexane respectively. In addition, we found that slower stirring rates create a less effective emulsion (see Supplementary Movie 1) and thus explored how this affects the FE towards chlorocyclohexane. In this case (Fig. S24), we observed that the FE towards chlorocyclohexane was reduced at the slower stirring speeds with an FE of 70.9% at 50 rpm as compared to 89.1% at 500 rpm. Similarly, we also observed an increased FE towards dichlorocyclohexane at the slower stirring speeds, with the highest value of 11.1% at 50 rpm. Hence, these experiments indicate that compromising the emulsion creates an imbalance between Cl free radicals and cyclohexane reactants, leading to double chlorination events and a reduced chlorocyclohexane FE.
The influence of the Cl− concentration was explored by varying the KCl concentrations from 0.2 M to 3.0 M. For each case, the K+ concentration was kept constant at 3 M by adding appropriate amounts of K2SO4 to the electrolyte. We found that the chlorocyclohexane FE is dependent on the KCl concentration. For instance, the FE increases from 32.5% with 0.2 M KCl to 94.8% with 3.0 M KCl using the IrOx electrode at 1000 mA (Fig. S25a, and S25b). However, once the KCl concentration reaches 1.0 M and above, increases in the chlorocyclohexane FE become less pronounced. For instance, the chlorocyclohexane FE was already 89.1% with 1.0 M KCl electrolyte at 1000 mA. Similar trends were also observed through a range of different applied currents (Fig. S25c). With 1.0 M KCl, we obtained FEs of 48.1%, 51.4%, 80.7% and 89.1% at applied currents of 400, 600, 800, and 1000 mA respectively (Fig. S26, Table S1).
We also evaluated the chemical yield of the process over a 120 min electrolysis duration (Fig. S27). For this experiment, we employed the IrOx electrode at a constant current of 1000 mA and the electrolyte consisted of 5 ml of cyclohexane added to 45 ml of 1.0 M KCl. The results show that the FE towards chlorocyclohexane decreases from 89% to 81% in 2 h for the IrOx based catalyst. (Fig. S27a). This is a result of competitive dichlorocyclohexane formation, which was observed to begin at the 75 min point with an FE of 1.2% (Fig. S27b). This value increases continuously and reaches an FE of 7.4% at the 120 min point. We also observed continuous consumption of cyclohexane and increasing chemical yields for chlorocyclohexane during this 120 min period (Fig. S27c). At the end of 120 min, we found that 65.8% of the total cyclohexane was consumed, while the conversion of cyclohexane to chlorocyclohexane was 60.3%.
Based on these observations, we reasoned that the formation of dichlorocyclohexane could be suppressed by separating out chlorocyclohexane from the unreacted cyclohexane. To do this, we carried out electrolysis at 1000 mA for 1 h and extracted the unreacted cyclohexane using a distillation setup (Fig. S28). The unreacted cyclohexane was then recycled and used for further chlorination. Through this strategy, we achieved an average FE towards chlorocyclohexane of 89.5% and a total conversion yield of 56% without dichlorocyclohexane generated. It is expected that through repeated distillation steps, the product yield can be further increased.
In sum, our experimental results conclusively show that electrogeneration of Cl free radicals is needed to perform C-H halogenation of cyclohexane, which is highly dependent on the anode material. We discovered that IrOx has the highest propensity for Cl free radical formation over the competing Cl2 evolution reaction, leading to the highest chlorocyclohexane FE amongst the various electrodes tested. We also found that rapid stirring is required to create an effective cyclohexane emulsion in the bulk electrolyte where majority of the reaction occurs. This enables operation at high current without being severely inhibited by mass transport issues. Importantly, this means that our system is not solely limited to reactants that have high solubility in aqueous media. For instance, this has restricted conventional systems to reactions such as glycerol48,49, hydroxymethylfurfural50,51 and methanol oxidation52,53. Hence, we show that a wider reactant pool can and should be considered for renewable energy-powered chemical transformations.
Catalyst design for enhanced chlorocyclohexane production
Following this, we further sought to optimize the electrocatalyst for achieving a higher chlorocyclohexane FE. Although we found that higher FEs could be achieved at the larger applied currents, the FE at the lower currents has room for significant improvement. Based on our findings, we reasoned that modification of the IrOx electrocatalyst could be used to tune the Cl binding energy, which likely controls whether a *Cl desorbs from the catalyst surface to form a free radical or undergoes the Heyrovsky step to form Cl2. This would in turn be a knob to tune the chlorocyclohexane FE. Hence, we built composite catalysts consisting of Co/IrOx, Au/IrOx and Pt/IrOx (characterization available in Fig. S29–S40), by mixing the appropriate metal salt precursor into the synthesis solution (see methods section in Supplementary Information)54. Notably Ir L3-edge X-ray absorption near edge structure (XANES) spectra of all three catalysts show similar results with IrOx (Fig. S41).
Transmission electron microscopy (TEM) images of Pt/IrOx show that the average particle size of Pt/IrOx is around 3-4 nm (Fig. 4d), which is similar to IrOx (Fig. S42). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of Pt/IrOx shows that the catalyst consists of discrete PtOx and IrOx components, with a high density of interfaces (Fig. 4c). This is consistent with our EDS mapping results, which show areas that are either rich in Pt or Ir (Figs. 4e, S43, and S44). We also employed in-situ X-ray absorption spectroscopy (XAS) to track and study the formation of Pt/IrOx at each stage (a to g) of the thermal annealing growth process (details in SI and Figs. 4a and S45). As expected, the extended X-ray absorption fine structure (EXAFS) results (Fig. 4b) show the gradual formation of Ir-O bonds and loss of Ir-Cl bonds during each stage of the process. Through Pt L3-edge XAS measurements, we also observed that Pt-Cl bonds are gradually lost during the thermal synthesis process, with the concomitant formation of Pt-O bonds (Fig. S46).
Fig. 4. Structural characterization of Ir-based electrocatalysts.

a In-situ Ir L3-edge XANES spectra of the thermal annealing process for formation of Pt/IrOx (stage a to g). Insert: temperature program used for the thermal annealing. b Ir-L3 edge Fourier-transform EXAFS spectra of the thermal annealing process from stage a to g. c HAADF-STEM and d TEM image of Pt/IrOx. e EDS mapping of the Pt/IrOx electrocatalyst. Source data are provided as a Source Data file.
Next, we performed electrolysis at 300, 400, 600, 800 and 1000 mA for all three catalysts in 1.0 M KCl electrolyte (Fig. 5a, b, and Table S1–S4). From the results, we observe that addition of Au has a detrimental impact on both the current and FE of IrOx. On the other hand, both Co and Pt can enhance the current and FE of IrOx. For instance, at 600 mA, Co/IrOx and Pt/IrOx display a higher FE of 82.6% and 67.5% respectively. However, at the higher applied currents (800 and 1000 mA), the FE of Co/IrOx becomes lower than that of IrOx. We posit that this is because CoOx species become unstable at higher currents and may dissolve into the electrolyte due to the induced higher local pH (calculations55 available in the Supplementary Information and Fig. S47).
Fig. 5. Electrochemical chlorocyclohexane production of Ir-based electrocatalysts.

a LSV curves in 1.0 M KCl electrolyte with cyclohexane with various electrocatalysts. (Pt could be oxidized into PtOx during the halogenation process) No IR correction was conducted. b Chlorocyclohexane FE under different applied currents for each of the Ir-based electrocatalysts. Note: quantification was based on GCMS and the geometric area of electrode used in all cases was 5 cm2. c FE of chlorocyclohexane under current of 1 A (200 mA/cm2) of different catalysts with different ratio of Ir and Pt. d Comparison of partial current density, product generation rate, yield over stability, Faradaic efficiency, and product specificity with previous literature reports. Product specificity refers to the percentage of all reacted substrate going towards the desired product. e Applied potential and chlorocyclohexane FE for 100 h at 1 A for Pt/IrOx. After each cycle (4 h), the electrolyte was removed for product quantification and refreshed (total 25 cycles). Note: for each cycle, 10 ml of fresh cyclohexane was used. No IR correction was conducted. Source data are provided as a Source Data file.
As for Pt/IrOx, this displays a higher chlorocyclohexane FE for all applied currents. For example, an average FE of 95.2% is achieved at a current of 1000 mA (Fig. 5a and b) with Pt/IrOx. In contrast, its individual components PtOx and IrOx display a lower FE of 43.5% and 89.1% respectively at 1000 mA. We also found that the mixed Pt/IrOx system exhibits a volcanic relationship as a function of composition whereby the highest FE of 95.2% was obtained using a Pt:Ir ratio of 1:3 (Fig. 5c). Since our catalysts are phase separated, we would expect a simple linear relationship if the PtOx and IrOx phases were non-interacting. We therefore reasoned that the IrOx phase is the main active site, with improved activity at the PtOx and IrOx interfaces. We also operated the system at a higher current, by performing electrolysis without a membrane to separate the cathode and anode chambers (Fig. S48). When KCl is used as the electrolyte, the gradual increase of the bulk electrolyte pH results in a significant decrease in the C-H chlorination efficiency. Hence, we employed 3 M HCl as the electrolyte to maintain a low electrolyte pH. The results show that C-H halogenation can still be achieved in this configuration at a current of 5 A (1 A/cm2), with a FE of 50% under such conditions with Pt/IrOx (Fig. 5b).
Additionally, our FTIR spectroscopy (Fig. S49–S51), 1H NMR (Fig. S52), 13C NMR (Fig. S53, and S54), GCMS (Fig. S55–S59), and HR-GCMS data (Fig. S60–S62) indicate that the product detected for these cases is chlorocyclohexane, consistent with the results obtained using IrOx. We also note there is always a non-negligible amount of missing FE. To account for this, we performed iodometric titration, which allows us to detect and quantify unreacted chlorine and hypochlorite species (Fig. S63 and Table. S5–S8). Furthermore, the system was operated over a 100 h period at a constant current of 1 A, with the electrolyte (10 ml cyclohexane) refreshed every 4 h (total 25 cycles) for product quantification (Fig. 5e). This generated a total of 1668 mmol (198 g) of chlorocyclohexane (Fig. S64), which we note is an unseparated mixture of chlorocyclohexane and unreacted cyclohexane. This has a corresponding turnover number of 74798 and the turnover frequency of 0.21 s−1 per gram of iridium (calculation details available in the supplementary information). System performance was stable throughout this period, maintaining a FE around 95% in every cycle (Fig. S65). Our process also substantially outperforms other reported anodic upgrading reactions in partial current density, product generation rate, product yield, FE and product specificity (Fig. 5d).
Next, we performed in-situ XAS to study IrOx and Pt/IrOx with 1.0 M KCl electrolyte using a custom electrochemical cell (Fig. S66). Ir L3-edge XANES spectra (Fig. 6c and S67a) shows that Ir in IrOx shifts towards a higher oxidation state upon application of a positive potential. On the other hand, the oxidation state of Ir in Pt/IrOx appears to remain unchanged even when positive potentials are applied (Figs. 6d and S67b). We postulate this is due to the presence of electronegative *Cl on IrOx which increases its oxidation state. In comparison, Pt/IrOx likely possess a weaker *Cl binding energy, which results in a lower *Cl coverage and hence no observed changes in the Ir oxidation state. This was supported by density functional theory (DFT) calculations56–61 using slab models of IrO2 as well as the PtO2/IrO2 interface (Fig. S68 and Fig. 6e). The results indicate that the *Cl binding energy of IrO2/PtO2 does indeed become weakened relative to IrO2 (Fig. 6f).
Fig. 6. Influence of the Cl binding energy for Ir-based electrocatalysts.

In-situ Raman spectroscopy result for chlorine evolution using (a) IrOx, and (b) Pt/IrOx as the catalyst using 1 M KCl as the solution. Operando Ir L3-edge XANES spectra of (c) IrOx and (d) Pt/IrOx collected at OCP, 1.6 V and 2.1 V vs. RHE in 1 M KCl electrolyte. Standard IrO2 sample is included as a reference. Insert figures in 6c and 6 d are the corresponding enlarged XANES spectra. e The IrO2/PtO2 slab model employed for DFT calculations. The red, gray, blue, and green spheres represent O, Ir, Pt, and Cl atoms respectively. Projected density of states (pDOS) are shown in Fig. S69. f Comparison of *Cl binding energy for IrO2 and IrO2/PtO2 respectively. Source data are provided as a Source Data file.
In-situ Raman spectroscopy was also carried out to reveal mechanistic insights into the differences between IrOx and Pt/IrOx. For both cases, we observed 3 peaks62,63 related to Cl adsorption on the catalyst for both IrOx and Pt/IrOx. Notably with Pt/IrOx, the positions of these 3 peaks are similar to that of IrOx, suggesting that the main active sites on Pt/IrOx are the IrOx regions. On the other hand, when the composition of PtOx is increased (Pt7Ir3Ox), these peak positions become similar to that of PtOx. This suggests that the PtOx regions on Pt7Ir3Ox are now the main active sites (Fig. S70) and is consistent with this electrode yielding a lower chlorocyclohexane FE compared to Pt/IrOx (Fig. 5c). The in-situ Raman spectroscopy heatmap (Figs. 6a, and 6b) also shows that the peak intensities change as a function of applied potential. For IrOx, the peak intensities tend to increase with an increasing anodic potential. However, for Pt/IrOx, the maximum peak intensity occurs at 1.5 V vs. RHE and thereafter decreases at increasing anodic potentials. We postulate that this decrease in intensity could be due to a lowered *Cl coverage on the catalyst surface due to an increased propensity for Cl free radical formation. This is consistent with why Pt/IrOx yields a higher chlorocyclohexane FE as compared to IrOx.
The scope of this process was also extended to other liquid alkanes such as hexane, methylcyclohexane, octane and cyclopentane. Using these different substrates, we performed electrolysis with IrOx based catalyst at a current of 800 mA or 1000 mA. FTIR, NMR, and GC-MS spectroscopy results confirm that electrochemical monochlorination can indeed be carried out with these substrates (Fig. S71–S77). From the product distribution, the selectivity of Cl addition towards alkyl hydrogens has probabilities according to tertiary (R3C-H)> secondary (R2CH-H)> primary (RCH2-H) for substrates containing different C-H bonds. Importantly, we note that all these substrates have low solubility in aqueous media. Hence, we show that our Cl free radical mediated process can overcome this limitation, enabling C-H halogenation of liquid alkanes at high applied currents.
We also carried out similar experiments for C-H bromination using 1 M KBr as the electrolyte with IrOx at a constant current of 1000 mA (200 mA/cm2) (Fig. S78a and b). Here, an electrolyte containing 5 ml cyclohexane and 45 ml 1 M KBr mixture was used, with the rotation speed set at 500 rpm. We show that the conversion of cyclohexane to bromocyclohexane is also possible, as confirmed by 1H-NMR spectroscopy (Fig. S78c) and FTIR spectroscopy (Fig. S78d). Using GCMS quantification (Fig. S79), the average Faradaic efficiency (FE) for bromocyclohexane was 21.5% for a 30 min electrolysis duration.
Solar-driven chlorocyclohexane production using seawater
Next, we evaluated the feasibility of using readily available seawater for our process. We obtained seawater from the Singapore Strait (Fig. S80) and directly used this as the electrolyte and Cl− source. We performed 1 h of electrolysis at 300 mA and confirmed the production of chlorocyclohexane (Fig. S81a). However, the required potential increases gradually with time (from 2.16 V to 2.31 V vs. RHE), indicating some form of system instability (Fig. S81b). At the same time, we observed the presence of a white precipitate on the cathode facing side of the membrane (Fig. S81c), which we posited to be due to the presence of Mg2+ and Ca2+. To resolve this, we pretreated the seawater by adding K2CO3, which precipitates these metal ions out of solution. The resulting electrolyte displays stable performance and comparable levels of productivity with 1.0 M KCl, without the appearance of precipitates on the membrane (Fig. S82).
Encouraged by this result, we sought to increase the system performance by increasing the concentration of Cl− ions in the seawater. To do this, we used a solar evaporation reverse osmosis process (see Supporting Information and Fig. S83) to concentrate the seawater from around 0.5 M to 3.3 M (6.6 times higher) (Fig. 7a). We then utilized the concentrated seawater in our electrochemical system and notably found an increase in the FE to chlorocyclohexane from 65.6% using seawater to 93.8% with concentrated seawater at a current of 1000 mA using the Pt/IrOx catalyst (Figs. 7b and S84).
Fig. 7. Solar-driven chlorocyclohexane production using seawater.

a Change in mass and Cl− concentration of the seawater during the solar evaporation reverse osmosis process. b Chlorocyclohexane FE as a function of Cl− concentration at a current of 1000 mA using Pt/IrOx as the anode in the H-type cell. c Schematic of solar-driven chlorocyclohexane production using seawater in the MEA cell. d I-V characteristics of the PV module and LSV of the MEA system with seawater and concentrated seawater. The MPP of the PV module and intersection operating points are marked out. No IR correction was conducted. e Chlorocyclohexane FE for the solar-driven system under 1-sun illumination over a 120 min operating period using either seawater or concentrated seawater. Ca2+ and Mg2+ ions were removed by addition of K2CO3. Source data are provided as a Source Data file.
Our final goal was to realize complete solar-driven production of chlorocyclohexane using seawater without any external bias. However, operation in the H-type cell requires very large full-cell voltages, leading to high energy consumption (Fig. S85). Such conditions reduce system practicality, especially if renewable energy sources are to be used. Hence, we translated the reaction to a flow system with a MEA type electrochemical cell (Fig. S86a) for lower full-cell voltage operation and continuous chlorocyclohexane production. In this configuration, the Pt/IrOx anode is directly contacted to the Nafion membrane, with a Pt cathode contacting the opposite side. The electrolyte with added cyclohexane was supplied from an external reservoir and was rapidly agitated before being introduced through a serpentine flow channel at the anode side. In this case, 2 M HCl was used instead of 1 M KCl to reduce the overall cell voltage for the reaction (Fig. S86b, c). At an applied current density of 200 mA/cm2, we found the average full-cell voltage to be 2.5 V over a 30 min period (Fig. S86d) and a chlorocyclohexane FE of 34.7% was obtained (Fig. S86e, f). Continuous operation over 120 h at a current density of 200 mA/cm2 was also achieved with no obvious decline in the performance (Fig. S87).
We then constructed a suitable photovoltaic (PV) module for powering our MEA system with seawater as the reactant. For this, we selected Si heterojunction based solar cells with an open circuit potential of 0.7 V and a short-circuit current density of 38 mA/cm2. To fabricate the PV module, we selected five individual cells each of around 6.6 cm2 in area and connected them in series to provide suitable voltage and current. The I−V curve of the Si PV module was then measured under simulated AM 1.5 G 1-sun illumination (Fig. 7c). Plotting this together with the LSV curves of our MEA system, we found intersection operating points of 197.5 mA/2.67 V using seawater and 225.4 mA/2.38 V with concentrated seawater. Notably, the operating point for concentrated seawater is a relatively good match with the maximum power point (MPP) output metrics of the solar panel at 220.7 mA/2.46 V. During 120 min of electrolysis powered by 1-sun solar illumination, the current was stable at 160.9 mA and a chlorocyclohexane FE of 21.6% was obtained (Fig. 7d and S88) using seawater as the electrolyte. With concentrated seawater, the current supplied to the electrolyzer was stable at 209.9 mA and an FE of 43.4% towards chlorocyclohexane was achieved. In addition, we also fabricated a perovskite-based PV module with an operating point of 150.3 mA/2.31 V using seawater and 162.7 mA/2.05 V with concentrated seawater (Fig. S89).
Discussion
In this work, our goal was to develop a solar-driven electrochemical process for monochlorination of alkanes at high current using seawater. We discovered that IrOx is well-suited towards the generation of Cl radicals as opposed to Cl2 from Cl− oxidation, which allows for the selective monochlorination of alkanes. We further identified Pt/IrOx as having an optimized Cl binding energy that allows for the monochlorination of cyclohexane with a Faradaic efficiency of chlorocyclohexane up to 95% and stable performance over 100 h at a current of 1 A (200 mA/cm2) with aqueous KCl and cyclohexane mixture as the electrolyte. Encouraged by these results, we then demonstrated that our system can directly utilize concentrated seawater, achieving a FE of 93.8% towards chlorocyclohexane at a current of 1 A. Notably, our concentrated seawater was derived from actual seawater using a solar evaporation reverse osmosis process. Finally, complete solar-driven production of chlorocyclohexane was achieved by using the concentrated seawater in a membrane electrode assembly flow system coupled to a Si PV module. Importantly, this simultaneously generates green hydrogen at the cathode. Our work presents a promising pathway towards the sustainable manufacturing of chlorinated chemicals with higher atom economy and reduction of HCl waste.
Methods
Materials
Potassium chloride (99.9%), hydrochloric acid (ACS reagent, 37%), potassium hydroxide (95.0%), potassium iodide (>99%), potassium carbonate (≥99%) Sodium thiosulfate (99%), cyclohexane (>99.0%), oxalic acid (≥98%), chloroplatinic acid (>99.9%), gold(III) chloride trihydrate (>99.9%), 2,2,6,6-tetramethylpiperidine oxide (TEMPO), and cobalt chloride hexahydrate were purchased from Sigma-Aldrich. Iridium(III) chloride (99.8%) was purchased from Alfa Aesar. Isopropanol (ACS reagent, 99.8%) was purchased from Fisher Chemicals. Sulfuric acid (98%) was purchased from J.T.Baker. Deionized (DI) water (18.2 MΩ) was obtained using an OmniaPure UltraPure Water System (Stakpure GmbH) and was used for all the experiments. Nafion exchange membrane (Nafion 117, size of 2.5 cm by 2.5 cm, with a thickness of 0.18 mm) and Ti screen mesh were purchased from Fuel Cell Store. The electrochemical H-type cell and Ag/AgCl (3 M KCl) reference electrodes were purchased from Tianjin Aida Hengsheng Technology Development Co., Ltd. All the chemicals used in this work were of analytical grade and used without further purification. The pretreatment and activation of Nafion 117 membrane was done by keeping the membrane for 1 h in 5 wt.% H2O2 solution under 80 °C, followed by washing with DI water. After that, the membrane was put in 0.5 M H2SO4 for another 1 h under 80 °C followed by again washing with DI water. After all process, the membrane was kept in a beaker filled with DI water. The seawater used in our experiments was acquired from East Coast Park, Singapore at location 1.2914 °N, 103.7667 °E.
Catalyst preparation
IrOx-based catalysts were synthesized on Ti mesh supports through a dip coating and thermal decomposition method, as previously described by Luc et al.54. Firstly, the Ti mesh was etched for 60 min in boiling 0.5 M oxalic acid. Subsequently, the mesh was dip coated into a 10 ml isopropanol solution with 10% volume concentrated HCl containing 30 mg of dissolved IrCl3·(H2O)x. This was followed by drying at 100 °C for 10 min and calcination at 500 °C for 10 min in air. This procedure was repeated 10 times to achieve a catalyst loading of 1 mg/cm2 and the resulting material was denoted as IrOx. The catalysts Pt/IrOx, Au/IrOx and Co/IrOx were prepared using the same procedure, with 30 mg of H2PtCl6·(H2O)6, HAuCl4·(H2O)3, or CoCl2·(H2O)6 added into the dipping solution for each respective case. Catalysts with different ratios of Ir and Pt were prepared using different precursor mixing ratios as shown below:
Electrochemical measurements
The cyclohexane halogenation experiments were conducted in a H-type cell configuration consisting of the anode catalyst, cation exchange membrane (Nafion 117), carbon rod as the cathode and Ag/AgCl as the reference electrode. 45 ml of 1 M KCl mixed with 5 ml of cyclohexane was used for the anolyte, while 50 ml 0.5 M H2SO4 was used for the catholyte. Electrochemical measurements in this work were carried out using an Autolab PGSTAT204. The reported current densities are based on the geometric surface area of 5 cm2 for the anode. Data in this work are all without IR compensation. After 30 min of testing (unless otherwise stated, all experiments were run for a duration of 30 min), the organic phase was separated from the aqueous electrolyte using a separating funnel and collected for product analysis and quantification. For tests conducted without using a membrane to separate the cathode and anode chambers, 50 ml 3 M HCl mixed with 5 ml of cyclohexane was used as the electrolyte. Bromination of cyclohexane was carried out using identical procedures, except that 1.0 M KBr electrolyte was used instead. The flow experiments were conducted with a membrane assembly electrode (MEA) reactor, which was purchased from Gaoss Union (Tianjin China). In this system, Pt/IrOx was used as the anode and Pt as the cathode. 2 M HCl mixed with cyclohexane was used as the anolyte and 0.5 M H2SO4 was used as the catholyte. All measurements were carried out at room temperature and pressure.
The potentials were rescaled to the potentials versus the reversible hydrogen electrode (RHE) according to Eq. (1):
| 1 |
Linear sweep voltammetry experiments
These tests were carried out in a three-electrode configuration cell using a carbon rod as the counter-electrode and Ag/AgCl (saturated KCl) as the reference electrode. The working electrodes were prepared by drop-casting catalyst ink onto a glassy carbon electrode (5 mm in diameter). The rotating disk electrode (RDE) system was from Pine Instrument Company, USA to control the electrode rotation speed. The scan rate was set at 10 mV/s from 0.5 to 1.8 V (versus Ag/AgCl) in 1.0 M KCl or KCl mixed with cyclohexane solutions using a potentiostat system (PARSTAT MC, Princeton Applied Research, US).
Product analysis and quantification
A combination of Fourier-transform infrared spectroscopy (FTIR), nuclear magnetic resonance (NMR) spectroscopy and gas chromatography mass spectrometry (GCMS) was used to analyse and quantify the products. FTIR was carried out on a Bruker Vertex 70 system equipped with a MIR-FIR ATR. The background (8 cm−1 resolution, 32 scans) was collected under ambient environmental conditions. After the baseline subtraction, the sample signal was collected after 32 scans. NMR spectroscopy was performed using a JEOL JNM-ECA500II 5 FT NMR. For 1H-NMR spectroscopy, 400 µL of the sample was mixed with 150 µL of D-Chloroform and 100 µL pure chloroform. For 13C-NMR spectroscopy, 500 µL of the sample mixed with 150 µL of D-Chloroform.
Quantification of the products was carried out using GCMS on an Agilent 5977 C GC/MSD equipped with a J&W HP-5ms capillary column (Part Number:19091S-433). Vials containing the liquid samples were placed into an autosampler holder, and 0.2 μL of sample was injected into the column with a split mode of 1:500. The GC used a temperature program during which the temperature was held at 50 °C for 5 min. Following this, the temperature was increased to 250 °C with a ramp rate of 15 °C/min. The temperature was then maintained at 250 °C for a further 7 min. All reported Faradaic efficiencies (FE) in this work were averaged from at least three different independent electrolysis experiments. The FE towards chlorocyclohexane was calculated according to the following Eq. (2):
| 2 |
Where F is the Faraday constant, na (a value of 2 for this work) is the number of the electron transfers required for 1 mol chlorocyclohexane production. Vorganic stands for the volume of the organic solution, cproduct is the concentration of the product in the organic solution, t is electrolysis duration (30 min in this work) and ioverall is the applied current.
HR-GCMS was carried out using the Agilent 7200 Q-TOF GCMS, which has a high-resolution MS coupled to an Agilent 7890B gas chromatography with NIST library.
Turnover number (TON) and turnover frequency (TOF) calculation
The turnover number (TON) for the 100 h experiment was calculated based on the total amount of chlorocyclohexane molecules generated divided by the total number of iridium atoms using the following Eq. (3):
| 3 |
Where nchlorocyclohexane is the total amount of chlorocyclohexane molecules generated and nIr is the number of Ir atoms present.
The turnover frequency (TOF) is calculated by dividing the TON by the total reaction time of the experiment (100 h in this case) based on the following Eq. (4).
| 4 |
Where t is the reaction time.
Materials characterization
The morphologies of the electrodes were investigated using scanning electron microscopy (SEM) with a JSM-7610FPlus (JEOL) at a 5 kV beam voltage. Transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDS) mapping was performed on a Hitachi HF-3300 equipped with a Bruker energy dispersive x-ray spectroscopy detector at an acceleration voltage of 300 kV. The X-ray photoelectron spectroscopy (XPS) measurements were conducted using a Kratos AXIS Supra+ with a monochromated Al Kα X-ray source. X-Ray powder diffraction (XRD) measurements were performed on a Bruker D8 Discover 3 diffractometer with Cu Kα radiation under 40 kV and 40 mA. Pt/IrOx based catalyst was characterized and identified with high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) using a FEI Themis Z scanning/transmission electron microscope operated at 300 kV, equipped with a probe spherical aberration corrector.
In-situ X-ray absorption spectroscopy
The in-situ Pt and Ir L3-edge X-ray absorption spectroscopy measurements were performed at the XAFCA beamline of Singapore Synchrotron Light Source (SSLS) under both fluorescence and transmission mode. The ex-situ Pt and Ir L3-edge X-ray absorption spectroscopy measurements were measured under transmission mode. The custom-made electrochemical cell used for in-situ XAS experiments is shown in Fig. S66, using IrOx/Ti mesh or Pt/IrOx/Ti mesh as the working electrode, Ag/AgCl electrode (KCl saturated) as the reference electrode, and carbon rod as the counter electrode. Energy calibrations were carried out using standard IrO2 powder and Pt foil.
The formation process of the Pt/IrOx-based catalyst was observed under transmission mode with a programmed temperature control system performed. 10 mg IrCl3·(H2O)x and 10 mg H2PtCl6·(H2O)6) was droped coated in a carbon paper for the testing. The normalization range was set to be 15-320 eV above the Ir L3 absorption edge, which was below the Pt L3-edge adsorption in this case. Furthermore, the normalization order is 3. The k-range for the Fourier transform of EXAFS for Fig. 5b was set from 2 to 9.2 Å. The K-space XAS spectra is given in Fig. S45.
In-situ Raman spectroscopy
In-situ Raman measurements were carried out using a LabRAM HR 800 Raman spectrometer with a custom electrochemical cell. Spectra were collected using an objective lens (×50) and a 532 nm laser (10% intensity). Pt wire and Ag/AgCl (KCl saturated) were used as the counter and reference electrodes respectively. PtOx, IrOx, and Pt/IrOx were synthesized onto a Au substrate through the same thermal decomposition method. Firstly, the Au substrate was electrochemically roughened in 0.1 M KCl. Subsequently, 40 µL of precursors with different ratio of Pt and Ir were added onto the Au substrate with catalyst loading ~0.25 mg/cm2. This was followed by drying at 100 °C for 10 min and calcination at 500 °C for 10 min in air. The resulting electrode was then allowed to cool to room temperature and directly used as the working electrode. For these experiments, 1.0 M KCl was used as the electrolyte. In-situ Raman spectra was then collected at various constant potentials (from 1.2 V to 2.1 V vs. RHE) for 15 consecutive scans and 10 s exposure time per scan.
Differential electrochemical mass spectroscopy (DEMS)
Oxygen and chlorine signals were collected using a Hiden HPR-40 DEMS Mass Spectrometer with a tube connected to the H-cell type cell (see schematic in Fig. S13). The oxygen signal was measured using a m/z value of 32, while the chlorine signal was set to a m/z value of 69.9. The reaction is started once the mass spectrometer baseline signals of oxygen and chorine have stabilized.
Electron paramagnetic resonance (EPR) testing
The EPR testing was conducted on a JEOL FA200 ESR. TEMPO was mixed with the KCl electrolyte and cyclohexane for EPR testing. A current of 400 mA was applied using IrOx and carbon rod as the anode catalyst for various reaction durations. Samples were collected and analyzed quickly using EPR (within 30 s). DMPO was also mixed with the KCl electrolyte for the EPR tests. A current density of 200 mA/cm2 was applied using IrOx as the electrode with a reaction duration of 4 min. Similarly, samples were then collected and analyzed quickly using EPR (within 30 s).
Iodometric titration
Iodometric titration of the anolyte was conducted by first adding an excess of 10% KI solution to react with the unreacted chlorine/hypochlorite species to form iodine. This was followed by addition of starch solution to form a dark blue starch-iodine complex. Next, this was then titrated with 1 M NaS2O3 solution until the solution turned clear again (pictures are shown in Fig. S63), and the amount of NaS2O3 was recorded and used to determine the FE of unreacted chlorine/hypochlorite species (results are shown in Table S1).
Boundary layer thickness calculation
Quantification of the hydrodynamic boundary layer thickness of the electrochemical cell with different stirring rates were conducted in the same H-type cell for the halogenation reaction using the methodology described by Bell and co-workers44. Specifically, we added 10 mM of K3Fe(CN)6 (Sigma-Aldrich 99%) to the 1 M KCl electrolyte and determined the limiting current density as a function of stirring rate. Based on the limiting current density, the boundary layer thickness can be calculated based on Fick’s law Eq. (5):
| 5 |
Where δBL is the boundary layer thickness, F is the Faraday constant, is the diffusion coefficient of Fe(CN)63−, is the concentration of Fe(CN)63− and JSS is the experimentally determined limiting current density.
Density function theory (DFT) calculations
Spin-polarized density functional theory (DFT) calculations were performed with periodic slab models by means of using Vienma ab initio simulation package (VASP)56,57. The electron exchange and correlation interaction were described within the generalized gradient approximation in the Perdew−Burke−Ernzerhof functional (GGA-PBE)58, while the electron-ion interaction was calculated using projector-augmented wave (PAW) method with a plane-wave basis set defined by a kinetic energy cutoff of 450 eV59. The long-range dispersion interactions between adsorbates and surface were treated applying DFT-D3 method developed by grimme et al.60. The k-point sampling of a (3 × 3 × 1) mesh within the Monkhorst−Pack scheme is utilized for optimization60. The geometry optimization and energy calculation are finished when the electronic self-consistent iteration and force reach 10–5 eV and 0.02 eV Å−1, respectively. The binding energy (ΔE) was calculated as follows Eq. (5): ΔE*A = E*A-E*-EA. Where the *A, *, and A represent adsorbed A species, catalysts, and isolated A species. For *Cl binding energy, we calculated using equation (6): ΔE*Cl = E*Cl - E* - . The atomic coordinates of the optimized models are provided in Supplementary Data 1.
Solar evaporation reverse osmosis process
A lab-scale setup solar evaporation was built to concentrate the seawater using a 3D-printed hierarchical chimney. The 3D evaporator stalk consisted of graphene oxide and cellulose acetate floating on the seawater feed by positioning it in a polyethylene foam. The top of the stalk was subjected to illumination with constant intensity of 1 kW/m2 using a solar simulator (CEL-PF300, Aulight) (Fig. S83). During the process, pure water was continuously removed via interfacial steam generation. The seawater mass change was continuously recorded using a mass balance (ME204E, Mettler Toledo) under one sun illumination. The process was stopped after 83.3% of water was removed, with a corresponding increase in the Cl– concentration of the seawater to 3.6 M (6.6 times).
Construction of the solar-driven system
The Si PV module was based on five Si heterojunction cells (HIT, LONGi Green Energy Technology Co., Ltd.) connected in series. Each cell was ~6.6 cm2 and were connected in series through soldering and fixed onto a glass slab.
Perovskite solar modules with three subcells connected in series were fabricated onto FTO glass with a size of 5.5 × 5.5 cm2. The series interconnection of the module was realized by P1, P2 and P3 lines, which were patterned using a laser scribing system with a wavelength of 1,064 nm (for P1) and 532 nm (for P2 and P3) with a power of 20 W. The FTO substrate was pre-patterned for P1 (20 μm wide) at 60% laser power and a speed of 200 mm/s with a frequency of 30 kHz and pulse width of 30 ns. Then the FTO glass substrates were wiped with soap water, then ultra-sonically washed with deionized water, acetone and isopropanol for 30 min each. After ultraviolet ozone treatment for 15 min, the substrates were then spin coated with ATOx (2.5 wt% in butanol) nanoparticles, followed by heating at 100 °C for 10 min in the open air. Then the Me-4PACz (1 mg/ml in IPA) was spin-coated on the substrates at 4000 rpm for 30 s and heated at 100 °C for 10 min in the nitrogen-filled glovebox. The 1.8 M perovskite (Cs0.1FA0.9PbI3) stock solution contains 52 mg CsI, 309.6 mg FAI, 922 mg PbI2 and 55.6 mg PbCl2 that were dissolved in 1.11 ml mixed solvent of DMF: NMP (V:V = 960:150). The 1.8 M perovskite precursor solution was shaken overnight to make sure all components were fully dissolved and then used to prepare the perovskite films. For the coating process, the substrate was spun at 5,000 rpm for 40 s with an acceleration of 5000 rpm/s and N2 gas was blown on top of the spinning substrates after 20 s. The perovskite films were pre-annealed at 70 °C for 2 min in the N2 glovebox to eliminate any residual solvent and then annealed at 150 °C for 10 min in 30% relative humidity to promote the crystallinity of perovskite with enhanced film quality and grain size. Then, C60 (20 nm)/BCP (8 nm) were deposited via evaporation. The P2 lines (40 μm wide) were patterned with an average laser power of 30% at a speed of 200 mm/s and frequency of 30 kHz. After deposition of 160 nm Ag, the the P3 lines (20 μm wide) were patterned with an average laser power of 31% at a speed of 200 mm/s and frequency of 30 kHz. The devices were masked with metal aperture masks (20 cm2) during the J–V measurement. The distance between the P1 and P3 lines is about 150 μm and the geometric fill factor is around 99%.
As for construction of the solar-driven system, the PV module was coupled directly to the MEA system. A potentiostat (Autolab PGSTAT204) was also connected to the system to monitor the current. The simulated solar illumination was provided using a 300 W Xenon arc lamp (Microsolar 300 UV, Beijing Perfectlight Technology Co. Ltd.) equipped with an air mass 1.5 global (AM 1.5 G) filter, and the power intensity of the incident light was calibrated to 100 mW/cm2 using a Si photodiode (FDS100, Thorlabs).
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
Y.L. acknowledges support and funding from the NUS Presidential Young Professorship (A-0009485-02-00). We acknowledge use of the XAFCA beamline of the Singapore Synchrotron Light Source (SSLS) for collection of the XAS data used in this work. We thank Gan Wang and Jie Wu for their assistance with preparing the distillation setup.
Author contributions
Y.L. supervised the project. Y.L. and B.Wu conceived the idea and designed the experiments. B.Wu carried out all the experimental work. R.L. and Z.W. performed and supervised the computational work respectively. B.Wu, C.W. and S.X. carried out the XAS experiments. B.Wu, Z.M., S.B.D., W.W.T., M.Z., B.Wang, and Z.A. performed the catalyst characterization and analysis. T. Y. and B. Z. carried out the in-situ Raman spectroscopy experiments. B.L. fabricated the Si photovoltaic module. X.W. and Y.H. fabricated the perovskite photovoltaic module. C.F. and S.Z. performed the solar evaporation reverse osmosis experiments. W.R.L contributed to data analysis and manuscript editing. Y.L and B.Wu. co-wrote the manuscript. All authors discussed the results and assisted during the manuscript preparation.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request. Source data are provided with this paper.
Competing interests
Yanwei Lum and Bo Wu are co-inventors on a patent application titled “Method for selective electrochemical halogenation of liquid alkanes at high current density under ambient conditions”. The application number is 10202303072 T and is now filed with the Registry of Patents, Singapore. The patent application covers the method for performing the electrochemical chlorination of liquid alkanes using Ir-based electrocatalysts. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Bo Wu, Ruihu Lu, Chao Wu.
Contributor Information
Wan Ru Leow, Email: wanru_leow@isce2.a-star.edu.sg.
Ziyun Wang, Email: ziyun.wang@auckland.ac.nz.
Yanwei Lum, Email: lumyw@nus.edu.sg.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-55283-x.
References
- 1.Lu, Z. et al. Regioselective aliphatic C–H functionalization using frustrated radical pairs. Nature619, 514–520 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang, S. et al. Cobalt-catalysed allylic fluoroalkylation of terpenes. Nat. Synth.2, 1202–1210 (2023). [Google Scholar]
- 3.Zhang, H. et al. Activation of light alkanes at room temperature and ambient pressure. Nat. Catal.6, 666–675 (2023). [Google Scholar]
- 4.Cao, X. et al. A photochromic composite with enhanced carrier separation for the photocatalytic activation of benzylic C–H bonds in toluene. Nat. Catal.1, 704–710 (2018). [Google Scholar]
- 5.Ibrahim, M. Y. S. et al. Electrochemical nickel -natalyzed C(sp3)-C(sp3) cross-coupling of alkyl halides with alkyl tosylates. J. Am. Chem. Soc.145, 17023–17028 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao, C., Rakesh, K. P., Ravidar, L., Fang, W. Y. & Qin, H. L. Pharmaceutical and medicinal significance of sulfur (S-VI)-Containing motifs for drug discovery: A critical review. Eur. J. Med. Chem.162, 679–734 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin, R., Amrute, A. P. & Perez-Ramirez, J. Halogen-mediated conversion of hydrocarbons to commodities. Chem. Rev.117, 4182–4247 (2017). [DOI] [PubMed] [Google Scholar]
- 8.U.S. Environmental Protection Agency (EPA), Hydrochloric acid supply chain – executive summary. EPA817-F-22-029 (2022).
- 9.Chen, C. P., Chen, C. C. & Han, T. H. Autoignition temperature data for isopropyl chloride, butyl chloride, isobutyl chloride, pentyl chloride, pentyl bromide, chlorocyclohexane, and benzoyl chloride. Ind. Eng. Chem. Res.52, 7986–7992 (2013). [Google Scholar]
- 10.Wu, J. R. & Yang, Y. W. Geminiarene: molecular scale dual selectivity for chlorobenzene and chlorocyclohexane fractionation. J. Am. Chem. Soc.141, 12280–12287 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Ingold, K. U., Lusztyk, J. & Raner, K. D. The unusual and the unexpected in an old reaction - the photochlorination of alkanes with molecular chlorine in solution. Acc. Chem. Res.23, 219–225 (1990). [Google Scholar]
- 12.Sun, N. J. & Klabunde, K. J. Nanocrystal metal oxide-chlorine adducts: selective catalysts for chlorination of alkanes. J. Am. Chem. Soc.121, 5587–5588 (1999). [Google Scholar]
- 13.Yuan, H. X., Xia, Q. H., Zhan, H. J., Lu, X. H. & Su, K. X. Catalytic oxidation of cyclohexane to cyclohexanone and cyclohexanol by oxygen in a solvent-free system over metal-containing ZSM-5 catalysts. Appl. Catal. a-Gen.304, 178–184 (2006). [Google Scholar]
- 14.Thomas, J. M., Raja, R., Sankar, G. & Bell, R. G. Molecular-sieve catalysts for the selective oxidation of linear alkanes by molecular oxygen. Nature398, 227–230 (1999). [Google Scholar]
- 15.Zhang, W., Cheng, G. H., Haller, G. L., Liu, Y. & Lercher, J. A. Rate enhancement of acid-catalyzed alcohol dehydration by supramolecular organic capsules. ACS Catal.10, 13371–13376 (2020). [Google Scholar]
- 16.Wu, W. F. et al. Light-triggered oxy-chlorination of cyclohexane by metal chlorides. Appl. Catal. a-Gen.469, 483–489 (2014). [Google Scholar]
- 17.Kozima, K. & Sakashita, K. The two stable isomers of chlorocyclohexane. Bull. Chem. Soc. Jpn.31, 796–801 (1958). [Google Scholar]
- 18.Shen, C., Dagnaw, W. M., Fong, C. W., Lau, K. C. & Chow, C. F. Selective functionalization of C(sp3)-H bonds: catalytic chlorination and bromination by Iron(III)-acacen-halide under ambient condition. Chem. Commun.58, 10627–10630 (2022). [DOI] [PubMed] [Google Scholar]
- 19.Lv, X. L. et al. A base-resistant metalloporphyrin metal-organic framework for C-H bond halogenation. J. Am. Chem. Soc.139, 211–217 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Zhao, M. D. & Lu, W. J. Visible light-induced oxidative chlorination of alkyl C(sp3)-H bonds with nacl/oxone at room temperature. Org. Lett.19, 4560–4563 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Li, Z. H. et al. Photoelectrocatalytic C-H halogenation over an oxygen vacancy-rich TiO2 photoanode. Nat. Commun.12, 6698 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee, D. et al. The impact of surface composition on the interfacial energetics and photoelectrochemical properties of BiVO4. Nat. Energy6, 287–294 (2021). [Google Scholar]
- 23.Andrei, V. et al. Long-term solar water and CO2 splitting with photoelectrochemical BiOI-BiVO4 tandems. Nat. Mater.21, 864–868 (2022). [DOI] [PubMed] [Google Scholar]
- 24.Kim, T. W. & Choi, K. S. Nanoporous BiVO4 photoanodes with dual-layer oxygen evolution catalysts for solar water splitting. Science343, 990–994 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Hintz, H. et al. Copper-catalyzed electrochemical C–H fluorination. Chem. Catal.3, 100491 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stangier, M., Scheremetjew, A. & Ackermann, L. Chemo‐and site‐selective electro‐oxidative alkane fluorination by C(sp3)− H cleavage. Chem.–A Eur. J.28, e202201654 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan, Y. et al. Electrochemical oxidative clean halogenation using HX/NaX with hydrogen evolution. Iscience12, 293–303 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou, Z. et al. Synergy of anodic oxidation and cathodic reduction leads to electrochemical C-H halogenation. Chin. J. Chem.37, 611–615 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Wang, Q. H. et al. Electrocatalytic methane oxidation greatly promoted by chlorine intermediates. Angew. Chem.-Int. Ed.60, 17398–17403 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Han, S. Y. et al. Preferential adsorption of ethylene oxide on Fe and chlorine on Ni enabled scalable electrosynthesis of ethylene chlorohydrin. Angew. Chem.-Int. Ed.62, e202216581 (2023). [DOI] [PubMed] [Google Scholar]
- 31.Marin, D. H. et al. Hydrogen production with seawater-resilient bipolar membrane electrolyzers. Joule7, 765–781 (2023). [Google Scholar]
- 32.Belyaeva, L. A., Fu, W., Arjmandi-Tash, H. & Schneider, G. G. F. Molecular caging of graphene with cyclohexane: transfer and electrical transport. ACS Cent. Sci.2, 904–909 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leow, W. R. et al. Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density. Science368, 1228–1233 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Kumar, A., Phillips, K. R., Thiel, G. P., Schroder, U. & Lienhard, J. H. Direct electrosynthesis of sodium hydroxide and hydrochloric acid from brine streams. Nat. Catal.2, 106–113 (2019). [Google Scholar]
- 35.Tong, W. M. et al. Electrolysis of low-grade and saline surface water. Nat. Energy5, 367–377 (2020). [Google Scholar]
- 36.Yang, J. R. et al. CO2-mediated organocatalytic chlorine evolution under industrial conditions. Nature617, 519–523 (2023). [DOI] [PubMed] [Google Scholar]
- 37.Wang, Y., Xue, Y. & Zhang, C. Rational surface and interfacial engineering of IrO2/TiO2 nanosheet arrays toward high‐performance chlorine evolution electrocatalysis and practical environmental remediation. Small17, 2006587 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Kim, S., Choi, S. K., Yoon, B. Y., Lim, S. K. & Park, H. Effects of electrolyte on the electrocatalytic activities of RuO2/Ti and Sb-SnO2/Ti anodes for water treatment. Appl. Catal. B-Environ.97, 135–141 (2010). [Google Scholar]
- 39.Sauer, G. S. & Lin, S. An electrocatalytic approach to the radical difunctionalization of alkenes. ACS Catal.8, 5175–5187 (2018). [Google Scholar]
- 40.Shao, D. et al. Decoupled photoelectrochemical cerium-catalyzed oxydichlorination of alkynes: slow releasing of chloride ions and chlorine radicals. ACS Sustain. Chem. Eng.10, 10294–10302 (2022). [Google Scholar]
- 41.Wang, H. M. et al. Dearomative ring expansion of thiophenes by bicyclobutane insertion. Science381, 75–81 (2023). [DOI] [PubMed] [Google Scholar]
- 42.Lian, P., Long, W., Li, J., Zheng, Y. & Wan, X. Visible‐light‐induced vicinal dichlorination of alkenes through LMCT excitation of CuCl2. Angew. Chem. Int. Ed.132, 23809–23814 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Bard, A. J., Faulkner, L. R. & White, H. S. Electrochemical methods: fundamentals and applications. (2022).
- 44.Clark, E. L. et al. Standards and protocols for data acquisition and reporting for studies of the electrochemical reduction of carbon dioxide. ACS Catal.8, 6560–6570 (2018). [Google Scholar]
- 45.Alegre, M. L. et al. Kinetic study of the reactions of chlorine atoms and Cl2•-radical anions in aqueous solutions. 1. Reaction with benzene. J. Phys. Chem. A104, 3117–3125 (2000). [Google Scholar]
- 46.Wang, W.-L., Wu, Q.-Y., Huang, N., Wang, T. & Hu, H.-Y. Synergistic effect between UV and chlorine (UV/chlorine) on the degradation of carbamazepine: influence factors and radical species. Water Res.98, 190–198 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Li, D. et al. Photocatalytic chlorination of methane using alkali chloride solution. ACS Catal.12, 7004–7013 (2022). [Google Scholar]
- 48.Yan, Y. F. et al. Electrocatalytic upcycling of biomass and plastic wastes to biodegradable polymer monomers and hydrogen fuel at high current densities. J. Am. Chem. Soc.145, 6144–6155 (2023). [DOI] [PubMed] [Google Scholar]
- 49.Sheng, H. Y. et al. Linear paired electrochemical valorization of glycerol enabled by the electro-fenton process using a stable NiSe2 cathode. Nat. Catal.5, 716–725 (2022). [Google Scholar]
- 50.Kar, S., Zhou, Q. Q., Ben-David, Y. & Milstein, D. Catalytic furfural/5-hydroxymethyl furfural oxidation to furoic acid/furan-2,5-dicarboxylic acid with H2 production using alkaline water as the formal oxidant. J. Am. Chem. Soc.144, 1288–1295 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeng, L. Y. et al. Cooperative Rh-O-5/Ni(Fe) site for efficient biomass upgrading coupled with H2 production. J. Am. Chem. Soc.145, 17577–17587 (2023). [DOI] [PubMed] [Google Scholar]
- 52.Li, S. L. et al. Coordination environment tuning of nickel sites by oxyanions to optimize methanol electro-oxidation activity. Nat. Commun.13, 2916 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu, J. X. et al. Ultrahigh stable methanol oxidation enabled by a high hydroxyl concentration on Pt clusters/mxene interfaces. J. Am. Chem. Soc.144, 15529–15538 (2022). [DOI] [PubMed] [Google Scholar]
- 54.Luc, W., Rosen, J. & Jiao, F. An Ir-based anode for a practical CO2 electrolyzer. Catal. Today288, 79–84 (2017). [Google Scholar]
- 55.Gupta, N., Gattrell, M. & MacDougall, B. Calculation for the cathode surface concentrations in the electrochemical reduction of CO2 in KHCO3 solutions. J. Appl. Electrochem.36, 161–172 (2006). [Google Scholar]
- 56.Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B54, 11169 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B49, 14251 (1994). [DOI] [PubMed] [Google Scholar]
- 58.Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett.77, 3865 (1996). [DOI] [PubMed] [Google Scholar]
- 59.Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B50, 17953 (1994). [DOI] [PubMed] [Google Scholar]
- 60.Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys.132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Monkhorst, H. J. & Pack, J. D. Special points for brillouin-zone integrations. Phys. Rev. B13, 5188 (1976). [Google Scholar]
- 62.Petruk, A. A. et al. Interaction between proteins and Ir based CO releasing molecules: Mechanism of adduct formation and CO release. Inorg. Chem.53, 10456–10462 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Yusenko, K. V. et al. Insight into the thermal decomposition of ammonium hexahalogenoiridates (IV) and hexachloroiridate (III). Phys. Chem. Chem. Phys.22, 22923–22934 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request. Source data are provided with this paper.